
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A call for action to the biomaterial community to tackle antimicrobial resistance
Thomas J.
Hall†
 a,
Victor M.
Villapún†
a,
Owen
Addison
b,
Mark A.
Webber
c,
Morgan
Lowther
a,
Sophie E. T.
Louth
a,
Sophie E.
Mountcastle
a,
Mathieu Y.
Brunet
a and
Sophie C.
Cox
*a
a,
Victor M.
Villapún†
a,
Owen
Addison
b,
Mark A.
Webber
c,
Morgan
Lowther
a,
Sophie E. T.
Louth
a,
Sophie E.
Mountcastle
a,
Mathieu Y.
Brunet
a and
Sophie C.
Cox
*a
aSchool of Chemical Engineering, University of Birmingham, Edgbaston B15 2TT, UK. E-mail: S.C.Cox@bham.ac.uk
bFaculty of Dentistry, Oral and Craniofacial Sciences, King's College London, London, SE1 9RT, UK
cQuadram Institute Bioscience, Norwich Research Park, Colney, NR4 7UQ, UK
First published on 21st August 2020
Abstract
The global surge of antimicrobial resistance (AMR) is a major concern for public health and proving to be a key challenge in modern disease treatment, requiring action plans at all levels. Microorganisms regularly and rapidly acquire resistance to antibiotic treatments and new drugs are continuously required. However, the inherent cost and risk to develop such molecules has resulted in a drying of the pipeline with very few compounds currently in development. Over the last two decades, efforts have been made to tackle the main sources of AMR. Nevertheless, these require the involvement of large governmental bodies, further increasing the complexity of the problem. As a group with a long innovation history, the biomaterials community is perfectly situated to push forward novel antimicrobial technologies to combat AMR. Although this involvement has been felt, it is necessary to ensure that the field offers a united front with special focus in areas that will facilitate the development and implementation of such systems. This paper reviews state of the art biomaterials strategies striving to limit AMR. Promising broad-spectrum antimicrobials and device modifications are showcased through two case studies for different applications, namely topical and implantables, demonstrating the potential for a highly efficacious physical and chemical approach. Finally, a critical review on barriers and limitations of these methods has been developed to provide a list of short and long-term focus areas in order to ensure the full potential of the biomaterials community is directed to helping tackle the AMR pandemic.
 Sophie Coxa | Dr Sophie C. Cox is a Lecturer in the School of Chemical Engineering and the Healthcare Technologies Institute at the University of Birmingham. Her vision is to improve patient quality of life by innovating new medical devices with unprecedented functionality. These translational activities are underpinned by basic science focused on understanding the biological response to biomaterials and unearthing mechanisms of action with particular attention on osteogenesis and infection. |
Introduction
Antibiotics are a precious resource capable of specifically targeting microbial infections with little impact on endogenous human cells. Since the discovery of penicillin, antibiotics have been coined ‘wonder drugs’, helping to save hundreds of millions of people.1,2 Today the use of antibiotics is broad and encompasses almost every aspect of our society, including healthcare, the food industry and agriculture. This makes it difficult to imagine a world devoid of such fundamental substances. Nevertheless, as microorganisms continue to develop resistance mechanisms to the use of antibiotics, termed antimicrobial resistance (AMR), this future may become a reality. This includes a rise in the spread of resistance to widely used antibiotics such as vancomycin in Enterococci and an increasing number of pan-drug resistant pathogens.3–5Predictions of the global socioeconomic impact caused by AMR have been estimated as $100 trillion per annum with a potential loss of 10 million lives annually by 2050.6 Although these predictions have been labelled as uncertain and pessimistic,7 AMR is certainly one of the great challenges of our era. The hazard posed by AMR has led to global action since 2001,8 however, it was not until the Global Action Plan on Antimicrobial Resistance was adopted at the 68th World Health Assembly in Geneva (2015) that positive developments have been felt.4,9 It is clear, that if we are to prevent a ‘post-antibiotic’ era then action must be taken at all levels and across multiple disciplines. This review focuses on steps that the biomaterials community, as a key field of innovation for antimicrobial substances, may take to help tackle AMR.
Bacteria have been present on this planet for 3.5 billion years, capable of resisting and flourishing in extreme conditions where more complex life is not able to prosper.10 This adaptability stems from their large populations, rapid division cycles, and the ability to transfer genetic information between species.11 As a consequence, bacteria have evolved a series of complex resistance mechanisms against toxic substances (Fig. 1a).12,13 These tools provide a natural resilience that can overcome antibiotic therapies, a problem noticed by Alexander Flemming shortly after the discovery of penicillin, which led to a warning on handling antibiotic dosages and treatment periods.14 Whilst the mechanisms of AMR demonstrate the adaptability of bacterial species, human overuse and misuse of antibiotics has been largely at fault for selection of resistance mutants and their expansion to high prevalence.
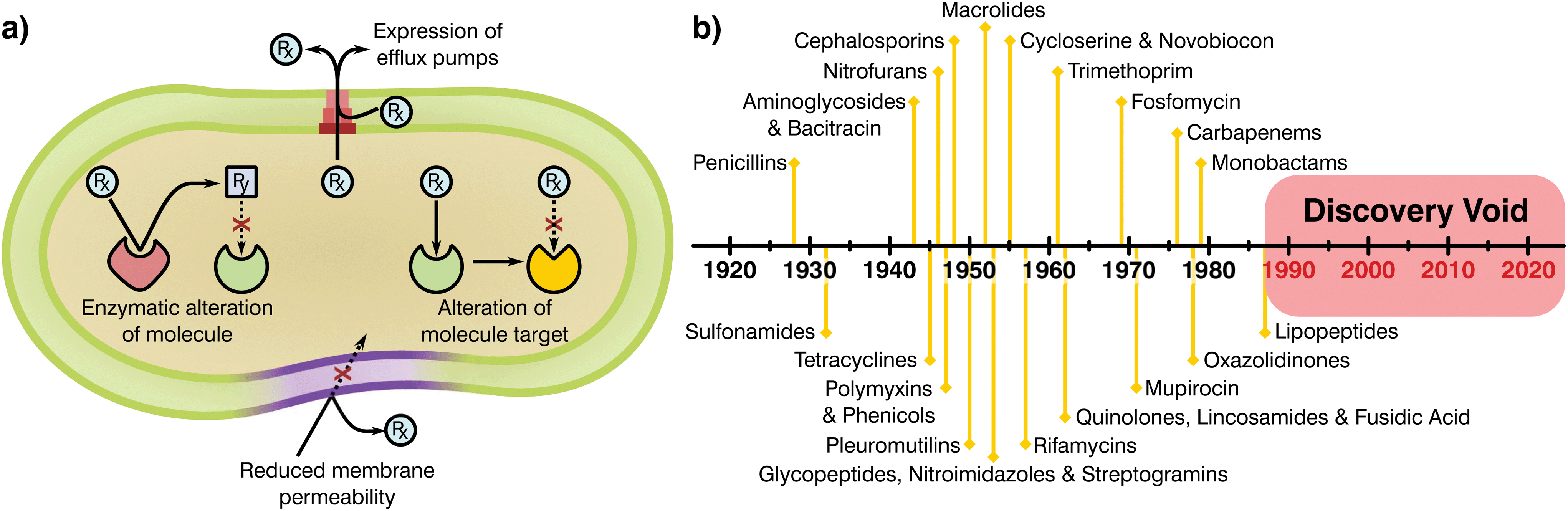 | ||
| Fig. 1 (a) Mechanisms attributed to antibiotic resistance, adapted from ReAct17 and (b) timeline of antibiotic class discovery showing the discovery void of marketed classes.17,18 | ||
Antimicrobials work by inhibiting various essential cellular processes and pathways. Specifically, antibiotics have class dependant modes of action that precisely target an aspect of a bacterial cell's ability to survive (Table 1).15 Most antibiotic classes were discovery between 1928 and 1987 with no new categories reaching the market since (Fig. 1b). In recent years, handling of antibiotics has been poorly managed, with these substances rapidly entering mass production in multiple industries and little control over availability, indications and dosage regimes. Due to this extensive availability and lack of oversight or control on application there has been a great deal of drug overuse which has exacerbated the global AMR problem.16 Understanding of the evolution of resistance in relation to selective pressures has improved since antibiotics were first employed. Specifically, the critical importance of delivering sufficient and lasting antibiotic concentrations to completely eradicate an infection and limiting resistance is today well appreciated. Now more than ever it is clear that the development of novel antimicrobial substances and delivery systems is vital to the containment and prevention of AMR, an area in which the biomaterials field is well placed to have a massive impact.
| Mechanism of action | Antibiotic classes |
|---|---|
| Inhibition of cell wall synthesis | Beta-lactams |
| Glycopeptides | |
| Cyclic lipopeptides | |
| Inhibition of protein synthesis | Tetracyclines |
| Aminoglycosides | |
| Oxazolidinones | |
| Streptogramins | |
| Ketolides | |
| Macrolides | |
| Lincosamides | |
| Inhibition of DNA housekeeping | Fluoroquinolones |
| Inhibition of RNA synthesis | Rifampin |
| Inhibition of folic acid synthesis | Sulphonamides |
| Trimethoprim | |
| Disorganisation of the cell membrane | Polymyxins |
Once novel antimicrobial technologies are discovered, it is necessary to begin the long and risky endeavour of gaining regulatory approval. The prospective damage that potentially hazardous antibiotics could cause to human health has led to a policy of safety first from the European Medicines Agency (EMA) and the Food and Drug Administration (FDA). As a result, the process of taking a newly discovered drug from the laboratory to the market requires extensive preclinical data and three phases of clinical trials before registration of the new product.20–22 Analysis of these pipelines shows that navigating these policies and obtaining regulatory approval can take more than a decade and cost up to $1 billion,20,21,23 generally making it unfeasible for small and medium R&D companies. Thus, antibiotic research has historically been driven by large medical corporations, however, today few incentives are in place to encourage their involvement.21 The main groups of novel antimicrobials were found early after the discovery of penicillin, with most antibiotic classes obtained by empirical screening more than 50 years ago24–26 for which oxazolidinones and cyclic polypeptides were discovered last in the late 70s and 80s, respectively (Fig. 1b).25,27 Thus, all “low hanging fruit” in the production of antimicrobial derivations and combinations may have been tapped. Since the discovery of new classes has slowed, progress has often been made by modifying existing ones to escape class-related resistances28 and the combination of several marketed drugs (i.e. “protecting drugs”)29 to prevent AMR while minimising the risks involved with the development of novel drugs. Although this approach has led to many new antibiotics entering the market, typically the ‘arms race’ continues with resistance emerging to each new derivative necessitating further development and refinement.29,30
The relatively low price and treatment period associated with antibiotics requires a significant, immediate demand of a new drug for a company to invest in its development. Although the number of patients requiring novel drugs for AMR is on the rise, currently the 5% increase in Health Acquired Infections (HAI) is not sufficient to make such an endeavour economically attractive.21 To put this into perspective, a study by the London School of Economics has indicated that the typical net present value (NPV) of an antibiotic is in the region of −$50 million compared, for example, to the development and marketing of drugs for musculoskeletal conditions, which have an NPV of $1.15 billion.31 These factors and statistics reveal that only 1 of 5 infectious disease products that start clinical trials will obtain regulatory approval.26,32,33 Thus, leading to a paradox where new antibiotic drugs are critically needed, while the antibiotic pipeline continues to run dry. As new antimicrobials are found, experts should be aware of the chasm between laboratory discovery and marketing to ensure that promising technologies are pushed forward. This requires building up a strong case to capture investors and relevant stakeholders, for which it is necessary to perform relevant tests to governmental approval and the correlation between in vitro and in vivo models.
The number of R&D teams experienced in discovery of antibiotics has been diminishing over the years as a direct consequence of mergers in large pharmaceutical companies.26,33 Currently only 4 large pharmaceutical companies (Merck & Co., Roche, GlaxoSmithKline and Pfizer) still have active antibiotic programmes with a limited presence of small-medium sized enterprises (SMEs).34 Consequently, there has been a slow and steady loss of knowledge and capacity in antimicrobial development that, coupled with the difficulties inherent to the development of novel antimicrobial substances, has pushed forward translation over innovation.
Sadly, this change in paradigm is not restricted to the antibiotic pipeline and has further been encouraged through modifications in the regulatory approval pipeline. Medical device regulations have changed over the years, leading to the appearance of simpler approval guidelines, such as the premarket notification (PMN) or 510k by the FDA.23 These regulatory pathways rely on the existence of predicate devices that are legally marketed, with similar intended uses, documented proof of substantial equivalences and no detrimental safety and effectiveness differences with the novel product. These normally require an investment between $1–5 million over 3–6 years,23 contrasting with the $45 to $150 million of capital spent over 5–8 years![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 for the more complex Premarket Approval (PMA) where no predicate can be found. This reduction in economic investment coupled with the risk of obtaining PMA approval (only 1% finally enter the market)23 has led the research community to focus their efforts on developing novel delivery systems of already approved substances or reworks of marketed products. This prevalence of translation has restricted innovation primarily to SMEs and universities, enhancing the need for collaboration between the biomaterials community and industrial partners.
35 for the more complex Premarket Approval (PMA) where no predicate can be found. This reduction in economic investment coupled with the risk of obtaining PMA approval (only 1% finally enter the market)23 has led the research community to focus their efforts on developing novel delivery systems of already approved substances or reworks of marketed products. This prevalence of translation has restricted innovation primarily to SMEs and universities, enhancing the need for collaboration between the biomaterials community and industrial partners.
Over the last two decades, the danger posed by AMR in society has been realised, making regulatory bodies take approaches to contain and control AMR drivers.16,36–39 Nevertheless, most changes are slow or must still deal with bureaucratic loops, which limits their application and contributes to the increasingly pressing hazard brought by antibiotic resistant bacteria.23 This slow pace of governance from international regulatory bodies further strengthens the importance of tackling AMR at all levels of the pipeline, including fields that may change the state of play such as biomaterials science. As the main source of innovation, the research community has taken a lead in understanding AMR, their mechanisms and developing novel therapies.
More than 70000 publications linked to AMR have been issued globally, with 8575 made available solely in 2019.40 More interestingly, roughly 8% of these can be correlated with the analysis and advancement of better antimicrobials and therapies by the biomaterials community, showcasing an increasing involvement (Fig. 2). Since the dissemination of the “Global strategy for containment of antimicrobial resistance” by the World Health Organization (WHO) in 2001,8 the number of research publications developed by the biomaterials science field on AMR has risen from 23 to 676 in the 2001 to 2019 period (Fig. 2a). Alongside this the diversity of fields that biomaterials science contributes to has evolved, with microbiology being the largest sector showcasing a willingness to support AMR solutions (Fig. 2b).
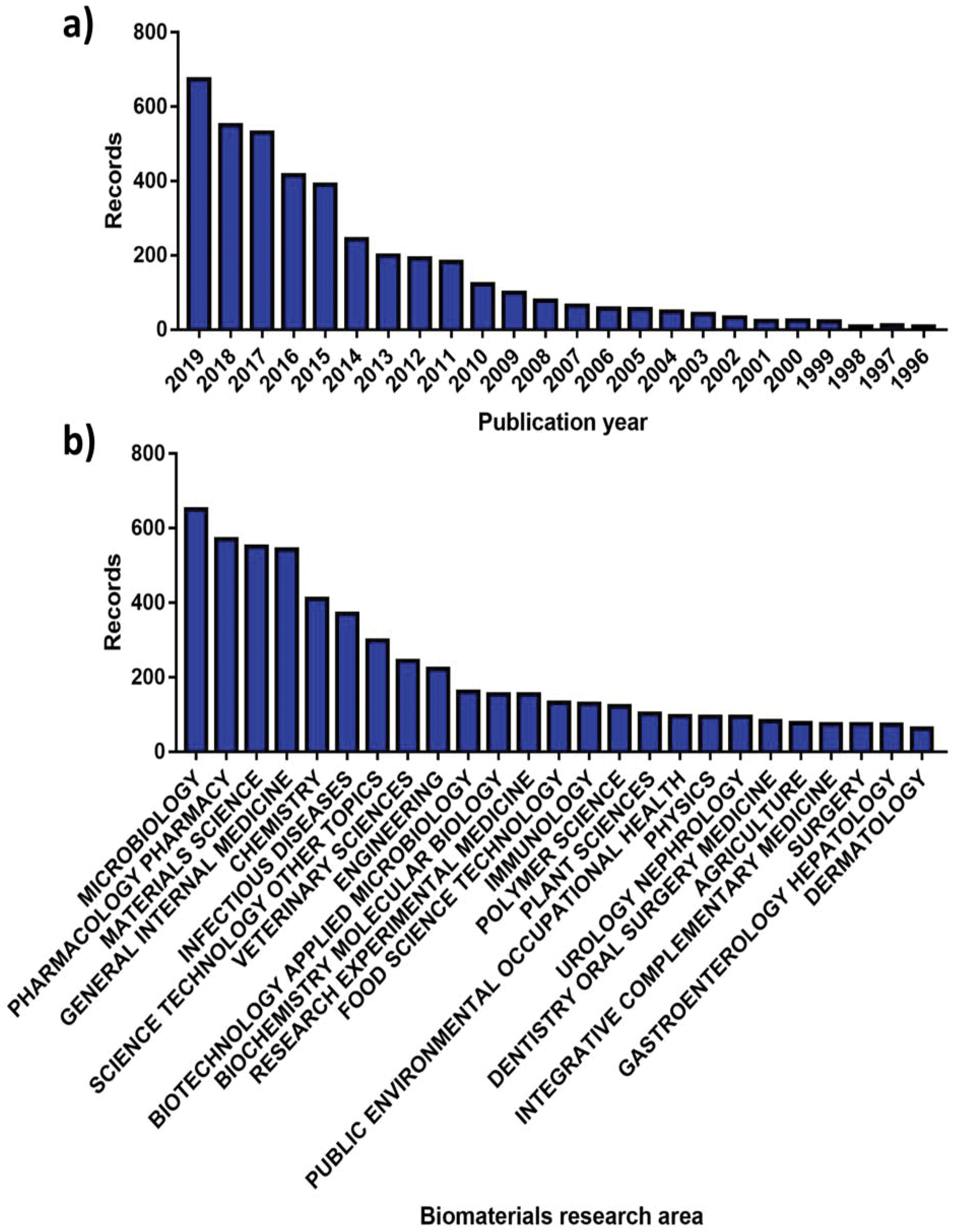 | ||
| Fig. 2 Number of publications for AMR materials ordered by (a) year and (b) research area. Source “Web of Science”. | ||
The aim of this review is to detail the current state of technologies developed by the biomaterials research community to tackle AMR. Two case studies are presented to highlight innovation that is being driven to improve topical and local delivery of antimicrobials. Finally, this work provides a concise list of recommendations to ensure that progress of novel technologies in the AMR field continues and that these game-changing approaches may effectively infiltrate the marketplace.
Novel antibiotics
Since the void of class discovery, numerous analogues of existing antibiotics have successfully been marketed, which has provided a short-term solution to the development of drug resistant bacteria.41 The limited effect of such strategies has already been highlighted by Watson,42 who found that 50% of European deaths associated with infection in 2008, may have been due to resistant bacteria. More recent studies suggest that this trend is on the rise with Cassini et al.43 stating that the burden of resistant infections is now closer to 75% of the total deaths caused by infection in Europe. There is also further concern surrounding the rise of last-line antibiotic resistant bacteria, with 35% of total deaths related to infection in 2019 caused by these species.42 Thus, it is clear that new antimicrobials are needed.Further study within medical services has shown that only 17 different microorganisms, including Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumonaie, Acinetobacter baumannii, Pseudomonas aeruginosa and Escherichia coli (ESKAPE pathogens) account for up to 87% of all HAI.44,45 With AMR increasing and the development of new antibiotics grinding to a halt, to help drive and guide development the WHO published a list of 12 priority pathogens in 2017 (Table 2).46 These are the species most likely to cause a significant impact on human health if resistance continues to develop on its present trajectory. The pathogens are graded from critical to medium with those in the critical category resistant to multiple antimicrobials, including carbapenem (a ‘last resort’ antibiotic), posing an imminent threat to health.46,47 In addition to the organisms prioritised by the WHO, the Centre for Disease Control and Prevention (CDC) also identifies Candida auris and Clostridioides difficile (C. difficile) as urgent threats to healthcare.
| Critical |
| Acinetobacter baumannii – carbapenem resistant |
| Pseudomonas aeruginosa – carbapenem resistant |
| Enterobacteriaceae – carbapenem resistant, extended-spectrum beta-lactamase producing (ESBL) |
| High |
| Enterococcus faecium – vancomycin resistant |
| Staphylococcus aureus – methicillin resistant, vancomycin intermediate and resistant |
| Helicobacter pylori – clarithromycin resistant |
| Campylobacter spp. – fluoroquinolone resistant |
| Salmonellae – fluoroquinolone resistant |
| Neisseria gonorrhoeae – cephalosporin resistant, fluoroquinolone resistant |
| Medium |
| Streptococcus pneumoniae – penicillin non susceptible |
| Haemophilus influenzae – ampicillin resistant |
| Shigella spp. – fluoroquinolone resistant |
According to The Pew Trust, as of December 2019 approximately 41 new antibiotics are in Phases 1–3 of development.48 This suggests that the drug development pipeline has been reinvigorated, although the number of substances that gain approval may of course be much lower. Historic data suggests only 20% of these new antibiotics will make it to market49 with only 11 of these being a novel class or targeting a new mechanism. Furthermore, only 6 of those have expected activity against WHO or CDC priority pathogens (Table 3),48 limiting the prospects of novel antibiotics available to tackle AMR.
| Drug name | Drug class | Target | Activity against ESKAPE pathogens? | Activity against WHO or CDC priority pathogens |
|---|---|---|---|---|
| Gepotidacin | Triazaacenaphthylene | Type II topoisomerase (novel A subunit site) | Yes: S. aureus | Yes: Drug-resistant |
| N. gonorrhoeae | ||||
| Possibly: ESBL | ||||
| Ridinilazole | Bis-benzimidazole | Inhibition of cell division and reduction of toxin production | No | Yes: C. difficile |
| Zoliflodacin | Spiropyrimidinetrione | Type II topoisomerase (GyrB) | Yes: S. aureus | Yes: Drug-resistant |
| N. gonorrhoeae | ||||
| MGB-BP-3 | Distamycin | DNA minor groove binder | Possibly: E. faecium, S. aureus | Yes: C. difficile |
| CRS3123 | Diaryldiamine | Methionyl-tRNA synthetase | Yes: E. faecium, | |
| S. aureus | Yes: C. difficile | |||
| Ibezapolstat (ACX-362E) | Dichlorobenzyl guanine (DCBG) | C. difficile DNA polymerase IIIC | No | Yes: C. difficile |
The discovery of penicillin later led to the creation of many derivatives, built with the intention of circumventing the issue of penicillin-resistant bacteria.50 Further advances were also made in the development of antibiotics delivered in conjunction with anti-bacterial resistance factors. Such an approach was taken by adding clavulanic acid, which inhibits beta-lactamase resistance, to amoxicillin.51 However, there is a limit to the number of different analogues that can be synthesised from a single chemical core and even more so on those that can counteract bacterial resistance mechanisms. Many other antibiotics have undergone analogue development, with some more amenable than others.52 Beta lactams have been the most successfully derived antibiotics with many analogues containing penicillin or cephalosporin cores, although the quinolone and aminoglycoside classes have also provided numerous alternatives.19 Unfortunately, rapid evolution and development of bacterial resistance has been shown in all cases, negating the effects of even the most ingenious medicinal chemistry solutions.19,52
Biomaterials strategies
As previously highlighted, there has been a global culture of misuse and overreliance on antibiotics, surrounded by a ‘magic bullet’ ethos where these compounds were viewed as the only solution to microbial infections. Nonetheless, the pipeline for novel classes of antibiotics is very poorly populated, limited by regulatory processes and the complex mechanisms behind antibiotic resistance. The development of next generation antibiotics is vital to the future of medicine; however, other alternatives and methodologies must be employed in order to both reduce the use of antibiotics and slow the development of resistance. In this regard, the biomaterial community has a long history of developing and innovating solutions to healthcare challenges. Current ongoing research is focused in various different areas including the development of alternative antimicrobials, modification of device surfaces and the improvement of delivery systems (Fig. 3), posing a prime example of game-changing research that this community has to offer against AMR emergence.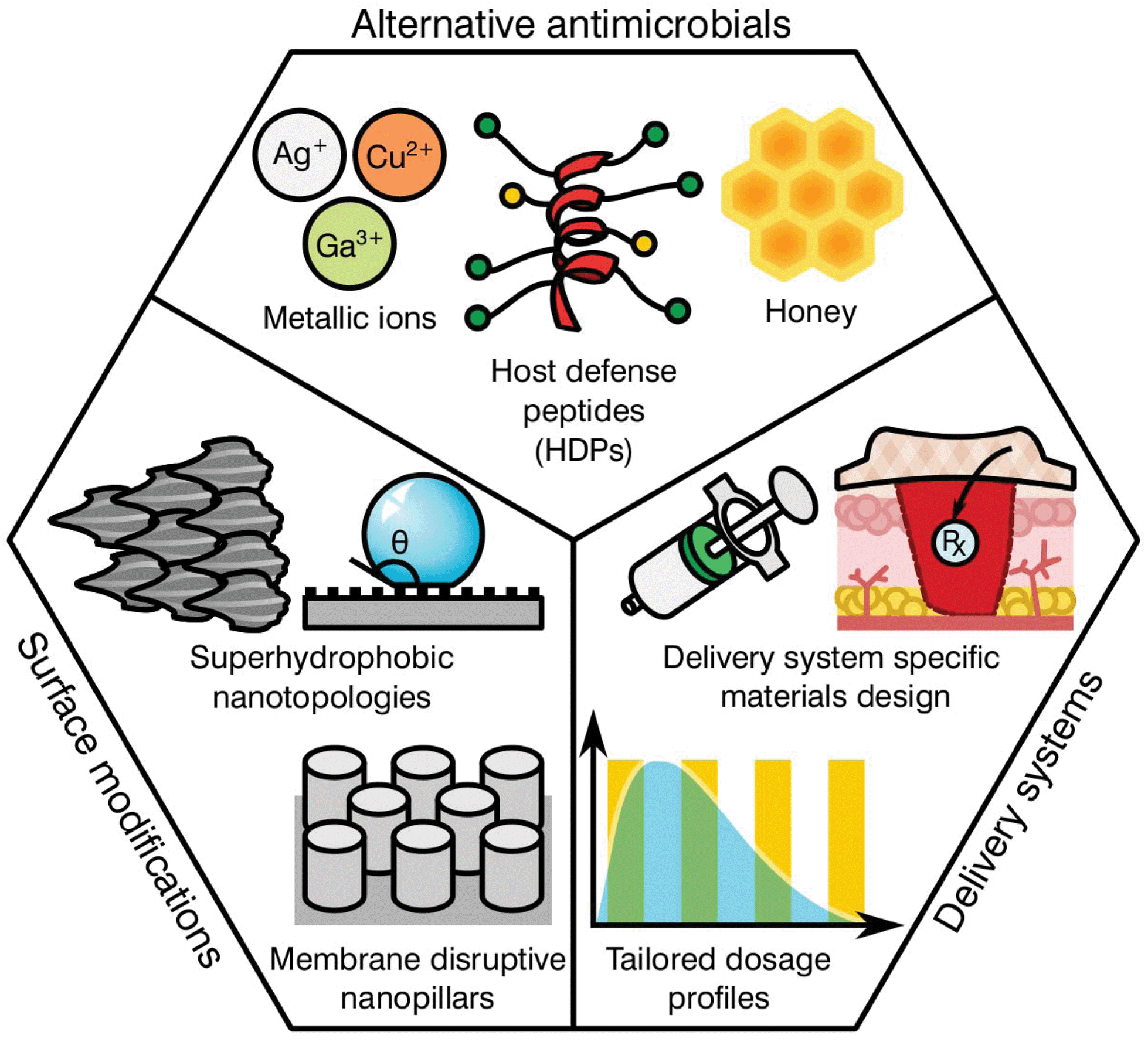 | ||
| Fig. 3 Biomaterial driven strategies for reduction of antimicrobial burden and AMR acquisition. | ||
Alternative antimicrobials
The goal of developing antimicrobial materials is to produce a biocompatible product adept at inhibiting or killing micro-organisms that may colonise the surface of a material or the surrounding area without the development of resistance. Before the discovery and introduction of antibiotics in medicine, natural antimicrobials such as metallic ions and honey were routinely used for the treatment and prevention of infection, with use dating back millennia.53–55 During the upcoming of the antibiotic era these substances where rapidly substituted, however, renewed interest in such materials is motivated by a hope to reduce the current burden of AMR on global healthcare.Unlike traditional antibiotics that have specific modes of action and provide an ease of progressive resistance, both antimicrobial metallic ions and honey target multiple cellular processes and lead to pleiotropic effects within the target organism.56–58 In the case of metallic ions, it has been shown that these certain elements, including silver, copper or arsenic, are capable of interfering with enzyme activities, disrupting membrane functions or damaging DNA, complicating resistance development.59,60 For most transition metals, this is further enhanced by involvement in reactive oxygen species (ROS) generation, which elicit its antimicrobial action through reactions with thiol groups, oxidation of cell walls and membranes, inhibition of protein synthesis and disruption of electron transport.61–65 These mechanisms have encouraged research on transition metals of the d-block, (i.e. Cu, Zn, Ag, Cd) and other metals and metalloids from groups 13–16 (i.e. Al, Ga, Ge, As).60 Nonetheless, the potential toxicity of such elements to human tissue has limited the applicability of most metals. This is the case for copper, with surfaces of the pure element capable of killing bacteria in mere minutes.66 However, its prospective cytotoxicity has reduced its wider implementation, limiting it to niche uses like touch surfaces.55 Other antimicrobial metals with high potency that are safer to the human body have been historically preferred, silver being the most prominently used in healthcare applications.67
More recent developments have seen the formulation of other antimicrobial compounds for which polymers and peptides have awakened the interest of the biomaterial community. Antimicrobial polymers are emerging within the biomedical field as a direct consequence of their wide applications in the treatment and prevention of infection, either alone or in conjunction with other active substances.68 The mechanism of action is dependent on formulation; however the majority of polymers interact with the charge on the surface of the microbial cell, disrupting transport, inhibiting protein synthesis and in some cases stripping the membrane completely causing contents leakage.69 Common antimicrobial polymers include chitosan, heparin, ε-polylysine, polyacrylamides, polyacrylates, polysiloxanes, polyionenes and polyoxazolines for which thorough reviews are available in the literature.70,71
Similar to the mechanisms of action for antimicrobial polymers, antimicrobial peptides (AMPs) tend to target the cell membrane of microbes, however, they may also disrupt cellular metabolism and target components within the cytoplasm.72 AMP's are usually amphiphilic consisting of both hydrophilic and hydrophobic groups. This grants the molecule solubility in aqueous environments and allows passage through lipophilic membranes73 with different formulations available (Table 4).
| Antimicrobial peptide | Antimicrobial activity | Ref. |
|---|---|---|
| Magainin | Bacteria, fungi, viruses | 79–81 |
| Cecropin | Bacteria, fungi, viruses | 82–84 |
| Brevinin-1 | Fungi, and viruses | 85 and 86 |
| PMAP-23 | Fungi | 79 and 87 |
| Protegrin | Bacteria and viruses | 80 and 81 |
| Dermaseptin | Viruses | 82 and 83 |
| Tachyplesin | Viruses | 84 and 85 |
| Polyphemusin | Viruses | 86 and 88 |
| Tenecin-3 | Fungi | 89 |
| PR-39 | Bacteria | 90 |
The attractiveness of AMP's as versatile antimicrobial agents has been further increased by their influence in various biological processes, including immune and anti-inflammatory responses, causing a recent shift in terminology to host defence peptides (HDPs) to recognise their multi-functional nature.74 As a consequence, the development of HDPs has progressed from simple contact killing or biocide releasing materials common in first generation polycations derived from quaternary ammonium compounds (QACs)75 to more complex multifunctional antimicrobials such as multifunctional hyperbranched polyaminoglycoside for improved antibacterial activity, biocompatibility, and gene delivery.76 A specific notable example of the flexibility of more modern HDPs can be seen in the nylon-3 polymers development by Liu et al.77 These multifunctional polymers are capable of fine tuning the hydrophobicity, charge density, and conformational propensity of the material to selectively target specific bacteria while providing low hemolysis and minimal cytotoxicity. Further broad advancements in HDPs may be found in reviews by Ding et al.,68 Haney et al.74 and Ge et al.78 Albeit promising, historically these types of antimicrobials have had significant disadvantages, such as short or limited periods of activity, tissue toxicity, high manufacturing costs and a poor ability to function in an ever changing healthcare environment.73 However, given the current global AMR problem, these technologies are re-emerging within the biomaterials community with hope that they offer a new generation of antimicrobials and antimicrobial devices.
To overcome the inherent issues of HDPs, recent advances have been made in synthetic forms that are able to emulate specific amino acid sidechains, overall charge, molecular weight and three dimensional structures, retaining the properties of their natural counterparts.91 Of particular utility is the ability to modify the properties of these synthetic HDPs to enhance therapeutic outcomes. Some examples are the helical, cationic polypeptides developed by Xiong et al.92 or the structurally nanoengineered antimicrobial peptide polymers (SNAPPs) reported by Lam et al.93 and Shirbin et al.,94 capable of selectively eliminating bacteria while reducing side effects to mammalian cells. Further research in this area has also demonstrated the ability of polypeptides to include alternative killing mechanisms to conventional HDP structures. Such is the case of poly(2-oxazoline) (POX) a mimicking peptide that enacts its antimicrobial effect by promoting reactive oxygen species (ROS), limiting the prospects of AMR development.95 These examples clearly highlight the novelty and growing potential of HDPs, for which more detailed information can be found in the literature.68,96,97
Surface modifications
Biomedical devices are a crucial part of modern medicine and the global use of for example stents, valves, artificial joints and other implantables is increasing yearly.98 However implanted device surfaces are susceptible to bacterial adhesion and biofilm growth, potentially compromising the materials effectiveness.99 Such infections frequently fail to respond to systemic antibiotics and require invasive revision surgeries consisting of debridement in conjunction with long-term antibiotic treatment. These revision procedures have a success rate between 30 to 50%100,101 with detrimental effects on the patient's recovery prospects alongside a heavy economic impact to the healthcare system. It has been found that the surface topography and degree of implant roughness has a significant effect on the attachment of micro-organisms, and therefore on biofilm formation.102,103 Other factors that further dictate colonisation may include electrostatic interaction, wettability, van der Waals forces and steric hindrance.104,105 Thus, the development of surfaces capable of preventing bacterial attachment and proliferation has become a relevant niche for biomaterial scientists.
Several studies have attempted to mimic the nano-textured topologies present in nature that demonstrate inherent anti-attachment properties leading to anti-biofouling or bactericidal surfaces.105 Some examples of plant and animal species exhibiting these characteristics can be found on the leaves of taro and lotus plants as well as shark skin.106,107 These surfaces prevent bacterial colonization through superhydrophobic structures and anisotropic flow patterns at both micro and nano scales, which allow for the repulsion of microbes on the surface.108,109 Consequently, these topologies confer a “self-cleaning” ability, reducing further proliferation of bacterial species. In contrast to antifouling, other natural topologies can interact with the cell membrane of the deposited bacteria, leading to its disruption and cell death. This is the case for the cicada wings and the skin of geckos110–113 that exhibit nano-scale pillar structures capable of piercing the cell walls of microorganisms.114,115 Interestingly, both antifouling and antibacterial properties can be present on the same surface. Some species of dragonfly combine both mechanisms on their wings for high anti-attachment and bactericidal effects.116 Nevertheless, microbial attachment is a complex process dependent on numerous physicochemical properties and differs between bacterial species with limited knowledge available in the literature. Research by Ginestra et al.117 and Villapún et al.118 both discuss the necessity of combining physical and chemical approaches to effectively prevent and treat medical device infections. The current research on metallic implants is pointing out the possibility of fine tuning the topology and chemistry of biomaterials with natural antimicrobial properties to increase effectiveness.118,119 Thus, surface alteration of inert and antibacterial surfaces is becoming more prominent in the biomaterial community.
Antimicrobial delivery systems
The complex problem of AMR is likely to require a multi-faceted approach from the biomaterials community with considerations taken at many different levels.120 One area of concern is the delivery of antimicrobials and how dose and material release kinetics are optimised to minimise resistance (Fig. 4). The mutant selection hypothesis121,122 theorises that antimicrobial resistant subpopulations present before the start of a treatment regime are enriched and amplified when dosing commences if the concentration falls within a specific range (Fig. 4a), known as the mutant selection window. The upper limit of this range is determined by the mutant prevention concentration (MPC), which represents the minimum inhibitory concentration (MIC) of the test antimicrobial against the least susceptible mutant present within the population.123 The lower limit of the mutant selection window is determined by the lowest concentration that is still able to exert selective pressure on the microorganism.124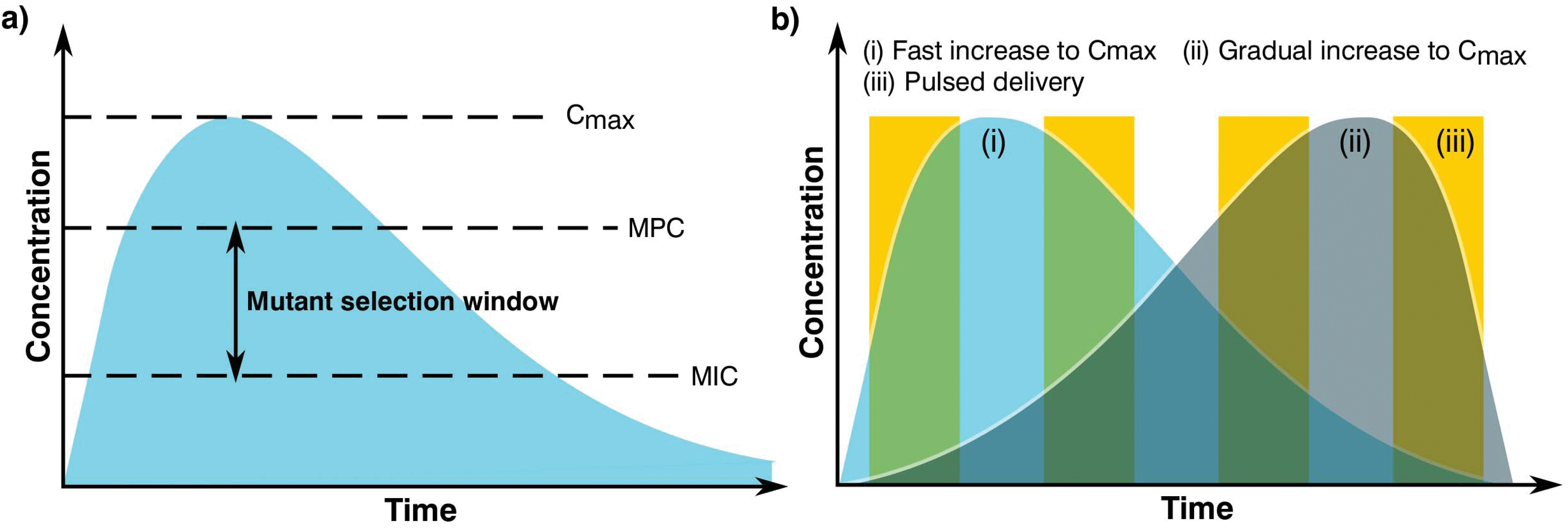 | ||
| Fig. 4 Graphs showing (a) definition of the mutant selection window for an antimicrobial dosage and (b) dosage regimes. | ||
From the mutant selection hypothesis, the biomaterials community can take two main principles. Firstly, traditional antimicrobial dosing strategies that aim to stop the development of resistance by killing susceptible cells still allow for the enrichment of resistant pathogens when the concentration of the treatment falls within that window.125 Secondly, if the maximum concentration (Cmax) of the antimicrobial exceeds and maintains above the mutant selection window, it should restrict the development of AMR for the treatment regime.124 These two principles are critical to the design of biomaterials and the dosing strategies they permit in order to treat infections not already resistant to the antimicrobial being used.
Although seemingly straightforward, application of the mutant selection hypothesis is not so simple due to the large doses required (>MPC). It is necessary to consider that the concentrations required to minimise resistance development are often far higher than what is needed to treat the infection. At these high doses, antimicrobials may also present a number of side effects to the patient with a trade-off between an increased risk of toxicity in exchange for the reduced likelihood of developing a resistant infection.124 Moreover, if the antimicrobial is dosed infrequently or the components are long lasting the concentration may end up falling within the mutant selection window. It is expected that AMR will develop faster in these instances than those microorganisms treated more regularly with concentrations maintained above the mutant selection window.126,127 As such, the selection of an optimal dosing profile for the biomaterial is crucial in AMR prevention.
Generally, there are two prevalent pharmacokinetic dosing profiles (Fig. 4b), those which begin at a high concentration and gradually decrease (Fig. 4b(i)) and those that start at a low concentration and gradually increase (Fig. 4b(ii)). The first profile is often preferred since it minimises the chance of developing microbial resistance, however as the concentration of the dose gradually begins to fall, the time spent close to or within the mutant selection window increases.125 An alternative solution of a short pulse kinetic profile has been proposed by Baker et al.,128 aiming to minimise the time that the organism has to select for resistance while simultaneously reducing pathogen abundance due to the high antimicrobial concentration (Fig. 4b(iii)). To further add to these dosing profiles, it may also be beneficial to use combination therapies involving two or more antimicrobials.129 This would allow high levels of protection to be maintained since when one concentration of antimicrobial falls below that of the MPC, another takes its place, providing good pharmacodynamic overlap. It is also worth noting that AMR can be selected during treatment, therefore this type of therapy may also be useful when antimicrobial-resistant genes are transmissible and can be gained by an infecting bacterial population.130–132 However, difficulties in joint dosing, toxicology monitoring and pharmacokinetics make combined administration complicated.
Based on the impact of AMR and the limited prospects of future antibiotics being marketed, it is critical to develop novel antimicrobial substances and remodel current delivery systems to prevent resistance acquisition. To showcase the promising solutions identified by the biomaterials field, two case studies have been prepared. The first highlights research into dermal systems, while the second focuses on bone cement, revealing differences in approach linked to specific body areas. At the same time, limitations in current methods and attitudes will be introduced to propose recommendations for the development of a united front within the biomaterials field against AMR.
Case study I: topical applications
Topical wound care has improved and evolved over many years with a shift in practise from drying the wound site using absorbent gauze to strategies that focus on maintaining a moist healing environment.133 This change in therapeutic process was brought about by observations made in 1962 by Winter134 and later supported by Cho et al.135 in 1998, where studies found that occluded wounds required less time for reepithelialisation than those allowed to air dry. By using a closed dressing, the wound is continuously exposed to growth factors, proteinases and chemotactic factors.136 This type of environment also preserves the electrical gradient across the wound site, allowing for the stimulation of fibroblasts and permitting epithelial cell migration.133Over the last decades, these134,135 findings have pushed forward the development of different biomaterials for wound healing. Today, dressings are often divided into two categories passive and interactive. Passive dressings, such as a gauze, deliver no clinically recognised effect on the wound itself other than providing protection from the environment.137 Interactive dressings are designed to actively stimulate the wound bed to support optimal healing conditions.133 The added functionality of interactive dressing have made it a focus point for the biomaterials community with different strategies being developed to manipulate the wound environment, which now extends beyond speeding up re-epithelialisation. Other promising functionalities that have been engineered include stimulation of collagen synthesis, formation of a hypoxic environment to promote angiogenesis and strategies to decrease topical pH, which may reduce infection incidence.138,139 In addition, modern dressing designs also factor in patient comfort, convenience of application and cosmetic outcomes, such as the reduction of scarring.140–142
With the realisation that maintaining a moist environment is beneficial for wound healing, some products have been designed to be moisture retentive, which may be quantified using the moisture vapour transmissions rate (MVTR). Dressings that have a MVTR of <840 g m−2 over a 24 hours period are described as moisture retentive.143,144 This has led to a multitude of products on the market; however, it is noted that none can be described as ideal for all wounds (Table 5).
| Dressing type | Commercially available products | Characteristics |
|---|---|---|
| Gauze | Curity, Vaseline Gause, Xeroform | Can dry wounds |
| Films | Bioclusive, Blisterfilm, Cutifilm, Flecigrid, OpSite, Tegaderm | Retains moisture |
| Hydrocolloids | Aquacel, Comfeel, DuoDERM, Granuflex, Tegasorb | Traps fluid |
| Hydrogels | Carrasyn, Curagel, Nu-gel, Purilon, Restore, SAF-gel, XCell | Rehydrates wound |
| Foams | 3 M Adhesive Foam, Allevyn, Lyofoam, Tielle | Moderately absorbent |
| Alginates | Algisite, Kaltostat, Sorbsan, Tegagen | Highly absorbent, acts as a haemostatic agent |
| Fibre mat | Aquacel Hydrofibre | Highly absorbent |
| Skin substitutes | Alloderm, Apligraf, Biobrane, Bioseed, Dermagraft, Epicel, EZ Derm, Hyalograft, Integra omnigraft, Laserskin, Myskin, TransCyte | Provides growth factors and cytokines |
Although modern occlusive dressings aim to reduce microbial colonisation of the wound by employing mechanisms to reduce pH and create unfavourable conditions for microbial growth, topical infection is still a huge clinical problem. Infections have historically, in the pre-antibiotic era, been one of the leading causes of death globally with dermal trauma (i.e. burns, insect bite, surgical wounds or even a simple scratch) often proving fatal if infected.146,147 After the discovery of penicillin, the number of individuals dying from topical infections significantly reduced, with current estimates suggesting that only 7–10% of hospitalised patients are affected with dermal infections.148 Within a healthcare environment one of the leading causes of topical infections can be found at a surgical site (surgical site infection – SSI). This can occur in up to 5% of patients undergoing ‘clean’ surgeries and up to 20% in those undergoing intra-abdominal surgeries, affecting upwards of 300000 people annually in the United States alone.149,150 Among the increased morbidity associated with an SSI, it also delays healing, which may prolong hospital stays and return to normal activities causing further knock on negative effects to healthcare resources, the economy and patient well-being.
One of the most common pathogens associated with SSIs is S. aureus, which can colonise and be carried by healthy individuals, often in the nasal cavities without detrimental effect.151,152 These bacteria can be found relatively easily, with 20% of healthy people designated as chronic carriers, 30% intermitted carriers and 50% not susceptible.152 More distressing is that carriers are up to 12 times more likely to contract an infection than non-carriers.148 To prevent and treat such infections, the biomaterials community have sought to functionalise topical dressing with various antibiotics, including ciprofloxacin (CIP), tetracycline, mupirocin, fusidic acid and gentamicin are commonly used153,154 (Table 6). This is exemplified by the work of García et al.155 whereby 1% chitosan films were modified using weisocyanate to provide a carrier for CIP. These films were found to inhibit bacterial growth with activity levels proportional to antibiotic content. Similar therapeutic responses had been reported by Li et al.156 who developed CIP loaded electrospun fibre mats using thermoresponsive polymers poly(N-isopropylacrylamide) and poly(l-lactic acid-co-ε-caprolactone)(PCL). Alternative approaches include cellulose scaffolds loaded with tetracycline hydrochloride with the ability to inhibit and reduce the growth of bacteria as tested by disc diffusion and plate count methodologies.157 Furthermore, hydrogel dressings have also been investigated for their ability to be functionalised to deliver tetracycline, as demonstrated by Chen et al.158 In this work, it was shown that alginate-chitosan hydrogel dressings with incorporated gelatin microspheres containing tetracycline hydrochloride provide a more sustained controlled release, fundamental for AMR prevention. Interestingly, a semi-derivative of tetracycline (tigecycline) was developed to treat skin and soft tissue infections, which avoids the efflux-mediated resistance mechanism exhibited by both Gram positive and Gram negative bacteria.159,160 Dhanalakshmi et al.161 formulated chitosan nanoparticles loaded with tigecycline and coated with lectin to provide a more sustained antimicrobial release profile. More recently Nimal et al.162 further developed this formulation by incorporating tigecycline-chitosan nano particles into a platelet rich plasma hydrogel. This system demonstrated improved antibacterial efficacy in comparison to other wound dressings. Thus, it is clear that wound dressings have been a particular medium used by the biomaterials community to deliver antimicrobial molecules.
| Antibiotic | Dressing type | Materials | Ref. |
|---|---|---|---|
| Ampicillin | Electrospun fibre mat | PCL | 163 |
| Hydrogel | PVA/alginate | 164 | |
| Cefazolin | Electrospun fibre mat | Gelatin | 165 |
| Ceftazidime | Electrospun fibre mat | Silk fibroin/gelatin | 166 |
| Film | Guar gum/ethylenediamine/collagen | 167 | |
| Ciprofloxacin | Electrospun fibre mat | Polyvinylpyrrolidone | 168 |
| Polyurethane/dextran | 169 | ||
| Doxycycline | Film | Collagen/gelatin | 170 |
| Gentamicin | Electrospun fibre mat | Chitosan | 171 |
| Moxifloxacin | Electrospun fibre mat | PVA/alginate | 172 |
| Neomycin | Electrospun fibre mat | Poly(styrene sulfonic acid-co-maleic acid)/PVA | 173 |
| Norfloxacin | Film | Chitosan | 174 |
| Streptomycin | Electrospun fibre mat | Polyurethane/cellulose acetate/zein | 175 |
| Hydrogel | PVA/cellulose | 176 | |
| Sulfadiazine | Film | Alginate/cellulose | 177 |
| Electrospun fibre mat | PCL/PVA | 178 | |
| Film | Alginate/cellulose | 177 | |
| Sulfanilamide | Fibre mat | Alginate | 179 |
| Tetracycline hydrochloride | Film | Cellulose | 157 |
| Hydrogel | Alginate/chitosan/gelatin | 158 | |
| Vancomycin | Hydrogel | Silk fibroin/gelatin | 180 |
| Film | Alginate/gelatin/halloysite nanotubes | 181 |
Delivering antibiotics locally to wound sites could be preferential to systemic deliver for a number of reasons. Antibiotics need to be dosed to achieve a sufficient level of systemic efficiency, which can often lead to toxic reactions such as that of cumulative organ and cell toxicity. Topical delivery can provide better tissue compatibility, a lower occurrence of resistance and less wound healing interference.153 In addition, the use of lower doses reduces the risks associated with systemic toxicity and can overcome problems related to poor blood circulation such as that found in wounds located in the body's extremities.182 However, there is increasing concern around the development and subsequent threat caused by AMR. As such the healthcare industry has seen a revived interest in promising natural topical wound care agents, such as silver and honey, for both prophylactic and therapeutic use.183
For millennia honey has been used to treat topical wound infections with AMR not reported in the available literature.184–187 The antimicrobial effects of honey are often attributed to a low pH (pH 3.2–4.5), high osmolarity and more notably the presence of peroxide or non-peroxide components.57,58 Non-peroxide honeys, such as Manuka are monofloral and dependant on one source of nectar, in this case from the Manuka tree.188 Increased levels of methylglyoxal (MGO), methylsyringate and bee defensin-1 are found within this type of honey, which results in a strong antimicrobial effect.189–191
Alternatively, there are other honey systems that instead of MGO utilise hydrogen peroxide (H2O2) and other ROS to produce an antimicrobial response. This compound arguably provides honey with its most potent antimicrobial mechanism.192 Similar to MGO in Manuka honey, ROS has also been found to support wound healing, specifically the process of angiogenesis, and to act as an anti-inflammatory.193–196 However, ROS is produced in the presence of water and therefore as a product makes it water sensitive and less stable than Manuka, further introducing batch to batch variation and limiting clinical adoption.
The bactericidal behaviour of Manuka and ROS producing honey, both in vitro and in vivo, has been extensively researched and against a diverse range of pathogenic organisms. This includes WHO priority pathogens such as MRSA and vancomycin-resistant Enterococcus faecium,197–203 increasing the interest in its application to tackle AMR. Further to testing efficacy, attempts were made to generate strains of bacteria resistant to Manuka honey, however it was unsuccessful with no reports of clinical isolates that acquired resistance by both Blair et al.204 and Cooper et al.187 In addition to inhibiting planktonic cells, Manuka and ROS honey was also found to disperse, disrupt and kill bacteria within forming and established biofilms. This included preventing the formation of biofilms of Staphylococcus and Streptococcus species, Pseudomonas aeruginosa, Escherichia coli, Acinetobacter baumannii and Klebsiella pneumonia, clearly indicating strong clinical potential.200,205–208
Albeit their antimicrobial properties, the advent of highly efficacious antibiotics in the 20th century led to a diminished interest in applying honey in wound care. A key explanation for the reduced use of honey as a wound care treatment, like most naturally occurring antimicrobials, is its physical properties and characteristics, which are not compatible with modern day medicine or suitable for delivery in a clinical setting. With the rapid emergence of AMR the biomaterials community has taken a step forward to formulate delivery systems for natural antimicrobials that exist in undesirable forms. Medical grade Manuka honeys, such as Activon and Medihoney, are commonly used to impregnate or coat wound dressings. This is exemplified through the work of Taher et al.209 and Kamaratos et al.,210 which utilise nanocellulose and tulle dressing respectively as a carrier for honey products. On the other hand, research such as that by Mancuso et al.,211 has taken this a step further through the development of biomimetic meshes containing Manuka honey. This work demonstrated the potential to develop a layer-by-layer assembly of nanocoated poly(ε-caprolactone) mesh, which elicited biocompatibility and antimicrobial behaviour.
Despite the water sensitivity of ROS producing honey biomaterial scientists have pushed forward the development of novel strategies to increase the presence of this type of honey in wound care. Research by Febriyenti et al.212 and Khounganian et al.213 explored formulating honey gels in order to ease delivery and stimulate wound healing. In both cases ROS producing honey gel shortened the wound healing time significantly and produced no negative side effects when compared with untreated and nondisclosed commercial products. However, the antimicrobial capacity of the formulation was not addressed. In order to realise the potential of ROS producing honey as a viable topical antimicrobial treatment, it must be capable of delivering specific and controlled amounts of the antimicrobial agent over a clinically relevant period. Surgihoney™ (SHRO), a bioengineered honey has been formulated to enable delivery of consistent antimicrobial doses.214
The antimicrobial effect of SHRO has also been demonstrated with clinical tests highlights its ability to prevent and treat SSIs as well as Hickman and Vascular line infections without the need for antibiotic administration.215,216 As aforementioned, SHRO is an engineered honey, allowing the potency to be tailored as required. This ‘dialling’ enabled SHRO to be modified such that it outperformed other medical grade honeys, including Activon and Medihoney, and two of the most commonly used antimicrobials dressings, silver and iodine.197 Although SHRO addresses the need to provide a medical grade honey product that can deliver a consistent dose of antimicrobial activity, its uptake clinically, much like Manuka honey, is hindered by its viscous and adherent properties. Therefore, in order for honey to become a more viable clinical treatment new delivery systems must be developed where the biomaterials community has significant expertise. To date research by Hall et al.,202 has demonstrated a clinically efficacious emulsion system capable of easing the delivery of SHRO using an inversion trigger. This showcases that by engineering biomaterial delivery systems with appropriate physicochemical properties we may be able to better harness the antimicrobial efficacy of honey.
Furthermore, as well as using honey as a sole antimicrobial agent, its interactions with conventional antibiotics has revealed some promising results. Particular value may be sought in the topical application of honey to a wound alongside systemic delivery of an antibiotic. Jenkins et al.217 noted synergistic effects between Manuka honey and a range of antibiotics, including oxacillin, tetracycline, imipenem and mupirocin against strains of meticillin-resistant Staphylococcus aureus (MRSA). Interestingly, it was found that the addition of honey caused the down regulation of mecR1, a gene that encodes for an MRSA specific penicillin binding protein (PBP2A) and in combination with oxacillin made MRSA once more susceptible to the antibiotic. Furthermore, Müller et al.218 discovered strong synergistic activity of Manuka honey and rifampicin against numerous Staphylococcus aureus strains, including clinical isolates of MRSA. It was also found that the presence of Manuka honey prevented the development of rifampicin resistance in vitro. However, despite displaying a strong clinical potential, inconsistency in efficacy due to the natural origin of Manuka honey and its physical characteristics make consistent application difficult and appear to limit healthcare adoption.
It is clear that the biomaterials community plays a critical role in developing more clinically useful systems that maintain the efficacy of natural antimicrobials, such as honey. Future considerations should focus on developing biomaterials reflecting on how dosage may be controlled to maintain concentrations above the MPC. With the aid of the biomaterial's community further development of efficacious treatments can be achieved while also endearing to learn from our previous use of antibiotics.
Case study II: bone cements
The prospect of an infected device caused by an antibiotic resistance species is of great relevance to implantable technologies. In topical applications, changes of antimicrobial agent can be easily performed. However, access to an implantable for modification of ongoing therapeutics is complex and costly. Alongside the biological and mechanical requirements of these systems, implantable devices require special considerations from the biomaterials community.Since the first successful long bone reconstructions performed in the 19th century, the field of orthopaedics has evolved through the implementation of novel techniques and biomaterials, nevertheless, infection is still a major concern in the 21st century.219 Between 1 to 5% of contemporary indwelling prostheses have been shown to develop an infection. Frequently removal of the implanted device and debridement of the surrounding tissue is required.220 During revision a patient's movement may be impaired or limited, which can impact long-term recovery, increase healthcare costs and reduce economic contributions. More worrying is the adaptability showcased by bacterial isolates present in these clinical settings. Pathogens recovered from implantable orthopaedic devices are often characterized by virulence factors that encourage adhesion and biofilm formation.221,222 Once such a community environment is formed, the effectiveness of antibiotic treatment is greatly reduced, leading to a higher infection risk with only 30 to 50% of revision surgeries successful in removing the infection source.100,223,224 The consequences of these hard to treat infections has resulted in the emergence of complementary therapies to antibiotic prophylaxis. The prime example of this approach is antibiotic-loaded bone cement that enables localised delivery of cargo to the implant surface and adjacent tissue.
Historically, the term bone cement has been used as a synonym for polymethyl methacrylate (PMMA) based formulations. These materials are commonly supplied as a liquid monomer and powder polymer with mixing resulting in the formation of a tacky substance that is easily manipulated and formed.225,226 Over the years, more complex formulations have been developed, generally including additives to prevent leaching of the liquid monomer, stabilization under light and heat, to facilitate cold curing, and increase imagining contrast or render the cement radiopaque. As a result, PMMA cements are a versatile biomaterial for prosthetic cementations, spacer manufacture and reconstructions with commercial products available since the early 70s.227,228 More recently, there has been a special interest in the incorporation of antibiotics during the mixing process. However, the inherent exothermic reaction of PMMA coupled with the impact of antibiotic selection and dosage on the physicochemical properties of the cement requires careful consideration. The selected antimicrobial molecule must be chemically and thermally stable to maintain its antibacterial efficacy after curing. Moreover, it has been suggested that the antibiotic should be provided in powder form to facilitate integration during mixing and minimise the influence on mechanical properties.226,229,230 This has led to a preference in the use of vancomycin, gentamicin, erythromycin, colistin, tobramycin, clindamycin or fusidic acid with an inclination for mixtures between different antibiotics for improved bacterial spectrum.227,228,231 Dosage selection must be high enough to ensure an effective therapy with minimal reduction in mechanical properties and limited detrimental effect on the human body. For this purpose, it is a common rule of thumb to incorporate more than 2 g per 40 g of cement (up to 6 or 8 g) for acute infections, while less than 2 g of antibiotic is acceptable for prophylaxis of first implants.226,227,232,233 The addition of such substances is commonly done in theatre, however the approval of low-dose antibiotic loaded bone cements by the FDA for second-stage reimplantation after infected arthroplasties in 2003 has led to commercially available pre-loaded products from Howmedica (Antibiotic Simplex), DePuy (CMW1 and CMW 3G), Merck (Copal and Palamed G) and Schering-Plough (Palacos R-G).226,234
Antibiotic loaded bone cements may be able to reduce resistance by the rapid elimination of pathogens through a mixture of antibiotics, nonetheless, inadequate elution profiles and concentrations can result in resistance acquisition. The rationale behind current antibiotic elution processes stems from the necessity to rapidly eliminate latent bacteria attached to the implant or introduced during surgery (exogenous).235 These early or delayed infections are developed during the initial 3 to 24 months after implantation and commonly require a high concentration of antibiotic in the following week after surgery. Thus, the use of elution profiles in the form of high dosage peaks during the first days, which rapidly decay in the coming week but with maintained antibiotic levels still present a month later has been encouraged.227,235 The current approach may be effective in the treatment of early infections, although the reduction in antibiotic load poses a problem for late stage infections occurring more than 24 months after implantation, where the reduction in antibiotic load may stimulate AMR. Statistics have shown that the likelihood of suffering a late stage infection after arthroplasty can be similar to early and delayed infections (30, 29 and 41%, respectively),236,237 suggesting that current elution profiles should be modified. To promote a higher release in the late stages after implantation, several novel antimicrobial delivery approaches are under development (Fig. 5).
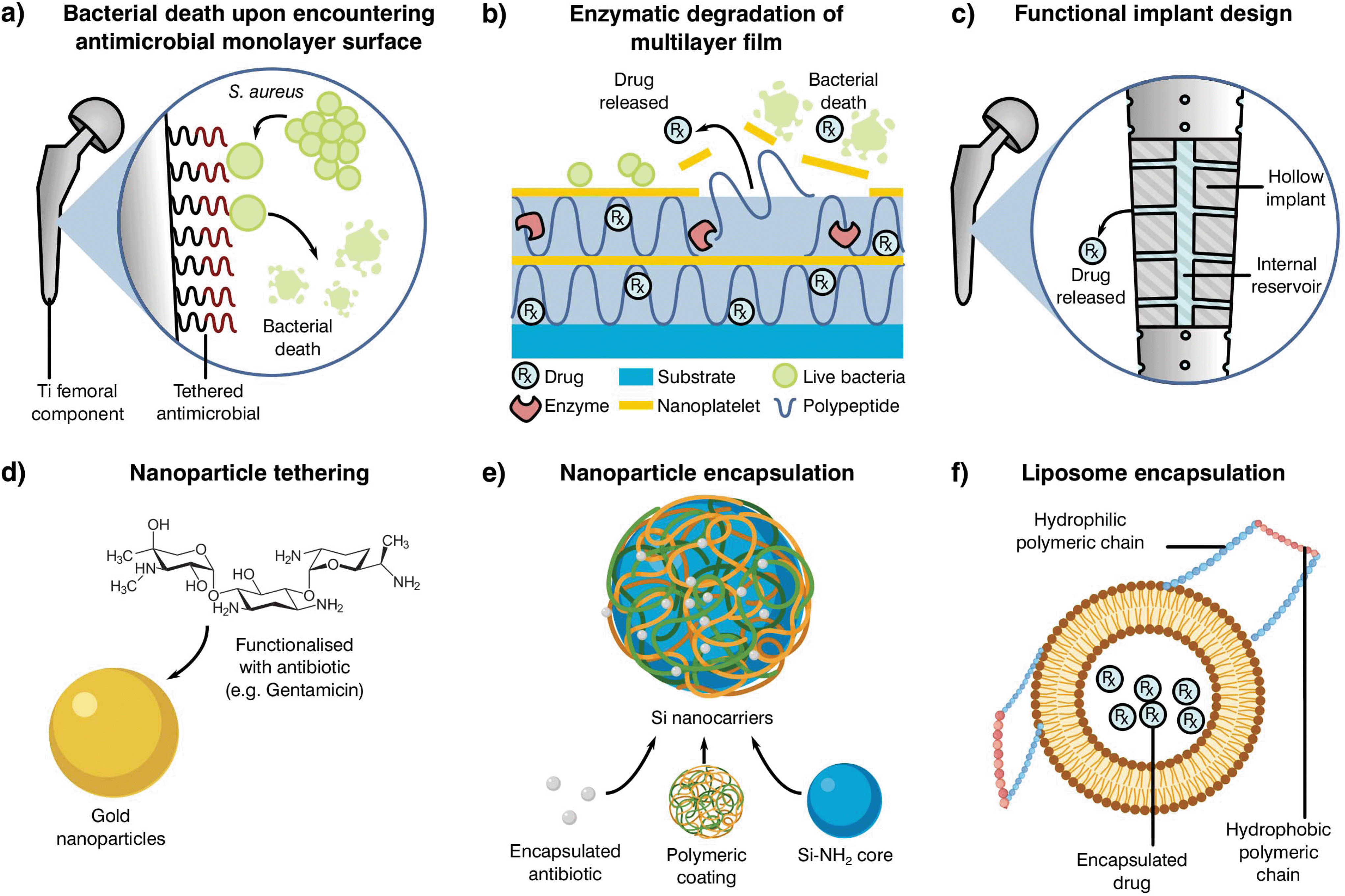 | ||
| Fig. 5 Examples of alternative delivery systems currently explored in the literature with examples for (a) implant antibiotic attachment as a monolayer,238 (b) multilayer,239 (c) direct implant modification,240 (d) particle tethering,241 antibiotic incorporation to (e) nanoparticles235 or (f) liposomes.242 Adapted from the source material. | ||
One such localised delivery approach is to anchor a desired antibiotic to the implanted device surface in a single (Fig. 5a)238 or multi-layered fashion (Fig. 5b).239 Other systems have focused on direct modification of implant surfaces and geometries to tailor antibiotic delivery, such as varying channel placement (Fig. 5c).240 Nevertheless, modification of implants may be difficult depending on the selected manufacturing and antimicrobial technologies, therefore the addition of carriers acting as antibiotic delivery systems is becoming of special interest to the field. These materials can be mixed into cement formulations and prepared with minimal changes to the current methodology, resulting in a myriad of possible configurations. Some technologies rely on the attachment of antibiotic molecules to particles in the nano to micro scale (Fig. 5d and e).241,243 In contrast to simple attachment, antibiotic encapsulation is an alternative approach (Fig. 5f), providing a more controlled form of elution and protection during curing in the form of metallic nanoparticles,235 liposomes242 or as a multilayer assembly for bone cement.244,245 Nevertheless, most of these approaches have been shown to have detrimental effects on the final product, with reduced mechanical properties or limited initial elution. This effect, and the poor understanding of the elution requirements needed to prevent AMR, indicate that there is a critical need to research antibiotic delivery systems and further our understanding of their influence on resistance development.231,235
Although the use of PMMA bone cements is highly widespread, reports have shown a significant persistence of infection associated with residual particles.246,247 Cement and implant debris can result in local inflammatory activity, leading to chronic complications and aseptic loosening.225 These potential negative outcomes and other disadvantages of PMMA bone cements, namely their lack of bioactivity or osseoconductivity, heat generation during curing, expansion due to water absorption and the potential allergic reaction to monomer remnants, has pushed forward the development of different formulations. One of the second-generation biomaterials employed in regenerative medicine is calcium and hydroxyapatite bone cement. These materials share a large similarity with natural bone where calcium and phosphate ions result in the promotion of osteoconduction and osteogenesis with antibiotic loading becoming an interesting research niche.248,249 Similarly, zinc polycarboxylate and glass polyalkenoate formulations have enhanced biological properties compared to common PMMA cements, making these materials especially popular in the dental field.250 Besides antibiotic loading (i.e. fusidic acid) it must be said that other antimicrobial agents are regularly used as antibacterial additives (i.e. Chlorhexidine diacetate or Benzalkonium chloride).251
The interest in developing antibiotic loaded cements to provide a sustained therapy and controlled dosage may result in a lower risk of infection, nonetheless, with AMR on the rise there is also a critical need to develop and implement novel antimicrobial substances. The natural antimicrobial activity of some metallic ions against bacteria252–254 has encouraged their use in medical devices, however, their potential toxic effects has restricted use with only a few candidates being granted governmental approval following successful marketing. One such element is silver for which the number of antimicrobial based products has elevated from 200 to nearly 1400 over the last decade.255 Examples include touch surfaces (“Safe Touch”, Dorset), hand sanitizers (Evolut Silver), textiles (Silver Plus®), dressings (Mepilex® Ag) and implant coatings (Agluna®), yet silver loaded cements are still not commercially accessible. Although the available literature presents research on cements loaded with different Ag donors, the detrimental effect that some silver salts have on the curing process of PMMA bone cement has moved the field onto a preferential use of silver nanoparticles (AgNPs).256–259 This form of silver presents a high surface area to volume ratio, conferring desirable chemical and physical properties to enhance antimicrobial efficacy.260 Specifically, AgNPs loaded PMMA bone cement tested in vitro reveal a broad antimicrobial spectrum against both Gram positive and Gram negative species, nevertheless of special interest is the high effectivity against antibiotic resistance strains showcased by Bistolfi et al.227 These promising results alongside the recent publishing of a clinical trial suggesting the safety of AgNPs loaded cements in vivo,261 solidify the rationale behind the use of this heavy metal to tackle AMR. That said, there is a limited understanding on the antimicrobial efficacy of silver particle loaded cements in vivo with only one animal model published to date.262 In this report, Moojen et al. carried out the analysis of AgNPs loaded cements in a rabbit infection model with no significant reduction of S. aureus colony forming units between loaded and plain cements after 14 days. Interestingly, available literature263–265 points to silver nanoparticles as an effective antimicrobial in vitro, indicating a disconnection between laboratory and animal methods. This detachment between in vitro and in vivo models in antimicrobial testing is not an isolated occurrence. Cytotoxicity and other cell work testing methods broadly used in the field have been shown to have a poor correlation with animal models.266
Alongside silver, other metals and non-organic species are being studied for their future application in antimicrobial loaded cements. Copper, gold, magnesium, zinc and their oxides have been successfully mixed with bone cements to confer antimicrobial properties, while graphene, paraben, quaternary amine monomers or chitosan are some of the more common non-metallic antimicrobial additives.227,267 Nevertheless, the clinical relevance of these technologies is diminished by the limited biological assessment available in vitro, with little immune response, hemocompatibility or genotoxicity studies available in most manuscripts. Thus, there is a need to develop more fundamental work on promising antimicrobial agents while revisiting old and over simplistic in vitro testing methods for the development, screening and implementation of new therapies.
The oversimplification of physiologically relevant processes is not only constrained to cell culture. To test the antibacterial effectiveness of antimicrobial materials it is common to employ a single pathogenic bacterial species generally found in clinical settings for the intended application of the biomaterial. Nevertheless, utilising single-species planktonic cultures for in vitro modelling does not compare to clinical reality. An extreme case is the oral microbiome where over 700 species have been identified in the human oral cavity, suggesting that single bacterium are not representative of the complex and mutable in vivo environment.268 Therefore, evaluating antimicrobial approaches to material design requires models of physiologically relevant environments that are likely to incorporate bacteria as a biofilm (possibly multispecies), a relevant implant material, and flow conditions.269 Several studies have incorporated these elements successfully to evaluate clinical implant materials such as titanium and zirconium, however they have not been frequently utilized in the development of antimicrobial biomaterials.269–271 Incorporating multiple disciplines, including biology, engineering, and mathematics is likely to become decisive to furthering the field due to the multifaceted nature of co-culture systems.272
As the regulatory pipeline becomes more time and cost consuming, it is important to develop a strong case to ensure present and future stakeholder participation. Silver is an attractive antimicrobial thanks to its status as an approved biomaterial, thus, it is possible to ease some of the requirements using predicate devices. Innovation and state of the art research should be a must to tackle AMR, nonetheless, the end purpose of novel technologies should be commercialization. Consequently, it is necessary to adapt the biomaterial community methodology to encompass the regulatory process and its application since the initial steps of promising technologies.
Future recommendations
At the start of the present manuscript, the complexity, risks and costs of developing and bringing to market a novel healthcare product were indicated. Over the following sections, different strategies to tackle AMR were emphasized and, through the use of two case studies, limitations on current biomaterials approaches were highlighted. As most global changes involving regulatory pathways can only receive a limited push forward from the biomaterials field, our role should be in the innovation and translation of new technologies. Thus, the principal aim of the following section is to clarify these limitations, identify clear areas of interest and put forward recommendations where biomaterial science may improve our response to AMR (Fig. 6).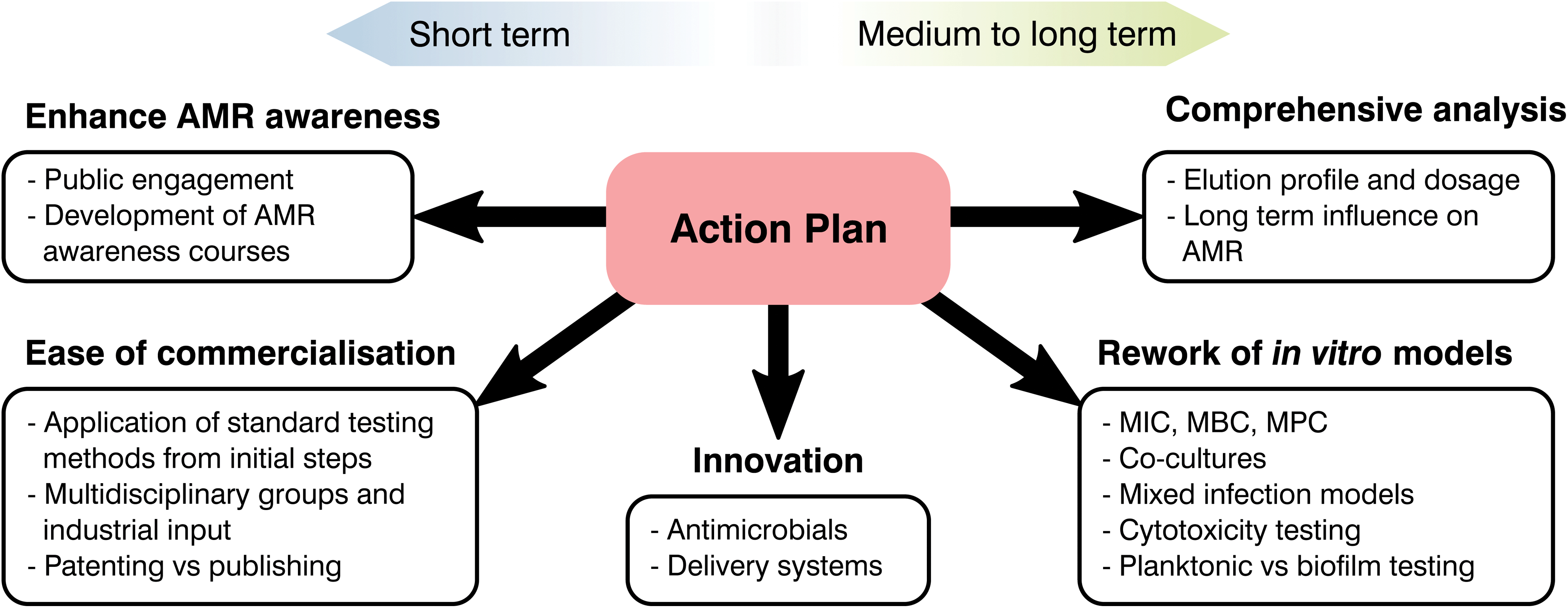 | ||
| Fig. 6 Diagram showcasing critical areas for the biomaterials community to tackle AMR. | ||
From the available literature it is becoming increasingly evident that promising materials tested in vitro may correlate poorly with in vivo analysis, leading to failures in clinical testing.266 Ethical and effectiveness concerns coupled with the extensive fiscal and time expenditures needed for animal testing has caused a decrease of such practices. Thus, special focus on in vitro testing as a screening method to assess and predict the host response to a biomaterial is being made.266 Despite this, as the number of novel healthcare technologies discovered over the decades has quickened, development of in vitro tests to ascertain their safety and effectiveness has stagnated.273–275 Susceptibility tests are of great relevance to analyse the interaction of antimicrobial agents and different microorganisms with numerous standards available depending on the base material and its application. Albeit methodologies differ, they commonly involve the contact or submersion of the desired material with a controlled bacterial density or plated culture for a specific time, followed by recovery and quantification or measurement of lack of growth on a plate.276 Comparison between controls and the antimicrobial agent tested will make it possible to ascertain if a material is inert, bacteriostatic or antimicrobial, helping to screen potential novel materials to tackle AMR. These tests are relatively simple with ordinary laboratory set ups required and timescales of days or weeks.276,277 Similarly to antimicrobial analysis, physicochemical tests rarely study biomaterials in a physiological relevant state. Few manuscripts ensure that samples are preconditioned with complex macromolecules or understand the ability of the added functionality to last in the long term, limiting the extrapolation of the obtained results to in vivo models. Nonetheless, the attractiveness driven by their simplicity and recognized status as standards in the field has led to a prevalence of such techniques without realising their potential downsides.
The composition of growth media is another factor that is rarely accounted for during testing and only changed depending on standards recommendations and the bacterial species used.277 However, elements present such as nutrients or salts to prevent osmotic shock can react with the desired molecule and cation to bias the obtained results (i.e. copper and chlorine).118 Similarly, timescales may be a misrepresentation of the reality. Touch surfaces are quickly becoming of interest to tackle HAI for which direct contact and recovery tests, such as JIS Z2801:2010 are the norm.55 These typically require the recovery of a deposited inoculum after 24 h of contact, however studies have shown that most surface to hand or hand to hand contact takes place in a few hours,55 demonstrating that long periods of time do not represent the reality of the intended material and application. Besides media and timescales, the recovery and quantification of survival bacterial populations may be another source of bias. This is commonly done either by spectroscopy (i.e. optical density) or by detachment of the bacterial biomass and plating, nevertheless, the former does not discriminate between live and dead microorganisms, while the latter requires complete recovery of bacterial species, which may be a challenge for porous or adhesive materials.55 Consequently, the biomaterials community should push forward the rework of such methods and encourage the broad use of more novel techniques for antimicrobial growth and AMR evaluation as suggested by Khan et al.278 and Belkum et al.279
Currently the biomedical field typically conducts testing on bacterial and mammalian cells separately, which disregards the complex interactions between cell types and infected devices. Similarly, human bacteria flora is multifaceted, with numerous species interacting and exchanging genetic information. Despite this, it is common to test for single bacterium, disregarding most Horizontal Gene Transfers (HGT) between bacterial species, commonplace for the exchange of resistance genes.12,13 Thus, there is an oversimplification of infection analysis in vitro in both bacterial and mammalian cell culture analysis.280 The prime example of such a scenario is the immune system, where white blood cells have been demonstrated to play a fundamental role in bone regeneration and infection control.281,282 Nonetheless, it is common to simply conduct separate analysis for inflammation, mineralization and antibacterial effectiveness of antimicrobial agents with different single cell types disregarding interactions. The selection of mammalian cells for each of these tests is also a notable point. Cytotoxicity analysis in vitro commonly entails the use of cells derived from tumours, more resilient and less expensive than more relevant cell lines, however, these do not necessary pose an accurate model of the real human tissue and biomaterial interaction.273,280 Similarly, most antimicrobial tests done in vitro to test the antibacterial efficacy of novel biomaterials involve laboratory strains, which may not be a real representation of the main clinical pathogens involved in infections.283 Consequently, as antimicrobial strategies continue to evolve so must our pre-clinical testing regimes, being the creation of a multicellular infection test, proper selection of bacteria and cell lines, and improved correlation between in vivo and in vitro models essential to progressing novel AMR prevention strategies.
Analysis of natural resistance acquisition is another aspect where the biomaterials community can help to challenge AMR. In drug development, it is usual to evaluate the possibility of microorganisms gaining resistance to novel and reworked molecules.20 Nevertheless, this trend is rarely followed during the development of biomaterials technologies where a focus on the initial efficacy of the system overrides long-term analyses. Such is the case for implantable devices functionalised with antimicrobial agents, however, these devices will need to halt infection for extended periods of time. Taking into account the adaptability and flexibility of bacteria, a lack of knowledge in the long-term effect of novel technologies poses a threat to treatment strategies focused on tackling AMR. This absence of long-lasting efficacy is especially critical for the implementation of novel molecules or antimicrobial metals, for which our understanding of the mechanisms driving AMR are still lacking. An example would be silver, microbiologist are aware of Ag resistant species and over the last decade genomic analysis of such bacteria has led to the discovery of the gene thought to encode silver resistance, the sil operon.284–286 Nevertheless, the natural acquisition of silver resistance is rarely studied with limited papers available dealing with long-term passages of controlled silver dosages to force resistance.285–287 The overuse of silver and the possibility of bacterial strains resistant to both antibiotics and heavy metals has awoken an interest in natural resistance development,288–291 exemplifying the importance of long-term analysis of antimicrobials. As a consequence, it is necessary to undertake such studies for novel biomaterials in vitro and perform a more complete analysis of resistance acquisition as suggested by Martínez et al.274
Moving forward, it is important that the way microorganisms develop resistance is understood so that appropriate treatments may be identified. It is well documented that microorganisms have what is known as intrinsic resistance, or the natural ability to resist an antimicrobials mechanism of action, which can occur at two stages: planktonic and community level.292 Resistance within an organism can occur due to gene mutation or by HGT293 when DNA is acquired from another resistant organism.294 This process can arise from the up-take of free DNA (transformation), plasma-mediated transfer (conjugation) or phage-mediated transfer (transduction).295 Community level organisms can develop further resistance and tolerate far greater stress than individual cells. This is demonstrated by the formation of biofilms that may increase antibiotic resistance up to 1000-fold.296 Due to the significant implication that the formation of a biofilm has on the ability of an organism to resist treatment, future developments may aim to provide approaches that disrupt or negate this effect.
Alongside this necessity to better understand resistance development, it is required to understand the limitations of current dosage analysis in resistance analysis and its prevention. The use of MIC to describe and classify an antimicrobial is a helpful tool for microbiologist and biomaterial scientist who are tackling AMR297–299 and is commonly analysed by subjecting controlled concentrations of the desired material to a specific density of a bacterial species either in liquid media or agar plated. After incubation, cultures are inspected visually or through spectroscopy to determine bacterial growth in the liquid media or to assess the size of the inhibition zone created in agar plates.297,299 For common drugs, standards are available with MIC breakpoints to determine if a specific bacteria species is susceptible, intermediate or resistant to the selected antibiotic which facilitates the following treatment.300 Thus, MIC has become one of the main tools available to select dosage in the biomaterials community. Nevertheless, by definition the MIC only inhibits the growth of bacteria, with cells still able to survive, but not proliferate, possibly still present in the culture. In contrast, plating of liquid cultures after antibiotic treatment and incubation provide the minimum bactericidal concentration (MBC), revealing the real amount of antimicrobial needed to kill the desired bacterial species.301 The range of antibiotic concentrations between MIC and MBC can led to a lower dosage being decided where resistant bacterial cells can be selected, enhancing AMR acquisition. This has been recognised in the mutant selection hypothesis where drug-resistant subpopulations present before treatment are enriched and amplified if the antimicrobial concentrations fall within a specific range.124 To prevent such a scenario, authors have shown that dosage selection should be complemented with parameters accounting for the mutant selection window, proposing the MPC (Fig. 4a).129,302 Selection of antimicrobial dosages taking the MPC into account will mean mutants fail to amplify124 and will provide a more robust dosage tool.
Further to the antimicrobial dosage being above the MPC, delivery systems should be fine-tuned in order to produce a pharmacokinetic release profile that enables this concentration to be maintained over the treatment period. This can be achieved by developing release kinetics that allow for the MPC to be exceeded shortly after the target site is reached.125 If the antimicrobial concentration is likely to fall below MPC values during treatment combination therapies or novel dosing profiles (Fig. 4b) should be exploited. In addition to the systemic administration of antimicrobials, localised delivery is highlighted as being preferential in order to prevent AMR. This is because it enables a more targeted approach that does not expose the rest of the body to systemic effects, including that of engaging with other microorganisms.303
As the crucial step to bring a novel technology to the market, regulatory approval should be considered as part of the initial steps of novel device research. The nature of contact between device and body, length of contact time and other parameters will decide the regulatory pathway, which will ultimately determine the economic and time investment required.22 As the main link between technical specifications and regulators, biomaterial researchers should be aware of the implications that the developed technology has in the future regulatory process and ensure that all preclinical data meets governmental requirements. This will require knowledge of the healthcare pipelines,304,305 however, the complexity of these processes makes it difficult to encompass all possible facets.22,275 Thus, it is necessary to focus on the two main aspects where one may have the highest impact in a multidisciplinary group: device categorization and preclinical testing. The first hurdle to be tackled consists in properly defining the technology and its intended purpose. If the novel technology intends to act as an implantable with added functionality, such as an antimicrobial coated implant, it will be recognised as a medical device which will follow the European Union Medical Device Regulations (MDR) 2017/745 and 2017/746. These divide medical devices into four categories (Class I, IIa, IIb and III) with increasingly difficult regulatory processes to be met. In contrast, if the developed material is mainly intended to correct or modify a physiological function by exerting a pharmacological, immunological or metabolic action, such as a novel molecule, this will mark the product as a medicinal agent, following Regulation (EC) No 726/2004 and Directive 2001/83/EC with extensive clinical trials required.306,307 During the inception of the novel biomaterials technology, proper knowledge of these classifications, requirements and similar marketed products alongside fundamental expertise can make it possible to modify the novel product and test it before further steps are taken. This fine-tuning prior to regulatory processes begin would ensure that costly reworks are prevented.308
The amount of preclinical data to demonstrate the effectiveness and safety of medical devices is dependent on the technology. In Europe, ISO 10993 2018 provides insight into the assays, data and quality control necessary for the in vitro testing of these devices (Fig. 7). Once the end category of the device is known, in vitro tests can be arranged following ISO 10993 recommendations. This will be done first through any “in house” capabilities available, limiting failures of future independent tests and early cheaper rework if necessary. It must be pointed out that there may be an apparent contradiction with the previous recommendation to develop and apply novel analytical methods. Nevertheless, tests mentioned and referenced in ISO 10993 are standardized methodologies, which in some cases have been shown to be outdated or poorly correlated with clinical outcomes.266,309–311 ISO 10993 takes into account the novelty of the field and the idiosyncrasies inherent to novel devices up to some extent, “It is not intended that ISO 10993 provide a rigid set of test methods, including pass/fail criteria, as this might result in either an unnecessary constraint on the development and use of novel medical devices, or a false sense of security in the general use of medical devices. Where a particular application warrants it, experts in the product or in the area of application concerned can choose to establish specific tests and criteria, described in a product-specific vertical standard.” However, technology progression still requires an approved methodology, limiting the current tests available to undertake. Consequently, it is still possible to encourage the use of better predictive analysis for future development of better standards.
 | ||
| Fig. 7 Considerations for biological assessment from ISO 10993-1:2018 where “X” means prerequisite information needed for a risk assessment; “E” means endpoints to be evaluated on the risk assessment (either through the use of existing data, additional endpoint-specific testing, or a rationale for why assessment of the endpoint does not require an additional data set assessment). | ||
Scientific work has become a competitive field since the early 2000s where a novel philosophy of “publish or perish” has taken hold.312 This change in mentality and all previously commented barriers to medical device approval and marketing led to a preference of publications over patents and, although this trend is changing, article writing is still the preferred outcome.313 Publishing instead of filing a patent ensures that the novel technology is broadly open to the public, which diminishes the prospects of future commercialization. Thus, it is necessary to continue exploring novel approaches to tackle AMR while ensuring that promising technologies are not lost. To achieve such an objective, greater involvement between academia and industry is needed. Finally, it has been shown that part of AMR emergence has been caused by poor use of antibiotics and lack of understanding of antibiotic resistance by the public.16 Academics have a privileged position from which to help by educating in the danger posed by bacterial resistance and the role of the public in tackling AMR. Consequently, enhancing public engagement, including AMR awareness courses would result in higher penetration of current policies.37
Conclusions
Antibiotic resistance in pathogenic bacteria is driving modern medicine to a bleak future where effective therapies against infection may become scarce. With prospects of a post-antibiotic era on the horizon, numerous organizations have focused their efforts on unearthing the causes behind such phenomena, with both bacterial and human causes identified. Over billions of years, bacterial cells have developed numerous mechanisms to survive and adapt to toxic substances, which coupled with their genomic plasticity, rapid life cycles and large populations, promotes development of antibiotic resistance. Nevertheless, human mismanagement of antibiotics has selected for high prevalence of resistant organisms, exacerbating AMR emergence. This coupled with the complexity inherent in their development and the high risk and low revenue associated with these substances has resulted in a paradox where novel antimicrobials are critically required, however, few products are currently in the development pipeline. This has led to action plans being developed and implemented by governmental bodies since the early 2000s. However, the complexity of AMR and the intricacy of moving large relevant associations forward calls for action at all levels. The biomaterials community, as one of the main innovation sources, has responded to the threat posed by AMR with increasing development of materials, substances and delivery systems conceived to halt the prospects of a post-antibiotic era. In this paper, we have conducted a review of available literature to showcase promising technologies under development and, through two case studies, pinpointed the following areas where special focus should be given to push forward a focused front against AMR:• Continue the current development of novel antimicrobials and delivery systems.
• Develop our understanding of the long-term influence of novel substances and delivery systems on AMR to complement current short-term analysis.
• Rework simplistic in vitro analysis broadly used for relevant correlation with in vivo analysis.
• Enhance the understanding of requirements and processes behind commercialization of these products to guide the initial research stages.
• Enhance AMR awareness at all levels.
Moving forward, a focus on these areas will make it possible to reduce prospects of bacteria gaining novel resistances, ensure that promising technologies in vitro will maintain their effectiveness in vivo, reduce costly reworks and failures during regulatory approval and increase the overall response to AMR. Consequently, novel therapies will retain their effectiveness with a smoother transition between laboratory and market, enhancing the critical role of the biomaterial community in confronting AMR.
Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This research was developed as part of grants EP/P02341X/1 (PREVENTION) and EP/V003356/1 (Invisible customisation). Authors Thomas J. Hall and Sophie C. Cox would like to thank the charitable donations given to the University of Birmingham for this research. We acknowledge the use of BioRender® during image preparation.References
- S. B. Zaman, M. A. Hussain, R. Nye, V. Mehta, K. T. Mamun and N. Hossain, Cureus, 2017, 9, e1403 Search PubMed.
- S. B. Levy, From tragedy the antibiotic age is born, Springer, Boston, 1992 Search PubMed.
- S. B. Levy, The antibiotic paradox: how miracle drugs are destroying the miracle, Springer, Boston, 2013 Search PubMed.
- D. Jasovský, J. Littmann, A. Zorzet and O. Cars, Upsala J. Med. Sci., 2016, 121, 159–164 CrossRef PubMed.
- A. P. Magiorakos, A. Srinivasan, R. B. Carey, Y. Carmeli, M. E. Falagas, C. G. Giske, S. Harbarth, J. F. Hindler, G. Kahlmeter, B. Olsson-Liljequist, D. L. Paterson, L. B. Rice, J. Stelling, M. J. Struelens, A. Vatopoulos, J. T. Weber and D. L. Monnet, Clin. Microbiol. Infect., 2012, 18, 268–281 CrossRef CAS PubMed.
- J. O′neill, Review on antimicrobial resistance: tackling a crisis for the health and wealth of nations, HM Government, London, 2016 Search PubMed.
- M. E. A. de Kraker, A. J. Stewardson and S. Harbarth, PLoS Med., 2016, 13, e1002184 CrossRef PubMed.
- W. H. Organization, WHO global strategy for containment of antimicrobial resistance, WHO, Geneva, 2001 Search PubMed.
- W. H. Organization, Global action plan on antimicrobial resistance, Geneva, 2015 Search PubMed.
- D. Chen and X. Qian, A Brief History of Bacteria: The Everlasting Game Between Humans and Bacteria, World Scientific Publishing Company Pte. Limited, Singapore, 2018 Search PubMed.
- A. Rodríguez-Rojas, J. Rodríguez-Beltrán, A. Couce and J. Blázquez, Int. J. Med. Microbiol., 2013, 303, 293–297 CrossRef PubMed.
- W. C. Reygaert, AIMS Microbiol., 2018, 4, 482 CAS.
- F. C. Tenover, Am. J. Med., 2006, 119, S3–S10 CrossRef CAS PubMed.
- R. I. Aminov, Front. Microbiol., 2010, 1, 134 Search PubMed.
- M. A. Kohanski, D. J. Dwyer and J. J. Collins, Nat. Rev. Microbiol., 2010, 8, 423–435 CrossRef CAS PubMed.
- J. Carlet, P. Collignon, D. Goldmann, H. Goossens, I. C. Gyssens, S. Harbarth, V. Jarlier, S. B. Levy, B. N'Doye, D. Pittet, R. Richtmann, W. H. Seto, J. W. M. van der Meer and A. Voss, Lancet, 2011, 378, 369–371 CrossRef.
- ReAct, Few antibiotics under development, https://www.reactgroup.org/toolbox/understand/how-did-we-end-up-here/few-antibiotics-under-development/, (accessed June 2020).
- L. L. Silver, Clin. Microbiol. Rev., 2011, 24, 71–109 CrossRef CAS PubMed.
- A. J. Alanis, Arch. Med. Res., 2005, 36, 697–705 CrossRef PubMed.
- M. A. Blaskovich, M. S. Butler and M. A. Cooper, Essays Biochem., 2017, 61, 103–114 CrossRef PubMed.
- A. Towse, C. K. Hoyle, J. Goodall, M. Hirsch, J. Mestre-Ferrandiz and J. H. Rex, Health Policy, 2017, 121, 1025–1030 CrossRef PubMed.
- R. M. Guerra-Bretaña and A. L. Flórez-Rendón, Rev. Bras. Eng. Biomed., 2018, 34, 356–367 Search PubMed.
- E. T. Pashuck and M. M. Stevens, Sci. Transl. Med., 2012, 4, 160sr4 Search PubMed.
- D. Jabes, Curr. Opin. Microbiol., 2011, 14, 564–569 CrossRef PubMed.
- L. D. Högberg, A. Heddini and O. Cars, Trends Pharmacol. Sci., 2010, 31, 509–515 CrossRef PubMed.
- L. J. Piddock, Lancet Infect. Dis., 2012, 12, 249–253 CrossRef PubMed.
- B. Spellberg, J. H. Powers, E. P. Brass, L. G. Miller and J. E. Edwards Jr., Clin. Infect. Dis., 2004, 38, 1279–1286 CrossRef CAS PubMed.
- U. Theuretzbacher, Nat. Rev. Drug Discovery, 2016, 15, 812–813 CrossRef CAS PubMed.
- U. Theuretzbacher, Clin. Microbiol. Infect., 2017, 23, 713–717 CrossRef CAS PubMed.
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS PubMed.
- D. M. Brogan and E. Mossialos, Global. Health, 2016, 12, 8 CrossRef PubMed.
- M. Hay, D. W. Thomas, J. L. Craighead, C. Economides and J. Rosenthal, Nat. Biotechnol., 2014, 32, 40–51 CrossRef CAS PubMed.
- R. Wise, Curr. Sci., 2008, 95, 181–187 CAS.
- Editorial, Nat. Biotechnol., 2018, 36, 555 CrossRef PubMed.
- J. Makower, A. Meer and L. Denend, FDA impact on US medical device technology innovation, 2010 Search PubMed.
- P. Beyer, V. Moorthy, S. Paulin, S. R. Hill, M. Sprenger, S. Garner, M. Simão, R. Guerra, N. Magrini and S. Swaminathan, Lancet, 2018, 392, 264–266 CrossRef.
- M. Courtenay, E. Castro-Sanchez, M. Fitzpatrick, R. Gallagher, R. Lim and G. Morris, J. Hosp. Infect., 2019, 101, 426–427 CrossRef CAS PubMed.
- J. Ogden, Prescriber, 2019, 30, 27–31 CrossRef.
- R. R. Uchil, G. S. Kohli, V. M. KateKhaye and O. C. Swami, J. Clin. Diagn. Res., 2014, 8, ME01–ME04 Search PubMed.
- Clarivate, Web of Science, https://apps.webofknowledge.com/, (accessed June 2020).
- A. R. M. Coates, G. Halls and Y. Hu, Br. J. Pharmacol., 2011, 163, 184–194 CrossRef CAS PubMed.
- R. Watson, BMJ, 2008, 336, 1266–1267 Search PubMed.
- A. Cassini, L. D. Högberg, D. Plachouras, A. Quattrocchi, A. Hoxha, G. S. Simonsen, M. Colomb-Cotinat, M. E. Kretzschmar, B. Devleesschauwer, M. Cecchini, D. A. Ouakrim, T. C. Oliveira, M. J. Struelens, C. Suetens, D. L. Monnet, R. Strauss, K. Mertens, T. Struyf, B. Catry, K. Latour, I. N. Ivanov, E. G. Dobreva, A. Tambic Andraševic, S. Soprek, A. Budimir, N. Paphitou, H. Žemlicková, S. Schytte Olsen, U. Wolff Sönksen, P. Märtin, M. Ivanova, O. Lyytikäinen, J. Jalava, B. Coignard, T. Eckmanns, M. Abu Sin, S. Haller, G. L. Daikos, A. Gikas, S. Tsiodras, F. Kontopidou, Á. Tóth, Á. Hajdu, Ó. Guólaugsson, K. G. Kristinsson, S. Murchan, K. Burns, P. Pezzotti, C. Gagliotti, U. Dumpis, A. Liuimiene, M. Perrin, M. A. Borg, S. C. de Greeff, J. C. M. Monen, M. B. G. Koek, P. Elstrøm, D. Zabicka, A. Deptula, W. Hryniewicz, M. Caniça, P. J. Nogueira, P. A. Fernandes, V. Manageiro, G. A. Popescu, R. I. Serban, E. Schréterová, S. Litvová, M. Štefkovicová, J. Kolman, I. Klavs, A. Korošec, B. Aracil, A. Asensio, M. Pérez-Vázquez, H. Billström, S. Larsson, J. S. Reilly, A. Johnson and S. Hopkins, Lancet Infect. Dis., 2019, 19, 56–66 CrossRef.
- D. M. Sievert, P. Ricks, J. R. Edwards, A. Schneider, J. Patel, A. Srinivasan, A. Kallen, B. Limbago and S. Fridkin, Infect. Control Hosp. Epidemiol., 2013, 34, 1–14 CrossRef PubMed.
- A. I. Hidron, J. R. Edwards, J. Patel, T. C. Horan, D. M. Sievert, D. A. Pollock and S. K. Fridkin, Infect. Control Hosp. Epidemiol., 2008, 29, 996–1011 CrossRef PubMed.
- W. H. Organization, WHO Priority Pathogens List for R&D of New Antibiotics, 2017 Search PubMed.
- C. Willyard, Nature, 2017, 543, 15 CrossRef CAS PubMed.
- T. P. Trust, Antibiotics Currently in Global Development, https://www.pewtrusts.org/en/research-and-analysis/data-visualizations/2014/antibiotics-currently-in-clinical-development, (accessed June 2020).
- V. L. Simpkin, M. J. Renwick, R. Kelly and E. Mossialos, J. Antibiot., 2017, 70, 1087–1096 CrossRef CAS PubMed.
- Z. Ashraf, A. Bais, M. M. Manir and U. Niazi, PLoS One, 2015, 10, e0135293 CrossRef PubMed.
- P. A. Todd and P. Benfield, Drugs, 1990, 39, 264–307 CrossRef CAS PubMed.
- R. J. Fair and Y. Tor, Perspect. Med. Chem., 2014, 6, 25–64 Search PubMed.
- S. Martinotti and E. Ranzato, J. Funct. Biomater., 2018, 9, 34 CrossRef PubMed.
- B. A. Lipsky and C. Hoey, Clin. Infect. Dis., 2009, 49, 1541–1549 CrossRef PubMed.
- V. M. Villapún, L. G. Dover, A. Cross and S. González, Materials, 2016, 9, 736 CrossRef PubMed.
- R. J. Turner, Microb. Biotechnol., 2017, 10, 1062–1065 CrossRef PubMed.
- N. F. Brady, P. C. Molan and C. G. Harfoot, Pharm. Pharmacol. Commun., 1996, 2, 471–473 CAS.
- L. Vandamme, A. Heyneman, H. Hoeksema, J. Verbelen and S. Monstrey, Burns, 2013, 39, 1514–1525 CrossRef CAS PubMed.
- J. A. Lemire, J. J. Harrison and R. J. Turner, Nat. Rev. Microbiol., 2013, 11, 371–384 CrossRef CAS PubMed.
- R. J. Turner, Microb. Biotechnol., 2017, 10, 1062–1065 CrossRef PubMed.
- T.-Y. Wang, M. D. J. Libardo, A. M. Angeles-Boza and J.-P. Pellois, ACS Chem. Biol., 2017, 12, 1170–1182 CrossRef CAS PubMed.
- J. Willi, P. Küpfer, D. Evéquoz, G. Fernandez, A. Katz, C. Leumann and N. Polacek, Nucleic Acids Res., 2018, 46, 1945–1957 CrossRef CAS PubMed.
- S. Kalghatgi, C. S. Spina, J. C. Costello, M. Liesa, J. R. Morones-Ramirez, S. Slomovic, A. Molina, O. S. Shirihai and J. J. Collins, Sci. Transl. Med., 2013, 5, 192ra85 Search PubMed.
- B. Dridi, A. Lupien, M. G. Bergeron, P. Leprohon and M. Ouellette, Antimicrob. Agents Chemother., 2015, 59, 5420–5426 CrossRef CAS PubMed.
- M. Kleczkowski, W. Kluciński, J. Sikora, R. Kasztelan and M. Zdanowicz, Pol. J. Vet. Sci., 2002, 5, 263 CAS.
- G. Grass, C. Rensing and M. Solioz, Appl. Environ. Microbiol., 2011, 77, 1541–1547 CrossRef CAS PubMed.
- K. Mijnendonckx, N. Leys, J. Mahillon, S. Silver and R. Van Houdt, BioMetals, 2013, 26, 609–621 CrossRef CAS PubMed.
- X. Ding, S. Duan, X. Ding, R. Liu and F. J. Xu, Adv. Funct. Mater., 2018, 28, 1802140 CrossRef.
- S. Mahira, A. Jain, W. Khan and A. J. Domb, in Antimicrobial Materials for Biomedical Applications, ed. A. J. Domb, K. R. Kunduru and S. Farah, The Royal Society of Chemistry, London, 2019, pp. 1–37, 10.1039/9781788012638-00001.
- E. R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- M. R. Santos, A. C. Fonseca, P. V. Mendonça, R. Branco, A. C. Serra, P. V. Morais and J. F. Coelho, Materials, 2016, 9, 599 CrossRef PubMed.
- O. G. Travkova, H. Moehwald and G. Brezesinski, Adv. Colloid Interface Sci., 2017, 247, 521–532 CrossRef CAS PubMed.
- A. K. Marr, W. J. Gooderham and R. E. W. Hancock, Curr. Opin. Pharmacol., 2006, 6, 468–472 CrossRef CAS PubMed.
- E. F. Haney, S. K. Straus and R. E. Hancock, Front. Chem., 2019, 7(43), 1–22 Search PubMed.
- M. Krishnamoorthy, S. Hakobyan, M. Ramstedt and J. E. Gautrot, Chem. Rev., 2014, 114, 10976–11026 CrossRef CAS PubMed.
- Y. Huang, X. Ding, Y. Qi, B. Yu and F. J. Xu, Biomaterials, 2016, 106, 134–143 CrossRef CAS PubMed.
- R. Liu, X. Chen, S. Chakraborty, J. J. Lemke, Z. Hayouka, C. Chow, R. A. Welch, B. Weisblum, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 4410–4418 CrossRef CAS PubMed.
- C. Ge, H. Ye, F. Wu, J. Zhu, Z. Song, Y. Liu and L. Yin, J. Mater. Chem. B, 2020, 8, 6530–6547 RSC.
- K. Park, D. Oh, S. Yub Shin, K.-S. Hahm and Y. Kim, Biochem. Biophys. Res. Commun., 2002, 290, 204–212 CrossRef CAS PubMed.
- W. T. Heller, A. J. Waring, R. I. Lehrer and H. W. Huang, Biochemistry, 1998, 37, 17331–17338 CrossRef CAS PubMed.
- L. Steinstraesser, B. Tippler, J. Mertens, E. Lamme, H.-H. Homann, M. Lehnhardt, O. Wildner, H.-U. Steinau and K. Überla, Retrovirology, 2005, 2, 2 CrossRef PubMed.
- A. Belaid, M. Aouni, R. Khelifa, A. Trabelsi, M. Jemmali and K. Hani, J. Med. Virol., 2002, 66, 229–234 CrossRef CAS PubMed.
- C. Lorin, H. Saidi, A. Belaid, A. Zairi, F. Baleux, H. Hocini, L. Bélec, K. Hani and F. Tangy, Virology, 2005, 334, 264–275 CrossRef CAS PubMed.
- M. Morimoto, H. Mori, T. Otake, N. Ueba, N. Kunita, M. Niwa, T. Murakami and S. Iwanaga, Chemotherapy, 1991, 37, 206–211 CrossRef CAS PubMed.
- T. Murakami, M. Niwa, F. Tokunaga, T. Miyata and S. Iwanaga, Chemotherapy, 1991, 37, 327–334 CrossRef CAS PubMed.
- H. Nakashima, M. Masuda, T. Murakami, Y. Koyanagi, A. Matsumoto, N. Fujii and N. Yamamoto, Antimicrob. Agents Chemother., 1992, 36(6), 1249–1255 CrossRef CAS PubMed.
- D. G. Lee, D.-H. Kim, Y. Park, H. K. Kim, H. N. Kim, Y. K. Shin, C. H. Choi and K.-S. Hahm, Biochem. Biophys. Res. Commun., 2001, 282, 570–574 CrossRef CAS PubMed.
- H. Tamamura, A. Otaka, T. Murakami, T. Ishihara, T. Ibuka, M. Waki, A. Matsumoto, N. Yamamoto and N. Fujii, Biochem. Biophys. Res. Commun., 1996, 219, 555–559 CrossRef CAS PubMed.
- D.-H. Kim, D. G. Lee, K. L. Kim and Y. Lee, Eur. J. Biochem., 2001, 268, 4449–4458 CrossRef CAS PubMed.
- H. G. Boman, B. Agerberth and A. Boman, Infect. Immun., 1993, 61, 2978–2984 CrossRef CAS PubMed.
- M. Hartlieb, E. G. Williams, A. Kuroki, S. Perrier and K. E. Locock, Curr. Med. Chem., 2017, 24, 2115–2140 CrossRef CAS PubMed.
- M. Xiong, Z. Han, Z. Song, J. Yu, H. Ying, L. Yin and J. Cheng, Angew. Chem., Int. Ed., 2017, 56, 10826–10829 CrossRef CAS PubMed.
- S. J. Lam, N. M. O'Brien-Simpson, N. Pantarat, A. Sulistio, E. H. Wong, Y. Y. Chen, J. C. Lenzo, J. A. Holden, A. Blencowe, E. C. Reynolds and G. G. Qiao, Nat. Microbiol., 2016, 1, 1–11 Search PubMed.
- S. J. Shirbin, I. Insua, J. A. Holden, J. C. Lenzo, E. C. Reynolds, N. M. O'Brien–Simpson and G. G. Qiao, Adv. Healthcare Mater., 2018, 7, 1800627 CrossRef PubMed.
- M. Zhou, Y. Qian, J. Xie, W. Zhang, W. Jiang, X. Xiao, S. Chen, C. Dai, Z. Cong, Z. Ji, N. Shao, L. Liu, Y. Wu and R. Liu, Angew. Chem., 2020, 59, 6412–6419 CrossRef CAS PubMed.
- S. N. Riduan, A. Armugam and Y. Zhang, Mater. Chem. B, 2020, 8(30), 6317–6321 RSC.
- A. R. Mazo, S. Allison-Logan, F. Karimi, N. J. A. Chan, W. Qiu, W. Duan, N. M. O'Brien-Simpson and G. G. Qiao, Chem. Soc. Rev., 2020, 49, 4737–4834 RSC.
- D. Sun, M. B. Shahzad, M. Li, G. Wang and D. Xu, Mater. Technol., 2014, 30(6), B90–B95 Search PubMed.
- S. Veerachamy, T. Yarlagadda, G. Manivasagam and P. K. D. V. Yarlagadda, Proc. Inst. Mech. Eng., Part H, 2014, 228, 1083–1099 CrossRef PubMed.
- E. Moran, S. Masters, A. R. Berendt, P. McLardy-Smith, I. Byren and B. L. Atkins, J. Infect., 2007, 55, 1–7 CrossRef CAS PubMed.
- S. Ferraris and S. Spriano, Mater. Sci. Eng., 2016, 61, 965–978 CrossRef CAS PubMed.
- L. Hsu, J. Fang, D. Borca-Tasciuc, R. Worobo and C. I. Moraru, Appl. Environ. Microbiol., 2013, 17, 2703–2712 CrossRef PubMed.
- K. A. Whitehead, J. Colligon and J. Verran, Colloids Surf., B, 2005, 41, 129–113 CrossRef CAS PubMed.
- A. Jaggessar, H. Shahali, A. Mathew and P. K. D. V. Yarlagadda, J. Nanobiotechnol., 2017, 15, 64 CrossRef PubMed.
- J. Hasan, R. J. Crawford and E. P. Ivanova, Trends Biotechnol., 2013, 31, 295–304 CrossRef CAS PubMed.
- J. Ma, Y. Sun, K. Gleichauf, J. Lou and Q. Li, Langmuir, 2011, 27, 10035–10040 CrossRef CAS PubMed.
- A. Kesel and R. Liedert, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2007, 146, S130 CrossRef.
- H. J. Ensikat, P. Ditsche-Kuru, C. Neinhuis and W. Barthlott, Beilstein J. Nanotechnol., 2011, 2, 152–161 CrossRef CAS PubMed.
- Y. Y. Yan, N. Gao and W. Barthlott, Adv. Colloid Interface Sci., 2011, 169, 80–105 CrossRef CAS PubMed.
- T. Diu, N. Faruqui, T. Sjöström, B. Lamarre, H. F. Jenkinson, B. Su and M. G. Ryadnov, Sci. Rep., 2014, 4, 7122 CrossRef CAS PubMed.
- J. Hasan, H. K. Webb, V. K. Truong, S. Pogodin, V. A. Baulin, G. S. Watson, J. A. Watson, R. J. Crawford and E. P. Ivanova, Appl. Microbiol. Biotechnol., 2013, 97, 9257–9262 CrossRef CAS PubMed.
- E. P. Ivanova, J. Hasan, H. K. Webb, V. K. Truong, G. S. Watson, J. A. Watson, V. A. Baulin, S. Pogodin, J. Y. Wang, M. J. Tobin, C. Löbbe and R. J. Crawford, Small, 2012, 8, 2489–2494 CrossRef CAS PubMed.
- G. S. Watson, D. W. Green, L. Schwarzkopf, X. Li, B. W. Cribb, S. Myhra and J. A. Watson, Acta Biomater., 2015, 21, 109–122 CrossRef CAS PubMed.
- M. N. Dickson, E. I. Liang, L. A. Rodriguez, N. Vollereaux and A. F. Yee, Biointerphases, 2015, 10, 21010 CrossRef PubMed.
- C. D. Bandara, S. Singh, I. O. Afara, A. Wolff, T. Tesfamichael, K. Ostrikov and A. Oloyede, ACS Appl. Mater. Interfaces, 2017, 9, 6746–6760 CrossRef CAS PubMed.
- C. M. Bhadra, V. Khanh Truong, V. T. H. Pham, M. Al Kobaisi, G. Seniutinas, J. Y. Wang, S. Juodkazis, R. J. Crawford and E. P. Ivanova, Sci. Rep., 2015, 5, 16817 CrossRef CAS PubMed.
- P. Ginestra, E. Ceretti, D. Lobo, M. Lowther, S. Cruchley, S. Kuehne, V. Villapun, S. Cox, L. Grover, D. Shepherd, M. Attallah, O. Addison and M. Webber, Procedia Manuf., 2020, 47, 1106–1112 CrossRef.
- V. M. Villapún, C. C. Lukose, M. Birkett, L. G. Dover and S. González, Surf. Coat. Technol., 2018, 350, 334–345 CrossRef.
- V. M. Villapún, B. Qu, P. A. Lund, W. Wei, L. G. Dover, J. R. Thompson, J. O. Adesina, C. Hoerdemann, S. Cox and S. González, Mater. Today Commun., 2020, 23, 101074 CrossRef.
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, S122–S129 CrossRef CAS PubMed.
- X. Zhao and K. Drlica, Clin. Infect. Dis., 2001, 33, S147–S156 CrossRef CAS PubMed.
- Z. Xilin and K. Drlica, J. Infect. Dis., 2002, 185, 561–565 CrossRef PubMed.
- Y. Dong, X. Zhao, J. Domagala and K. Drlica, Antimicrob. Agents Chemother., 1999, 43(7), 1756–1758 CrossRef CAS PubMed.
- K. Drlica and X. Zhao, Clin. Infect. Dis., 2007, 44, 681–688 CrossRef PubMed.
- C. W. Stratton, Emerging Infect. Dis., 2003, 9, 10–16 CrossRef CAS PubMed.
- F. Baquero, J. Chemother., 1999, 11, 35–43 CrossRef CAS PubMed.
- A. Vernon, W. Burman, D. Benator, A. Khan and L. Bozeman, Lancet, 1999, 353, 1843–1847 CrossRef CAS.
- C. M. Baker, M. J. Ferrari and K. Shea, Sci. Rep., 2018, 8, 5866 CrossRef PubMed.
- X. Zhao and K. Drlica, Clin. Infect. Dis., 2001, 33, 147–156 CrossRef PubMed.
- R. Davidson, R. Cavalcanti, J. L. Brunton, D. J. Bast, J. C. S. de Azavedo, P. Kibsey, C. Fleming and D. E. Low, N. Engl. J. Med., 2002, 346, 747–750 CrossRef PubMed.
- K. Sieradzki, T. Leski, J. Dick, L. Borio and A. Tomasz, J. Clin. Microbiol., 2003, 41, 1687–1693 CrossRef CAS PubMed.
- K. Sieradzki, R. B. Roberts, S. W. Haber and A. Tomasz, N. Engl. J. Med., 1999, 340, 517–523 CrossRef CAS PubMed.
- S. Sarabahi, Indian J. Plast. Surg., 2012, 45, 379–387 CrossRef PubMed.
- G. D. Winter, Nature, 1962, 193, 293–294 CrossRef CAS PubMed.
- C. Y. Cho and J. S. Lo, Dermatol. Clin., 1998, 16, 25–47 CrossRef CAS.
- C. H. Moon and T. G. Crabtree, Curr. Treat. Options Infect. Dis., 2003, 5, 251–260 Search PubMed.
- S. Dhivya, V. V. Padma and E. Santhini, Biomedicine, 2015, 5, 22 CrossRef PubMed.
- M. C. Varghese, A. K. Balin, D. M. Carter and D. Caldwell, Arch. Dermatol., 1986, 122, 52–57 CrossRef CAS.
- O. M. Alvarez, P. M. Mertz and W. H. Eaglstein, J. Surg. Res., 1983, 35, 142–148 CrossRef CAS PubMed.
- P. A. Rubio, Int. Surg., 1991, 76, 253–254 CAS.
- A. M. Jones and L. San Miguel, J. Wound Care, 2006, 15, 65–69 CrossRef CAS PubMed.
- A. J. Singer and R. A. Clark, N. Engl. J. Med., 1999, 341, 738–746 CrossRef CAS PubMed.
- C. K. Field and M. D. Kerstein, Am. J. Surg., 1994, 167, S2–S6 CrossRef.
- L. L. Bolton, K. Monte and L. A. Pirone, Ostomy Wound Manag., 2000, 46, 51S–62S CAS.
- G. Han and R. Ceilley, Adv. Ther., 2017, 34, 599–610 CrossRef PubMed.
- M. Society, The History of Antibiotics, https://microbiologysociety.org/members-outreach-resources/outreach-resources/antibiotics-unearthed/antibiotics-and-antibiotic-resistance/the-history-of-antibiotics.html, (accessed June 2020).
- L. Zaffiri, J. Gardner and L. H. Toledo-Pereyra, J. Invest. Surg., 2012, 25, 67–77 CrossRef PubMed.
- S. Banerjee and C. Argáez, Topical Antibiotics for Infection Prevention: A Review of the Clinical Effectiveness and Guidelines, Canadian Agency for Drugs and Technologies in Health, 2017 Search PubMed.
- B. De Simone, M. Sartelli, F. Coccolini, C. G. Ball, P. Brambillasca, M. Chiarugi, F. C. Campanile, G. Nita, D. Corbella, A. Leppaniemi, E. Boschini, E. E. Moore, W. Biffl, A. Peitzmann, Y. Kluger, M. Sugrue, G. Fraga, S. Di Saverio, D. Weber, B. Sakakushev, O. Chiara, F. M. Abu-Zidan, R. ten Broek, A. W. Kirkpatrick, I. Wani, R. Coimbra, G. L. Baiocchi, M. D. Kelly, L. Ansaloni and F. Catena, World J. Emerg. Surg., 2020, 15, 10 CrossRef PubMed.
- M. J. Mukagendaneza, E. Munyaneza, E. Muhawenayo, D. Nyirasebura, E. Abahuje, J. Nyirigira, J. D. D. Harelimana, T. Z. Muvunyi, F. Masaisa, J. C. Byiringiro, T. Hategekimana and C. M. Muvunyi, Patient Saf. Surg., 2019, 13, 10 CrossRef PubMed.
- S. Pal, A. Sayana, A. Joshi and D. Juyal, J. Family Med. Prim. Care, 2019, 8, 3600–3606 CrossRef PubMed.
- A. Sakr, F. Brégeon, J. L. Mège, J. M. Rolain and O. Blin, Front. Microbiol., 2018, 9, 2419 CrossRef PubMed.
- R. Edge and C. Argáez, Topical Antibiotics for Impetigo: A Review of the Clinical Effectiveness and Guidelines, Canadian Agency for Drugs and Technologies in Health, Ottawa, 2017 Search PubMed.
- D. Simões, S. P. Miguel, M. P. Ribeiro, P. Coutinho, A. G. Mendonça and I. J. Correia, Eur. J. Pharm. Biopharm., 2018, 127, 130–141 CrossRef PubMed.
- M. C. García, A. A. Aldana, L. I. Tártara, F. Alovero, M. C. Strumia, R. H. Manzo, M. Martinelli and A. F. Jimenez-Kairuz, Carbohydr. Polym., 2017, 175, 75–86 CrossRef PubMed.
- H. Li, G. R. Williams, J. Wu, H. Wang, X. Sun and L. M. Zhu, Mater. Sci. Eng., C, 2017, 79, 245–254 CrossRef CAS PubMed.
- W. Shao, H. Liu, S. Wang, J. Wu, M. Huang, H. Min and X. Liu, Carbohydr. Polym., 2016, 145, 114–120 CrossRef CAS PubMed.
- H. Chen, X. Xing, H. Tan, Y. Jia, T. Zhou, Y. Chen, Z. Ling and X. Hu, Mater. Sci. Eng., C, 2017, 70, 287–295 CrossRef CAS PubMed.
- L. R. Peterson, Int. J. Antimicrob. Agents, 2008, 32, S215–S222 CrossRef CAS PubMed.
- P. Montravers, M. Bassetti, H. Dupont, C. Eckmann, W. R. Heizmann, X. Guirao, M. S. García, M. R. Capparella, D. Simoneau and K. F. Bodmann, J. Antimicrob. Chemother., 2013, 68, ii15–ii24 CrossRef CAS PubMed.
- V. Dhanalakshmi, T. R. Nimal, M. Sabitha, R. Biswas and R. Jayakumar, J. Biomed. Mater. Res., Part B, 2016, 104, 797–807 CrossRef CAS PubMed.
- T. R. Nimal, G. Baranwal, M. C. Bavya, R. Biswas and R. Jayakumar, ACS Appl. Bio Mater., 2016, 8, 22074–22083 CrossRef CAS PubMed.
- H. Liu, K. K. Leonas and Y. Zhao, J. Eng. Fibers Fabr., 2010, 5(4), 10–19 CAS.
- E. A. Kamoun, E. R. S. Kenawy, T. M. Tamer, M. A. El-Meligy and M. S. M. Eldin, Arabian J. Chem., 2015, 8, 38–47 CrossRef CAS.
- G. Rath, T. Hussain, G. Chauhan, T. Garg and A. K. Goyal, Mater. Sci. Eng., C, 2016, 58, 242–253 CrossRef CAS PubMed.
- M. Safdari, E. Shakiba, S. H. Kiaie and A. Fattahi, Fibers Polym., 2016, 17, 744–750 CrossRef CAS.
- P. Jana, T. Mitra, T. K. R. Selvaraj, A. Gnanamani and P. P. Kundu, Carbohydr. Polym., 2016, 153, 573–581 CrossRef CAS PubMed.
- M. Contardi, J. A. Heredia-Guerrero, G. Perotto, P. Valentini, P. P. Pompa, R. Spanò, L. Goldoni, R. Bertorelli, A. Athanassiou and I. S. Bayer, Eur. J. Pharm. Sci., 2017, 104, 133–144 CrossRef CAS PubMed.
- A. R. Unnithan, N. A. Barakat, P. T. Pichiah, G. Gnanasekaran, R. Nirmala, Y. S. Cha, C.-H. Jung, M. El-Newehy and H. Y. Kim, Carbohydr. Polym., 2012, 90, 1786–1793 CrossRef CAS PubMed.
- N. Adhirajan, N. Shanmugasundaram, S. Shanmuganathan and M. Babu, J. Pharm. Pharmacol., 2009, 61, 1617–1623 CrossRef CAS PubMed.
- N. Monteiro, M. Martins, A. Martins, N. A. Fonseca, J. N. Moreira, R. L. Reis and N. M. Neves, Acta Biomater., 2015, 18, 196–205 CrossRef CAS PubMed.
- R. Fu, C. Li, C. Yu, H. Xie, S. Shi, Z. Li, Q. Wang and L. Lu, Drug Delivery, 2016, 23, 818–829 CrossRef CAS PubMed.
- T. Nitanan, P. Akkaramongkolporn, T. Rojanarata, T. Ngawhirunpat and P. Opanasopit, Int. J. Pharm., 2013, 448, 71–78 CrossRef CAS PubMed.
- N. J. M. Sebri and K. A. M. Amin, Orient. J. Chem., 2017, 33, 628–636 CrossRef CAS.
- A. R. Unnithan, G. Gnanasekaran, Y. Sathishkumar, Y. S. Lee and C. S. Kim, Carbohydr. Polym., 2014, 102, 884–892 CrossRef CAS PubMed.
- R. I. Baron, M. E. Culica, G. Biliuta, M. Bercea, S. Gherman, D. Zavastin, L. Ochiuz, M. Avadanei and S. Coseri, Materials, 2019, 12, 1569 CrossRef CAS PubMed.
- W. Shao, H. Liu, X. Liu, S. Wang, J. Wu, R. Zhang, H. Min and M. Huang, Carbohydr. Polym., 2015, 132, 351–358 CrossRef CAS PubMed.
- M. Mohseni, A. Shamloo, Z. Aghababaei, M. Vossoughi and H. Moravvej, Artif. Organs, 2016, 40, 765–773 CrossRef CAS PubMed.
- C. Ma, L. Liu, W. Hua, Y. Cai and J. Yao, Fibers Polym., 2015, 16, 1255–1261 CrossRef CAS.
- Y. Lan, W. Li, R. Guo, Y. Zhang, W. Xue and Y. Zhang, J. Biomater. Sci., Polym. Ed., 2014, 25, 75–87 CrossRef CAS PubMed.
- J. Kurczewska, P. Pecyna, M. Ratajczak, M. Gajęcka and G. Schroeder, Saudi Pharm. J., 2017, 25, 911–920 CrossRef PubMed.
- J. S. Boateng, K. H. Matthews, H. N. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97, 2892–2923 CrossRef CAS PubMed.
- D. A. Carter, S. E. Blair, N. N. Cokcetin, D. Bouzo, P. Brooks, R. Schothauer and E. J. Harry, Front. Microbiol., 2016, 7, 569 Search PubMed.
- A. Zumla and A. Lulat, J. R. Soc. Med., 1989, 82, 384–385 CrossRef CAS.
- R. Cooper, GMS Hyg. Infect. Control., 2007, 2, 1–3 Search PubMed.
- S. E. Maddocks and R. E. Jenkins, Future Microbiol., 2013, 8, 1419–1429 CrossRef CAS PubMed.
- R. A. Cooper, L. Jenkins, A. F. M. Henriques, R. S. Duggan and N. F. Burton, Eur. J. Clin. Microbiol. Infect. Dis., 2010, 29, 1237–1241 CrossRef CAS PubMed.
- J. Alvarez-Suarez, M. Gasparrini, T. Forbes-Hernández, L. Mazzoni and F. Giampieri, Foods, 2014, 3, 420–432 CrossRef PubMed.
- J. M. Packer, J. Irish, B. R. Herbert, C. Hill, M. Padula, S. E. Blair, D. A. Carter and E. J. Harry, Int. J. Antimicrob. Agents, 2012, 40, 43–50 CrossRef CAS PubMed.
- J. Majtan, J. Bohova, E. Prochazka and J. Klaudiny, J. Med. Food, 2014, 17, 290–293 CrossRef CAS PubMed.
- P. H. S. Kwakman and S. A. J. Zaat, IUBMB Life, 2012, 64, 48–55 CrossRef CAS PubMed.
- F. C. Bizerra, P. I. Da Silva Jr. and M. A. F. Hayashi, Front. Microbiol., 2012, 3, 398 Search PubMed.
- M. Ushio-Fukai and R. W. Alexander, Mol. Cell. Biochem., 2004, 264, 85–97 CrossRef CAS PubMed.
- C. Dunnill, T. Patton, J. Brennan, J. Barrett, M. Dryden, J. Cooke, D. Leaper and N. T. Georgopoulos, Int. Wound J., 2017, 14, 89–96 CrossRef PubMed.
- S. Samarghandian, T. Farkhondeh and F. Samini, Pharmacogn. Res., 2017, 9, 121–127 CAS.
- S. A. Meo, S. A. Al-Asiri, A. L. Mahesar and M. J. Ansari, Saudi J. Biol. Sci., 2017, 24, 975–978 CrossRef CAS PubMed.
- M. Dryden, G. Lockyer, K. Saeed and J. Cooke, J. Glob. Antimicrob. Resist., 2014, 2, 168–172 CrossRef PubMed.
- M. Dryden, Int. J. Antimicrob. Agents, 2017, 51, 299–303 CrossRef PubMed.
- M. Dryden, J. Cooke, R. J. Salib, R. Holding, S. Pender and J. Brooks, J. Glob. Antimicrob. Resist., 2017, 8, 194–198 CrossRef PubMed.
- F. D. Halstead, M. A. Webber, M. Rauf, R. Burt, M. Dryden and B. A. Oppenheim, J. Wound Care, 2016, 25, 93–102 CrossRef CAS PubMed.
- M. Dryden, A. Dickinson, J. Brooks, L. Hudgell, K. Saeed and K. F. Cutting, J. Wound Care, 2016, 25, 140–146 CrossRef CAS PubMed.
- T. J. Hall, J. M. A. Blair, R. J. A. Moakes, E. G. Pelan, L. M. Grover and S. C. Cox, Mater. Sci. Eng., 2019, 103, 1–10 Search PubMed.
- W. H. Organization, Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug resistant bacterial infection, including tuberculosis, WHO Press, 2017, pp. 1–87 Search PubMed.
- S. E. Blair, N. N. Cokcetin, E. J. Harry and D. A. Carter, Eur. J. Clin. Microbiol. Infect. Dis., 2009, 28, 1199–1208 CrossRef CAS PubMed.
- R. Cooper, J. Anal. At. Spectrom., 2009, 1, 6–10 Search PubMed.
- S. E. Maddocks, R. E. Jenkins, R. S. Rowlands, K. J. Purdy and R. A. Cooper, Future Microbiol., 2013, 8, 1523–1536 CrossRef CAS PubMed.
- J. Lu, L. Turnbull, C. M. Burke, M. Liu, D. A. Carter, R. C. Schlothauer, C. B. Whitchurch and E. J. Harry, PeerJ, 2014, 2, e326 CrossRef PubMed.
- J. Majtan, J. Bohova, M. Horniackova, J. Klaudiny and V. Majtan, Phytother. Res., 2014, 28, 69–75 CrossRef CAS PubMed.
- A. Taher, K. A. Zahan, M. A. Rajaie, C. R. Leong, S. A. Rashid, N. S. M. N. Hamin, W. N. Tan and W. Y. Tong, Mater. Today: Proc., 2020 Search PubMed.
- A. V. Kamaratos, K. N. Tzirogiannis, S. A. Iraklianou, G. I. Panoutsopoulos, I. E. Kanellos and A. I. Melidonis, Int. Wound J., 2014, 11, 259–263 CrossRef PubMed.
- E. Mancuso, C. Tonda-Turo, C. Ceresa, V. Pensabene, S. D. Connell, L. Fracchia and P. Gentile, Front. Bioeng. Biotechnol., 2019, 7, 344 CrossRef PubMed.
- F. Febriyenti, H. Lucida, A. Almahdy, I. Alfikriyah and M. Hanif, J. Pharm. BioAllied Sci., 2019, 11, 176 CrossRef PubMed.
- R. Khounganian, S. Auda, R. Al-Zaqzouq, R. Al-Zaqzouq, H. Al-Semari and F. Shakeel, J. Food Sci. Technol., 2020 DOI:10.1007/s13197-020-04459-6.
- M. Dryden, G. Milward and K. Saeed, J. Hosp. Infect., 2014, 88, 121–122 CrossRef CAS PubMed.
- M. Dryden, C. Goddard, A. Madadi, M. Heard, K. Saeed and J. Cooke, Br. J. Midwifery, 2014, 22, 23–27 CrossRef.
- M. Dryden, C. Tawse, J. Adams, A. Howard, K. Saeed and J. Cooke, J. Wound Care, 2014, 23(6), 338–341 CrossRef CAS PubMed.
- R. Jenkins and R. Cooper, PLoS One, 2012, 7, e45600 CrossRef CAS PubMed.
- P. Müller, D. G. Alber, L. Turnbull, R. C. Schlothauer, D. A. Carter, C. B. Whitchurch and E. J. Harry, PLoS One, 2013, 8, e57679 CrossRef PubMed.
- K. Markatos, G. Tsoucalas and M. Sgantzos, Acta Med. Hist. Adriat., 2016, 14, 161–176 Search PubMed.
- P. Brouqui, M. C. Rousseau, A. Stein, M. Drancourt and D. Raoult, Antimicrob. Agents Chemother., 1995, 39, 2423–2425 CrossRef CAS PubMed.
- N. Cerca, G. B. Pier, M. Vilanova, R. Oliveira and J. Azeredo, Res. Microbiol., 2005, 156, 506–514 CrossRef CAS PubMed.
- J. M. Higashi, I. W. Wang, D. M. Shlaes, J. M. Anderson and R. E. Marchant, J. Biomed. Mater. Res., 1998, 39, 341–350 CrossRef CAS PubMed.
- P. H. Hsieh, M. S. Lee, K. Y. Hsu, Y. H. Chang, H. N. Shih and S. W. Ueng, Clin. Infect. Dis., 2009, 49, 1036–1043 CrossRef PubMed.
- I. Uækay and L. Bernard, Clin. Infect. Dis., 2010, 50, 795 CrossRef PubMed.
- R. Vaishya, M. Chauhan and A. Vaish, J. Clin. Orthop. Trauma, 2013, 4, 157–163 CrossRef PubMed.
- J. G. E. Hendriks, J. R. Van Horn, H. C. Van Der Mei and H. J. Busscher, Biomaterials, 2004, 25, 545–556 CrossRef CAS PubMed.
- A. Bistolfi, G. Massazza, E. Verné, A. Massè, D. Deledda, S. Ferraris, M. Miola, F. Galetto and M. Crova, ISRN Orthop., 2011, 2011, 1–8 CrossRef PubMed.
- S. Breusch and H. Malchau, The Well-Cemented Total Hip Arthroplasty: Theory and Practice, Springer, Verlag Heidelberg, 2005 Search PubMed.
- L. Frommelt and K. D. Kuhn, Properties of bone cement: antibiotic loaded cement, Springer, Berlin, 2006 Search PubMed.
- C. P. Scott, P. A. Higham and J. H. Dumbleton, J. Bone Jt. Surg., 1999, 81, 440–443 CrossRef CAS.
- J. Martínez-Moreno, V. Merino, A. Nácher, J. L. Rodrigo, M. Climente and M. Merino-Sanjuán, Orthop. Surg., 2017, 9, 331–341 CrossRef PubMed.
- H. van de Belt, D. Neut, W. Schenk, J. R. van Horn, H. C. van der Mei and H. J. Busscher, Acta Orthop. Scand., 2011, 72, 557–571 CrossRef PubMed.
- D. J. F. Moojen, B. Hentenaar, H. C. Vogely, A. J. Verbout, R. M. Castelein and W. J. Dhert, J. Arthroplasty, 2011, 23, 1152–1156 CrossRef PubMed.
- A. C. McLaren, M. Nugent, K. Economopoulos, H. Kaul, B. L. Vernon and R. McLemore, Clin. Orthop. Relat. Res., 2009, 406, 1693–1698 CrossRef PubMed.
- S. Perni, S. Caserta, R. Pasquino, S. A. Jones and P. Prokopovich, ACS Appl. Bio Mater., 2019, 2, 1850–1861 CrossRef CAS.
- W. Zimmerli, A. Trampuz and P. E. Ochsner, N. Engl. J. Med., 2004, 351, 1645–1654 CrossRef CAS PubMed.
- S. G. Giulieri, P. Graber, P. E. Ochsner and W. Zimmerli, Infection, 2004, 32, 222–228 CrossRef CAS PubMed.
- N. J. Hickok and I. M. Shapiro, Adv. Drug Delivery Rev., 2012, 64, 1165–1176 CrossRef CAS PubMed.
- W. Ahmed, Z. Zhai and C. Gao, Mater. Today Bio., 2019, 2, 100017 CrossRef CAS PubMed.
- S. C. Cox, P. Jamshidi, N. M. Eisenstein, M. A. Webber, H. Hassanin, M. M. Attallah, E. T. Duncan, D. E. T. Shepherd, O. Addison and L. M. Grover, Mater. Sci. Eng., 2016, 64, 407–415 CrossRef CAS PubMed.
- A. Ahangari, M. Salouti, Z. Heidari, A. R. Kazemizadeh and A. A. Safari, Drug Delivery, 2013, 20, 34–39 CrossRef CAS PubMed.
- W. N. Ayre, J. C. Birchall, S. L. Evans and S. P. Denyer, J. Biomed. Mater. Res., Part B, 2016, 104, 1510–1524 CrossRef CAS PubMed.
- M. Salouti, Z. Heidari, A. Ahangari and S. Zare, Drug Delivery, 2016, 23, 49–54 CrossRef CAS PubMed.
- J. S. Moskowitz, M. R. Blaisse, R. E. Samuel, H. P. Hsu, M. B. Harris, S. D. Martin, J. C. Lee, M. Spector and P. T. Hammond, Biomaterials, 2011, 31, 6019–6030 CrossRef PubMed.
- S. Ikeda, K. Uchiyama, Y. Minegishi, K. Ohno, M. Nakamura, K. Yoshida, K. Fukushima, N. Takahira and M. Takaso, J. Orthop. Surg. Res., 2018, 13, 322 CrossRef PubMed.
- K. Anagnostakos, J. Bone Jt. Infect., 2017, 2, 29 CrossRef PubMed.
- D. J. McDonald, R. H. Fitzgerald and D. M. Ilstrup, J. Bone Jt. Surg., 1989, 71, 828–834 CrossRef CAS.
- A. M. Yousefi, J. Appl. Biomater. Funct. Mater., 2019, 17, 2280800019872594 Search PubMed.
- S. M. Rabiee, Bull. Mater. Sci., 2013, 36, 171–174 CrossRef CAS.
- S. M. Kenny and M. Buggy, J. Mater. Sci.: Mater. Med., 2003, 14, 923–938 CrossRef CAS PubMed.
- T. Tüzüner, A. Dimkov and J. W. Nicholson, Acta Biomater. Odontol. Scand., 2019, 5, 9–21 CrossRef PubMed.
- D. H. Nies, Appl. Microbiol. Biotechnol., 1999, 51, 730–750 CrossRef CAS PubMed.
- J. J. Harrison, H. Ceri, C. A. Stremick and R. J. Turner, Environ. Microbiol., 2004, 6, 1220–1227 CrossRef CAS PubMed.
- G. M. Teitzel and M. R. Parsek, Appl. Environ. Microbiol., 2013, 69, 2313–2320 CrossRef PubMed.
- W. Sim, R. T. Barnard, M. A. T. Blaskovich and Z. M. Ziora, Antibiotics, 2018, 7, 93 CrossRef CAS PubMed.
- C. Fan, L. Chu, H. R. Rawls, B. K. Norling, H. L. Cardenas and K. Whang, Dent. Mater., 2011, 27, 322–328 CrossRef CAS PubMed.
- D. R. Monteiro, L. F. Gorup, A. S. Takamiya, A. C. Ruvollo-Filho, E. R. de Camargo and D. B. Barbosa, Int. J. Antimicrob. Agents, 2009, 34, 103–110 CrossRef CAS PubMed.
- E. Sudmann, H. Vik, M. Rait, K. Todnem, K. J. Andersen, K. Julsham, O. Flesland and J. Rungby, Med. Prog. Technol., 1994, 20, 179–184 CAS.
- J. Slane, J. Vivanco, W. Rose, H. L. Ploeg and M. Squire, Mater. Sci. Eng., 2015, 48, 188–196 CrossRef CAS PubMed.
- M. K. Rai, S. D. Deshmukh, A. P. Ingle and A. K. Gade, J. Appl. Microbiol., 2012, 112, 841–852 CrossRef CAS PubMed.
- V. Alt, M. Rupp, K. Lemberger, T. Bechert, T. Konradt, P. Steinrücke, R. Schnettler, S. Söder and R. Ascherl, Bone Joint Res., 2019, 8, 387–396 CrossRef PubMed.
- D. J. F. Moojen, H. C. Vogely, A. Fleer, A. J. Verbout, R. M. Castelein and W. J. Dhert, J. Orthop. Res., 2009, 27, 1002–1007 CrossRef CAS PubMed.
- S. A. Brennan, C. Ní Fhoghlú, B. M. Devitt, F. J. O′mahony, D. Brabazon and A. Walsh, Bone Joint J., 2015, 97, 582–589 CrossRef PubMed.
- P. Prokopovich, M. Köbrick, E. Brousseau and S. Perni, J. Biomed. Mater. Res., Part B, 2015, 103, 273–281 CrossRef PubMed.
- S. Marin, G. M. Vlasceanu, R. E. Tiplea, I. R. Bucur, M. Lemnaru, M. M. Marin and A. M. Grumezescu, Curr. Trends Med. Chem., 2015, 15, 1596–1604 CrossRef CAS PubMed.
- G. Hulsart-Billström, J. I. Dawson, S. Hofmann, R. Müller, M. J. Stoddart, M. Alini, H. Redl, A. El Haj, R. Brown, V. Salih, J. Hillborn, S. Larsson and R. O. Oreffo, Eur. Cell Mater., 2016, 31, 312–322 CrossRef PubMed.
- V. Athans, M. P. Veve and S. L. Davis, Pharmacotherapy, 2017, 37, 1565–1577 CrossRef PubMed.
- J. A. Aas, B. J. Paster, L. N. Stokes, I. Olsen and F. E. Dewhirst, J. Clin. Microbiol., 2005, 43, 5721–5732 CrossRef PubMed.
- N. Kommerein, K. Doll, N. S. Stumpp and M. Stiesch, PLoS One, 2018, 13, e0196967 CrossRef PubMed.
- M. Astasov-Frauenhoffer, O. Braissant, I. Hauser-Gerspach, A. U. Daniels, R. Weiger and T. Waltimo, FEMS Microbiol. Lett., 2012, 337, 31–37 CrossRef CAS PubMed.
- S. Roehling, M. Astasov-Frauenhoffer, I. Hauser-Gerspach, O. Braissant, H. Woelfler, T. Waltimo, H. Kniha and M. Gahlert, J. Periodontol., 2017, 88, 298–307 CrossRef CAS PubMed.
- S. E. Mountcastle, S. C. Cox, R. L. Sammons, S. Jabbari, R. M. Shelton and S. A. Kuehne, J. Oral Microbiol., 2020, 12, 1773122 CrossRef.
- J. Sjollema, S. A. Zaat, V. Fontaine, M. Ramstedt, R. Luginbuehl, K. Thevissen, J. Li, H. C. van der Mei and H. J. Busscher, Acta Biomater., 2018, 70, 12–24 CrossRef CAS PubMed.
- J. L. Martínez, F. Baquero and D. I. Andersson, Curr. Opin. Pharmacol., 2011, 11, 439–445 CrossRef PubMed.
- R. Masaeli, K. Zandsalimi and L. Tayebi, Ther. Innov. Regul. Sci., 2019, 53, 120–127 CrossRef PubMed.
- L. B. Reller, M. Weinstein, J. H. Jorgensen and M. J. Ferraro, Clin. Infect. Dis., 2009, 49, 1749–1755 CrossRef PubMed.
- R. W. Bauman, E. Machunis-Masuoka and I. R. Tizard, Microbiology: with diseases by taxonomy, Benjamin Cummings, California, 2011 Search PubMed.
- Z. A. Khan, M. F. Siddiqui and S. Park, Diagnostics, 2019, 9, 49 CrossRef CAS PubMed.
- A. van Belkum, C. A. D. Burnham, J. W. Rossen, F. Mallard, O. Rochas and W. M. Dunne, Nat. Rev. Microbiol., 2020, 18, 299–311 CrossRef CAS PubMed.
- J. M. Anderson, Regener. Biomater., 2016, 3, 73–77 CrossRef CAS PubMed.
- Y. Jiang, C. K. Mehta, T. Y. Hsu and F. F. Alsulaimani, Infect. Immun., 2002, 70, 3143–3148 CrossRef CAS PubMed.
- D. Seybold, T. A. Schildhauer, J. Geßmann, G. Muhr, M. Köller and B. Roetman, Langenbecks Arch. Surg., 2010, 395, 719–726 CrossRef PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. CélinePulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini and W. P. P. L. W. Group, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef PubMed.
- N. Gugala, J. Lemire, K. Chatfield-Reed, Y. Yan, G. Chua and R. J. Turner, Genes, 2018, 9, 344 CrossRef PubMed.
- C. P. Randall, L. B. Oyama, J. M. Bostock, I. Chopra and A. J. O'Neill, J. Antimicrob. Chemother., 2013, 68, 131–138 CrossRef CAS PubMed.
- C. P. Randall, A. Gupta, N. Jackson, D. Busse and A. J. O'Neill, J. Antimicrob. Chemother., 2015, 70, 1037–1046 CAS.
- J. L. Graves Jr., M. Tajkarimi, Q. Cunningham, A. Campbell, H. Nonga, S. H. Harrison and J. E. Barrick, Front. Genet., 2015, 6, 42 Search PubMed.
- S. Sütterlin, PhD Thesis, Acta Universitatis Upsaliensis, 2015.
- E. Elkrewi, C. P. Randall, N. Ooi, J. L. Cottell and A. J. O'Neill, J. Antimicrob. Chemother., 2017, 72, 3043–3046 CrossRef CAS PubMed.
- A. Panáček, L. Kvítek, M. Smékalová, R. Večeřová, M. Kolář, M. Röderová, F. Dyčka, M. Šebela, R. Prucek, O. Tomanec and R. Zbořil, Nat. Nanotechnol., 2018, 13, 65–71 CrossRef PubMed.
- M. B. Massani, J. Klumpp, M. Widmer, C. Speck, M. Nisple, R. Lehmann and M. Schuppler, BioMetals, 2018, 31, 1101–1114 CrossRef PubMed.
- G. Cheng, M. Dai, S. Ahmed, H. Hao, X. Wang and Z. Yuan, Front. Microbiol., 2016, 7, 470 Search PubMed.
- F. De la Cruz and J. Davies, Trends Microbiol., 2000, 8, 128–133 CrossRef CAS PubMed.
- J. W. Lee, MethodsX, 2019, 6, 1564–1574 CrossRef PubMed.
- A. R. Burmeister, Evol. Med. Public Health, 2015, 2015, 193–194 CrossRef PubMed.
- D. Lebeaux, J.-M. Ghigo and C. Beloin, Microbiol. Mol. Biol. Rev., 2014, 78, 510–543 CrossRef CAS PubMed.
- R. J. W. Lambert and J. Pearson, J. Appl. Microbiol., 2000, 88, 784–790 CrossRef CAS PubMed.
- F. Sepandj, H. Ceri, A. Gibb, R. Read and M. Olson, Peritoneal Dial. Int., 2004, 24, 65–67 CrossRef CAS.
- Y. Kanazawa, J. Antibiot., 1966, 19, 175–189 CAS.
- Clinical and Laboratory Standards Institute, Performance Standards for Antimicrobial Susceptibility Testing, 2018 Search PubMed.
- C. I. Owuama, J. Microbiol. Res., 2017, 11, 977–980 CAS.
- X. Zhao and K. Drlica, J. Infect. Dis., 2002, 185(4), 561–565 CrossRef PubMed.
- D. Campoccia, L. Montanaro, P. Speziale and C. R. Arciola, Biomaterials, 2010, 31, 6363–6377 CrossRef CAS PubMed.
- J. C. Schuh and K. A. Funk, Toxicol. Pathol., 2019, 47, 344–357 CrossRef PubMed.
- A. Purnama, FDA regulatory pathways for medical devices, TOPRA, 2019 Search PubMed.
- S. Rauh, L. Mavroeidis, P. Ntellas, I. Gazouli, S. Gkoura, A. Papadaki, D. Mauri, Y. Metaxas, J. Y. Douillard and G. Pentheroudakis, ESMO Open, 2020, 5, e000615 CrossRef.
- D. B. Jefferys, Br. J. Clin. Pharmacol., 2001, 52, 229–235 CrossRef CAS PubMed.
- I. Chaston, Lack of data problems, SAGE Publications Ltd, London, 2009 Search PubMed.
- E. Mariani, G. Lisignoli, R. M. Borzì and L. Pulsatelli, Int. J. Med. Microbiol., 2019, 20, 636 CAS.
- A. F. Widmer, R. Frei, Z. Rajacic and W. Zimmerli, J. Infect. Dis., 1990, 162, 96–102 CrossRef CAS PubMed.
- J. A. Washington II, Diagn. Microbiol. Infect. Dis., 1983, 1, 25–31 CrossRef.
- M. De Rond and A. N. Miller, J. Manag. Inq., 2005, 14, 321–329 CrossRef.
- B. Van Looy, J. Callaert and K. Debackere, Resour. Policy, 2006, 35, 596–608 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |