
Recent advances in photocatalytic C–S/P–S bond formation via the generation of sulfur centered radicals and functionalization
Wei
Guo†
 *,
Kailiang
Tao†
,
Wen
Tan
,
Mingming
Zhao
,
Lvyin
Zheng
* and
Xiaolin
Fan
*
*,
Kailiang
Tao†
,
Wen
Tan
,
Mingming
Zhao
,
Lvyin
Zheng
* and
Xiaolin
Fan
*
Key Laboratory of Organo-pharmaceutical Chemistry of Jiangxi Province, Gannan Normal University, Ganzhou 341000, China. E-mail: guoweigw@126.com; zhenglvyin@126.com; fanxl2008@gnnu.edu.cn
First published on 31st January 2019
Abstract
Thioyl and sulfonyl radicals are usually produced from various thiols and sulfonyl derivatives in high efficiency by single-electron-transfer (SET) oxidation. The generated sulfur (thioyl/sulfonyl) radicals are also highly reactive intermediates having various applications in the construction of organosulfur compounds in the field of organic synthesis. Recently, photoredox-catalyzed C–S/P–S bond formation via the generation of sulfur centered radicals has been studied extensively. In the photoredox catalytic process, a variety of S–H, S–S, S–C, S–N, and S–X (F, Cl, Br, I) bonds, and even active sulfone-containing skeletons can be easily transformed into the corresponding thioyl/sulfonyl radicals. Some of these transformations are achieved by a combination of photoredox catalysts (i.e., TiO2, Bi2O3, eosin Y, fac-[Ir(ppy)3], [Ru(bpy)3]2+) and other catalysts such as strong bases, Lewis acids, organocatalysts and transition metal catalysts. Compared with previous methods, photoredox catalysis is inexpensive and features the advantages of high efficiency and easy utilization in addition to being environmentally-benign. In this review, we have focused on the research on photoredox-catalyzed C–S/P–S bond formation via the generation of thioyl/sulfonyl radicals and further functionalization in the past few years. We hope to offer chemists the tools to open the door for further progress in organsulfur chemistry.
 Wei Guo | Wei Guo received his Ph.D. degree under the guidance of Dr Yuguo Guo from the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), Beijing, P. R. China. Subsequently, he was a postdoctoral fellow with Prof. Huanfeng Jiang at the School of Chemistry and Chemical Engineering, South China University of Technology (SCNT). In March 2016, he began his independent research career at Gannan Normal University, focusing on the development of green organic chemistry and their applications in medicinal chemistry. |
 Kailiang Tao | Kailiang Tao received his B.S. degree from Gannan Normal University in 2017. Currently, he is a M.S. student in the research group of Dr W. Guo. His research focuses on the synthesis chemistry of organosulfur compounds. |
 Wen Tan | Wen Tan was born in 1995 in Bazhong, Sichuan, China. She received her B.S. degree from Qufu Normal University in 2016. Currently, she is a M.S. student in the research group of Dr W. Guo. Her research focuses on green organic synthesis chemistry. |
 Mingming Zhao | Mingming Zhao received her B.S. degree from Shandong University of Traditional Chinese Medicine in 2016. Currently, she is a M.S. student in the research group of Dr W. Guo. Her research focuses on N-heterocycle synthesis. |
 Lvyin Zheng | Lvyin Zheng completed her M.S. studies in 2006 under the direction of Prof. Zhaoyang Wang at South China Normal University. Currently she is a member of the Key Laboratory of Organo-pharmaceutical Chemistry of Jiangxi Province, Gannan Normal University. She is mainly interested in new synthetic methodologies and biologically active compounds synthesis. |
 Xiaolin Fan | Professor Xiaolin Fan is the director of the Key Laboratory of Organo-pharmaceutical Chemistry of Jiangxi Province. He is the president of the Gannan Normal University. He obtained a Ph.D. degree in 1999 from the National University of Defense Technology. His research interest focuses on developing advanced medicinal chemistry. He has more than 70 peer-reviewed papers published in leading scientific journals, and more than 20 granted patents. |
1. Introduction
Organosulfur compounds have been receiving increasing attention in medicinal chemistry, pharmaceuticals, chemical biology, and advanced functional materials.1,2 Previous studies on the synthesis of organosulfur compounds require strong bases, and/or stoichiometric oxidants, or rely on high reaction temperatures, or expensive and unavailable materials.2e–h Recently, sulfur centered radicals have served as important intermediates for the construction of organosulfur compounds.3 Cahard's group also disclosed the redox potential of some sulfur precursors, which could help us understand the details.4a It is noted that the oxidation potential of some excited photocatalysts ([Ru(bpz)32+]* = 1.4 V; rose bengal = 0.99 V; eosin Y = 0.83 V) are significantly more positive than that of the sulfur precursors.4b–d For example, the oxidation potential of benzyl thiol is +0.50 V,4b,e and for sodium benzenesulfinate it is −0.37 V.4f,g Thus, the direct photooxidation of thiol/sulfone-containing skeletons by photocatalysts is thermodynamically very favourable. Generally, there are many strategies to generate sulfur centered radicals for the synthesis of organosulfur compounds by the addition of peroxides, azo compounds, and organic catalysts (e.g., PhCOCO2H).5 These transformations can be carried out under conditions of high temperature, UV irradiation or ultrasound, etc.5,6 The functionalization of sulphur radicals in those strategies involve the addition of thioyl radicals to unsaturated bonds (alkenes, alkynes, etc.) or substitution with R–X (X = halides/H, etc.).3d,5,6 For the past several years, photoredox catalysis has emerged as a useful tool for radical reactions for the synthesis of organosulfur compounds via visible-light induced processes.7 Compared with previous methods, photoredox catalysis is inexpensive and features the advantage of being environmentally-benign in addition to high efficiency and ease of utilization. Commonly, visible-light photocatalysts mainly include metal-oxide-semiconductors, metal-ligand complexes, metal-organic frameworks, organic photosensitizers and so on. Among these photocatalysts, such as fac-[Ir(ppy)3] (bpy = 2,2′-bipyridine; ppy = 2-pyridylphenyl), [Ru(bpy)3]2+, eosin Y, eosin B, and rose bengal can be used to trigger the photooxidation or photoreduction process.This review mainly highlights the recent advances in visible light induced C–S/P–S bond formation for the synthesis of organosulfur compounds via the generation of sulfur centered radicals. There are two major approaches for the construction of these organosulfur compounds including the highly reactive thioyl radical species (Scheme 1, path a), or the sulfonyl radical species in a controllable way (Scheme 1, path b). In the photoredox catalytic process, a variety of S–H, S–S, S–C, S–N, S–X (F, Cl, Br, I) bonds, and even active sulfone-containing skeletons can be easily transformed into the corresponding thioyl/sulfonyl radicals by single-electron-transfer (SET) oxidation. Further transformations could be divided into two types: radical substitutions and radical addition reactions. In this context, such catalytic strategies have also proved to be mild and general tools for the conversion of thioyl/sulfonyl radicals, thus enabling the achievement of a wide variety of new chemical reactions towards the synthesis of diversely functionalized organosulfur molecules. Elegant achievements from the most recent literature also demonstrate the significance of this fast developing area of research.
 | ||
| Scheme 1 Two major approaches for the generation of sulfur centered radicals and their functionalization. | ||
2. Visible-light induced C–S/P–S bond formation via the generation of thioyl radicals
2.1. C(sp3)–thioyl radicals coupling reactions
The majority of research studies on the thiolation of unactivated C(sp3)–H bonds have focused on the transition-metal-catalyzed process.8 However, those transformations could demonstrate excellent functional group tolerances. It is noted that the use of transition metals would limit the suitability of its substrates in organic synthesis. To the best of our knowledge, visible-light photoredox-controlled C(sp3)–H thiolations are particularly rare, especially the selective introduction of sulfur.9 Wang and co-workers presented a protocol to access α-arylthioethers via visible-light induced direct thiolation at α-C(sp3)–H of ethers with diaryl disulfides in the presence of acridine red (Scheme 2).10 The reactions exhibited advantages including ambient conditions (room temperature and air), eco-energy sources, and good functional group compatibilities. It is the first example of a coupling reaction through the energy transfer method under visible light irradiation. In this case, the authors proposed that the formed excited state acridine red (AR)* interacted with tBuOOH (TBHP) via an energy transfer method to generate the tBuO˙ species. Then, the trapping of tBuO˙ with ethers resulted in the propagation and formation of the alkoxyalkyl radical. Finally, the alkoxyalkyl radical could couple with the thioyl radical to give the desired product. | ||
| Scheme 2 Direct thiolation at α-C(sp3)–H of ethers. | ||
In 2017, our group also succeeded in the utilization of thioyl radicals derived from thioureas for the construction of 2-iminothiazolidin-4-ones scaffolds via C(sp3)–thioyl radical coupling reactions under visible-light promoted conditions (Scheme 3).11 The reaction was also found to tolerate a range of functional groups, such as monosubstituted, disubstituted, trisubstituted, and heterocyclic benzylamines, various aliphatic amines, and even 3-morpholinopropan-1-amine, which provided potential applications in drug screening. Moreover, UV–vis spectroscopy revealed that an in situ-generated H-bonding electron donor–acceptor (EDA) complex probably acted as the photocatalyst, promoting the reaction process. On the basis of several control experimental results, we proposed a plausible reaction mechanism. First, the amine reacts with isothiocyanate to form intermediate 1. Next, 1 couples with α-bromoester to deliver the H-bonding EDA complex 2. Then, 2 releases radicals 3 and 4 through hemolytic dissociation. Furthermore, intermediate 1 can also be converted into intermediate 5 by isomerization, which is further oxidized to intermediate 6. Subsequently, the radical coupling of thioyl radical 6 with radical 4 gives intermediate 7. Finally, intermediate 7 can afford the desired product by intramolecular cyclization.
 | ||
| Scheme 3 Synthesis of 2-iminothiazolidin-4-ones. | ||
2.2. C(sp2)–thioyl radical coupling reactions
Glorius's group developed a visible-light photoredox-controlled C(sp2)–H direct trifluoromethylthiolation with SCF3 radicals to access various vinyl-SCF3 products under mild conditions (Scheme 4).12 This approach allowed for the efficient addition of the SCF3 group to aryl-, alky- and heteroaryl substituted alkenes with yields up to 96%. The proposed mechanism for the formation of vinyl-SCF3 products is shown in Scheme 4. SCF3-halide species A initially interacts with the excited fac-Ir(ppy)3 and undergoes a single-electron oxidation process to form species B. Then, the species B affords the SCF3 radical and regenerates the halide via the mesolytic process. Subsequently, the SCF3 radical is directly added to the alkene and further gives the alkyl radical 8. Next, the alkyl radical 8 is successfully converted into the corresponding aryl-stabilized cation 9, affording the desired product via deprotonation.
 | ||
| Scheme 4 Trifluoromethylthiolation of styrenes. | ||
Scanlan et al. reported the intramolecular reactions of acyl-thioyl radicals with alkene or alkyne moieties to access thiolactones in the presence of visible light (Scheme 5).18 The present methodology tolerated a wide range of functionalities, including alkyl, aromatic, and alkyne-containing groups. Moreover, fused and spiro-bicyclic thiolactones could be prepared successfully in good yields.
 | ||
| Scheme 5 Synthesis of thiolactone derivatives. | ||
Besides the intramolecular reactions, Robinson et al. developed a visible-light driven photocatalytic initiation system of hydrothiolation of alkenes using Bi2O3 as the photoredox catalyst (Scheme 6a).19 This transformation facilitated widely functional group tolerances. Alkyl, benzyl, acyl, various Boc-protected cysteines, free hydroxyl, and even cyclohexyl tertiary thiols, all served good to excellent yields. Furthermore, the sulfur atoms have been successfully introduced to various complex biologically active relevant scaffolds and complex drug-like molecules, affording the corresponding active products in high yields.
 | ||
| Scheme 6 Reactions of thiols and alkenes. | ||
Yoon and co-workers have also succeeded in the intermolecular reaction of radical thiol–ene conjugation from varied thiols and alkenes in the presence of photoredox catalysts (Scheme 6b).4b Not only aliphatic thiols, secondary thiols, and thiols with free hydroxyl could perform smoothly, but more importantly, glutathiones also reacted with a variety of coupling partners of potential biologically active molecules and gave the corresponding products in good yields.
Simultaneously, Greaney's group disclosed a titania photoredox route for the thiol–ene reaction via a visible-light induced transformation (Scheme 6c).20 The reaction proceeded at room temperature for all kinds of aromatic and aliphatic thiols and alkenes, and tolerated various functionalized substrates (alcohols, esters, silanes, and nitriles). However, the tert-butyl thiol failed to react with the alkene and no desired product was detected. The proposed mechanism for the photocatalytic initiation of the radical thiol–ene reactions are clearly depicted in Scheme 6b. Thiol initially interacts with the excited organophotocatalyst D and undergoes a single-electron-transfer oxidation process to form intermediate F. Then the deprotonation of F gives thioyl radical G. Subsequently, the thioyl radical can initiate the thiol–ene cycle through the addition to an alkene and the generation of an alkyl radical, which propagates the reaction and affords the desired product by abstracting a hydrogen atom from the thiol starting material.
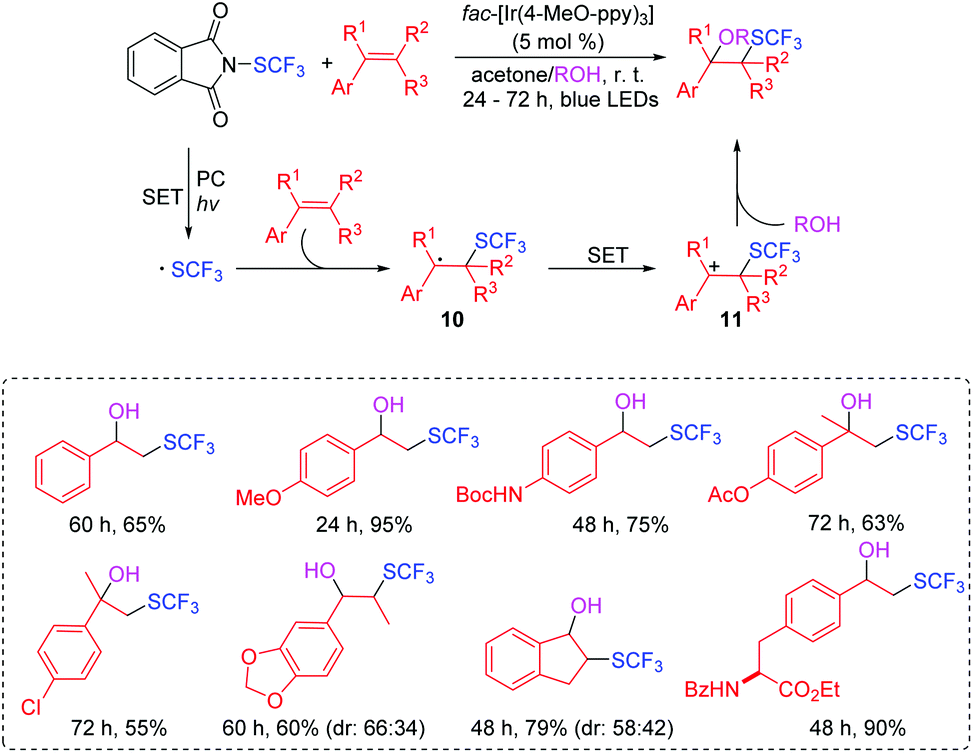
Organofluorine compounds have found increasing applications in the medicinal, pharmaceutical, agricultural, and materials sciences.22,23 In 2016, Akita's group disclosed a photocatalytic radical oxy-trifluoromethylthiolation of aromatic alkenes under mild and simple reaction conditions at room temperature without the addition of any activators for the SCF3 reagent (Scheme 7).24 During the course of trifluoromethylthiolation of alkenes, the selective incorporation of oxygen-nucleophilic reagents such as water and alcohol were compatible, affording the corresponding products with good to excellent efficiencies. The proposed reaction mechanism is outlined in Scheme 7. The generated SCF3 radical reacts with aromatic alkene to form product 10, which is oxidized to give the carbocationic intermediate 11. Intermediate 11 is attacked by water/or alcohol to afford the oxytrifluoromethylthiolated product.
 | ||
| Scheme 7 Trifluoromethylthiolation of aromatic alkenes. | ||
 | ||
| Scheme 8 Thioacetalization of aldehydes. | ||
2.3. Aryl carbon–thioyl radicals coupling reactions
 | ||
| Scheme 9 Visible-light induced aryl sulfide reactions. | ||
Aryl sulfoxides and aryl sulfones play a significant role in numerous organic compounds and drug candidates as building blocks27 and exhibit many important biological activities.28 In 2017, Lee's group developed a visible-light driven one-pot approach for the synthesis of diaryl sulfoxides and diaryl sulfones from aryl thiols and aryl diazonium salts using silver catalysis in the absence of a photocatalyst (Scheme 10).29 As for the reaction mechanism, the thioyl radical and the aryl radical are initially generated by the silver catalyst through visible light irradiation. Then, the formed thioyl radical couples with the aryl radical to deliver the thioether intermediate. Finally, the thioether intermediate is oxidized to the desired product. The transformation does not require the pre-activation of aryl thiols. Therefore, the method facilitates the rapid access to various aryl sulfoxides and aryl sulfones.
 | ||
| Scheme 10 Synthesis of diaryl sulfoxides and diaryl sulfones. | ||
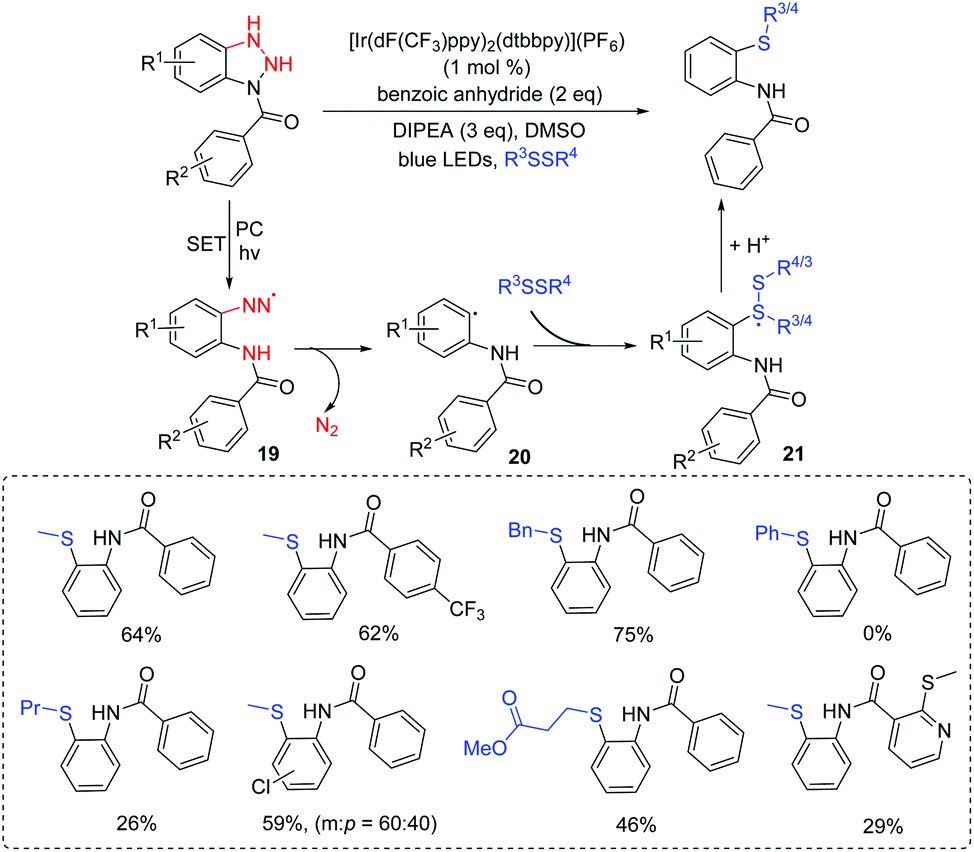
Glorius and co-workers reported a protocol for direct C–S bond-forming denitrogenative transformations to access o-thiolated N-arylbenzamide derivatives. The strategy facilitated a novel disconnection process via visible-light irradiation under mild conditions (Scheme 11).30 Benzotriazoles with alkyl, halides, and heterocyclic groups were suitable coupling partners. Moreover, disulfide derivatives with alkyl and benzyl groups could react with benzotriazoles smoothly. However, aryl disulfides failed to give the corresponding products. For the reaction mechanism, the highly active intermediate 19 is generated via visible light irradiation under standard conditions, which immediately extrudes nitrogen to afford the aryl radical 20. Intermediate 20 can add to the disulfide coupling partner to give the stabilized radical 21. Trapping of the intermediate 21 with another proton affords the desired o-thiolated N-arylbenzamide product.
 | ||
| Scheme 11 Denitrogenative thiolation of benzotriazoles. | ||
Noël's group developed a simple photoredox catalytic Stakler–Ziegler reaction to prepare arylsulfides from arylamines and aryl/alkylthiols at room temperature with minimum formation of diazosulfides (Scheme 12).31 In this case, thiophenol with an active free hydroxy group gave the corresponding product in good yields, which could be subject to further modification. Notably, the generated diazonium salt intermediates were not isolated and afforded the aryl radicals. Subsequently, the radical coupling reactions of the generated thioyl radicals with the aryl radicals produced the desired products.
 | ||
| Scheme 12 Reactions of arylamines and aryl/alkylthiols. | ||
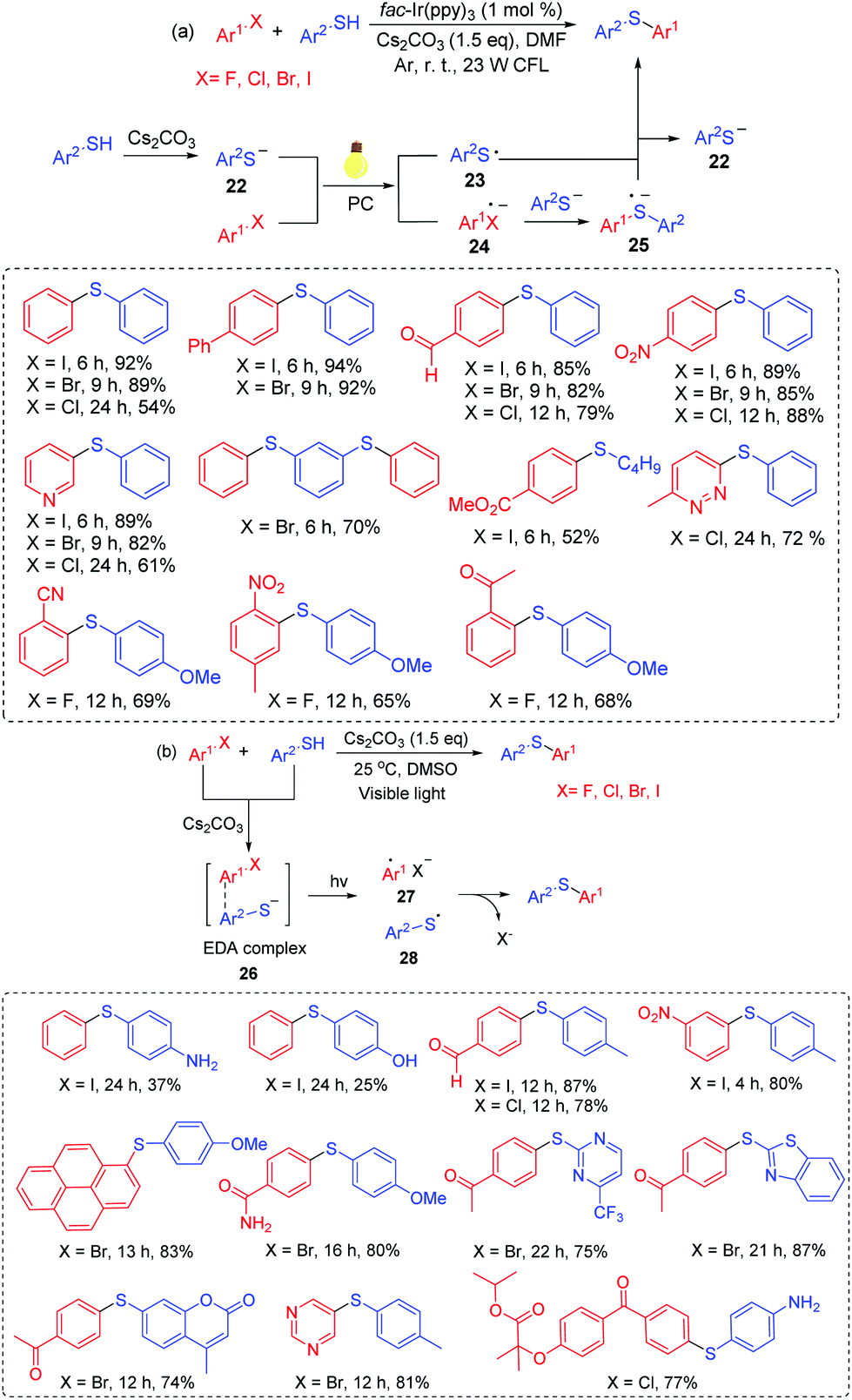
Fu and co-workers developed the representative arylation of thiols with aryl halides via a visible-light photoredox reaction at room temperature (Scheme 13a).32a This transformation explored readily available aryl halides as the arylating reagents and led to various desired products in good to high yields. In this process, the arylation of thiols tolerated various functional groups, including ether, ketone, aldehyde, ester, nitro, trifluoromethyl and an N-heterocycle. For the reaction mechanism, thiolate anion 22 is generated in the presence of Cs2CO3. Then, 22 can be converted into thioyl radical 23 through single-electron transfer (SET) oxidation. The reduction of aryl halides affords the radical anion 24. Subsequently, the transformation of 24 with another Ar2S− anion affords the sulfur-centered radical anion 25. Finally, the reaction of 25 with thioyl radical 23 gives the desired product with the release of Ar2S− anion via the single-electron transfer process.
 | ||
| Scheme 13 Arylation of thiols with aryl halides. | ||
Miyake's group further managed to expand the strategy for the C–S cross-coupling of thiols and aryl halides without the use of either transition metals or photoredox catalysts (Scheme 13b).32b A wide range of organosulfur compounds were constructed under mild, efficient visible-light irradiation. Especially, a number of pharmaceutical ingredients and biologically active molecules have been modified, affording the desired products in high yields. In this case, an EDA complex 26 is generated from a thiolate anion and an aryl halide under Cs2CO3 conditions. Next, intermediates 27 and 28 are generated via electron transfer under visible-light induced conditions. Finally, the coupling reaction of 27 with 28 affords the target products.
 | ||
| Scheme 14 Synthesis of 2-substituted benzothiazoles. | ||
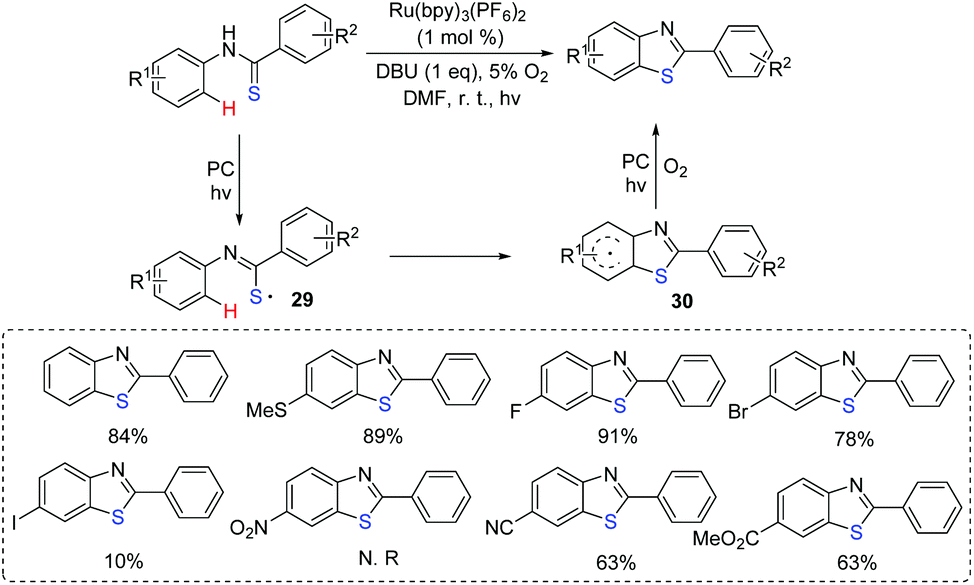
3-Substituted indoles compose an important skeleton in natural products, agrochemicals, and functional materials.34 The development of simple and efficient procedures for the acquisition of 3-sulfenylindoles from easily available starting materials under mild conditions continues to attract the interest of organic chemists because of the remarkable application values of the targeting products. Photocatalytic direct construction of the C–S bond for the preparation of 3-sulfenylindoles is still an appealing process.
In 2017, our group reported the visible-light induced transformation of indoles into 3-sulfenylindoles (Scheme 15).35 It is noted that the reaction system employed rose bengal as the photocatalyst, which provided a more convenient and environmentally friendly process compared with previous studies. Various indoles with electron-donating and electron-withdrawing groups were good coupling partners. Control experiments showed that the generated thioyl radical addition to the indole to form the radical intermediate 31 was a key step. The oxidation of 31 afforded intermediate 32, which further underwent the deprotonation process to deliver the desired 3-substituted indole product. Subsequently, Barman's group also reported a similar strategy for the C-3 sulfenylation of imidazopyridine derivatives from the reaction of indoles with thiols (Scheme 16).36
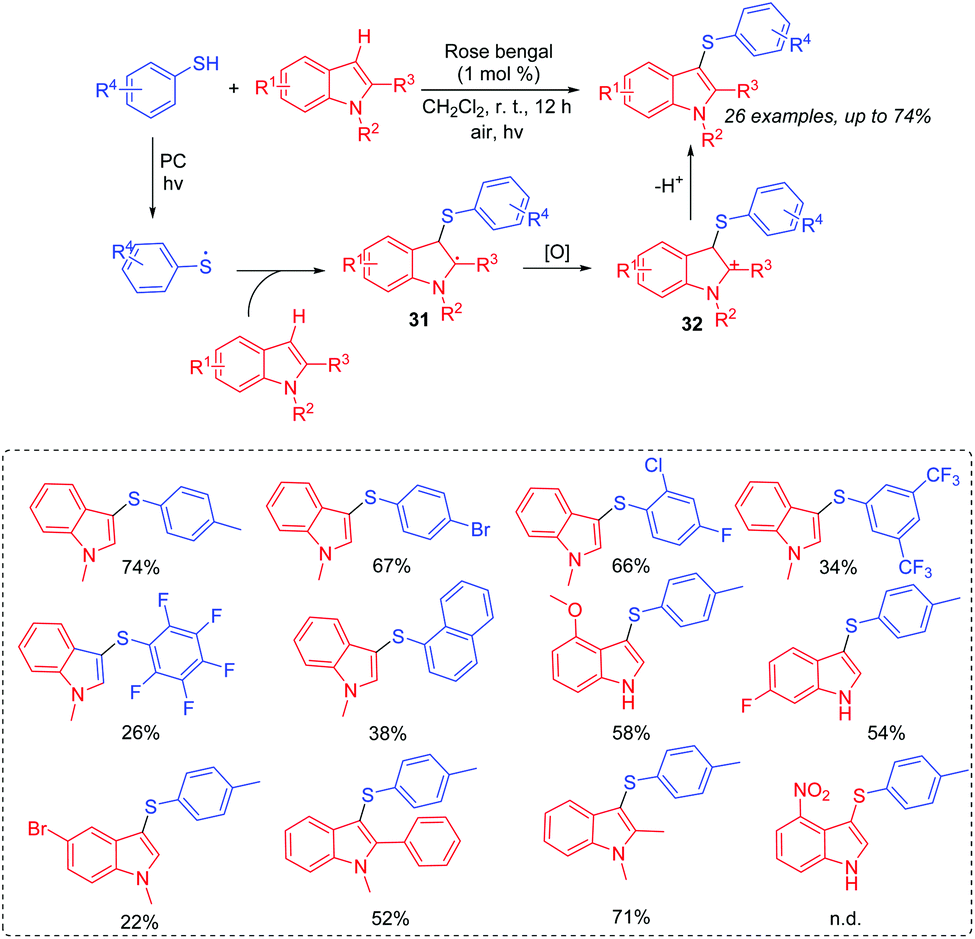 | ||
| Scheme 15 Synthesis of 3-thioindoles. | ||
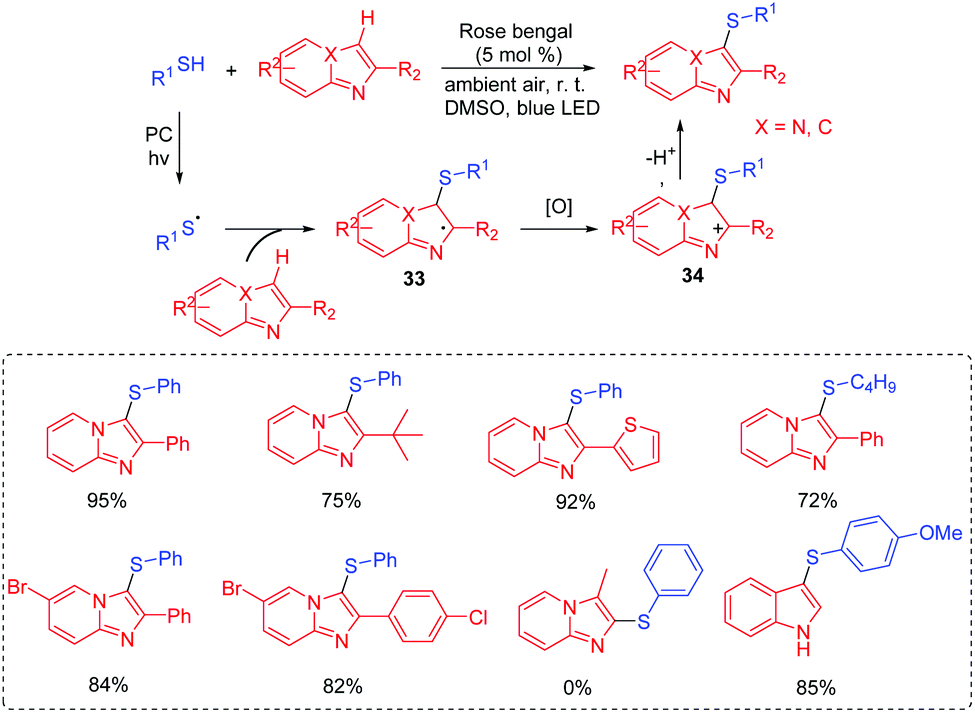 | ||
| Scheme 16 Regioselective sulfenylation of imidazopyridines. | ||
Additionally, Wang's group also developed a protocol for the regioselective C-3 sulfenylation of heteroaryl compounds to synthesize heteroaryl sulfides at room temperature using eosin B as the photocatalyst (Scheme 17).37 Generally, the desired products were obtained in moderate to good yields with a wide range of imidazopyridines, including those derived from the naphthalene group and the tertiary butyl group. Unfortunately, 1H-indole failed to react with sulfinic acids and no desired product was detected. A proposed mechanism for the formation of heteroaryl sulfide derivatives is shown in Scheme 17. Firstly, the generated sulfonyl radical 35 is converted into thioyl radical 36via a sequence of reduction reactions. Then, the addition of thioyl radical 36 to a heteroaryl compound produces the radical intermediate 37, which can be further transformed into intermediate 38via the SET process. Finally, the attack of the sulfonic acid anion by the β-H of intermediate 38 affords the desired product.
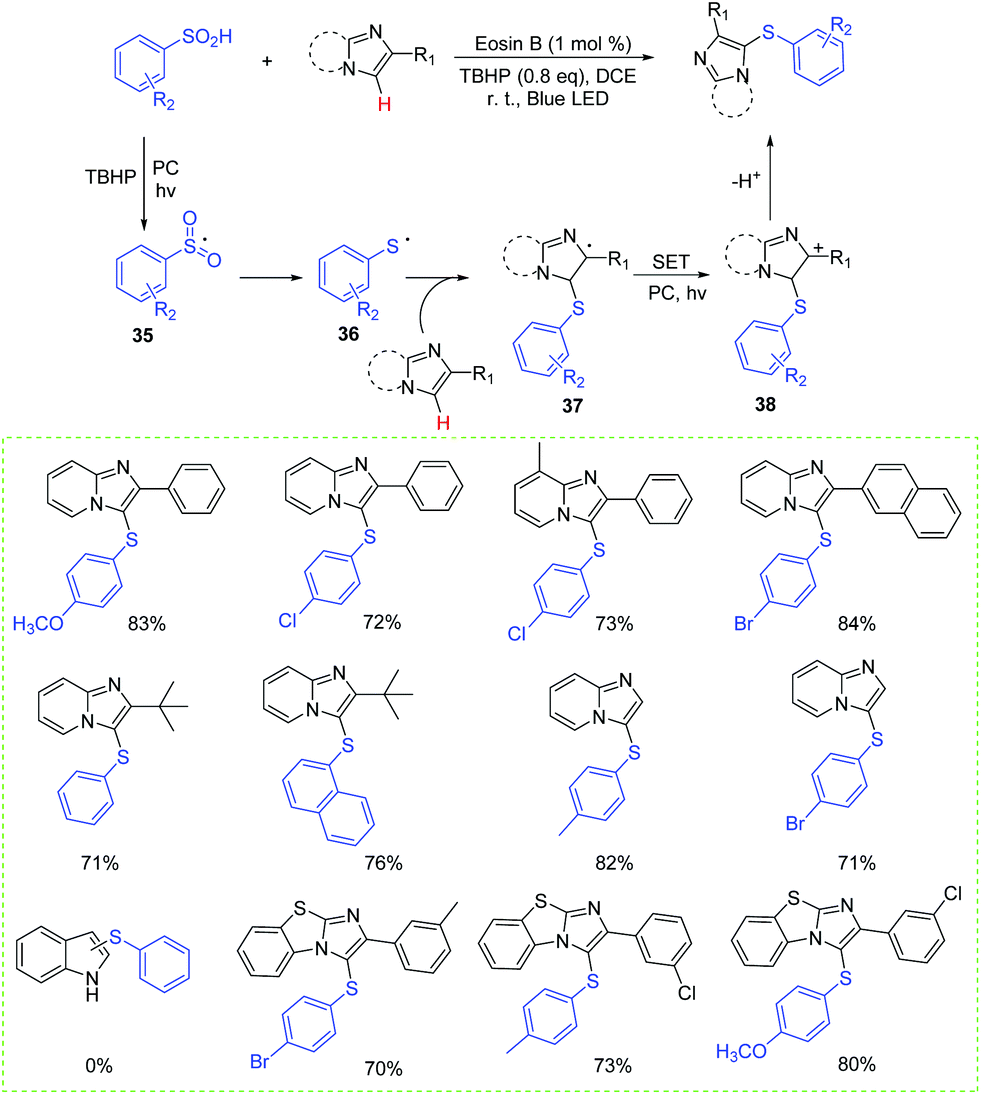 | ||
| Scheme 17 Synthesis of heteroaryl sulphides. | ||
In 2017, Wang et al. reported an improved strategy for the synthesis of 3-substituted indoles from 2-alkynylanilines with disulfides in the presence of H2O2 under transition-metal-free and photocatalyst-free conditions (Scheme 18).38 In this process, the in situ generated indoles from 2-alkynylaniline deliver the aromatic carbon radical 39. Finally, the formed intermediate 39 interacts with the disulfide/or thioyl radical to afford the desired product.
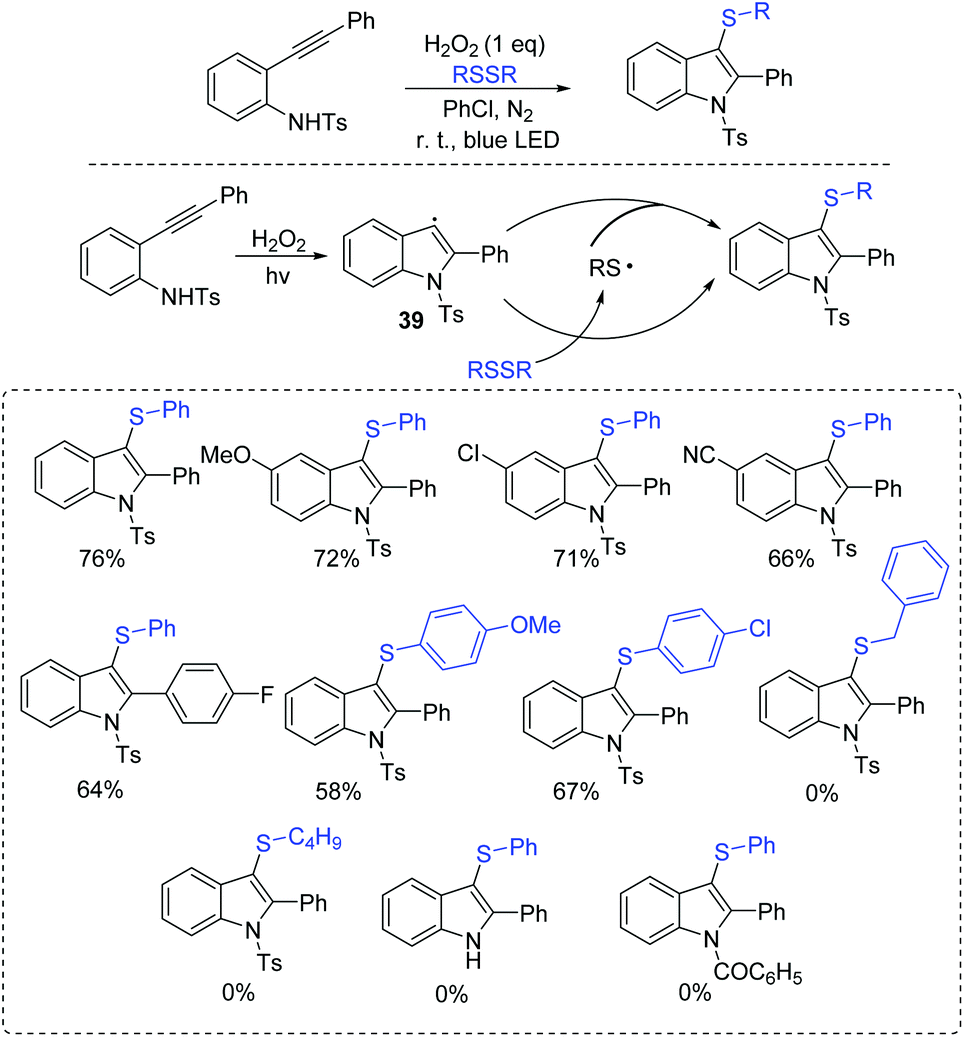 | ||
| Scheme 18 Reaction of 2-alkynylanilines with disulfides. | ||
Thiocyanates are usually transformed into trifluoromethyl sulfides, sulfonyl chlorides, thiols, thiocarbamates, heterocycles, disulfides, and thioethers.39–41 In 2014, Li and co-workers developed a direct C-3 thiocyanation of indoles in the presence of rose bengal at room temperature (Scheme 19).4c Generally, the reaction worked well either with weak electron-withdrawing group or with electron-donating group substituted indoles, affording the corresponding 3-thiocyanoindoles in good to excellent yields. A possible explanation for this transformation is illustrated in Scheme 18. Firstly, the formed SCN radical reacts with indole to afford intermediate 40. Then, intermediate 40 is converted into intermediate 41 by oxidization, which further gives the desired product by losing a proton.
 | ||
| Scheme 19 C-3 Thiocyanation of indoles. | ||
Additionally, Hajra's group also developed a similar protocol for the C-3 thiocyanation of imidazoheterocycles at room temperature via visible-light promoted by using eosin Y as the photocatalyst (Scheme 20).4d The reaction exhibited a wide range of functional group tolerances, such as imidazopyridines with the hydroxy group also afforded the desired product in a good yield. The transformation initiates the formation of the SCN radical by the SET mechanism. Then, the resulting SCN radical interacts with imidazoheterocycle to give intermediate 42, which is oxidized to intermediate 43. Finally, intermediate 43 is converted into the desired product via the aromatization process.
 | ||
| Scheme 20 Thiocyanation of imidazoheterocycles. | ||
2.4. C(sp)–thioyl radical coupling reactions
 | ||
| Scheme 21 Synthesis of alkynyl sulfides. | ||
 | ||
| Scheme 22 Photocatalytic S–H bond direct cyanation. | ||
A possible mechanism is shown in Scheme 22. The generated thioyl radical under the irradiation of visible light couples with SCN− to form intermediate 46. Trapping of intermediate 46 with another thiol can result in the propagation and formation of intermediate 47. With the release of HS−, 47 is further converted into the desired thiocyanate product.
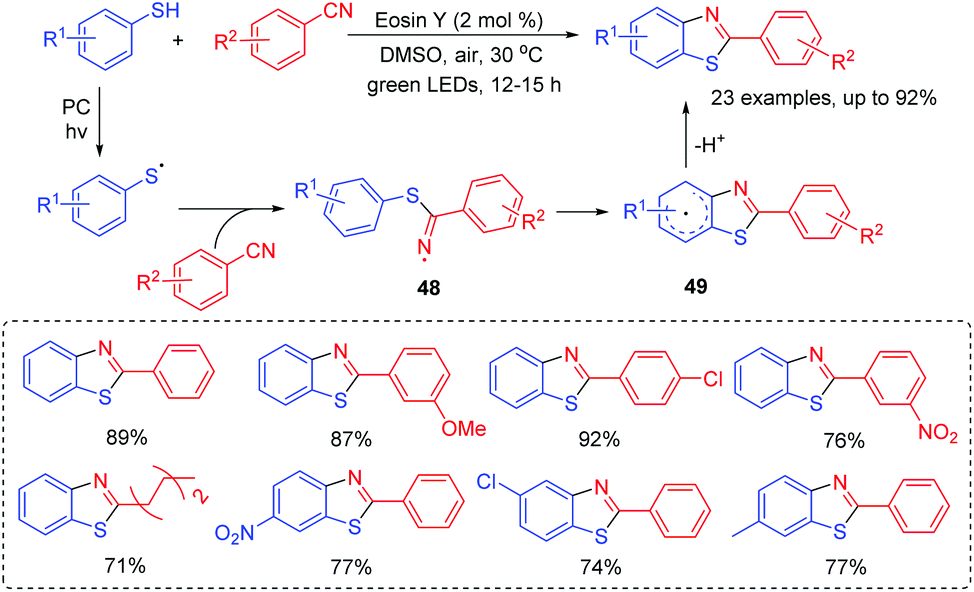 | ||
| Scheme 23 Synthesis of 2-substituted benzothiazoles. | ||
2.5. P(O)–H substitution reactions
Thiophosphates present important structural scaffolds, and usually serve as therapeutic molecules and agrochemicals.47 Zhang and co-workers demonstrated an efficient S–P(O) bond formation protocol through the direct cross-coupling of thiols with P(O)H compounds using rose bengal as the photoredox catalysts (Scheme 24).48 Various S–P(O) coupling products were easily constructed in moderate to excellent yields. Moreover, thiols with the active groups such as the free amine group and the hydroxy group were also performed smoothly, which could be further modified for more applications. In this process, 1O2 is generated in the presence of rose bengal under visible light irradiation. Subsequently, the generated 1O2 abstracts the hydrogen atom from thiol and affords the thioyl radical. At the same time, the P-centered radical is also generated from the P(O)H compound by a similar oxidation process. Finally, the coupling of the thioyl radical with P-centered radical produces the desired compound. | ||
| Scheme 24 Construction of S–P(O) bonds. | ||
Wu's group also succeeded in the visible-light catalyzed dehydrogenative coupling reaction of thiols with phosphonates using methylene blue as a promoter to directly construct the S–P(O) bonds (Scheme 25).49 The method provided a green route for the synthesis of thiophosphates under mild conditions. Moreover, thiols with different functional groups, including the halogen, amide, trifluoromethyl, free amine and the hydroxyl group, were successfully coupled with dialkyl phosphonates/or alkyl phosphine oxides, giving the desired products in good to excellent yields. In this photoredox catalysis process, the formed thioyl radical couples with the P-centered radical to produce the desired product.
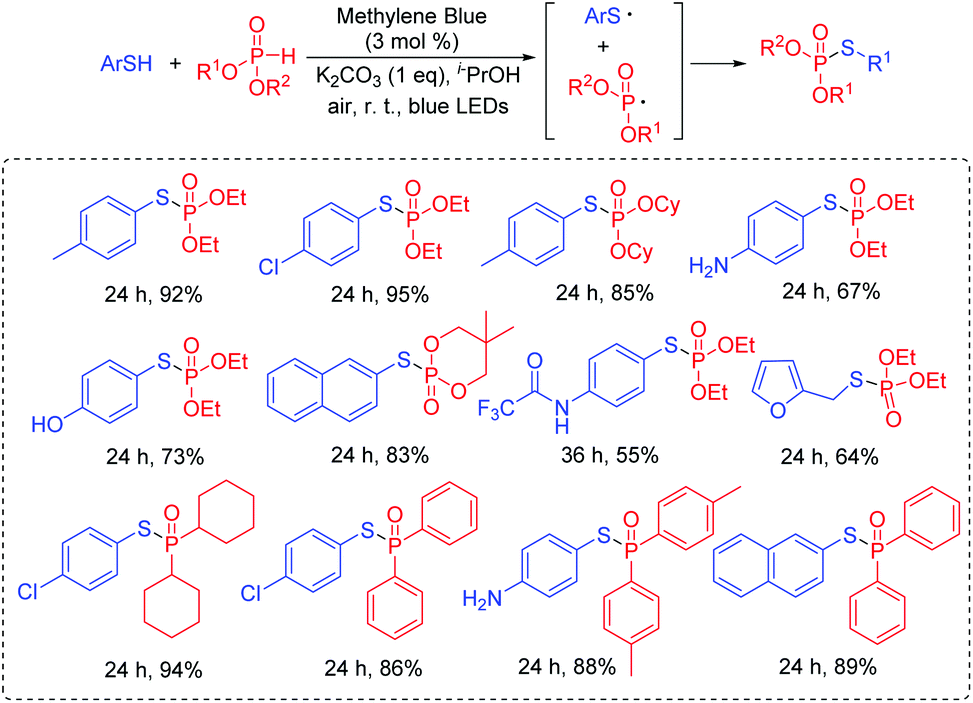 | ||
| Scheme 25 Synthesis of thiophosphates. | ||
3. Visible-light induced C–S bond formation via the generation of the sulfonyl radical
3.1. C(sp2)–sulfonyl radicals coupling reactions
 | ||
| Scheme 26 Synthesis of α-substituted vinyl sulfones. | ||
β-Amido sulfones are privileged scaffolds found in many natural products and biologically active compounds.53 So far, the preparation of β-acetylamino acrylosulfone substrates relies on the several known methods, such as rearrangement reactions, the reduction of nitro alkenes or ketoximes, the acylation of imines, and the direct condensation of ketones with amides.54 However, visible-light photoredox catalysis processes to access β-acetylamino acrylosulfones have been reported rarely. Zhang's group reported a visible-light induced oxidative cross-coupling strategy to access β-acetylamino acrylosulfones from enamides and sodium sulfinates using rose bengal as the photocatalyst at room temperature (Scheme 27).55 In the transformation, the radical addition of the generated sulfonyl radical to enamide produces the carbon-centered radical 53. Then, intermediate 53 is oxidated to the iminium ion 54. Finally, 54 delivers the β-acetylamino acrylosulfone product via deprotonation.
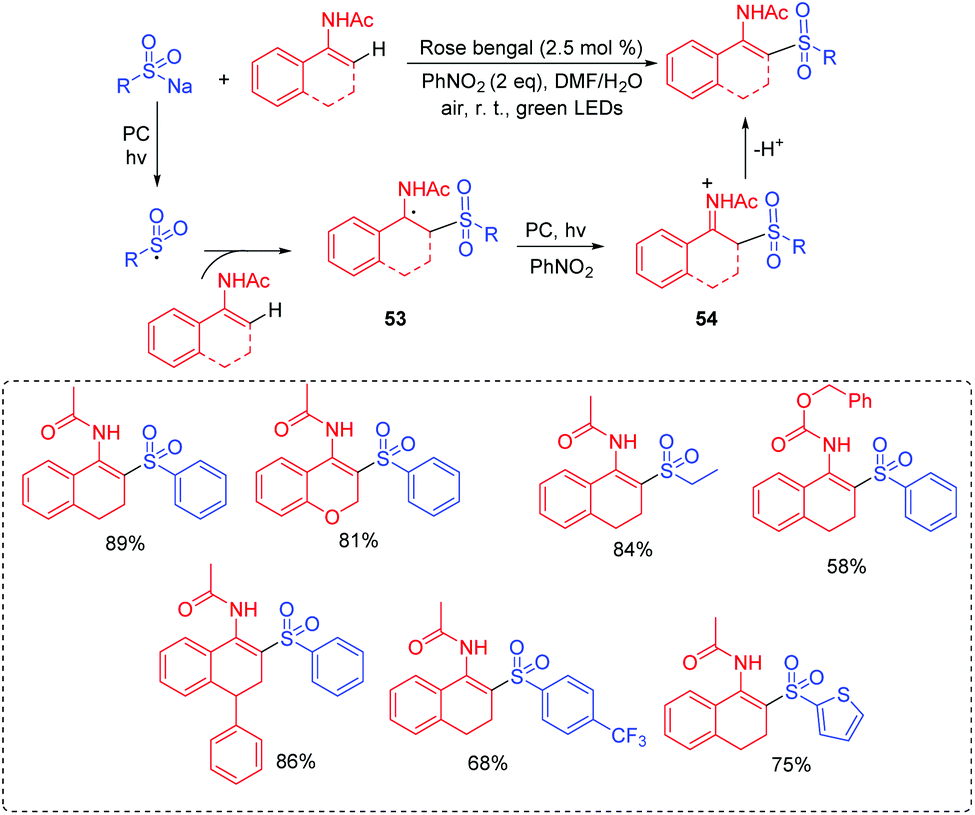 | ||
| Scheme 27 Synthesis of β-acetylamino acrylosulfones. | ||
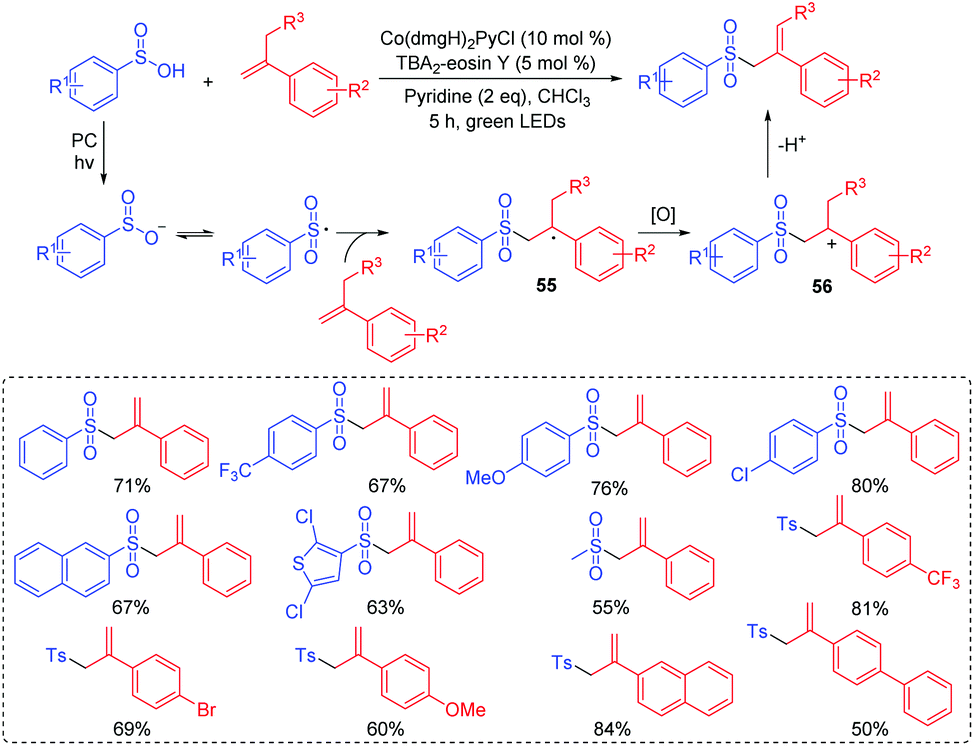 | ||
| Scheme 28 Synthesis of allylic sulfones. | ||
The importance of alkylsulfonyl compounds is known since some compounds with alkylsulfonyl units are marketed as drugs or biologically active molecules.60 In 2018, Wu's group successfully explored a three-component reaction to access alkylsulfonyl compounds via photocatalysis at room temperature (Scheme 29).61 This reaction worked efficiently with the use of potassium alkyltrifluoroborates, sulfur dioxide surrogate of DABCO·(SO2)2, and alkenes as the starting materials, affording diverse sulfones in good to excellent yields. Furthermore, sulfur dioxide was also successfully inserted into biologically active molecules. Mechanistic studies show that the alkylsulfonyl radical is a key intermediate. After that, the radical addition of an alkylsulfonyl radical to alkene produces the radical intermediate 57, which is further transformed into intermediate 58. Subsequently, the expected alkylsulfonyl product is generated by the protonation reaction.
 | ||
| Scheme 29 Synthesis of alkylsulfonyl compounds. | ||
The β-hydroxysulfone is an important scaffold for the synthesis of pharmaceutical drugs and various biologically active molecules.62 Traditional methods for the preparation of β-hydroxysulfones are based on the opening of epoxides with sulfinate salts, the reduction of β-oxosulfones, or the hydroxylation of α,β-unsaturated sulfones.63,64 In 2016, Reiser's group described a dual-catalyst-catalyzed reaction through a visible-light promoted process to synthesize β-hydroxysulfone derivatives from alkenes and sulfonyl chlorides in the presence of water (Scheme 30).65 The reaction featured a wide range of functional group tolerances such as strong/weak electron-withdrawing/donating groups, alkyl and aryl groups, affording corresponding products in high yields. The authors proposed that the key step for the reaction was the radical addition of sulfonyl radicals to alkenes under visible-light irradiation.
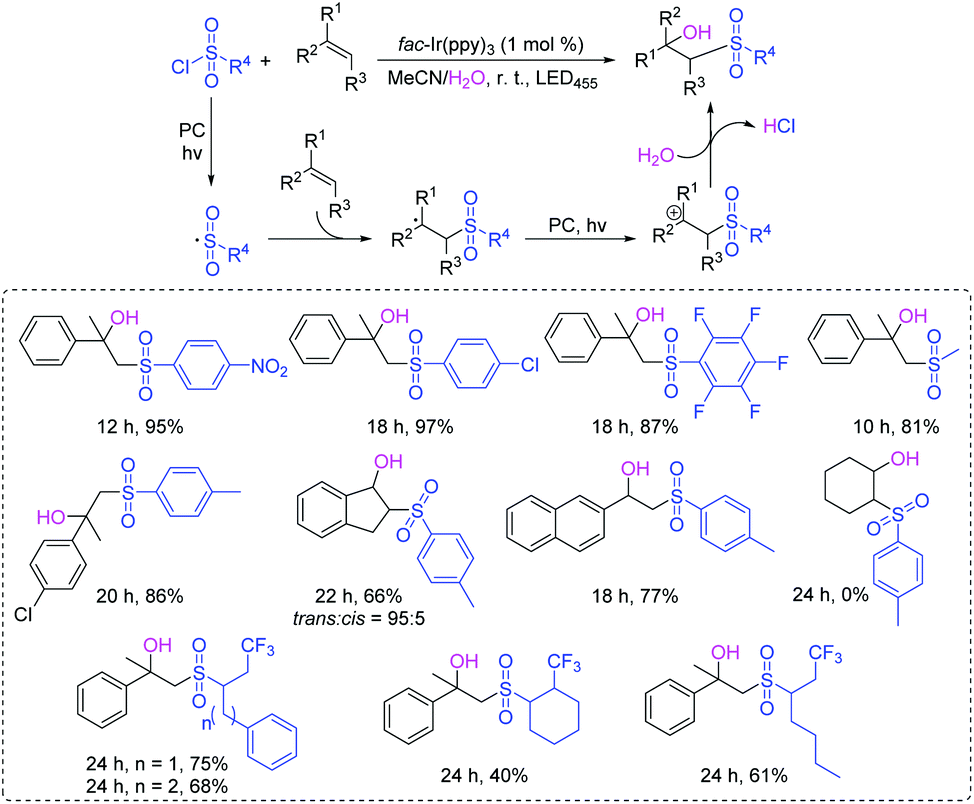 | ||
| Scheme 30 Synthesis of β-hydroxysulfones from sulfonyl chlorides and alkenes. | ||
Dimethyl sulfoxide is the one of important organosulfur compounds. Due to its low cost and stability,66 DMSO is usually utilized as a solvent and a versatile oxidant reagent in the organic synthesis reaction.67 Furthermore, DMSO could be used as a common precursor for the methyl, formyl, cyano, methylmercapto, and so on.68 In 2018, an elegant work for the synthesis of benzo[a]fluoren-5-one derivatives via the visible-light photocatalytic methylsulfonylation of 1,7-enynes under mild reaction conditions was reported by the group of Jiang (Scheme 31).69 This method employed the DMSO/H2O system as the methylsulfonyl synthon and afforded a broad range of functionalized benzo[a]fluoren-5-ones from readily available materials. The reaction of the generated sulfonyl radical with 1,7-enynes 59 delivers the radical intermediate 60, which is further converted into the radical intermediate 61via intramolecular 6-exo-dig/5-endo-trig bicyclization. The final benzo[a]fluoren-5-one product is obtained from 61via the SET process.
 | ||
| Scheme 31 Photocatalytic methylsulfonylation of 1,7-enynes. | ||
3.2. Alkynes addition reactions
Wang's group developed a radical tandem process to synthesize (E)- and (Z)-C6-(vinyl sulfone) phenanthridines (Scheme 32).70 The reaction was operationally simple using biaryl isocyanides, alkynes, and arylsulfinic acids as the starting materials via the light-controlled process under mild conditions. This approach exhibited a broad substrate scope, and provided the corresponding (E)- and (Z)-C6-(vinyl sulfone)phenanthridine derivatives in good yields. In this reaction, the novel electron donor–acceptor (EDA) complex 62 is generated from the reaction of arylsulfinic acid and biaryl isocyanide. Then, the generated sulfonyl radical reacts with the alkyne to afford vinyl radical 63, which further performs an addition into the biaryl isocyanide to produce the imidoyl radical 64. Finally, the desired product can be obtained via the intramolecular homolytic aromatic substitution and oxidation processes.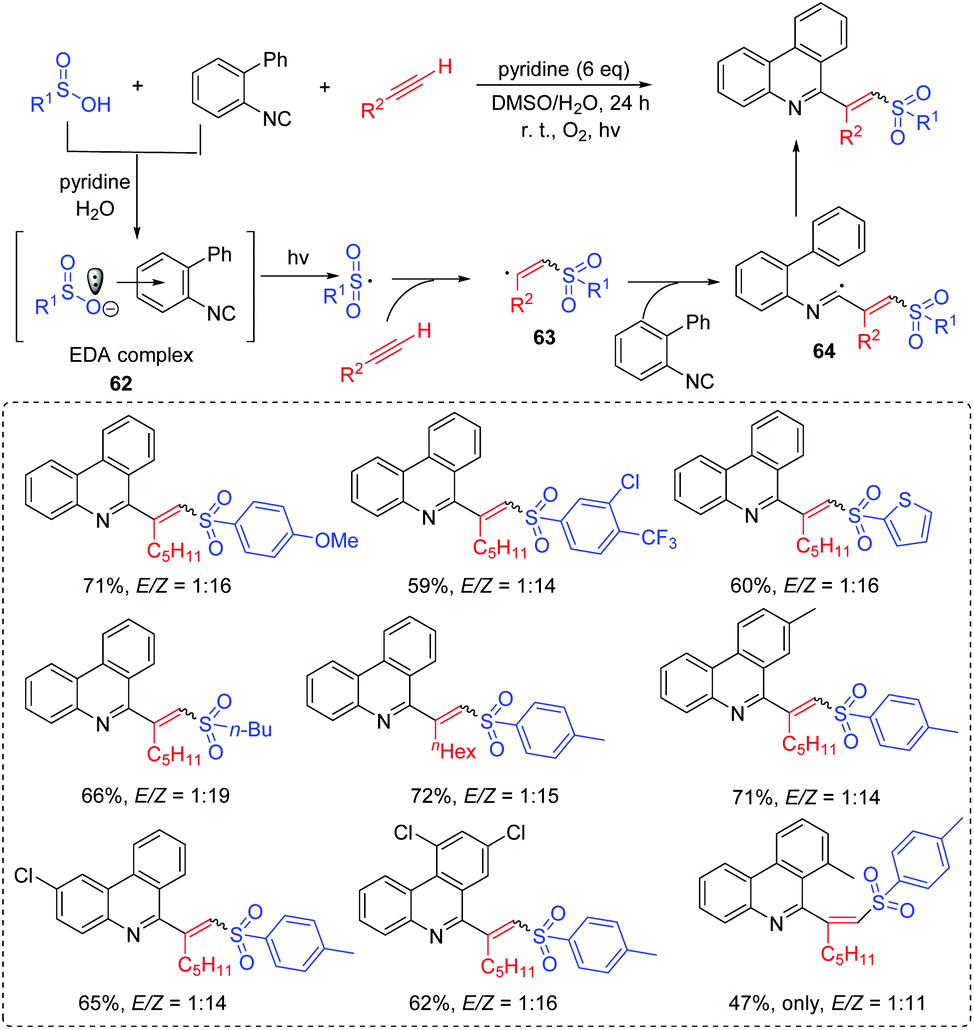 | ||
| Scheme 32 Synthesis of (E)- and (Z)-C6-(vinyl sulfone)phenanthridines. | ||
The coumarin skeleton plays a significant role in natural products, biologically active molecules, and it can serve as anti-HIV, and anti-diabetic drugs, and so on.71 In 2015, Wang's group developed a visible-light initiated arylsulfonylation of alkynes with arylsulfinic acids to access 3-sulfonated coumarins (Scheme 33).72 The method was compatible with a wide range of electron-rich aromatic compounds. More importantly, the halogens (F, Cl, Br and I) attached to the phenyl group of alkynes were well tolerated, which could be further modified in organic and medicinal chemistry. In this study, the addition of the generated sulfonyl radical to alkyne 65 delivers the radical intermediate 66. Then, 66 is transformed into the carbon-cation intermediate 67 by the oxidation reaction. Finally, 67 is converted into a 3-sulfonated coumarin product by the deprotonation and rearomatization processes.
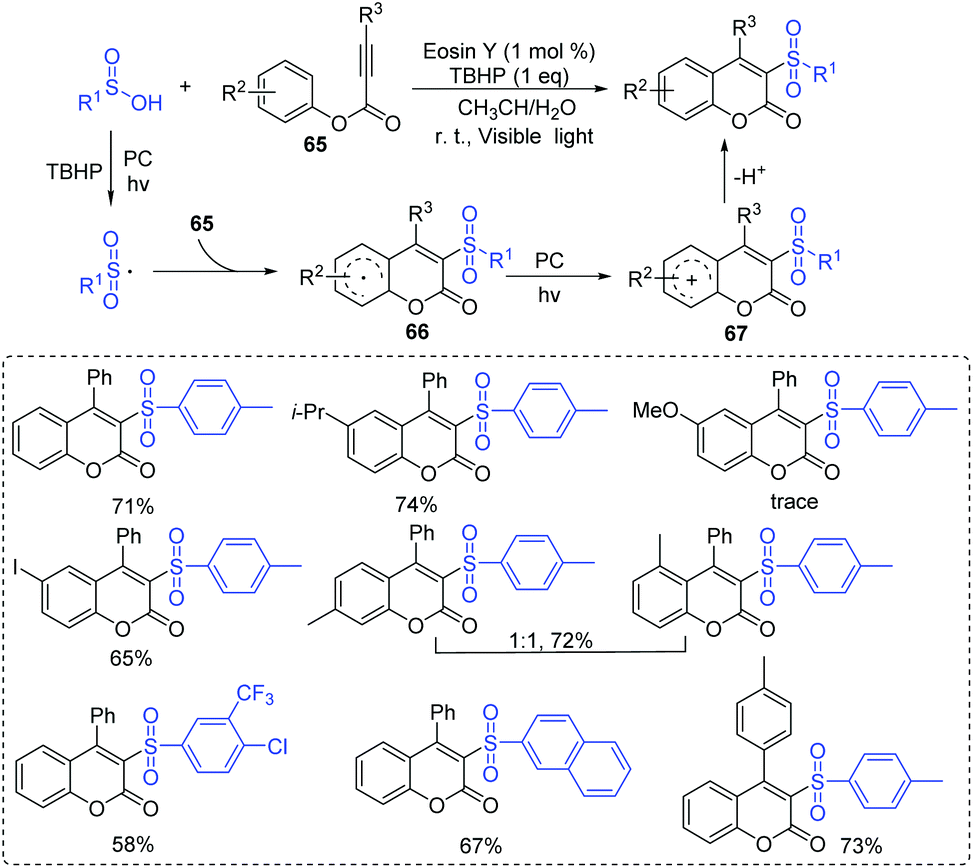 | ||
| Scheme 33 Arylsulfonylation of alkynes. | ||
Vinyl sulfones are known to be highly versatile encountered motifs, which have found widespread applications in biological research studies.73 In 2016, Lei and co-workers reported a visible-light mediated Markovnikov-selective radical addition of sulfinic acids to terminal alkynes via the α-substituted vinyl radical/sulfonyl radical cross-coupling process (Scheme 34).74 The transformation exhibited highly regioselective and broad functional-groups tolerance. It is noteworthy that a complex estrone derivative was also successfully generated. A plausible mechanism is illustrated in Scheme 34. The generated sulfinate 68 is generated from sulfinic acid with a base via an acid–base neutralization reaction. Then, the reaction of the formed species 68 with the excited state of eosin Y affords intermediate 69 along with the formation of eosin Y˙−, which further resonates to intermediate 70. Subsequently, the alkyne reacts with eosin Y˙− to give the α-vinyl carbon radical 71. Finally, radical cross-coupling of 70 with 71 produces the desired Markovnikov product via the proton transfer process.
 | ||
| Scheme 34 Synthesis of α-substituted vinyl sulfones. | ||
3.3. Aryl substitution reactions
Aryl sulfones could be served as a new class of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) inhibitors acting at the non-nucleoside binding site of this enzyme.75 In 2017, Manolikakes and co-workers reported the reactions of sulfinates with aryl iodides to give aryl sulfones under photoredox/nickel dual catalysts system conditions (Scheme 35).76 Various sulfinates and aryl iodides were suitable for this transformation. The drug-like scaffolds also successfully realized diversification. In the same year, Rueping and coworkers developed a similar strategy for the synthesis of sulfones via the photoredox/nickel dual catalysis at room temperature (Scheme 36).77 Coupling reactions of sodium sulfinates with various aryl halogen (Cl, Br, I) compounds were performed to synthesize the sulfones derivatives, giving the desired products in good to high yields. | ||
| Scheme 35 Synthesis of aryl sulfones from sulfinic acid salts and aryl iodides. | ||
 | ||
| Scheme 36 Synthesis of aryl sulfones from sulfinates and halides. | ||
The proposed catalytic pathway is initiated by the electron transfer process to form a sulfonyl radical. After that, a series of addition and oxidative addition reactions with nickel catalysts gives the NiIII intermediate 72 (or 73). Finally, the reductive elimination of the NiIII intermediate 72 (or 73) affords sulfone products.
Furthermore, Reiser and co-workers demonstrated a visible-light induced photocatalytic strategy for the direct sulfonylation of heteroaromatic compounds at room temperature without pre-activating a heterocyclic ring and a sulfonyl source (Scheme 37).78 All kinds of aryl and alkyl sulfonyl chlorides were good coupling partners for this transformation. Furthermore, indoles and pyrroles were also suitable substrates. A proposed mechanism is shown in Scheme 37. The key process for the reaction is the formation of the sulfonyl radical by the oxidative quenching cycle of the Ir(III)-catalyst. The sulfonyl radical is sufficiently stable at room temperature to effectively undergo coupling with heteroarenes and then affords intermediate 74. Finally, the desired product is obtained with the release of an electron and a proton from 74.
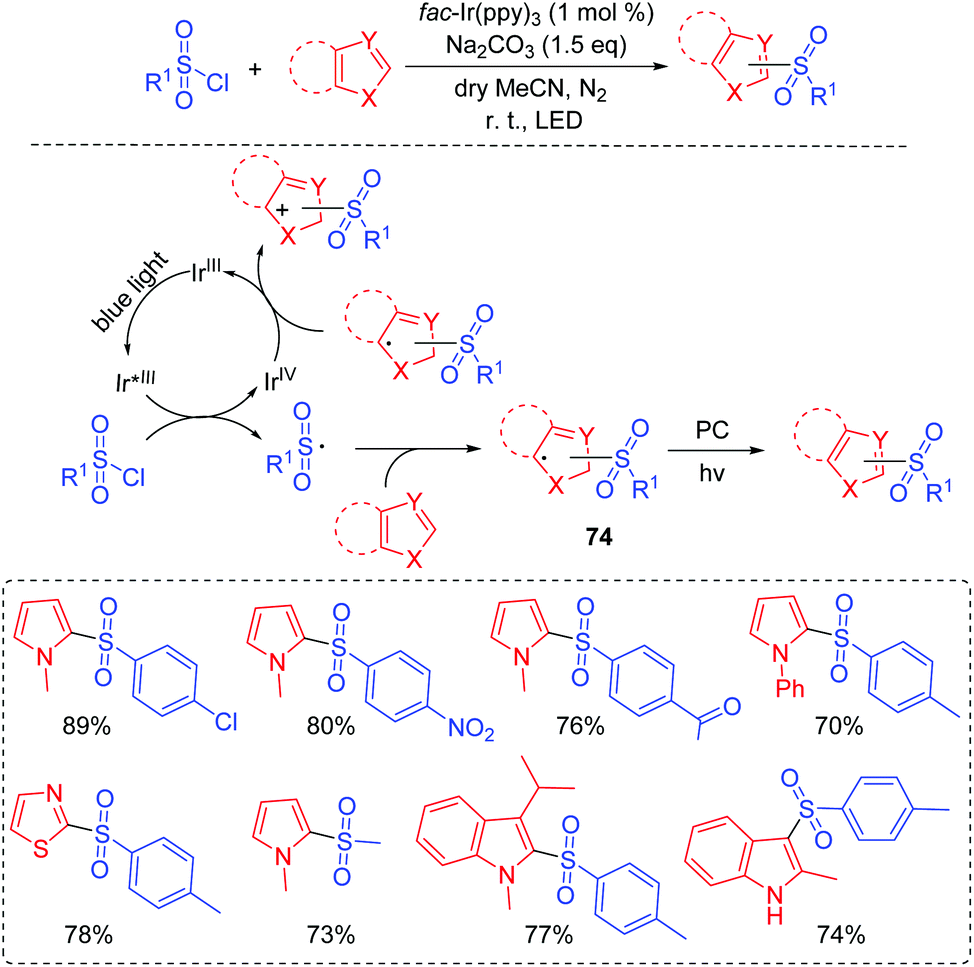 | ||
| Scheme 37 Sulfonylation of unactivated heterocycles. | ||
4. Summary
In conclusion, visible-light photoredox catalysis has very rapidly been established as a powerful tool for the construction of organosulfur compounds. The reactions exhibited advantages including ambient conditions (room temperature or air atmosphere), eco-energy source, good functional group compatibility, and in some cases, the use of organic dyes as photocatalysts (i.e., transition metal free conditions). In this review, we have described the two major approaches for the construction of these organosulfur compounds, including highly reactive thioyl/sulfonyl radical species substitution reactions and addition reactions in a controllable way. The transformation successfully provides diversely functionalized organosulfur compounds, which will be applied to the elaboration of pharmaceutical molecules and skeletons of natural products.Although with unprecedented achievements of the C–S/P–S bond formation in hand, sulfur centered radicals chemistry still remains a fascinating hot topic.79 However, the generated by-products (such as disulfide compounds) from the self-coupling of sulfur radicals are still challenging issues.35,36 The exploration of novel and efficient photocatalysts, advanced reaction systems and the corresponding theoretical research represent an essential development field to improve the performance of sulfur centered radicals. The scale-up application for visible-light photoredox catalyzed transformations is another limitation, which could be adapted to continuous flow technology.80 On the other hand, there is no doubt that some C–S/P–S bond formation and functionalization involving sulfur centered radicals mechanism have been carried out with the addition of transition-metal, I2 or external oxidants.3,81,82 Considering its great potential for synthetic applications, it is believed that the above strategies could be performed via the photoredox catalytic process. We envision that this strategy will help to promote continued interest in the visible light photoredox catalysis-controlled reactions involving the formation and further functionalization of sulfur centered radicals, which can be envisaged in the near future.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for the generous financial support provided by the National Natural Science Foundation of China (21602031, 21662002 and 21867001) and the Jiangxi Natural Science Foundation (20171BAB203010).References
- (a) D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed; (b) J. Liu, J. Yang, Q. Yang, G. Wang and Y. Li, Adv. Funct. Mater., 2005, 15, 1297–1302 CrossRef CAS; (c) A. Dondoni, Angew. Chem., Int. Ed., 2008, 120, 9133–9135 CrossRef.
- (a) A. Natarajan, Y. Guo, F. Harbinski, Y.-H. Fan, H. Chen, L. Luus, J. Diercks, H. Aktas, M. Chorev and J. A. Halperin, J. Med. Chem., 2004, 47, 4979–4982 CrossRef CAS PubMed; (b) D. C. Cole, W. J. Lennox, S. Lombardi, J. W. Ellingboe, R. C. Bernotas, G. J. Tawa, H. Mazandarani, D. L. Smith, G. Zhang, J. Coupet and L. E. Schechter, J. Med. Chem., 2005, 48, 353–356 CrossRef CAS PubMed; (c) M. Banerjee, A. Poddar, G. Mitra, A. Surolia, T. Owa and B. Bhattacharyya, J. Med. Chem., 2005, 48, 547–555 CrossRef CAS PubMed; (d) E. Feng, H. Huang, Y. Zhou, D. Ye, H. Jiang and H. Liu, J. Comb. Chem., 2010, 12, 422–429 CrossRef CAS PubMed; (e) G.-Y. Gao, A. J. Colvin, Y. Chen and X. P. Zhang, J. Org. Chem., 2004, 69, 8886–8892 CrossRef CAS PubMed; (f) K. Inamoto, C. Hasegawa, K. Hiroya and T. Doi, Org. Lett., 2008, 10, 5147–5150 CrossRef CAS PubMed; (g) M. Jegelka and B. Plietker, Org. Lett., 2009, 11, 3462–3465 CrossRef CAS PubMed; (h) P. Niu, J. Kang, X. Tian, L. Song, H. Liu, J. Wu, W. Yu and J. Chang, J. Org. Chem., 2015, 80, 1018–1024 CrossRef CAS PubMed.
- (a) N. B. Heine and A. Studer, Org. Lett., 2017, 19, 4150–4153 CrossRef CAS PubMed; (b) G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691–705 RSC; (c) F. Teng and J. Cheng, Chin. J. Chem., 2017, 35, 289–298 CrossRef CAS; (d) R. S. Glass, Top. Curr. Chem., 2018, 376, 22 CrossRef PubMed.
- (a) H. Chachignon and D. Cahard, Chin. J. Chem., 2016, 34, 445–454 CrossRef CAS; (b) E. L. Tyson, Z. L. Niemeyer and T. P. Yoon, J. Org. Chem., 2014, 79, 1427–1436 CrossRef CAS PubMed; (c) W. Fan, Q. Yang, F. Xu and P. Li, J. Org. Chem., 2014, 79, 10588–10592 CrossRef CAS PubMed; (d) S. Mitra, M. Ghosh, S. Mishra and A. Hajra, J. Org. Chem., 2015, 80, 8275–8281 CrossRef CAS PubMed; (e) F. G. Bordwell, X.-M. Zhang, A. V. Satish and J.-P. Cheng, J. Am. Chem. Soc., 1994, 116, 6605–6610 CrossRef CAS; (f) A. U. Meyer, S. Jäger, D. P. Hari and B. König, Adv. Synth. Catal., 2015, 357, 2050–2054 CrossRef CAS; (g) G. Zhang, L. Zhang, H. Yi, Y. Luo, X. Qi, C.-H. Tung, L.-Z. Wu and A. Lei, Chem. Commun., 2016, 52, 10407–10410 RSC.
- (a) J. Gorges and U. Kazmaier, Eur. J. Org. Chem., 2015, 8011–8017 CrossRef CAS; (b) B. D. Fairbanks, D. M. Love and C. N. Bowman, Macromol. Chem. Phys., 2017, 218, 1700073 CrossRef; (c) D. Limnios and C. G. Kokotos, Adv. Synth. Catal., 2017, 359, 323–328 CrossRef CAS; (d) K. Griesbaum, Angew. Chem., Int. Ed. Engl., 1970, 9, 273–287 CrossRef CAS.
- E. K. Skinner, F. M. Whiffen and G. J. Price, Chem. Commun., 2012, 48, 6800–6802 RSC.
- (a) Y.-Y. Gui, L. Sun, Z.-P. Lu and D.-G. Yu, Org. Chem. Front., 2016, 3, 522–526 RSC; (b) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC; (c) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed; (d) L.-H. Wu, J.-K. Cheng, L. Shen, Z.-L. Shen and T.-P. Loh, Adv. Synth. Catal., 2018, 360, 3894–3899 CrossRef CAS.
- (a) W.-H. Rao, B.-B. Zhan, K. Chen, P.-X. Ling, Z.-Z. Zhang and B.-F. Shi, Org. Lett., 2015, 17, 3552–3555 CrossRef CAS PubMed; (b) P.-F. Wang, X.-Q. Wang, J.-J. Dai, Y.-S. Feng and H.-J. Xu, Org. Lett., 2014, 16, 4586–4589 CrossRef CAS PubMed.
- (a) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- X. Zhu, X. Xie, P. Li, J. Guo and L. Wang, Org. Lett., 2016, 18, 1546–1549 CrossRef CAS PubMed.
- W. Guo, M. Zhao, W. Tan, L. Zheng, K. Tao, L. Liu, X. Wang, D. Chen and X. Fan, J. Org. Chem., 2018, 83, 1402–1413 CrossRef CAS PubMed.
- R. Honeker, R. A. Garza-Sanchez, M. N. Hopkinson and F. Glorius, Chem. – Eur. J., 2016, 22, 4395–4399 CrossRef CAS PubMed.
- S. Aubry, K. Sasaki, L. Eloy, G. Aubert, P. Retailleau, T. Cresteil and D. Crich, Org. Biomol. Chem., 2011, 9, 7134–7143 RSC.
- D. V. Ferraris, P. Majer, C. Ni, C. E. Slusher, R. Rais, Y. Wu, K. M. Wozniak, J. Alt, C. Rojas, B. S. Slusher and T. Tsukamoto, J. Med. Chem., 2014, 57, 243–247 CrossRef CAS PubMed.
- L. Kremer, J. D. Douglas, A. R. Baulard, C. Morehouse, M. R. Guy, D. Alland, L. G. Dover, J. H. Lakey, W. R. Jacobs Jr., P. J. Brennan, D. E. Minnikin and G. S. Besra, J. Biol. Chem., 2000, 275, 16857–16864 CrossRef CAS PubMed.
- (a) P. Espeel and F. E. Du Prez, Eur. Polym. J., 2015, 62, 247–272 CrossRef CAS; (b) F. Goethals, S. Martens, P. Espeel, O. van den Berg and F. E. Du Prez, Macromolecules, 2014, 47, 61–69 CrossRef CAS.
- (a) M. Langlais, I. Kulai, O. Coutelier and M. Destarac, Macromolecules, 2017, 50, 3524–3531 CrossRef CAS; (b) R. S. Varma and D. Kumar, Org. Lett., 1999, 1, 697–700 CrossRef CAS.
- R. O. McCourt, F. Dénes, G. Sanchez-Sanz and E. M. Scanlan, Org. Lett., 2018, 20, 2948–2951 CrossRef CAS PubMed.
- O. O. Fadeyi, J. J. Mousseau, Y. Feng, C. Allais, P. Nuhant, M. Z. Chen, B. Pierce and R. Robinson, Org. Lett., 2015, 17, 5756–5759 CrossRef CAS.
- V. T. Bhat, P. A. Duspara, S. Seo, N. S. B. Abu Bakar and M. F. Greaney, Chem. Commun., 2015, 51, 4383–4385 RSC.
- M.-Y. Cao, X. Ren and Z. Lu, Tetrahedron Lett., 2015, 56, 3732–3742 CrossRef CAS.
- (a) M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 31–42 RSC; (b) M. Salwiczek, E. K. Nyakatura, U. I. M. Gerling, S. Ye and B. Koksch, Chem. Soc. Rev., 2012, 41, 2135–2171 RSC; (c) T. Nakajima, J. Fluorine Chem., 2013, 149, 104–111 CrossRef CAS.
- (a) F. Yin and X.-S. Wang, Org. Lett., 2014, 16, 1128–1131 CrossRef CAS PubMed; (b) L. Zhu, G. Wang, Q. Guo, Z. Xu, D. Zhang and R. Wang, Org. Lett., 2014, 16, 5390–5393 CrossRef CAS PubMed; (c) J. Luo, Z. Zhu, Y. Liu and X. Zhao, Org. Lett., 2015, 17, 3620–3623 CrossRef CAS PubMed; (d) C. Xu and Q. Shen, Org. Lett., 2015, 17, 4561–4563 CrossRef CAS PubMed; (e) Q. Xiao, Q. He, J. Li and J. Wang, Org. Lett., 2015, 17, 6090–6093 CrossRef CAS PubMed; (f) T. Yang, L. Lu and Q. Shen, Chem. Commun., 2015, 51, 5479–5481 RSC.
- Y. Li, T. Koike and M. Akita, Asian J. Org. Chem., 2017, 6, 445–448 CrossRef CAS.
- K. Du, S.-C. Wang, R. S. Basha and C.-F. Lee, Adv. Synth. Catal. DOI:10.1002/adsc.201800999.
- M. Majek and A. J. von Wangelin, Chem. Commun., 2013, 49, 5507–5509 RSC.
- (a) G. A. Doherty, T. Kamenecka, E. McCauley, G. Van Riper, R. A. Mumford, S. Tong and W. K. Hegmann, Bioorg. Med. Chem. Lett., 2002, 12, 729–731 CrossRef CAS PubMed; (b) R. A. Hartz, A. G. Arvanitis, C. Arnold, J. P. Rescinito, K. L. Hung, G. Zhang, H. Wong, D. R. Langley, P. J. Gilligan and G. L. Trainor, Bioorg. Med. Chem. Lett., 2006, 16, 934–937 CrossRef CAS PubMed.
- M. Artico, R. Silvestri, E. Pagnozzi, B. Bruno, E. Novellino, G. Greco, S. Massa, A. Ettorre, A. G. Loi, F. Scintu and P. La Colla, J. Med. Chem., 2000, 43, 1886–1891 CrossRef CAS PubMed.
- D. H. Kim, J. Lee and A. Lee, Org. Lett., 2018, 20, 764–767 CrossRef CAS PubMed.
- M. Teders, A. Gómez-Suárez, L. Pitzer, M. N. Hopkinson and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 902–906 CrossRef CAS PubMed.
- X. Wang, G. D. Cuny and T. Noël, Angew. Chem., Int. Ed., 2013, 52, 7860–7864 CrossRef CAS PubMed.
- (a) M. Jiang, H. Li, H. Yang and H. Fu, Angew. Chem., Int. Ed., 2017, 56, 874–879 CrossRef CAS PubMed; (b) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS PubMed.
- Y. Cheng, J. Yang, Y. Qu and P. Li, Org. Lett., 2012, 14, 98–101 CrossRef CAS PubMed.
- (a) R. Ragno, A. Coluccia, G. La Regina, G. De Martino, F. Piscitelli, A. Lavecchia, E. Novellino, A. Bergamini, C. Ciaprini, A. Sinistro, G. Maga, E. Crespan, M. Artico and R. Silvestri, J. Med. Chem., 2006, 49, 3172–3184 CrossRef CAS PubMed; (b) G. La Regina, M. C. Edler, A. Brancale, S. Kandil, A. Coluccia, F. Piscitelli, E. Hamel, G. De Martino, R. Matesanz, J. F. Diaz, A. I. Scovassi, E. Prosperi, A. Lavecchia, E. Novellino, M. Artico and R. Silvestri, J. Med. Chem., 2007, 50, 2865–2874 CrossRef CAS PubMed.
- W. Guo, W. Tan, M. Zhao, K. Tao, L.-Y. Zheng, Y. Wu, D. Chen and X.-L. Fan, RSC Adv., 2017, 7, 37739–37742 RSC.
- R. Rahaman, S. Das and P. Barman, Green Chem., 2018, 20, 141–147 RSC.
- P. Sun, D. Yang, W. Wei, M. Jiang, Z. Wang, L. Zhang, H. Zhang, Z. Zhang, Y. Wang and H. Wang, Green Chem., 2017, 19, 4785–4791 RSC.
- Q. Shi, P. Li, Y. Zhang and L. Wang, Org. Chem. Front., 2017, 4, 1322–1330 RSC.
- (a) T. B. Johnson and I. B. Douglass, J. Am. Chem. Soc., 1939, 61, 2548–2550 CrossRef CAS; (b) N. L. Brock, A. Nikolay and J. S. Dickschat, Chem. Commun., 2014, 50, 5487–5489 RSC; (c) V. Aureggi and G. Sedelmeier, Angew. Chem., Int. Ed., 2007, 46, 8440–8444 CrossRef CAS PubMed; (d) M. D′hooghe, A. Waterinckx and N. De Kimpe, J. Org. Chem., 2005, 70, 227–232 CrossRef PubMed.
- K. R. Prabhu, A. R. Ramesha and S. Chandrasekaran, J. Org. Chem., 1995, 60, 7142–7143 CrossRef CAS.
- I. W. J. Still and F. D. Toste, J. Org. Chem., 1996, 61, 7677–7680 CrossRef CAS PubMed.
- (a) G. Hilt, S. Lüers and K. Harms, J. Org. Chem., 2004, 69, 624–630 CrossRef CAS PubMed; (b) H. Ishitani, S. Nagayama and S. Kobayashi, J. Org. Chem., 1996, 61, 1902–1903 CrossRef CAS.
- J. Santandrea, C. Minozzi, C. Cruché and S. K. Collins, Angew. Chem., Int. Ed., 2017, 56, 12255–12259 CrossRef CAS PubMed.
- W. Guo, W. Tan, M. Zhao, L. Zheng, K. Tao, D. Chen and X. Fan, J. Org. Chem., 2018, 83, 6580–6588 CrossRef CAS PubMed.
- (a) C. Chen and Y. J. Chen, Tetrahedron Lett., 2004, 45, 113–115 CrossRef CAS; (b) I. Hutchinson, M. F. G. Stevens and A. D. Westwell, Tetrahedron Lett., 2000, 41, 425–428 CrossRef; (c) N. Siddiqui, A. Rana, S. A. Khan, M. A. Bhat and S. E. Haque, Bioorg. Med. Chem. Lett., 2007, 17, 4178–4182 CrossRef CAS PubMed; (d) A. Mori, A. Sekiguchi, K. Masui, T. Shimada, M. Horie, K. Osakada, M. Kawamoto and T. Ikeda, J. Am. Chem. Soc., 2003, 125, 1700–1701 CrossRef CAS PubMed; (e) C. J. Lion, C. S. Matthews, G. Wells, T. D. Bradshaw, M. F. G. Stevens and A. D. Westwell, Bioorg. Med. Chem. Lett., 2006, 16, 5005–5008 CrossRef CAS PubMed.
- P. Natarajan, M. Manjeet, M. Muskan, N. K. Brar and J. Jot Kaur, Org. Chem. Front., 2018, 5, 1527–1531 RSC.
- (a) N. S. Li, J. K. Frederiksen and J. A. Piccirilli, Acc. Chem. Res., 2011, 44, 1257–1269 CrossRef CAS PubMed; (b) R. Xie, Q. Zhao, T. Zhang, J. Fang, X. Mei, J. Ning and Y. Tang, Bioorg. Med. Chem., 2013, 21, 278–282 CrossRef CAS PubMed; (c) T. S. Kumar, T. Yang, S. Mishra, C. Cronin, S. Chakraborty, J.-B. Shen, B. T. Liang and K. A. Jacobson, J. Med. Chem., 2013, 56, 902–914 CrossRef CAS PubMed; (d) J. A. Fraietta, Y. M. Mueller, D. H. Do, V. M. Holmes, M. K. Howett, M. G. Lewis, A. C. Boesteanu, S. S. Alkan and P. D. Katsikis, Antimicrob. Agents Chemother., 2010, 54, 4064–4073 CrossRef CAS PubMed; (e) A. M. Lauer and J. Wu, Org. Lett., 2012, 14, 5138–5141 CrossRef CAS PubMed; (f) A. M. Lauer, F. Mahmud and J. Wu, J. Am. Chem. Soc., 2011, 133, 9119–9123 CrossRef CAS PubMed; (g) D. Wang, J. Zhao, W. Xu, C. Shao, Z. Shi, L. Li and X. Zhang, Org. Biomol. Chem., 2017, 15, 545–549 RSC.
- J.-G. Sun, H. Yang, P. Li and B. Zhang, Org. Lett., 2016, 18, 5114–5117 CrossRef CAS PubMed.
- H. Zhang, Z. Zhan, Y. Lin, Y. Shi, G. Li, Q. Wang, Y. Deng, L. Hai and Y. Wu, Org. Chem. Front., 2018, 5, 1416–1422 RSC.
- (a) B. A. Frankel, M. Bentley, R. G. Kruger and D. G. McCafferty, J. Am. Chem. Soc., 2004, 126, 3404–3405 CrossRef CAS PubMed; (b) Y. Fang, Z. Luo and X. Xu, RSC Adv., 2016, 6, 59661–59676 RSC; (c) R. Mao, Z. Yuan, R. Zhang, Y. Ding, X. Fan and J. Wu, Org. Chem. Front., 2016, 3, 1498–1502 RSC; (d) Y. Liu, P. Xie, Z. Sun, X. Wo, C. Gao, W. Fu and T.-P. Loh, Org. Lett., 2018, 20, 5353–5356 CrossRef CAS PubMed.
- (a) R. Ettari, E. Nizi, M. E. Di Francesco, M.-A. Dude, G. Pradel, R. VičÍk, T. Schirmeister, N. Micale, S. Grasso and M. Zappalà, J. Med. Chem., 2008, 51, 988–996 CrossRef CAS PubMed; (b) J. Morales-Sanfrutos, A. Megia-Fernandez, F. Hernandez-Mateo, M. D. Giron-Gonzalez, R. Salto-Gonzalez and F. Santoyo-Gonzalez, Org. Biomol. Chem., 2011, 9, 851–864 RSC; (c) V. P. Sandanayaka, A. S. Prashad, Y. Yang, R. T. Williamson, Y. I. Lin and T. S. Mansour, J. Med. Chem., 2003, 46, 2569–2571 CrossRef CAS PubMed; (d) Q. Zhu and Y. Lu, Org. Lett., 2009, 11, 1721–1724 CrossRef CAS PubMed; (e) S. Mao, Y. R. Gao, X. Q. Zhu, D. D. Guo and Y. Q. Wang, Org. Lett., 2015, 17, 1692–1695 CrossRef CAS PubMed; (f) W. Wei, J. Li, D. Yang, J. Wen, Y. Jiao, J. You and H. Wang, Org. Biomol. Chem., 2014, 12, 1861–1864 RSC.
- S. Cai, Y. Xu, D. Chen, L. Li, Q. Chen, M. Huang and W. Weng, Org. Lett., 2016, 18, 2990–2993 CrossRef CAS PubMed.
- (a) S. I. Shirokawa, M. Kamiyama, T. Nakamura, M. Okada, A. Nakazaki, S. Hosowaka and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 13604–13605 CrossRef CAS PubMed; (b) J. P. Vidal, R. Escale, J. P. Girard, J. C. Rossi, J. M. Chantraine and A. Aumelas, J. Org. Chem., 1992, 57, 5857–5860 CrossRef CAS; (c) Y. M. Zhang, S. Cockerill, S. B. Guntrip, D. Rusnak, K. Snith, D. Vanderwall, E. Wood and K. Lackey, Bioorg. Med. Chem. Lett., 2004, 14, 111–114 CrossRef CAS; (d) J. Morales-Sanfrutos, A. Megia-Fernandez, F. Hernandez-Mateo, M. D. Giron-Gonzalez, R. Salto-Gonzalez and F. Santoyo-Gonzalez, Org. Biomol. Chem., 2011, 9, 851–864 RSC.
- (a) P. A. S. Smith and J. P. Horwitz, J. Am. Chem. Soc., 1950, 72, 3718–3722 CrossRef CAS; (b) N. M. Laso, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 1996, 37, 1605–1608 CrossRef CAS; (c) M. J. Burk, G. Casy and N. B. Johnson, J. Org. Chem., 1998, 63, 6084–6085 CrossRef CAS PubMed; (d) Z. Zhang, G. Zhu, Q. Jiang, D. Xiao and X. Zhang, J. Org. Chem., 1999, 64, 1774–1775 CrossRef CAS PubMed; (e) H. Zhao, C. P. Vandenbossche, S. G. Koenig, S. P. Singh and R. P. Bakale, Org. Lett., 2008, 10, 505–507 CrossRef CAS PubMed.
- D. Sun and R. Zhang, Org. Chem. Front., 2018, 5, 92–97 RSC.
- (a) A. El-Awa, M. N. NoShi, X. M. du Jourdin and P. L. Fuchs, Chem. Rev., 2009, 109, 2315–2349 CrossRef CAS PubMed; (b) A. N. R. Alba, X. Companyó and R. Rios, Chem. Soc. Rev., 2010, 39, 2018–2033 RSC.
- I. Churcher, D. Beher, J. D. Best, J. L. Castro, E. E. Clarke, A. Gentry, T. Harrison, L. Hitzel, E. Kay, S. Kerrad, H. D. Lewis, P. Morentin-Gutierrez, R. Mortishire-Smith, P. J. Oakley, M. Reilly, D. E. Shaw, M. S. Shearman, M. R. Teall, S. Williams and J. D. J. Wrigley, Bioorg. Med. Chem. Lett., 2006, 16, 280–284 CrossRef CAS PubMed.
- (a) X. Chen, S. Hussain, S. Parveen, S. Zhang, Y. Yang and C. Zhu, Curr. Med. Chem., 2012, 19, 3578–3604 CrossRef CAS PubMed; (b) F. Reck, F. Zhou, M. Girardot, G. Kern, C. J. Eyermann, N. J. Hales, R. R. Ramsay and M. B. Gravestock, J. Med. Chem., 2005, 48, 499–506 CrossRef CAS PubMed; (c) C. L. Percicot, C. R. Schnell, C. Debon and C. Hariton, J. Pharmacol. Toxicol. Methods, 1996, 36, 223–228 CrossRef CAS PubMed; (d) J. D. Buynak, V. R. Doppalapudi, A. S. Rao, S. D. Nidamarthy and G. Adam, Bioorg. Med. Chem. Lett., 2000, 10, 847–851 CrossRef CAS PubMed.
- (a) H.-H. Li, D.-J. Dong, Y.-H. Jin and S.-K. Tian, J. Org. Chem., 2009, 74, 9501–9504 CrossRef CAS PubMed; (b) K. Xu, V. Khakyzadeh, T. Bury and B. Breit, J. Am. Chem. Soc., 2014, 136, 16124–16127 CrossRef CAS PubMed; (c) E. F. Altenhofer and M. Harmata, J. Org. Chem., 2015, 80, 8168–8174 CrossRef CAS PubMed; (d) P.-X. Zhou, Y. Zhang, C. Ge, Y.-M. Liang and C. Li, J. Org. Chem., 2018, 83, 4762–4768 CrossRef CAS PubMed.
- N. K. Konduru, S. Dey, M. Sajid, M. Owais and N. Ahmed, Eur. J. Med. Chem., 2013, 59, 23–30 CrossRef CAS PubMed.
- T. Liu, Y. Li, L. Lai, J. Cheng, J. Sun and J. Wu, Org. Lett., 2018, 20, 3605–3608 CrossRef CAS PubMed.
- G. Solladié, C. Fréchou, G. Demailly and C. Greck, J. Org. Chem., 1986, 51, 1912–1914 CrossRef.
- A. L. Moure, R. G. Arrayás and J. C. Carretero, Chem. Commun., 2011, 47, 6701–6703 RSC.
- (a) G. Sarakinos and E. J. Corey, Org. Lett., 1999, 1, 1741–1744 CrossRef CAS PubMed; (b) J. K. Crandall and C. Pradat, J. Org. Chem., 1985, 50, 1327–1329 CrossRef CAS.
- S. K. Pagire, S. Paria and O. Reiser, Org. Lett., 2016, 18, 2106–2109 CrossRef CAS PubMed.
- (a) X. Gao, X. Pan, J. Gao, H. Huang, G. Yuan and Y. Li, Chem. Commun., 2015, 51, 210–212 RSC; (b) G. Yuan, J. Zheng, X. Gao, X. Li, L. Huang, H. Chen and H. Jiang, Chem. Commun., 2012, 48, 7513–7515 RSC.
- K. Omura, A. K. Sharma and D. Swern, J. Org. Chem., 1976, 41, 957–962 CrossRef CAS.
- (a) W. W. Epstein and F. W. Sweat, Chem. Rev., 1967, 67, 247–260 CrossRef CAS; (b) L. Chu, X. Yue and F. L. Qing, Org. Lett., 2010, 12, 1644–1647 CrossRef CAS PubMed; (c) F.-L. Liu, J.-R. Chen, Y.-Q. Zou, Q. Wei and W.-J. Xiao, Org. Lett., 2014, 16, 3768–3771 CrossRef CAS PubMed; (d) X. Ren, J. Chen, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 6725–6727 RSC; (e) Z. Zhang, Q. Tian, J. Qian, Q. Liu, T. Liu, L. Shi and G. Zhang, J. Org. Chem., 2014, 79, 8182–8188 CrossRef CAS PubMed; (f) G. A. Russell and S. A. Weiner, J. Org. Chem., 1966, 31, 248–251 CrossRef CAS; (g) H. Cao, S. Lei, N. Li, L. Chen, J. Liu, H. Cai, S. Qiu and J. Tan, Chem. Commun., 2015, 51, 1823–1825 RSC; (h) X. Gao, X. Pan, J. Gao, H. Jiang, G. Yuan and Y. Li, Org. Lett., 2015, 17, 1038–1041 CrossRef CAS PubMed.
- M.-H. Huang, C.-F. Zhu, C.-L. He, Y.-L. Zhu, W.-J. Hao, D.-C. Wang, S.-J. Tu and B. Jiang, Org. Chem. Front., 2018, 5, 1643–1650 RSC.
- Y. Li, T. Miao, P. Li and L. Wang, Org. Lett., 2018, 20, 1735–1739 CrossRef CAS PubMed.
- (a) A. Murakami, G. Gao, M. Omura, M. Yano, C. Ito, H. Furukawa, D. Takahashi, K. Koshimizu and H. Ohigashi, Bioorg. Med. Chem. Lett., 2000, 10, 59–62 CrossRef CAS PubMed; (b) J. F. Vasconcelos, M. M. Teixeira, J. M. Barbosa-Filho, M. F. Agra, X. P. Nunes, A. M. Giulietti, R. Ribeiro-dos-Santos and M. B. P. Soares, Eur. J. Pharmacol., 2009, 609, 126–131 CrossRef CAS PubMed; (c) K. V. Sashidhara, A. Kumar, M. Chatterjee, K. B. Rao, S. Singh, A. K. Verma and G. Palit, Bioorg. Med. Chem. Lett., 2011, 21, 1937–1941 CrossRef CAS PubMed.
- W. Yang, S. Yang, P. Li and L. Wang, Chem. Commun., 2015, 51, 7520–7523 RSC.
- (a) T. G. Back, K. N. Clary and D. Gao, Chem. Rev., 2010, 110, 4498–4553 CrossRef CAS PubMed; (b) J. Choi, P. Martín-Gago and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 12161–12165 CrossRef CAS PubMed; (c) A.-N. R. Alba, X. Companyó and R. Rios, Chem. Soc. Rev., 2010, 39, 2018–2033 RSC; (d) R. Ettari, L. Tamborini, I. C. Angelo, N. Micale, A. Pinto, C. De Micheli and P. Conti, J. Med. Chem., 2013, 56, 5637–5658 CrossRef CAS PubMed; (e) E. Dunny, W. Doherty, P. Evans, J. P. G. Malthouse, D. Nolan and A. J. S. Knox, J. Med. Chem., 2013, 56, 6638–6650 CrossRef CAS PubMed; (f) P. Chauhan, C. Hadad, A. H. López, S. Silvestrini, V. La Parola, E. Frison, M. Maggini, M. Prato and T. Carofiglio, Chem. Commun., 2014, 50, 9493–9496 RSC; (g) R. van der Westhuyzen and E. Strauss, J. Am. Chem. Soc., 2010, 132, 12853–12855 CrossRef CAS PubMed.
- H. Wang, Q. Lu, C.-W. Chiang, Y. Luo, J. Zhou, G. Wang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 595–599 CrossRef CAS PubMed.
- M. Artico, R. Silvestri, S. Massa, A. G. Loi, S. Corrias, G. Piras and P. La Colla, J. Med. Chem., 1996, 39, 522–530 CrossRef CAS PubMed.
- N.-W. Liu, K. Hofman, A. Herbert and G. Manolikakes, Org. Lett., 2018, 20, 760–763 CrossRef CAS PubMed.
- H. Yue, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 1371–1375 CrossRef CAS.
- S. K. Pagire, A. Hossain and O. Reiser, Org. Lett., 2018, 20, 648–651 CrossRef CAS.
- (a) J. Zhou, G.-L. Zhang, J.-P. Zou and W. Zhang, Eur. J. Org. Chem., 2011, 3412–3415 CrossRef CAS; (b) H. Huang, J. Ash and J. Y. Kang, Org. Biomol. Chem., 2018, 16, 4236–4242 RSC; (c) Y. Moon, Y. Moon, H. Choi and S. Hong, Green Chem., 2017, 19, 1005–1013 RSC; (d) J.-G. Sun, W.-Z. Weng, P. Li and B. Zhang, Green Chem., 2017, 19, 1128–1113 RSC; (e) L. Wang, S. Yang, L. Chen, S. Yuan, Q. Chen, M.-Y. He and Z.-H. Zhang, Catal. Sci. Technol., 2017, 7, 2356–2361 RSC.
- (a) B. Pieber, M. Shalom, M. Antonietti, P. H. Seeberger and K. Gilmore, Angew. Chem., Int. Ed., 2018, 57, 9976–9979 CrossRef CAS PubMed; (b) A. Hu, Y. Chen, J.-J. Guo, N. Yu, Q. An and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 13580–13585 CrossRef CAS PubMed.
- C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. Andy Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC.
- (a) J. Xu, Z. Wu, H. Wan, G. Deng, B. Lu, A. K. Eckhardt, P. R. Schreiner, T. Trabelsi, J. S. Francisco and X. Zeng, J. Am. Chem. Soc., 2018, 140, 9972–9978 CrossRef CAS PubMed; (b) G. Tan and X. Wang, Chin. J. Chem., 2018, 36, 573–586 CrossRef CAS; (c) Z. Wu, J. Xu, G. Deng, X. Chu, L. Sokolenko, T. Trabelsi, J. S. Francisco, A. K. Eckhardt, P. R. Schreiner and X. Zeng, Chem. – Eur. J., 2018, 24, 1505–1508 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © the Partner Organisations 2019 |