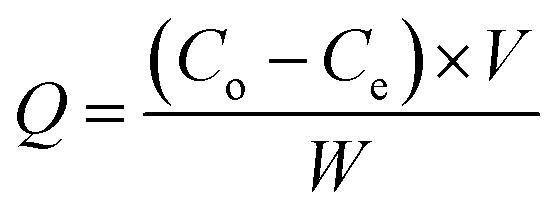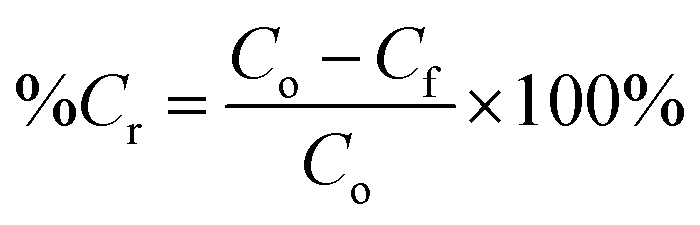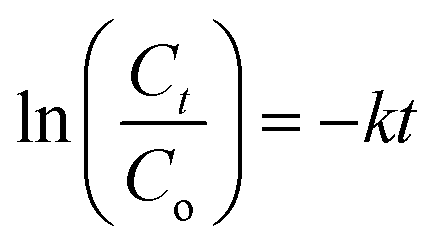DOI:
10.1039/C9RA01209E
(Paper)
RSC Adv., 2019,
9, 18076-18086
Effective photoreduction of graphene oxide for photodegradation of volatile organic compounds
Received
16th February 2019
, Accepted 27th May 2019
First published on 10th June 2019
Abstract
Nowadays, humans spend most of their time indoors and are frequently exposed to volatile organic compounds (VOCs) from various sources. The photocatalytic oxidation (PCO) method is a relatively more efficient method than the adsorption method for removing VOCs from the environment. In this work, graphene oxide (GO) was partially reduced via photoreduction under ultraviolet light (UV-A) irradiation and then used as a photocatalyst to degrade VOCs. After photoreduction, the band gap of the partially reduced graphene oxide (PRGO) decreased from 3.5–4.5 eV to 3.1–4.0 eV. Methanol vapour, which acts as a model VOC, was photodegraded using the PRGO. The effectiveness of the PRGO was mainly due to the removal of oxygen functional groups and restoration of the sp2 domain. This lowered the band gap and slowed down the electron recombination rate, which resulted in a higher photocatalytic activity. The photocatalytic activity of PRGO followed pseudo-first order kinetics, with a rate constant of 0.0025 min−1, and it could be reused for five cycles without any significant loss in the photocatalytic activity. This study demonstrates the potential of PRGO as a versatile and stable metal-free photocatalyst to remove indoor pollutants.
1. Introduction
In recent years, a great deal of attention has been paid to understanding and improving indoor air quality. There has been substantial research in determining personal exposure to pollutants, as people today spend more than 70% of their time indoors.1,2 Although indoor pollution is not seen as being as hazardous as outdoor pollution, concentrations of indoor contaminants are often higher than those outdoors and most of them can be attributed to human activities, furniture, and building materials.3 Concern has been raised over possible health effects, such as the “sick-building syndrome”, from exposure to indoor air pollutants, especially to volatile organic compounds (VOCs).1,4 One study found that people exposed to a mixture of indoor VOCs exceeding 3 ppb can start to experience feelings of discomfort.3 Methanol was used as a model VOC compound for this study of photodegradation, as it is one of the most abundant VOCs found indoors and has widespread use.5,6 It can be emitted from surface coatings,4 furniture,7 human breath,8 and detergents.9 Methanol is colourless, mild in odour, and has a relatively high vapour pressure. Furthermore, inhalation of methanol vapour can cause headaches, eye damage, and dermatitis.10
Generally, there are two ways to treat VOCs, namely non-destructive and destructive methods. Non-destructive methods are preferred in some industries, where VOCs can be captured and reused to lowers costs.11 Meanwhile, the destructive method is suitable for the indoor environment to limit the exposure of occupants to VOCs. Photocatalytic oxidation (PCO) is one of the most widely studied destructive technologies.12–14 In the PCO process, reactive oxygen species (ROS) are generated from the photocatalyst upon light irradiation. Then, the ROS mineralize VOCs into simpler, harmless compounds.11 Commonly, metal oxides such as titanium dioxide and zinc oxide are used to carry out PCO studies.15–17 For instance, methanol PCO was previously explored in a few studies with metal oxide-based photocatalysts.18–21
In contrast to metal oxide-based photocatalysis, a carbon-based catalyst is a metal-free option that is low in cost and uses carbon, which has high abundance in the earth.22–24 In addition, the adsorption capacity and specificity of carbon-based photocatalysts can be controlled by surface functionalization,25 altering the amount of π-conjugated binding,26 and creating pore structures.27 Carbon-based catalysts are highly versatile in carrying out various photocatalytic activities, as their band gaps are tunable28 and can be tailored into n-type or p-type photocatalysts.29 Recently, graphene oxide (GO) has arisen as a potential eco-friendly photocatalyst.24 GO can be reduced to produce partially reduced graphene oxide (PRGO) or reduced graphene oxide (rGO) through various reduction methods, such as thermal, microwave, photoreduction, chemical, electrochemical, and multistep reduction processes.30,31 Photoreduction of GO is a more recent reduction technique in which different magnitudes of photon energy produce different degrees of reduction.32,33 GO reduced by irradiation shows a higher efficiency as a conductor than PRGO reduced via chemical and hydrothermal techniques.34 Substantial amounts of research on GO with different oxidation levels and band gaps have been carried out to explore its use as a photocatalyst in dye degradation,28,35–37 water splitting,38–40 photoreduction of CO2,41 and organic pollutant removal from water.24,42 Moreover, GO has also been studied as a support to improve the adsorption capability of photocatalysts for pollutant removal.43,44 Nevertheless, there is lack of studies on photoreduced GO as a photocatalyst for the photodegradation of VOCs.
In this study, PRGO was synthesized via photoreduction and demonstrated high efficiency in the photodegradation of a VOC, namely methanol, under UV light irradiation. The physicochemical properties and photodecomposition activity of the PRGOs were investigated.
2. Experimental section
2.1. Materials
Graphite flakes (100 mesh size) were purchased from XFNANO Ltd. Sulfuric acid (H2SO4, 95–97%), phosphoric acid (H3PO4, 85%), potassium permanganate (KMnO4, >99%), hydrogen peroxide (H2O2, 30%), hydrochloric acid (HCl, 37%), isopropanol (IPA, >99.5%), and magnesium nitrate hexahydrate salt (Mg(NO3)2·6H2O, 99%) were all purchased from Sigma-Aldrich. The stainless-steel 304 wire mesh (100 mesh size) was purchased from Audew Ltd.
2.2. Synthesis of GO
In a typical synthesis, 3 g of graphite flakes was dispersed in a mixture of 300 ml of H2SO4 and H3PO4 (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio) under constant stirring. Then, 18 g of KMnO4 was slowly added into the suspension and heated to 50 °C for 12 hours before being transferred into an ice bath to stop the reaction. The reaction was stopped by adding 50 ml of deionized (DI) water dropwise into the suspension, followed by the pouring of 250 ml of DI water rapidly into the solution. Then, 6 ml of H2O2 was added dropwise to the suspension and the colour changed from purplish brown to yellowish brown, indicating the end point. The suspension was washed with 1 M HCl and DI water alternately via centrifugation until a pH of 3–4 was reached. The GO was dispersed in DI water for storage after the washing process.
:1 volume ratio) under constant stirring. Then, 18 g of KMnO4 was slowly added into the suspension and heated to 50 °C for 12 hours before being transferred into an ice bath to stop the reaction. The reaction was stopped by adding 50 ml of deionized (DI) water dropwise into the suspension, followed by the pouring of 250 ml of DI water rapidly into the solution. Then, 6 ml of H2O2 was added dropwise to the suspension and the colour changed from purplish brown to yellowish brown, indicating the end point. The suspension was washed with 1 M HCl and DI water alternately via centrifugation until a pH of 3–4 was reached. The GO was dispersed in DI water for storage after the washing process.
2.3. Synthesis of PRGO via photoreduction
A solvent exchange technique was performed, where water was separated from the GO suspension via centrifugation, followed by redispersion in IPA repeatedly to transfer GO from water into the IPA medium. IPA was used as the electrophoretic deposition (EPD) medium to prevent the bubbling effect (simultaneous water splitting) during EPD.45 Next, the GO/IPA suspension was diluted to a concentration of 0.20 mg ml−1, and then a small amount of magnesium salt (0.00025 mg ml−1) was added. A 100-mesh stainless-steel 304 wire mesh was cut into 11 cm × 5 cm size as the substrate electrode, while another piece was cut into 12 cm × 6 cm as the counter electrode. The EPD process was carried out at 110 V, with a constant electrode distance of 2 cm for 60 min on both sides of the wire mesh. After that, the substrate was dried in an oven for 60 min at 70 °C and the weight of GO loaded was measured. The electrophoretic-deposited GO was then photoreduced by irradiation with UV-A (4W) for specific times. The prepared PRGOs were denoted as PRGO-X, with X representing the UV-A irradiation times of 0, 2, 4, 6, 8, and 10 hours. After photoreduction, the PRGO was dried in the oven (60 min at 70 °C). Changes in colour of the PRGO were monitored throughout the photoreduction process.
2.4. Characterization
Ultraviolet-visible (UV-vis) spectra were collected using a PerkinElmer Lambda 35 UV/Vis spectrophotometer (Massachusetts, USA) from 200–800 nm. The Tauc plot linear extrapolation technique was used to determine the optical band gaps (EBg), where the square of the absorbance energy (αE) was plotted against the photon energy (hν) to determine the direct band gap (π → π*) transition. Raman and photoluminescence (PL) analysis were performed using a Renishaw inVia Raman microscope (Gloucestershire, UK) with 514 nm Ar-ion laser (1000–3200 cm−1) and a 325 nm Ar-ion laser (400–750 nm). Thin film X-ray diffraction (XRD) analysis was carried out using a PANalytical Empyrean X-ray diffractometer (Almelo, The Netherlands) equipped with CuKα radiation (λ = 1.5406 Å) at 45 kV and 40 mA, at a scanning rate of 0.1° s−1 for 2θ values from 5° to 80°. X-ray photoelectron spectroscopy (XPS) measurements were performed using a JPS9030-JEOL (Tokyo, Japan) with a MgKα X-ray source (1253.6 eV) and an aperture diameter of 6 mm (correction factor of carbon: 284.5 eV). Attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectra in the region 500–4000 cm−1 were obtained with a PerkinElmer FTIR-Spectrum 400 spectrometer (Massachusetts, USA).
2.5. Adsorption and PCO of methanol
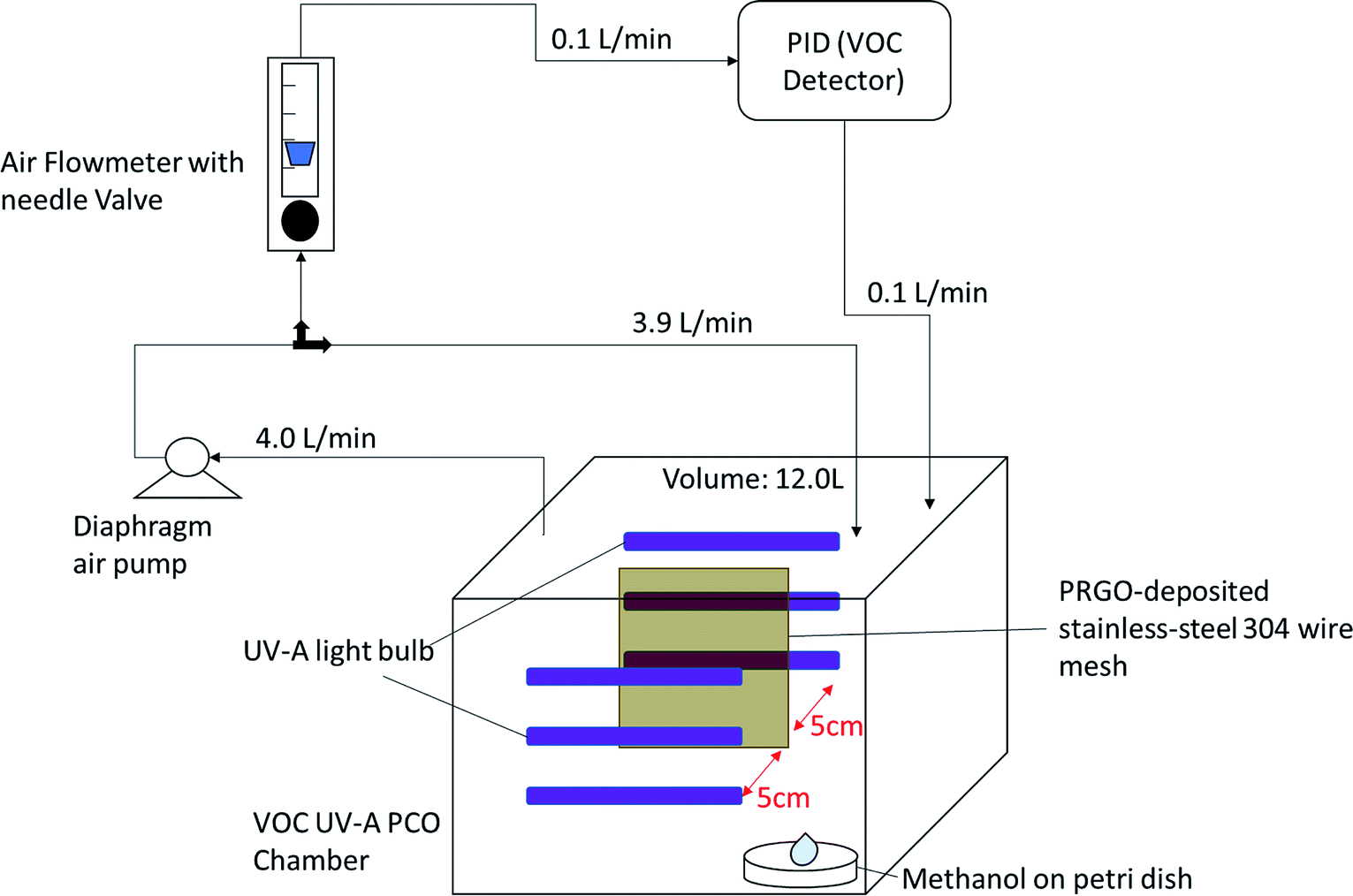
A custom-made VOC PCO chamber with a total volume of 12 L was designed to simulate an enclosed indoor environment under ambient conditions (Fig. 1). A dark adsorption experiment was carried out to determine the adsorption capacity of methanol (1 ppm) of each PRGO sample. Methanol concentration was measured by a Gastech photoionization detector (PID) for VOC. The VOC concentration was measured in units of composite index of air quality (CIAQ). A linear calibration curve was constructed to relate CIAQ to ppm.
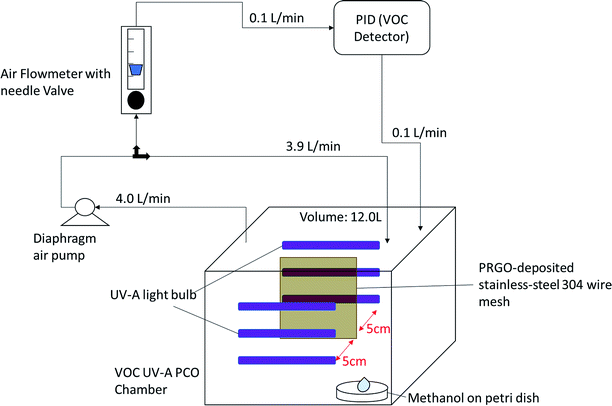 |
| | Fig. 1 Self-built ambient-condition VOC UV-A PCO chamber. | |
The adsorption capacity, Q (mg g−1) was calculated using eqn (1):
| |
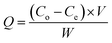 | (1) |
where
Co and
Ce are the methanol concentration (ppm) at initial and equilibrium,
V is the internal volume of the photoreactor (L), and
W is the weight of the loaded PRGO (g).
For the PCO process, 50 mg of PRGO was placed in the middle of the PCO chamber located at 5 cm from the 4W UV-A lamp (peak wavelength at 365 nm, equivalent to a photon energy of 3.39 eV). Methanol (1 ppm) was introduced into the chamber and the chamber was quickly sealed to be air tight, allowing the VOC to vapourize inside the chamber. A diaphragm pump was connected to the chamber to provide air circulation, and to dose a portion of the circulated air into the PID. After adsorption equilibrium was achieved, the UV-A light bulbs were switched on to begin the PCO test. The PCO experiment was carried out at room temperature (25 °C) with an indoor humidity of 65% relative humidity (RH). The PID value was monitored continuously to measure the methanol concentration for 100 min. The removal photodegradation percentage of methanol, %Cr was calculated using eqn (2):
| |
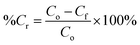 | (2) |
where
Cf (ppm) is the residual concentration of methanol after 100 min. The methanol removal
versus time was fitted to pseudo-first order kinetics as described by the Langmuir–Hinshelwood kinetic model given in
eqn (3):
| |
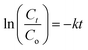 | (3) |
where
Ct is the concentration of methanol (ppm) at time
t (min) and
k (min
−1) is the pseudo-first order rate constant.
2.6. Photocatalyst recyclability tests
To determine the best PCO performance the stability of the PRGO was examined by five consecutive PCO cycles under the same reaction conditions with 2 ppm initial methanol concentration. After each cycle, the PRGO was dried at 70 °C to use for the next cycle.
3. Results and discussion
3.1. Synthesis of PRGOs
Fig. 2 shows the colour of PRGO changed from brown to black with longer photoreduction time. The change of colour suggested the occurrence of photoreduction. This colour darkening effect was commonly observed in several GO photoreduction studies.46,47
 |
| | Fig. 2 Colour appearance of PRGOs deposited on the surface of a 100-mesh stainless-steel wire mesh. | |
3.2. Characterization of GO and PRGOs
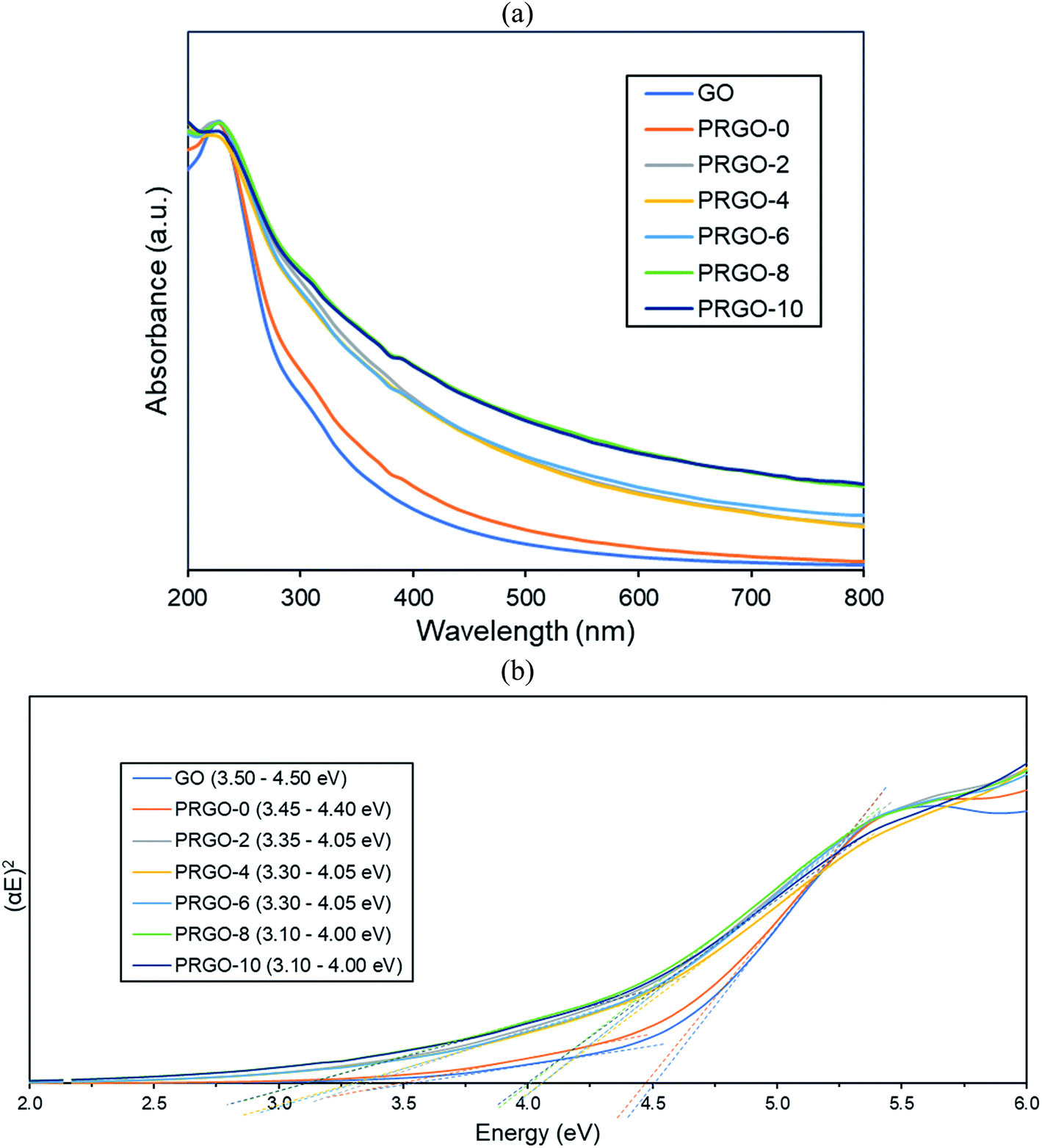
3.2.1. UV-vis analysis. UV-vis absorbance analysis was carried out to investigate the optical properties of the as-synthesized GO and PRGOs (Fig. 3a). All of them showed a peak at around 230 nm (π–π* transition of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and the appearance of a small shoulder near 300 nm (n–π* transition of CO). After photoreduction, the peak was not significantly shifted, while the shoulder at 300 nm was broadened. The non-shifting peak is in contrast with some GO reduction studies where the peak is gradually red-shifted to 260–270 nm after reduction.48,49 This probably implies that the UV-A photoreduction method is a milder reduction method, where GO is only partially reduced. This is important as PRGO is a semiconductor with a finite band gap, while a greatly reduced GO (rGO) will have a near-zero band gap and behave more like a conductor.50 In addition, the broadened area between 300 and 800 nm indicated that the sp2 hybridization carbon atom fraction was partially recovered and the π electron concentration was increased after photoreduction.51,52 As shown in Fig. 3b, the Tauc plot linear extrapolation technique was used to determine the band gaps of the GO and PRGOs. GO-based materials have a cluster of collective band structures due to graphene of different oxygenation levels, giving them a range of band gaps instead of an absolute value.26,53 After photoreduction the band gap was reduced and stabilized, with PRGO-8 and PRGO-10 having the lowest band gap at 3.10–4.00 eV. The band gap energy was sufficient to overcome the theoretical energy requirement of 2.71 eV to produce the ROS pair (superoxide, ·O−2 and hydroxyl radical, OH˙) under UV-A excitation for VOC photodegradation.54
C) and the appearance of a small shoulder near 300 nm (n–π* transition of CO). After photoreduction, the peak was not significantly shifted, while the shoulder at 300 nm was broadened. The non-shifting peak is in contrast with some GO reduction studies where the peak is gradually red-shifted to 260–270 nm after reduction.48,49 This probably implies that the UV-A photoreduction method is a milder reduction method, where GO is only partially reduced. This is important as PRGO is a semiconductor with a finite band gap, while a greatly reduced GO (rGO) will have a near-zero band gap and behave more like a conductor.50 In addition, the broadened area between 300 and 800 nm indicated that the sp2 hybridization carbon atom fraction was partially recovered and the π electron concentration was increased after photoreduction.51,52 As shown in Fig. 3b, the Tauc plot linear extrapolation technique was used to determine the band gaps of the GO and PRGOs. GO-based materials have a cluster of collective band structures due to graphene of different oxygenation levels, giving them a range of band gaps instead of an absolute value.26,53 After photoreduction the band gap was reduced and stabilized, with PRGO-8 and PRGO-10 having the lowest band gap at 3.10–4.00 eV. The band gap energy was sufficient to overcome the theoretical energy requirement of 2.71 eV to produce the ROS pair (superoxide, ·O−2 and hydroxyl radical, OH˙) under UV-A excitation for VOC photodegradation.54
 |
| | Fig. 3 (a) UV-vis absorbance spectra and (b) band gaps derived by Tauc plots of GO and PRGOs. | |
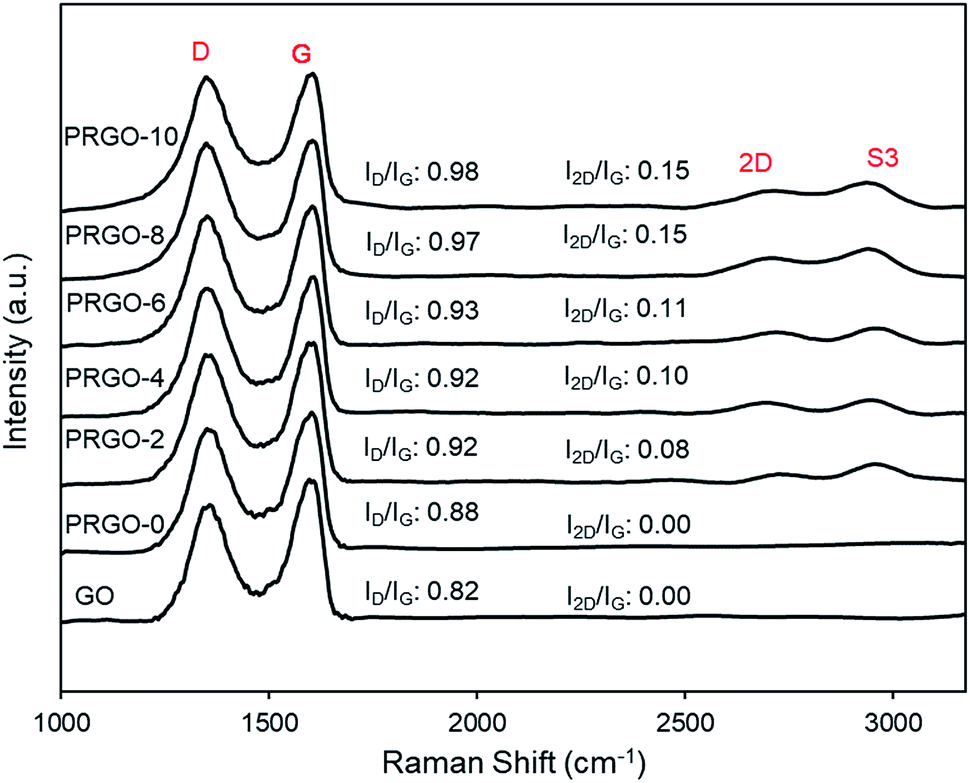
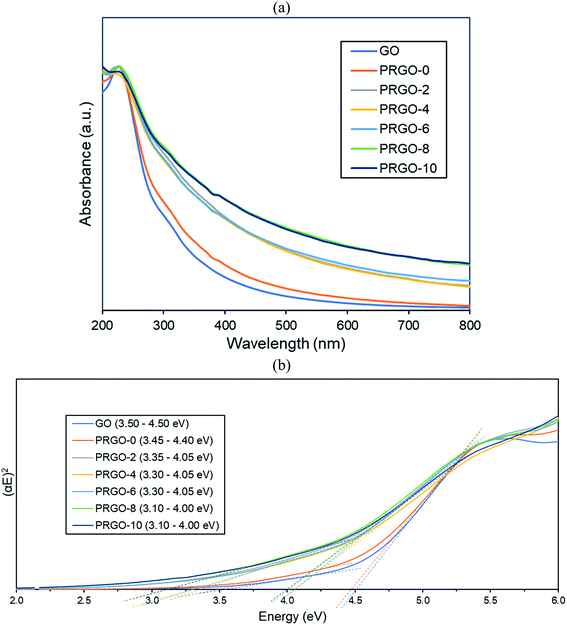
3.2.2. Raman analysis. Fig. 4 presents the Raman spectra of the GO and PRGOs and all have two strong peaks at the D (∼1350 cm−1) and G (∼1600 cm−1) modes. Two smaller peaks at 2D (∼2722 cm−1) and S3 (∼2930 cm−1) appeared after photoreduction. The D band represents the disordered structure of graphene; the G band is from the scattering of the E2g phonon of sp2 carbon atoms; 2D is the second-order of the D band, which is used to evaluate the stacking order of the c-axis orientation; while the S3 band is from imperfect activated grouping of phonons.55 Before photoreduction, the 2D peak was not observed (GO and PRGO-0). This could be due to the heavily oxidized surface causing high disorder and multilayers overlapping at the c-axis.56–58 The peak intensity ratio of D to G (ID/IG ratio) and 2D to G (I2D/IG) increased steadily after photoreduction, from 0.88 and 0.00 (PRGO-0) to 0.98 and 0.11 (PRGO-10). The increase of ID/IG ratio indicated the formation of new smaller graphitic domains upon photoreduction, which reduce the average size of the sp2 fraction.59,60 Meanwhile, the increase of I2D/IG ratio was due to reinstallation of the sp2 domain.61 The Raman analysis agreed well with the UV-vis results, in which the sp2 domain was partially recovered after photoreduction and there is a reduced disorder-induced fraction.
 |
| | Fig. 4 Raman spectra of GO and PRGOs. | |
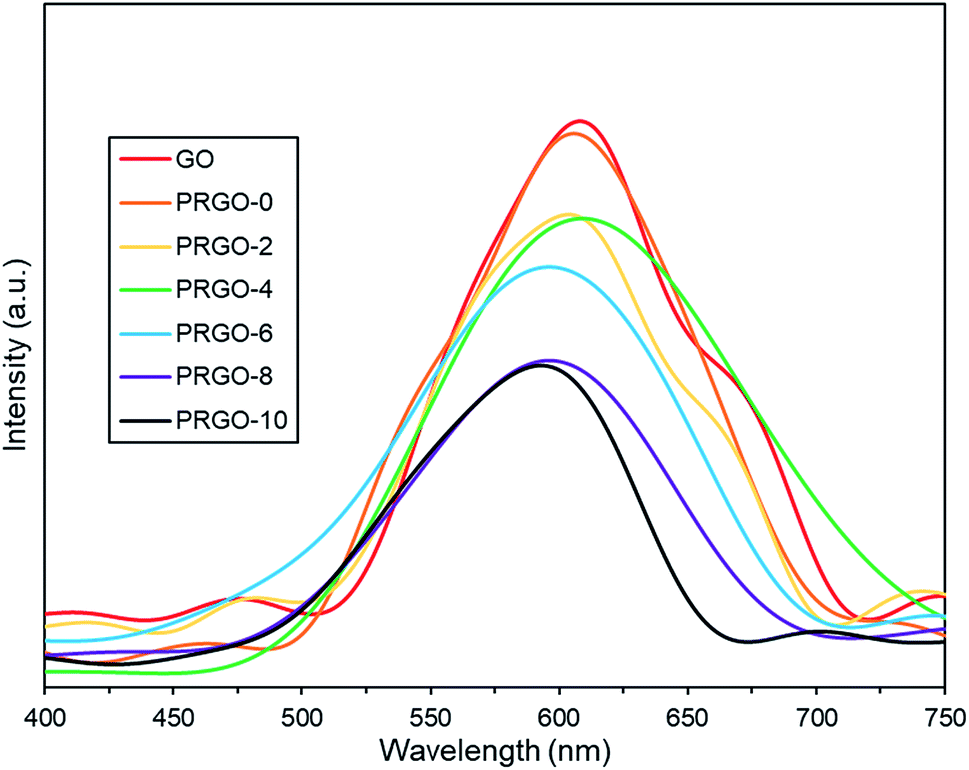
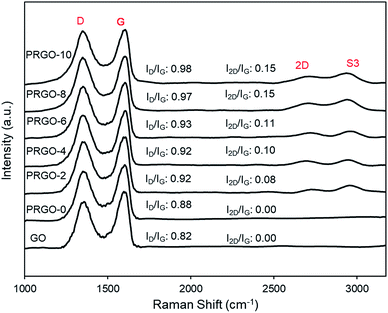
3.2.3. Photoluminescence analysis. Photoluminescence (PL) spectra are shown in Fig. 5. All GO and PRGOs exhibited a broad peak between 400 and 750 nm, suggesting a wide band gap structure,62 which supports the band gap results derived from the Tauc plots. The relative PL peak intensity decreased with longer photoreduction time. PRGO-8 and PRGO-10 have the lowest peak intensity, indicating that the rate of electron and hole recombination was possibly reduced with longer photoreduction time. This could be due to partial restoration of the sp2 domain during photoreduction leading to better electrical conductivity and charge carrier mobility. This is in agreement with previous studies,63,64 where higher conductivity led to better charge separation, hence lowering the rate of electron recombination. In addition, the PL peak location was shifted slightly from 600–610 nm to 590–600 nm as photoreduction time increases. These findings agree with others,65,66 where the blue-shift in PL was caused by the partial deoxygenation of GO, which led to more sp2 clusters and less disorder-induced fraction within the π–π* gap.
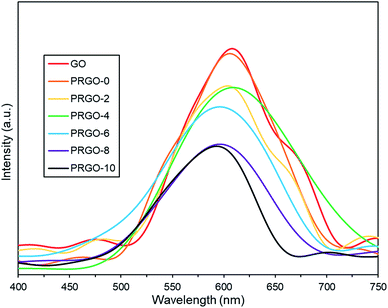 |
| | Fig. 5 Photoluminescence spectra of GO and PRGOs. | |
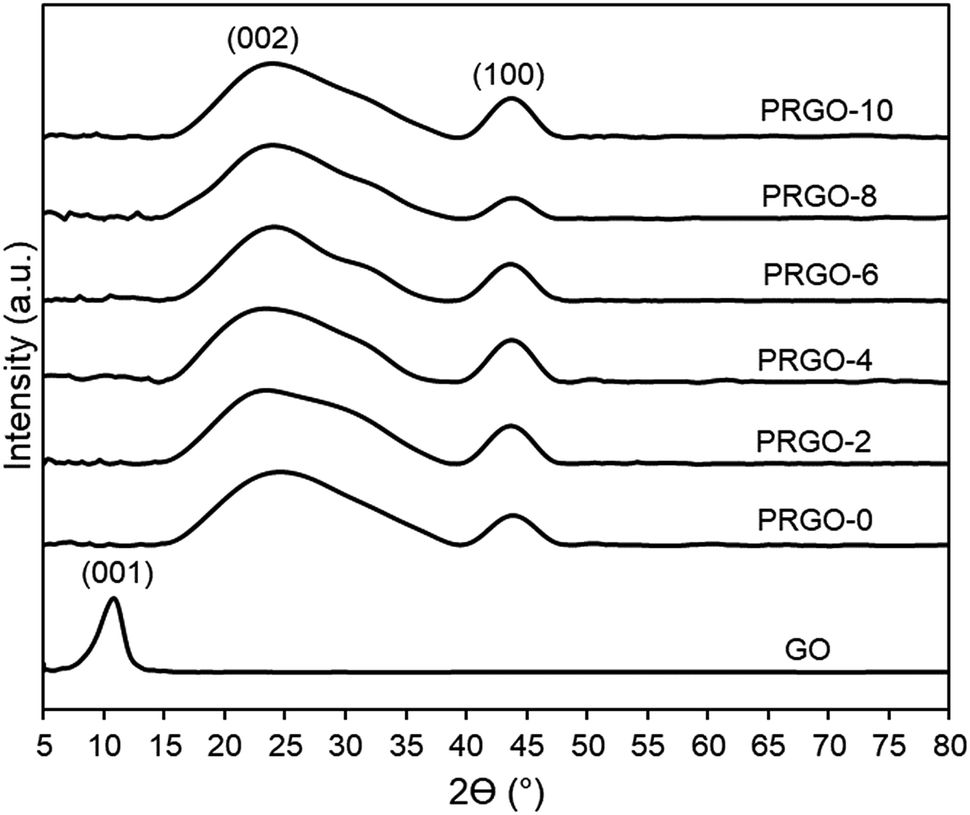
3.2.4. XRD analysis. Fig. 6 shows that the XRD pattern of GO has a diffraction peak (001) at 2θ = 10.04°, which is typical for exfoliated GO. After EPD, the XRD diffraction (001) peak disappeared, while a broad peak (002) with a centre at 2θ = 24.5° appeared. This was due to GO deposited and agglomerated on the substrate; hence part of the interlayer spacing had collapsed. The broad (002) peak is a typical pattern of lowered stacking order between the graphene layers.67,68 XRD patterns of PRGO-0 to PRGO-10 were similar, indicating that the interlayer spacing structure of the PRGOs remained the same after photoreduction. In addition, peak (100) was present at 2θ = 44° for all PRGOs, indicating a short-range order of stacked graphene layers.69
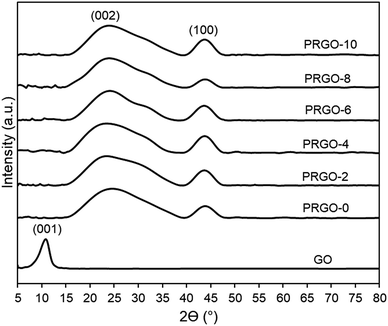 |
| | Fig. 6 XRD patterns of GO and PRGOs. | |
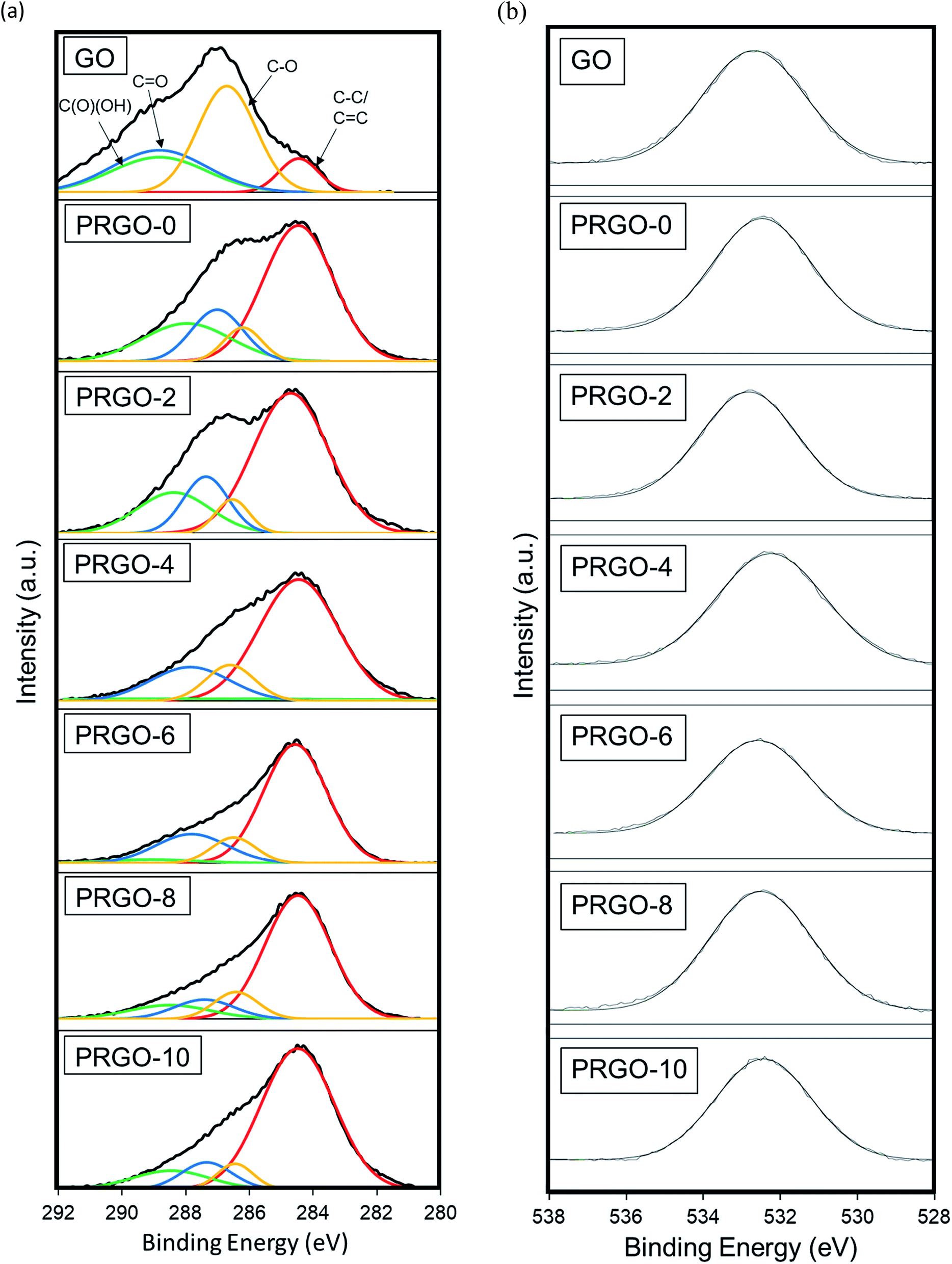
3.2.5. XPS analysis. An XPS study was used to analyse the elemental composition and oxygen functionalities of the GO and PRGOs. Fig. 7(a and b) displays the high-resolution scan of the XPS spectra C 1 s (284.5 eV) and O1s (532.4 eV) results. In Fig. 7a, the C1s spectra are deconvoluted into four peaks C–C/CC (284.5 eV), C–O (286.6 eV), CO (287.7 eV), and C(O)(OH) (289.6 eV), and then fitted using a symmetric Gaussian function.69 Table 1 summarizes the composition of the functionalities analysed from the C1s spectra and the O:C atomic ratios. The proportion of the C–C/CC group showed a gradual intensity increase with photoreduction time, where PRGO-10 has the largest composition of 73%. In contrast, the other three oxygen-containing functional groups became less intense, but with a fluctuating trend, with photoreduction. The fluctuating reducing trend suggested that the UV-A irradiation photoreduction method was random and did not target a specific carbon-oxygen group. From Fig. 7b, it was found that the O1s peak relative area reduced with photoreduction. Quantitative analysis was carried out to determine the O and C concentrations, the atomic ratio of O:C gradually decreased from 0.44 to 0.25 with longer photoreduction. These results reflect that the reduction of PRGO is responsible for the decrease in oxygen content with photoreduction time. In addition, the O:C ratios of PRGO-8 to PRGO-10 were similar, even after further light irradiation. This indicated that the material might have resisted photoreduction and drastic oxygen functional group reduction after 8 hours. This is in agreement with a previous study where PRGO was shown to resist further light-induced reduction unless a higher amount of energy is used.52
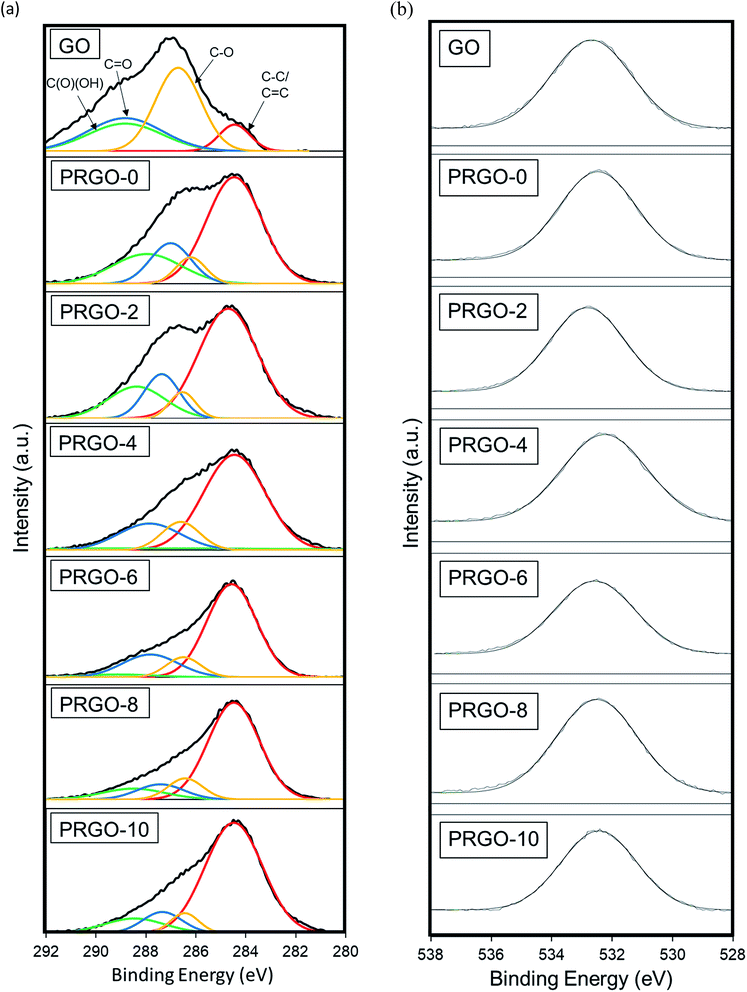 |
| | Fig. 7 (a) C1s and (b) O1s XPS spectra of GO and PRGOs. | |
Table 1 XPS carbon bonding composition and O:C atomic ratio of GO and PRGOs
| Material |
Carbon bonding composition (%) |
O:C ratio |
| C–C/CC |
C–O |
CO |
C(O)(OH) |
| GO |
32 |
23 |
24 |
20 |
0.44 |
| PRGO-0 |
57 |
8 |
16 |
20 |
0.42 |
| PRGO-2 |
61 |
7 |
15 |
17 |
0.38 |
| PRGO-4 |
66 |
12 |
18 |
4 |
0.37 |
| PRGO-6 |
68 |
10 |
19 |
3 |
0.31 |
| PRGO-8 |
70 |
10 |
10 |
10 |
0.26 |
| PRGO-10 |
73 |
7 |
10 |
10 |
0.25 |
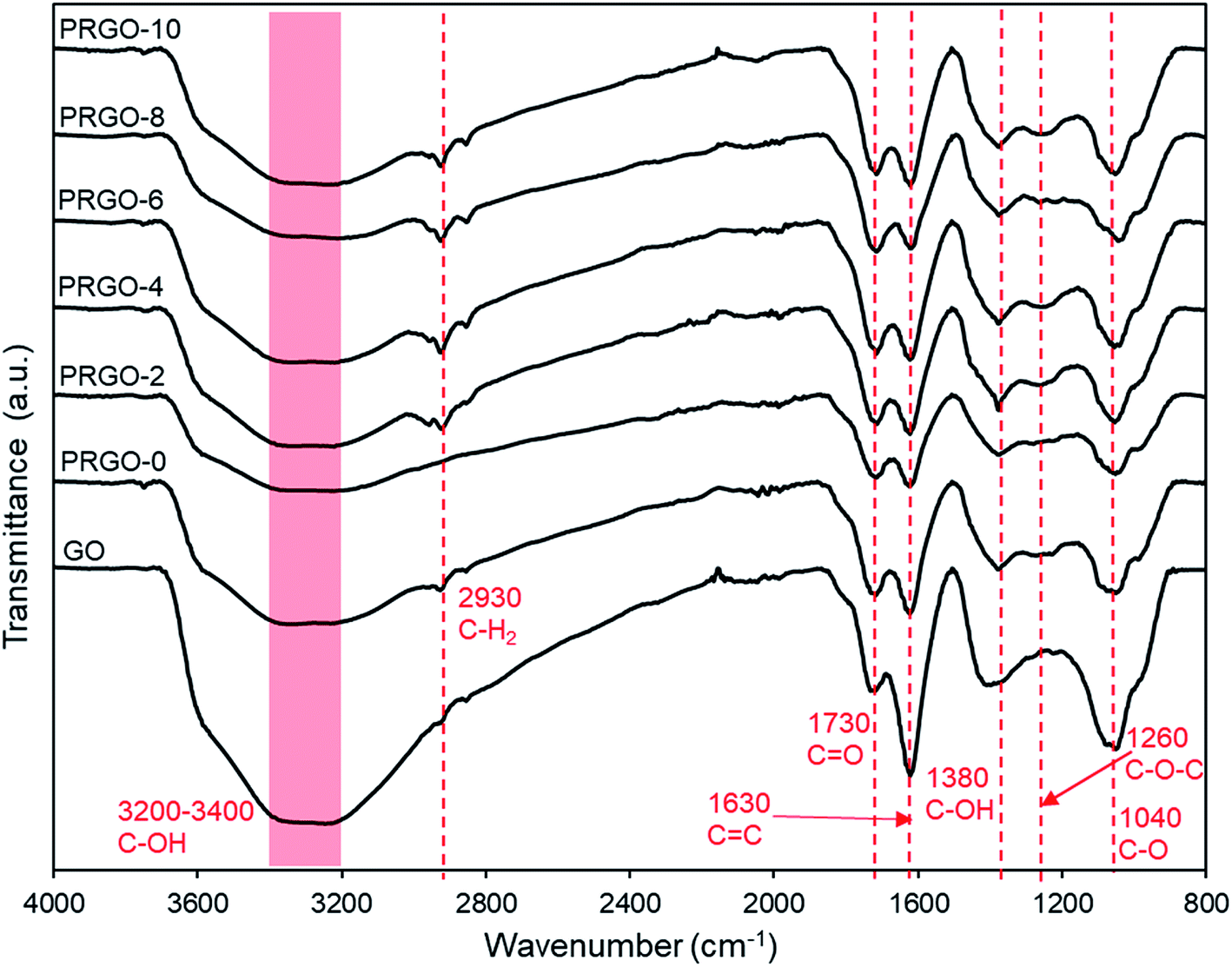
3.2.6. ATR-FTIR analysis. The ATR-FTIR results for GO and PRGOs are shown in Fig. 8. It was found that all the major oxygen functional groups were present despite photoreduction. The peaks for CO stretching, CC (sp2 of aromatic ring), C–OH group, C–O–C (epoxy), C–O stretching at 1730, 1630, 1380, 1260, and 1040 cm−1 respectively were observed, and only the C–OH group showed a relatively significant reduction in intensity. These results agreed well with the XPS analysis, where the oxygen groups were still present even after photoreduction. The C–H2 group at 2930 cm−1 appeared after photoreduction, which might be due to the reaction of carbon atoms with the hydrogen ions produced during photoreduction. The formation of hydrogen during photoreduction of GO was also observed in previous studies.34
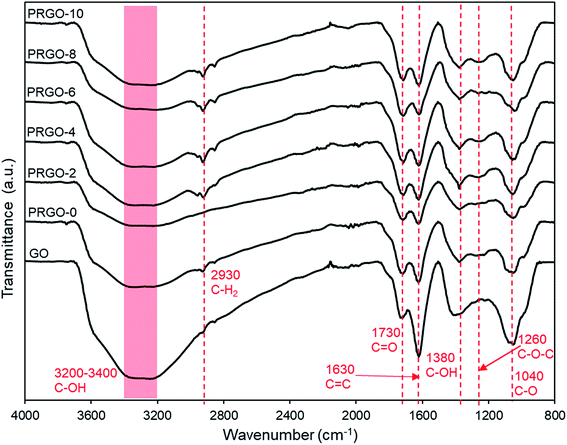 |
| | Fig. 8 ATR-FTIR spectra of GO and PRGOs. | |
3.3. PCO and reusability of PRGO
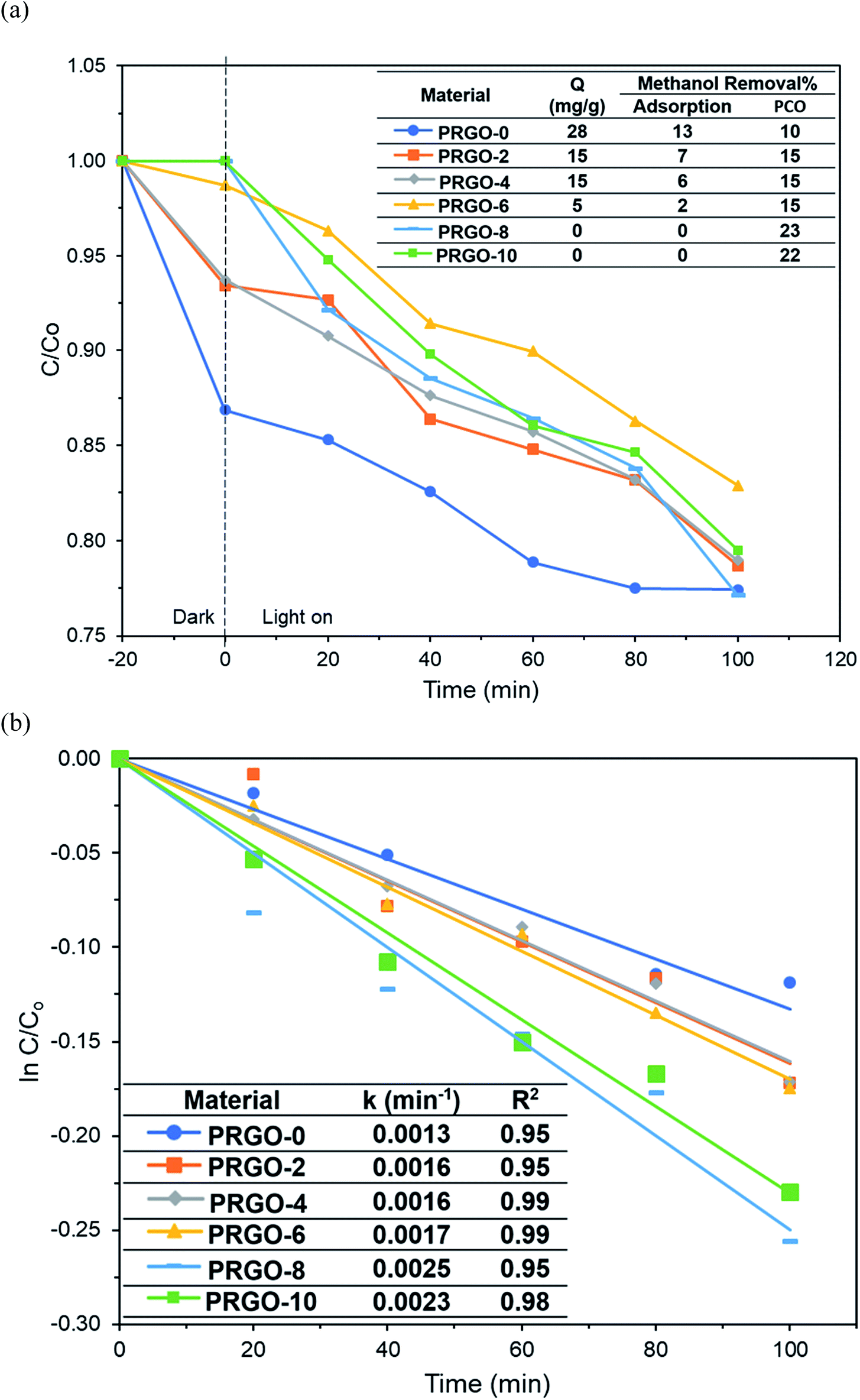
Methanol was successfully removed by the PRGOs via adsorption and PCO and it was found that the adsorption capacity decreased with increase of the photoreduction time (Fig. 9a). The adsorption mechanism of methanol on the carbon surface is mainly due to the dipole–dipole interaction between methanol and the oxygen functional groups on the carbon material.70 A previous study also stated that the elimination of oxygen functional groups on the carbon material reduces its hydrophilicity and decreases the adsorption of polar chemicals.71 From the XPS results, the concentration of oxygen functional groups on the PRGO was reduced after photoreduction, leading to lower electronegativity and weaker dipole–dipole interactions between the PRGO and methanol, and so resulting in a lower adsorption.
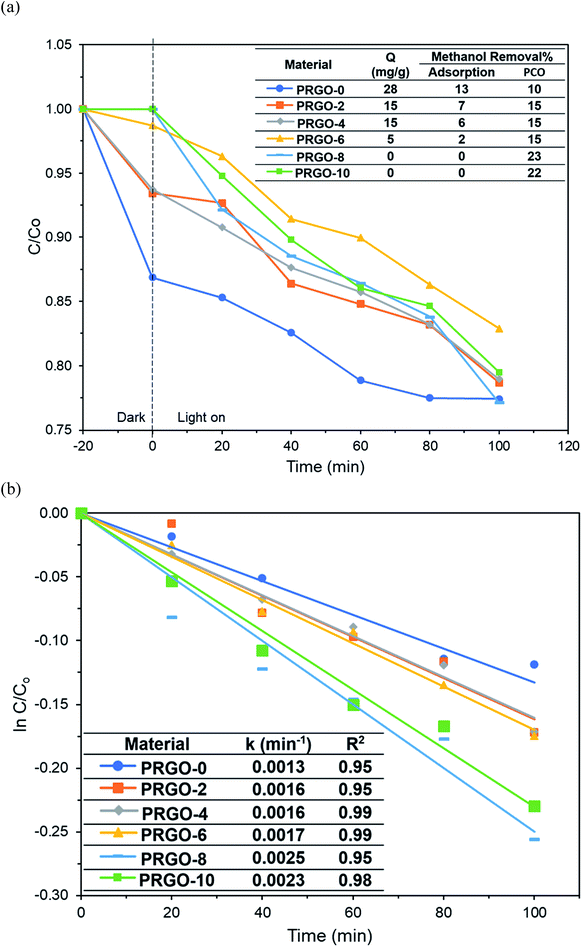 |
| | Fig. 9 (a) Adsorption and PCO of methanol and (b) pseudo-first order kinetics of methanol PCO by PRGOs. | |
The photocatalytic activity of methanol was found to follow the order PRGO-8 > PRGO-10 > PRGO-6 > PRGO-4 = PRGO-2 > PRGO-0 (Fig. 9a and b). After photoreduction, the PCO performance was improved by more than twofold, where PRGO-8 had the highest methanol PCO at 23% and the highest pseudo-first order rate constant, k, of 0.0025 min−1.
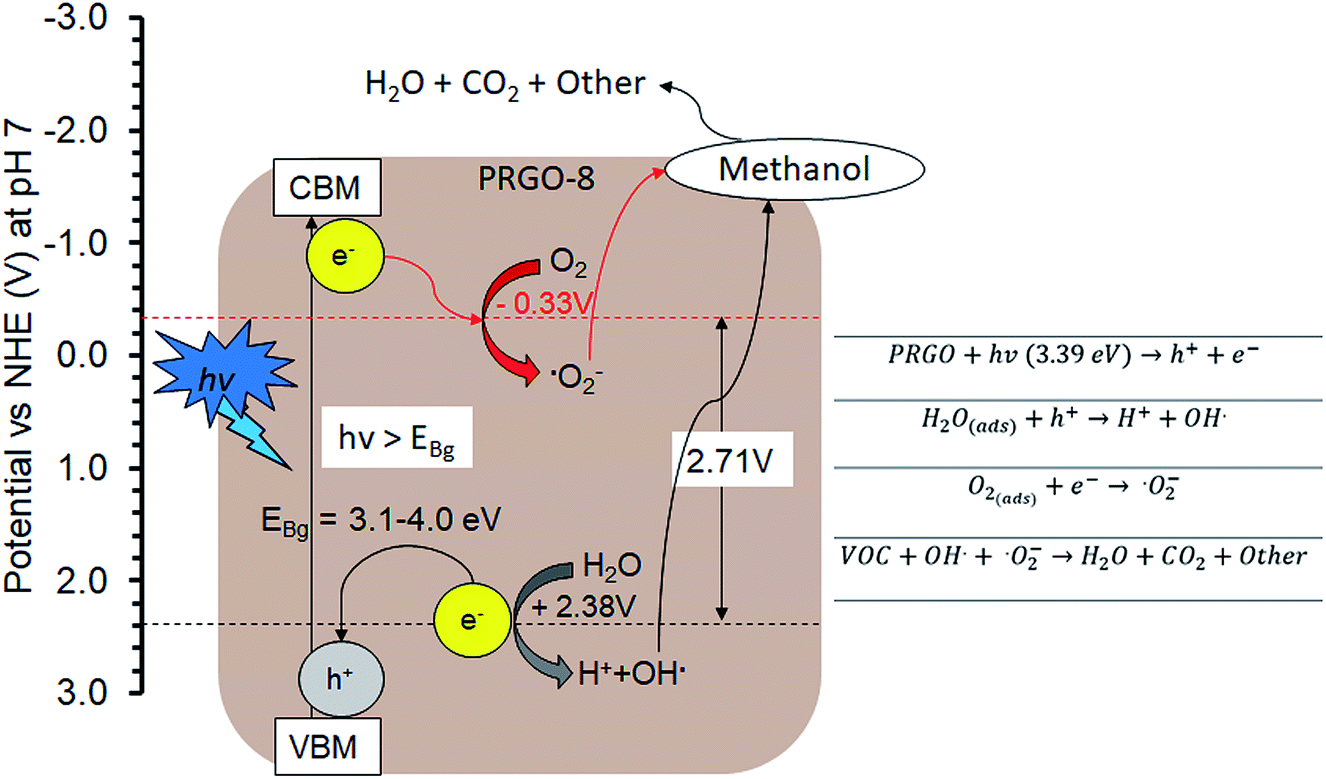
The PCO activity of methanol increased with photoreduction time despite fewer oxygen functional groups and lower adsorption capacity. This is in contrast with some studies that stated that abundant surface hydroxyl groups and high adsorption capacity of photocatalysts (such as TiO2) led to better photoactivity.72,73 The involvement of surface hydroxyl groups on the photoactivity of GO is unlikely, as the photoexcited electrons mainly originate from the π → π* transition within the sp2 orbital.74,75 Moreover, there are other studies that have found that adsorption capacity is not the only factor affecting photoactivity.76,77 The plausible PCO mechanism on PRGO is depicted in Fig. 10. First, the UV-A photon energy is absorbed by PRGO because the band gap is less than 3.39 eV. Then the electrons (e−) and holes (h+) are generated and act as the oxidizing and reducing sites. The reduction potential of e− in the PRGO conduction band must be more negative than the potential of O2/·O2− (−0.33 V vs. normal hydrogen electrode (NHE))54 to donate an electron and facilitate the production of superoxide from adsorbed oxygen. Whereas the oxidation potential of the h+ in the PRGO valence band needs to be more positive than H2O/H+ + OH˙ (+2.38 V vs. NHE)54 to accept an electron and generate hydroxyl radical from adsorbed water. The potential difference between the generation of superoxide and hydroxyl radical is 2.71 eV. The two ROS attack methanol and decompose it into simpler compounds, such as H2O and CO2. With longer photoreduction time and less adsorbed methanol on the PRGO, this possibly led to less competition for adsorption sites.78 Therefore, more O2 and H2O can be adsorbed to generate ROS and thus achieve a higher effiency for PCO.
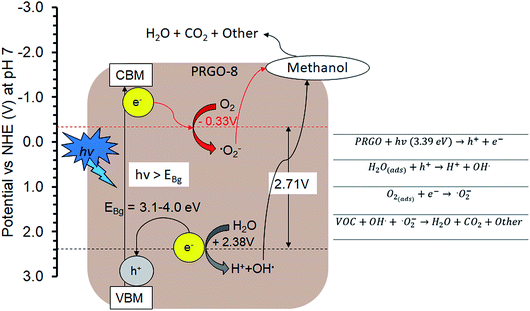 |
| | Fig. 10 Plausible methanol photodegradation pathway by PRGO-8. VBM: valence band maximum; CBM: conduction band minimum. | |
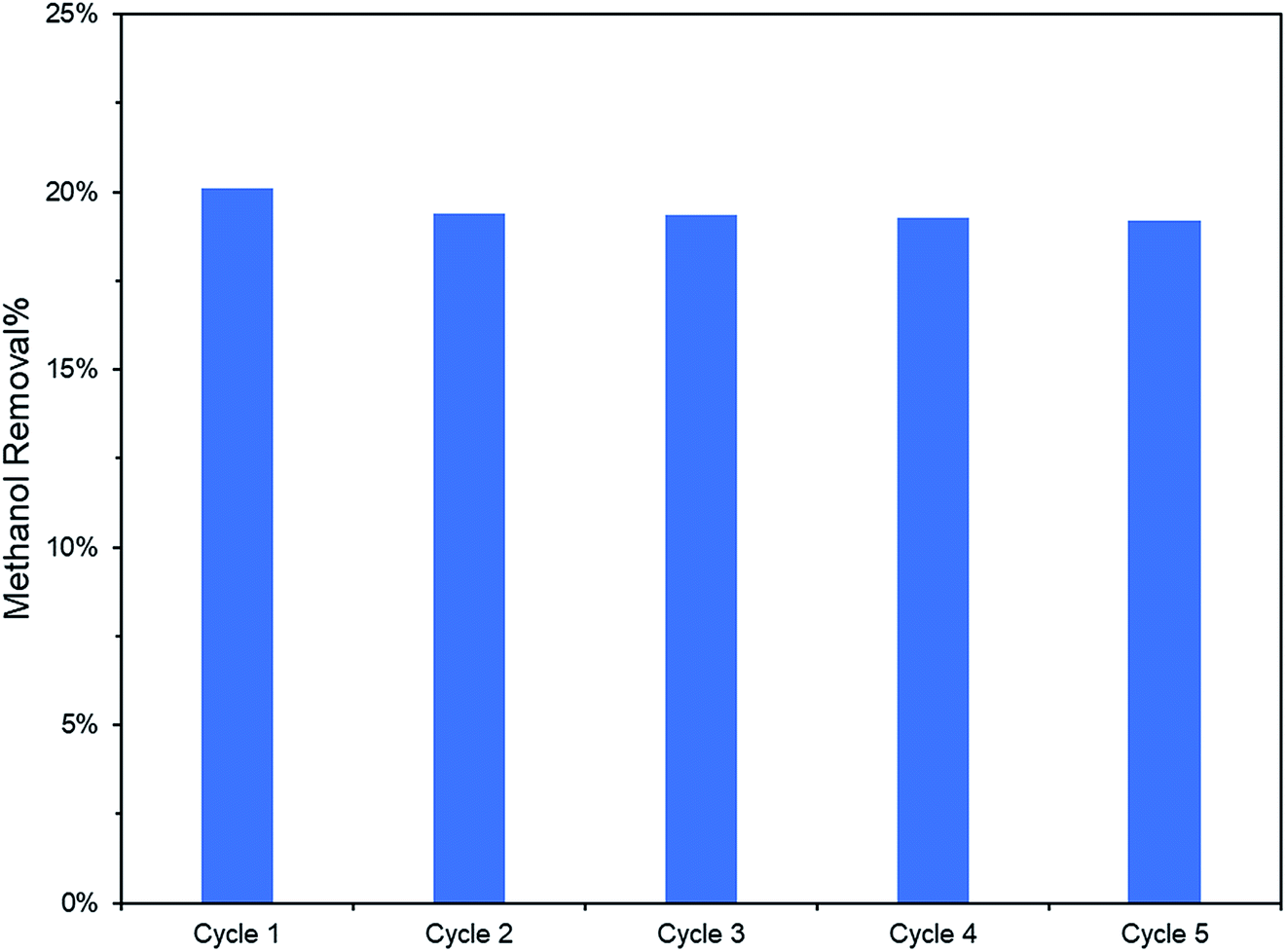
Additionally, the lower PL peak intensity of PRGO-8 and PRGO-10 (Fig. 5) indicated a lower charge recombination rate. This could also lead to better photocatalytic activity65 as more electron and hole pairs can be utilized for ROS production. Another possible factor affecting methanol PCO is the band gap of the PRGO. The band gap before photoreduction was too wide to be fully photoexcited by UV-A light, but after photoreduction the band gap was reduced. Hence, more PRGO molecules would become photoactive under UV-A, thus contributing to higher photoactivity. This finding is in agreement with a previous study, where the band gap of GO was found to be a factor affecting its photoactivity for water splitting.40 Fig. 11 shows that after five cycles, the performance of PRGO-8 was almost the same, without any drastic decrease. This result indicates that PRGO-8 is a stable photocatalyst with good reusability.
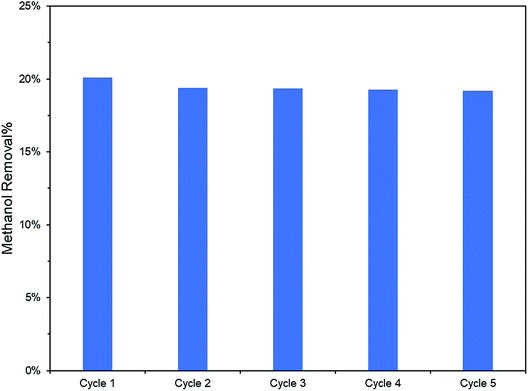 |
| | Fig. 11 PCO recyclability test (2 ppm methanol in 100 min) of PRGO-8. | |
4. Conclusions
In conclusion, PRGO photocatalysts were successfully synthesized via a photoreduction technique without using any harmful reducing agent or solvents. This study shows that PRGO is a potential low cost, eco-friendly, and metal-free photocatalyst to carry out indoor PCO of methanol under UV-A irradiation. Methanol photodegradation was boosted up to 23%, with a rate constant of 0.0025 min−1, using PRGO-8. This enhancement is mainly due to the smaller band gap and slower electron recombination. The photocatalyst was stable and no significant loss of performance was observed even after five cycles of PCO.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge financial support from Transdisciplinary Research Grant Scheme (TR001B-2015A) and SATU Grant (ST010-2017 and RU018M-2016).
References
- J. Jaakkola, P. Tuomaala and O. Seppänen, Am. J. Public Health, 1994, 84, 422–428 CrossRef CAS PubMed.
- J. C. Lerner, E. Sanchez, J. Sambeth and A. Porta, Atmos. Environ., 2012, 55, 440–447 CrossRef.
- L. Mølhave, Indoor Air, 1991, 1, 357–376 CrossRef.
- C. Yu and D. Crump, Build Environ., 1998, 33, 357–374 CrossRef.
- S. Solomon, G. Schade, J. Kuttippurath, A. Ladstätter-Weissenmayer and J. Burrows, Indoor Built Environ., 2008, 17, 260–268 CrossRef CAS.
- C. W. Babbitt, A. Pacheco and A. S. Lindner, Bioresour. Technol., 2009, 100, 6207–6216 CrossRef CAS PubMed.
- S. Brown, Indoor air, 1999, 9, 209–215 CrossRef CAS PubMed.
- J. D. Fenske and S. E. Paulson, J. Air Waste Manage. Assoc., 1999, 49, 594–598 CrossRef CAS.
- A. Carrière, C. Kaufmann, J. Shapiro, P. Paine and J. Prinsen, SAE Trans., 2000, 227–234 Search PubMed.
- A. Mirzaei, S. Leonardi and G. Neri, Ceram. Int., 2016, 42, 15119–15141 CrossRef CAS.
- Z. Zhang, Z. Jiang and W. Shangguan, Catal. Today, 2016, 264, 270–278 CrossRef CAS.
- L. Lin, Y. Chai, B. Zhao, W. Wei, D. He, B. He and Q. Tang, Open J. Inorg. Chem., 2013, 3, 14 CrossRef CAS.
- Y. Huang, S. Ho, Y. Lu, R. Niu, L. Xu, J. Cao and S. Lee, Molecules, 2016, 21, 56 CrossRef PubMed.
- A. H. Mamaghani, F. Haghighat and C.-S. Lee, Appl. Catal., B, 2017, 203, 247–269 CrossRef CAS.
- L. D. Alencar, A. Mesquita, C. A. Feitosa, R. Balzer, L. F. Probst, D. C. Batalha, M. G. Rosmaninho, H. V. Fajardo and M. I. Bernardi, Ceram. Int., 2017, 43, 4462–4469 CrossRef CAS.
- F. He, F. Ma, J. Li, T. Li and G. Li, Ceram. Int., 2014, 40, 6441–6446 CrossRef CAS.
- C. Wang and T. Wu, Ceram. Int., 2015, 41, 2836–2839 CrossRef CAS.
- S. W. Verbruggen, J. Photochem. Photobiol., C, 2015, 24, 64–82 CrossRef CAS.
- T. Tsuru, T. Kan-no, T. Yoshioka and M. Asaeda, Catal. Today, 2003, 82, 41–48 CrossRef CAS.
- J. Taranto, D. Frochot and P. Pichat, Ind. Eng. Chem. Res., 2007, 46, 7611–7614 CrossRef CAS.
- J. M. Stokke, D. W. Mazyck, C. Wu and R. Sheahan, Environ. Prog., 2006, 25, 312–318 CrossRef CAS.
- D. S. Su, J. Zhang, B. Frank, A. Thomas, X. Wang, J. Paraknowitsch and R. Schlögl, ChemSusChem, 2010, 3, 169–180 CrossRef CAS PubMed.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- K. Bustos-Ramirez, C. E. Barrera-Diaz, M. De Icaza, A. L. Martínez-Hernández and C. Velasco-Santos, J. Chem., 2015, 2015, 254631 Search PubMed.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Nano Energy, 2015, 13, 757–770 CrossRef CAS.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015 CrossRef CAS PubMed.
- X. Zhang, B. Gao, A. E. Creamer, C. Cao and Y. Li, J. Hazard. Mater., 2017, 338, 102–123 CrossRef CAS PubMed.
- C. P. P. Wong, C. W. Lai, K. M. Lee and S. B. A. Hamid, Materials, 2015, 8, 7118–7128 CrossRef CAS PubMed.
- J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Fu and X. Wang, Energy Environ. Sci., 2011, 4, 675–678 RSC.
- F. W. Low, C. W. Lai and S. B. A. Hamid, Ceram. Int., 2015, 41, 5798–5806 CrossRef CAS.
- V. L. Siong, C. W. Lai, J. C. Juan, K. M. Lee, B. F. Leo and C. S. Khe, Curr. Nanosci., 2019, 15, 157–162 CrossRef.
- A. Stroyuk, N. Andryushina, V. Il’in, V. Efanov, I. Yanchuk, S. Y. Kuchmii and V. Pokhodenko, Theor. Exp. Chem., 2012, 48, 2–13 CrossRef CAS.
- N. S. Andryushina, O. L. Stroyuk, I. B. Yanchuk and A. V. Yefanov, Colloid Polym. Sci., 2014, 292, 539–546 CrossRef CAS.
- M. Mohandoss, S. S. Gupta, A. Nelleri, T. Pradeep and S. M. Maliyekkal, RSC Adv., 2017, 7, 957–963 RSC.
- K. Krishnamoorthy, R. Mohan and S.-J. Kim, Appl. Phys. Lett., 2011, 98, 244101 CrossRef.
- L. Guardia, S. Villar-Rodil, J. Paredes, R. Rozada, A. Martínez-Alonso and J. Tascón, Carbon, 2012, 50, 1014–1024 CrossRef CAS.
- E. E. Pérez-Ramírez, G. de la Rosa-Alvarez, P. Salas, C. Velasco-Santos and A. L. Martínez-Hernández, Environ. Eng. Sci., 2015, 32, 872–880 CrossRef.
- T. F. Yeh, J. M. Syu, C. Cheng, T. H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef CAS.
- Y. Matsumoto, M. Koinuma, S. Ida, S. Hayami, T. Taniguchi, K. Hatakeyama, H. Tateishi, Y. Watanabe and S. Amano, J. Phys. Chem. C, 2011, 115, 19280–19286 CrossRef CAS.
- T.-F. Yeh, F.-F. Chan, C.-T. Hsieh and H. Teng, J. Phys. Chem. C, 2011, 115, 22587–22597 CrossRef CAS.
- H.-C. Hsu, I. Shown, H.-Y. Wei, Y.-C. Chang, H.-Y. Du, Y.-G. Lin, C.-A. Tseng, C.-H. Wang, L.-C. Chen and Y.-C. Lin, Nanoscale, 2013, 5, 262–268 RSC.
- K. Bustos-Ramírez, C. E. Barrera-Díaz, M. De Icaza-Herrera, A. L. Martínez-Hernández, R. Natividad-Rangel and C. Velasco-Santos, J. Environ. Health Sci. Eng., 2015, 13, 33 CrossRef PubMed.
- V. Kumar, K.-H. Kim, J.-W. Park, J. Hong and S. Kumar, Chem. Eng. J., 2017, 315, 210–232 CrossRef CAS.
- W. Zou, B. Gao, Y. S. Ok and L. Dong, Chemosphere, 2018, 845–859 Search PubMed.
- A. Hajizadeh, M. Aliofkhazraei, M. Hasanpoor and E. Mohammadi, Ceram. Int., 2018, 44, 10951–10960 CrossRef CAS.
- S. R. Kim, M. K. Parvez and M. Chhowalla, Chem. Phys. Lett., 2009, 483, 124–127 CrossRef CAS.
- B. Xue, Y. Zou and Y. Yang, J. Mater. Sci., 2017, 52, 12742–12750 CrossRef CAS.
- K. Muthoosamy, R. G. Bai, I. B. Abubakar, S. M. Sudheer, H. N. Lim, H.-S. Loh, N. M. Huang, C. H. Chia and S. Manickam, Int. J. Nanomed., 2015, 10, 1505 CAS.
- S. Türk, I. Altınsoy, G. Ç. Efe, M. Ipek, M. Özacar and C. Bindal, Vacuum, 2018, 148, 1–10 CrossRef.
- A. Mathkar, D. Tozier, P. Cox, P. Ong, C. Galande, K. Balakrishnan, A. Leela Mohana Reddy and P. M. Ajayan, J. Phys. Chem. Lett., 2012, 3, 986–991 CrossRef CAS PubMed.
- Y. Ding, P. Zhang, Q. Zhuo, H. Ren, Z. Yang and Y. Jiang, Nanotechnology, 2011, 22, 215601 CrossRef CAS PubMed.
- W.-C. Hou, I. Chowdhury, D. G. Goodwin Jr, W. M. Henderson, D. H. Fairbrother, D. Bouchard and R. G. Zepp, Environ. Sci. Technol., 2015, 49, 3435–3443 CrossRef CAS PubMed.
- Z. Gan, S. Xiong, X. Wu, C. He, J. Shen and P. K. Chu, Nano Lett., 2011, 11, 3951–3956 CrossRef CAS PubMed.
- T. Saison, P. Gras, N. Chemin, C. Chanéac, O. Durupthy, V. Brezova, C. Colbeau-Justin and J.-P. Jolivet, J. Phys. Chem. C, 2013, 117, 22656–22666 CrossRef CAS.
- Z. Li, S. C. Xu, C. Zhang, X. Y. Liu, S. S. Gao, L. T. Hu, J. Guo, Y. Ma, S. Z. Jiang and H. P. Si, Sci. Rep., 2016, 6, 38539 CrossRef CAS PubMed.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S.-J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- A. Kaniyoor and S. Ramaprabhu, AIP Adv., 2012, 2, 032183 CrossRef.
- K. Bustos-Ramírez, A. Martínez-Hernández, G. Martínez-Barrera, M. Icaza, V. Castaño and C. Velasco-Santos, Materials, 2013, 6, 911–926 CrossRef PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- E. J.-C. Amieva, R. Fuentes-Ramirez, A. Martinez-Hernandez, B. Millan-Chiu, L. M. Lopez-Marin, V. Castaño and C. Velasco-Santos, J. Alloys Compd., 2015, 643, S137–S143 CrossRef.
- S. Mortazavi, M. Mollabashi, R. Barri, K. Jones, J. Q. Xiao, R. L. Opila and S. I. Shah, RSC Adv., 2018, 8, 12808–12814 RSC.
- Z. Luo, P. M. Vora, E. J. Mele, A.
C. Johnson and J. M. Kikkawa, Appl. Phys. Lett., 2009, 94, 111909 CrossRef.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
- J. Liu, H. Bai, Y. Wang, Z. Liu, X. Zhang and D. D. Sun, Adv. Funct. Mater., 2010, 20, 4175–4181 CrossRef CAS.
- C. T. Chien, S. S. Li, W. J. Lai, Y. C. Yeh, H. A. Chen, I. S. Chen, L. C. Chen, K. H. Chen, T. Nemoto and S. Isoda, Angew. Chem., Int. Ed., 2012, 51, 6662–6666 CrossRef CAS PubMed.
- C.-H. Chuang, Y.-F. Wang, Y.-C. Shao, Y.-C. Yeh, D.-Y. Wang, C.-W. Chen, J. Chiou, S. C. Ray, W. Pong and L. Zhang, Sci. Rep., 2014, 4, 4525 CrossRef PubMed.
- A. A. Amer, S. Reda, M. Mousa and M. M. Mohamed, RSC Adv., 2017, 7, 826–839 RSC.
- N. Díez, A. Śliwak, S. Gryglewicz, B. Grzyb and G. Gryglewicz, RSC Adv., 2015, 5, 81831–81837 RSC.
- L. Stobinski, B. Lesiak, A. Malolepszy, M. Mazurkiewicz, B. Mierzwa, J. Zemek, P. Jiricek and I. Bieloshapka, J. Electron Spectrosc. Relat. Phenom., 2014, 195, 145–154 CrossRef CAS.
- F. Meng, M. Song, Y. Wei and Y. Wang, Environ. Sci. Pollut. Res., 2019, 1–10 CAS.
- B. Pan and B. Xing, Environ. Sci. Technol., 2008, 42, 9005–9013 CrossRef CAS.
- Y. Xu, W. Wen and J.-M. Wu, J. Hazard. Mater., 2018, 343, 285–297 CrossRef CAS PubMed.
- S. H. Szczepankiewicz, A. Colussi and M. R. Hoffmann, J. Phys. Chem. B, 2000, 104, 9842–9850 CrossRef CAS.
- T.-F. Yeh, J. Cihlář, C.-Y. Chang, C. Cheng and H. Teng, Materials Today, 2013, 16, 78–84 CrossRef CAS.
- J. Ito, J. Nakamura and A. Natori, J. Appl. Phys., 2008, 103, 113712 CrossRef.
- Q. Wang, D. Gao, C. Gao, Q. Wei, Y. Cai, J. Xu, X. Liu and Y. Xu, Int. J. Photoenergy, 2012, 2012, 680419 Search PubMed.
- M. Moztahida, J. Jang, M. Nawaz, S.-R. Lim and D. S. Lee, Sci. Total Environ., 2019, 741–750 CrossRef CAS PubMed.
- C.-y. Wang, H. Groenzin and M. J. Shultz, J. Am. Chem. Soc., 2004, 126, 8094–8095 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kian Mun Leea,
Thomas Chung Kuang Yangb,
Siewhui Chong
a,
Kian Mun Leea,
Thomas Chung Kuang Yangb,
Siewhui Chong