
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeighborhood watch: tools for defining locale-dependent subproteomes and their contextual signaling activities
Marcus J. C.
Long
 a,
Yi
Zhao
b and
Yimon
Aye
*b
a,
Yi
Zhao
b and
Yimon
Aye
*b
aBeverley, East Riding of Yorkshire HU17 8QH, UK
bSwiss Federal Institute of Technology Lausanne (EPFL), Institute of Chemical Sciences and Engineering, 1015, Lausanne, Switzerland. E-mail: yimon.aye@epfl.ch
First published on 27th May 2020
Abstract
Transient associations between numerous organelles—e.g., the endoplasmic reticulum and the mitochondria—forge highly-coordinated, particular environments essential for cross-compartment information flow. Our perspective summarizes chemical–biology tools that have enabled identifying proteins present within these itinerant communities against the bulk proteome, even when a particular protein's presence is fleeting/substoichiometric. However, proteins resident at these ephemeral junctions also experience transitory changes to their interactomes, small-molecule signalomes, and, importantly, functions. Thus, a thorough census of sub-organellar communities necessitates functionally probing context-dependent signaling properties of individual protein-players. Our perspective accordingly further discusses how repurposing of existing tools could allow us to glean a functional understanding of protein-specific signaling activities altered as a result of organelles pulling together. Collectively, our perspective strives to usher new chemical–biology techniques that could, in turn, open doors to modulate functions of specific subproteomes/organellar junctions underlying the nuanced regulatory subsystem broadly termed as contactology.
 Marcus J. C. Long | Marcus J. C. Long was born in Yorkshire, UK. He was a chemistry scholar at Oxford University, and performed his PhD thesis research at Brandeis University, USA. He subsequently performed postdoctoral research in the Aye laboratory. |
 Yi Zhao | Yi Zhao received his BSc In Chemistry from Sichuan University in 2009. In the same year, he was enrolled in the National University of Singapore (NUS) under the supervision of Prof. Yeung Ying-Yeung to study novel methods in C–H activation and catalytic enantioselective halocyclization. To pursue his interests in chemical biology, he decided to leave his comfort zone and joined Prof. Yimon Aye's group as a postdoc in late 2014, Cornell University. He then moved to EPFL, Switzerland, with Prof. Yimon Aye in late 2018. He has helped to developed G-REX method to profile kinetically-privileged sensor (KPS) proteins. |
 Yimon Aye | Born and raised in Burma, Yimon Aye read chemistry at the University of Oxford (UK) (2000–2004), and achieved her PhD degree in organic chemistry with Prof. David Evans at Harvard University (USA) (2004–2009). She then switched research discipline and pursued her postdoctoral training in life sciences with Prof. JoAnne Stubbe. As a Damon Runyon cancer research fellow at MIT (USA) (2009–2012), she established the mechanisms-of-action of therapeutics targeting the enzyme ribonucleotide reductase. In her independent career that began mid-2012, Yimon Aye set out to understand the detailed mechanisms of electrophile signaling. This impetus culminated in the development of “REX” technologies (T-REX™ delivery and G-REX™ profiling). In a parallel research program, she studies pathways involved in genome maintenance and nucleotide signaling, including the mechanisms of anticancer drugs in clinical use. In Autumn 2018, she and her team members established the Laboratory of Electrophiles and Genome Operation (LEAGO) at EPFL https://leago.epfl.ch/. |
The eukaryotic cell is a surprisingly diverse environment, consisting of myriad different locales, or “neighborhoods”. In particular, there are manifold membrane-bound structures defining clear precincts within the cell, such as specialized districts within membranes where specific lipids congregate.1 There are also interface regions where different membrane-bound structures form transient contacts.2–4 The conservation of a core number of organelles across all eukaryotes and a similarly-shared set of genes associated with these organelles, imply that all eukaryotes are derived from a single common ancestor. This ancestral cell appears to have been a fully functional eukaryotic entity. Such conserved importance of organelles holds great promise for engineering generalizable chemical–biology methods with which to map organellar/sub-organellar proteins, and critically, to unearth the biological function of residents in those locales. However, in reality, tools capable of canvassing individual proteins active in specific neighborhoods, particularly in intact cells and animals, remain limited. Notably, beyond subproteomics target-ID methods, tools to focus-in on proteins that have locale-dependent signaling functions are underdeveloped. This is despite the fact that proteins with such context-dependent functions are likely critical nexuses/coordinators of signaling cascades across organelles/organellar contacts,5 not unakin to town criers.
In this perspective, we put in a nutshell emerging chemical–biology tools that can identify neighborhood subpopulations and also pinpoint locale-specific functions. We first provide case examples, primarily limited to the past 5 years, but also drawing on historical landmark discoveries where relevant, of proteins undertaking locale-dependent switches in functions/properties. We build on these examples to examine the latest tools that have been applied to perform a census of proteins resident in specific neighborhoods. We next discuss tools that interrogate the functional role each protein plays in specific neighborhoods, even if the protein were to spend a relatively small percentage of its time there. As we limit our perspective to spatiotemporally-controllable approaches that function directly in living models, chemoproteomics target-ID methods that map locale-specific protein targets through organelle isolation/fractionation are omitted.
Neighborhood-dependent switches in protein function
Given that membrane-bound structures are cornerstones within the infrastructure of eukaryotes, it is unsurprising that many proteins were at one point believed to localize exclusively to specific organelles. The stereotypical functions of these proteins were, in turn, associated with a specific function performed by the organelle. Mitochondria, for instance, are the seat of oxidative phosphorylation and the citric acid cycle in eukaryotes.6,7 Enzymes associated with these processes were very early in the history of life sciences shown to reside in the mitochondria. It was not originally questioned what could happen if these proteins were to leave their traditional haunts. However, it is now appreciated that several proteins from the mitochondria are functional in other regions of the cell. One well-known example is fumarase,8 a protein that plays an important role in DNA damage response in the nucleus. Below we delve further into select recent examples whereby neighborhood swaps, specifically between the cytosol and nucleus, result in alteration of proteins’ functions, including chemical and biological cues that regulate such switches.Regulation of RNAs: Dicer and FUS
It is now known that most proteins start off life in the cytosol and then are directed to specific organelles using complex sorting processes.9,10 This aspect alone offers considerable scope for migration across suborganellar neighborhoods. In many instances, itinerant proteins perform the same function in each different locale in which they are found: the proteasome is present in the nucleus and cytosol, and is responsible for targeted degradation in each region; several isoforms of glutathione-S-transferase also localize to different subcellular compartments where they are principally involved in glutathione conjugation to xenobiotics;11 ubiquitin also lives up to its name quite aptly, performing a variety of signaling processes all involving conjugation to specific proteins. Alternatively, changes in locale can bring about related functions, where, for instance, the protein's substrate is changed. This can often happen when a substrate akin to the canonical substrate exists in the locale to which the protein translocates, but this substrate variant is absent in the protein's original or canonical locale. For instance, Dicer—the protein responsible for formation of microRNA duplexes from pre-microRNA in the cytoplasm of higher eukaryotes—can translocate to the nucleus upon DNA damage (Fig. 1A). In the nucleus, Dicer processes double-stranded RNA formed as a result of DNA damage.12,13 Thus, Dicer serves a DNA-damage protection role beyond microRNA processing by switching neighborhoods, and in turn hosting different, yet related, substrates (guests).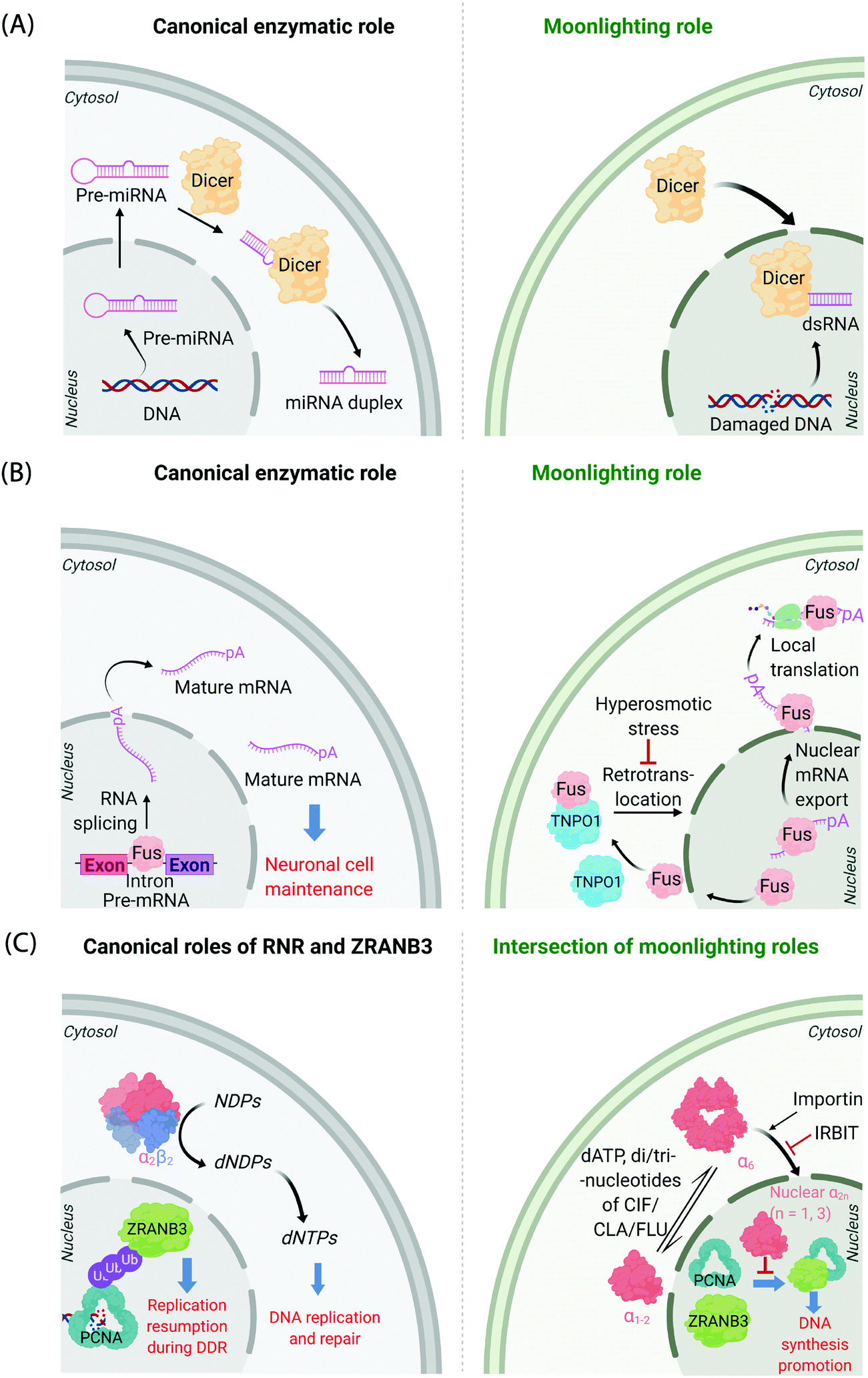 | ||
| Fig. 1 Canonical and moonlighting functions of Dicer, Fus, and RNR-α/ZRANB3. (A) Dicer's canonical function is to trim double-stranded pre-microRNA, to form mature microRNA in the cytosol (left panel). Dicer's moonlighting role occurs in the nucleus (right panel), where Dicer processes double-stranded RNA, formed from damaged DNA, as part of the DNA Damage Response (DDR).12,13 (B) Fus splices pre-mRNA in the nucleus to facilitate the maturation of mRNA (left panel). During moonlighting (right panel), Fus exports nuclear mRNA into the cytosol, and itself exits the nucleus passively. mRNA so exported may undergo locale-specific translation. Retrotranslocation/nuclear import of Fus by Transportin1 (TNPO1) clears cytosol-accumulated Fus. This nuclear translocation pathway is inactivated by hyperosmolarity14 in a cell-type dependent manner consistent with pathology of FTD. (C) Canonically, minimally-reductase-active quaternary state of RNR-enzyme necessitates RNR-α forming a heterotetramer with RNR-β (α2β2). The resulting complex catalyzes the reduction of NDPs (N = A, U, G, and C) to dNDPs. This pathway is the sole means to generate dNTPs de novo in mammalian cells, although there are salvage pathways. ZRANB3's canonical role is in DDR, whereby polyubiquitylated-PCNA recruits ZRANB3 to damage sites, allowing stressed cells to bypass/manage stalled replication forks17 (left panel). Binding of triphosphates of adenosine, and adenosine-analog drugs to RNR-α results in the formation of hexameric states (RNR-α6), which are involved in moonlighting (right panel). (RNR-α)6 is ushered into the nucleus by importin-α1. This process is suppressed by the (RNR-α)6-specific-binding cytosolic protein, IRBIT. Nuclear RNR-α (irrespective of its quaternary state) directly interacts with ZRANB3, preventing the ubiquitin-independent binding of ZRANB3 to PCNA, and ultimately suppressing DNA-synthesis.15,16 This ubiquitin-independent ZRANB3–PCNA interaction was recently discovered to promote DNA synthesis.15 | ||
A further example is the protein Fus, a ribonucleoprotein expressed in many cell types. The canonical role of Fus lies in the regulation of splicing events of particular importance in neuronal cell maintenance. These events occur in the nucleus and are important for mRNA maturation and translocation of mature mRNA to the cytosol (Fig. 1B). Fus is also found in the cytosol due to passive diffusion through the nuclear pore. In the cytosol, Fus regulates mRNA export and local translation. In several neurodegenerative diseases, including frontotemporal dementia (FTD), Fus is a hallmark component present in inclusions in the cytosol in diseased neurons. It was recently shown that these inclusions occur under hyperosmotic shock due to export of transportin 1 (TNPO1) into the cytosol, which prevents TNPO1-dependent Fus retrotranslocation into the nucleus from the cytosol.14 Such inclusions were not found in astrocytes, which do not build up Fus or TNPO1 in the cytosol under hyperosmotic shock. This difference between neurons and astrocytes mimics FTD pathology, as astrocytes do not show these inclusions.
Nucleotide pools maintenance: RNR
Mechanisms of disease associated with multifunctional/multi-locale-associated proteins are particularly hard to unravel, often because the function of the protein(s) of interest in the disease pathway is(are) not related to its(their) “ascribed” behavior(s). A recent discovery from our laboratory highlights this complexity because the non-canonical roles of multiple proteins intersect as a function of subcellular locale.15,16 The large subunit of ribonucleotide reductase (RNR-α) is canonically cytosolic, where it associates with the small subunit RNR-β. The resulting heterodimeric complex catalyzes the rate-limiting step in the chemical synthesis of all deoxynucleotides (Fig. 1C). This heterodimeric RNR–enzyme complex is thus a driver of DNA synthesis, cell growth, and its activity is often stimulated in cancer.18 However, several unexplained reports18–22 have strongly implied that RNR-α may function as a tumor suppressor, with data showing that knockdown of RNR-α can promote transformation.During our studies to unravel this conundrum, we found that in the presence of activity-downregulating nucleotides (such as dATP, or dATP-like drugs, such as clofarabine triphosphate, ClFTP) RNR-α translocates to the nucleus.15 This translocation is stimulated by an ordered series of steps including, ligand-induced formation of RNR-α hexamers (independent of RNR-β); and association of (some of) these RNR-α-hexamers with importins. There is also a negative regulator of this nuclear import mechanism, namely IRBIT, that acts as a cytosolic anchor of RNR-α-hexamers, by competing with importins for (some of) the hexamer(s).
In the nucleus, RNR-α interacts with ZRANB3. ZRANB3 is a multifunctional protein that canonically plays an important role in regulating how collapsed/stalled replication forks are processed/restarted.17 This function occurs principally through a strong interaction with K63-linked poly-ubiquitinated proliferating cell nuclear antigen (PCNA), a form of PCNA that occurs during DNA damage.23–25 ZRANB3 also interacts with non-ubiquitinated PCNA,25 but no role was ascribed to this process until our studies.15 We found that nuclear RNR-α displaces ZRANB3 from non-ubiquitinated PCNA, but likely cannot displace ZRANB3 from K63-linked-ubiquitinated PCNA. Building on these data, we discovered a moonlighting role of ZRANB3,26 which is dependent on ZRANB3's interaction with non-ubiquitinated PCNA. This new role involves promotion of DNA synthesis and proliferation of numerous cell types, by around 40–50%. This function occurs in the absence of DNA damage stimulation. Unsurprisingly, RNR-α in the nucleus suppresses DNA synthesis only in cells expressing ZRANB3. Furthermore, nuclear RNR-α's DNA-synthesis inhibition (in cells expressing ZRANB3) is suppressed when PCNA is overexpressed. We went on to show that ZRANB3 is important for assisting transformation induced by transfection of RASG12V16 in NIH-3T3 cells. A contemporary report reported a similar relationship between Myc and ZRANB3.26,27 Critically, ZRANB3 is not an essential gene and its knockdown does not appear to elevate DNA damage in unstressed cells.15 Thus, we have proposed that nuclear-specific RNR-α-mediated ZRANB3 inhibition of a non-canonical function of ZRANB3 may be the mechanism of the long-elusive RNR-α tumor suppression properties.
Neighborhood twinning: life on the limits
It is not just proteins that can change their environment; organelle surfaces have emerged to be particularly dynamic. Membrane contact sites have arisen as unique zones that are hives of cellular communication and regulation, essential for maintaining fitness.28 These junctions are somewhat makeshift, but can persist for some time, likely minutes at least.3,29 Recently, it has emerged that specific proteins are essential for maintenance of integrity of such junctions, and many of these proteins are performing non-canonical roles.2,7,29 Such environments, that are highly localized, transient and require proteins to adopt diverse and unfamiliar roles are ripe for scientific discovery. However, such investigations require development of methods that can peer into, and hopefully specifically perturb, this cloistered, ephemeral world with spatiotemporal resolution that matches the lifetime of these junctions. But these are far-removed territories, where the proteins essential for stability make up only a fraction of the total of that protein expressed in the whole cell,30 and where there could be significant functional redundancy between different proteins. In such neighborhoods, genetic methods, such as knockout and RNAi, often are not ideal to interrogate protein function. We now discuss methods that have been used to dissect protein localization and enact controlled perturbation that could be suited to study function of proteins at these contact sites, with an eye toward developing ways to interfere with such locale-specific protein functions.Methods of investigating subcellular localization
One aspect of chemistry that has been appreciated by biologists for almost 100 years, is that chemical compounds offer the ability to rewire cellular processes rapidly. For instance, even drugs that require metabolic activation can exert their effects on cells on the order of minutes.15,16 In contrast, classical genetic methods to modulate protein activity, such as RNAi or overexpression, can take several hours or likely more to be impactful, and may also be more perturbing to the overall system than small-molecule treatment. If we move forward in time to foundational chemical biology experiments using chemical-induced dimerization (CID, pioneered in the 1990s),31,32 we see that using CID, protein associations can be modulated on a time scale that is inaccessible by genetic means, at least those that were available at the time of publication. Such experiments were possible because chemistry allowed scientists to fashion structures that otherwise would not exist in nature. These structures can have properties that are orthogonal from the gamut of signaling molecules in cells, and can hence turn on cellular signaling processes by modulating interactomes. But chemistry has many guises, and its reach in biology over the intervening years has broadened. Much of modern chemical–biology effort has been expended at developing assays for specific events/modifications. We thus now have chemical–biology-driven approaches probing for sulfur oxidation (of various oxidation states, all with likely some redundancy),33 various metabolic signals and activities,34 and membrane tension that function in real time,35 to name but a few. Chemists have also discovered molecular zip codes that can broadly speaking, direct reagents to specific neighborhoods.36 Coupling molecular probes with these zip codes can give insights into the environments in specific locales.37Biotinylation methods to census proteins resident in specific organelles
Chemical localization tags, such as triarylphosphonium motifs targeting mitochondria, have also been utilized to investigate proteins localizing to specific organelles. It is worth noting that deployment of such strategies requires no genetic manipulation or ectopic protein expression, which may ultimately mitigate some of the concerns we voice below and certainly could contribute to generality of such methods. All-small-molecule-based censusing methods for protein localization have, to date, typically used a trifunctional molecular design. These trefoil molecules consist of an alkyne for biotin tagging post protein labeling, a small-molecule localization tag (SMLT), and an affinity agent. Most strategies have used unprotected, reactive pharmacophores, such as acyl chlorides38 or epoxides39 as the affinity reagents. Unsurprisingly, such strategies have proven to be overall low resolution and have suffered from issues with poor localization and temporal control. Photoactivatable pharmacophores have been investigated, although these were eschewed over uncaged, intrinsically-reactive motifs. Nevertheless, from these early studies, it has become clear that SMLT strategies can semi-selectively label proteins localizing to the target organelle. More intriguingly, chemically-different pharmacophores label only partially overlapping fractions of proteins congregating in the target locale.40 Assuming that (sub)organellar localization is dominated by the SMLT, the result from SMLT is consistent with the observation that, at the “global” level, reactive pharmacophores have non-overlapping preferences41 for protein targets. Such chemoselectivity was proposed to be a pathway to fragment-based drug design and molecular probes. However, in terms of developing organelle-specific profiling agents, chemoselectivity restricts the number of proteins that can be profiled and may be considered a limitation.42 Conversely, gleaning understanding of target selectivity across numerous chemotypes could form a basis for performing a detailed census as well as gleaning a more refined understanding of how to achieve locale- and protein-specific perturbation. Such knowledge has translational applications. We will discuss these issues and opportunities in the backdrop of more focused techniques below.Nevertheless, overall, clearly many issues need to be surmounted in the SMLT-based profiling arena. Aside from what appear to be so far not well addressed design issues, SMLTs, are likely not as effective as genetically-encoded tags localizing proteins to specific organelles, and certainly suborganelles (e.g., outer mitochondrial membrane43). It has also been reported that uptake differs significantly among probes, and further that fidelity of subcellular targeting can depend on numerous factors, including positively correlating with cLopP.42 Such concerns limit spatial resolution and hence confidence in accuracy of the census produced. Our overall understanding of SMLTs is also hampered because subcellular localization of the SMLT-functionalized molecule is often assayed at low resolution, post fixation and permeabilization of cells. Furthermore, these tags likely do not offer the added contextual/spatial control that, for instance, split proteins can provide. The use of SMLTs in whole organisms is also not well described and still poorly understood. Finally, how SMLTs affect innate biological systems of interest is not always known, and is difficult to assess or control for.44
Cognizant of the issues above that are admittedly common in many areas of modern chemical biology, some chemical biologists have turned to hybrid techniques. These strategies blend protein engineering and chemical insight to dual yoke novel chemical behaviors, and the control/selectivity offered by proteins/enzymes. Although fluorescent sensors of specific metabolites/chemical species based on engineered fluorescent proteins are good examples of this strategy, one can argue that proteins that generate reactive species for proteome/subproteome mapping are the current apogee. Such strategies are mostly associated with defining interactomes/localization/reactivity. In terms of defining localization/interactomes, two related but different systems are most commonly used: APEX(2)45 and BioID46 (Fig. 2A). Both systems release chemically-reactive biotin species on demand, to their surrounding locale. It should be noted that the crux of these methods relies on the fact that the reactivity of the molecules generated is folds higher than that of the SMTL-tagged molecules deployed above. Thus APEX(2) and BioID are often assumed to label proteins indiscriminately. APEX(2) uses an engineered soybean ascorbate peroxidase that generates a biotin-functionalized phenoxy radical, formed as a result of exposure to peroxide and a biotin phenol, both of which are administered from outside of cells. The resultant radical species is not believed to be able to traverse biological membranes,47 allowing for very high facial selectivity when studying membrane-bound structures.48 However, it is noteworthy that biological membranes are complex, dynamic, and sensitive to oxidation (see below). APEX(2) is also able to be used as a contrast agent for electron microscopy, which can readily afford a high-resolution understanding of APEX(2)'s localization.49 BioID, on the other hand, uses a mutant biotin ligase from E. coli. This generates a biotinyl adenosine monophosphate (biotin-AMP), following whole-cell stimulation with biotin. Both the biotin phenoxy radical and the biotin-AMP have short diffusion distances, which confers high spatial control to each technique per unit molecule. There are similar systems that release other reactive species. However, these are not as widely used, and their principals are similar to APEX(2) and BioID, so we focus on APEX(2) and BioID.
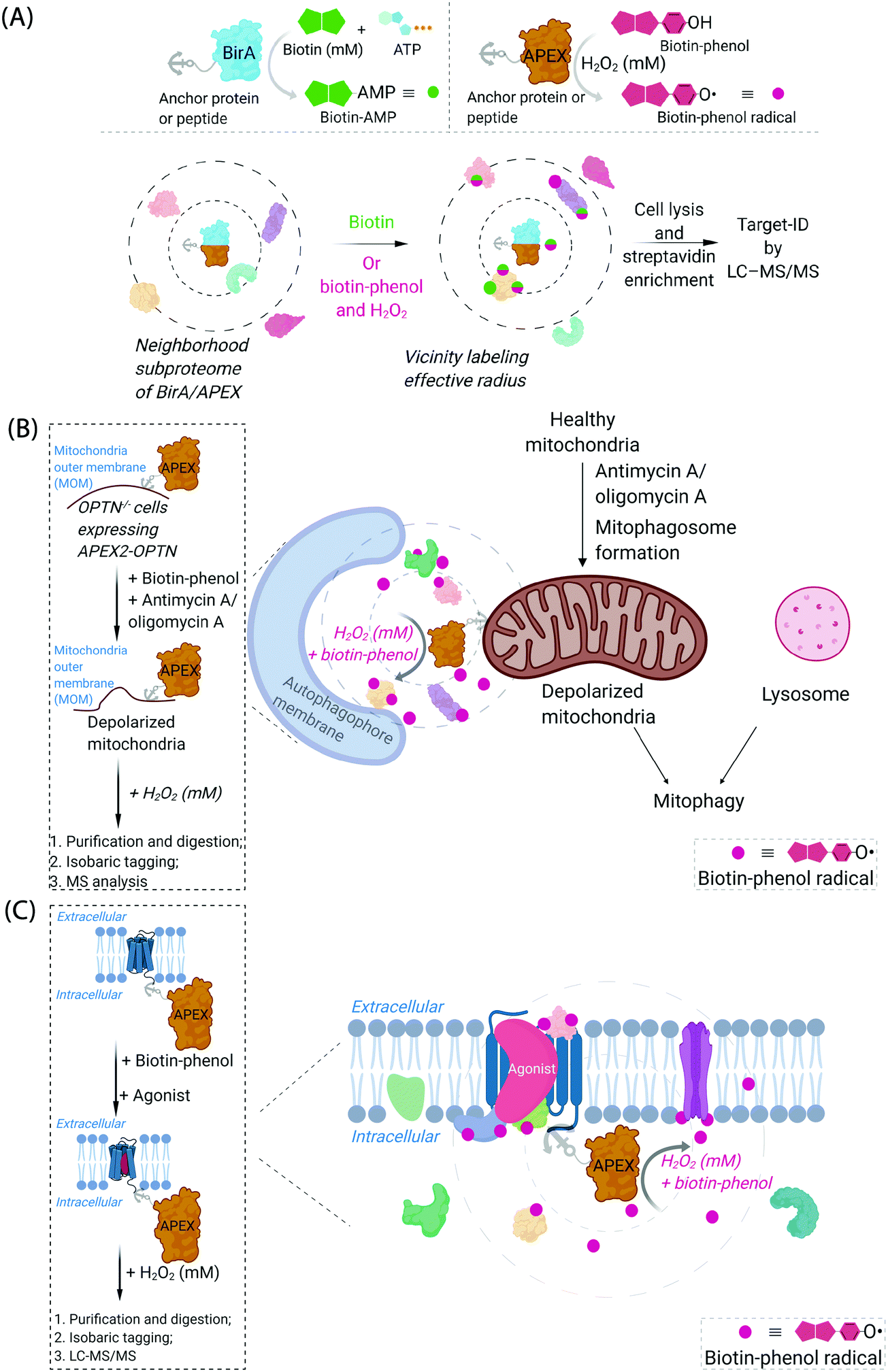 | ||
| Fig. 2 Proximity subproteomics mapping techniques, BioID, and APEX. (A) Top left panel: BioID profiling is based on the expression of an anchor peptide or protein fused to BirA, a mutant biotin ligase catalyzing the conversion of biotin to biotinyl adenosine monophosphate (biotin-AMP) in the presence of ATP.46 Top right panel: APEX is based on the expression of an anchor peptide or protein fused to APEX-peroxidase, which catalyzes the conversion of biotin-phenol to biotin-phenoxyl radical in the presence of millimolar H2O2.45 Bottom panel: Both BioID and APEX generate reactive molecules (biotin-AMP or biotin-phenoxyl radical) that bind covalently to proximal and accessible nucleophilic amino acid residues within neighborhood proteins. The neighboring subproteome in the vicinity of BirA/APEX, is potentially mobile, making the duration of the experiment also of the essence. LC-MS/MS (post streptavidin enrichment) is used to identify the targeted proteome. (B) Proteome characterization of a depolarized mitochondrial-autophagosome synapse during mitophagy using APEX2.55 OPTN−/− cells expressing APEX2-OPTNwt were treated with biotin-phenol, and mitochondrial depolarization-inducing agent, either antimycin or oligomycin A. Cells were then treated briefly with 1 mM H2O2, prior to harvest. Following cell lysis, labelled proteins were enriched using streptavidin. The purified biotinylated proteins were analyzed by digest MS allowing quantitative proteomics target-ID of proteins in the vicinity of OPTN during mitophagy. Inset: The flow chart of the experimental procedure. The figure shows an expanded view of the autophagophore membrane and depolarized mitochondrial proteome in the APEX2-assisted vicinity profiling experiment during mitophagy. (C) GPCR-APEX profiling method to capture GPCR protein interaction networks.57 HEK293 cells expressing GPCR-fused APEX-peroxidase were treated with biotin-phenol, and GPCR was activated with several agonists. Cells were then treated with 1 mM H2O2 for 30 seconds. Cells were then harvested, lysed, and labelled proteins were enriched using streptavidin. The purified biotinylated proteins were analyzed as in Fig. 2B, to identify proteins in the vicinity of APEX-peroxidase. Inset: The flow chart of the experimental procedure. The figure shows an expanded view of APEX-peroxidase-assisted locale-specific proteomic mapping in this setup. | ||
Given that BioID and APEX(2) generate different chemical entities, it is important to note that BioID and APEX(2) show different proclivities to label specific proteins. In direct comparisons, often strikingly different results are gained from the two approaches. Based on these results, it has been suggested that it is best to use these proteins in tandem.50,51 Variables contributing to these differences are not well understood.
Aside from the chemical difference in released chemical probes, other differences between BioID and APEX(2) could contribute to differences in labeling outputs between these methods. APEX(2) is considerably more active than BioID. Consequentially, APEX is usually activated (i.e., present in cells treated with peroxide) over a period of not much more than minutes during an experiment. Conversely, BioID is a relatively slow enzyme, meaning that profiling experiments with this enzyme are run typically for long periods of time (hours or longer). This sluggishness can limit spatial resolution of BioID relative to APEX(2) as labeled proteins have the opportunity to move around the cell during the time of the experiment for BioID, whereas this is less likely in experiments using APEX(2). However, BioID may have more likelihood of catching proteins translocating only under a specific set of circumstances, not considered during experimental planning. The main limitation of APEX-based methods, highlighted some years ago,32 is the reliance on bulk treatment of cells with high dosage of peroxide to stimulate intracellular production of the biotin phenoxy radical species. BioID does not have this issue. With the recognition of these limitations to APEX(2), recent efforts to improve the kinetics of BioID have produced TurboID. In terms of target ID in cell culture, the improvement of TurboID over traditional BioID was not particularly marked.52 However, TurboID was more effective than BioID in whole organisms,53,54 and plants.53 Both BioID and APEX(2) are applicable to split protein technology,53 potentially offering much greater contextual control, although current applications in this line of research need further development.
One recent example of application of APEX2 to studying membrane interfaces investigated the mitochondrial-autophagosome synapse during mitophagy,55 post treatment with antimycin A/oligomycin A. This represents a particularly challenging system because it is highly context-dependent and it is sub-stoichiometric relative to each organelle. In these well-controlled experiments, APEX was fused to several proteins believed to be critical for mitophagy, namely, optineurin (OPTN) or TAX1-binding protein 1 (TAX1BP1) (Fig. 2B). These fusions were expressed in respective knockout cells at close to endogenous levels and were shown to rescue mitophagy defects in the respective knockout cells. Data from the APEX screens showed several interesting proteins not previously considered to be involved in mitophagy, that were enriched at these regions relative to their general abundance in mitochondria. These experiments were performed sequentially with data-derived from high-throughput genetics screens assaying for changes in mitophagic flux upon knockout of a specific gene. From both screens, several interesting overlapping proteins were uncovered, including HK2, a protein previously associated with recruitment of ubiquitin ligases to mitochondria,56 an early step in mitophagy. However, on the whole, overlap was relatively low between these data sets (∼8%), indicating, among numerous possibilities, that proximity to OPTN or TAX1BP1 may not be the biggest determinant of functional importance in mitophagy, that the genetic screen focuses on steps of the pathway not covered by the proximity screen (i.e., not involved in synapse formation), or that there is redundancy in the system (leading to the genetic screen not identifying hits from proximity experiment). Redundancy could arise through there being multiple proteins in the ground state performing the same function, or during knockout line generation (which typically requires a week or more), another protein could have been upregulated to compensate for loss of the protein of interest. Furthermore, the effects of peroxide treatment during APEX were also not extensively investigated. BioID was not compared in this study.
Another recent example investigated surface-expressed proteins involved in neuronal rewiring during development (which used a membrane-localized horseradish peroxidase coupled with a cell-impermeable form of biotin-phenol) (Fig. 2C). This specific experimental approach is interesting as it uses a developmental time course as a variable to extract changes in cell-surface proteomes. Several identified proteins appeared to be particularly important for neuronal wiring.58
We take from the above examples that peering deeper into the subcellular world to build a high-resolution blueprint of biological processes can also be met with mixed fortunes and that different approaches will likely lead to different outputs. General issues may be somewhat mitigated when tackling spatially- and numerically-restricted proteomes, such as surface interactomes.59 Thus, in general, the field would hugely benefit from further optimizations and development of new methods, especially to probe intact intracellular/subcellular systems, particularly because of the implicitly-complex intracellular (micro)environments, and because of several limitations in APEX/BioID.
Precision electrophile delivery methods to roll-call functional subproteomes
Generation of reactive ectopic chemical entities is a generally useful strategy. For instance, several methods have deployed free,60 or tethered61 photoactivatable crosslinkers to study macromolecular interactomes.62 But generation of ectopic molecules is not the only feat chemical biology is capable of. There are also examples of controlling reagents that can deliver a burst of a specific biomolecule at a given time, allowing locale-specific signaling subsystems to be uncovered;63 and nanoparticles that localize high concentration of a specific ligand to modulate endogenous proteins to enable control of cellular adhesion, actin polymerization, or sorting among others.64 These methods and others alike seek to control cell signaling processes with a view to reprogramming or perturbing specific decision-making steps enacted by the cell.In our case, we first developed a chemical-genetic method that could deliver a specific electrophile to a specific protein of interest (POI) in vivo, dubbed targetable reactive electrophiles and oxidants (T-REX).65–67 T-REX is compatible with applications in several living models. The method uses a photocaged electrophile that is non-toxic, bioinert, and permeable to cultured cells,65–70C. elegans,67,71 and zebrafish.66,70 Electrophiles compatible with the T-REX-platform include any α,β-unsaturated ketone, or α,β-unsaturated aldehyde72 (Fig. 3A). We are working toward further extending the chemical space available. The photocaged electrophile contains an alkyl chloride motif, which reacts stoichiometrically and specifically with Halo protein in vivo. This setup side-steps one of the issues from the SMLT-based platforms above, as the electrophile available in cells is determined by intracellular concentration of Halo-POI (Fig. 3B), which is quantifiable and controlable. When a specific POI is expressed as a Halo fusion in vivo, administration of the photocaged electrophile from outside the cells/animals, followed by washout of excess probe, leads to stoichiometric and irreversible binding of Halo with the specific photocaged electrophile of interest. Light-driven electrophile uncaging (t1/2 < 1 min72) can liberate an electrophile in the specific subcellular neighborhood where the Halo-fused POI resides. At this point, there are two possible outcomes:
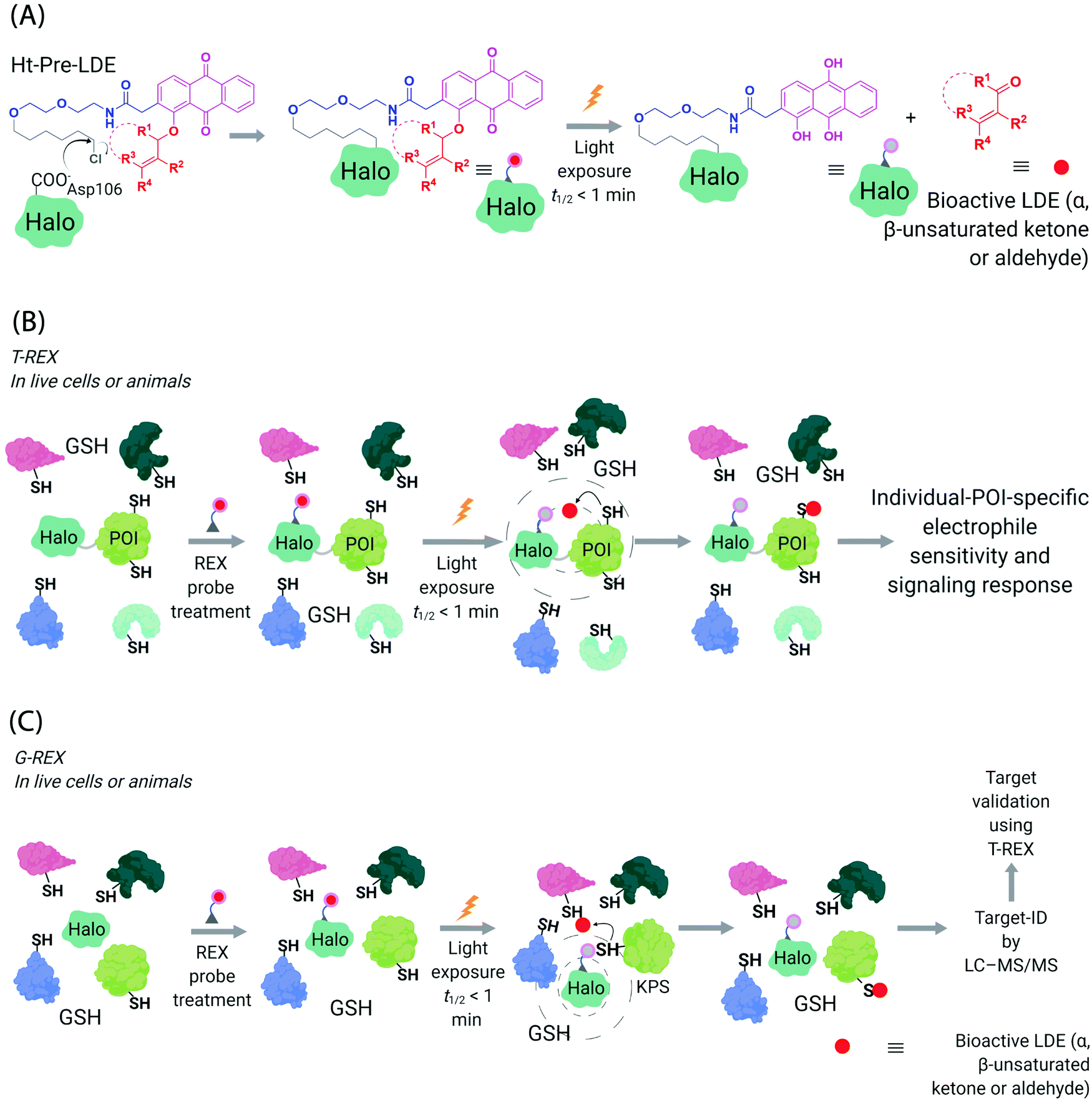 | ||
| Fig. 3 Functional subproteomics mapping and on-target signaling-interrogation techniques, G-REX and T-REX. (A) G-REX maps functional subproteomes primed to sense lipid-derived electrophiles (LDEs) and related covalent drugs, whereas T-REX functionally validates and evaluates individual-POI-specific covalent-ligand sensitivity and signaling responses.82 The figure shows chemistry underlying photocaged electrophiles (Ht-Pre-LDEs) common to both G-REX and T-REX methods. Halo in G-REX (or Halo-POI in T-REX) expressed in cells or animals covalently binds Ht-Pre-LDE, following cell/animal incubation with designated Ht-Pre-LDE in culture media (typically at 1 μM overnight, or at 10 μM over 2–3 h). Following washout of excess, unbound Ht-Pre-LDE (step not shown in the figure), exposure of live cells/animals to light (365 nm, 5 mW cm−2 over 1–5 min) results in rapid liberation of LDE (t1/2 of photouncaging <1 min) within proximity of Halo in G-REX (or Halo-POI in T-REX). Concentration of LDE released is maximally stoichiometric to intracellular concentration of Halo/Halo-POI (which has been quantified to be <5 μM).70 (B) In T-REX, the protein of interest (POI) is fused to Halo. Cells/animals expressing Halo-POI are treated with designated Ht-Pre-LDE. Following rinsing cycles to remove the excess/unbound Ht-Pre-LDE, the system is exposed to low-energy UV light (see (A) legend). Providing the POI is a kinetically-privileged sensor (KPS) of the LDE in the vicinity of the POI in limited amounts, the LDE can be captured by the POI before it irreversibly diffuses away beyond the solvent shell of Halo-POI. See also Fig. 4B. Percentage LDE-occupancy of POI and the identity of LDE-sensing residue are assessed using previously published protocols. In parallel, T-REX set-up allows functional consequences of POI-specific covalent-ligand modifications to be assayed directly and precisely in intact cells/animals.65,66,70,71 (C) In G-REX, cells/animals expressing Halo (not fused to any protein) are treated with designated Ht-Pre-LDE and the subsequent steps remain the same as in T-REX (see Fig. 3B legend). The LDE (in alkyne-functionalized version) rapidly released in limited dosage in G-REX covalently tags the most kinetically-privileged native LDE sensors within the microenvironment of Halo in cells/animals. Following cell/animal lysis, endogenous KPSs covalently bound to the released ligand are enriched following Click-coupling to biotin-azide and streptavidin pulldown, and protein-ID is achieved using standard quantitative proteomics methods.70 | ||
(I) the first, which is more common, is that the electrophile diffuses away from the Halo-POI and the POI is not labeled by the electrophile. The liberated electrophile is then averaged over the rest of the available proteome and/or metabolically/enzymatically degraded.73 It is noteworthy that due to genetic-based intracellular delivery, the amount of unreacted electrophile that diffuses away is minimal and in the range of low μM to nM,66,70,74 depending on cells vs. animals, and has been shown not to affect cellular glutathione pools.68,69
In the second scenario (II), the POI is reactive to the specific electrophile of interest, and hence the POI can intercept the electrophile prior to diffusion away from the encounter complex. These POIs have been dubbed privileged first responders. A large amount of experimentation indicates that POIs able to intercept electrophiles under T-REX are genuinely reactive to electrophiles:63,67 most of the proteins so identified have higher than expected second-order reaction kinetics with their preferred electrophile in vitro and in cells.63,75 Many of the top-enriched proteins captured following bolus-electrophile dosing procedures failed to be labeled when the same electrophile is delivered using T-REX against appropriate controls.63,65–67 Critically, most of the proteins that are inherently reactive to electrophiles (i.e., those labeled under electrophile-limited conditions in T-REX) are also acutely sensitive to changes in protein activity/downstream function upon electrophile labeling74 (Fig. 4A).
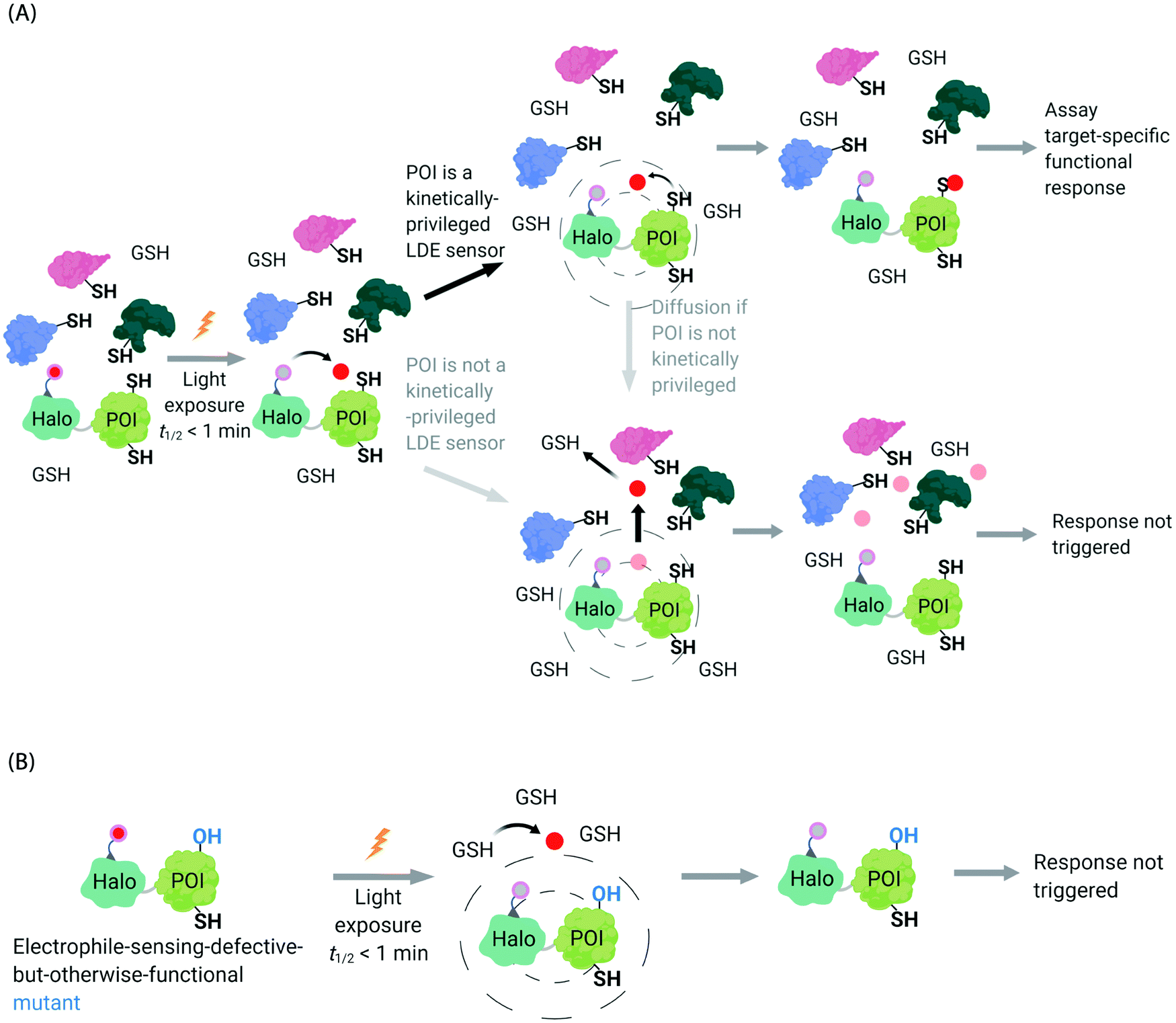 | ||
| Fig. 4 Concepts underlying REX techniques: competition between reactivity vs. native diffusion propensity of electrophiles or covalent ligands, and associated controls. (A) In T-REX, after the stoichiometric anchoring of Ht-Pre-LDEs, washing away of excess Ht-Pre-LDEs, and subsequent low-energy UV light-induced photo-uncaging (365 nm, 5 mW cm−2 over 1–5 min), LDEs are liberated. This liberation forms a transient “encounter complex” (dotted circle). When POI is a kinetically privileged sensor for the liberated LDE, the POI can intercept the LDE before its diffusion, leading to POI-specific fractional labeling and (likely) a functional response. When POI is not a kinetically privileged sensor of the specific LDE, the liberated LDE (<5 μM)70 is not intercepted and diffuses into the cellular environment. The POI is not labelled, and no response is triggered. (B) The electrophile-sensor cysteine within the POI can be mutated into serine (shown) or alanine to generate a sensing-defective mutant-POI (that is otherwise validated to be functional). This functional mutant-POI fails to capture the LDE when T-REX is replicated under otherwise identical conditions. In this scenario, the LDE released diffuses from the POI (see below) is averaged across the cell, leading to mimimal perturbation of the cell. For instance, the low (<5 μM)70 amount of LDE released does not affect the overall cellular GSH/GSSG pools (present at mM levels).69,72 Generally, such a mutation silences downstream signaling changes that are otherwise measured by T-REX using wild-type POI that senses electrophiles during T-REX.65,66,70,71 | ||
Recognizing that even for the most electrophile-sensitive proteins, not 100% of the electrophile released during T-REX, labels the POI, we have developed numerous controls to prove that phenotypes are due to on target labeling. One of the most functionally-relevant controls that we have established is replication of T-REX procedure on electrophile-sensing-defective-but-otherwise-functional mutant POI, whereby sensing Cys is mutated to Ala/Ser, and confirm that the response measured is consequently ablated (Fig. 4B).66,70 These data have led us to propose that identifying electrophile-sensitive proteins can be used in drug discovery programs.74,76,77 We recently made the first step to realizing this translational promise by using data from T-REX to design a first in class Akt3-selective covalent inhibitor.78
It became clear to us that interrogation into POI-specific electrophile sensing and signaling by T-REX could be adapted as a method to rapidly identify native, and also locale-specific, electrophile sensors. This is because ideal protein sensors react rapidly with (specific) electrophiles. As our method can: (i) release a predetermined dose of an electrophile with a half-life of uncaging of <1 min;65 and (ii) this electrophile will have a relatively short half-life/diffusion distance due to natural detoxification processes, only rapidly-reacting sensors (i.e., privileged first responders, vide supra) will be labeled. In a variant termed G-REX70 (Fig. 3C), which enables ostensibly unbiased profiling of electrophile-responsive proteins, Halo protein alone without any POI fused to it, is expressed and the rest of the T-REX procedure replicated. An initial application of G-REX integrating ubiquitous expression of Halo in cells, identified several novel electrophile sensors, including Ube2V2, a defunct E2-conjugating enzyme that has evolved to be an effector allosterically promoting activity of the E2 ubiquitin-conjugating enzyme, Ube2N.70 Intriguingly, Ube2N was not a hit protein, even though Ube2N houses a catalytically-active cysteine that reacts with electrophilic inhibitors.79,80 By contrast, Ube2V2 possesses what, at least according to the crystal structure, is a seemingly-buried cysteine.80,81 Ube2V2 was also unknown at the time to be electrophile sensitive (Fig. 5).70 Both Ube2N and Ube2V2 have similar folds, although Ube2V2 has shed several helices that are present in most E2-conjugating enzymes. Subsequent analysis, under electrophile-limited conditions via T-REX in cells or in vitro, showed that Ube2V2 was an electrophile sensor, whereas Ube2N was not a sensor, under electrophile-limited conditions via T-REX or in vitro. Strikingly, electrophile modification specifically at Ube2V2 promoted Ube2N ubiquitination activity and this elevation in activity primed several DNA-damage responses, including increase in γ-H2AX in developing zebrafish.70 Replicating T-REX using a functional Ube2V2-mutant housing Ser/Ala in place of Cys muted electrophile-sensing and ablated elevation of Ube2N activity. These data show that functional information can be readily read out using controlled electrophile-delivery experiments. Furthermore, electrophile-sensing behavior is unpredictable and requires actual activity/functional assays to understand it.
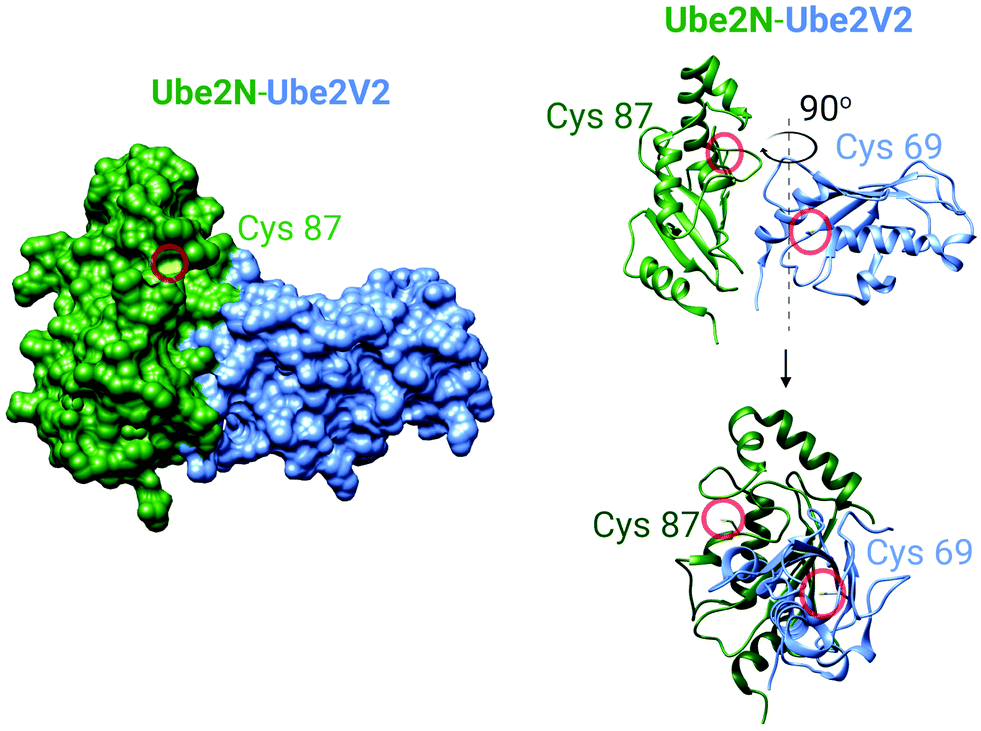 | ||
| Fig. 5 The crystal structure of the human Ube2N and Ube2V2 heterodimer (PDB: 1J7D).81 Left panel: The surface-accessible model of the heterodimer. Cysteine sulfur atoms in both Ube2N(C87) and Ube2V2(C69) are colored yellow, but only Ube2N(C87) is visible (see text for discussion). Right panel: The ribbon structure of the heterodimer featuring Cys87 of Ube2N and electrophile-sensing Cys69 of Ube2V2.70 | ||
Taking our data, and that of others as a whole,63 we conclude that specific—often what may be considered to be unreactive—proteins are readily controllable (either in terms of stimulation or inhibition), using electrophiles. Critically, proteins that rapidly interact with electrophilic small molecules are usually functionally affected by electrophile modifications, even at low ligand occupancy. Thus, electrophile responsivity and function, and potentially nature of the subcellular environments in which sensing occurs, are linked together.
Neighborhood planning: what do we need to do next?
It may be helpful here to draw a parallel between the 1930s and now. In the 1920s–1930s, scientists were trying to understand the chemical roles played by the larger neighborhoods, which were assumed to be unchanging. There were relatively few techniques available, and many of these were chemical/biochemical, or biophysical in nature, allowing our comprehension of biochemistry to grow. Today we live in a protein, gene, and target-rich world, but our spheres of interest are getting smaller and smaller and more and more dynamic. We further have tools that are uniquely sensitive to ask penetrating questions as to which proteins: overall change expression; change association with other proteins, DNA or RNA; or exist in which locale, under specific conditions. Huge data sets integrating changes in protein localization, and association/interactome are now available, not to mention software to analyze genomic data to predict localization, activity, etc. In the sense that more is more, we have come a long way during that 100 years. However, if we look at these data from another perspective, we see that a large proportion of these data is not functionally validated, nor are the data linked strongly to functional changes. We actually possess few chemical- or activity-based means to test the locale-specific data we have amassed on the same timeframe/with the same resolution as which the large-scale data sets are generated. These issues critically limit the spatiotemporal resolution with which we can investigate specific proteins’ involvements in specific locales, and preclude gaining understanding of which proteins are critically involved in transient, context-specific processes. Of course, gaining such nuanced information is far from easy. Mostly we use genetic methods to complement our chemical biology-derived data. However, these methods are still largely limited in several critical dimensions (e.g., temporal resolution, low background proteome perturbation, and spatial control) that we sought to understand further through creating chemical–biology methods in the first place. Genetic manipulations can be supplemented with global chemical perturbation, such as bolus treatment of cells, but this strategy is not greatly different from what was available in the 1930s. Thus, not for the first time in science, we live in an age where we have gained resolution in one dimension (namely spatial resolution); however, our ability to probe the ramifications of these data has not largely improved. Hence a one-time strength has become a limitation.Encouragingly, there are signs that we are turning the page again, and adapting some old tricks to these new problems. Examples of such approaches include GPCR-APEX (Fig. 2C), which was able to uncover several aspects of how global activation and deactivation affect the GPCR interactome.57 However, non-targeted inhibitors/chemical modulators will likely prove to be inadequate for use in locale-specific investigations as they interfere with target proteins in numerous locales, which could cause artifactual results, and many proteins are not able to be targeted selectively. This predicament highlights that the emerging innovative tools that can map locale-specific subproteome such as APEX(2) and BioID, are not applicable to inform on specific tools for downstream functional investigations. This is because the indiscriminately reactive species generated, admittedly ideal to perform profiling, cannot be used to regulate function of identified proteins. Thus, interrogation of an identified protein's role specifically in these cloistered environments remains a significant task.
Should neighborhood canvassing get active?
It is our belief that current and future effort, that could be complementary to APEX-type approaches, should invest more heavily in using biologically-relevant labeling technologies to functionally canvass specific neighborhoods to provide a census of reactive proteins in specific locales. Particularly, the use of specific bioactive molecules, such as electrophiles (either native lipid-derived electrophiles housing drug-like electrophilic units, or covalently-reactive drugs/candidates) to identify locale-specific sensing (through G-REX or SMLT), would offer a means to probe locale-specific biological functionality. This second step could be carried out using T-REX to deliver the specific matched electrophile to the identified POI. Indeed, coupled together, the REX systems could determine who are the key players in a specific neighborhood, and then assess the functions of this protein directly by leveraging the ligand-promoted signaling behavior of each resident protein. Such a tandem analysis also does not rely on orthogonal assays, which often target poorly-overlapping sets of players, or could require the use of techniques that have poor resolution and are prone to error, diminishing the relevance of the initial screening effort. Critically, the technique is versatile, as genetic manipulation of Halo allows a faithful choice of neighborhood (organelle/interorganelle) specificity. The chemical reactome, can also be altered by changing the electrophile chemical architecture, similar to how BioID and APEX have been proposed to be used, but with unprecedented ability to vary the chemotype and ligandable units.Outlook
We strongly believe that the revolution that was started through the employment of chemical genetics methods to probe locales or reactivity,83 solved key issues that exist with previous all chemical or all biological approaches. The manifold systems to which these hybrid techniques have been successfully applied clearly show that such approaches are of great utility. We thus see that the field is going in the right direction, and that this approach is paying dividends. However, we are mindful that the roots of chemical application to biology are grounded in the spatiotemporal perturbation of specific proteins (or locales) with specific reagents, and we hope that we can see a return to that spirit. We thus advocate a more holistic investigation strategy that can potentially uncover key players and means to perturb them through simple shifts in conditions or constructs. We believe that such an experimental protocol readily offers hypotheses and a means to test them. We thus continue to advocate the implication of such stratagems with genetics and other classically biological techniques.84,85 We also believe that there are data available showing that such combinations can uncover aspects of signaling that are otherwise hidden. However, the onus is on the chemical biology community to create such systems, and with rigorous and careful examinations, to expand them to their full potential.Abbreviations
| APEX | An engineered ascorbate peroxidase |
| dATP | Deoxyadenosine triphosphate |
| biotin-AMP | Biotinyl adenosine monophosphate |
| CLFTP | Clofarabine 5′-triphosphates |
| CID | Chemical-induced dimerization |
| FUS | Fused in sarcoma (RNA-binding protein FUS) |
| GPCR | G protein-coupled receptor |
| G-REX | Genome-wide profiling of reactive-electrophile and -oxidant sensors |
| γ-H2AX | Phosphorylated form (Serine-140) of H2A histone family member X |
| HK2 | Hexokinase 2 |
| Halo-POI | Halotag fused protein of interested |
| Ht-Pre-LDEs | Halotag targetable photocaged lipid-derived electrophiles |
| KPS | Kinetically privileged sensor |
| LDEs | Lipid-derived electrophiles |
| cLogP | Logarithm of 1-octanol/water partition coefficient |
| OPTN | Optineurin |
| PCNA | Proliferating cell nuclear antigen |
| POI | Protein of interesting |
| RASG12V | HRAS G12V mutant |
| RNR-α/-β | Ribonucleotide reductase subunit-α/-β |
| SMLT | Small-molecule localization tag |
| TAX1BP1 | TAX1-binding protein 1 |
| TNPO1 | Transportin 1 |
| T-REX | Targetable reactive electrophiles and oxidants |
| Ube2N | Ubiquitin conjugating enzyme E2 N |
| Ube2V2 | Ubiquitin-conjugating enzyme E2 variant 2 |
| ZRANB3 | Zinc finger RANBP2-type containing 3 |
Author contributions
Concept and outline: Y. A.; writing: M. J. C. L., Y. A.; figures: Y. Z.; figure legends: Y. Z., M. J. C. L.; final proof-editing: all authors.Conflicts of interest
REX-technologies and inhibitors derived from their applications have been filed for US-patent applications by the authors' former institution, Cornell University.Acknowledgements
Swiss National Science Foundation (SNSF) NCCR Chemical Biology for personnel support (Y. Z.); Swiss Federal Institute of Technology (EPFL), SNSF Project Funding 184729, SNSF Spark Funding 190192, NIH 1DP2GM114850 (PI: Y. A.) for shared instruments and supplies supporting the Aye Lab's electrophile signaling research program.References
- T. Harayama and H. Riezman, Nat. Rev. Mol. Cell Biol., 2018, 19, 281 CrossRef CAS PubMed.
- W. A. Prinz, A. Toulmay and T. Balla, Nat. Rev. Mol. Cell Biol., 2020, 21, 7 CrossRef CAS PubMed.
- G. Csordás, D. Weaver and G. Hajnóczky, Trends Cell Biol., 2018, 28, 523 CrossRef PubMed.
- A. A. Rowland and G. K. Voeltz, Nat. Rev. Mol. Cell Biol., 2012, 13, 607 CrossRef CAS PubMed.
- N. E. Hynes, P. W. Ingham, W. A. Lim, C. J. Marshall, J. Massague and T. Pawson, Nat. Rev. Mol. Cell Biol., 2013, 14, 393 CrossRef CAS PubMed.
- V. Eisner, M. Picard and G. Hajnóczky, Nat. Cell Biol., 2018, 20, 755 CrossRef CAS.
- A. Murley and J. Nunnari, Mol. Cell, 2016, 61, 648 CrossRef CAS PubMed.
- M. Leshets, Y. B. H. Silas, N. Lehming and O. Pines, Front. Mol. Biosci., 2018, 5, 68 CrossRef PubMed.
- R. Dalbey and G. v. Heijne, Protein Targeting, Transport, and Translocation, Academic Press, 1st edn, 2002 Search PubMed.
- I. Mellman and W. J. Nelson, Nat. Rev. Mol. Cell Biol., 2008, 9, 833 CrossRef CAS.
- R. E. Thomson, A. L. Bigley, J. R. Foster, I. R. Jowsey, C. R. Elcombe, T. C. Orton and J. D. Hayes, J. Histochem. Cytochem., 2004, 52, 653 CrossRef CAS.
- K. Burger and M. Gullerova, PLoS Genet., 2018, 14, e1007151 CrossRef PubMed.
- K. Burger, M. Schlackow, M. Potts, S. Hester, S. Mohammed and M. Gullerova, J. Cell Biol., 2017, 216, 2373 CrossRef CAS PubMed.
- E.-M. Hock, Z. Maniecka, M. Hruska-Plochan, S. Reber, F. Laferriere, S. S. M K, H. Ederle, L. Gittings, L. Pelkmans and L. Dupuis, Cell Rep., 2018, 24, 987 CrossRef CAS PubMed.
- Y. Fu, M. J. C. Long, S. Wisitpitthaya, H. Inayat, T. M. Pierpont, I. M. Elsaid, J. C. Bloom, J. Ortega, R. S. Weiss and Y. Aye, Nat. Chem. Biol., 2018, 14, 943 CrossRef CAS PubMed.
- M. J. C. Long, Y. Zhao and Y. Aye, Cell Chem. Biol., 2020, 27, 122 CrossRef CAS PubMed.
- L. A. Poole and D. Cortez, Crit. Rev. Biochem. Mol. Biol., 2017, 52, 696 CrossRef CAS PubMed.
- Y. Aye, M. Li, M. Long and R. Weiss, Oncogene, 2015, 34, 2011 CrossRef CAS PubMed.
- M.-Y. Cao, Y. Lee, N.-P. Feng, K. Xiong, H. Jin, M. Wang, A. Vassilakos, S. Viau, J. A. Wright and A. H. Young, Clin. Cancer Res., 2003, 9, 4553 CAS.
- G. Bepler, S. Sharma, A. Cantor, A. Gautam, E. Haura, G. Simon, A. Sharma, E. Sommers and L. Robinson, J. Clin. Oncol., 2004, 22, 1878 CrossRef CAS PubMed.
- A. Gautam, Z.-R. Li and G. Bepler, Oncogene, 2003, 22, 2135 CrossRef CAS.
- H. Qi, M. Lou, Y. Chen, X. Liu, N. Chen, J. Shan, Z. Ling, J. Shen, L. Zhu and Y. Yen, Tumor Biol., 2015, 36, 4833 CrossRef CAS.
- R. Weston, H. Peeters and D. Ahel, Genes Dev., 2012, 26, 1558 CrossRef CAS.
- J. Yuan, G. Ghosal and J. Chen, Mol. Cell, 2012, 47, 410 CrossRef CAS PubMed.
- A. Ciccia, A. V. Nimonkar, Y. Hu, I. Hajdu, Y. J. Achar, L. Izhar, S. A. Petit, B. Adamson, J. C. Yoon, S. C. Kowalczykowski, D. M. Livingston, L. Haracska and S. J. Elledge, Mol. Cell, 2012, 47, 396 CrossRef CAS.
- M. J. C. Long, A. Van Hall-Beauvais and Y. Aye, Curr. Opin. Chem. Biol., 2020, 54, 10 CrossRef CAS PubMed.
- M. V. Puccetti, C. M. Adams, S. Kushinsky and C. M. Eischen, Cancer Res., 2019, 79, 1612 CAS.
- S. Paillusson, R. Stoica, P. Gomez-Suaga, D. H. W. Lau, S. Mueller, T. Miller and C. C. J. Miller, Trends Neurosci., 2016, 39, 146 CrossRef CAS PubMed.
- M. J. Phillips and G. K. Voeltz, Nat. Rev. Mol. Cell Biol., 2016, 17, 69 CrossRef CAS PubMed.
- R. Rizzuto, P. Pinton, W. Carrington, F. S. Fay, K. E. Fogarty, L. M. Lifshitz, R. A. Tuft and T. Pozzan, Science, 1998, 280, 1763 CrossRef CAS PubMed.
- C. J. Gerry and S. L. Schreiber, Nat. Chem. Biol., 2020, 16, 369 CrossRef CAS.
- M. J. C. Long, J. R. Poganik and Y. Aye, J. Am. Chem. Soc., 2016, 138, 3610 CrossRef CAS PubMed.
- H. Xiao, M. P. Jedrychowski, D. K. Schweppe, E. L. Huttlin, Q. Yu, D. E. Heppner, J. Li, J. Long, E. L. Mills, J. Szpyt, Z. He, G. Du, R. Garrity, A. Reddy, L. P. Vaites, J. A. Paulo, T. Zhang, N. S. Gray, S. P. Gygi and E. T. Chouchani, Cell, 2020, 180, 968 CrossRef CAS PubMed.
- W. Wang, C. K. Kim and A. Y. Ting, Nat. Chem. Biol., 2019, 15, 101 CrossRef CAS.
- R. Loewith, H. Riezman and N. Winssinger, Curr. Opin. Chem. Biol., 2019, 50, 19 CrossRef CAS PubMed.
- S. Wisnovsky, Eric K. Lei, Sae R. Jean and Shana O. Kelley, Cell Chem. Biol., 2016, 23, 917 CrossRef CAS.
- M. J. C. Long, X. Liu and Y. Aye, Front. Chem., 2019, 7, 125 CrossRef CAS PubMed.
- J. Dong, D. Hong, W. Lang, J. Huang, L. Qian, Q. Zhu, L. Li and J. Ge, ChemBioChem, 2019, 20, 1155 CrossRef CAS PubMed.
- S. D. Wiedner, L. N. Anderson, N. C. Sadler, W. B. Chrisler, V. K. Kodali, R. D. Smith and A. T. Wright, Angew. Chem., Int. Ed., 2014, 53, 2919 CrossRef CAS PubMed.
- Y. Yasueda, T. Tamura, A. Fujisawa, K. Kuwata, S. Tsukiji, S. Kiyonaka and I. Hamachi, J. Am. Chem. Soc., 2016, 138, 7592 CrossRef CAS PubMed.
- C. G. Parker, A. Galmozzi, Y. Wang, B. E. Correia, K. Sasaki, C. M. Joslyn, A. S. Kim, C. L. Cavallaro, R. M. Lawrence and S. R. Johnson, Cell, 2017, 168, 527 CrossRef CAS PubMed.
- A. Fujisawa, T. Tamura, Y. Yasueda, K. Kuwata and I. Hamachi, J. Am. Chem. Soc., 2018, 140, 17060 CrossRef CAS PubMed.
- S. Feng, T. Harayama, D. Chang, J. T. Hannich, N. Winssinger and H. Riezman, Chem. Sci., 2019, 10, 2253 RSC.
- Y. Yasueda, T. Tamura and I. Hamachi, Chem. Lett., 2016, 45, 265 CrossRef CAS.
- S. S. Lam, J. D. Martell, K. J. Kamer, T. J. Deerinck, M. H. Ellisman, V. K. Mootha and A. Y. Ting, Nat. Methods, 2015, 12, 51 CrossRef CAS.
- D. I. Kim, B. Kc, W. Zhu, K. Motamedchaboki, V. Doye and K. J. Roux, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E2453 CrossRef CAS.
- H.-W. Rhee, P. Zou, N. D. Udeshi, J. D. Martell, V. K. Mootha, S. A. Carr and A. Y. Ting, Science, 2013, 339, 1328 CrossRef CAS.
- V. Hung, S. S. Lam, N. D. Udeshi, T. Svinkina, G. Guzman, V. K. Mootha, S. A. Carr and A. Y. Ting, eLife, 2017, 6, e24463 CrossRef PubMed.
- S. S. Lam, J. D. Martell, K. J. Kamer, T. J. Deerinck, M. H. Ellisman, V. K. Mootha and A. Y. Ting, Nat. Methods, 2015, 12, 51 CrossRef CAS.
- N. Zuzow, A. Ghosh, M. Leonard, J. Liao, B. Yang and E. J. Bennett, Mol. Biol. Cell, 2018, 29, 1258 CrossRef CAS PubMed.
- E. Coyaud, M. Mis, E. M. N. Laurent, W. H. Dunham, A. L. Couzens, M. Robitaille, A.-C. Gingras, S. Angers and B. Raught, Mol. Cell. Proteomics, 2015, 14, 1781 CrossRef CAS.
- T. C. Branon, J. A. Bosch, A. D. Sanchez, N. D. Udeshi, T. Svinkina, S. A. Carr, J. L. Feldman, N. Perrimon and A. Y. Ting, Nat. Biotechnol., 2018, 36, 880 CrossRef CAS.
- Y. Zhang, G. Song, N. K. Lal, U. Nagalakshmi, Y. Li, W. Zheng, P.-j. Huang, T. C. Branon, A. Y. Ting and J. W. Walley, Nat. Commun., 2019, 10, 3252 CrossRef PubMed.
- I. M. Schopp, C. C. Amaya Ramirez, J. Debeljak, E. Kreibich, M. Skribbe, K. Wild and J. Béthune, Nat. Commun., 2017, 8, 15690 CrossRef CAS PubMed.
- J.-M. Heo, N. J. Harper, J. A. Paulo, M. Li, Q. Xu, M. Coughlin, S. J. Elledge and J. W. Harper, Sci. Adv., 2019, 5, eaay4624 CrossRef PubMed.
- M. K. McCoy, A. Kaganovich, I. N. Rudenko, J. Ding and M. R. Cookson, Hum. Mol. Genet., 2014, 23, 145 CrossRef CAS.
- J. Paek, M. Kalocsay, D. P. Staus, L. Wingler, R. Pascolutti, J. A. Paulo, S. P. Gygi and A. C. Kruse, Cell, 2017, 169, 338 CrossRef CAS.
- J. Li, S. Han, H. Li, N. D. Udeshi, T. Svinkina, D. Mani, C. Xu, R. Guajardo, Q. Xie, T. Li, D. J. Luginbuhl, B. Wu, C. N. McLaughlin, A. Xie, P. Kaewsapsak, S. R. Quake, S. A. Carr, A. Y. Ting and L. Luo, Cell, 2020, 180, 373 CrossRef CAS.
- D. Bausch-Fluck, A. Hofmann, T. Bock, A. P. Frei, F. Cerciello, A. Jacobs, H. Moest, U. Omasits, R. L. Gundry, C. Yoon, R. Schiess, A. Schmidt, P. Mirkowska, A. Härtlová, J. E. Van Eyk, J.-P. Bourquin, R. Aebersold, K. R. Boheler, P. Zandstra and B. Wollscheid, PLoS One, 2015, 10, e0121314 CrossRef.
- A. F. Gomes and F. C. Gozzo, J. Mass Spectrom., 2010, 45, 892 CrossRef CAS.
- D. C. McCutcheon, G. Lee, A. J. Carlos, J. E. Montgomery and R. E. Moellering, J. Am. Chem. Soc., 2019, 142, 146 CrossRef.
- D. D. Licatalosi, A. Mele, J. J. Fak, J. Ule, M. Kayikci, S. W. Chi, T. A. Clark, A. C. Schweitzer, J. E. Blume and X. Wang, Nature, 2008, 456, 464 CrossRef CAS.
- S. Parvez, M. J. C. Long, J. R. Poganik and Y. Aye, Chem. Rev., 2018, 118, 8798 CrossRef CAS.
- M. J. Mitchell, R. K. Jain and R. Langer, Nat. Rev. Cancer, 2017, 17, 659 CrossRef CAS.
- S. Parvez, M. J. C. Long, H. Y. Lin, Y. Zhao, J. A. Haegele, V. N. Pham, D. K. Lee and Y. Aye, Nat. Protoc., 2016, 11, 2328 CrossRef CAS PubMed.
- M. J. C. Long, H. Y. Lin, S. Parvez, Y. Zhao, J. R. Poganik, P. Huang and Y. Aye, Cell Chem. Biol., 2017, 24, 944 CrossRef CAS.
- S. L. Surya, M. J. C. Long, D. A. Urul, Y. Zhao, E. J. Mercer, I. M. EIsaid, T. Evans and Y. Aye, ACS Chem. Biol., 2018, 13, 1824 CrossRef CAS PubMed.
- X. Fang, Y. Fu, M. J. C. Long, J. A. Haegele, E. J. Ge, S. Parvez and Y. Aye, J. Am. Chem. Soc., 2013, 135, 14496 CrossRef CAS PubMed.
- S. Parvez, Y. Fu, J. Li, M. J. C. Long, H. Y. Lin, D. K. Lee, G. S. Hu and Y. Aye, J. Am. Chem. Soc., 2015, 137, 10 CrossRef CAS PubMed.
- Y. Zhao, M. J. C. Long, Y. Wang, S. Zhang and Y. Aye, ACS Cent. Sci., 2018, 4, 246 CrossRef CAS PubMed.
- A. V. Hall-Beauvais, Y. Zhao, D. A. Urul, M. J. C. Long and Y. Aye, Curr. Protoc. Chem. Biol., 2018, 10, e43 CrossRef PubMed.
- H. Y. Lin, J. A. Haegele, M. T. Disare, Q. Lin and Y. Aye, J. Am. Chem. Soc., 2015, 137, 6232 CrossRef CAS PubMed.
- J. R. Poganik, M. J. C. Long and Y. Aye, BioEssays, 2018, 40, 1700240 CrossRef PubMed.
- M. J. C. Long, X. Liu and Y. Aye, Curr. Opin. Chem. Biol., 2019, 51, 48 CrossRef CAS.
- M. J. C. Long, D. A. Urul and Y. Aye, in Methods Enzymol., ed. A. K. Shukla, Academic Press, 2020, vol. 633, p. 203 Search PubMed.
- Y. Aye, X. Liu and M. J. C. Long, US Pat., WO2018226794A1, 2018 Search PubMed.
- J. R. Poganik and Y. Aye, Front. Aging Neurosci., 2020, 12, 1 CrossRef PubMed.
- X. Liu, M. J. C. Long, B. Hopkins, C. Luo, L. Wang and Y. Aye, ACS Cent. Sci., 2020 DOI:10.1021/acscentsci.9b00893.
- J. Cheng, Y. H. Fan, X. Xu, H. Zhang, J. Dou, Y. Tang, X. Zhong, Y. Rojas, Y. Yu, Y. Zhao, S. A. Vasudevan, H. Zhang, J. G. Nuchtern, E. S. Kim, X. Chen, F. Lu and J. Yang, Cell Death Dis., 2014, 5, e1079 CrossRef CAS PubMed.
- C. D. Hodge, R. A. Edwards, C. J. Markin, D. McDonald, M. Pulvino, M. S. Y. Huen, J. Zhao, L. Spyracopoulos, M. J. Hendzel and J. N. M. Glover, ACS Chem. Biol., 2015, 10, 1718 CrossRef CAS PubMed.
- T. F. Moraes, R. A. Edwards, S. McKenna, L. Pastushok, W. Xiao, J. M. Glover and M. J. Ellison, Nat. Struct. Biol., 2001, 8, 669 CrossRef CAS PubMed.
- J. R. Poganik, M. J. C. Long and Y. Aye, Trends Biochem. Sci., 2019, 44, 380 CrossRef CAS PubMed.
- C. H. Foyer, A. Baker, M. Wright, I. A. Sparkes, A. Mhamdi, J. H. M. Schippers and F. Van Breusegem, J. Exp. Bot., 2019, 71, 620 CrossRef PubMed.
- I. K. Khan and D. Kihara, Bioinformatics, 2016, 32, 2281 CrossRef CAS PubMed.
- P. J. Thul, L. Åkesson, M. Wiking, D. Mahdessian, A. Geladaki, H. A. Blal, T. Alm, A. Asplund, L. Björk, L. M. Breckels, A. Bäckström, F. Danielsson, L. Fagerberg, J. Fall, L. Gatto, C. Gnann, S. Hober, M. Hjelmare, F. Johansson, S. Lee, C. Lindskog, J. Mulder, C. M. Mulvey, P. Nilsson, P. Oksvold, J. Rockberg, R. Schutten, J. M. Schwenk, Å. Sivertsson, E. Sjöstedt, M. Skogs, C. Stadler, D. P. Sullivan, H. Tegel, C. Winsnes, C. Zhang, M. Zwahlen, A. Mardinoglu, F. Pontén, K. von Feilitzen, K. S. Lilley, M. Uhlén and E. Lundberg, Science, 2017, 356, eaal3321 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |