
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile one-pot synthesis of sulfonyl fluorides from sulfonates or sulfonic acids†
Ying Jianga,
Njud S. Alharbib,
Bing Sun*a and
Hua-Li Qin *a
*a
aSchool of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: bing.sun@whut.edu.cn; qinhuali@whut.edu.cn
bBiotechnology Research Group, Deportment of Biological Sciences, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia. E-mail: njud_alharbi@yahoo.com
First published on 7th May 2019
Abstract
A facile cascade process for directly transforming the abundant and inexpensive sulfonates (or sulfonic acids) to the highly valuable sulfonyl fluorides was developed. This new protocol features mild reaction conditions using readily available and easy-to-operate reagents. A diverse set of sulfonyl fluorides was prepared facilitating the enrichment of the sulfonyl fluoride library.
Sulfonyl fluorides (SFs) have been identified and utilized in the realms of biology, pharmaceuticals and functional molecules for their unique stability-reactivity balance.1 Owing to the relatively low reactivity toward nucleophilic substitution and the exclusive heterolytic property, SF electrophiles are privileged motifs in the selective covalent interaction with context-specific amino acids or proteins for diverse applications (Fig. 1).2 For example, Murthy and co-workers disclosed that 2-nitrobenzenesulfonyl fluoride (NBSF, Fig. 1) was effective at killing Gram-negative bacteria, where the SF group could possibly react with target proteins directly or via an intermediate.3 By holding a compatible electrophilicity, SFs have found remarkable utility as covalent probes in chemical biology, which enable the efficient targeting of active-site amino acid residues.2,4 Meanwhile, through the reaction of SFs with active site amino acids to inactivate these enzymes, the corresponding SF-type protease inhibitors could be developed.5 The commonly used serine protease inhibitors include (2-aminoethyl)benzenesulfonyl fluoride (AEBSF, Fig. 1), of which the hydrochloride salt is called Pefabloc®.5a,c In addition, the diagnostic value of SF in 18F-labelled biomarkers in positron emission tomography has also drawn much attention, such as the application of [18F]4-formylbenzenesulfonyl fluoride ([18F]FBSF, Fig. 1) as a radio labeling synthon.6
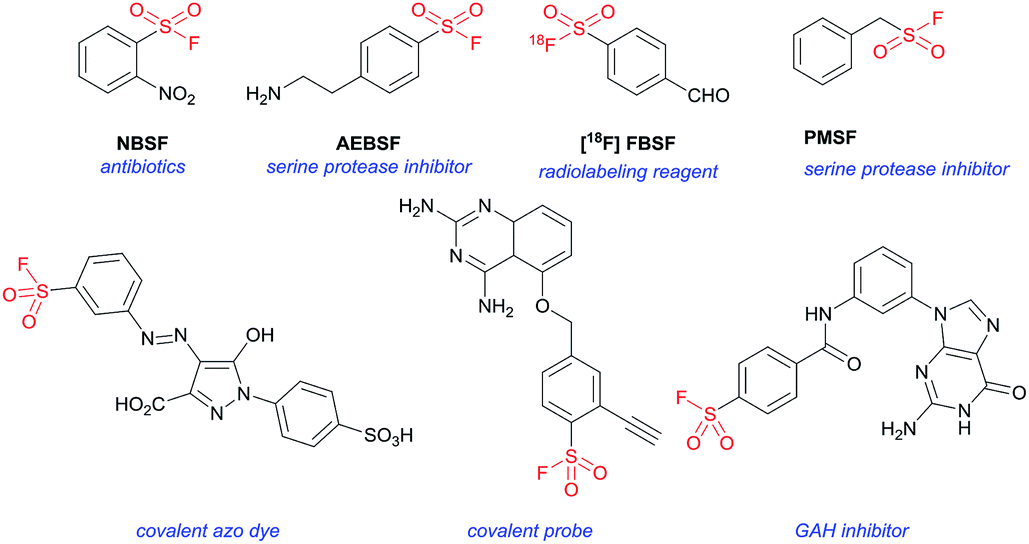 | ||
| Fig. 1 Representative sulfonyl fluoride-containing molecules with significant biological values. | ||
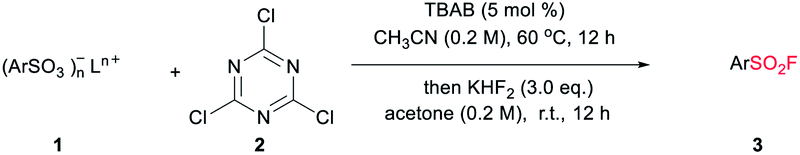
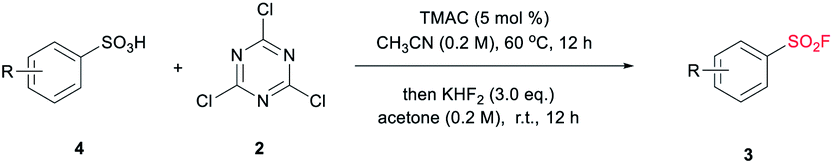
Despite the versatile findings on the utility of SFs, the efforts toward their synthesis often encountered sluggishness. The most common method involves a chlorine–fluorine exchange of arenesulfonyl chloride in the presence of aqueous solution of KF7 or KHF2 (ref. 8) (Scheme 1a). A more efficient system could be achieved with phase transfer catalyst including KF and 18-crown-6-ether in acetonitrile.9 However, the generated sulfonyl chloride intermediates as very reactive electrophiles, are easily subjected to nucleophilic attack without selectivity thus are considered as poor candidates in the synthetic sequence of complex molecules. To alleviate this issue, a pd-catalysed strategy through the insertion of SO2 to Ar–X for assembly of ArSO2F was introduced (Scheme 1b).10 As a continuation of our search for general and practical protocols for the synthesis of sulfonyl fluoride motifs,11 we attempted to use the stable and readily available sulfonates as starting material. In fact, a successful exploration on sulfonate as raw material has been conducted by Sharpless and co-workers, unveiling a two-step synthesis of SFs comprising the initial preparation of sulfonyl chloride by reacting sulfonate with chlorosulfuric acid, and subsequent chlorine–fluorine exchange to form SVI-F,1a whereas the use of toxic chlorosulfuric acid inevitably raises the safety risk. On the other hand, DAST (diethyl amino sulfur trifluoride), XtalFluor-M (difluoro-4-morpholinylsulfonium'tetrafluoroborate) and related reagents were also reported for fluorination reactions, however, these reagents have rarely been successfully applied for the synthesis of sulfonyl fluorides.12a,b Comparing with DAST, KHF2 is an inexpensive, stable, and industrially applicable chemical acting as both nucleophile and buffer for the preparation of sulfonyl fluorides.12c,d Herein, we report the transition-metal-free one-pot synthesis of SFs from sulfonates in the presence of cyanuric chloride and KHF2 (Scheme 1c). Phase transfer catalyst tetra-n-butylammonium bromide (TBAB) was selected for the transformation of sulfonates, while tetramethylammonium chloride (TMAC) was used for the case of sulfonic acid.
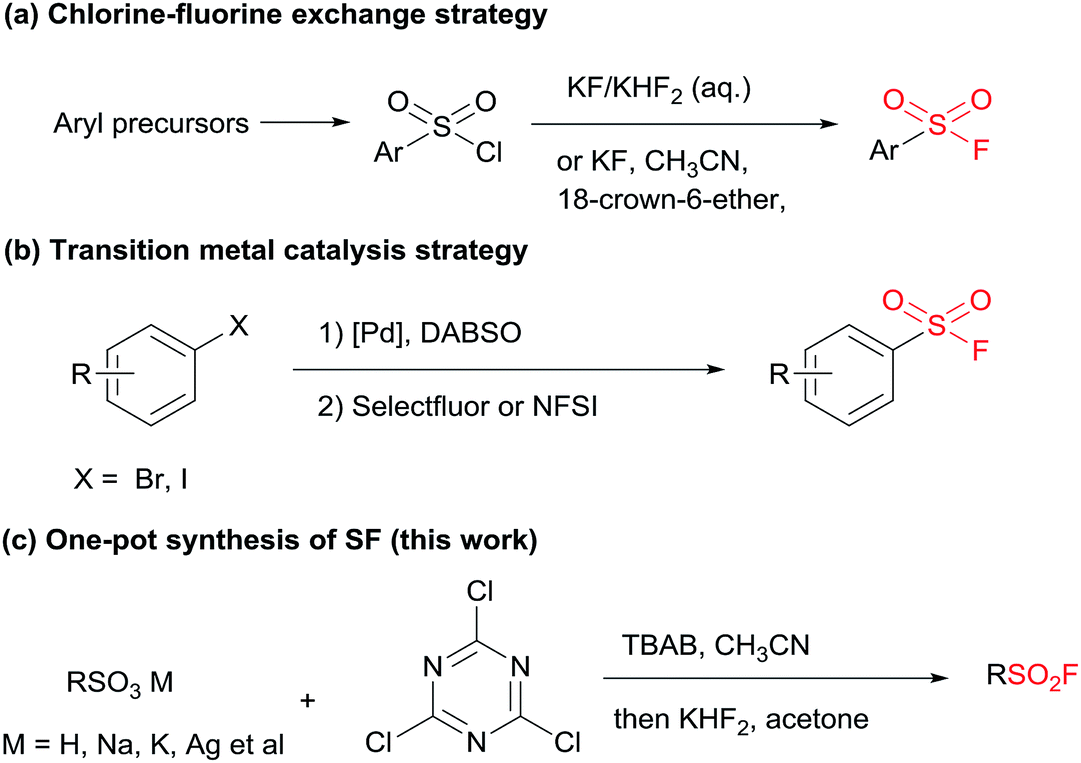 | ||
| Scheme 1 Strategies for the assembly of sulfonyl fluorides. | ||
The preparations of SFs from sulfonate salts1a,13,14 are rarely reported and could date back to Kulka's work over sixty years ago (or even earlier), and therein chlorosulfuric acid (occasionally oxalyl chloride) is essential for the provisional forming of SVI-Cl, followed by chlorine-fluorine transformation in a separated step to produce SVI-F. The chlorination process is regarded as the pivotal step, which normally requires harsh conditions. From a practical point of view, the convenient handling of the stable and nontoxic raw materials is particularly advantageous. Therefore, our focus is addressed on the efficient transformations of sulfonate with readily available and easy-handling reagents to install SF group. Cyanuric chloride (2) is usually used as an alternative to oxalyl chloride in the Swern oxidation,15 which may also be effective in the preparation of sulfonyl chloride. To verify this hypothesis, our preliminary study was set out for reaction condition screening (Table 1). Various quaternary ammonium salts, such as tetrabutylammonium iodide (TBAI), tetrabutylammonium acetate (TBAA), tetramethylammonium chloride (TMAC) and TBAB, were tested as catalysts for the reaction of sodium 4-methylbenzenesulfonate (1a) with 2 in acetonitrile (entries 1–7, Table 1). The addition of reagents was divided into two steps according to the reaction nature. Notably, the fluorine source was introduced directly after quenching the system to room temperature without any additional separation or purification. The maximum HPLC yield of 74% were observed for TBAB with a broad range of amount (5.0% to 20%). Interestingly, when the single solvent acetonitrile was replaced with dual-solvent of acetonitrile and acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v:v), the yield of 3a was improved to 86% (entry 8, Table 1). In spite of the significant promotion effect of “on water” phenomenon for bifluoride-involved fluorination proposed by Sharpless,16 it seems that the present system is also feasible (the detailed study of solvent screening can be found in ESI†). Then, a series of fluoride salts as F− source were screened. To be expected, potassium bifluoride showed superior activity compared to other fluoride salts (entries 8–11, Table 1), which may be attributed to the strong nucleophilicity of the bifluoride anion [F–H–F]−, but a minimum amount of 5 mol% was required (entries 12–14, Table 1). Thus, it can be concluded that the optimal condition refers to 5 mol% of TBAB, 3.0 equivalent of KHF2, acetonitrile/acetone (v:v 1:1), 1.1 equivalent of 2 and a grading temperature system (60 °C for chlorination step and r.t. for fluorination step).
:1 v:v), the yield of 3a was improved to 86% (entry 8, Table 1). In spite of the significant promotion effect of “on water” phenomenon for bifluoride-involved fluorination proposed by Sharpless,16 it seems that the present system is also feasible (the detailed study of solvent screening can be found in ESI†). Then, a series of fluoride salts as F− source were screened. To be expected, potassium bifluoride showed superior activity compared to other fluoride salts (entries 8–11, Table 1), which may be attributed to the strong nucleophilicity of the bifluoride anion [F–H–F]−, but a minimum amount of 5 mol% was required (entries 12–14, Table 1). Thus, it can be concluded that the optimal condition refers to 5 mol% of TBAB, 3.0 equivalent of KHF2, acetonitrile/acetone (v:v 1:1), 1.1 equivalent of 2 and a grading temperature system (60 °C for chlorination step and r.t. for fluorination step).
|
|||
|---|---|---|---|
| Entry | Cat. (mol%) | F− (eq.) | Yield (%) |
| a Reaction conditions: 1a (0.2 mmol), 2 (0.22 mmol, 1.1 eq.), CH3CN (1.0 mL), acetone (1.0 mL). Yields were determined by HPLC using 4-methylbenzenesulfonyl fluoride (3a) as the external standard.b CH3CN (2.0 mL) as the single solvent. | |||
| 1b | TBAI (20) | KHF2 (5.0) | 12 |
| 2b | TBAA (20) | KHF2 (5.0) | 17 |
| 3b | TMAC (20) | KHF2 (5.0) | 70 |
| 4b | TBAB (20) | KHF2 (5.0) | 74 |
| 5b | TBAB (10) | KHF2 (5.0) | 74 |
| 6b | TBAB (5.0) | KHF2 (5.0) | 74 |
| 7b | TBAB (3.0) | KHF2 (5.0) | 60 |
| 8 | TBAB (5.0) | KHF2 (5.0) | 86 |
| 9 | TBAB (5.0) | CsF (5.0) | <10 |
| 10 | TBAB (5.0) | KF (5.0) | 82 |
| 11 | TBAB (5.0) | CuF2 (5.0) | Trace |
| 12 | TBAB (5.0) | KHF2 (8.0) | 87 |
| 13 | TBAB (5.0) | KHF2 (3.0) | 86 |
| 14 | TBAB (5.0) | KHF2 (2.0) | 58 |
With the optimized conditions in hand, we next examined the substrate scope of sodium sulfonate (Table 2). To be delighted, both the electron-donating (3a, 3c, 3d, 3e) and electron-withdrawing groups (3f) were tolerated with the present strategy, however, the existence of nitro group obviously suppressed the yield (3f, 45%). It has to be mentioned that the arylsulfonate containing electron withdrawing groups such as 3,5-bis(methoxycarbonyl), 3-cyano and 2-trifluoromethyl failed to generate the corresponding products (3k, 3l and 3m) under this condition. The steric hindrance effect was not obvious, even for trimethyl-substituted sulfonate (3d).
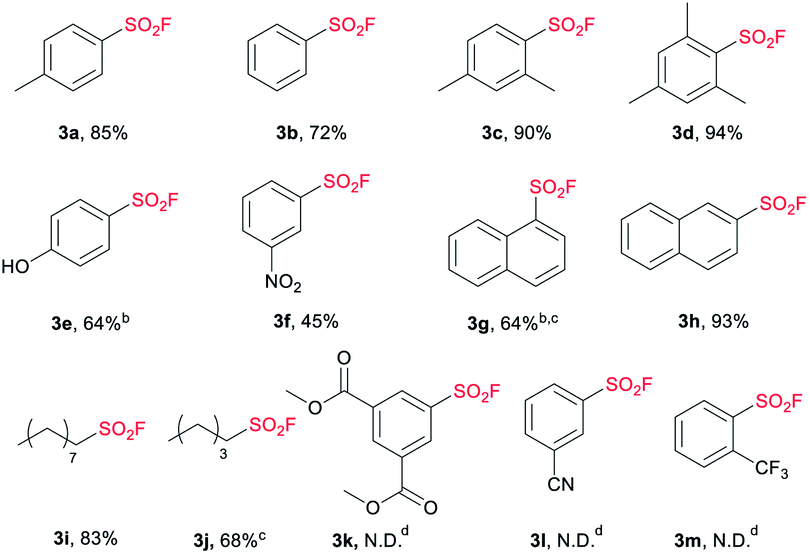
Naphthalene sulfonates were successfully converted to the corresponding SFs with moderate to good yields (3g, 3h), but the transformation of naphthalene-1-sulfonate required more catalyst and higher reaction temperature in the chlorination process. Importantly, the synthesized pentane-1-sulfonyl fluoride (3j) was found to be an efficient inhibitor of lipoprotein lipase.14 The potency of the inhibitors decreased as the chain length decreased,14,17 which means nonane-1-sulfonyl fluoride (3i) bearing a longer chain can also be a potential candidate as lipoprotein lipase inhibitor. Moreover, the present strategy shows superior efficiency compared with Kokotos' work14 wherein 10-fold molar excess of anhydrous sodium fluoride was required under refluxing acetone.
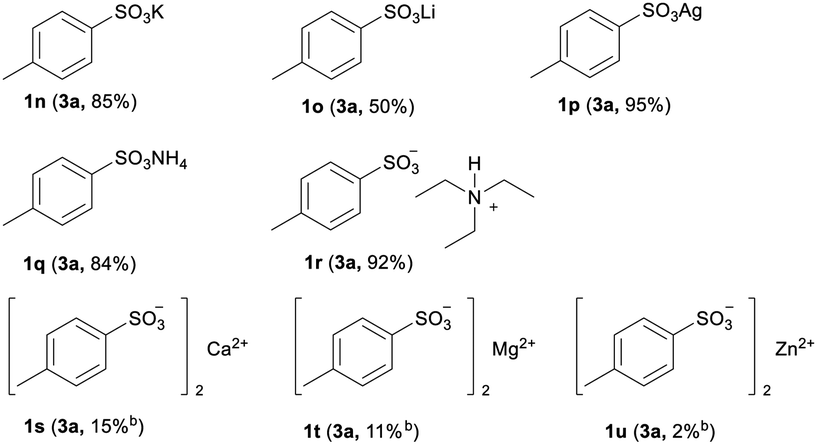
To evaluate the influence of different cations for the efficiency of this new protocol, a series of sulfonate salts containing various cations were examined. As shown in Table 3, the common sulfonate salts of monovalent potassium, lithium and silver were smoothly converted to 3a with moderate to good yields (1k–1m). Meanwhile, the nonmetallic sulfonate salts of ammonium and tertiary ammonium, patented for internal and external antistatic agents for non-conducting organic materials,18 were also successfully transformed to 3a with good yields (1n, 1o). Nevertheless, the sulfonate salts of divalent metals, such as calcium, magnesium and zinc, were only slightly reacted, resulting in poor yields (1p–1r). The lower yields of the corresponding sulfonyl fluorides from the sulfonate salts of divalent metals may be attributed to the stronger coordination ability of the divalent salts comparing to their monovalent conterparties,19 and the stronger coordination ability is not favourable for the preliminary formation of arylsulfonyl chlorides prior to generating sulfonyl fluorides.
|
|---|
| a General conditions: 1n–1r (2.0 mmol), 2 (2.2 mmol), TBAB (32.2 mg, 5 mol%), CH3CN (10 mL, 0.2 M); KHF2 (468 mg, 3.0 eq.), acetone (10 mL, 0.2 M); 1s–1u (0.2 mmol), 2 (0.44 mmol), TBAB (6.4 mg, 10 mol%), CH3CN (1 mL, 0.2 M); KHF2 (93.6 mg, 6.0 eq.), acetone (1 mL, 0.2 M). In the bracket shows the corresponding product and its isolated yield.b Yields were determined by HPLC using 4-methylbenzenesulfonyl fluoride (3a) as the external standard. |
 |
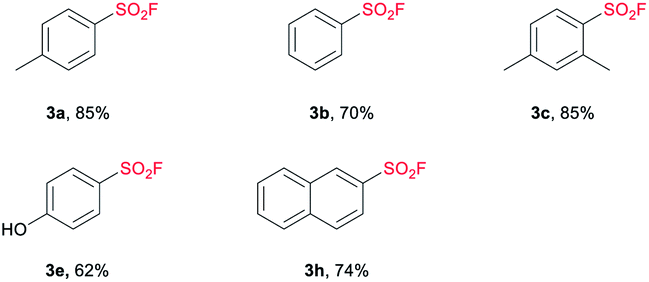
In our attempt to expand the substrate scope, we observed that TBAB as catalyst was not able to give satisfactory yield of SF from sulfonic acid under standard conditions (the detailed catalyst screening can be found in ESI†). Upon extensive study, we were pleased to find that the use of 5 mol% of tetramethylammonium chloride (TMAC) allowed the transformation of sulfonic acid to the corresponding sulfonyl fluorides effectively and efficiently. As demonstrated in Table 4, a series of aryl SFs bearing electron-donating groups were obtained with moderate to good yields (3a–3c, 3e). The naphthalene moiety could also be tolerated, achieving a yield of 74% (3 h). Compared with the reported methods,9,20 this system shows higher efficiency and mildness, providing a beneficial complement to the current synthetic strategy toward the installation of SF.
|
|---|
| a General conditions: 4 (2.0 mmol), 2 (2.2 mmol), TMAC (10.9 mg, 5 mol%), CH3CN (10 mL, 0.2 M), then KHF2 (468 mg, 3.0 eq.), acetone (10 mL, 0.2 M), isolated yields. |
 |
In conclusion, we have developed a new transition-metal-free one-pot method for the concise preparation of sulfonyl fluorides from sulfonates or sulfonic acids. The high efficiency and compatibility were demonstrated under mild reaction conditions from readily available and easy-handling reagents. This new protocol provides a favourable alternative to the current synthetic toolbox for the preparation of diverse SFs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (Grant No. 21703162, and Grant No. 21772150), the Wuhan Applied Fundamental Research Program of Wuhan Science and Technology Bureau (Grant No. 2017060201010216), the 111 Project (Grant No. B18038) and Wuhan University of Technology.Notes and references
- (a) J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430 CrossRef CAS PubMed; (b) P. K. Chinthakindi and P. I. Arvidsson, Eur. J. Org. Chem., 2018, 2018, 3648 CrossRef CAS; (c) T. Abdul Fattah and A. Saeed, J. Fluorine Chem., 2018, 213, 87 CrossRef CAS.
- (a) A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650 RSC; (b) M. Gehringer and S. A. Laufer, J. Med. Chem., 2018 DOI:10.1021/acs.jmedchem.8b01153.
- C. Sadlowski, B. Park, C. A. Borges, S. Das, D. Lucas Kerr, M. He, H. Han, L. Riley and N. Murthy, Mol. Syst. Des. Eng., 2018, 3, 599 RSC.
- (a) N. P. Grimster, S. Connelly, A. Baranczak, J. Dong, L. B. Krasnova, K. B. Sharpless, E. T. Powers, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 5656 CrossRef CAS PubMed; (b) E. C. Hett, H. Xu, K. F. Geoghegan, A. Gopalsamy, R. E. Kyne, C. A. Menard, A. Narayanan, M. D. Parikh, S. Liu, L. Roberts, R. P. Robinson, M. A. Tones and L. H. Jones, ACS Chem. Biol., 2015, 10, 1094 CrossRef CAS PubMed; (c) C. Dubiella, H. Cui and M. Groll, Angew. Chem., Int. Ed., 2016, 55, 13330 CrossRef CAS PubMed; (d) Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680 CrossRef CAS PubMed; (e) T. E. J. Chavas, M. J. Fuchter and P. A. DiMaggio, ACS Chem. Biol., 2018, 13, 2897 CrossRef CAS PubMed.
- (a) D. E. Fahrney and A. M. Gold, J. Am. Chem. Soc., 1963, 85, 997 CrossRef CAS; (b) A. E. Annamalai and R. F. Colman, J. Biol. Chem., 1981, 256, 10276 CAS; (c) J. C. Powers, J. L. Asgian, Ö. Doǧan Ekici and K. E. James, Chem. Rev., 2002, 102, 4639 CrossRef CAS PubMed; (d) I. M. Serafimova, M. A. Pufall, S. Krishnan, K. Duda, M. S. Cohen, R. L. Maglathlin, J. M. McFarland, R. M. Miller, M. Frödin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471 CrossRef CAS PubMed; (e) D. Manvar, K. Singh and V. N. Pandey, Biochemistry, 2013, 52, 432 CrossRef CAS PubMed; (f) H. Mukherjee, J. Debreczeni, J. Breed, S. Tentarelli, B. Aquila, J. E. Dowling, A. Whitty and N. P. Grimster, Org. Biomol. Chem., 2017, 15, 9685 RSC.
- (a) J. A. H. Inkster, K. Liu, S. Ait-Mohand, P. Schaffer, B. Guérin, T. J. Ruth and T. Storr, Chem.–Eur. J., 2012, 18, 11079 CrossRef CAS PubMed; (b) L. Matesic, N. A. Wyatt, B. H. Fraser, M. P. Roberts, T. Q. Pham and I. Greguric, J. Org. Chem., 2013, 78, 11262 CrossRef CAS PubMed.
- (a) A. R. Beauglehole, S. P. Baker and P. J. Scammells, J. Med. Chem., 2000, 43, 4973 CrossRef CAS PubMed; (b) J. W. Clader, W. Billard, H. Binch III, L.-Y. Chen, G. Crosby Jr, R. A. Duffy, J. Ford, J. A. Kozlowski, J. E. Lachowicz, S. Li, C. Liu, S. W. McCombie, S. Vice, G. Zhou and W. J. Greenlee, Bioorg. Med. Chem., 2004, 12, 319 CrossRef CAS PubMed.
- A. Talko and M. Barbasiewicz, ACS Sustainable Chem. Eng., 2018, 6, 6693 CrossRef CAS.
- T. A. Bianchi and L. A. Cate, J. Org. Chem., 1977, 42, 2031 CrossRef CAS.
- (a) A. T. Davies, J. M. Curto, S. W. Bagley and M. C. Willis, Chem. Sci., 2017, 8, 1233 RSC; (b) A. L. Tribby, I. Rodríguez, S. Shariffudin and N. D. Ball, J. Org. Chem., 2017, 82, 2294 CrossRef CAS PubMed.
- (a) H.-L. Qin, Q. Zheng, G. A. L. Bare, P. Wu and K. B. Sharpless, Angew. Chem., Int. Ed., 2016, 55, 14155 CrossRef CAS PubMed; (b) G.-F. Zha, Q. Zheng, J. Leng, P. Wu, H.-L. Qin and K. B. Sharpless, Angew. Chem., Int. Ed., 2017, 56, 4849 CrossRef CAS PubMed; (c) G.-F. Zha, G. Bare, J. Leng, Z.-P. Shang, Z. Luo and H.-L. Qin, Adv. Synth. Catal., 2017, 359, 3237 CrossRef CAS; (d) S.-M. Wang, C. Li, J. Leng, S. N. A. Bukhari and H.-Li. Qin, Org. Chem. Front., 2018, 5, 1411 RSC; (e) C. Li, S.-M. Wang and H.-L. Qin, Org. Lett., 2018, 20, 4699 CrossRef CAS PubMed; (f) S.-M. Wang, B. Moku, J. Leng and H.-L. Qin, Eur. J. Org. Chem., 2018, 32, 4407 CrossRef; (g) J. Leng and H.-L. Qin, Chem. Commun., 2018, 54, 4477 RSC; (h) L. Ravindar, S. N. A. Bukhari, K. P. Rakesh, H. M. Manukumar, H. K. Vivek, N. Mallesha, Z.-Z. Xie and H.-L. Qin, Bioorg. Chem., 2018, 81, 107 CrossRef CAS PubMed; (i) G.-F. Zha, S.-M. Wang, K. P. Rakesh, S. N. A. Bukhari, H. M. Manukumar, H. K. Vivek, N. Mallesha and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 364 CrossRef CAS PubMed; (j) X. Zhang, B. Moku, J. Leng, K. P. Rakesh and H.-L. Qin, Eur. J. Org. Chem., 2019, 1763 CrossRef CAS.
- (a) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed; (b) M. Nonn, L. Kiss, M. Haukka, S. Fustero and F. Fulop, Org. Lett., 2015, 17, 1074 CrossRef CAS PubMed; (c) A. G. Beaman and R. K. Robins, J. Am. Chem. Soc., 1961, 83, 4038 CrossRef CAS; (d) S. W. Wright and K. N. Hallstrom, J. Org. Chem., 2006, 71, 1080 CrossRef CAS PubMed.
- M. Kulka, J. Am. Chem. Soc., 1950, 72, 1215 CrossRef CAS.
- G. Kokotos, S. Kotsovolou, V. Constantinou-Kokotou, G. Wu and G. Olivecrona, Bioorg. Med. Chem. Lett., 2000, 10, 2803 CrossRef CAS PubMed.
- L. De Luca, G. Giacomelli and A. Procheddu, J. Org. Chem., 2001, 66, 7907 CrossRef CAS PubMed.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS PubMed.
- D. P. O'Connell, D. F. LeBlanc, D. Cromley, J. Billheimer, D. J. Rader and W. W. Bachovchin, Bioorg. Med. Chem. Lett., 2012, 22, 1397 CrossRef PubMed.
- C. Govindan, US Pat, 5 053 531, 1991.
- F. Horkay, I. Tasaki and P. J. Basser, Biomacromolecules, 2001, 2, 195 CrossRef CAS.
- (a) V. A. Petrov, S. Swearingen, W. Hong and W. Chris Petersen, J. Fluorine Chem., 2001, 109, 25 CrossRef CAS; (b) J.-G. Kim and D. O. Jang, Synlett, 2010, 20, 3049 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02531f |
| This journal is © The Royal Society of Chemistry 2019 |