
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSymmetry breaking and the turn-on fluorescence of small, highly strained carbon nanohoops†
Terri C.
Lovell‡
 a,
Curtis E.
Colwell‡
a,
Lev N.
Zakharov
b and
Ramesh
Jasti
*a
a,
Curtis E.
Colwell‡
a,
Lev N.
Zakharov
b and
Ramesh
Jasti
*a
aDepartment of Chemistry & Biochemistry, Materials Science Institute, University of Oregon, Eugene, Oregon 97403, USA. E-mail: rjasti@uoregon.edu
bCAMCOR – Center for Advance Materials Characterization in Oregon, University of Oregon, Eugene, Oregon 97403, USA
First published on 25th February 2019
Abstract
[n]Cycloparaphenylenes, or “carbon nanohoops,” are unique conjugated macrocycles with radially oriented π-systems similar to those in carbon nanotubes. The centrosymmetric nature and conformational rigidity of these molecules lead to unusual size-dependent photophysical characteristics. To investigate these effects further and expand the family of possible structures, a new class of related carbon nanohoops with broken symmetry is disclosed. In these structures, referred to as meta[n]cycloparaphenylenes, a single carbon–carbon bond is shifted by one position in order to break the centrosymmetric nature of the parent [n]cycloparaphenylenes. Advantageously, the symmetry breaking leads to bright emission in the smaller nanohoops, which are typically non-fluorescent due to optical selection rules. Moreover, this simple structural manipulation retains one of the most unique features of the nanohoop structures—size dependent emissive properties with relatively large extinction coefficients and quantum yields. Inspired by earlier theoretical work by Tretiak and co-workers, this joint synthetic, photophysical, and theoretical study provides further design principles to manipulate the optical properties of this growing class of molecules with radially oriented π-systems.
Introduction
Carbon nanomaterials have been intensely studied in materials science, physics, and biology due to their outstanding strength, enhanced conductivity, biocompatibility, and near-infrared absorption and emission.1–5 The arrangement of the carbon atoms in these materials along with the size of the structure play a key role in the observed properties. As such, achieving atomic-level control in the preparation of carbon nanomaterials is a grand challenge in the field nanoscience. Over the last several years, in an effort to prepare carbon nanotubes with precise structure, the syntheses of the [n]cycloparaphenylenes (CPPs) (Fig. 1a) were developed.6–10 These strained macrocyclic structures, often referred to as carbon nanohoops, are composed of all para-linked phenylenes and can be thought of as a short slice of a carbon nanotube (Fig. 1a). These structures, unlike typical carbon nanomaterials, can be manipulated with atomic precision since they are prepared using stepwise organic synthesis. Moreover, these materials and related derivatives have shown advantageous optoelectronic properties for potential applications ranging from organic electronics,11,12 to supramolecular sensing,13–15 to bioimaging.16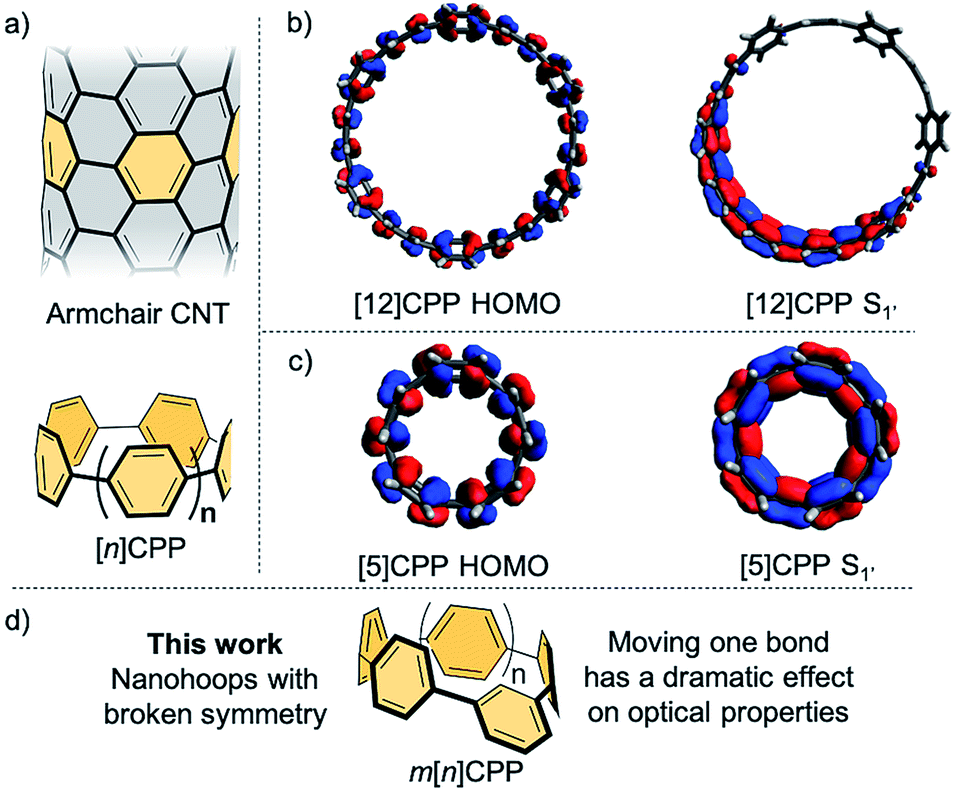 | ||
| Fig. 1 (a) Structure of an armchair carbon nanotube (CNT) and its relation to [n]cycloparaphenylenes; (b) HOMO (left) and excited state (right) S1′ orbitals of [12]CPP and (c) HOMO (left) and excited state (right) S1′ orbitals of [5]CPP. Orbitals have been calculated using CAM-B3LYP/STO-3G level of theory. (d) meta[n]CPPs with broken symmetry in this work. | ||
Interestingly, the nanohoops possess size dependent optical properties that stand in stark contrast to related materials such as acyclic oligophenylenes or even semiconducting quantum dots. Whereas most materials show a red-shifting fluorescence emission with increasing size, the CPPs have a red-shifting fluorescence with decreasing size. For example, [12]CPP emits at 450 nm whereas [8]CPP emits at 533 nm.17 Concomitant with this red-shifting fluorescence is an accompanying decreasing quantum yield as the nanohoop size decreases. For example, [12]CPP has a quantum yield of 81% whereas the smallest CPPs, [5]- and [6]CPP, are completely non-emissive.18–20 Another very unique feature of the CPP optics is that the major absorption is entirely independent of the diameter with a maximum at 340 nm for all CPPs.21,22
These unique photophysical properties spurred investigation into theoretical explanations of these phenomena. The absorption phenomena has been explained by Yamago and co-workers21 wherein the major absorption is dominated by transitions that are similar in energy (i.e. HOMO → LUMO+1 or LUMO+2 and HOMO−1 or HOMO−2 → LUMO) amongst all sized CPPs and the HOMO → LUMO transition is forbidden due to symmetry. Similarly, detailed theoretical work by Tretiak and co-workers suggested that CPPs with more than seven phenyl rings are emissive due to exciton localization in an S1′ excited state in which the centrosymmetry is broken, seen in Fig. 1b for [12]CPP.23 Since this localization and symmetry breaking does not happen in the smaller sizes (Fig. 1c), these structures become non-emissive as the transition is forbidden by symmetry. These works suggest that disrupting the centrosymmetric nature of the molecular orbitals is a strategy that could be employed to alter the photophysical properties of the nanohoops. This basic concept was theoretically explored by Tretiak wherein they postulated that inserting different acenes into the CPP backbone would break the excited state symmetry.24
Inspired by these works, we report the synthesis, characterization, and analysis of a new class of carbon nanohoops wherein one phenyl ring is linked in the meta-position (Fig. 1d). This minor change in linkage, or “kink”, acts to break the conjugation of the nanohoop, therefore altering the symmetry of the molecular orbitals without significantly decreasing the inherent strain in the molecule. The meta-nanohoops, termed meta[n]CPPs (m[n]CPPs), are compared to the [n]CPP series to further understand what effect this small structural perturbation of the hoop has on the photophysical properties and to provide experimental evidence corroborating Tretiak's theoretical prediction.23 Additionally, tuning the photophysical properties of this growing class of structures is critical for exploiting them as novel scaffolds in biological imaging,16 supramolecular sensing13–15 as well as novel optoelectronic materials.11,12 Herein, we report the general synthesis of an entire series of m[n]CPPs, carbon nanohoops with broken symmetry, and a detailed study of their photophysical properties.
Results and discussion
Synthesis
The preparation of these fully conjugated and highly bent macrocycles is a synthetic challenge due to the large amount of intrinsic strain in the target molecules. The most strained target compound, m[5]CPP, is calculated to have 102 kcal mol−1 of strain (vide infra). Fortunately, methods for the synthesis of [n]CPPs can be adapted, wherein the strain is incorporated using cyclohexadienes as curved masked phenylenes. Building blocks 1–7 can easily be accessed on the gram scale using methods previously developed (Scheme 1).10,25 By combining these building blocks through selective lithiations followed by diastereoselective additions, or Suzuki Miyaura cross couplings, advanced intermediates 8–12 were readily prepared (see Section 1 of ESI† for more detail). Following this, relatively unstrained macrocycles 13–17 were prepared via Suzuki–Miyaura cross coupling of intermediates 8–12 and 1,3-dibromobenzene or 1,3-benzenediboronic acid bis(pinacol) ester in moderate yields ranging from 10–45%. The triethylsilyl protecting groups were removed and the cyclohexadienes were unmasked via reductive aromatization to yield m[6]-, m[7]-, m[8]-, m[10]-, and m[12]CPP in fairly good yields. As proposed, upon synthesis of m[6]CPP, we immediately noticed bright green fluorescence, which is not observed in the parent [6]CPP. Characterization by NMR (1H and 13C), IR, mass spectrometry, and X-ray crystallography (for m[6]CPP) confirmed structural assignment. A telling piece of characterization data for the product is the chemical shift of the inward pointing proton present on the meta-connected phenylene. As the nanohoop shrinks, the proton is forced further into the shielding cones of the flanking phenylenes. This results in the signal shifting upfield from 7.12 ppm for m[12]CPP to 5.62 ppm for m[6]CPP. Characterization by cyclic voltammetry resulted in redox chemistry similar to that of [n]CPPs (page S36–S39†).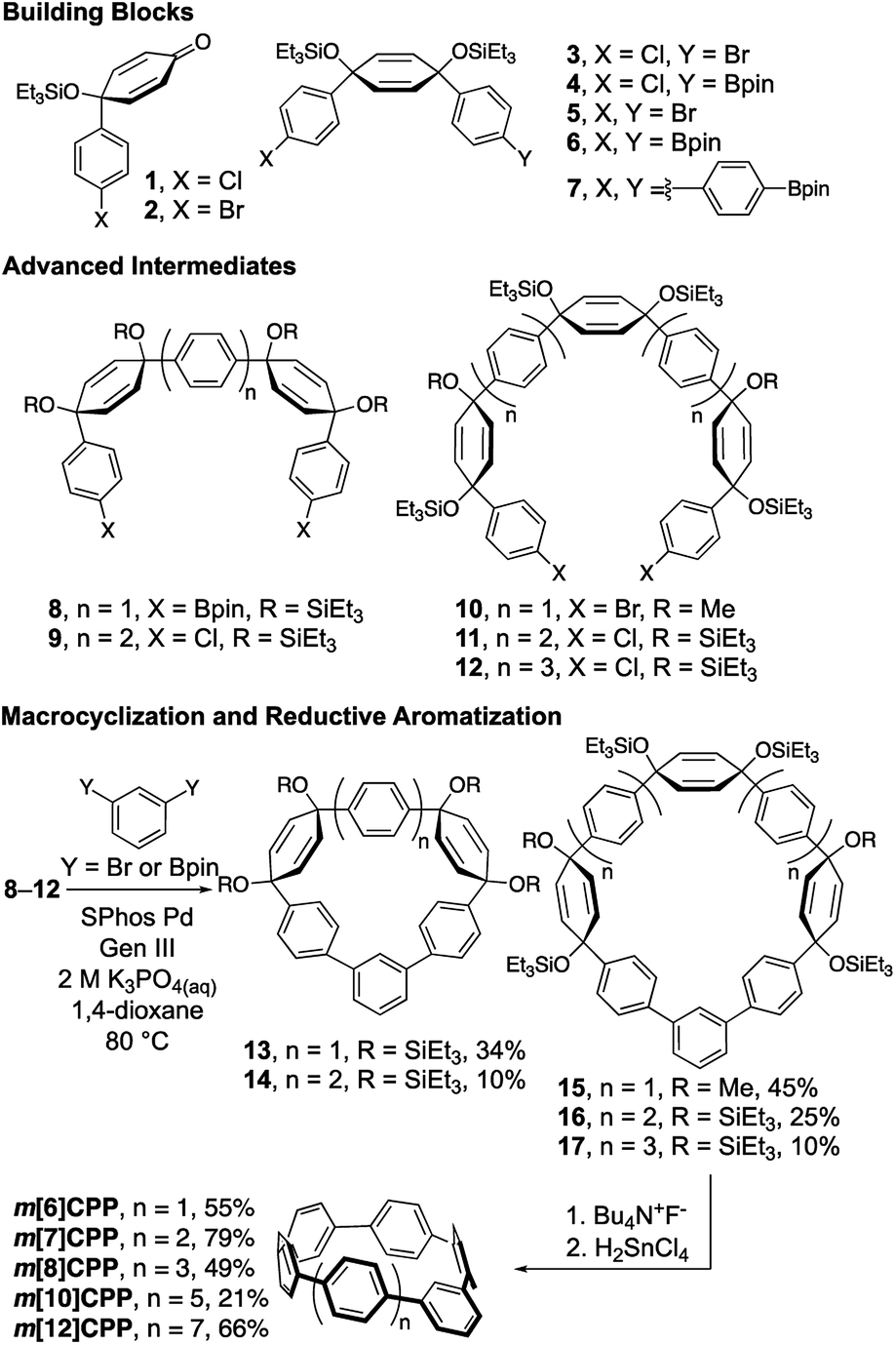 | ||
| Scheme 1 Building block synthetic approach to m[6]-, m[7]-, m[8]-, m[10]- and m[12]CPP. | ||
The synthesis of the most strained m[5]CPP required a slightly different strategy (Scheme 2). Here, the meta-functionalized benzene was incorporated into ketone precursor 18. Lithiation of 3 and addition to ketone 18, followed by protection with triethylsilyl chloride affords advanced intermediate 19. Miyaura borylation gives the bisboronate 20 in good yield. Oxidative homocoupling10 then smoothly transforms 20 to the challenging macrocycle 21 in 42% yield under mild conditions. Deprotection and reductive aromatization yielded m[5]CPP. Again, we noticed immediately that this very strained meta-nanohoop is fluorescent whereas the parent [5]CPP is non-emissive. With a series of these highly strained CPP analogues in hand, the influence of symmetry breaking on the properties of these materials was explored.
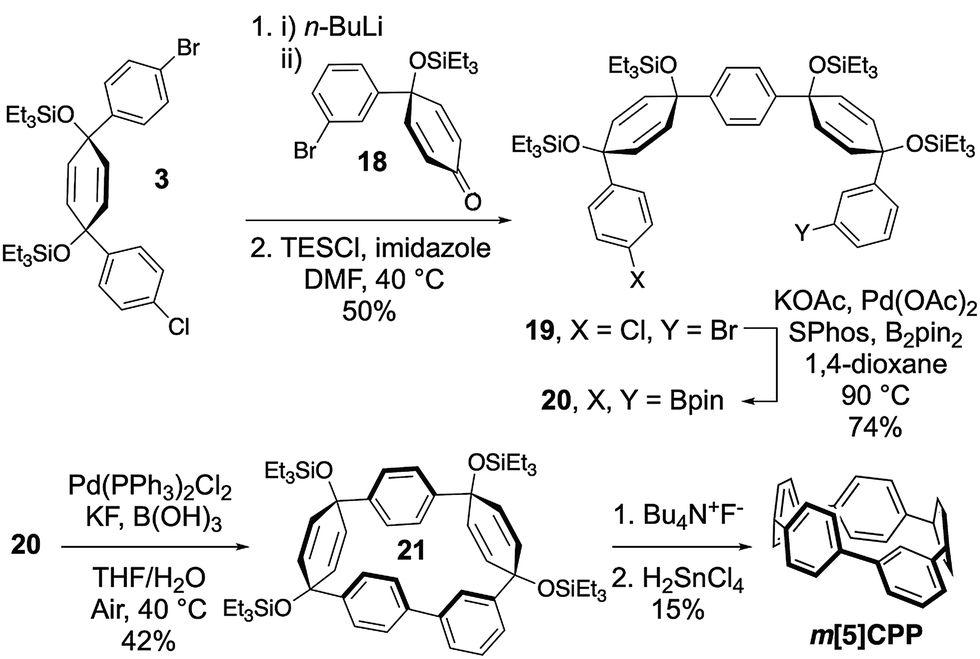 | ||
| Scheme 2 Modified synthetic strategy for m[5]CPP. | ||
Structural features
When discussing carbon nanohoops, strain is a critical quality that endows them with atypical optical properties22,26 and reactivity.27,28 As the diameter of these carbon nanohoops decreases, the inherent strain of the molecule increases and changes the geometry and optical properties. To gain some insight into the strain of these new nanohoop molecules, single crystals of m[6]CPP were obtained by slow evaporation of dichloromethane solutions. From inspection of the crystal structure, the dihedral angle decreases from 49° at the meta-phenylene to 19° at the opposite side of the hoop, seen in Fig. 2a. This suggests that the part of the nanohoop opposing the meta-phenyl unit has even more dihedral strain than [6]CPP, which has an average dihedral angle of 26°.18 In order to quantify the strain energy of the entire m[n]CPP series, theoretical homodesmotic reactions were performed at B3LYP/6-31G(d,p) level of theory.29 The inherent strain for the series ranges from 43–102 kcal mol−1. Compared to conventional CPPs with the same amount of phenylenes, m[n]CPPs are less strained by about 20%, as seen in Fig. 2b. The most strained macrocycle of our series, m[5]CPP, possesses 102 kcal mol−1 of strain. This places it below [5]CPP (119 kcal mol−1),18 and above [6]CPP (97 kcal mol−1)21 in terms of strain. The calculated structures show an expected gradual decrease in dihedral angle and increase in ipso carbon deviation from planarity as the size of the nanohoop decreases, similar to that seen in the parent [n]CPPs (Table S9†). Interestingly, the dihedral angle next to the meta-phenylene widens with increasing strain. Although the strain can only be inferred from dihedral angles between phenylenes and calculated homodesmotic reactions, it is clear the strain plays a central role in defining the properties of these molecules.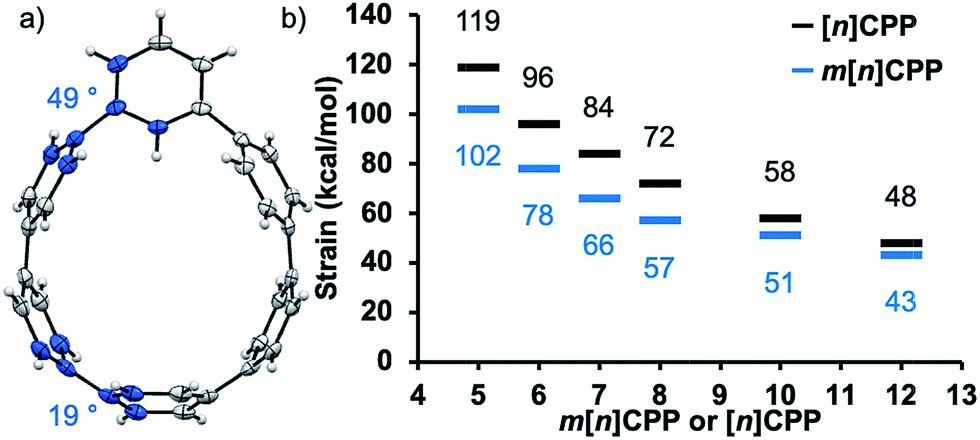 | ||
| Fig. 2 (a) Molecular structure of m[6]CPP determined by X-ray crystallography (thermal ellipsoids shown at 50% probability). (b) Calculated strain using homodesmotic reactions of [n]CPPs (black) and m[n]CPPs (blue) at B3LYP/6-31G(d,p) level of theory. | ||
Photophysical properties
The photophysical properties of these molecules are particularly exciting. Similar to CPPs, the m[n]CPPs have a common absorption maximum around 328 nm (Fig. 3a) from HOMO−1 → LUMO and HOMO → LUMO+1 transitions (Fig. S13–S18†). However, in this series there is a red-shifting second absorption as the size of the hoop decreases (visible as a peak for m[6]–m[8]CPP and a shoulder to the main absorption at 328 nm for m[10]- and m[12]CPP), which is the HOMO → LUMO absorption. The extinction coefficient of the higher energy transition is larger than that of the lower energy transition in all cases (Fig. 3c and Table S3†). The series shows decreasing, but never vanishing, fluorescence ranging from 429–534 nm and quantum yields ranging from 0.01 for m[5]CPP to 0.77 for m[12]CPP (Fig. 3a and b). Fluorescent lifetimes of all m[n]CPPs are around 3 ns (Table S4†), which is different than the [n]CPP series with lifetimes ranging from 2–18 ns.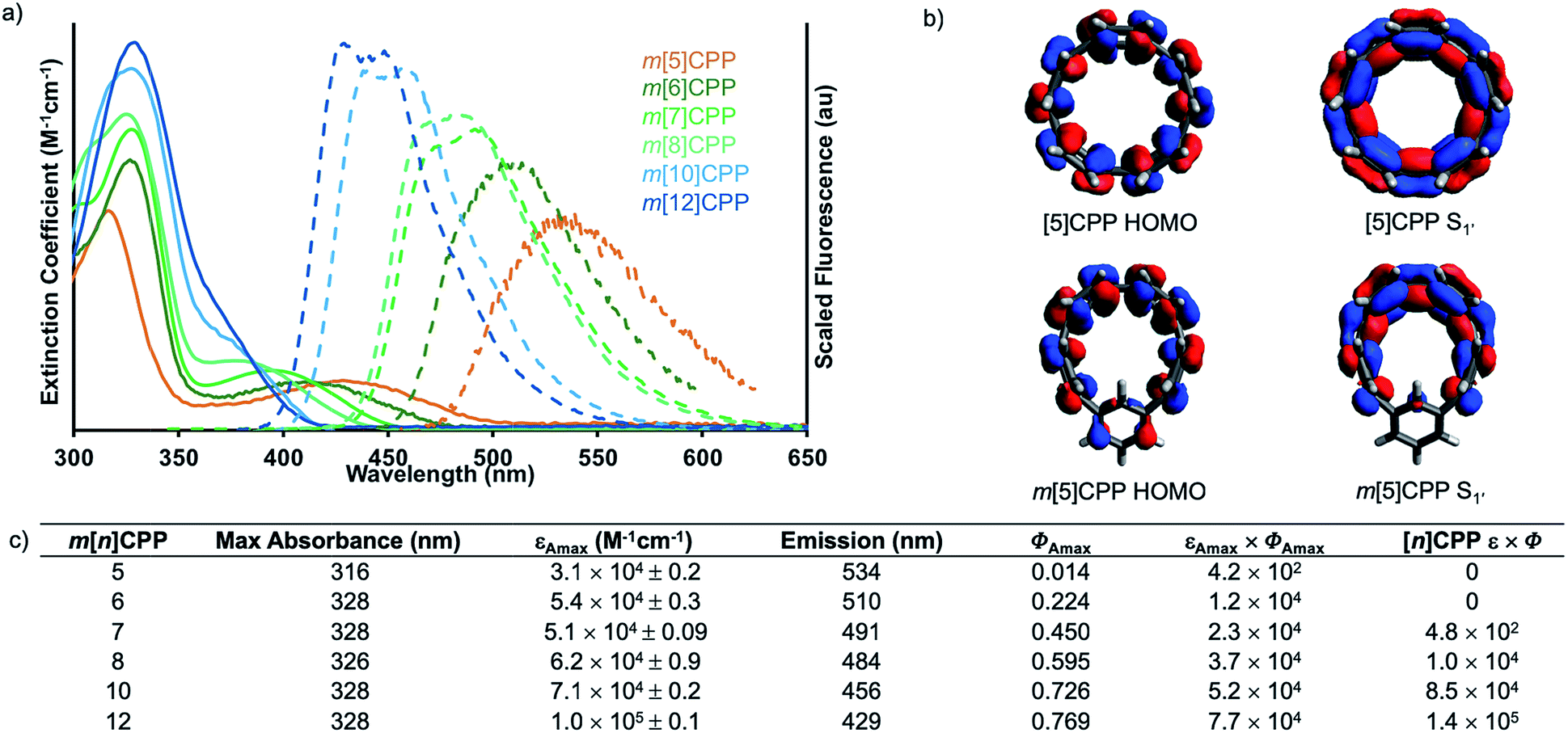 | ||
| Fig. 3 (a) Absorbance and emission spectra of m[n]CPPs; (b) HOMO (left) and S1′ (right) orbital depiction of [5]CPP and m[5]CPP, demonstrating change in orbital symmetry. Calculated using CAM-B3LYP/STO-3G level of theory; (c) absorbance, extinction coefficient (ε), emission and quantum yield (Φ) of m[5]–m[8]-, m[10]- and m[12]CPP and brightness comparison of m[n]CPPs and [n]CPPs. | ||
Density functional theory calculations and a comparison to the [n]CPPs were used to explain the photophysical phenomena further. As mentioned earlier, HOMO → LUMO transition of [n]CPPs is Laporte forbidden due to conservation of orbital symmetry of the ground and excited state. The CPPs are therefore excited through HOMO → LUMO+1 and HOMO → LUMO+2 or HOMO−1 → LUMO and HOMO−2 → LUMO. From these states, internal conversion to a spatially localized S1′ state occurs. Here, the larger [n]CPPs (n ≥ 8) exhibit exciton localization over about seven of the phenylenes (Fig. 1b). When exciton localization occurs, the symmetry is different than the ground state, allowing the S1′ → HOMO transition. When n ≤ 7 there is complete orbital delocalization over the whole S1′ excited state structure (Fig. 1c), therefore the ground state symmetry is conserved. In these cases, the S1′ → HOMO transition is Laporte forbidden, resulting in undetectable fluorescence for [5]CPP and [6]CPP and only weak fluorescence for [7]CPP.
Our calculations show that changing a single phenylene from para to meta does in fact change the orbital symmetry of the π-system. Fig. 3b demonstrates the difference in orbital symmetry between the HOMO and relaxed excited state of m[5]CPP compared to [5]CPP. The symmetry broken nanohoops show a dramatic increase in intensity for both the HOMO → LUMO and S1′ → HOMO transitions. This is apparent from a tenfold increase in the extinction coefficient and oscillator strength of the HOMO → LUMO transition when comparing m[5]CPP with an extinction coefficient of 6.0 × 103 M−1 cm−1 and oscillator strength of 0.1217 to [5]CPP possessing an extinction coefficient of 4.5 × 102 M−1 cm−1 and oscillator strength of 0.0015.18 The change in orbital symmetry also results in a “turn on” in fluorescence of the smaller sizes.
Like [n]CPPs, the quantum yield of m[n]CPPs decreases with decreasing size. However, the transition is at no point forbidden by symmetry as is the case for [n]CPPs. As such, the reduction in quantum yield is attributed to strain effects. It has been reported previously that curving a conjugated system, such as p-phenylenes30 or pyrene,31 reduces the quantum yield respective to the increase in strain. For m[n]CPPs, the decrease in quantum yield indicates an increase in the rate of non-radiative decay (knr) as the fluorescence lifetime was found to be relatively constant across all m[n]CPPs measured (Table S4†). In CPPs, the lifetime increases as the diameter decreases and the S1′ → HOMO transition is forbidden due to centrosymmetry. In contrast, introducing a meta phenylene allows S1′ → HOMO transitions across the entire series of m[n]CPPs.
To truly assess the aptitude of the m[n]CPPs to serve as enhanced fluorophores compared to their [n]CPPs analogues, we turn to their brightness, which is the product of the extinction coefficient and quantum yield. Nanohoops m[5]–m[8]CPPs have an obvious increase in brightness over their para-counterparts, seen in Fig. 3c. For example, [8]CPP was previously used as a fluorescent probe with a brightness of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.16 Now, m[6]CPP has a comparable brightness of 12000 M−1 cm−1, but is synthetically far easier to access. This edge is lost at larger sizes wherein m[10]- and m[12]CPPs are still brighter than many commercial fluorophores like DAPI,32–34 AMC,35 and rhodamine 110,32,36,37 but not quite as bright as [10]-38 and [12]CPP.38,39 We anticipate that this is relevant to the biological applications of nanohoops as new types of biocompatible fluorophores and novel fluorescent sensing materials.16
000 M−1 cm−1.16 Now, m[6]CPP has a comparable brightness of 12000 M−1 cm−1, but is synthetically far easier to access. This edge is lost at larger sizes wherein m[10]- and m[12]CPPs are still brighter than many commercial fluorophores like DAPI,32–34 AMC,35 and rhodamine 110,32,36,37 but not quite as bright as [10]-38 and [12]CPP.38,39 We anticipate that this is relevant to the biological applications of nanohoops as new types of biocompatible fluorophores and novel fluorescent sensing materials.16
Conclusions
The connectivity of carbon atoms, size, and symmetry all play critical roles in determining the properties of carbon nanomaterials. Rarely can these variables be systematically probed in a precise manner. Bottom-up synthetic strategies allow for the examination of these fundamental questions in an unambiguous manner. By rational design, a series of m[n]CPPs were prepared wherein a single carbon–carbon bond is moved over by one position from the parent carbon nanohoop structures ([n]CPPs). Shifting a CPP's phenylene from para to meta was proven as an efficient means to activate the previously forbidden absorption and emission transitions by breaking orbital symmetry, resulting in a fluorescence turn-on of the smaller nanohoops. The enhancement of fluorescence was accompanied by a blue-shift of these transitions proportional to a decrease in strain of about 20%. Advantageously, smaller nanohoops, which are more easily accessed by synthesis, can now be rendered fluorescent. Moreover, the smaller binding pockets of the smaller nanohoops provide an opportunity for fluorescence sensing of analytes that are not possible with the larger hoops. In relation to this possible application, fortuitously, these m[n]CPPs have a C–H group directed to the interior of the structure which could be exchanged in a second-generation design to a coordinating group. Further studies of these new nanohoop structures will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Science Foundation (RJ, CHE-1800586), UO OHSU Seed Grant Program, the Camille and Henry Dreyfus foundation and generous start-up funds from the University of Oregon. Mass spectroscopy support was provided by NSF (CHE-1625529). This work benefited from access to the University of Oregon high performance computer, Talapas. We would also like to acknowledge Thermo Fisher Scientific (Eugene, OR) for the use of their instrumentation.Notes and references
- Z. Spitalsky, D. Tasis, K. Papagelis and C. Galiotis, Prog. Polym. Sci., 2010, 35, 357–401 CrossRef CAS.
- G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816–10906 CrossRef CAS PubMed.
- M. M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. Del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824–2860 RSC.
- L. Wen, F. Li and H. M. Cheng, Adv. Mater., 2016, 28, 4306–4337 CrossRef CAS PubMed.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS PubMed.
- S. Yamago, Y. Watanabe and T. Iwamoto, Angew. Chem., Int. Ed., 2010, 49, 757–759 CrossRef CAS PubMed.
- H. Omachi, S. Matsuura, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2010, 49, 10202–10205 CrossRef CAS PubMed.
- V. K. Patel, E. Kayahara and S. Yamago, Chem.–Eur. J., 2015, 21, 5742–5749 CrossRef CAS PubMed.
- E. R. Darzi, B. M. White, L. K. Loventhal, L. N. Zakharov and R. Jasti, J. Am. Chem. Soc., 2017, 139, 3106–3114 CrossRef CAS PubMed.
- M. Ball, Y. Zhong, B. Fowler, B. Zhang, P. Li, G. Etkin, D. W. Paley, J. Decatur, A. K. Dalsania, H. Li, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2016, 138, 12861–12867 CrossRef CAS PubMed.
- E. Kayahara, L. Sun, H. Onishi, K. Suzuki, T. Fukushima, A. Sawada, H. Kaji and S. Yamago, J. Am. Chem. Soc., 2017, 139, 18480–18483 CrossRef CAS PubMed.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Angew. Chem., Int. Ed., 2011, 50, 8342–8344 CrossRef CAS PubMed.
- P. Della Sala, C. Talotta, A. Capobianco, A. Soriente, M. De Rosa, P. Neri and C. Gaeta, Org. Lett., 2018, 20, 37 CrossRef PubMed.
- P. Della Sala, C. Talotta, T. Caruso, M. De Rosa, A. Soriente, P. Neri and C. Gaeta, J. Org. Chem., 2017, 82, 9885–9889 CrossRef CAS PubMed.
- B. M. White, Y. Zhao, T. E. Kawashima, B. P. Branchaud, M. D. Pluth and R. Jasti, ACS Cent. Sci., 2018, 4, 1173–1178 CrossRef CAS PubMed.
- E. R. Darzi and R. Jasti, Chem. Soc. Rev., 2015, 44, 6401–6410 RSC.
- P. J. Evans, E. R. Darzi and R. Jasti, Nat. Chem., 2014, 6, 404–408 CrossRef CAS PubMed.
- E. Kayahara, V. K. Patel and S. Yamago, J. Am. Chem. Soc., 2014, 136, 2284–2287 CrossRef CAS PubMed.
- J. Xia and R. Jasti, Angew. Chem., Int. Ed., 2012, 51, 2474–2476 CrossRef CAS PubMed.
- T. Iwamoto, Y. Watanabe, Y. Sakamoto, T. Suzuki and S. Yamago, J. Am. Chem. Soc., 2011, 133, 8354–8361 CrossRef CAS PubMed.
- Y. Segawa, A. Fukazawa, S. Matsuura, H. Omachi, S. Yamaguchi, S. Irle and K. Itami, Org. Biomol. Chem., 2012, 10, 5979–5984 RSC.
- L. Adamska, I. Nayyar, H. Chen, A. K. Swan, N. Oldani, S. Fernandez-Alberti, M. R. Golder, R. Jasti, S. K. Doorn and S. Tretiak, Nano Lett., 2014, 14, 6539–6546 CrossRef CAS PubMed.
- R. Franklin-Mergarejo, D. O. Alvarez, S. Tretiak and S. Fernandez-Alberti, Sci. Rep., 2016, 6, 31253 CrossRef CAS PubMed.
- E. P. Jackson, T. J. Sisto, E. R. Darzi and R. Jasti, Tetrahedron, 2016, 72, 3754–3758 CrossRef CAS.
- E. R. Darzi, T. J. Sisto and R. Jasti, J. Org. Chem., 2012, 77, 6624–6628 CrossRef CAS PubMed.
- E. Kayahara, R. Qu and S. Yamago, Angew. Chem., Int. Ed., 2017, 56, 10428–10432 CrossRef CAS PubMed.
- E. Kayahara, T. Hayashi, K. Takeuchi, F. Ozawa, K. Ashida, S. Ogoshi and S. Yamago, Angew. Chem., Int. Ed., 2018, 57, 11418–11421 CrossRef CAS PubMed.
- Y. Segawa, H. Omachi and K. Itami, Angew. Chem., Int. Ed., 2010, 12, 2262–2265 CAS.
- P. Li, T. J. Sisto, E. R. Darzi and R. Jasti, Org. Lett., 2014, 16, 182–185 CrossRef CAS PubMed.
- R. Y. Lai, J. J. Fleming, B. L. Merner, R. J. Vermeij, G. J. Bodwell and A. J. Bard, J. Phys. Chem. A, 2004, 108, 376–383 CrossRef CAS.
- R. P. Haugland, Handbook of fluorescent probes and research chemicals, Molecular Probes, Eugene, 1992 Search PubMed.
- G. Cosa, K.-S. Focsaneanu, J. R. N. McLean, J. P. McNamee and J. C. Scaiano, Photochem. Photobiol., 2001, 73, 585 CrossRef CAS PubMed.
- T. Hard, P. Fan and D. R. Kearns, Photochem. Photobiol., 1990, 51, 77–86 CrossRef CAS PubMed.
- I. Johnson, Histochem. J., 1998, 30, 123–140 CrossRef CAS PubMed.
- L. D. Lavis, T.-Y. Chao and R. T. Raines, ACS Chem. Biol., 2006, 1, 252–260 CrossRef CAS PubMed.
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1982, 27, 455–462 CrossRef.
- E. R. Darzi, T. J. Sisto and R. Jasti, J. Org. Chem., 2012, 77, 6624–6628 CrossRef CAS PubMed.
- T. J. Sisto, M. R. Golder, E. S. Hirst and R. Jasti, J. Am. Chem. Soc., 2011, 133, 15800–15802 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1883238. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00169g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |