
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and applications of peptide-based injectable hydrogels
Chang Liu
a,
Qingguo Zhang
a,
Song Zhu
*a,
Hong Liu
 *a and
Jie Chen
*b
*a and
Jie Chen
*b
aSchool and Hospital of Stomatology, Jilin University, Changchun 130021, P. R. China. E-mail: zhusong1965@163.com; jdliuhong@163.com
bKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: chenjie@ciac.ac.cn
First published on 9th September 2019
Abstract
In situ injectable hydrogels have shown tremendous potential application in the biomedical field due to their significant drug accumulation at lesion sites, sustained release and markedly reduced systemic side effects. Specifically, peptide-based hydrogels, with unique biodegradation, biocompatibility, and bioactivity, are attractive molecular skeletons. In addition, peptides play a prominent role in normal metabolism, mimicking the natural tissue microenvironment and responding to stimuli in the lesion environment. Their advantages endow peptide-based hydrogels with great potential for application as biomedical materials. In this review, the fabrication and production of peptide-based hydrogels are presented. Several promising candidates, which are smart and environment-sensitive, are briefly reviewed. Then, the recent developments of these hydrogels for biomedical applications in tissue engineering, as drug/gene vehicles, and anti-bacterial agents are discussed. Finally, the development of peptide-based injectable hydrogels for biomedical applications in the future is surveyed.
 Chang Liu | Chang Liu is an attending doctor of College and Hospital of Stomatology Jilin University. She obtained her master degree in Stomatology at Jilin University in 2009. Now she focuses on the dental materials and prosthodontics. |
 Qingguo Zhang | Qingguo Zhang has been working as an associate professor in Department of Comprehensive Treatment, College and Hospital of Stomatology, Jilin University, since 2004. Currently he is studying in the field of tissue engineering and biological antibacterial materials. |
 Song Zhu | Song Zhu is a professor of Prosthodontics and the director of Department of Prosthetic Dentistry at Hospital of Stomatology, Jilin University. An expert reviewer for National Natural Science Foundation of China, he also holds the position of vice-chairman of Dental Material Committee of Chinese Stomatological Association. Dr Zhu has authored more than 30 SCI/EI papers, mainly on dental antibacterial materials, prosthetic polymer materials and dental restorative materials. Previously he was a visiting scholar at School of Dentistry, Indiana University. Dr Zhu holds a B.S. degree in Stomatology and a PhD degree in Prosthodontics from Jilin University. |
 Hong Liu | Hong Liu has been working as a professor in Department of Comprehensive Treatment, College and Hospital of Stomatology, Jilin University, since 2010. Currently she is studying in the field of tissue engineering and biological antibacterial materials. |
 Jie Chen | Jie Chen is an associate professor in Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. His research interests are focused on polymeric gene/drug carriers and hydrogels for biomedical applications. He has published more than 60 papers in SCI journals and 13 Chinese invention patents. |
1. Introduction
Hydrogels, 3-dimensional (3D) network structures containing hydrophilic groups or chain segments, are insoluble polymers that swell after absorbing water. They can combine with water molecules in the environment to form hydrogel structures. Injectable hydrogels, a new type of hydrogel, have become a hot topic in research due to their unique advantages. Injectable hydrogels are flowing liquids before injection but form solids at the injection site after injection into subcutaneous or muscle tissue.1 The polymers that form injectable hydrogels can change their state or molecular conformation to undergo a sol–gel transformation under different physiological conditions, such as different temperatures, pH, magnetic fields, electric fields, ionic strength, and chemical conditions.2 Injectable hydrogels can be used as biological carriers and injected into a lesion site after simply mixing with various biological products. This allows, for the maintenance of biological product properties and long periods of sustained drug release, as well as a reduction in the initial burst release and side effect of drugs.1 In tissue engineering, injectable hydrogels can achieve minimally invasive and rapid recovery, avoiding the problems associated with anastomoses of pre-plastic materials and implantation trauma.3Peptide-based hydrogels are particularly fascinating because of their unique biocompatibility, biodegradability, low toxicity, and bioactivity with structures that mimic natural proteins.4,5 They are prepared by a physical assembly that involves non-covalent interactions, including hydrogen bonding, electrostatic interactions, hydrophobic interactions, and π–π interactions.6 Peptide-based hydrogels can also be formed by chemical and enzymatic cross-linking.7,8 These hydrogels have 3D networks that can encapsulate large biologics, which provides them with a stimuli-triggered and drug controlled release capacities. Peptides themselves can also regulate physiological functions in vivo and participate in the circulation and metabolism of active substances, including biological identification,9 antigen–antibody reactions,10 and signal transduction,11 In addition, peptide-based hydrogels are readily modified and functionalized through side-chain and backbone modification to satisfy their specific applications.4 Therefore, peptide-based hydrogels hold great promise in biomedical applications including tissue engineering,12 as drug/gene vehicles,13 as anti-bacterial agents,14,15 and in wound healing.16
In this review, we highlighted the preparation and biomedical application of peptide-based injectable hydrogels (Fig. 1). The fabrication and construction of these hydrogels were presented and several smart and promising peptide-based hydrogels with environmental sensitivity were reviewed. More importantly, the recent development of hydrogels for biomedical applications was identified and extensively summarized. Finally, the development of peptide-based injectable hydrogels for biomedical applications in the future was prospected.
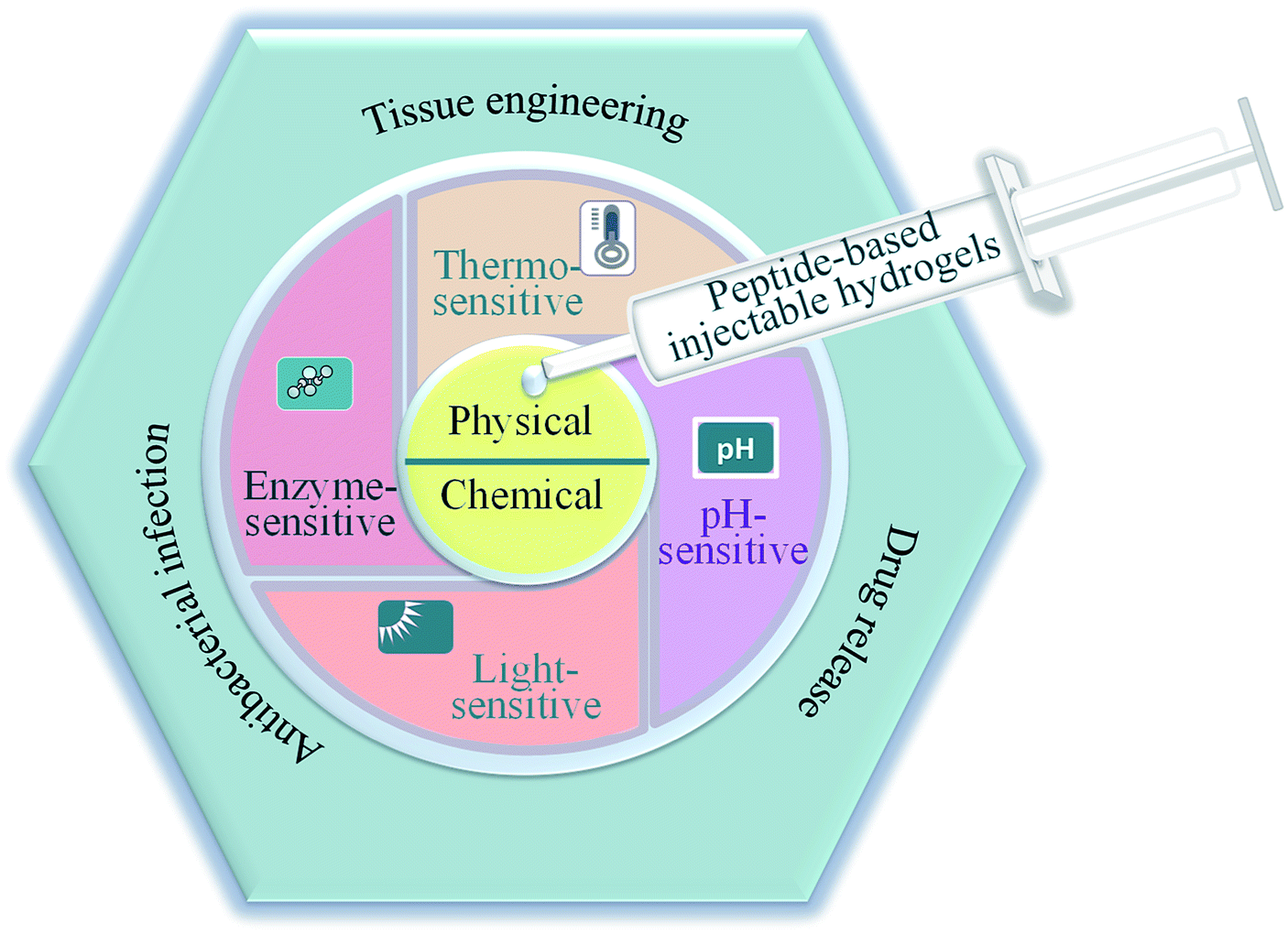 | ||
| Fig. 1 Schematic illustration of the preparation and applications of peptide-based injectable hydrogels. | ||
2. Formation of peptide-based injectable hydrogel
Injectable implant hydrogels are a class of fluid hydrogels that can be injected into the body directly to easily fill irregular shapes and form freestanding solids in defective parts.17,18 Compared to systemic injections, injectable hydrogel-based local treatments have great potential biomedical applications due to their significant drug accumulation at lesion sites, sustained release and markedly reduced systemic side effects.18 Hydrogels are mainly constructed by physical and chemical cross-linking (Table 1).19| Cross-linking mechanisms | Peptide | Functions or applications | References |
|---|---|---|---|
| a HRP: horseradish peroxidase; POSS-R: polyhedral oligomeric silsesquioxane (POSS) core-based generation one (L-arginine) dendrimer (POSS-R); Me: L-methionine; D-1MT: dextro-1-methyl tryptophan; CMP: collagen mimetic peptides; PCLA: poly[(ε-caprolactone)-co-lactide]; hMSCs: human mesenchymal stem cells; PELG: poly(γ-ethyl-L-glutamate); HLT2: VLTKVKTKVDPPTKVEVKVLV-NH2. | |||
| Self-assemble | KYFIL pentapeptides | 3D cell culture | 12 |
| Self-assemble | P(Me-D-1MT)-PEG-P(Me-D-1MT) | Checkpoints | 22 |
| Self-assemble | VKVKVKTKVDPPTKVKTKVKV-NH2 | None | 37 |
| Self-assemble | PEG-CMP | Tissue engineering scaffolds | 38 |
| Self-assemble | KRGDKK-PCLA-PEG-PCLA-KRGDKK | Drug delivery | 39 |
| Self-assemble | IKIKIKIK-IDPPT-OIKIKIKI-NH2 | Protein delivery | 40 |
| Self-assemble | Fmoc-FF/PLL-SH | Anticancer | 41 |
| Self-assemble | mPEG-b-PELG | Anticancer | 42 |
| Self-assemble | HLT2 | Tumor cells (Plk1) | 43 |
| HRP and H2O2 | PLG-g-TA/PEG | Tissue engineering scaffolds drug delivery carriers | 7 |
| HRP and H2O2 | PASP-g-TA/AP | 3D scaffolds | 44 |
| Electrophilic cross-linking | [(Val-Pro-Gly-Val-Gly)4(Val-Pro-Gly-Lys-Gly)] | None | 34 |
| Photopolymerization | PEG-peptide | hMSCs delivery | 45 |
2.1. Physically cross-linked hydrogels
Physically cross-linked hydrogels are hydrogels that are cross-linked by hydrogen bonds,20 electrostatic interactions,21 hydrophobic interactions,22 and π–π stacking.6 Peptide-based physical hydrogels are mainly formed by molecular self-assembly based on several secondary structural motifs that include α-helices, β-sheets, β-hairpins, and coiled-coils, which are usually affected by charge and ions.23,24 Among these secondary structures, hydrogels based on α-helices and β-sheets have been extensively studied. A common α-helix structure exhibits a strong propensity for forming repeated residues, including hydrophobic residues for stabilizing helix–helix interactions and polar residues for constituting the helical structure.25 Yang and coworkers developed peptide-based hydrogels into α-helical conformation by enzyme-instructed self-assembly, which significantly inhibited cancer cells.26 The α-helical structure played a key role in endocytosis,27 thus demonstrating important prospects in the controlled drug release field. β-sheets represent lower energy due to enthalpy contributions from extensive electrostatic and H-bonding interactions.28 β-sheet bilayers were initially stack together via H-bonding and electrostatic interaction, and then packed in parallel and assembled into hydrogels. Scherman et al. explored and screened a pentapeptide hydrogel (IDIDI) with tunable morphology and mechanical properties produced through reassembly of the β-sheet interactions, which showed potential applications in delivery systems and tissue engineering.29 The primary effect of physically cross-linked hydrogels is the formation of physical crosslinking points inside the gels. In 1993, Zhang et al.30 reported that a 16-residue peptide [(Ala-Glu-Ala-Glu-Ala-Lys-Ala-Lys)2] (EAK16) could spontaneously assemble into a hydrogel structure in the presence of salt, which opened up the new field of self-assembly technology of polypeptide molecules. The formation of self-assembled structures was attributed to their molecular structure, which consisted of both hydrophilic and hydrophobic amino acid residues that could form folding structures β-sheets. In addition, the hydrophilic amino acid residues had positive or negative charges, allowing the hydrophilic surface to form complementary ionic bonds. In the process of self-assembly, the side chains of the hydrophobic alanine residues were hidden inside, while the hydrophilic groups, lysine and glutamate, formed ionic bonds with each other due to the alternating positive and negative charges. To improve resistance to enzymatic degradation and improve the long-term stability of peptide-based hydrogels, Yang and co-workers31 developed D-peptide-based hydrogels, which were insensitive to most endogenous enzymes and retained long-term stability in vivo (Fig. 2). This work illustrated a potential strategy to generate supramolecular hydrogels with outstanding biological stability. Overall, compared to chemically cross-linked hydrogels, physically cross-linked hydrogels can achieve mild sol–gel phase transition without additional solvents or chemical reactions.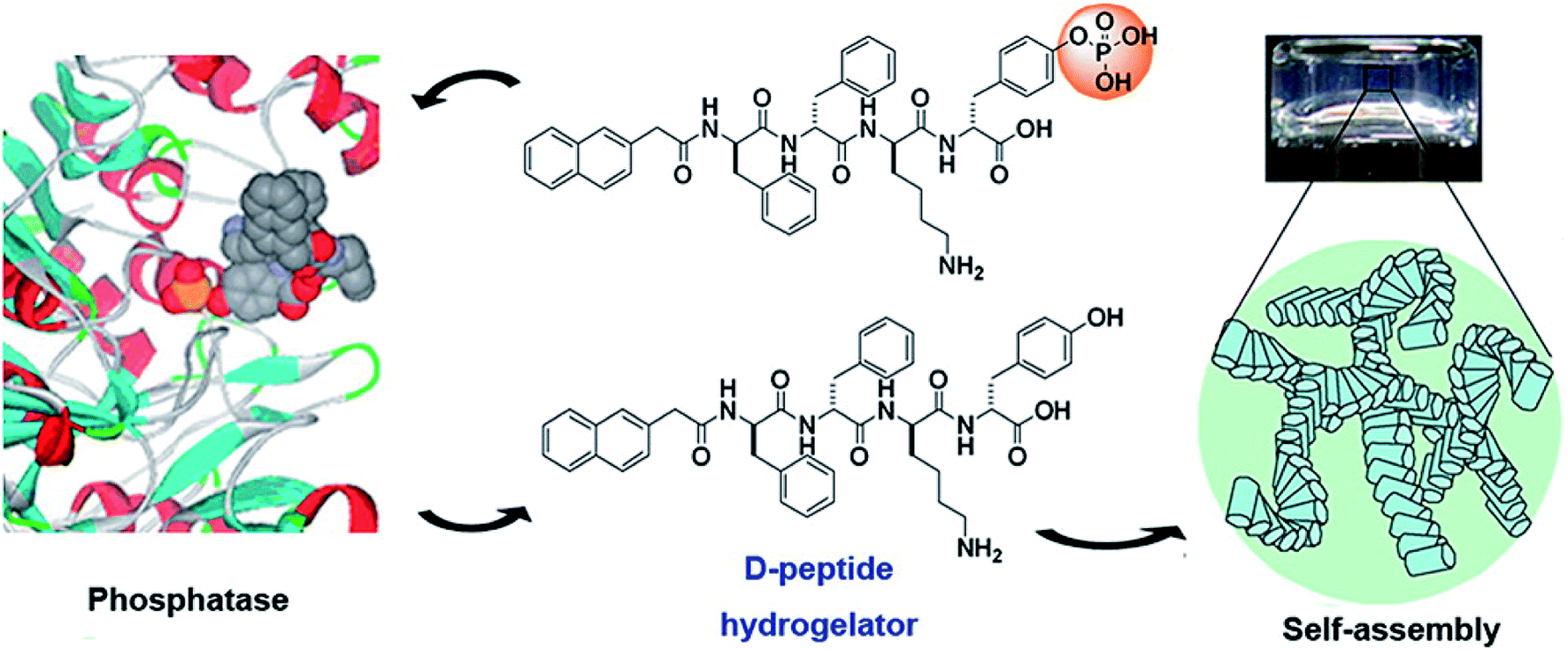 | ||
| Fig. 2 Dephosphorylation of D-peptide derivatives to form supramolecular hydrogels.31 | ||
2.2. Chemically cross-linked hydrogels
Chemically cross-linked hydrogels are covalently cross-linked networks formed by covalent bonds generated by copolymerization or condensation polymerization initiated by traditional synthesis methods, photo-polymerization,32 enzyme polymerization,7 radiation polymerization,33 and other technologies.6,18 Compared to physically cross-linked hydrogels, chemically cross-linked hydrogels have stable structures, high strength, slow degradation, good reaction controllability, and easy operation, which are beneficial to their biomedical applications. However, due to the introduction of reactive cross-linking agents or functional groups with reactivity, the reaction between the active reactive group in the polypeptide chain and the cross-linking agent creates the gel network, which accompanies by chemical reaction in the process of gel formation. Therefore, it will take potential toxic and have a certain impact on normal tissues and cells. McMillan et al.34 prepared an elastin-mimetic peptide (Lys-25) using genetic engineering and microbial protein expression. The regular lysine residues in Lys-25 were used as the junction to react with the cross-linking agents, affording solvent-swollen networks. The hydrogels showed reversible, temperature-dependent swelling and shrinkage and the phase transition temperature was estimated at 35 °C. These properties indicate that Lys-25 might be suitable as an in situ injectable. Chemically cross-linked peptide hydrogels can also be prepared by ligand chemistry. Cysteine terminal lysine dendritic compound was prepared using polyethylene glycol (PEG) with an aldehyde ester as the cross-linking agent. These hydrogels exhibited extremely good stability and kept their shape and size unchanged for a long time.35 Peptide-based hydrogels can also be prepared by introducing photosensitive groups into the polypeptide chain.33 In addition, radiation can promote the formation of hydrogels. This strategy introduces no cross-linking agents or groups, and can avoid the influence of other components on the biocompatibility of the hydrogels. Therefore, this method shows great advantages in the preparation of biomedical hydrogels. Moreover, peptides can also be introduced into other hydrogels in the form of branched chains to form hybrid or compound hydrogels. The introduction of polypeptide components can improve the cellular compatibility of these biogels.363. Types of peptide-based injectable hydrogels
The injectable hydrogels are a class of fluid hydrogels that can be directly injected into the body and exhibit sol–gel phase transitions in response to environmental conditions.46 Peptide-based hydrogels have attracted great promise due to their biocompatibility, biodegradability, controllable spatial structure, ease of modification, and specific biological activity. The self-assembly ability of peptide-based hydrogel is related to intramolecular folding of the peptide chains. When the polypeptides are induced to fold by external stimulation, this leads to amphiphilic folding.28,47,48 Therefore, these intelligent hydrogels are a kind of macromolecular material that can be sensed and processed and has executive function.49 They are sensitive to stimuli including temperature, pH, light, enzymes, and numerous other conditions in the external environment.50 Therefore, they could be used as a new type of intelligent material with good prospects for scientific research and biomedical applications.3.1. Thermo-sensitive peptide-based hydrogels
Thermo-sensitive peptide-based hydrogels are highly sensitive to changes in temperature. Their structures contain hydrophobic and hydrophilic groups in different quantities and proportions and these groups can interact with each other under changes in temperature, resulting in a sol–gel phase transition. This feature can be used to exploit temperature changes at injection sites.51 These hydrogels have low critical solution temperatures (LCST), and the volume of the hydrogels present as sudden shrinkage and expansion at LCST. This characteristic makes it more suitable for biomedical application by local injection. Furthermore, thermo-sensitive hydrogels are quick and convenient to produce, and are minimally invasive, qualities that make them topics of active research.Pochan et al.37 designed a peptide (MAX3)-based hydrogel that exhibited thermoreversible self-assembly capacity. MAX3 was an amphiphilic polypeptide that consisted of a hydrophilic lysine residue and a hydrophobic valine residue providing β-sheet capacity. In this sequence, both hydrogen bond interaction and hydrophobic interaction existed. With increasing temperature, the peptides in the solution were folded and self-assembled into a hydrogel network, indicating that the increased temperature tended to dehydrate the nonpolar residues and trigger folding via hydrophobic collapse. When the temperature decreased to room temperature, the hydrogen bond and hydrophobic interaction made the self-assembled hydrogel completely dissolve. The sol–gel transition temperature could be controlled by altering the hydrophobicity of the MAXs. Therefore, MAXs was a typical thermo-reversible peptide-based hydrogel and could be used as an injectable hydrogel.
Collagen mimetic peptides (CMPs) are another kind of synthetic collagen. CMPs consists of repeating tripeptide templates that self-assemble into a triple helix of three single strands wrapped around each other.52 CMPs exists as single strands at temperatures above its melting point and reassembles into triple-helix structures upon cooling. The melting temperature is usually 21–39 °C and varies with the number of triplets.53 Therefore, CMP has significant research value as an injectable hydrogel. Hartgerink and co-workers54 prepared a CMP by incorporating lysine and aspartate residues into proline–hydroxyproline–glycine repeating units by salt-bridging hydrogen bonds between lysine and aspartate to stabilize its triple helix according to a sticky-ended assembly. The melting point of this CMP was 40–41 °C, indicating that it could assemble into hydrogel at temperatures over 41 °C. In addition, this hydrogel could be also degraded by collagenase, a natural collagen.
Moreover, in order to increase the stiffness and thermo-responsive behavior of peptide-based hydrogels, synthetic polymers, such as poly(ethylene glycol) (PEG),38,53 poly[(ε-caprolactone)-co-lactide]-poly(ethylene-glycol)-poly[(ε-caprolactone)-co-lactide] (PCLA–PEG–PCLA),39 poly(DL-lactide-co-glycolide-b-ethylene glycol-b-DL-lactide-co-glycolide) (PLGA–PEG–PLGA),55 polyacrylic acid (PAA)56 and others,57 have also been conjugated to these hydrogels.
3.2. pH-sensitive peptide-based hydrogels
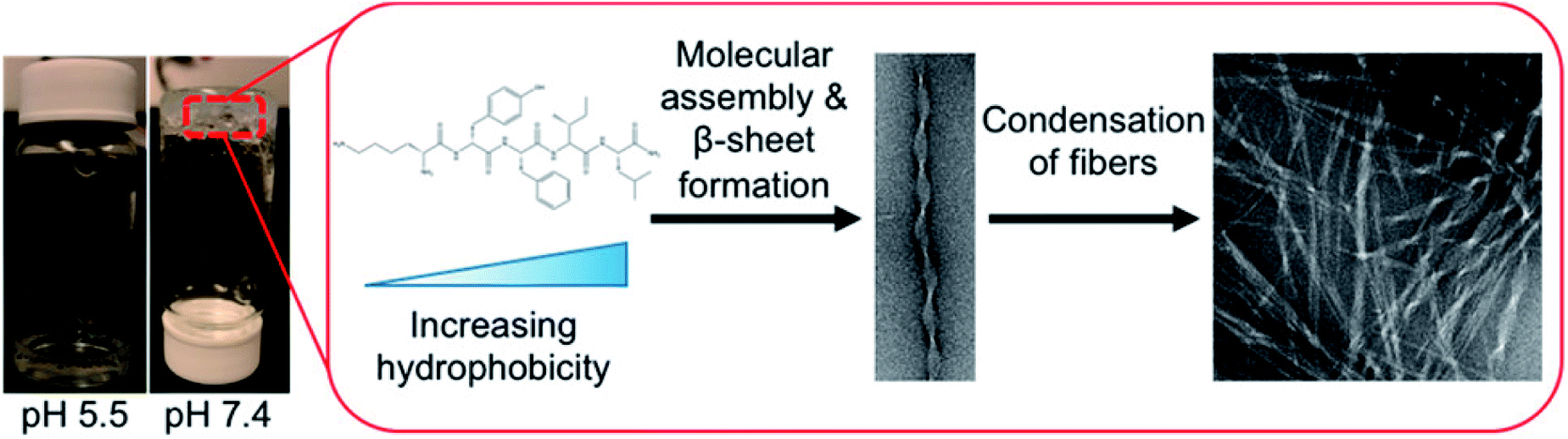
Hydrogels with pH-responsiveness refer to their swelling or shrinking that varies with pH. These hydrogels can form macromolecular networks through cross-linking. The networks contain acidic or alkaline groups. With a change in pH and ionic strength of the medium, these groups ionize, leading to the dissociation of hydrogen bonds between the macromolecular chain segments in the network, causing discontinuous swelling volume changes.Ge and coworkers demonstrated a pH-sensitive self-assembling peptide hydrogel with a peptide sequence of IKIKIKIK-IDPPT-KIOIKIKI.40 This hydrogel was able to self-assemble into hydrogel and was extremely stable without noticeable changes over 12 h at pH 7.4. The solubility of this peptide increased significantly below pH 6.0 due to the protonation of lysine/ornithine side chains, leading to charge repulsion between the side chains of neighboring amino acids. Subsequently, the hairpin structure of the peptide was unfolded, which prompted its dissociation from hydrogel to solution. Inspired by silk fibroin, Guo et al.58 constructed an amphiphilic peptide, which could form hydrogel by the pH-induced assembly of C12-GAGAGAGY (Fig. 3). This hierarchically structured robust hydrogel was prepared under shear force by the aggregation of peptide-based nanofibers. The hydrogel exhibited unique pH-sensitive reversible sol–gel transition when the pH decreased, thus displaying potential applications as tissue engineering scaffolds.
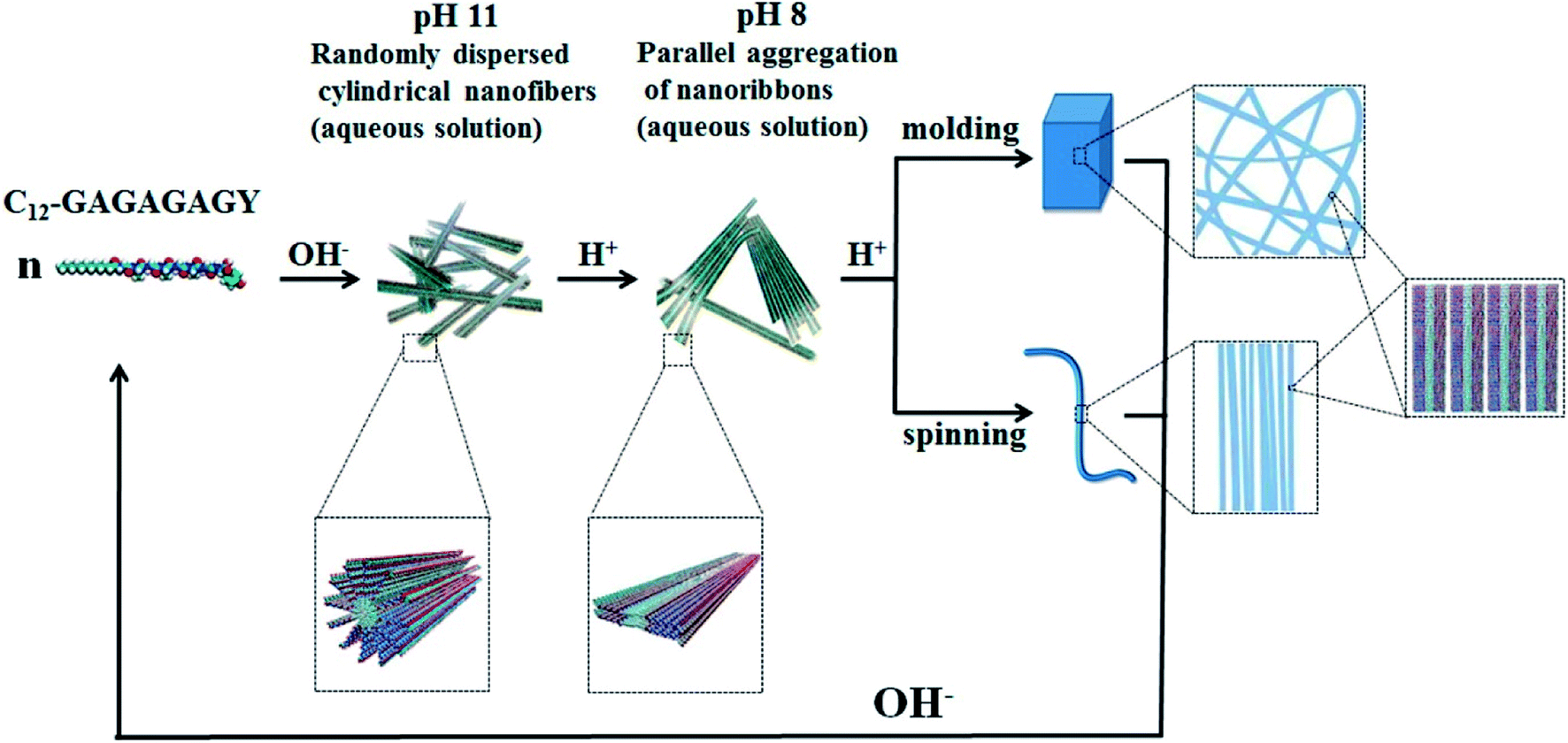 | ||
| Fig. 3 pH-induced hierarchical self-assembly of peptide-based hydrogels.58 | ||
3.3. Light-sensitive peptide-based hydrogels
The light-sensitivity property of hydrogel polymer materials refers to a phenomenon whereby the volume of hydrogel changes under light stimulation.59 To construct the hydrogel microenvironment, light is applied to control the hydrogel construction and regulate the microenvironmental properties.60,61 By adjusting the illumination time, excitation wavelength or light intensity, the number of photons absorbed in a specific area of the material can be precisely controlled to achieve control of time and space. Optical control has unique advantages, including spatio-temporal control, indirect invasion, and low biological toxicity.62 Thus, based on the influence of the cell microenvironment on cell fate, the physicochemical properties of these materials can be changed in time and space by light regulation technology. This allows cell culture on the surface or inside of biological materials and provides a way to observe and guide cell physiological processes in real time, which has become a new trend in modern biological research.63–65 Therefore, the accurate construction of hydrogels can be achieved by photochemical reactions.Fairbanks et al.45 prepared proteolytically degradable polyethylene glycol (PEG) peptide hydrogels, which were synthesized by a radically mediated thiol-norbornene photopolymerization. These strategy exhibited many advantages including homogeneous network structure, facile incorporation of peptides without further modification, and spatio-temporal control. This hydrogels were cytocompatible and allowed for the encapsulation of living cells with viability over 95%. To enhance the mechanical stability of peptide-based hydrogels, Ding et al.66 employed a light cross-linking strategy to construct a novel hydrogel by improving the storage modulus by four orders of magnitude (Fig. 4). This hydrogel was established by the ruthenium-complex-catalyzed conversion of tyrosine to dityrosine within two minutes through photo cross-linking. Importantly, this strategy was versatile for improving the mechanical stability of tyrosine-containing peptide-based hydrogels. Therefore, it is anticipated to have potential applications in drug delivery and tissue engineering.
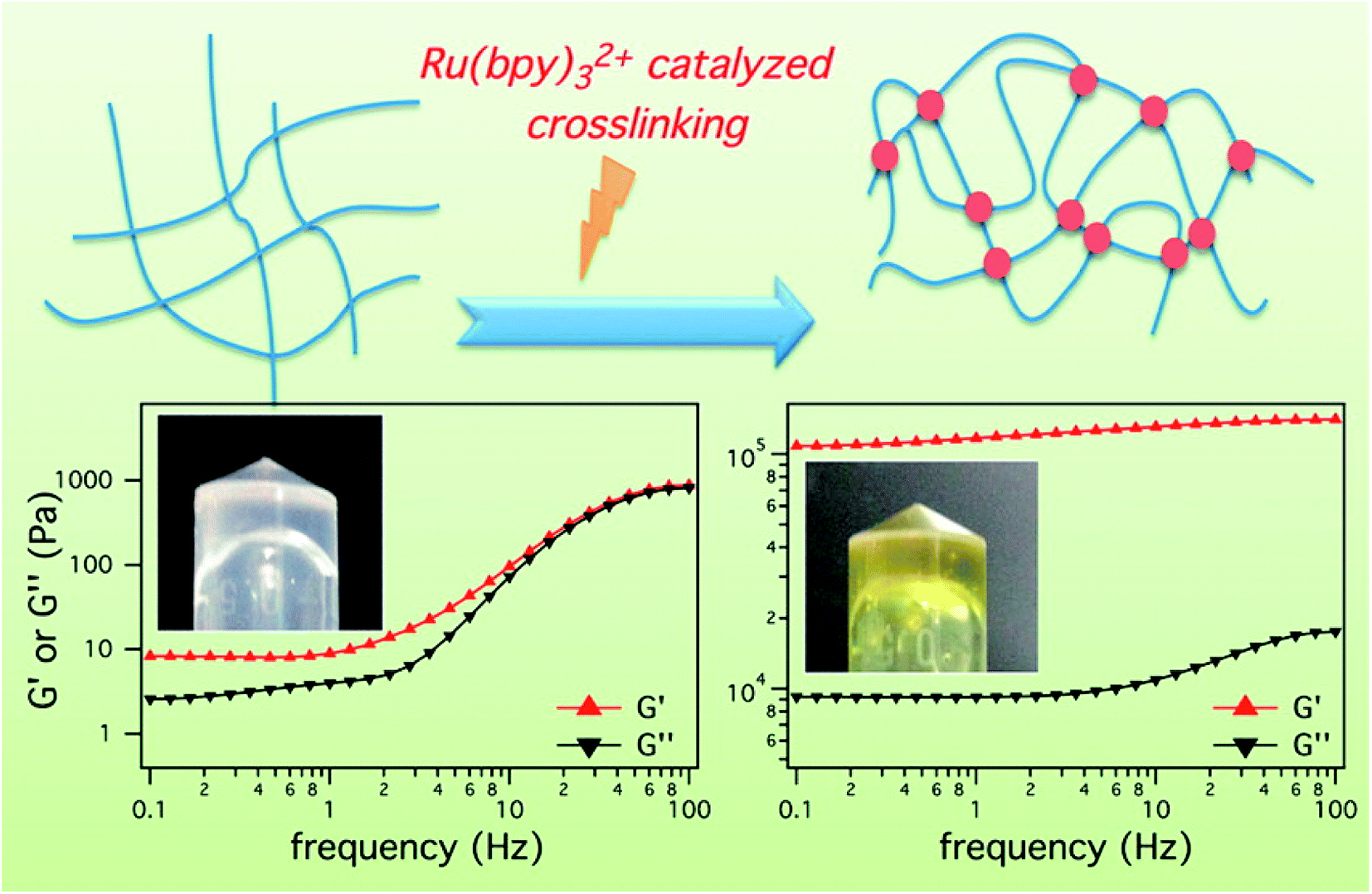 | ||
| Fig. 4 Ru(bpy)32+ catalyzed photo crosslinking to enhance the mechanical stability of the peptide-based hydrogels.66 | ||
3.4. Enzyme-sensitive peptide-based hydrogels
Enzymatically crosslinked peptide-based hydrogels are biocompatible, highly sensitive, easily degradable, and have few side effects. They are attractive materials for potential applications in biomaterials and drug delivery.67 Enzyme-catalyzed reactions have many advantages, including high efficiency, specificity, and gentleness.68 Biological enzymes capable of inducing the self-assembly of peptides mainly include cross-linking enzymes to control the self-assembly of peptide conjugates,44,69 dephosphorylase to trigger β-sheets,70 and protease-catalyzed hydrolysis.71Recently, enzymatically crosslinked peptide-based hydrogels have been paid worldwide attention due to their good biocompatibility, tunable mechanical properties, and efficient gelation process. Ulijn and colleagues69 demonstrated the formation of amphiphilic peptide hydrogels by using proteases. These proteases selectively triggered the self-assembly of peptides via reverse hydrolysis and facilitated the in situ formation of hydrogel scaffolds in the body. Ren et al.7 described an injectable hydrogel based on poly(L-glutamic acid) grafted with tyramine and PEG (PLG-g-TA/PEG) under physiological conditions in the presence of hydrogen peroxide and horseradish peroxidase via an amidation reaction (Fig. 5). The injected hydrogels in the subcutaneous sites of rats could maintain for 10 weeks without generating an inflammatory response. PLG-g-TA/PEG hydrogels showed promise as candidates for tissue engineering scaffolds to achieve the long-term release of bioactive molecules. Enzymatic phosphorylation/dephosphorylation reactions were performed to regulate the β-sheet assembly of peptides. The alcohol group of serine was phosphorylated, inhibiting the formation of β-sheets due to the combination of increased charge–charge repulsion. The inhibition of β-sheet structures formations improved the solubility of peptides and the formation of hydrogels.70 Moreover, several hydrolases, such as tryptase, chymotrypsin, and matrix metalloproteinase (MMP), could specifically cleave peptide bonds, resulting in the enzyme-mediated digestion and collapse of the peptide-based hydrogels. These specific interactions were able to promote the targeted release of bioactive medicines.72 Enzymes are widely present in living organisms and show significant differences among different tissues, while enzyme-sensitivity is a typical property of polypeptides. Therefore, for different disease types and tissues, the development of enzyme-sensitive peptide-based hydrogels indicates theoretical significance and application value.
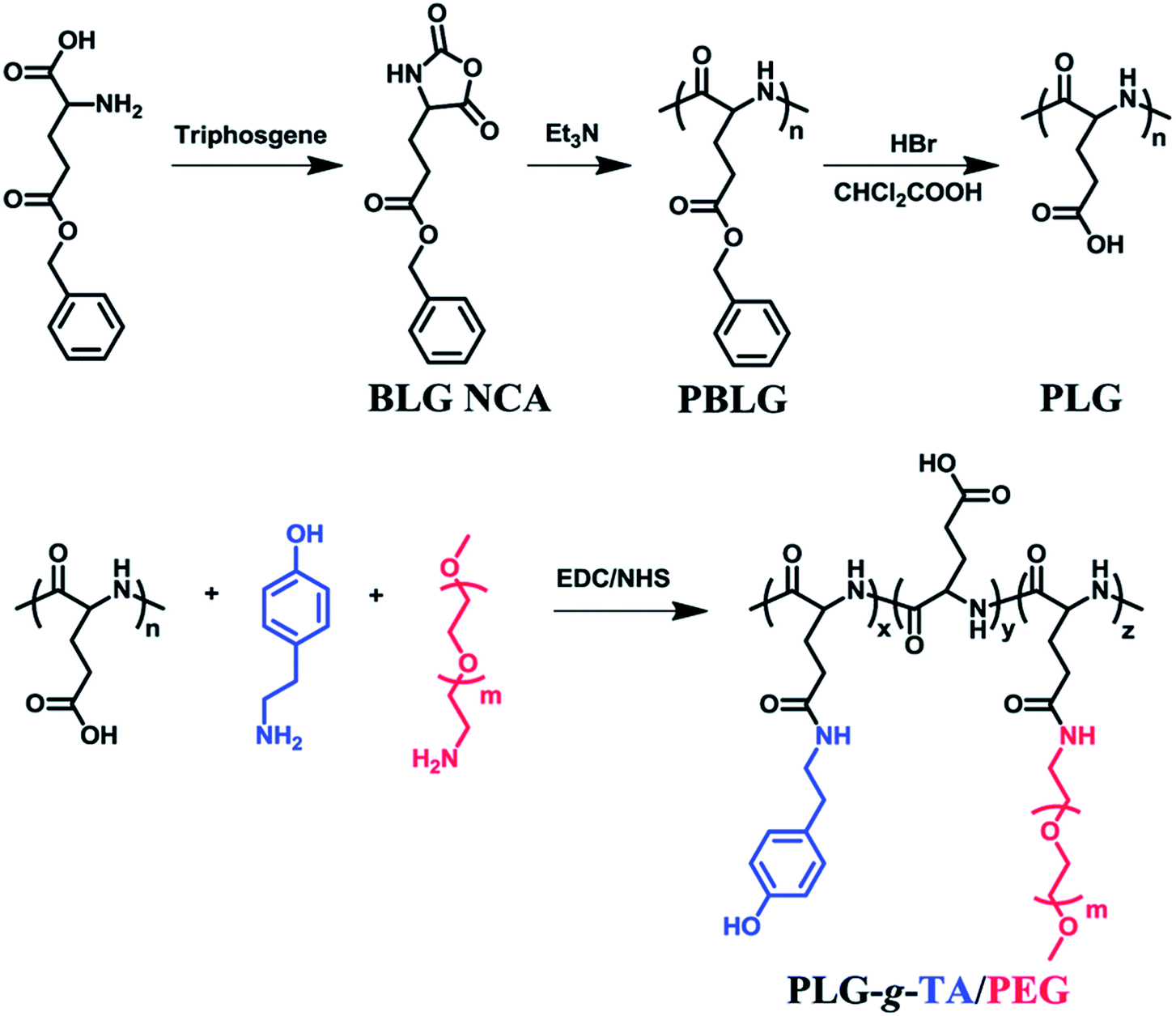 | ||
| Fig. 5 Tyramine and poly(ethylene glycol) grafted poly(L-glutamic acid) was prepared under physiological conditions in the presence of horseradish peroxidase (HRP) and hydrogen peroxide (H2O2).7 | ||
4. Biomedical applications of peptide-based injectable hydrogel
Peptide-based injectable hydrogels can be used as biological carriers and injected into a lesion site after simply mixing with various biological products. This can not only maintain the properties of the biological products and release drugs over long periods but also reduce the irritation of drugs. Injectable hydrogels can achieve minimally invasive and rapid recovery, avoiding the problems of anastomosis and implantation trauma of preplastic materials.4.1. Tissue engineering
Tissue engineering is a discipline that can repair or replace damaged or diseased tissues with natural or synthetic tissue mimics. These biological substitutes are used for repairing, maintaining, and promoting the function and morphology of various tissues or organs after injury.73 Tissue engineering repair involves the adsorption of normal tissue cells cultured onto a biological material with excellent cellular compatibility and biodegradable properties to form a cell/biomaterial complex, which is then implanted onto the damaged parts of human tissues and organs.74,75 As the biological materials of the cell growth scaffolds are gradually degraded and absorbed by the body, the cells are constantly proliferated and differentiated to form new tissues. These scaffolds are consistent with the corresponding tissues and organs in terms of morphology and function, thereby repairing trauma and rebuilding functions.76 The 3D structure of biomaterial scaffolds can provide a good environment for cells to acquire nutrients, grow and metabolize.77 Furthermore, the development of tissue engineering provides a technical means of tissue regeneration, which will change the traditional treatment mode of “repairing trauma with trauma” in surgery and step into a new stage of non-traumatic repair.78 Successful tissue engineering mainly include four elements, cells, scaffold materials, the integration of cells and scaffold materials, and the integration of implants and microenvironment in vivo. It is particularly important to develop novel tissue engineering scaffold materials with excellent comprehensive properties.Peptide-based hydrogels have several potential advantages in tissue engineering. They are biocompatible and can provide nutrient environments suitable for endogenous cell growth and the accumulation of extracellular matrices. They can also mimic the natural extracellular matrix (ECM) and present a prospective capacity to encapsulate cells or bioactive molecules. Moreover, only amino acids are formed during their degradation, which will not induce adverse effects, immune or inflammatory reactions in the human body.79 Koutsopoulos et al.80 encapsulated neural stem cells into 3D matrices of self-assembling peptide hydrogels. The neural cells in the peptide hydrogels could proliferate and differentiate effectively in the 2 week culture periods and they were able to thrive for at least five months. In addition, these hydrogels provided growing space and necessary nutrients for cell proliferation without using any medium, allowing for more realistic applications in tissue engineering. Tang et al.12 demonstrated pH-sensitive hydrogel biomaterials consisting of pentapeptides (Fig. 6). The gelation behavior of the peptides corroborated their rapid gelling ability by increasing β-sheet in response to variations in solvent accessibility, which protected the inner cells from acute membrane disruption. These hydrogels could achieve cytocompatible encapsulation of oligodendrocyte progenitor cells in vitro and could be delivered as injectables by syringes, inducing the proliferation and 3D process extension of cells in vivo. Peptide-based hydrogel scaffold materials have also been used in bone tissue engineering. Martin and coworkers81 developed TGF-β3 or BMP-2 functionalized PEI-peptide hydrogels, encapsulated human bone marrow-derived mesenchymal stromal cells (BMSCs), and nasal chondrocytes (NCs) by enzymatical polymerization in vivo. Afterward, BMSCs were induced to form ossicles containing bone and marrow by endochondral ossification, while NCs formed cartilage tissue to maintain phenotypic stability with the assistance of BMP-2 and TGF-β3. Therefore, peptide based hydrogels have attracted more and more attention due to their excellent properties and are expected to have broad prospects for development in the field of tissue engineering.
 | ||
| Fig. 6 Peptide-based pH-responsive hydrogels for tissue engineering.12 | ||
4.2. Drug controlled release
Conventional drug delivery strategies are restricted by systemic toxicity, repeated dosing, rapid metabolism, undesirable drug absorption, and easy degradation under physiological conditions. Injectable hydrogels, with tunable physical properties, controllable degradability, and sustained drug release properties, are promising smart drug delivery vehicles to minimize these disadvantages and optimize the therapeutic benefits.82–85 Peptide-based hydrogels are suitable to form hydrogels for use as drug delivery vehicles because of their good biocompatibility, mild working conditions, and responsiveness to enzymatic, hydrolytic or environmental stimuli.86 Specifically, injectable hydrogels have attracted worldwide attention due to their unique advantages that include good syringeability and minimal invasiveness.19,87Yan and coworkers41 constructed injectable peptide-based hydrogels by combining positively charged poly-L-lysines (PLL) and self-assembling N-fluorenylmethoxycarbonyl diphenylalanine (Fmoc-FF) by electrostatic interactions (Fig. 7). Fmoc-FF self-assembled nanofibers had helical structures, which were similar to fimbrial antigens and showed antigenic activities. The fabricated helical fibrous structures of Fmoc-FF self-assembled fibers were enhanced after introducing PLL. The prepared hydrogel itself served as a vaccine to activate powerful antitumor immune responses without any antigen and adjuvant. Karavasili et al.88 reported a self-assembling peptide-based hydrogel ac-(RADA)4-CONH2 that combined doxorubicin and curcumin for head and neck cancer therapy. These hydrogels enabled precise control of the drug release rate in vitro. Significant antitumor effect was achieved after treating with the dual-drug-loaded hydrogels compared to water-soluble drugs. The synergistic antitumor effect was attributed to efficient cell cycle blockage and apoptosis at dose levels below the half-maximal inhibitory concentration of both drugs, which was facilitated by the formation of peptide hydrogels. Yang and coworkers13 demonstrated a melittin-RADA32 peptide hydrogel loaded with doxorubicin for combination chemotherapy and immunotherapy against melanoma. This hydrogel exhibited a nanofiber structure, biocompatibility, drug controlled release capacities, and enhanced antitumor effects. Furthermore, this strategy could not only effectively kill the tumor in situ but also provide a strong immunological memory effect against metastatic tumor cells.
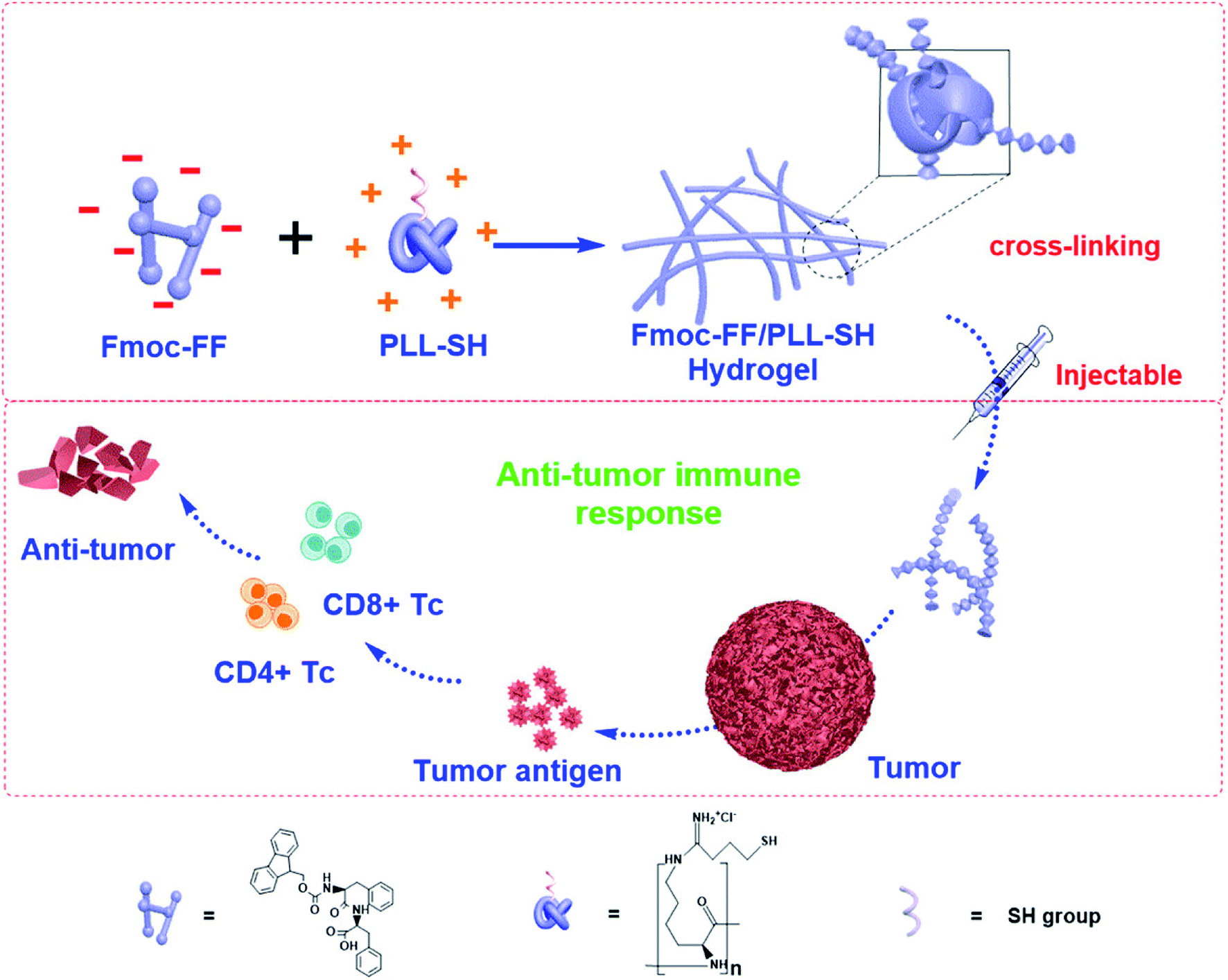 | ||
| Fig. 7 Peptide-based hydrogels themselves as immune stimulants for antitumor therapy.41 | ||
Overview, peptide-based hydrogels have made great progress as drug carriers and can not only carry small molecule drugs, but also effectively carry large molecule drugs such as growth factors and genetic medicines.22,42,43 Peptide-based hydrogels can significantly improve the pharmacokinetic parameters of drugs in vivo, improve the curative effects, and reduce their toxicity and side effects through drug encapsulation and targeted delivery. However, the distribution, degradation, toxicity and large-scale production as drug carriers in vivo still need further research and strengthening.
4.3. Antibacterial infection
Bacterial infection has become a common and intractable problem in the biomedical field. If a durable antibacterial and biocompatible material can be developed and applied clinically, it is expected to greatly reduce bacterial-related infections and patient' pain, as well as the burden on medical resources and the social economy.89 However, the development and application of antibacterial materials are still restricted by the problems of drug resistance effect, poor biocompatibility, and short antibacterial effects.14,90 Therefore, it is meaningful to develop strategies to minimize the dosage of antibiotics needed to effectively treat an infection. Currently, peptide-based hydrogels with an antibacterial function have attracted broad attention.15 Two forms of peptide-based hydrogels are used for antimicrobial activity. One is a hydrogel-coated active material with an antimicrobial-agent, such as ciprofloxacin, gentamicin, vancomycin, cephalosporin, or other synthetic antibacterial drugs. The other is peptide hydrogels with inherent antimicrobial activity.Local antibiotic administration through peptide-based hydrogels can deliver adequate antibiotics into the infected sites without causing significant systemic toxicity.91,92 Marchesan et al.92 demonstrated antibacterial hydrogels by self-assembling the tripeptide D-Leu-Phe-Phe and the antibiotic ciprofloxacin. The hydrogels exhibited high drug loading efficiency and sustained drug release behavior, triggering significant antimicrobial activity against Staphylococcus aureus and Escherichia coli. No major cytotoxicity was detected either in red blood cell hemolysis assays or in L929 fibroblast cells. Furthermore, the hydrogels alone showed mild antibacterial activity against Gram-negative bacteria.
Antibacterial peptides (AMPs) have been identified as a major defense mechanism against a broad spectrum of microorganisms, especially for antibiotic-resistant bacterial infections.93 Usually, AMPs have amphiphilic structures with hydrophobic core and polycationic surfaces, facilitating interactions with negatively-charged bacterial membranes. It is widely believed that AMPs disrupt bacterial membranes, inhibit cell-wall and nucleic acid synthesis, alter cytoplasmic membrane septum formation, and inhibit protein synthesis, leading to the death of bacteria.94 Schneider and coworker95 designed a peptide-based β-hairpin hydrogel scaffold whose surface exhibited inherent antibacterial activity. The hydrogels revealed the ability to disrupt membranes and cause bacterial death upon contact. Furthermore, the hydrogels showed selective toxicity for bacterial, rather than mammalian cells, and did not hemolyze human erythrocytes. The hydrogels contained a high arginine residue content and showed an enhanced interaction with the phospho-rich bacterial membrane by forming strong salt bridges and hydrogen bonds.96 Schneider and coworkers97 constructed arginine-rich self-assembling peptide hydrogels (VKVRVRVRVDPPTRVRVRVKV), which effectively killed both Gram-negative and Gram-positive bacteria, especially multi-drug resistant Pseudomonas aeruginosa. Nandi et al.98 prepared a series of peptide-based hydrogels by changing the chain length of alkyl groups (Fig. 8). Although the hydrogels did not contain any lysine or arginine residues, they exhibited effective antimicrobial activity against Staphylococcus aureus, Bacillus subtilis, and Escherichia coli. Meanwhile, they were noncytotoxic for human lung carcinoma cells and showed little hemolysis of human red blood cells at the minimum inhibitory concentration of the bacteria. The effective antibacterial properties were mainly due to the amphiphilic structure of the cationic and hydrophobic residues, which mimicked the structure of natural antibacterial peptides.
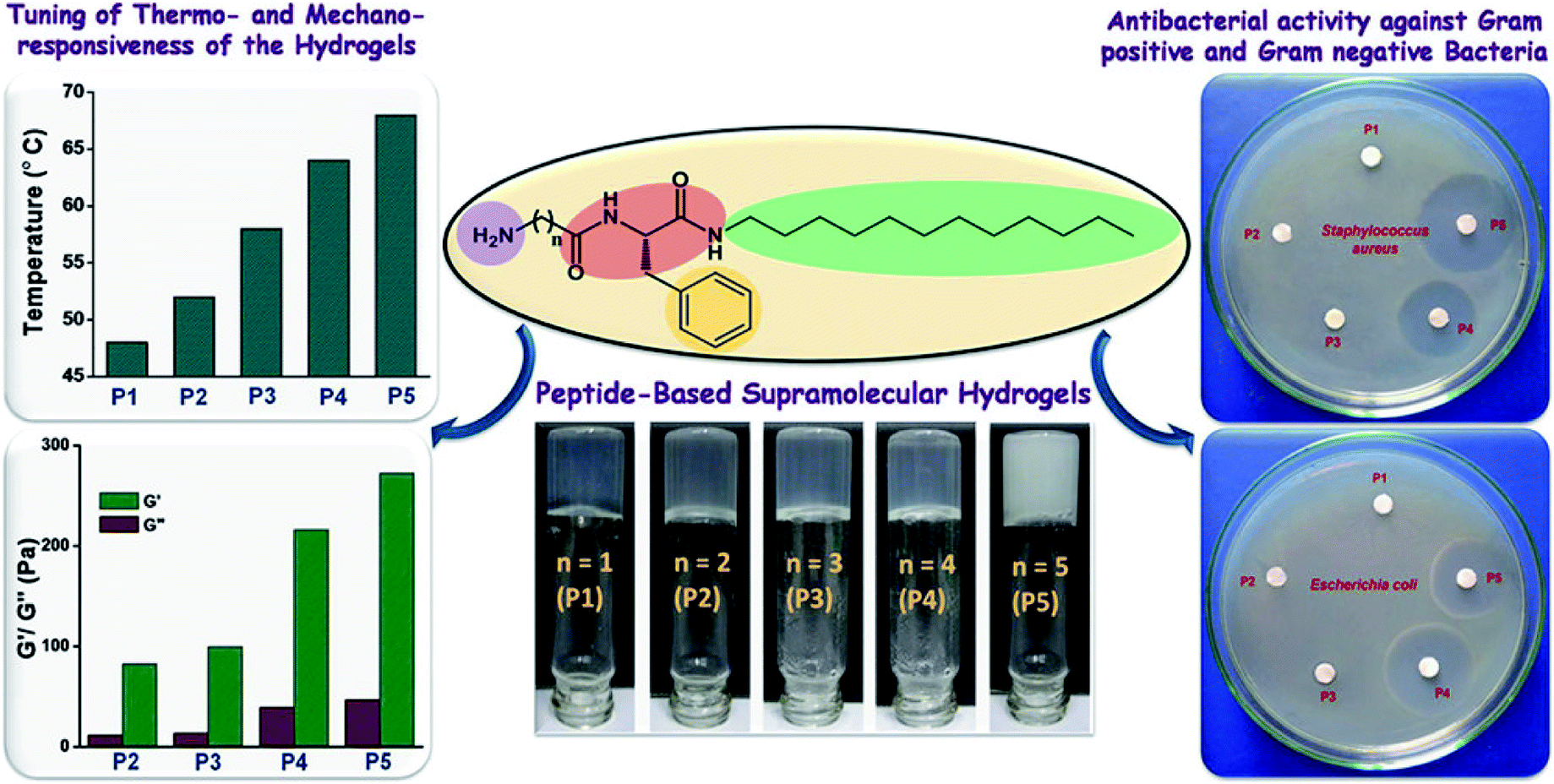 | ||
| Fig. 8 Amphiphilic peptide-based hydrogels for antibacterial.98 | ||
5. Conclusions and prospects
Over the previous two decades, peptide-based injectable hydrogels have been constructed for biomedical applications due to their unique biodegradation, biocompatibility, and bioactivity.99 This review highlighted the fabrication and formation of peptide-based hydrogels.100 Several promising candidates for biomedical application with smart and environmentally sensitive capacities were summarized. Peptide-based injectable hydrogels have important application value in the field of wound healing, 3D bioprinting, dressings, immune adjuvants, and oral materials. The recent development of peptide-based hydrogels for biomedical applications in tissue engineering, drug/gene vehicles, and antibacterial infections was discussed. However, the clinical application of peptide-based injectable hydrogels is hindered by the challenges associated with their relatively low mechanical strength and stability. The biologic' release profile of hydrogels is also a critical point for further application. The future trend in hydrogel development will be to design the structure and function of peptide-based hydrogels according to the needs of different applications in different-fields. Therefore, it is necessary to design the rational two-dimensional and 3D peptides structures by controlling their hydrophobic interactions, π–π stacking, and hydrogen bonding. Moreover, the enhanced mechanical strength of peptide-based hydrogels can be achieved through covalently linking polymers to construct peptide/polymer hybrid hydrogels.50 Many strategies have been introduced to improve the intelligence of peptide-based hydrogels by generating thermo-sensitive, pH-sensitive, enzyme-sensitive and PEGylated hydrogels, as well as those modified with other active molecules.Overall, peptide-based injectable hydrogels have made great progress not only in basic research, but also in several biomedical fields. We firmly believe that the construction of hydrogel materials with the advantage of peptides has a very broad prospective application potential. There are two main types of peptide-based hydrogels, synthetic hydrogels and natural biological hydrogels. The latter has better biocompatibility and is frequently utilized in the fabrication of peptide-based injectable hydrogels. However, synthetic peptides have several unique advantages, including variety, easy functionalization, controllable structure and functions, thus indicating potential values in biomedical applications. Based on the problems encountered in the above analysis of peptide-based hydrogels for biomedical application, future research can focus on the following aspects: (1) the preparation process should be convenient, “green” and low cost; (2) intelligent hydrogels should be prepared with environmental responsiveness to improve the biological value of hydrogels; (3) biofunctional peptide-based hydrogels should be constructed based on theoretical knowledge of structural biology and bioinformatics; and (4) bioactive hydrogels prepared by genetic engineering will be the key direction of future development. We believe that peptide-based injectable hydrogels will be an important research topic with great vitality and practical potential in biomedical applications.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge the financial support from National Natural Science Foundation of China (51973217, 81671033 and 81771123).References
- J. A. Yang, J. Yeom, B. W. Hwang, A. S. Hoffman and S. K. Hahn, Prog. Polym. Sci., 2014, 39, 1973–1986 CrossRef CAS.
- L. Yu and J. D. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- X. M. Li, Q. Q. Sun, Q. Li, N. Kawazoe and G. P. Chen, Front. Chem., 2018, 6, 499 CrossRef CAS PubMed.
- X. W. Du, J. Zhou, J. F. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- Y. Guo, R. J. Wang, Y. Z. Shang and H. L. Liu, RSC Adv., 2018, 8, 10633 RSC.
- Y. Li, F. H. Wang and H. G. Cui, Bioeng. Transl. Med., 2016, 1, 306–322 CrossRef CAS PubMed.
- K. X. Ren, C. L. He, Y. L. Cheng, G. Li and X. S. Chen, Polym. Chem., 2014, 5, 5069–5076 RSC.
- M. Sheikhpour, L. Barani and A. Kasaeian, J. Controlled Release, 2017, 253, 97–109 CrossRef CAS PubMed.
- T. Serizawa, H. Fukuta, T. Date and T. Sawada, Chem. Commun., 2016, 52, 2241–2244 RSC.
- G. Sharifzadeh and H. Hosseinkhani, Adv. Healthcare Mater., 2017, 6, 1700810 Search PubMed.
- M. Y. Kwon, S. L. Vega, W. M. Gramlich, M. Kim, R. L. Mauck and J. A. Burdick, Adv. Healthcare Mater., 2018, 7, 1701199 CrossRef PubMed.
- J. D. Tang, C. Mura and K. J. Lampe, J. Am. Chem. Soc., 2019, 141, 4886–4899 CrossRef CAS PubMed.
- H. L. Jin, C. Wan, Z. W. Zou, G. F. Zhao, L. L. Zhang, Y. Y. Geng, T. Chen, A. Huang, F. G. Jiang, J. P. Feng, J. F. Lovell, J. Chen, G. Wu and K. Y. Yang, ACS Nano, 2018, 12, 3295–3310 CrossRef CAS PubMed.
- B. H. Hu, C. Owh, P. L. Chee, W. R. Leow, X. Liu, Y. L. Wu, P. Z. Guo, X. J. Loh and X. D. Chen, Chem. Soc. Rev., 2018, 47, 6917–6929 RSC.
- S. Q. Li, S. J. Dong, W. G. Xu, S. C. Tu, L. S. Yan, C. W. Zhao, J. X. Ding and X. S. Chen, Adv. Sci., 2018, 5, 1700527 CrossRef PubMed.
- R. Dimatteo, N. J. Darling and T. Segura, Adv. Drug Delivery Rev., 2018, 127, 167–184 CrossRef CAS PubMed.
- Y. L. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- S. J. Yu, C. L. He and X. S. Chen, Macromol. Biosci., 2018, 18, 1800240 CrossRef PubMed.
- D. Y. Ko, U. P. Shinde, B. Yeon and B. Jeong, Prog. Polym. Sci., 2013, 38, 672–701 CrossRef CAS.
- B. G. Xing, C. W. Yu, K. H. Chow, P. L. Ho, D. G. Fu and B. Xu, J. Am. Chem. Soc., 2002, 124, 14846–14847 CrossRef CAS PubMed.
- X. Q. Lian, D. X. Shi, J. Ma, X. J. Cai and Z. W. Gu, Chin. Chem. Lett., 2018, 29, 501–504 CrossRef CAS.
- S. J. Yu, C. Wang, J. C. Yu, J. Q. Wang, Y. Lu, Y. Q. Zhang, X. D. Zhang, Q. Y. Hu, W. J. Sun, C. L. He, X. S. Chen and Z. Gu, Adv. Mater., 2018, 30, 1801527 CrossRef PubMed.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822–835 RSC.
- H. Green, G. Ochbaum, A. Gitelman-Povimonsky, R. Bitton and H. Rapaport, RSC Adv., 2018, 8, 10072–10080 RSC.
- H. M. Wang, Y. Z. Wang, X. L. Zhang, Y. W. Hu, X. Y. Yi, L. S. Ma, H. Zhou, J. F. Long, Q. Liu and Z. M. Yang, Chem. Commun., 2015, 51, 14239–14242 RSC.
- C. H. Liang, D. B. Zheng, F. Shi, T. Y. Xu, C. H. Yang, J. F. Liu, L. Wang and Z. M. Yang, Nanoscale, 2017, 9, 11987–11993 RSC.
- H. P. Fang, Z. P. Guo, L. Lin, J. Chen, P. J. Sun, J. Y. Wu, C. N. Xu, H. Y. Tian and X. S. Chen, J. Am. Chem. Soc., 2018, 140, 11992–12000 CrossRef CAS PubMed.
- Z. Q. Yu, Z. Cai, Q. L. Chen, M. H. Liu, L. Ye, J. Y. Ren, W. Z. Liao and S. W. Liu, Biomater. Sci., 2016, 4, 365–374 RSC.
- D. E. Clarke, C. D. J. Parmenter and O. A. Scherman, Angew. Chem., Int. Ed. Engl., 2018, 57, 7709–7713 CrossRef CAS PubMed.
- S. G. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS PubMed.
- J. Y. Li, Y. Gao, Y. Kuang, J. F. Shi, X. W. Du, J. Zhou, H. M. Wang, Z. M. Yang and B. Xu, J. Am. Chem. Soc., 2013, 135, 9907–9914 CrossRef CAS PubMed.
- C. C. Lin and K. S. Anseth, Adv. Funct. Mater., 2009, 19, 2325–2331 CrossRef CAS PubMed.
- L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025–17029 CrossRef CAS PubMed.
- R. A. McMillan and V. P. Conticello, Macromolecules, 2000, 33, 4809–4821 CrossRef CAS.
- M. Wathier, C. S. Johnson, T. Kim and M. W. Grinstaff, Bioconjugate Chem., 2006, 17, 873–876 CrossRef CAS PubMed.
- J. A. Burdick and K. S. Anseth, Biomaterials, 2002, 23, 4315–4323 CrossRef CAS PubMed.
- D. J. Pochan, J. P. Schneider, J. Kretsinger, B. Ozbas, K. Rajagopal and L. Haines, J. Am. Chem. Soc., 2003, 125, 11802–11803 CrossRef CAS PubMed.
- P. J. Stahl, N. H. Romano, D. Wirtz and S. M. Yu, Biomacromolecules, 2010, 11, 2336–2344 CrossRef CAS PubMed.
- W. Xun, D. Q. Wu, Z. Y. Li, H. Y. Wang, F. W. Huang, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Macromol. Biosci., 2009, 9, 1219–1226 CrossRef CAS PubMed.
- X. Li, M. Fu, J. Wu, C. Y. Zhang, X. Deng, A. Dhinakar, W. L. Huang, H. Qian and L. Ge, Acta Biomater., 2017, 51, 294–303 CrossRef CAS PubMed.
- R. R. Xing, S. K. Li, N. Zhang, G. Z. Shen, H. Mohwald and X. H. Yan, Biomacromolecules, 2017, 18, 3514–3523 CrossRef CAS PubMed.
- X. L. Wu, Y. D. Wu, H. B. Ye, S. J. Yu, C. L. He and X. S. Chen, J. Controlled Release, 2017, 255, 81–93 CrossRef CAS PubMed.
- S. H. Medina, S. Li, O. M. Z. Howard, M. Dunlap, A. Trivett, J. P. Schneider and J. J. Oppenheim, Biomaterials, 2015, 53, 545–553 CrossRef CAS PubMed.
- X. Cheng, J. Liu, L. Wang, R. L. Wang, Z. L. Liu and R. X. Zhuo, RSC Adv., 2016, 6, 101334–101346 RSC.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005 CrossRef CAS PubMed.
- C. L. He, X. L. Zhuang, Z. H. Tang, H. Y. Tian and X. S. Chen, Adv. Healthcare Mater., 2012, 1, 48–78 CrossRef CAS PubMed.
- S. Kuroyanagi, N. Shimada, S. Fujii, T. Furuta, A. Harada, K. Sakurai and A. Maruyama, J. Am. Chem. Soc., 2019, 141, 1261–1268 CrossRef CAS PubMed.
- T. Masuda, N. Shimada and A. Maruyama, Biomacromolecules, 2018, 19, 1333–1339 CrossRef CAS PubMed.
- Z. X. Deng, T. L. Hu, Q. Lei, J. K. He, P. X. Ma and B. L. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 6796–6808 CrossRef CAS PubMed.
- E. Radvar and H. S. Azevedo, Macromol. Biosci., 2019, 19, 1800221 CrossRef PubMed.
- J. Wang, K. Liu, R. R. Xing and X. H. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- F. W. Kotch and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3028–3033 CrossRef CAS PubMed.
- X. Mo, Y. J. An, C. S. Yun and S. M. Yu, Angew. Chem., Int. Ed., 2006, 45, 2267–2270 CrossRef CAS PubMed.
- L. E. R. O'Leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821–828 CrossRef PubMed.
- M. X. Qiao, D. W. Chen, T. N. Hao, X. L. Zhao, H. Y. Hu and X. C. Ma, Int. J. Pharm., 2007, 345, 116–124 CrossRef CAS PubMed.
- A. Gandhi, A. Paul, S. O. Sen and K. K. Sen, Asian J. Pharm. Sci., 2015, 10, 99–107 CrossRef.
- C. Chassenieux and C. Tsitsilianis, Soft Matter, 2016, 12, 1344–1359 RSC.
- H. Guo, J. M. Zhang, T. Xu, Z. D. Zhang, J. R. Yao and Z. Z. Shao, Biomacromolecules, 2013, 14, 2733–2738 CrossRef CAS PubMed.
- M. B. Charati, I. Lee, K. C. Hribar and J. A. Burdick, Small, 2010, 6, 1608–1611 CrossRef CAS PubMed.
- J. W. Ko, W. S. Choi, J. Kim, S. K. Kuk, S. H. Lee and C. B. Park, Biomacromolecules, 2017, 18, 3551–3556 CrossRef CAS PubMed.
- M. T. He, J. B. Li, S. Tan, R. Z. Wang and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 18718–18721 CrossRef CAS PubMed.
- M. E. Roth-Konforti, M. Comune, M. Halperin-Sternfeld, I. Grigoriants, D. Shabat and L. Adler-Abramovich, Macromol. Rapid Commun., 2018, 39, 1800588 CrossRef PubMed.
- A. Assion, T. Baumert, M. Bergt, T. Brixner, B. Kiefer, V. Seyfried, M. Strehle and G. Gerber, Science, 1998, 282, 919–922 CrossRef CAS PubMed.
- X. Q. Dou and C. L. Feng, Adv. Mater., 2017, 29, 1604062 CrossRef PubMed.
- B. L. Guo and P. X. Ma, Sci. China: Chem., 2014, 57, 490–500 CrossRef CAS.
- Y. Ding, Y. Li, M. Qin, Y. Cao and W. Wang, Langmuir, 2013, 29, 13299–13306 CrossRef CAS PubMed.
- Y. Li, Y. Ding, M. Qin, Y. Cao and W. Wang, Chem. Commun., 2013, 49, 8653–8655 RSC.
- L. Q. Wu and G. F. Payne, Trends Biotechnol., 2004, 22, 593–599 CrossRef CAS PubMed.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS PubMed.
- S. Winkler, D. Wilson and D. L. Kaplan, Biochemistry, 2000, 39, 12739–12746 CrossRef CAS PubMed.
- Z. M. Yang, H. W. Gu, D. G. Fu, P. Gao, K. J. K. Lam and B. Xu, Adv. Mater., 2006, 18, 545 CrossRef CAS.
- K. M. Galler, L. Aulisa, K. R. Regan, R. N. D'Souza and J. D. Hartgerink, J. Am. Chem. Soc., 2010, 132, 3217–3223 CrossRef CAS PubMed.
- B. L. Guo, J. Qu, X. Zhao and M. Y. Zhang, Acta Biomater., 2019, 84, 180–193 CrossRef CAS PubMed.
- J. F. Zheng, F. J. Zhao, W. Zhang, Y. F. Mo, L. Zeng, X. Li and X. F. Chen, Mater. Sci. Eng., C, 2018, 89, 119–127 CrossRef CAS PubMed.
- T. L. Gao, M. H. Jiang, X. Q. Liu, G. J. You, W. Y. Wang, Z. H. Sun, A. G. Ma and J. Chen, Polymers, 2019, 11, 171 CrossRef PubMed.
- L. Wang, Y. B. Wu, T. L. Hu, B. L. Guo and P. X. Ma, Acta Biomater., 2017, 59, 68–81 CrossRef CAS PubMed.
- K. X. Ren, H. T. Cui, Q. H. Xu, C. L. He, G. Li and X. S. Chen, Biomacromolecules, 2016, 17, 3862–3871 CrossRef CAS PubMed.
- P. Hitscherich, P. K. Nguyen, A. Kannan, A. Chirayath, S. Anur, B. Sarkar, E. J. Lee and V. A. Kumar, J. Biomed. Nanotechnol., 2018, 14, 802–807 CrossRef CAS PubMed.
- R. Silva, B. Fabry and A. R. Boccaccini, Biomaterials, 2014, 35, 6727–6738 CrossRef CAS PubMed.
- S. Koutsopoulos and S. G. Zhang, Acta Biomater., 2013, 9, 5162–5169 CrossRef CAS PubMed.
- C. Studle, Q. Vallmajo-Martin, A. Haumer, J. Guerrero, M. Centola, A. Mehrkens, D. J. Schaefer, M. Ehrbar, A. Barbero and I. Martin, Biomaterials, 2018, 171, 219–229 CrossRef CAS PubMed.
- R. Narayanaswamy and V. P. Torchilin, Molecules, 2019, 24, 603 CrossRef PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- A. Shah, M. S. Malik, G. S. Khan, E. Nosheen, F. J. Iftikhar, F. A. Khan, S. S. Shukla, M. S. Akhter, H. B. Kraatz and T. M. Aminabhavi, Chem. Eng. J., 2018, 353, 559–583 CrossRef CAS.
- Z. Mhlwatika and B. A. Aderibigbe, Molecules, 2018, 23, 2527 CrossRef PubMed.
- C. Karavasili, E. Panteris, I. S. Vizirianakis, S. Koutsopoulos and D. G. Fatouros, Pharm. Res., 2018, 35, 166 CrossRef PubMed.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- C. Karavasili, D. A. Andreadis, O. L. Katsamenis, E. Panteris, P. Anastasiadou, Z. Kakazanis, V. Zoumpourlis, C. K. Markopoulou, S. Koutsopoulos, I. S. Vizirianakis and D. G. Fatouros, Mol. Pharm., 2019, 16, 2326–2341 CrossRef CAS.
- J. Qu, X. Zhao, Y. P. Liang, Y. M. Xu, P. X. Ma and B. L. Guo, Chem. Eng. J., 2019, 362, 548–560 CrossRef CAS.
- Y. P. Liang, X. Zhao, T. L. Hu, B. J. Chen, Z. H. Yin, P. X. Ma and B. L. Guo, Small, 2019, 15, 1900046 CrossRef PubMed.
- E. De Giglio, S. Cometa, M. A. Ricci, D. Cafagna, A. M. Savino, L. Sabbatini, M. Orciani, E. Ceci, L. Novello, G. M. Tantillo and M. Mattioli-Belmonte, Acta Biomater., 2011, 7, 882–891 CrossRef CAS PubMed.
- S. Marchesan, Y. Qu, L. J. Waddington, C. D. Easton, V. Glattauer, T. J. Lithgow, K. M. McLean, J. S. Forsythe and P. G. Hartley, Biomaterials, 2013, 34, 3678–3687 CrossRef CAS PubMed.
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117–9149 RSC.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- D. A. Salick, J. K. Kretsinger, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2007, 129, 14793–14799 CrossRef CAS.
- N. W. Schmidt, A. Mishra, G. H. Lai, M. Davis, L. K. Sanders, D. Tran, A. Garcia, K. P. Tai, P. B. McCray, A. J. Ouellette, M. E. Selsted and G. C. L. Wong, J. Am. Chem. Soc., 2011, 133, 6720–6727 CrossRef CAS PubMed.
- A. S. Veiga, C. Sinthuvanich, D. Gaspar, H. G. Franquelim, M. A. R. B. Castanho and J. P. Schneider, Biomaterials, 2012, 33, 8907–8916 CrossRef CAS PubMed.
- N. Nandi, K. Gayen, S. Ghosh, D. Bhunia, S. Kirkham, S. K. Sen, S. Ghosh, I. W. Hamley and A. Banerjee, Biomacromolecules, 2017, 18, 3621–3629 CrossRef CAS.
- T. Thambi, V. H. G. Phan and D. S. Lee, Macromol. Rapid Commun., 2016, 37, 1881–1896 CrossRef CAS PubMed.
- D. Tesauro, A. Accardo, C. Diaferia, V. Milano, J. Guillon, L. Ronga and F. Rossi, Molecules, 2019, 24, 351 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |