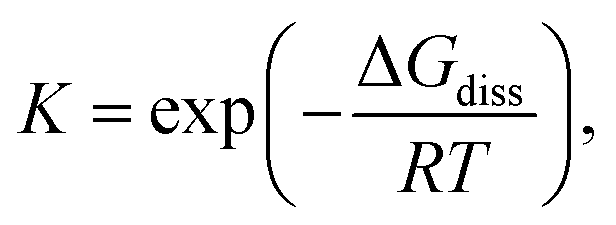DOI:
10.1039/C9QM00488B
(Review Article)
Mater. Chem. Front., 2019,
3, 2270-2282
Dynamic covalent bonds: approaches from stable radical species
Received
29th July 2019
, Accepted 6th September 2019
First published on 9th September 2019
Abstract
Reversible covalent bond formation/scission systems, referred to as Dynamic Covalent Chemistry (DCC), have received significant interest in view of the molecular systems, which offer feasible “error-correction” of the targeted chemical structures and the rearrangement of chemical bonds into the proper manner during the synthetic processes. DCC has been widely designed and developed with molecules in chemical equilibria where a set of reactants and a product are both in closed shell molecules. Within a few years, the concept of DCC systems has been extended to the formation of a covalent bond between a set of stable radicals, utilizing a common feature of radical species: the ease of the bond cleavage and formation. Generally, the coupling reactions among radical species are thermodynamically favorable in a down-hill manner with no or extremely small energetic barriers, and the barrier of the dissociation reactions can be minimized by properly designing the radical species. This review highlights the examples of the radicals showing reversible oligomerization–dissociation behavior, which have been or will potentially be utilized as building blocks in DCC, and the recent development of molecular self-assembly based on reversible radical coupling and homolytic cleavage reactions.

Daisuke Sakamaki
| Daisuke Sakamaki received his Master's degree in 2010 and PhD degree in 2013 under the supervision of Prof. Kazuyoshi Tanaka from Kyoto University. After completing his post-doctoral research at the group of Prof. Shu Seki, he became an assistant professor at the same group. In 2018, he moved to Osaka Prefecture University as an assistant professor of the group of Prof. Hideki Fujiwara. His research interests involve the synthesis of functional π-conjugated molecules and novel organic radicals. |

Samrat Ghosh
| Samrat Ghosh obtained his Master's degree from Banaras Hindu University, India in 2011 and subsequently PhD degree under the supervision of Prof. Ayyappanpillai Ajayaghosh in 2018 from CSIR-NIIST, India. After that he joined the research group of Prof. Shu Seki as a program-specific researcher and is currently continuing his research as a JSPS post-doctoral fellow in the same laboratory. His research interest now focuses on the design and synthesis of π-conjugated covalent organic frameworks and studying their charge dynamics. |

Shu Seki
| Shu Seki received his Bachelor's from the University of Tokyo in 1993, and his PhD in 2001 from Osaka University. He has worked as a researcher at the Argonne National Laboratory, USA (1993), and the Delft University of Technology, Netherlands (2001). He joined Osaka University as Assist. Prof. (1995–2001), Assoc. Prof. (2002–2009) and full professor (2009–2015). He was appointed as a fellow of Royal Society of Chemistry in 2017. Since 2015 he has been appointed as full professor in Kyoto University. He focuses his research on physical chemistry of condensed matter and functional organic materials. |
1. Introduction
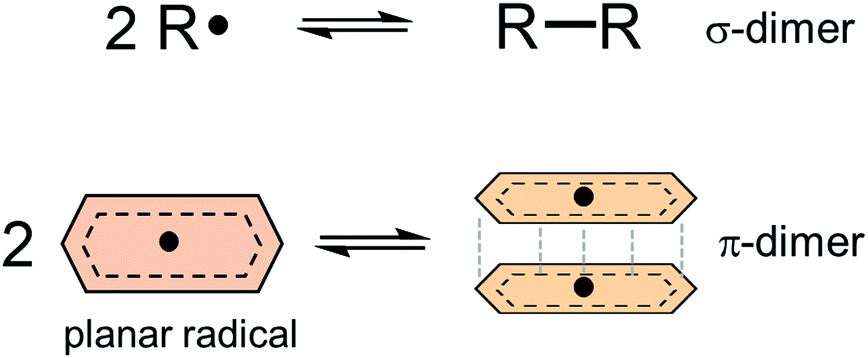
Dynamic covalent chemistry (DCC) is a field of research that treats a molecular system constituted through reversible exchange of components by repeating the chemical bond cleavage and formation of covalent bonds.1 DCC has attracted wide interest in recent years because of its potential for selective construction of the thermodynamically most stable product by self-assembly processes.2–5 Various types of reversible reactions have been utilized as a motif of DCC, such as aldol reaction,2,3 ester/amide/imine formation,3,5 olefin- or alkyne-metathesis,6,7 and boronic acid condensation.8 Most of the DCC systems require catalysts to achieve rapid equilibrium due to the intrinsically high activation energies coming from the robustness of covalent bonds compared to supramolecular systems derived from non-covalent interactions.5 As a new class of DCC motifs, organic radicals9–13 showing reversible association/dissociation reactions are very promising. Association of a set of radicals can be categorized into the following two types: σ-dimerization and π-dimerization14 (Fig. 1). The former involves the formation of a covalent (σ) bond between two radicals and is most commonly observed in the oligomerization of radicals. The latter gives a closely stacked pair of radicals via π-orbitals and is typically observed in the association of planar π-conjugated radicals (or radical ions15). Radical-based DCC has been implemented by such reversible inter-radical associations. A major advantage of the radical-based DCC is the facility and simplicity of the radical–radical association/dissociation reactions without the need for catalysts and formation of byproducts. In addition, DCC systems based on radicals can be reached at a reversible equilibrium under mild conditions compared with other DCC systems because of the relatively small energy of the bond formation (bond dissociation enthalpy, BDE, ΔHdiss) between the radicals. Of course, most radicals easily react with molecular oxygen, moisture, and other surrounding molecules, and this instability of radicals hampers the application of radicals in DCC. However, properly designed radicals can act as building blocks of DCC even under ambient conditions. For the design of DCC radicals, BDE is a primary parameter for the equilibrium of a set of DCC radicals, as well as the bond dissociation entropy (ΔSdiss). A typical value of ΔHdiss ranges from several tens of kJ mol−1 to a hundred and several tens of kJ mol−1 for a DCC system, and the values are very small compared with ΔHdiss of the usual C–C single bond (∼350 kJ mol−1). The standard Gibbs free energy for the dissociation (ΔGdiss) and the equilibrium constant (K) are given as follows:
| ΔGdiss = ΔHdiss − TΔSdiss |
and
where R is the gas constant. The most common method for evaluating ΔHdiss and ΔSdiss is the van’t Hoff plot based on the temperature dependence of K. The values of K at each temperature can be calculated from the concentration of the dissociated radical at equilibrium. Electron spin resonance (ESR) is the most straightforward way to measure the concentration of radical species quantitatively. Because the dissociated radical species show characteristic electronic absorption bands in many cases, electronic absorption measurements are often a good choice.
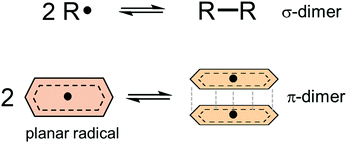 |
| | Fig. 1 Schematic representations of σ- and π-dimerization of radicals. | |
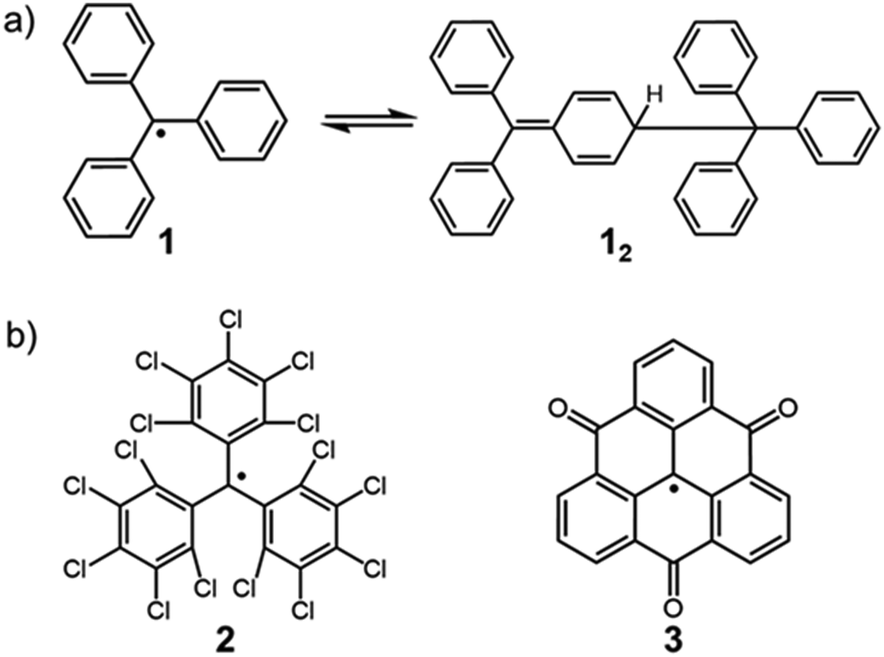
The chemistry of stable organic radicals dates to the discovery of the triphenylmethyl radical (1, Fig. 2a) by Gomberg in 1900,16 and 1 was recognized to be in equilibrium with the σ-dimer in solution. In that context, this radical could be regarded also as the first DCC radical. However, most radicals including 1 easily undergo irreversible reactions other than the reversible self-association, and such radicals are unfavorable for practical use in DCC. It should be noted that most of the established radicals in a monomer–dimer equilibrium have been found accidentally, and not as the product of intended design. We can find some common characteristics for such radicals from a retrospective viewpoint. As a general strategy for stabilizing radicals, the following two approaches have been adopted: the first is the introduction of bulky substituents by protecting the spin center (kinetic stabilization); the second is the facilitation of the spin delocalization by expanding the π-system and/or introducing heteroatoms that can participate in the spin delocalization (thermodynamic stabilization). Until now, a number of studies aimed at isolation of radicals and hence revealing their physico-chemical nature have been carried out based on these strategies. One of the guiding principles for designing radicals, that are suitable for DCC should have “moderate” stabilization. Herein, we explain this concept using triphenylmethyl radicals as an example. The structure of the dimer of triphenylmethyl radical 1 is the head-to-tail structure as shown in Fig. 2a.17,18 Because 1 is absolutely reactive toward molecular oxygen or other chemicals, a great deal of research has been carried out for preparing more stable derivatives of 1. It was found that the stability of the radical increased dramatically via kinetic stabilization by introducing substituents on the phenyl groups at the ortho- and para-positions to the central methyl radical, and various derivative radicals such as tris(pentachlorophenyl)methyl radical (2) were successfully isolated as pure chemical entities (Fig. 2b).19 For radicals contributing DCC, thermodynamic stabilization of the dissociated radical is a key factor to facilitate the dissociation of the dimer and shift the equilibrium to the radical form. Recently, Morita et al. synthesized trioxoriangulene without substituents (3) and proved that this radical is an intrinsically stable radical that does not undergo σ-dimerization in solution and the solid state (Fig. 2b).20 Both 2 and 3 could be regarded as extreme cases of kinetically and thermodynamically stabilized triphenylmethyl radicals, respectively; these radicals lost the habit of σ-dimerization of 1. In order to impart DCC properties to a radical, the prerequisite is not only to suppress the undesired side reactions but also to facilitate the reversible oligomerization reaction. These requirements can be fulfilled and achieved rationally by designing the structure of the dissociated radical. This review highlights the examples of the radicals showing reversible σ-bond formation/dissociation behavior, which have been or potentially can be utilized as building blocks in DCC. The radicals are classified according to the type of covalent bonds formed on the association. In particular, we focus on the radical units whose thermodynamic parameters of the equilibrium have been elucidated to date. This review also includes examples of self-assembly of DCC radicals forming macrocycles or other complex superstructures. The systems involving bond dissociation triggered by photo-irradiation or redox stimuli, and the reactions of radical ions are postponed in the other review discussion on the properties of DCC systems.
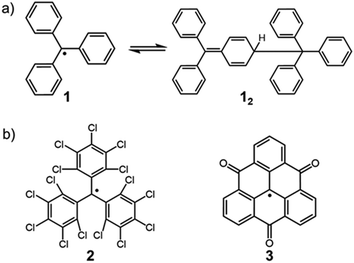 |
| | Fig. 2 (a) Triphenylmethyl radical (1) and the equilibrium between its dimer (12). (b) Extreme examples of a kinetically stabilized (2) and thermodynamically stabilized (3) triphenylmethyl radical. | |
2. Examples of radicals showing DCC character
2.1 Carbon-centered radicals
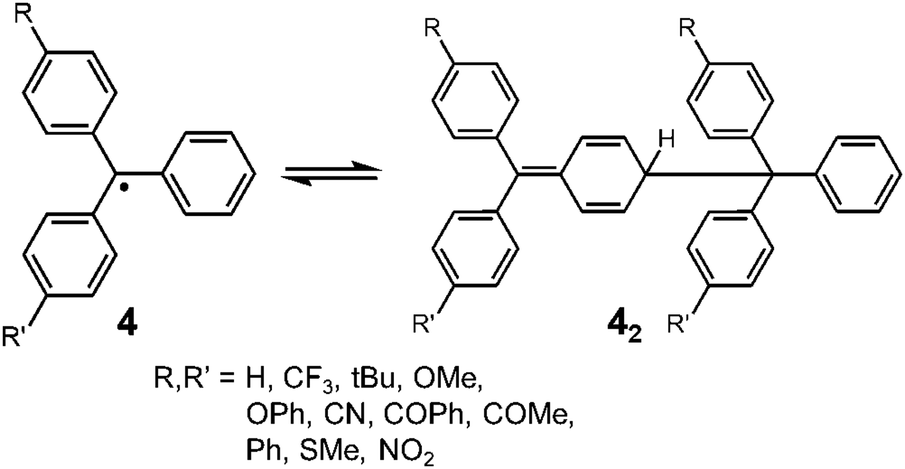
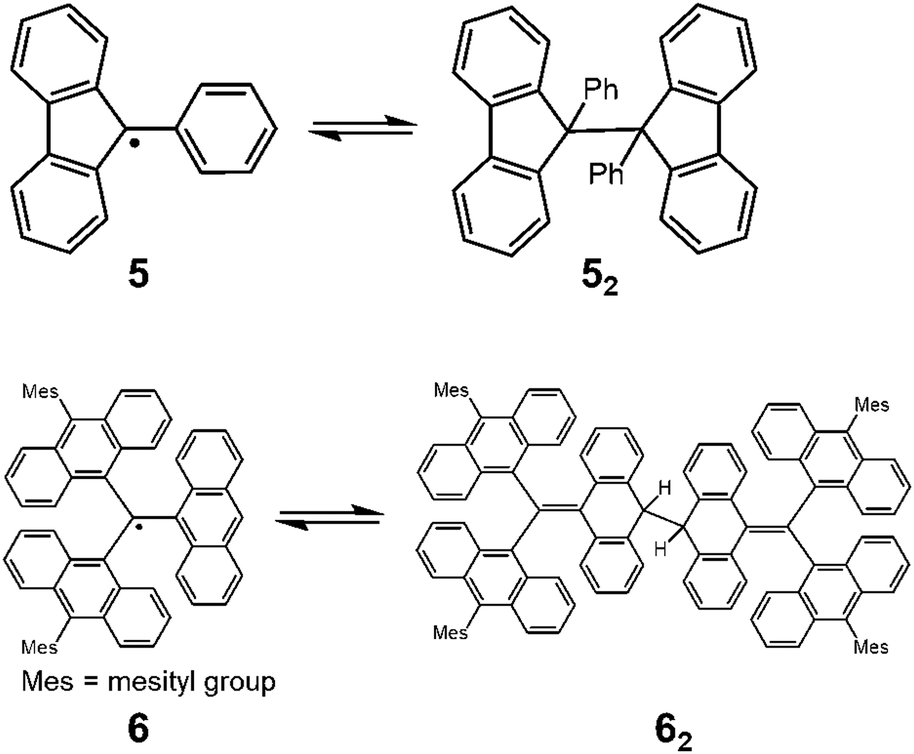
Since the discovery of Gomberg's radical 1, numerous carbon-centered radicals have been prepared and among them, the dynamic properties of the radicals being in monomer–dimer equilibrium have been continuously studied. Neumann et al. carried out an exhaustive research study to investigate the effect of para-substitution of 1 on its monomer–dimer equilibrium.21 They have synthesized 10 compounds of 4-mono-substituted and 24 compounds of 4,4′-disubstituted 1 (4), and evaluated the monomer–dimer equilibrium constants of them in benzene solutions by ESR measurements (Fig. 3). The following para-substituents: CF3, tBu, OMe, OPh, CN, COPh, COMe, Ph, SMe, and NO2, were examined comprehensively in their work. They found that all substituents caused facilitated dissociation of the dimer, hence stabilizing the radical form. Among the substituents, Ph and SMe groups most effectively facilitated dissociation to the corresponding radical forms. The BDE of the dimer of 4 ranged from 31 to 39 kJ mol−1. Fluorenyl radicals have been investigated as other examples of hydrocarbon radicals existing in a reversible monomer–dimer equilibrium.22–24 The BDE of the dimer of 9-phenylfluorene radical (52, Fig. 4) was evaluated to be 63.6 kJ mol−1 by Scaiano et al.24 Recently, Kubo and co-workers reported novel dibenzo-fluorenyl radicals25 and a tri(anthryl)methyl radical (6)26 forming σ-dimers reversibly, being regarded as π-extended analogues of 1. Hydrocarbon radicals including these radicals and phenalenyl radicals were discussed in detail in a recent review by Kubo.27
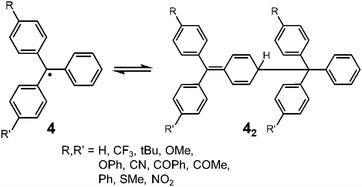 |
| | Fig. 3 Equilibrium between 4-substituted and 4,4′-disubstituted triphenylmethyl radicals and their dimers. | |
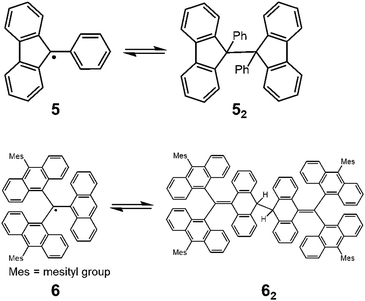 |
| | Fig. 4 Equilibria of 9-phenylfluorenyl radicals (5) and tri(9-anthryl)methyl radicals (6) between their dimers. | |
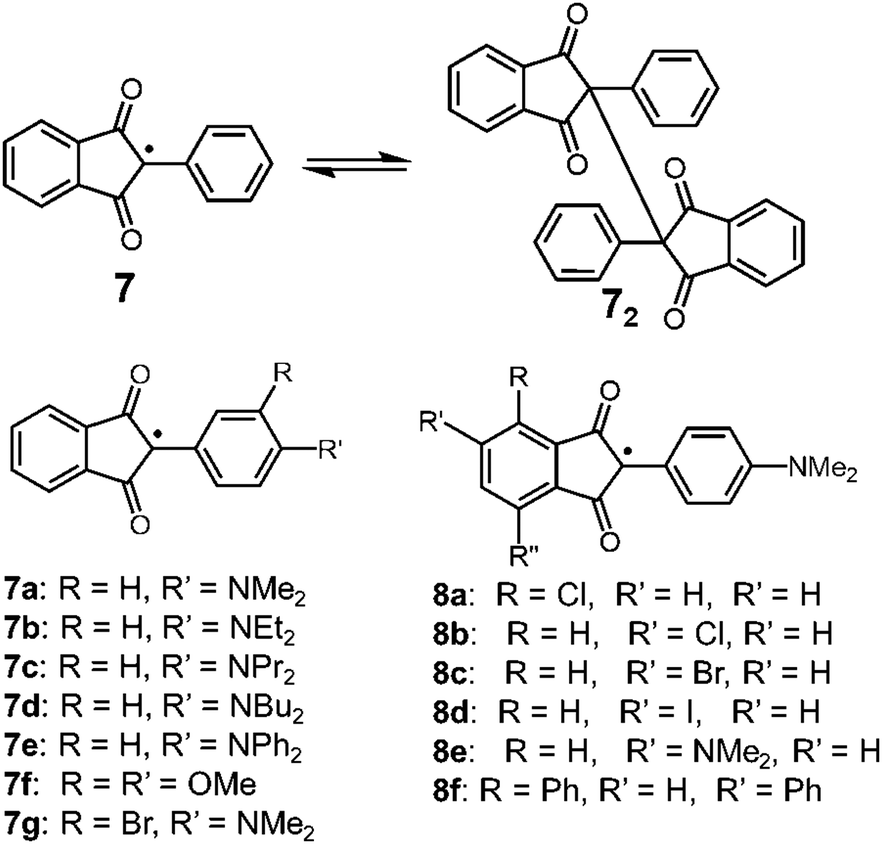
Carbon-centered radicals with adjacent carbonyl groups have relatively higher stability due to the spin delocalization over the carbonyl groups. Among this class, radicals with an arylindandione skeleton are known to show reversible dimerization–dissociation behavior (Fig. 5). In 1963, Beringer et al. reported the thermal homolysis of the dimer of 2-phenyl-1,3-indandione (72) to give two 2-phenyl-1,3-indandione radicals (7).28 However, the homolysis of the dimer only occurred above 373 K and the dissociated radical easily decomposed by abstracting hydrogen from the solvent or recombining in a different manner irreversibly. In 1979, Khudyakov, Yasmenko, and Kuzmin reported a comprehensive work on the thermodynamic parameters of the monomer–dimer equilibria of seven derivatives of 7 with substituents on the aryl unit (7a–7g).29 The chlorobenzene solutions of the dimers of 7a–7d having a 4-dialkyllamino substituent showed electronic absorption bands around 700 nm derived from the dissociated radicals even at room temperature along with the ESR signals, and the intensities of the absorption and ESR signals increased upon heating. The dimer of 7e having a 4-diphenylamino substituent also dissociated in solution when the temperature was increased, but the equilibrium was strongly shifted toward the dimer form compared to 7a–7d. On the other hand, the dimer with 3,4-dimethoxyphenyl groups (7f2) did not show either radical absorption bands or ESR signals even at 393 K. Furthermore, the introduction of a bromine atom to 7a at the ortho-position of the dimethylamino group (7g) completely inhibited the dissociation of the dimer. The robustness of the dimer of 7g was explained by the steric hindrance form the bromine atom decreasing the coplanarity between the phenyl ring and the dimethylamino group, resulting in destabilization of the dissociated form 7g. The effect of the substitution at the phthaloyl ring of 7a was also investigated (8a–8f), and it was found that the introduction of electron accepting substituents on the phthaloyl ring facilitates the dissociation, and the introduction of electron donating substituents stabilizes the dimer forms.30,31 The solid samples of the dimers of 7a and 8a–8f exhibited mechanochromism by applying shear stress, resulting from the dissociation of the intermonomer C–C bond by mechanical stimuli.32 The yellow crystalline powder of these dimers turned green after grinding in a mortar and pestle or a ball mill, and the ESR intensity increased by a hundred times relative to the initial powder containing a small amount of free radicals. Interestingly, these dimers did not show mechanochromism by applying hydrostatic pressure, whereas they responded to shear and grinding stress.
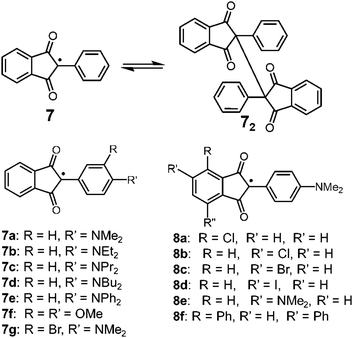 |
| | Fig. 5 2-Phenyl-1,3-indandione radical (7) and its derivatives. | |
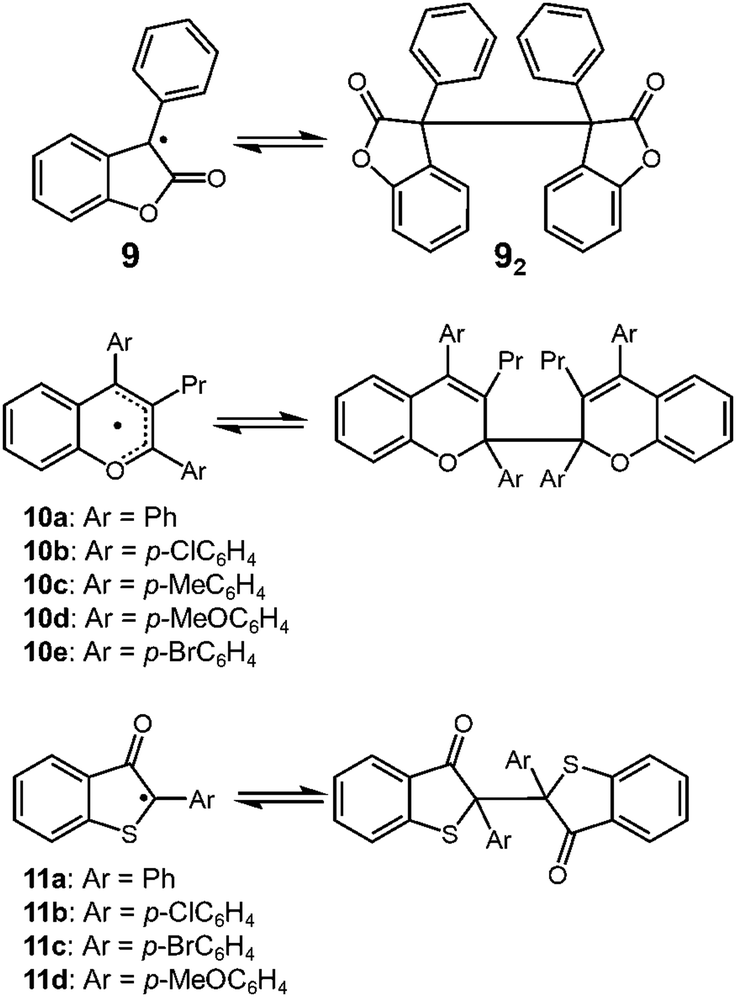
Lactone-derived radicals, such as 3-phenyl-2-coumaranone (3-phenyl-2(3H)-benzofuranone) 9 also show reversible dimerization–dissociation behavior, and have been investigated by some different research groups in the past (Fig. 6). The thermochromism of the dimers of 9 (92) and its derivatives has been reported in the 1920's.33 Toluene or chloroform solution of 92 showed a pale-blue color at room temperature, and when cooled in an ice-bath, it turned colorless.34 In 2004, Scaiano and co-workers reported the thermodynamic parameters of the monomer–dimer equilibrium of 9, and they evaluated the BDE of 72 in toluene to be 98.8 kJ mol−1.24 Some other examples of heterocyclic carbon-centered radicals showing DCC behavior, 2,2′-bis(2,3,4-triarlychromeny)s (10a–10e)35,36 and 2,2′-bis(2-arly-3-benzothiophenonyl)s (11)37–40 have been reported and these dimers also dissociate into radicals by mechanical stimuli.
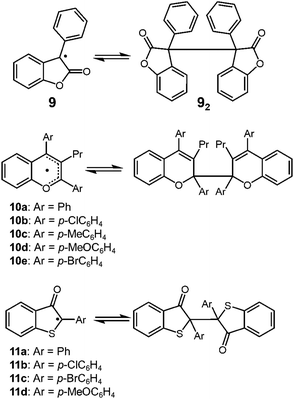 |
| | Fig. 6 Carbon-centered radicals with an adjacent carbonyl group or oxygen atom being in reversible equilibrium with the dimers. | |
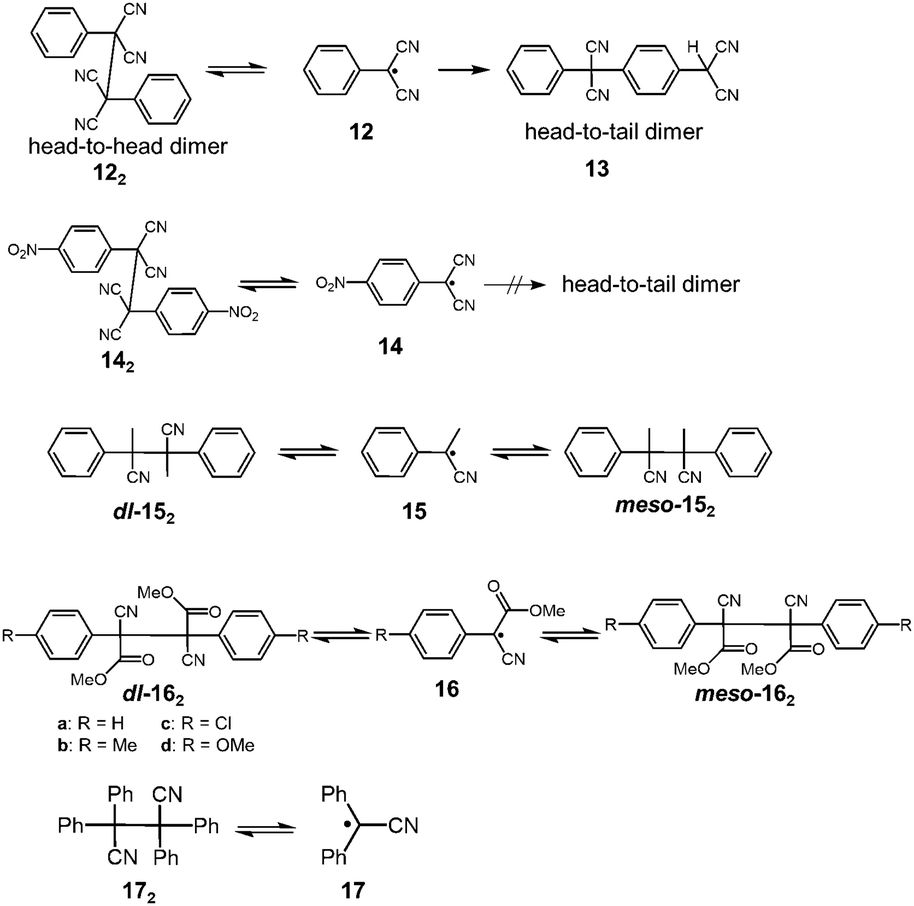
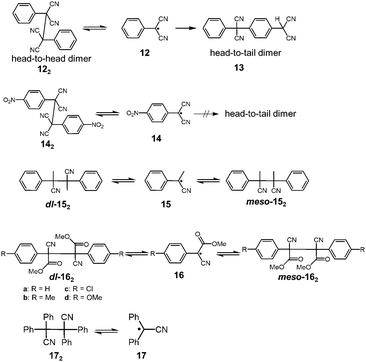
It has been a long-known fact that the radicals substituted with cyano groups are relatively unreactive toward molecular oxygen or other chemicals. Until now, several cyano-substituted radicals showing reversible dimerization have been reported. In 1966, Hartzler reported that the oxidation of phenylmalononitrile with potassium ferricyanide gave the dicyanomethylphenyl radical 12, and the generated radical immediately dimerized to afford 1,2-diphenyl-1,1,2,2,-tetracyanoetane (122) nearly quantitatively (Fig. 7).41 The acetonitrile solution of 122 showed a weak ESR signal and a pink coloration of the solution was observed42 when heated at 343 K, indicating the partial dissociation of 122 into 12 by heating. However, 122 completely isomerized to 13 in solution within a day at room temperature. The formation of 13 is irreversible due to the head-to-tail coupling of 12. The unfavored “dead-end“ coupling such as the isomerization of 12 to 13 should be prohibited when applied in DCC. Hartzler also synthesized 4-nitro substituted analogue 14, which does not undergo head-to-tail coupling. However, the equilibrium between 14 and the head-to-head dimer 142 strongly favored the dimeric form as well as 12. Furthermore, 12 and 14 abstracted a hydrogen atom from the solvent (e.g. tetrahydrofuran) and readily reacted with other organic radicals such as 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical. In the following year, Peterson reported the mechanistic study of the dl–meso isomerization of 2,3-dimethyl-2,3-diphenylsuccinonitrile (152) by thermal activation.43 Peterson observed that heating of either the dl or meso isomer of 152 in o-dichlorobenzene quickly gave the mixture of dl and meso isomers at 423 K, and this isomerization proceeded by homolytic scission of 152 to the radical 15, followed by recombination of 15. In the early 1970's, De Jongh et al. reported a systematic study on the dissociation behaviors of the products of oxidative coupling of various α-substituted benzyl cyanides, and in their work, they also demonstrated the thermal interconversion between dl- and meso-162 in refluxing toluene or acetic acid.44 The activation enthalpies for the dissociation of 16a and 16b were evaluated to be ca. 96 kJ mol−1. Tetraphenylsuccinonitrile (172) is also known to be in equilibrium with the dissociated radical (17). Scaiano et al. calculated the BDE of 172 to be 110 kJ mol−1.24
 |
| | Fig. 7 Cyanomethyl radicals being in reversible equilibrium with the dimers. | |
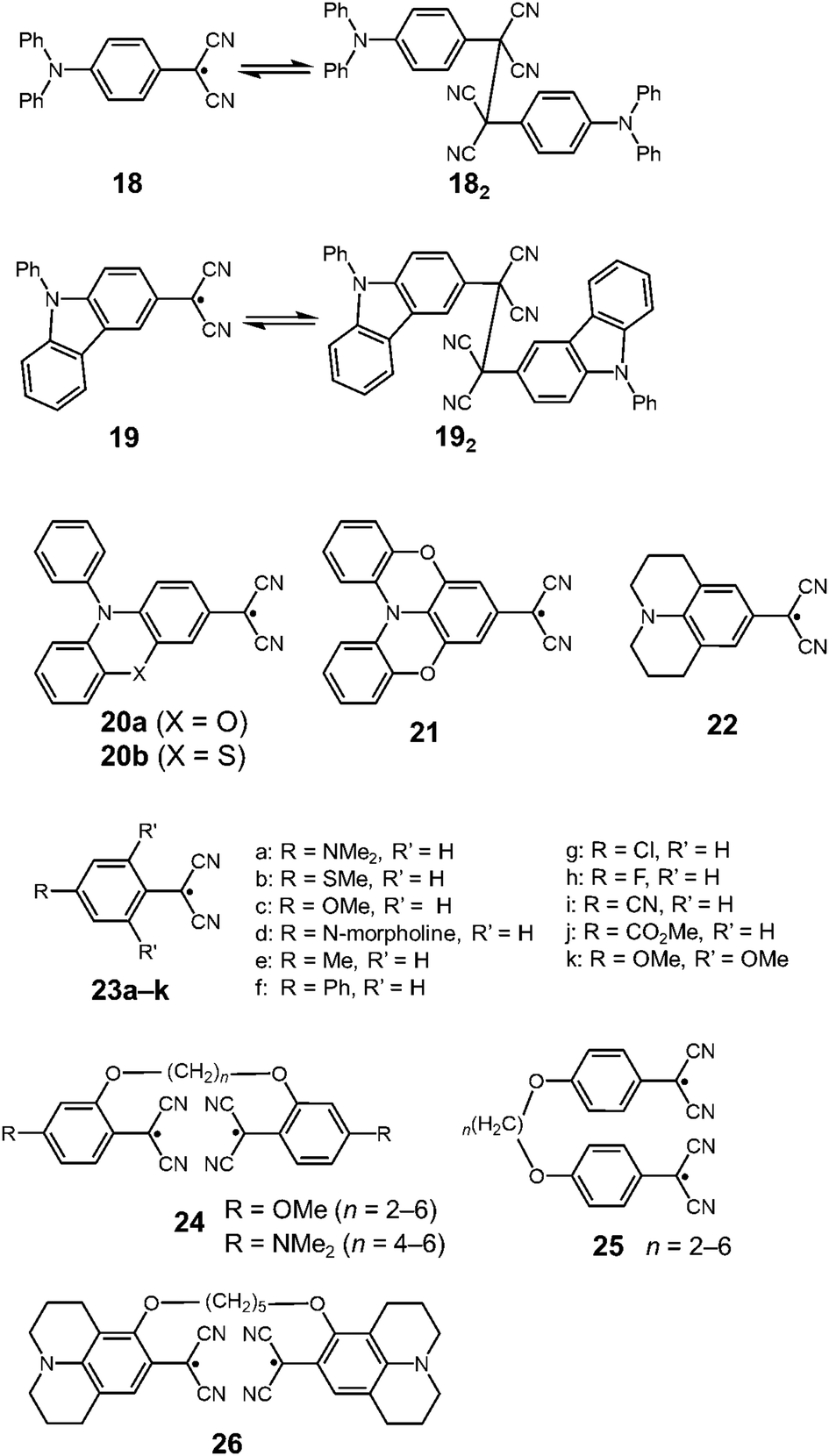
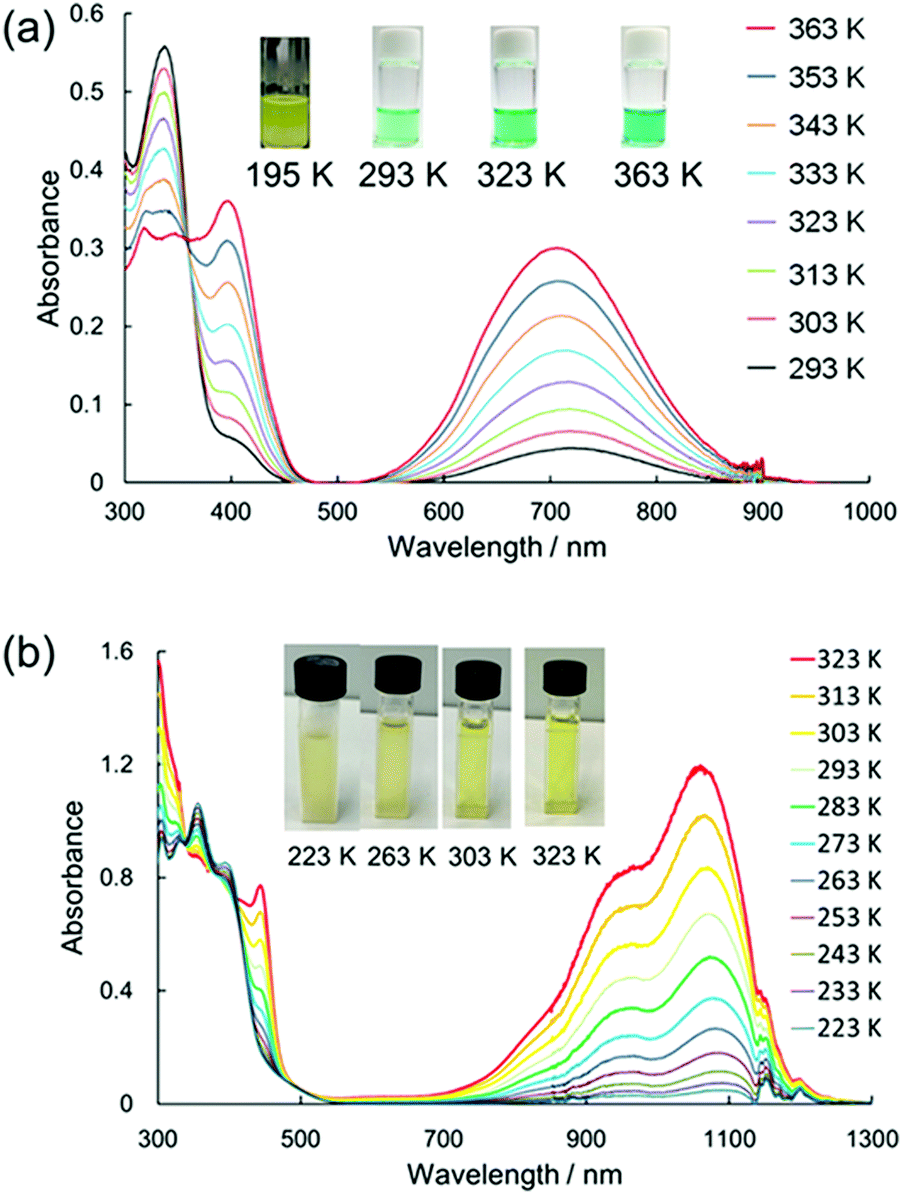
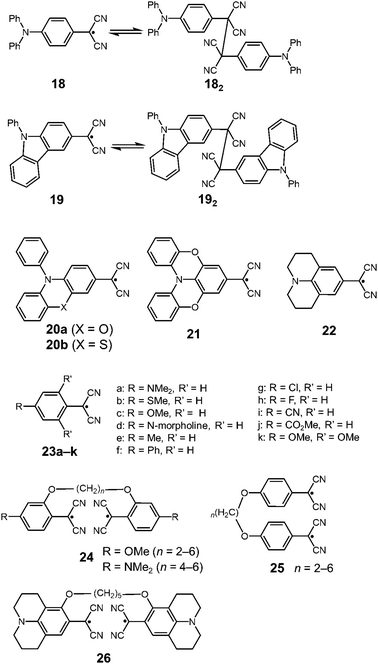
In 2016, Sakamaki and Seki et al. demonstrated that the introduction of an amino nitrogen at the para-position of 12 can significantly stabilize the dissociated form and facilitate the dissociation of the dimer.45 In this work, two dicyanomethyl radicals conjugated with triphenyl amine (18) and carbazole (19) were synthesized (Fig. 8). These radicals existed as the σ-bonded dimers (182 and 192) in the solid state, and the structures of the dimers were revealed by X-ray single crystal analysis. The solutions of 182 and 192 showed clear ESR signals, and had green and blue colors, respectively, resulting from the dissociated radicals even at 293 K. These solutions exhibited drastic thermochromism from almost colorless to deep green or blue over a wide temperature range (Fig. 9a). In contrast to the previously reported dicyanomethyl radicals, 18 and 19 do not react with DPPH. The increased stabilities of these radicals could be attributed to the thermodynamic stabilization by the spin-delocalization onto the amino nitrogen atom. The BDEs of 182 and 192 were calculated to be 55 and 75 kJ mol−1, respectively, and these values were significantly lower than those of the previously reported this class of radicals. The same research group tested the DCC properties of the various para-amino substituted dicyanomethyl radicals, and found that the DFT-calculated spin density on the central carbon of the dicyanomethyl group (ρC) correlated with the BDE; a radical with a lower ρC tends to have a smaller BDE.46,47 Among the radicals tested, the dicyanomethyl radicals with a partially planarized triphenylamine skeleton by insertion of sulfur or oxygen atoms (20a, 20b, and 21) exhibited interesting near-infrared thermochromism.47 The absorption maxima of these radicals were observed at 1050 nm (20a), 934 nm (20b), and 1059 nm (21), whereas other dicyanomethyl radicals had their radical absorption bands in the visible region (Fig. 9b). Due to the remarkable bathochromic shifts of the radical bands, the solutions of these radicals had a wide transparent range in the visible region, and therefore the solutions showed little color changes by temperature variation, whereas the NIR absorption intensities drastically changed. Furthermore, a series of works by the same research group revealed that the dicyanomethyl radical with a julolidine skeleton (22) instead of forming the σ-dimer both in solution and the solid state forms the π-stacked dimer.46 In 2017, Winter and co-workers published a systematic investigation of the substituent effect on the dicyanomethyl radicals.48 They introduced 10 different functional groups to the para-position of the dicyanomethylphenyl radical (23a–k), and also synthesized 2,4,6-trimethoxyphenyldicyanomethyl radicals to test the effect of the additional substituents at the ortho-positions. All these radicals were in equilibrium with the σ-dimers in solution, and characteristic thermochromism depends on the para-substituent. The thermodynamic parameters for self-dimerization were evaluated for all the radicals, and they concluded that the Hammett parameter of the para-substituent linearly correlated with the binding constant (Ka); electron-donating substituents weaken the σ-dimer, whereas electron-withdrawing substituents strengthen the σ-dimer. Among the 11 radicals, the 2,4,6-trimethoxyphenyldicyanomethyl radical (23k) had the smallest Ka value and BDE of 1.9 × 105 and 48 kJ mol−1, respectively due to the steric repulsions between the ortho-substituents in the σ-dimer. They also prepared alkyl-tethered dimers of dicyanobenzyl radicals (24–26) and revealed that 24 and 25 undergo intramolecular σ-dimerization and 26 undergoes intramolecular π-dimerization, rather than forming higher oligomers.49
 |
| | Fig. 8 Recent examples of dicyanomethyl radicals. | |
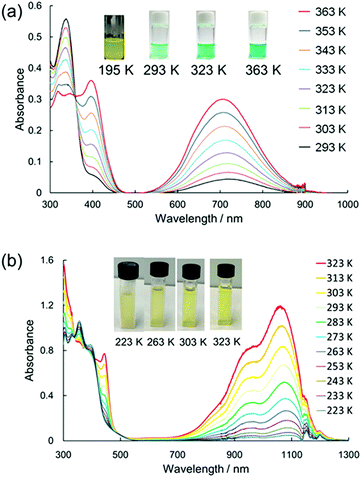 |
| | Fig. 9 Temperature dependence of the absorption spectra and the colour changes of (a) 18 and (b) 21 in toluene. Reprinted with permission from ref. 47. Copyright 2019 American Chemical Society. | |
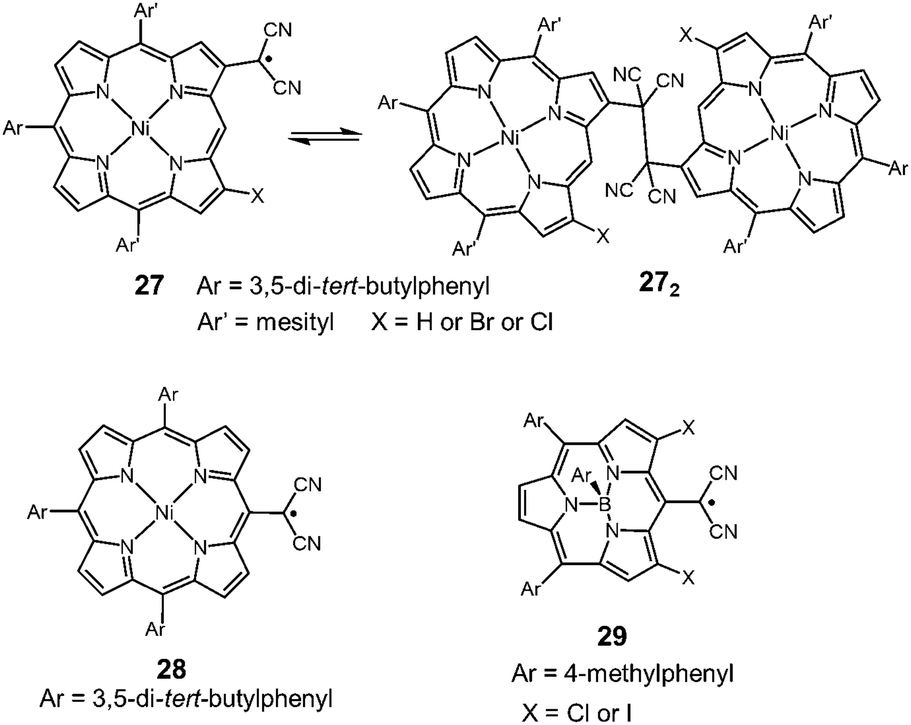
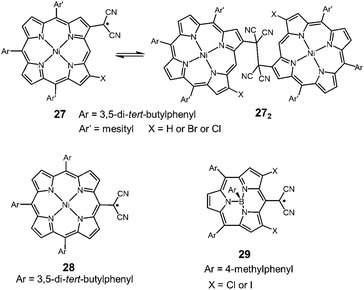
Very recently, Osuka and co-workers synthesized two types of porphyrins bearing a dicyanomethyl radical at the β-position (27) or meso-position (28) of the porphyrin ring (Fig. 10).50 The β-substituted porphyrin 27 was in equilibrium with the σ-dimers in solution, and the σ-dimer formation in the solid state was unequivocally confirmed by X-ray single crystal analysis. On the other hand, the meso-substituted porphyrin 28 did not show any dimerization behaviors and existed as a monomeric radical both in solution and the solid state. The difference in the dynamic character was explained by the spin density distribution. The spin on 28 was fully delocalized over the porphyrin skeleton, leading to the stabilization of the dissociated radical form, whereas the spin on 27 was relatively localized on the dicyanomethylene moiety and a part of the porphyrin skeleton. This is probably the first example showing that the difference of the substitution pattern qualitatively changed the self-association behavior of dicyanomethyl radicals with a common backbone. Osuka et al. also synthesized subporphyrins bearing a dicyanomethyl radical at the meso-position (29).51 The dicyamomethyl-substituted subporphyrins underwent neither σ- nor π-dimerization in solution due to the efficient spin delocalization over the entire subporphyrin skeleton.
 |
| | Fig. 10 Dicyanomethyl radical-substituted (sub)porphyrins. | |
2.2 Nitrogen-centered radicals
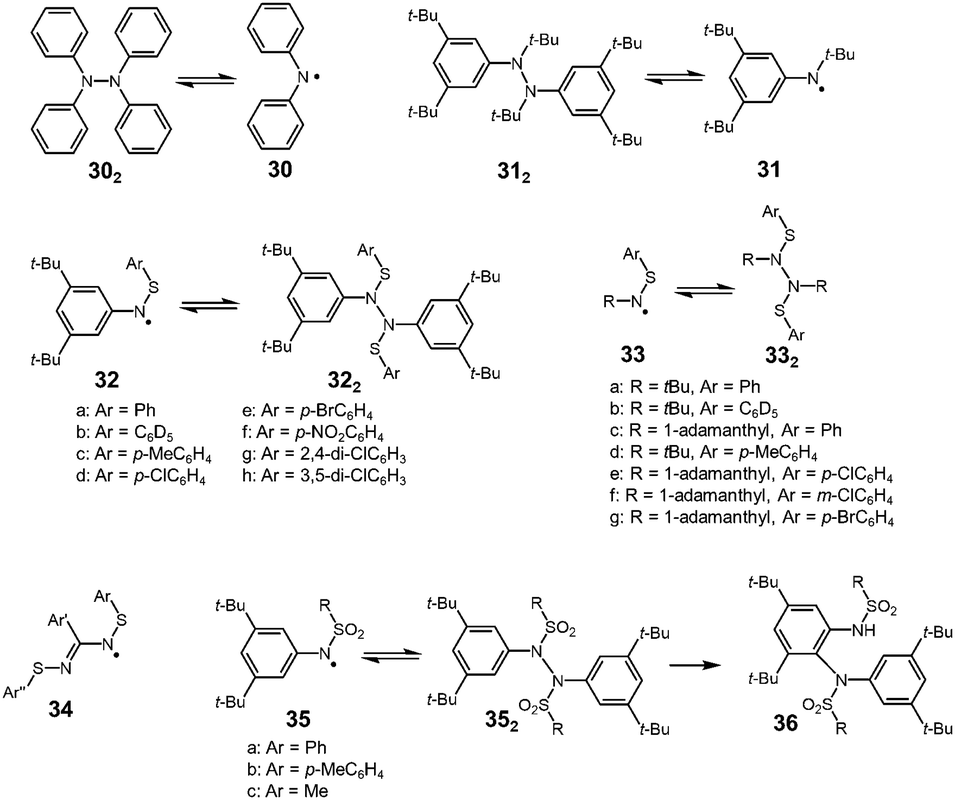
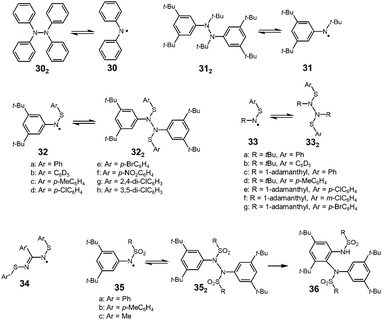
It is well known that the bond strength of N–N single bonds is relatively weak among other covalent bonds, and there have been various examples of nitrogen-centered radicals showing reversible N–N bond formation and dissociation (Fig. 11). In 1911, Wieland found that a colorless solution of tetraphenylhydrazine (302) turned green upon heating and then decolorized on cooling, and from this observation he concluded that 302 is in equilibrium with diphenylamino radical 30.52 After this report, Wieland synthesized and investigated many tetraarylhydrazines and found that the ease of dissociation increased by introducing dimethylamino or methoxy groups and decreased with the introduction of a nitro group at the para-positions compared with the unsubstituted 302.9,53 However, the dimerization of these diarylamino radicals to the corresponding tetraarylhydrazines was not completely reversible and they gradually converted to oligomeric arylamines or 9,10-diaryldihydrophenazine via C–C or C–N bond formations.54 In 1972, Nelsen revealed that an N,3,5-tri-tert-butylamino radical (31) was in a highly reversible equilibrium with the dimer (312) in a carefully deoxygenated methylcyclohexane solution showing no decomposition even at 338 K for 72 hours.55 However, the dissociated radical 31 easily reacted with molecular oxygen to form the nitroxide under air. The BDE of 312 was evaluated to be 131 kJ mol−1 by the ESR measurements, and this value is not so small compared with that of 1,2-diphenylhydrazine (158 kJ mol−1) obtained from the heats of combustion.56 As another nitrogen-centered radical, thioaminyl radicals [R–N–S–R′]˙ (R, R′ = aryl or alkyl), which have been intensively studied by Miura since the 1970's,57 were inert to molecular oxygen, and some of them were in a stable monomer–dimer equilibrium. The dimers of thioaminyl radicals easily dissociate compared to the hydrazine derivatives. The increased stability of thioaminyl radicals is attributed to the spin delocalization over the sulfur atom next to the nitrogen. The g-value of the observed ESR signal for [PhNSPh]˙ was 2.0059, which was large for a nitrogen-centered radical, indicating the considerable spin delocalization on the sulfur atom.57 The solutions of 32a2–h2 were deeply colored even at room temperature due to the presence of dissociated radicals. The characteristic colors disappeared upon cooling to 195 K, and reappeared on raising to room temperature. The calculated BDEs of 32c2 and 32d2 were only about 54–59 kJ mol−1.58 In the solid state, these radicals were proved to exist as the dimer. The analogues of 32 with a tert-alkyl group (tert-butyl or 1-adamanthyl) on the nitrogen atom (33) also showed similar monomer–dimer equilibria in solution, but the BDEs were larger than those of the N-aryl analogues 32 (about 85 kJ mol−1).59 The smaller BDE of 32 compared with those of 33 could be explained by the spin delocalization over the aryl group. Actually, the spin densities on the nitrogen atoms of [PhNSPh]˙ and [tBuNSPh]˙ (33a) were calculated to be 0.384 and 0.535, respectively, from the hyperfine coupling constants of the observed ESR spectra. The N,N′-bis(arylthiol)benzamidyl radicals (34), which are the analogues of thioaminyl radicals with an extended conjugation pathway were also highly persistent to oxygen and dimerized reversibly to form hydrazine-like dimers.60 The BDEs of the dimers were very small (∼25 kJ mol−1) because of the resonance stabilization of the radicals owing to the two equivalent N–S–Ar units. Miura also reported the dimerization–dissociation behaviors of the dimers of N-sulfonylaminyl radicals [RNSO2R′]˙ (35).61,62 The benzene solution of the dimers showed no ESR signal at room temperature, but when being heated over 333 K, the ESR signal derived from the dissociated radical was observed. The BDE of 35a2 was estimated to be 121 kJ mol−1, which was significantly higher than the dimers of the thioaminyl radicals.62 Furthermore, the dimers of 35 gradually isomerized to 36 irreversibly in the hot benzene solutions. The decreased stabilities of N-sulfonylaminyl radicals compared with the thioaminyl radicals were attributed to the lack of the spin delocalization on the sulfur atom due to the loss of the lone pairs.
 |
| | Fig. 11 Examples of nitrogen-centered radicals being in reversible equilibrium with the dimers. | |
2.3 Oxygen-centered radicals
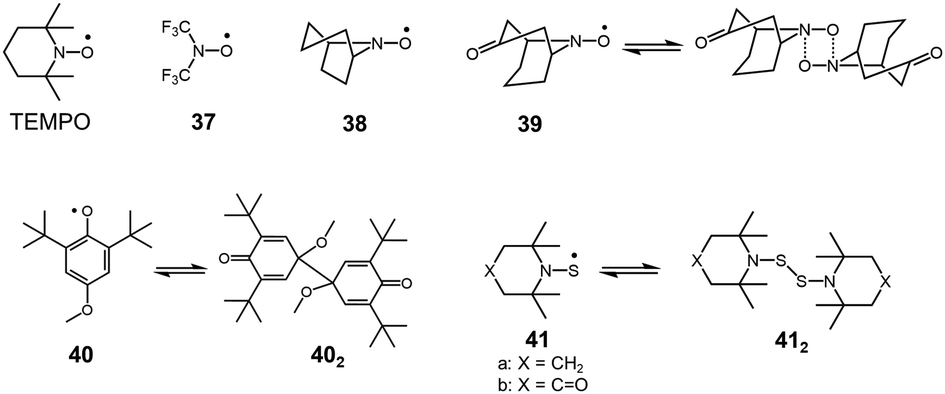
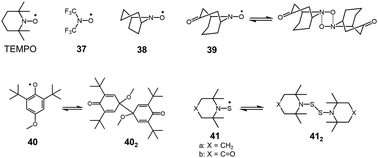
Organic peroxides containing O–O bonds are a very common class of chemicals widely utilized as initiators for polymerization or bleaching agents. In general, the O–O bond of peroxides is easily cleaved to form two oxygen-centered radicals. However, there have not been many peroxides showing a stable dissociation–dimerization behavior relative to vast examples of peroxides. This is the case of the intrinsic weakness of O–O bonds, and the dissociated RO˙ radicals tend to prefer reactions other than self-dimerization such as a hydrogen abstraction or addition reactions to other chemicals. Actually, sterically hindered nitroxide radicals such as 2,2,6,6,-tetramethylpiperidinyl-1-oxy (TEMPO) show no tendency to dimerize. Although most of the sterically unhindered nitroxide radicals easily decompose,63 some nitroxide radicals such as bis(trifluoromethyl)nitroxide64 (37) or azabicycloalkane N-oxyl derivatives65,66 (38) exist in monomer–dimer equilibrium (Fig. 12). However, the heats of dimerization of these radicals in CF2Cl2 solutions were just ∼10 kJ mol−1 and the monomer–dimer equilibria could only be observed at lower temperature (<∼200 K). The structure of the dimers of nitroxide radicals is still not fully understood.63 It is assumed that the dimers of nitroxide radicals are bound by a four-center bond instead of an O–O bond, as confirmed in a few cases such as the dimer of 39 by X-ray crystal analyses.67,68 In 2013, Mayer and co-workers reported that 2,6-di-tert-butyl-4-methoxyphenoxyl radical (40), that dimerized in solution and solid states.69 The X-ray crystal analysis revealed the structure of the dimer 402, whose monomers coupled between the 4-positions. The BDE of 402 from van’t Hoff analysis was evaluated to be only 26 ± 2 kJ mol−1, indicating the exceptional weakness of the intermonomer bond in 402 for a C–C bond.
 |
| | Fig. 12 TEMPO radical and examples of oxygen-centered and sulfur-centered radicals being in reversible equilibrium with the dimers. | |
2.4 Sulfur-centered radicals
S–S single bonds are also known to be relatively weaker covalent bonds but much stronger than those of O–O single bonds. The bond strength of a typical S–S bond of a disulfide is about 250–300 kJ mol−1, and this value is too large to utilize in DCC.70 On the other hand, the S–S bonds of bis(dialkylamino)disulfides (R2NS–SNR2) are much weaker and some bis(dialkylamino)disulfides showing reversible dissociation–dimerization behavior have been reported. In 1967, Bennett, Sieper, and Tavs reported that bis[2,2,6,6,-tetramethylpiperidyl-1]disulfide (41a) and bis[4-oxo-2,2,6,6,-tetramethylpiperidyl-1]disulfide (41b), which are the sulfur analogues of TEMPO radicals, dissociate to thionitroxide radicals in solution upon heating (Fig. 12).71 The solutions of these disulfides (412) showed ESR signals when heated (>363 K for 41a2, >413 K for 41b2). The reversible increase and decrease of ESR intensities, upon raising or lowering the temperature over a limited range, suggested that the radicals were formed reversibly. The reversible dimerization–dissociation behaviors of the sulfur-analogues of TEMPO radicals are contrasting to the original TEMPO radicals showing no dimerization. Bennett et al. hypothesized that the difference was due to the insufficient steric hindrances by the methyl groups in the sulfur analogs, which are too small to restrict the dimerization due to the larger atomic radius of sulfur. These disulfides were further investigated by Danen et al. and Ingold et al. independently, and the details of the radical–dimer equilibria have been published as back-to-back papers in 1976.72,73 The BDEs of the S–S bond of these disulfides were estimated to be about 130 kJ mol−1 regardless of the nature of the alkyl groups substituted on the nitrogen atoms by both research groups. Ingold et al. confirmed that the ESR spectra of the dissociated radicals were unchanged in the presence of oxygen, 1-decene, 1,1′-di-tert-butylethylene, triethyl phosphite, and triethylphosphine, indicating the inertness of the radicals to oxygen and addition to olefins and phosphorous atoms.73 They also concluded that the radicals do not abstract hydrogen of 2,4,6-teri-tert-butylphenol.
3. Self-assembly and applications of DCC radicals
3.1 Self-assembly of DCC multi radicals
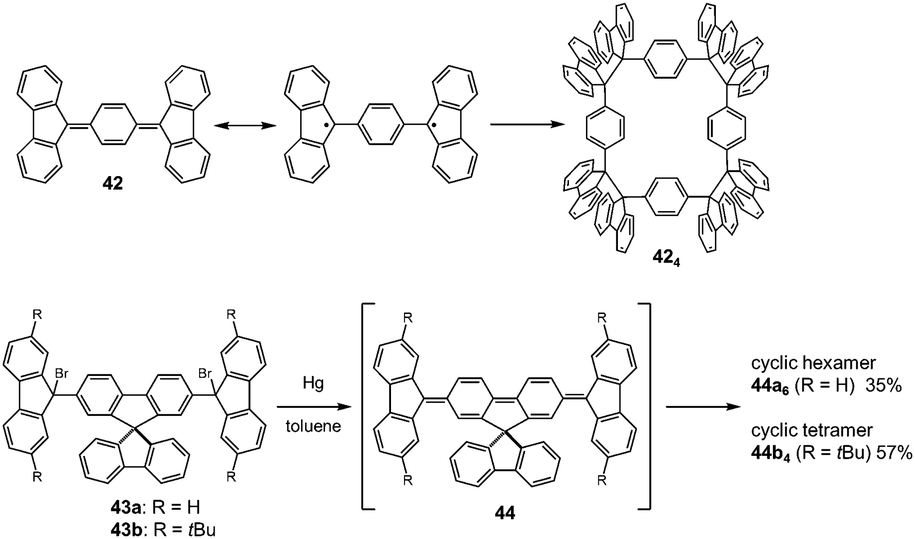
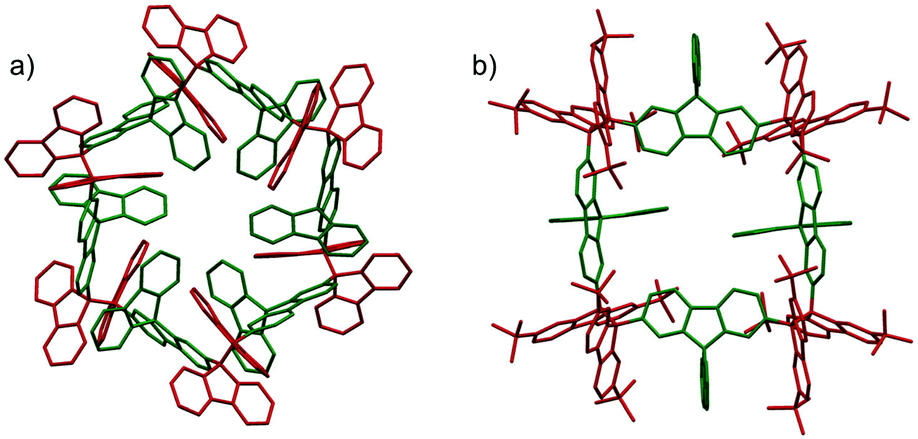
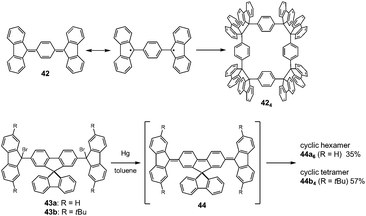
The molecules bearing more than two DCC radical units as reaction sites can form either linear or cyclic oligomers. An early work on the oligomerization of diradicals has been published by Wittig et al. in 1969.74 In that work, Wittig et al. intended to prepare a para-quinodimethane 42 and their derivatives were expected to have diradical characters, and they found that these quinodimethanes oligomerized to form cyclic tetramers (424) in solution (Fig. 13). The cleavage of the tetramers by applying heat or mechanical pressure has already been observed in this work. Nearly 30 years after Wittig's work, Ipaktschi and co-workers investigated the substituted derivatives of Wittig's hydrocarbon, and they unambiguously determined the cyclic tetramer structures by X-ray single crystal analyses.75,76 In 2015, Beaudoin and Wuest et al. demonstrated the construction of giant macrocyclic hydrocarbons using π-extended analogs of Wittig's hydrocarbon with spirobifluorenyl cores.77 The reduction of dibromides (43a and 43b) with Hg in hot toluene gave green supernatant solutions, indicating the generation of the π-extended Wittig's hydrocarbons (44a and 44b). After purification, colorless solid products were obtained and the structures of the obtained products were determined by single crystal X-ray analyses. Interestingly, the reduction of 43b gave a macrocyclic tetramer 44b4 similar to the case of Wittig's hydrocarbons, whereas 43a gave a macrocyclic hexamer 44a6 (Fig. 14). In both cases C–C bond lengths between the monomers in 44a6 and 44b4 were significantly elongated (>1.61 Å) compared to usual C(sp3)–C(sp3) bonds. The cyclic hexamer 44a6 is the largest hydrocarbon whose structure has been characterized by X-ray crystal analysis. The reason of the selective formation of the cyclic hexamer was attributed to an intramolecular C–H⋯π network among the peripheral fluorene units as donors and the spirobifluorenyl units as acceptors in 44a6. The net stabilization energy by the C–H⋯π interactions in 44a6 was estimated to be 42–84 kJ mol−1, and due to such high stabilization energy, the macrocyclic hexamer was obtained as the enthalpically favored product. The tBu groups at the peripheral fluorene units in 44b disturb intramolecular C–H⋯π interactions, leading to the cyclic tetramer. The results demonstrated the importance of the weak interactions for determining the self-assembled structures of DCC radicals.
 |
| | Fig. 13 Macrocycle formation of π-extended Wittig's hydrocarbons. | |
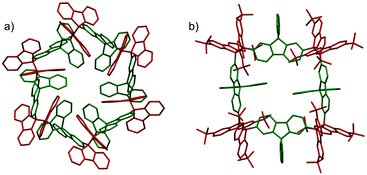 |
| | Fig. 14 X-ray crystal structures of (a) 44a6 and (b) 44b4. Reprinted with permission from ref. 77. Copyright 2015 Wiley-VCH. | |
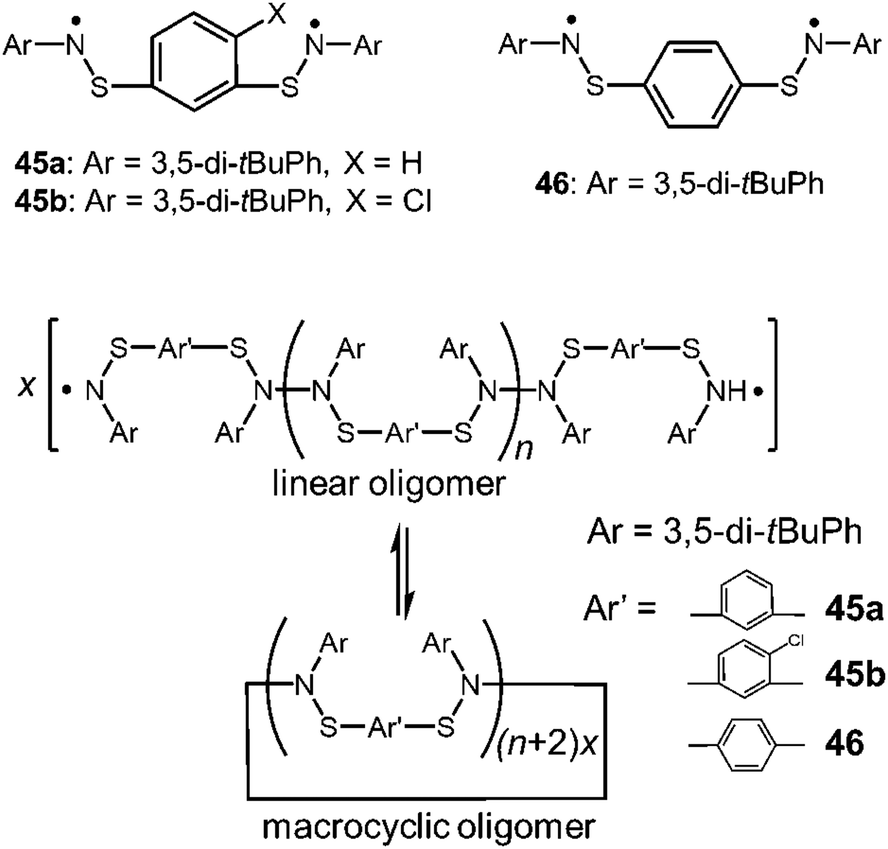
As another early work of the macrocyclic formation with DCC radicals, Miura et al. reported the reversible cyclization of thioaminyl diradicals connected through a meta- or para-phenylene linker (45a,b and 46), which were synthesized in the course of searching for organic ferromagnetic materials (Fig. 15).78 When the precursors of these radicals were oxidized with PbO2 in benzene, the clear solutions immediately turned dark blue or dark greenish blue resulting from the appearance of the radical species with absorption around 600 nm, and the solutions gave intense ESR signals. Interestingly, the observed ESR signal patterns suggested no or negligibly weak spin-exchange interaction between two radical spins, indicating the formation of linear dimers or longer oligomers bearing two spins at their ends. The intensities of the radical absorption bands and the ESR signals reversibly increased and decreased by heating and cooling, suggesting the equilibria between the linear oligomers and the diamagnetic macrocyclic oligomers (Fig. 15). The removal of the solvent and subsequent recrystallization gave light-blue micro crystals, which were attributed to the cyclic oligomers of the diradicals by the IR spectra and the elemental analyses. The thermodynamic parameters of the equilibria between the linear and the cyclic oligomers could not be determined because a number of the monomers were unknown. However, it was shown that the solution of the cyclic oligomer of the para-substituted diradical gave a much higher ESR signal than those of the oligomers of the meta-substituted diradicals, suggesting the ease of dissociation of the para-oligomer.
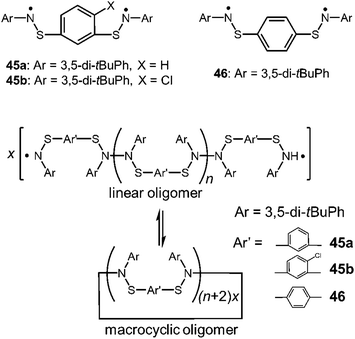 |
| | Fig. 15 Structures of thioaminyl diradicals and their oligomers. | |
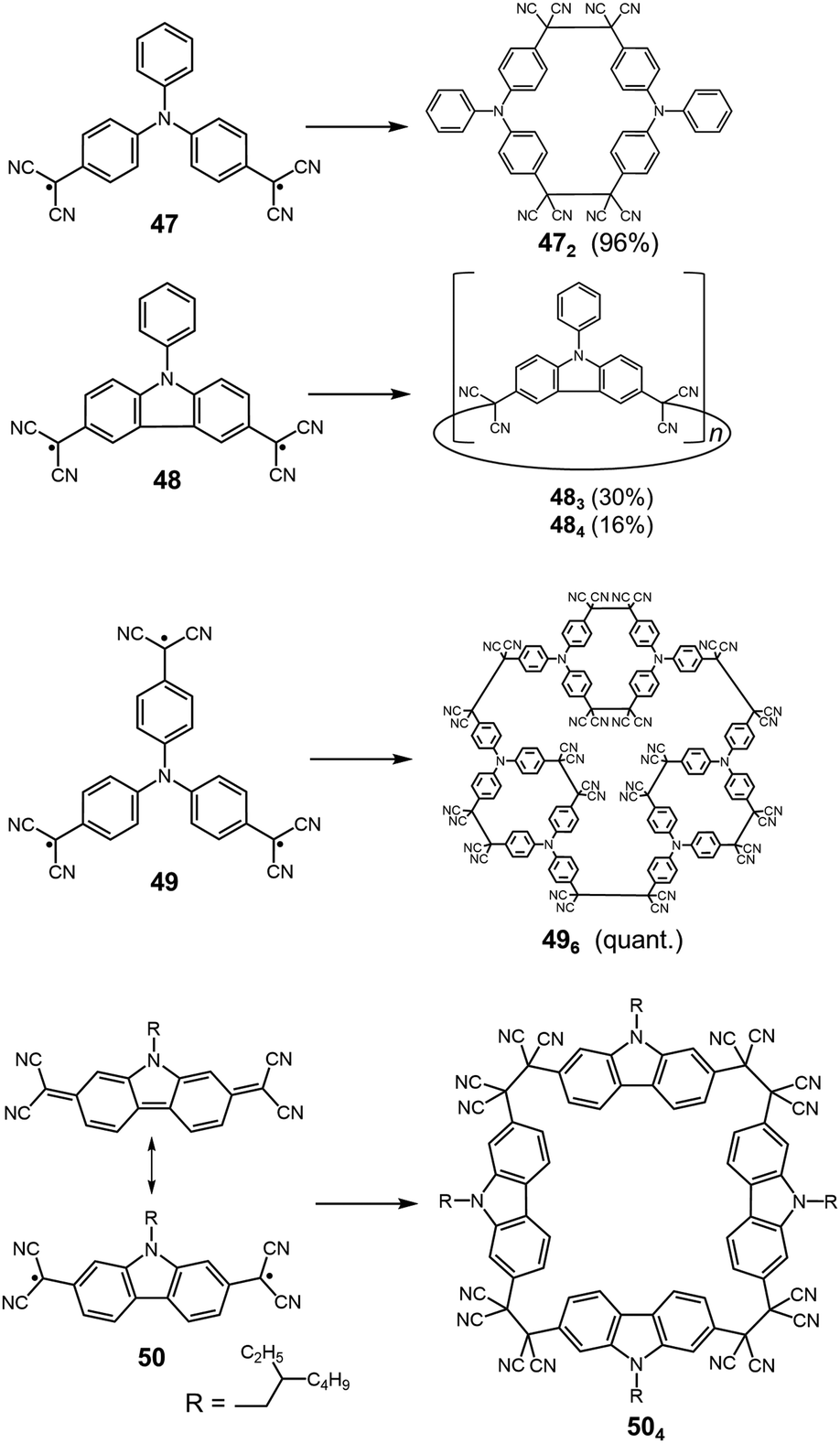
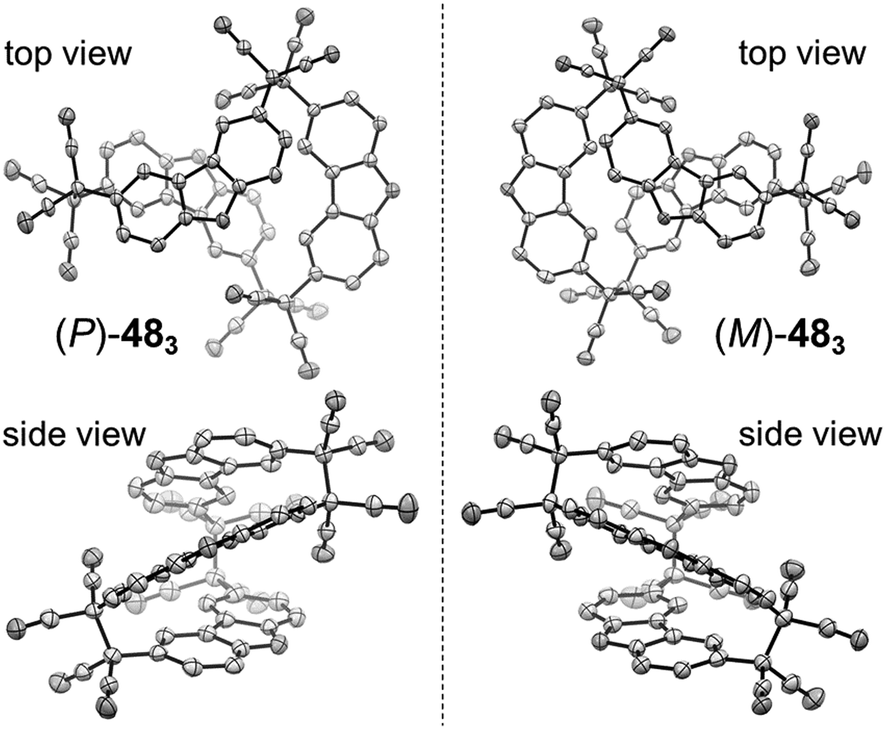
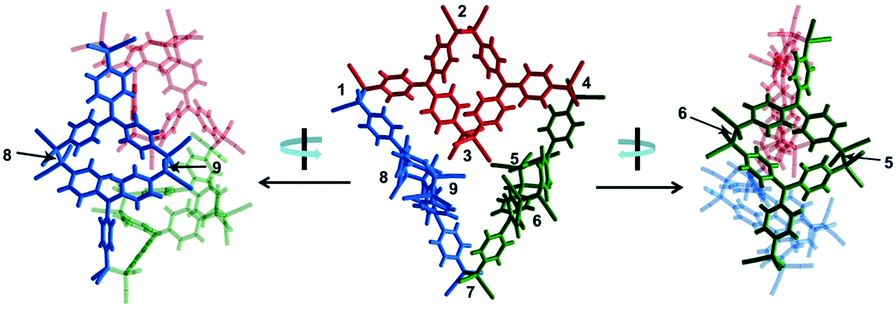
Quite recently, several examples of the formation of macrocycles by self-assembly of molecules bearing multiple dicyanomethyl radicals have been continuously reported. Sakamaki and Seki et al. reported the formation of macrocycles using amine-conjugated dicyanomethylene diradicals with triphenylamine and carbazole skeleton 47 and 48, which are the diradical analogues of 18 and 19 (Fig. 16).45 When 47 and 48 were generated in dichloromethane at room temperature, these radicals immediately formed macrocycles with particular ring sizes. The triphenylamine diradical 47 selectively formed a cyclic dimer almost quantitatively. On the other hand, the carbazole diradical 48 gave a mixture of a cyclic trimer (30%), cyclic tetramer (16%), and insoluble precipitates probably composed of higher oligomers and polymers. X-ray single crystal analyses revealed the structures of 472 and 483, and the elongated C–C bonds between the monomers (1.61–1.64 Å) were observed. Interestingly, the macrocycle of the carbazole trimer 483 were helically twisted, and a crystal of 483 contained two enantiomers originating from the helicity (Fig. 17). The absence of 482 could be attributed to the rigidness of the carbazole moiety. The DFT optimized geometry of 482 exhibited slightly bent carbazole planes and unusually long intermonomer C–C bonds (1.686 Å), suggesting that 482 may be thermodynamically disfavored. The cyclophanes 472 and 483 showed thermo- and mechanochromic behavior in the solid state resulting from the generation of radical species by intermonomer bond cleavage, and the recovery of their colors by dropping solvent and drying were also observed. Following this report, Chi and co-workers reported the self-assembly of the triphenylamine substituted with three dicyanomethyl radicals at all the para-positions (49).79 When the precursor of 49 was oxidized with DDQ in acetone at room temperature, the generated triradical species immediately self-assembled to give a solo product, which was isolated as a light yellow powder. The structure of the product was confirmed to be the macrocyclic hexamer 496 with a unique macrocycle-in-macrocycle structure with helical chirality, which could be also regarded as the cyclic trimer of 472 (Fig. 18). The macrocyclic hexamer 496 was confirmed to be a thermodynamic product not a kinetic product as follows. When 49 was generated at 233 K, a dark green powder showing an intense ESR signal was obtained as a product, and this colored product could be attributed to the randomly-linked kinetic polymer of 49 containing a large number of unreacted radicals. Interestingly, this kinetic product was quantitatively converted to 496 within one minute by dissolving in solvent and stirring at room temperature. Li, Ruiz Delgado, and Hartl et al. prepared a carbazole substituted with two dicyanomethyl radicals at the 2,7-positions (50) having a different substitution pattern from the 3,6-substituted carbazole diradical 48.80 As shown in Fig. 16, 50 can take a closed-shell quinoid structure. The evaporation of the deep violet solution of 50 selectively gave a cyclic tetramer 504 as a white powder, and the structure of the tetramer 504 was explicitly determined by X-ray analysis. When the white powder of pure tetramer 504 was dissolved in chloroform at room temperature, the colorless solution gradually turned purple, indicating the dissociation of 504 to 50. This is in stark contrast to the result that the solutions of the white powders of 484 and 483 remain colorless and show no radical absorption bands.45 These results tell the ease of dissociation of 2,7-disubstituted carbazole diradicals compared to 3,6-diradicals. This difference could be understood qualitatively from the contribution of the closed-shell quinoid structure of 50 increasing the relative stability of the dissociated form. Actually, DFT calculations estimated the larger diradical character y (0 < y < 1, where y = 0 represents a pure closed-shell and y = 1 represents a pure diradical) for 48 (y = 0.80) compared with 50 (y = 0.51).81
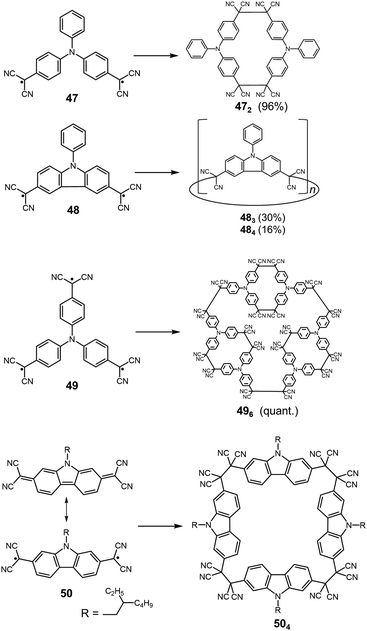 |
| | Fig. 16 Structures of dicyanomethyl diradicals and multi radicals and their cyclic oligomers. | |
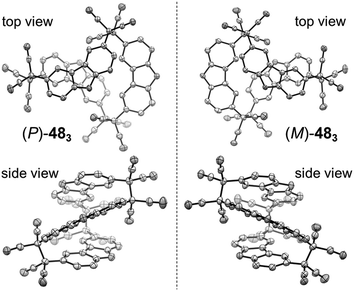 |
| | Fig. 17 X-ray crystal structures of the (P)- and (M)-isomers of 483. Phenyl groups and hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at 50% probability. Reprinted with permission from ref. 45. Copyright 2016 Wiley-VCH. | |
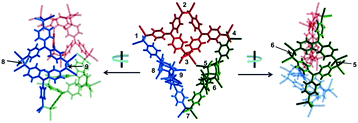 |
| | Fig. 18 X-ray crystallography structure of 496 (P-hexamer) viewed from different angles. The structure is presented as three dimeric components (depicted in red, blue, and green) linked together for clarity. All C(sp3)–C(sp3) bonds formed by radical association are labeled by numerals beside them. Reprinted with permission from ref. 79. Copyright 2018 Wiley-VCH. | |
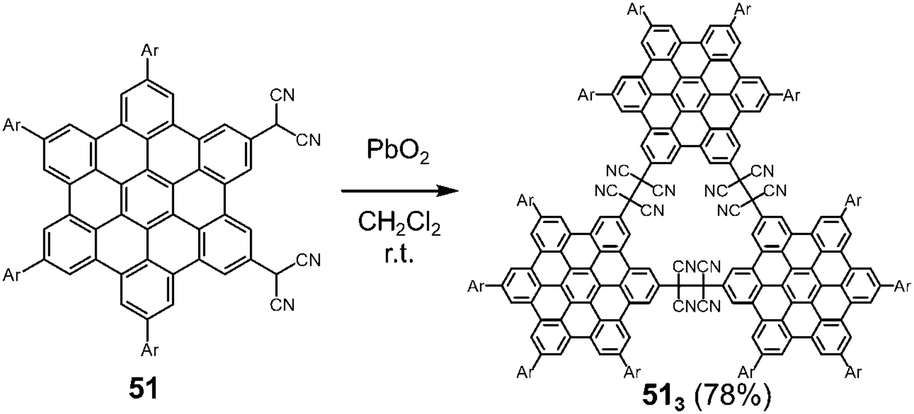
Hiroto and Shinokubo et al. reported the self-assembly of a hexa-peri-hexabenzocoronene (HBC) with two dicyanomethyl radicals at the 2,5-positions, which could be regarded as a π-extended analog of ortho-quinodimethane (Fig. 19).82 The oxidation of 51 with PbO2 in dichloromethane at room temperature afforded the cyclic trimer 513 in a good yield via trimerization of the generated diradicals. The structure of the trimer 513 was confirmed by high-resolution mass spectroscopy, NMR spectra, and preliminary X-ray diffraction analysis. The solid sample of 513 also exhibited a mechanochromic behavior coming from the C–C bond dissociation by shearing. The characteristic absorption band of the dissociated radicals appeared in the NIR region (λmax = 953 nm) by grinding, demonstrating the unique NIR mechanochromic character of 513.
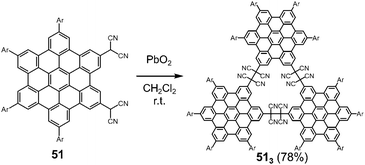 |
| | Fig. 19 HBC with two dicyanomethylene units and its cyclic trimer. | |
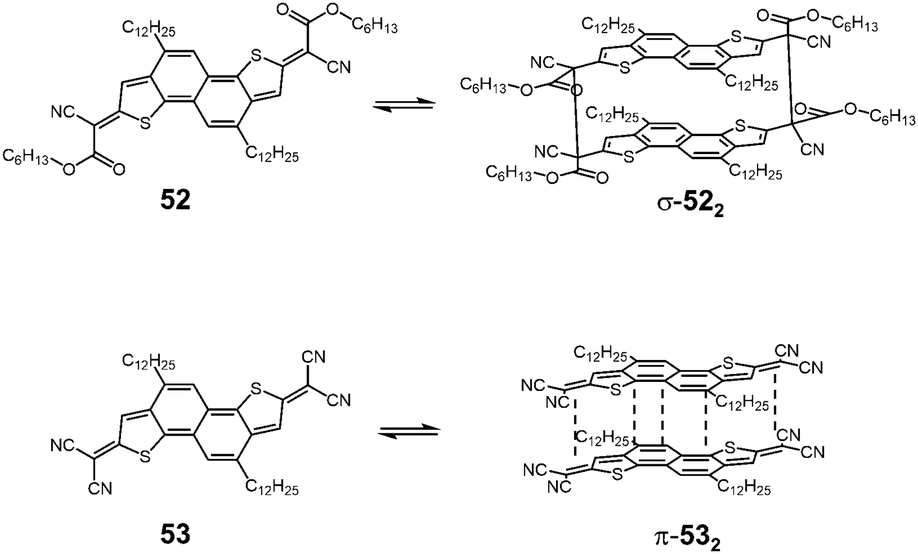
Takimiya and Casado et al. synthesized two kinds of naphthodithiophenes bearing two cyanoalkyloxycarbonyl-methyl radicals (52) and two dicyanomethyl radicals (53) being the resonance hybrids between closed-shell quinoids and open-shell diradicals (Fig. 20).83 These two molecules showed different self-assembly behaviors; 52 formed the cyclic dimer or higher oligomers bound via σ-bonds (σ-522), whereas 53 did not form σ-bonded aggregates but a π-dimer (π-532). Furthermore, in the mixed solution of 52 and 53, two types of hetero-dimers, σ-(52 + 53) and π-(52 + 53) were also observed.
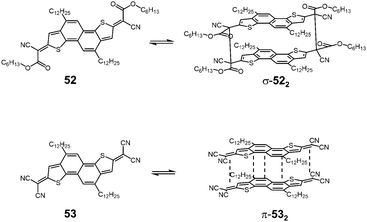 |
| | Fig. 20 Structures of quinoidal naphthodithiophenes and their dimer formation. | |
3.2 Polymer materials bearing DCC radical units
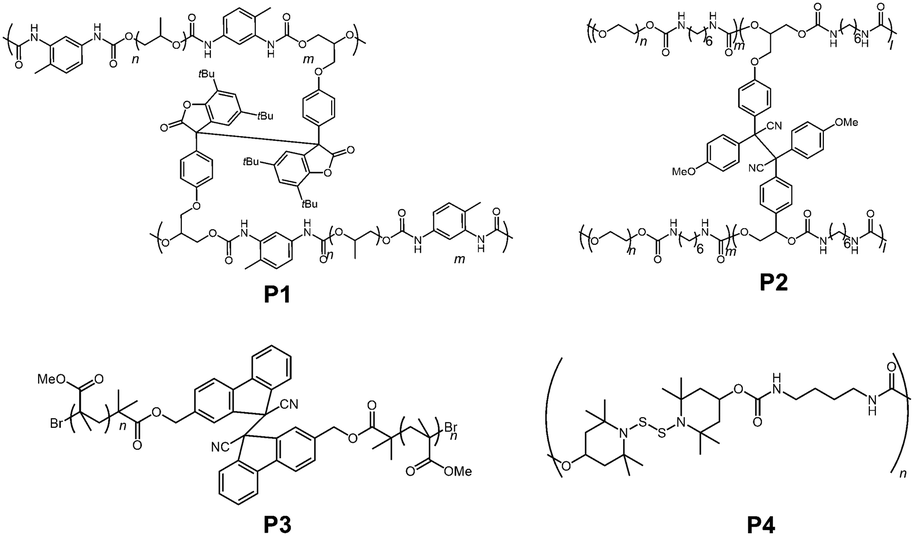
Not only in small molecules or oligomers, DCC radical units are also introduced into polymer chains to impart stimuli responsiveness and self-healing properties. The very first example came from the group of Otsuaka in 2012.84 In their work, they prepared a self-healable polymeric gel by incorporating arylbenzofranone radicals as a cross linker (P1 in Fig. 21). Since then, various polymer materials bearing DCC radical units such as diphenylcyanomethyl radicals (P2),85,86 9-cyanofluorenyl radicals (P3),87 4-aminophenylthiyl radicals,88 and thionitroxide radicals (P4)89 have been reported. The sensitivity of bond dissociation of dynamic bonds by mechanical stimuli could be enhanced by incorporating the radical units into the polymer chains or dendrimers.90 Recently, Otsuka et al. published a comprehensive review article about reorganizable and stimuli-responsive polymers based on DCC properties of carbon-centered radicals,91 and therefore we will not go into detail about this topic in this review.
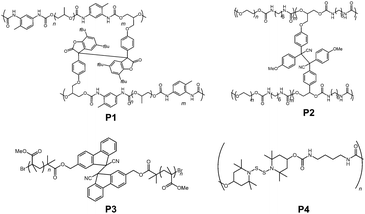 |
| | Fig. 21 Examples of polymers bearing DCC radicals. | |
4. Summary and outlook
In this review, we summarize organic radicals showing reversible association–dissociation behavior from the viewpoint of dynamic covalent chemistry (DCC). Almost 120 years have already passed since Gomberg's discovery of the triphenylmethyl radical, and an enormous number of organic radicals have been found and synthesized to date. Compared with the long history of radical chemistry, DCC is a new but rapidly growing area of research, and it is only in recent years that organic radicals have been considered in relation to DCC. Because the fundamental properties of radicals (ease of self-association and dissociation) are a great fit for the components of DCC, it would be very informative to look back at the library of organic radicals with the prospect of searching for possible building blocks of DCC. Some key factors for designing new radicals with DCC characters are induced from the past examples of DCC radicals that work well: (1) precise steric protection to avoid irreversible side reactions, and (2) spin delocalization by introducing heteroatoms or π-extension to tune the reactivity of DCC radicals. As described in this review, molecular self-assembly using a reversible bond formation–dissociation process of organic multi radicals is a rapidly growing research topic. However, the structures constructed by self-assembly of DCC radicals were limited to closed structures such as macrocycles, and construction of two- or three-dimensional periodic structures has not yet been achieved. Highly crystalline covalent organic frameworks (COFs) and self-healable materials would be the future prospects of the radical-based DCC via their unique features of high reaction barrier tunability and error correction capability. The unique switchable chemical and opto-electronic properties by external stimuli such as mechanical or thermal ones will gain more and more importance of radical-based DCC as a novel materials platform toward wide-range dyes, sensing, or spintronic applications. We hope that this review provides useful suggestions for developing the field of radical-based DCC.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (No. 26810023, 17H04874, 18H03918, 15K21721, 26102011, and 19F19044) from the Japan Society for the Promotion of Science (JSPS). D. S. and S. G. are thankful for JSPS Research fellowships.
Notes and references
-
W. Zhang and Y. Jin, Dynamic Covalent Chemistry: Principles, Reactions, and Applications, John Wiley & Sons, New York City, USA, 2017 Search PubMed
 .
.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed .
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed .
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC .
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC .
- K. Jyothish and W. Zhang, Angew. Chem., Int. Ed., 2011, 50, 8478–8480 CrossRef CAS PubMed .
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed .
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1124–1150 RSC .
-
A. R. Forrester, J. M. Hay and R. H. Thomsom, Organic Chemistry of Stable Free Radicals, Academic Press, New York, NY, 1968 Search PubMed .
-
Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, ed. R. G. Hicks, Wiley, Chichester, 2010 Search PubMed .
- D. Grille and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13–18 CrossRef .
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC .
- K. Kato and A. Osuka, Angew. Chem., Int. Ed., 2019, 58, 8978–8986 CrossRef CAS PubMed .
- M. Kertesz, Chem. – Eur. J., 2019, 25, 400–416 CrossRef CAS PubMed .
- D.-W. Zhang, J. Tian, L. Chen, L. Zhang and Z.-T. Li, Chem. – Asian J., 2015, 10, 56–68 CrossRef CAS PubMed .
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef .
- H. Lankamp, W. Th. Nauta and C. MacLean, Tetrahedron Lett., 1968, 2, 249–254 CrossRef .
- J. M. McBride, Tetrahedron, 1974, 30, 2009–2022 CrossRef CAS .
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC .
- Y. Morita, T. Murata, A. Ueda, C. Yamada, Y. Kanzaki, D. Shiomi, K. Sato and T. Takui, Bull. Chem. Soc. Jpn., 2018, 91, 922–931 CrossRef CAS .
- W. P. Neumann, A. Penenory, U. Stewen and M. Lehnig, J. Am. Chem. Soc., 1989, 111, 5845–5851 CrossRef CAS .
- K. Rakus, S. P. Verevkin, J. Schätzer, H.-D. Beckhaus and C. Rüchardt, Ber. Dtsch. Chem. Ges., 1994, 127, 1095–1103 CrossRef CAS .
- E. Font-Sanchis, C. Aliaga, K.-S. Focsaneanu and J. C. Scaiano, Chem. Commun., 2002, 1576–1577 RSC .
- M. Frenette, C. Aliga, E. Font-Sanchis and J. C. Scaiano, Org. Lett., 2004, 6, 2579–2582 CrossRef CAS PubMed .
- Y. Tian, K. Uchida, H. Kurata, Y. Hirao, T. Nishiuchi and T. Kubo, J. Am. Chem. Soc., 2014, 136, 12784–12793 CrossRef CAS PubMed .
- T. Nishiuchi, S. Aibara and T. Kubo, Angew. Chem., Int. Ed., 2018, 57, 16516–16519 CrossRef CAS PubMed .
- T. Kubo, Molecules, 2019, 24, 665–677 CrossRef .
- F. M. Beringer, S. A. Galton and S. J. Huang, Tetrahedron, 1963, 19, 809–816 CrossRef CAS .
- I. V. Khudyakov, A. I. Yasmenko and V. A. Kuzmin, Int. J. Chem. Kinet., 1979, 11, 621–633 CrossRef CAS .
- L. M. Pisarenko, V. I. Nikulin and A. B. Gagarina, Izv. Akad. Nauk SSSR, Ser. Khim., 1987, 6, 1237–1243 Search PubMed .
- L. M. Pisarenko, V. I. Nikulin and I. V. Khudyakov, Izv. Akad. Nauk SSSR, Ser. Khim., 1987, 8, 1734–1738 Search PubMed .
- L. M. Pisarenko, A. B. Gagarina and V. A. Roginskii, Izv. Akad. Nauk SSSR, Ser. Khim., 1987, 12, 2861–2863 Search PubMed .
- A. Löwenbein and W. Folberth, Ber. Dtsch. Chem. Ges., 1925, 58, 601–609 CrossRef .
- H. H. Wasserman, T.-C. Liu and E. R. Wasserman, J. Am. Chem. Soc., 1953, 75, 2056–2058 CrossRef CAS .
- A. Löwenbein and B. Rosenbaum, Justus Liebigs Ann. Chem., 1926, 448, 223–248 CrossRef .
- J. Ohkanda, Y. Mori, K. Maeda and E. Osawa, J. Chem. Soc., Perkin Trans. 2, 1992, 59–63 RSC .
- L. Kalb and J. Bayer, Ber. Dtsch. Chem. Ges., 1913, 46, 3879–3885 CrossRef .
- R. W. Baldock, P. Hudson and A. R. Katritzky, J. Chem. Soc., Perkin Trans. 1, 1974, 1422–1427 RSC .
- L. A. Margulis, I. V. Khudyakov, V. A. Kuzmin, A. I. Prokof’ev, A. I. Yasmenko and G. Smets, Int. J. Chem. Kinet., 1985, 17, 735–747 CrossRef CAS .
- Y. Mori, N. Yamada, M. Kanazawa, Y. Horikoshi, Y. Watanabe and K. Maeda, Bull. Chem. Soc. Jpn., 1996, 69, 2355–2359 CrossRef CAS .
- H. D. Hartzler, J. Org. Chem., 1966, 31, 2654–2658 CrossRef CAS .
- H. A. P. de Jongh, C. R. H. I. de Jonge, H. J. M. Sinnige, W. J. de Klein, W. G. B. Huysmans and W. J. Mijs, J. Org. Chem., 1972, 37, 1960–1966 CrossRef CAS .
- L. I. Peterson, J. Am. Chem. Soc., 1967, 89, 2677–2681 CrossRef CAS .
- H. A. P. de Jongh, C. R. H. I. de Jonge and W. J. Mijs, J. Org. Chem., 1971, 36, 3160–3168 CrossRef CAS .
- T. Kobashi, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2016, 55, 8634–8638 CrossRef CAS PubMed .
- K. Okino, S. Hira, Y. Inoue, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2017, 56, 16597–16601 CrossRef CAS PubMed .
- K. Okino, D. Sakamaki and S. Seki, ACS Mater. Lett., 2019, 1, 25–29 CrossRef CAS .
- J. P. Peterson, M. R. Geraskina, R. Zhang and A. H. Winter, J. Org. Chem., 2017, 82, 6497–6501 CrossRef CAS .
- R. Zhang, J. P. Peterson, L. J. Fischer, A. Ellern and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 14308–14313 CrossRef CAS .
- B. Adinarayana, D. Shimizu, K. Furukawa and A. Osuka, Chem. Sci., 2019, 10, 6007–6012 RSC .
- B. Adinarayana, D. Shimizu and A. Osuka, Chem. – Eur. J., 2019, 25, 1706–1710 CrossRef CAS PubMed .
- H. Wieland, Liebigs Ann. Chem., 1911, 381, 200–216 CrossRef CAS .
- H. Wieland, Ber. Dtsch. Chem. Ges., 1915, 48, 1078–1095 CrossRef CAS .
- H. Wieland and A. H. Lecher, Liebigs Ann. Chem., 1912, 392, 156–169 CrossRef .
- S. F. Nelsen and R. T. Landis, II, J. Am. Chem. Soc., 1973, 95, 8707–8713 CrossRef CAS .
- L. G. Cole and E. C. Gilbert, J. Am. Chem. Soc., 1951, 73, 5423–5427 CrossRef CAS .
- Y. Miura, Y. Katsura and M. Kinoshita, Chem. Lett., 1977, 409–412 CrossRef CAS .
- Y. Miura, A. Yamamoto, Y. Katsura and M. Kinoshita, J. Org. Chem., 1980, 45, 3875–3880 CrossRef CAS .
- Y. Miura and M. Kinoshita, J. Org. Chem., 1984, 49, 2724–2728 CrossRef CAS .
- Y. Miura, T. Kunishi, M. Isogai and M. Kinoshita, J. Org. Chem., 1985, 50, 1627–1632 CrossRef CAS .
- Y. Miura, Y. Nakamura and M. Kinoshita, Bull. Chem. Soc. Jpn., 1981, 54, 3217–3218 CrossRef CAS .
- Y. Miura and T. Ohnishi, J. Org. Chem., 1988, 53, 3012–3016 CrossRef CAS .
- A. Nilsen and R. Braslau, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 697–717 CrossRef CAS .
- W. D. Blackley and R. R. Reinhard, J. Am. Chem. Soc., 1965, 87, 802–805 CrossRef CAS .
- D. F. Bowman, T. Gillan and K. U. Ingold, J. Am. Chem. Soc., 1973, 93, 6555–6561 CrossRef .
- G. D. Mendenhall and K. U. Ingold, J. Am. Chem. Soc., 1973, 95, 6390–6394 CrossRef CAS .
- R. A. Howie, L. S. D. Glasser and W. Moser, J. Chem. Soc. A, 1968, 3043–3047 RSC .
- P. A. Capiomont, B. Chion and J. Lajzérowicz, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 322–326 CrossRef .
- J. M. Wittman, R. Hayoun, W. Kaminsky, M. K. Coggins and J. M. Mayer, J. Am. Chem. Soc., 2013, 135, 12956–12959 CrossRef CAS PubMed .
- S. Sunner, Acta Chem. Scand., 1955, 9, 837–846 CrossRef CAS .
- J. E. Bennett, H. Sieper and P. Tavs, Tetrahedron, 1967, 23, 1697–1699 CrossRef CAS .
- W. C. Danen and D. D. Newlirk, J. Am. Chem. Soc., 1976, 98, 516–520 CrossRef CAS .
- B. Maillard and K. U. Ingold, J. Am. Chem. Soc., 1976, 98, 520–523 CrossRef CAS .
- G. Wittig, E. Dreher, W. Reuther, H. Weidinger and R. Steinmetz, Justus Liebigs Ann. Chem., 1969, 726, 188–200 CrossRef .
- J. Ipaktschi, R. Hosseinzadeh, P. Schlaf, E. Dreiseidler and R. Goddard, Helv. Chim. Acta, 1998, 81, 1821–1834 CrossRef CAS .
- J. Ipaktschi, R. Hosseinzadeh and P. Schlaf, Angew. Chem., Int. Ed., 1999, 38, 1658–1660 CrossRef CAS PubMed .
- D. Beaudoin, O. Levasseur-Grenon, T. Maris and J. D. Wuest, Angew. Chem., Int. Ed., 2015, 55, 894–898 CrossRef .
- Y. Miura and T. Ohana, J. Org. Chem., 1988, 53, 5770–5772 CrossRef CAS .
- L. Yuan, Y. Han, T. Tao, H. Phan and C. Chi, Angew. Chem., Int. Ed., 2018, 57, 9023–9027 CrossRef CAS .
- D. Wang, C. C. Ferrón, J. Li, S. Gámez-Valenzuela, R. P. Ortiz, J. T. López Navarrete, V. H. Jolín, X. Yang, M. P. Ávarez, V. G. Baonza, F. Hartl, M. C. R. Delgado and H. Li, Chem. – Eur. J., 2017, 23, 13776–13783 CrossRef CAS PubMed .
- I. Badía-Domínguez, A. Pérez-Guardiola, J. C. Sancho-García, J. T. L. Navarrete, V. H. Jolín, H. Li, D. Sakamaki, S. Seki and M. C. R. Delgado, ACS Omega, 2019, 4, 4761–4769 CrossRef .
- K. Oda, S. Hiroto and H. Shinokubo, J. Mater. Chem. C, 2017, 5, 5310–5315 RSC .
- J. L. Zafra, L. Qiu, N. Yanai, T. Mori, M. Nakano, M. P. Alvarez, J. T. L. Navarrete, C. J. Glmez-Garc, M. Kertesz, K. Takimiya and J. Casado, Angew. Chem., Int. Ed., 2016, 55, 14563–14568 CrossRef CAS PubMed .
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed .
- T. Sumi, R. Goseki and H. Otsuka, Chem. Commun., 2017, 53, 11885–11888 RSC .
- S. Kato, K. Ishizuki, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 1087–1091 CrossRef CAS .
- H. Sakai, T. Sumi, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 1359–1363 CrossRef CAS .
- A. Rekondo, R. Martin, A. R. de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC .
- A. Takahashi, R. Goseki and H. Otsuka, Angew. Chem., Int. Ed., 2017, 56, 2016–2021 CrossRef CAS PubMed .
- T. Watabe, K. Ishizuki, D. Aoki and H. Otsuka, Chem. Commun., 2019, 55, 6831–6834 RSC .
- K. Imato and H. Otsuka, Polymer, 2018, 137, 395–413 CrossRef CAS .
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
Shu
Seki
a and
Shu
Seki