
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining enzymes and organometallic complexes: novel artificial metalloenzymes and hybrid systems for C–H activation chemistry
Carlos
Perez-Rizquez
 ,
Alba
Rodriguez-Otero
and
Jose M.
Palomo
*
,
Alba
Rodriguez-Otero
and
Jose M.
Palomo
*
Department of Biocatalysis, Institute of Catalysis (CSIC), Marie Curie 2, Cantoblanco, Campus UAM, 28049 Madrid, Spain. E-mail: josempalomo@icp.csic.es; Fax: +34-91-585-4760
First published on 4th July 2019
Abstract
This review describes the recent advances in the design of novel artificial metalloenzymes and their application in C–H activation reactions. The combination of enzymes and metal or organometallic complexes for the creation of new artificial metalloenzymes has represented a very exciting research line. In particular, the development of proteins with the ability to perform C–H functionalization presents a significant challenge. Here we discuss the development of these processes on natural metalloenzymes by using directed evolution, biotin-(strept)avidin technologies, photocatalytic hybrids or reconstitution of heme-protein technology.
 Carlos Perez-Rizquez | Carlos Pérez Rízquez was born in Madrid, Spain (1994). He obtained his bachelor's degree in Biotechnology in 2016 from the Technical University of Madrid. Next, he obtained his MSc in Plant Biotechnology from Complutense University of Madrid. After that, he joined J. M. Palomo's group and started working on C–H activation and biocatalysis. |
 Alba Rodriguez-Otero | Alba Rodríguez Otero was born in Madrid, Spain (1996). She is currently finishing her degree in Chemical Engineering from Universidad Autónoma de Madrid (UAM). She completed her bachelor's thesis with J. M. Palomos's group at the Institute of Catalysis (ICP, CSIC) and continued with an internship working on nano- and bio-catalysis. |
 Jose M. Palomo | Jose M. Palomo was born in Coin, Malaga (Spain) (1976). He received his Ph.D. degree (summa cum laude) in 2003 from Madrid Universidad Autonoma (Spain). Then, Jose moved to the Max Planck Institute in Dortmund, Germany (2004–2006) as an EMBO postdoctoral researcher. In 2006, he began his appointment as an Associate Research Scientist in the Biocatalysis Department at the ICP-CSIC. From 2009, he has been a Tenured Scientist (Associate Professor) at the ICP-CSIC. Jose has published more than 140 articles in high-impact journals, with more than 7000 citations (H-index of 42). His current research interests are in protein chemistry, nanocatalysis, nanobiotechnology, glycochemistry, chemical biology and biocatalysis. |
1. Introduction
In the last few years, the creation of new artificial metalloenzymes by the combination of metal or complex organometallic systems and enzymes has represented a very exciting line of research. This field is expanding not only in terms of its applications but also its design.1–8 The success of these kinds of hybrids is based on the advantages offered by the combination of these two domains (Fig. 1). The enzyme provides a tridimensional structure, excellent for generating a coordination sphere, that has shown high selectivity in different processes. In addition, the use of metal catalysts (a coordinated metal or an organometallic structure) offers a wide range of catalytic possibilities. Therefore, this concept has allowed obtaining improved activity and selectivity of natural metalloenzymes and metalloenzymes with catalytic activities not existing in nature.1–8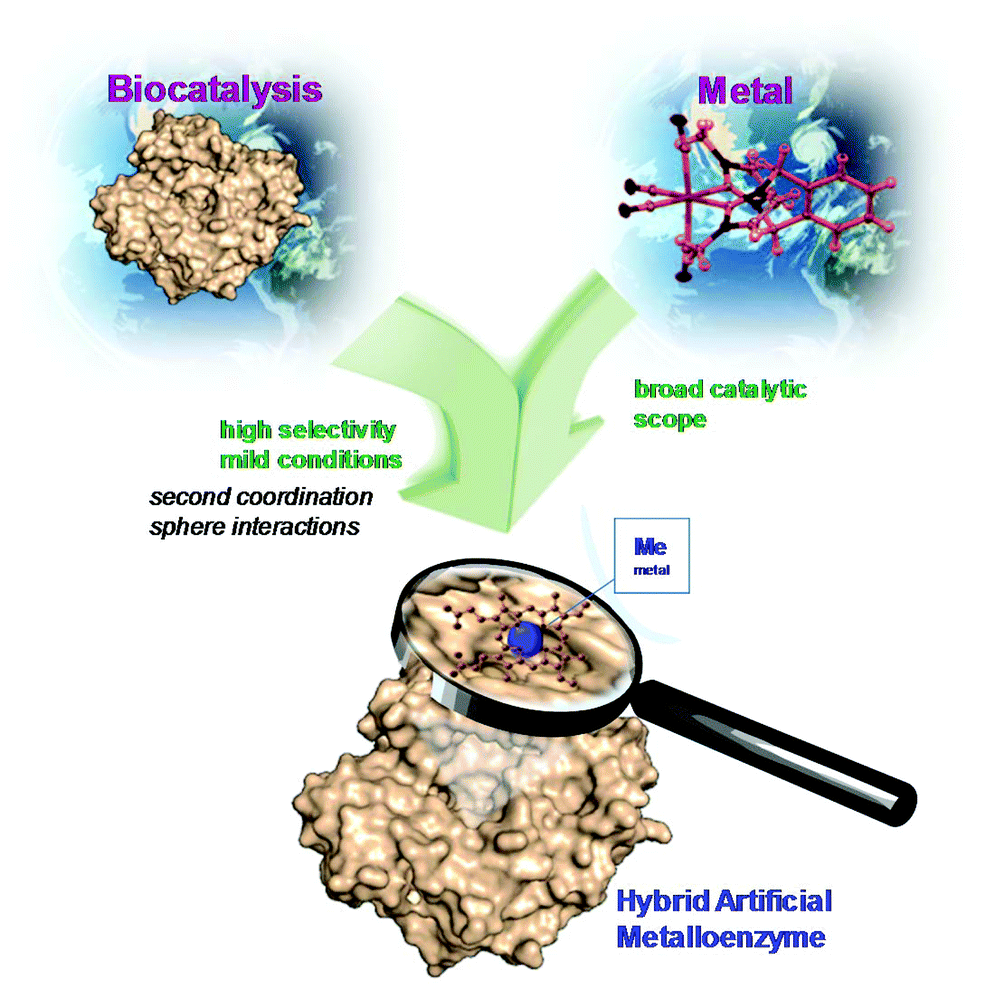 | ||
| Fig. 1 Concept of the preparation of artificial metalloenzymes. | ||
Different approaches have been described for the preparation of artificial metalloenzymes (Fig. 2).9 Mainly, we have emphasized three different strategies: (I) creation of metalloenzymes with new catalytic activities or improved activity from a natural one. A directed evolution technique to modify the metal-coordinated amino acid environment in natural metalloenzymes has been applied to produce enzymes with tuned or enhanced reactivity and selectivity towards non-natural substrates.9,10
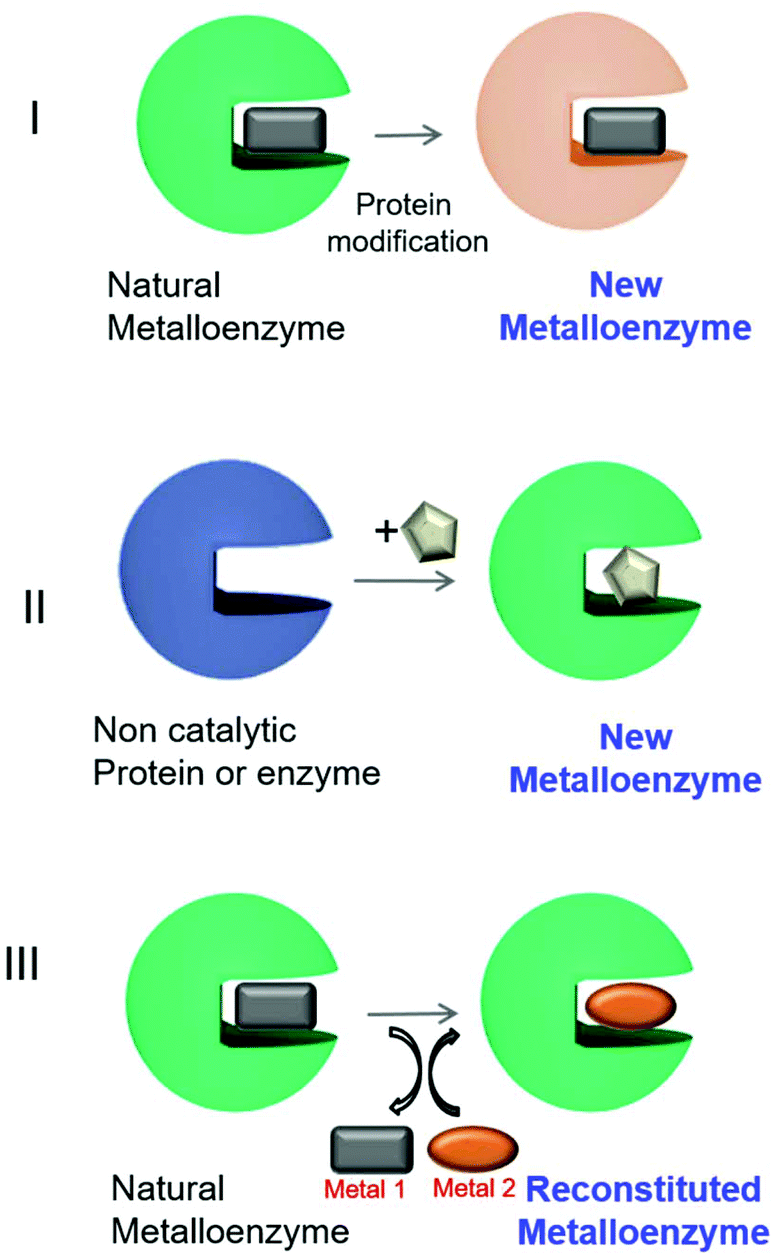 | ||
| Fig. 2 Different approaches for artificial metalloenzyme synthesis. The square, circle and pentagons represent organometallic complexes. | ||
A second approach (II, Fig. 2) has been focused on the creation of a completely new metalloenzyme by insertion strategies of an organometallic complex in a specific area of three-dimensional space within the protein cavity6,9,11,12 with the potential to confer selectivity to chemical reactions or completely new reactivities.13,14 This selectivity would be difficult to achieve using metal catalysts alone.6,9,12,15
The third strategy consists of the replacement of the existing metal ion or organometallic complex inside the natural metalloenzyme with an alternative one (III, Fig. 2), mainly using engineered heme-containing proteins.16,17
The application of these biocatalysts in organic chemistry has been a major challenge in the last few years. In particular, the C–C bond formation represents one of the most useful synthetic tools, since the generation of this bond allows obtaining innumerable “building blocks”, which is a necessary synthetic step in most of the pharmaceutical compounds.18,19 This type of bond can be obtained in several ways. One of the most advantageous ways is the C–H bond activation reaction since it uses molecules not previously activated, which is important for the atom economy and the synthesis process. This is, however, a complex reaction, much more difficult than other C–C bond formations such as cycloaddition, Suzuki–Miyaura or Heck reactions. Therefore, the creation of active and selective artificial metalloenzymes in this C–H functionalization process is a challenge necessary to move towards green chemistry.
This mini-review focuses on describing the few examples that up to now exist in the literature about the creation of artificial metalloenzymes with C–H activation activity.
2. New metalloenzymes by directed evolution
The application of enzymes and microorganisms in synthetic chemistry is the main focus of biocatalysis. This is primarily based on the high specificity and selectivity of enzymes in a particular process. However, these good properties normally correspond to natural substrates and normally they show very poor or no reactivity or selectivity towards non-natural compounds. Thus, innovation is mandatory to exploit their industrial capacity. Currently, DNA sequencing and gene synthesis together with bioinformatics are key for engineering proteins. The modification of biocatalysts using in vitro Darwinian evolution is commonly called directed evolution. This technique requires a method for generating variations in a protein, followed by the screening and selection of variants with improved characteristics. Frances Arnold is the pioneer in this field, which she started exploring in the 1990s.20 Arnold's efforts in this field bore fruit and she was awarded the Nobel Prize in Chemistry (2018).The Arnold group used directed evolution to improve biocatalysis, in particular creating enzymes for reactions that neither synthetic chemistry nor biology had exploited.10,21 Regarding C–H bond activation, they devoted their effort to engineering iron-containing proteins, making iron a suitable catalyst for these reactions. This strategy consisted of altering the coordination of the iron metal in the catalytic core of the enzyme by modifying the protein amino acids (type I class of metalloenzymes, Fig. 2).21
Thus, a highly enantioselective intramolecular amination of benzylic C–H bonds was obtained using an engineered modified cytochrome P450 monooxygenase (Cytocrome P411CHA).22 Particular mutations were located at the active sites (Fig. 3a) impacting positively the selectivity and activity when compared with another variant that did not have them. This variant showed excellent results in the enantioselective amination of alkane 1 (Fig. 3b) obtaining, under a particular set of conditions, the benzylic amine 2 at >99% enantiomeric excess (ee).
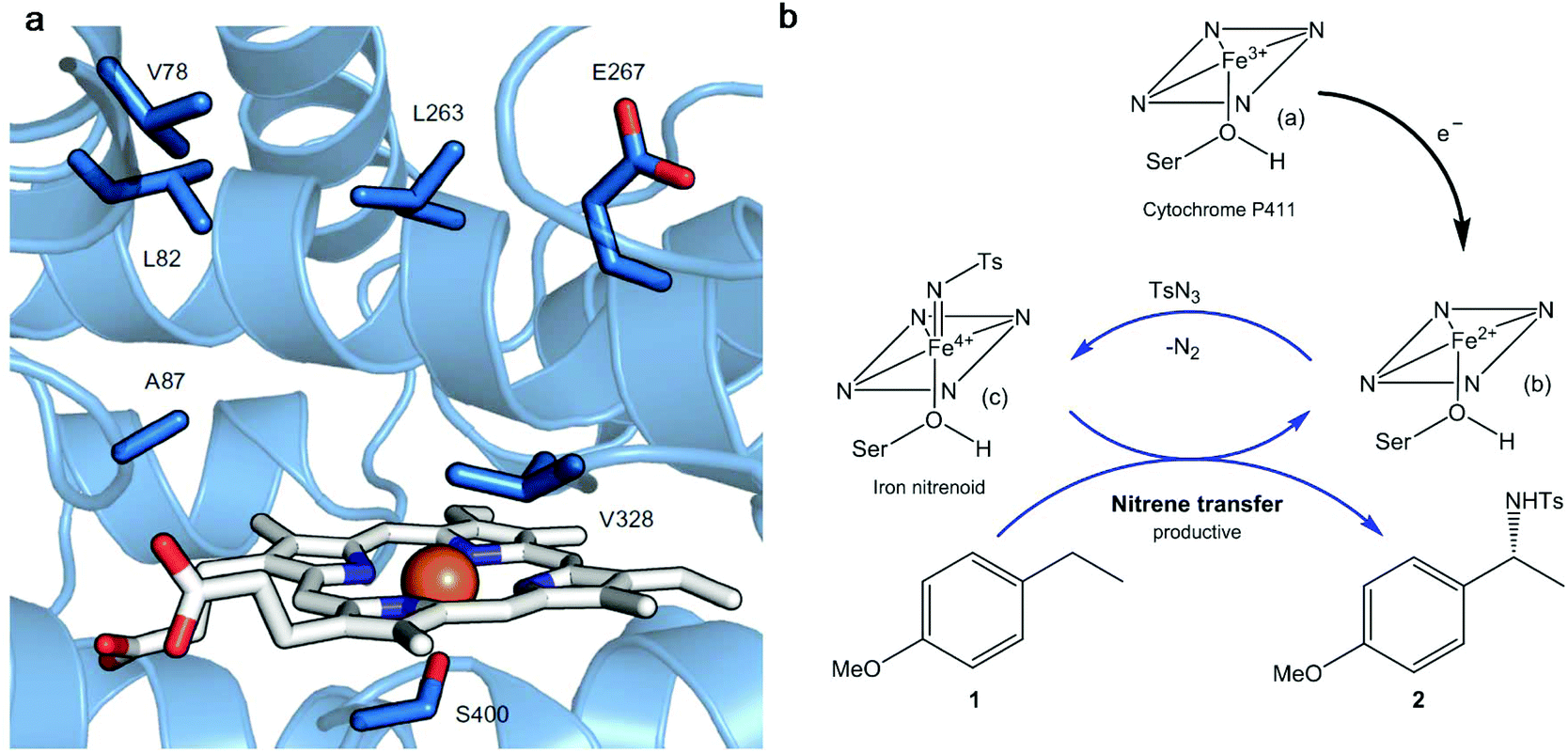 | ||
| Fig. 3 (a) Active site of the cytochrome P411 mutant (P-4 A82L A78V F263L). The haem group is presented in white and in orange is the iron atom. The key active site residues are shown as sticks in blue and labelled. (b) Cytocrome P411-catalysed C–H amination proposed mechanism. Nitrene transference is achieved thanks to the haem group reduction. Ts: p-toluenesulfonyl; Ser: serine. Figure adapted from ref. 22. | ||
The mechanism proposed for the intermolecular amination catalyzed by the enzyme consists, first of all, of the reduction of the ferric state of the haem cofactor ((a), Fig. 3b), with electrons derived from NADPH, giving the ferrous state (b). The reaction with tosyl azide (TsN3), a source of nitrene, provides the iron nitrenoid (c) (Fig. 3b). A subsequent reaction of this intermediate with 4-ethylanisole (1) would yield the amination product C–H 2 and regenerate the ferrous state of the catalyst (b).
Indeed, using 10 mM of both substrates (1 and TsN3), P411CHA artificial enzyme can support up to 1300 turnovers, far exceeding the highest turnover number (TON) reported with any chiral transition metal complex for intermolecular enantioselective C–H amination (the best reported is 85 TON using a chiral manganese porphyrin).23
Optimization of the reaction allowed the synthesis of product 2 with a yield of 86%, with >99% ee and 670 turnovers in water at room temperature in contrast to the chemical protocols reported (Fig. 4).
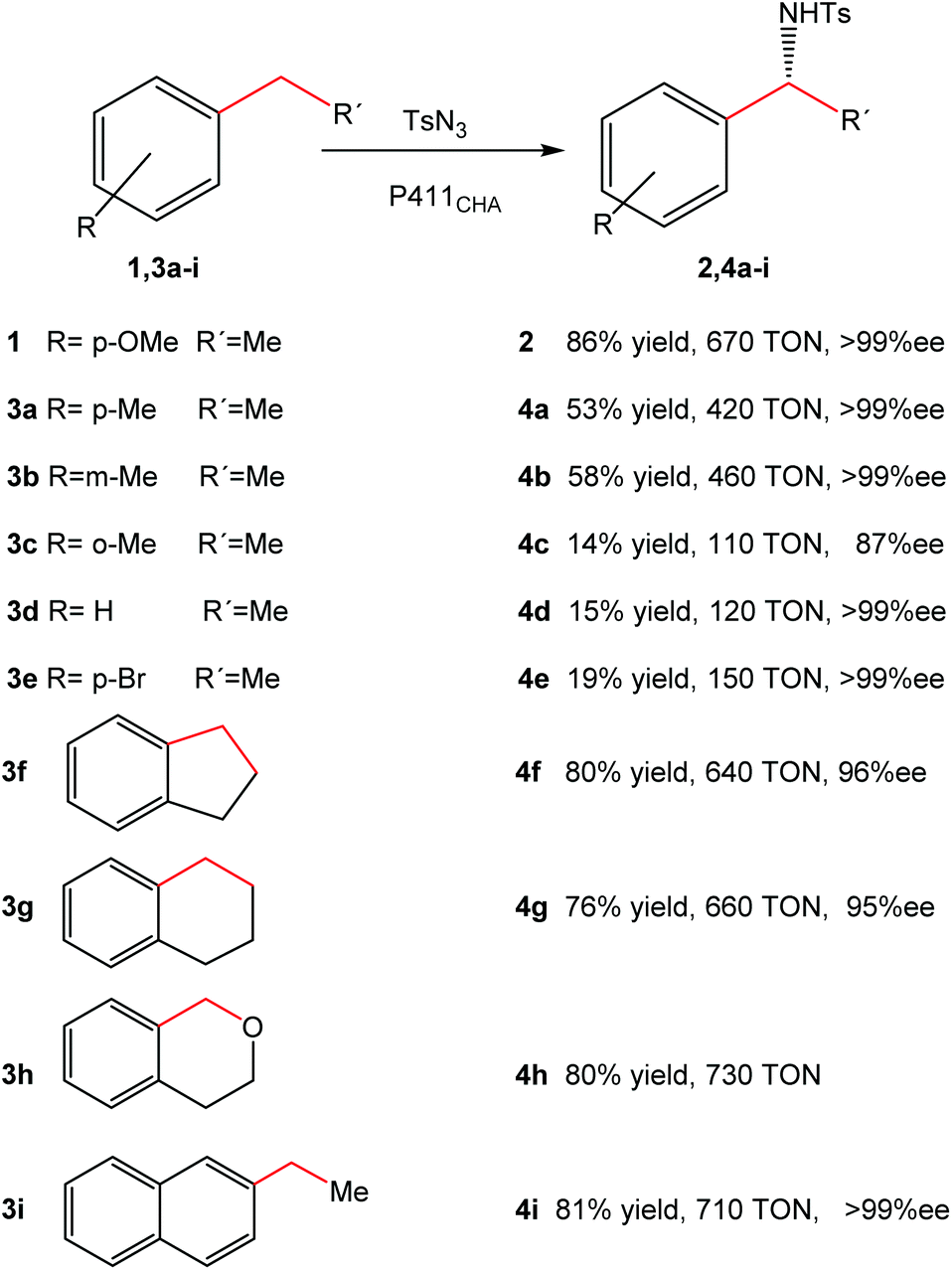 | ||
| Fig. 4 Substrate range of intramolecular C–H amination catalyzed by cells expressing P411CHA. The figure is adapted from ref. 22. | ||
Substrate range studies revealed that this artificial metalloenzyme recognized a wide variety of alkanes (Fig. 4), which makes this iron-based catalyst suitable for synthetic chemistry. The enantiomeric excesses for all substrates were excellent in most cases (>95% ee) (Fig. 4). The yields, on the other hand, depended on the substituents and the position in the aromatic ring, e.g. with the methyl substituent at the para or meta position (4a–4b), more than 50% yield was obtained (Fig. 4) while it was less than 15% when the group was at the ortho position (4c).22
The intramolecular activation of C–H produced good overall yields (>70%) (4f–4i) (Fig. 4).
A very recent work by the Arnold group also showed an outstanding application of another version of the evolved cytochrome P450 enzyme (Cytochrome P411CHF) in catalytic enantio-, regio- and chemoselective intermolecular alkylation of sp3 C–H bonds through carbene C–H insertion with high turnover and excellent selectivity (Fig. 5).24
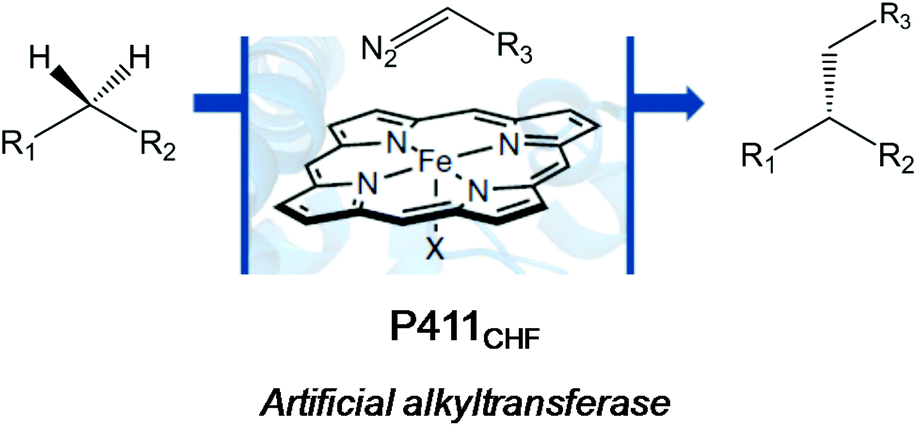 | ||
| Fig. 5 C–H alkylation reaction catalyzed by an artificial haem-protein. | ||
They created more than 70 different haem proteins tested in the C–H alkylation reaction at room temperature and under anaerobic conditions using p-methoxybenzyl methyl ether (5) and ethyl diazoacetate (6) (Fig. 6).24 Product 7 was synthesized using the cytochrome P411CHF variant with an excellent stereoselectivity 96.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.3 enantiomeric ratio (er) and 2020 total turnover number (TTN). Even, the enzymatic C–H activation was successfully extended using 1.0 mmol 5, obtaining an isolated product yield of 82%, 1060 TTN, and 98.0:2.0 er.24
:3.3 enantiomeric ratio (er) and 2020 total turnover number (TTN). Even, the enzymatic C–H activation was successfully extended using 1.0 mmol 5, obtaining an isolated product yield of 82%, 1060 TTN, and 98.0:2.0 er.24
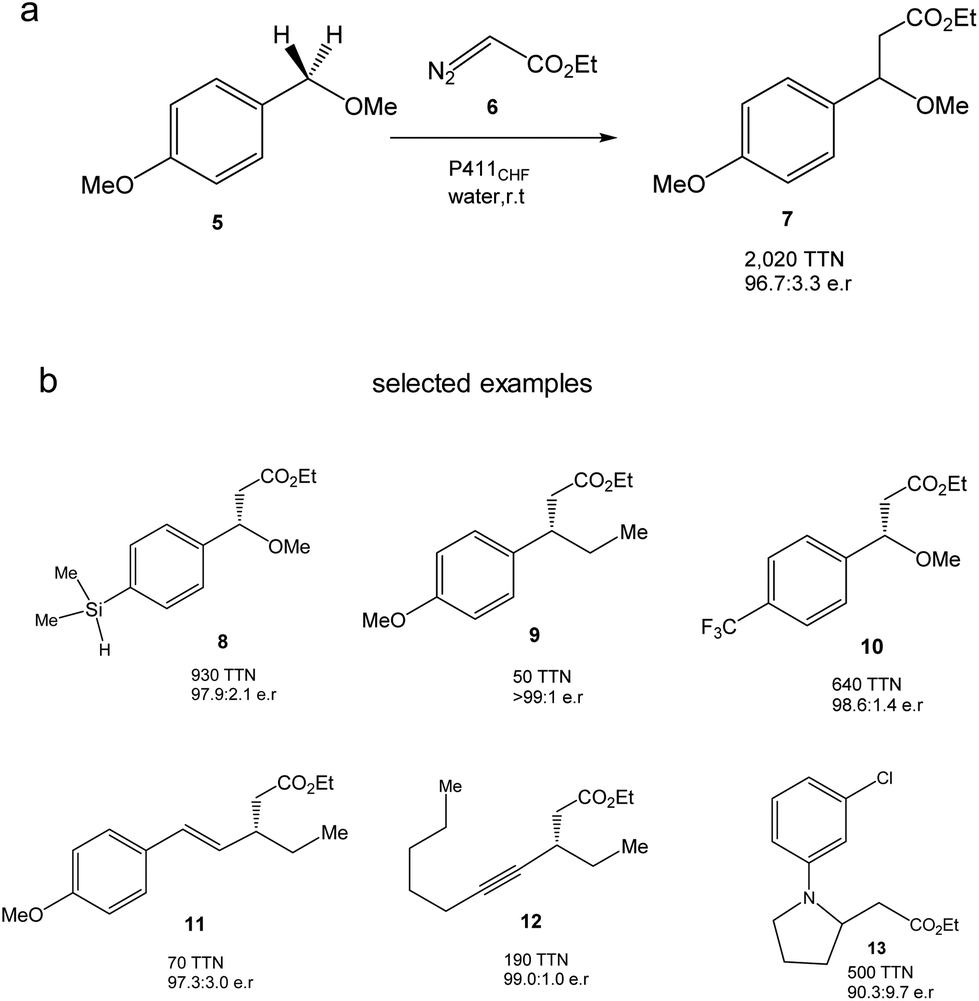 | ||
| Fig. 6 C–H alkylation catalyzed by the P411CHF enzyme variant. (a) Alkylation of 5. (b) Selected examples of synthesized products by this strategy. | ||
Finally, the authors demonstrated the applicability of this methodology to extend the enzymatic C–H alkylation in different benzylic, allylic, propargylic, alkylamine and diazo compounds, with up to 30 different compounds being successfully synthesized (Fig. 6b).24
C–H alkylation represents a very interesting chemical reaction. In this term, recently, the Fasan group has developed different evolved myoglobin variants for site-specific C–H cyclopropanation25–27 or functionalization of unprotected indoles.28
Functionalized indoles are very interesting molecules which have received significant attention.29 They are structural motifs found in many biologically active molecules.30
Initially, the Fasan group prepared a set of different variants of sperm whale myoglobin (Mb) by amino acid modifications at the active site.28 Then, they performed the first test of C3-functionalization directly on the reaction of an unprotected indole 14 and ethyl α-diazoacetate (6) to synthesize 15 (Fig. 7). They compared the new variants with the native Mb and the best result was found at pH 9, where 85% conversion of a completely C–H activation product was found using the Mb variant (H64V,V68A). The catalytic efficiency of this enzyme was more than 3 times higher than that reported using metal based catalysts for the C3 functionalization of N-protected indoles.31
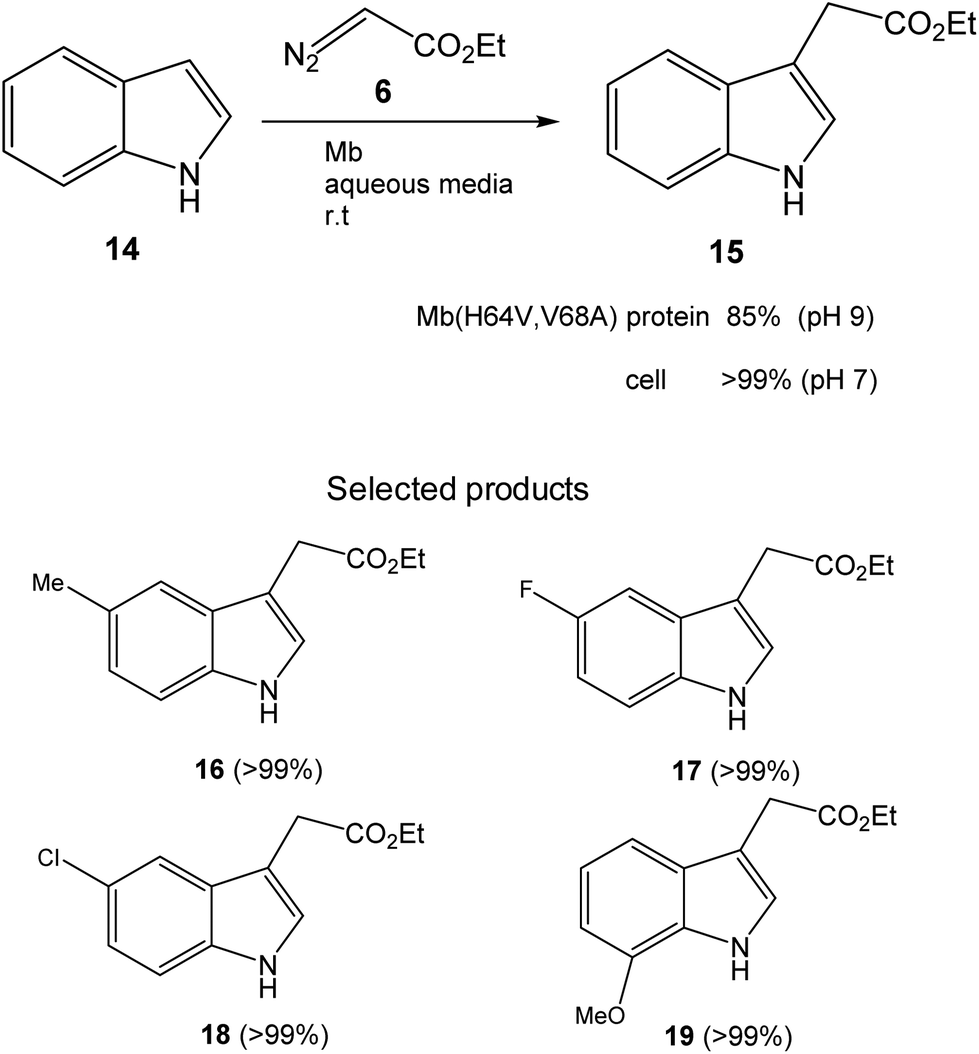 | ||
| Fig. 7 C–H functionalization of indole alkylation with ethyl α-diazoacetate catalyzed by the Mb(H64V,V68A) variant. | ||
However, they obtained a complete conversion in the process by using a whole cell biotransformation, using E. coli cells expressing the Mb(H64V,V68A) variant (Fig. 7). The scope of the reaction was carried out using whole cells as a catalyst using different substituted-indole substrates with excellent results in many cases (Fig. 7).
Engineered variants of cytochrome P45032 or myoglobin33 have also been created for synthetic C–H oxidation processes, with a clear application in the total enantioselective synthesis of the norditerpenoid alkaloid nigelladine A.32
The expansion in the C–H reactivity of the evolved cytochrome P450 has been recently demonstrated in the tunable enantioselective C–H intramolecular amidation reactions of divergently constructed β-, γ- and δ-lactams.34
In this case, directed evolution was used to obtain the LSsp3 variant that catalyzed the amidation reaction of 20 with a TTN (total turnover number) value of >200000 and an enantiomeric excess (ee) of 96% (Fig. 8). The application of this variant to other substrates once again demonstrated the potential of this technique.34
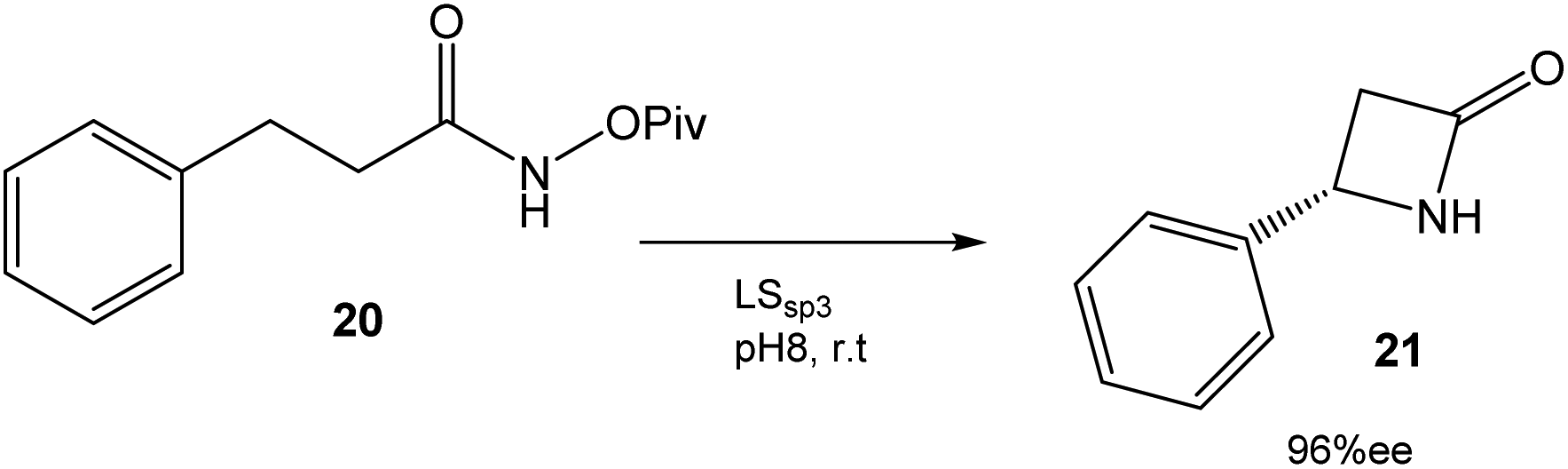 | ||
| Fig. 8 Synthesis of β-lactams by an enantioselective C–H reaction catalyzed by P411 variants.34 | ||
3. Artificial metalloprotein hybrids
A second approach to produce artificial metalloenzymes is based on anchoring a metal cofactor within a protein, combining characteristics that typically are from homogeneous catalysts with those from enzymes (II, Fig. 2).14,17 There are different strategies to achieve this. They differ in how the metal is incorporated into the protein and in how the purpose of the protein was redesigned.35 Artificial metalloenzymes can be synthesized by modifying the abiotic cofactor that is inserted or by genetic engineering of the protein.9,10 This extends their possibilities since chemistry and biotechnology can work together to improve the catalytic properties.6In this area, a successful strategy has been developed in recent years by the Ward group, using the biotin-(strept)avidin technology to locate biotinylated metal derivatives within (strept)avidin.6,9,11,35–38 This methodology has allowed the synthesis of different artificial metalloenzymes with very efficient catalytic properties in different reactions such as reduction, oxidation and C–C bond formation.6,35–38
In particular, one of the most successful complex applications of this technology has been the activation of C–H bonds. In this term, the conjugation of streptavidin with biotin site-specifically modified with an organometallic rhodium(III) molecule generated a highly active and selective catalyst for an asymmetric C–H activation (Fig. 9).
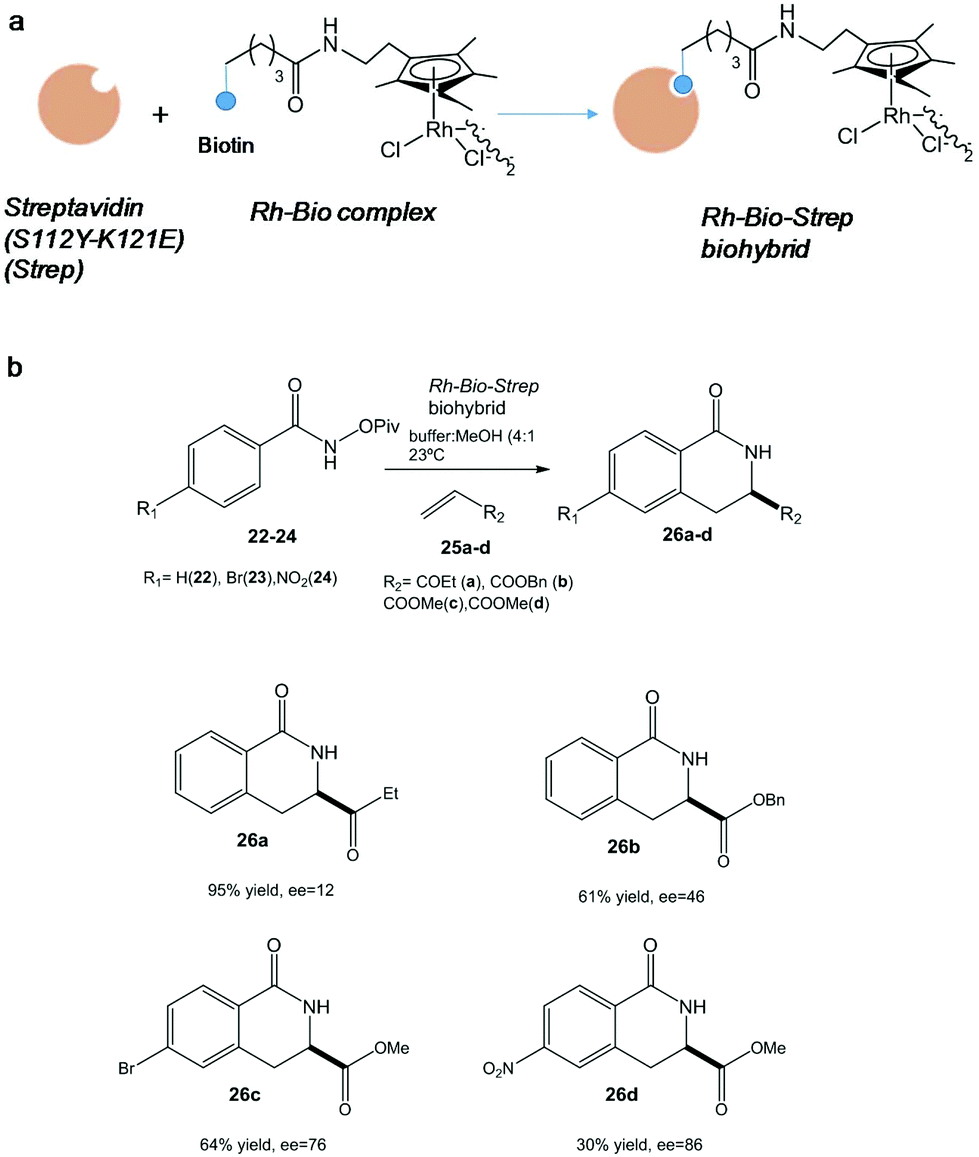 | ||
| Fig. 9 (a) Artificial metalloenzyme preparation. (b) Benzannulation reaction of different substrates catalyzed by the Rh-Bio-Strep biohybrid. | ||
Interesting results were found depending on the substituent on the aromatic ring in the pivaloyl-protected benzhydroxamic acid (22–24) or on the alkene moiety (25a-d) (Fig. 9b). In particular, isoquinolone 26d was synthesized using this artificial metalloenzyme with a very high enantiopreference and enantioselectivity with >85% enantiomeric excess (ee), showing a 100-fold rate acceleration compared to the isolated Rh(III) complex.38
Following this previous approach, the Rovis group has recently performed the asymmetric synthesis of α,β-unsaturated-δ-lactams by a tandem C–H activation and [4 + 2] annulation reaction using a monomeric streptavidin-Rh(III)-complex hybrid.39
In this case the Rh cyclopentadienyl embedded on monomeric streptavidin, in comparison with the tetrameric protein employed by the previous group, was an excellent catalyst for the preparation of different δ-lactams, via a direct enantioselective coupling of acrylamide hydroxamate esters and styrenes, with an excellent yield (99%) and enantiomeric excess (>96%) under aqueous conditions at room temperature.39
A different approach focused on the creation of an artificial Ru-P450 hybrid for a photocatalytic C–H activation (Fig. 10).40,41
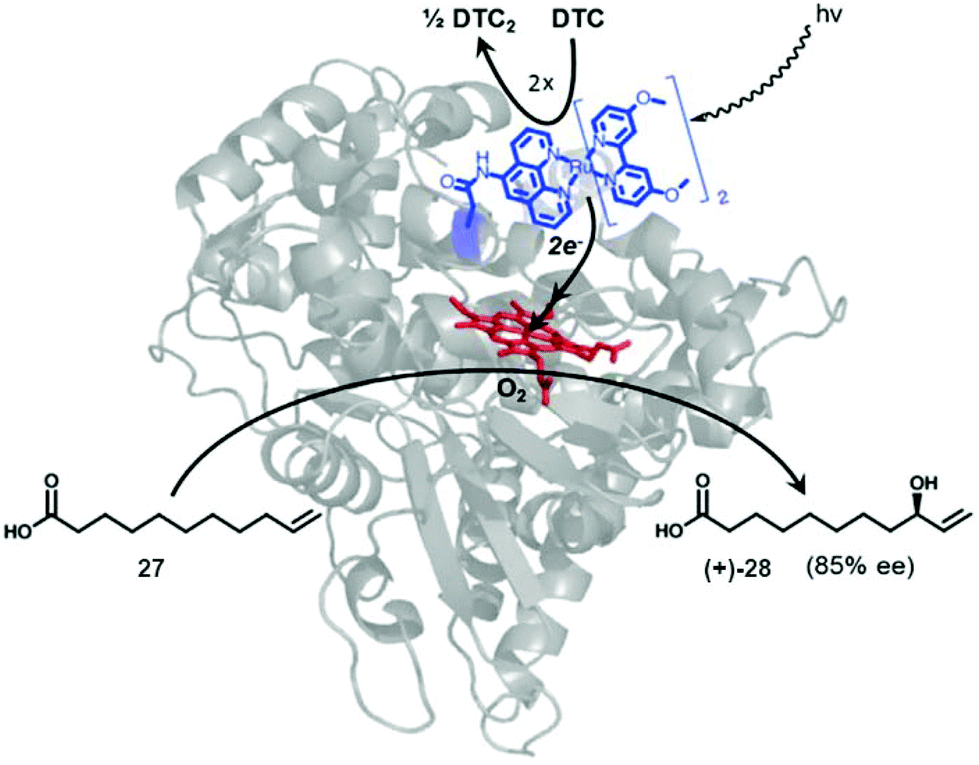 | ||
| Fig. 10 Representation of a photocatalytic C–H activation by a novel Ru-P450 hybrid enzyme. Reprinted with permission from ref. 41. Copyright 2014 Elsevier. | ||
A set of different Ru(ii) complexes as photosensitizers were covalently attached near the heme active site of the P450 BM3 enzyme, in order to evaluate the effect of the substituent in photocatalytic applications. The mechanism of action of the new hybrid system is based on the fact that upon light activation, the Ru(II) photosensitizer is quenched by sodium diethyldithiocarbamate (DTC), generating highly reductive species capable of rapidly delivering electrons to the heme active site to initiate the C–H activation reaction.
One of these hybrid catalysts was successfully applied in the C–H activation under visible light by selective monohydroxylation of 10-undecenoic acid (27) producing the enantiomer R-28 in 85% ee. Hydroxylation occurs exclusively at the allylic position (Fig. 10).41
This result opens a future line of research into the generation of hybrid enzymes activated by light for photocatalytic C–C bond reactions.
4. Heme-reconstituted artificial metalloenzymes
The obtaining of artificial metalloenzymes through the replacement of a naturally existing metal ion or organometallic complex inside the enzyme with a different one (III, Fig. 2) has been described as a very useful strategy for creating new abiological catalysts, expanding its applicability to a particular enzyme for a wide range of non-natural reactions.42–45In this case, the incorporation of the metal by using this strategy could overcome some limitations that could exist in terms of spatial arrangement of the metal complex at the protein active sites in the case of the preparation of artificial metalloproteins from un-natural metalloenzymes.
The methodology has been clearly demonstrated in oxidation reactions, and C–C bond processes46 and, recently, some examples confirmed its applicability in the activation of C–H in water.42–45
The Hartwig group pioneered the development of new artificial enzymes with new reactivities by formally replacing iron in Fe-porphyrin IX (Fe-PIX) proteins with non-biological metals (Fig. 11), with a particular focus on C–H activation enzymes.42–44
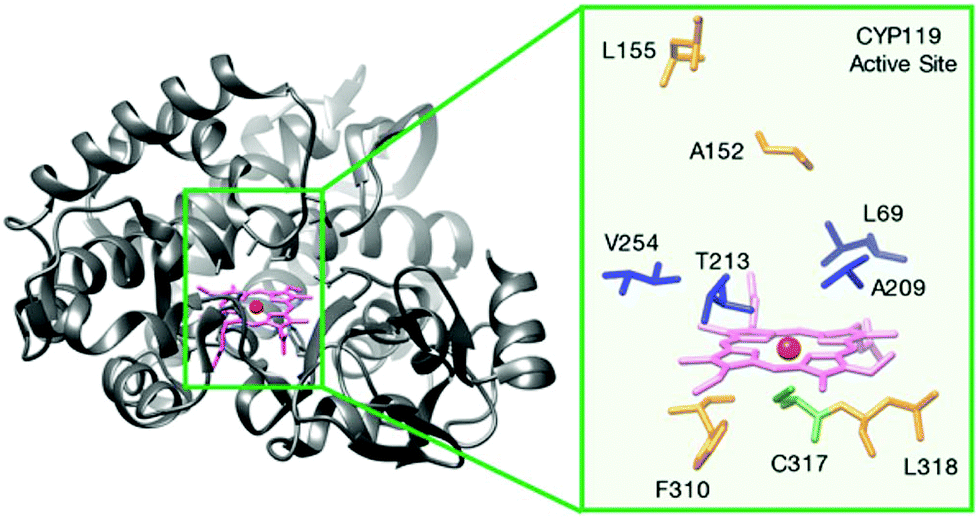 | ||
| Fig. 11 Structure of a cytochrome P450, Fe-CYP119 and its active centre residues. Reprinted with permission from ref. 44. Copyright 2017 American Chemical Society. | ||
They selected cytochrome P450 enzymes (P450s) that are native metalloproteins containing Fe-protoporphyrin IX (Fe-PIX) as a model for the creation of new reconstituted enzymes. This was a good example because, in principle, the native enzymes could catalyze some C–H oxidation and halogenation47 but not less reactive C–H bond functionalization.
In the process of creating robust artificial metalloenzymes, they designed an artificial reconstituted enzyme from an engineered P450 enzyme variant (CYP119)44 (Fig. 11), which is a thermally stable P450 from the archaeon Sulfolobus solfataricus.
This CYP119 P450 enzyme variant was used for the creation of a reconstituted metalloenzyme in which the heme was exchanged for an iridium porphyrin cofactor (Ir(Me)-PIX) that successfully catalyzed the enantioselective intramolecular C–H bond amination reaction of sulfonyl azides (Fig. 12).43
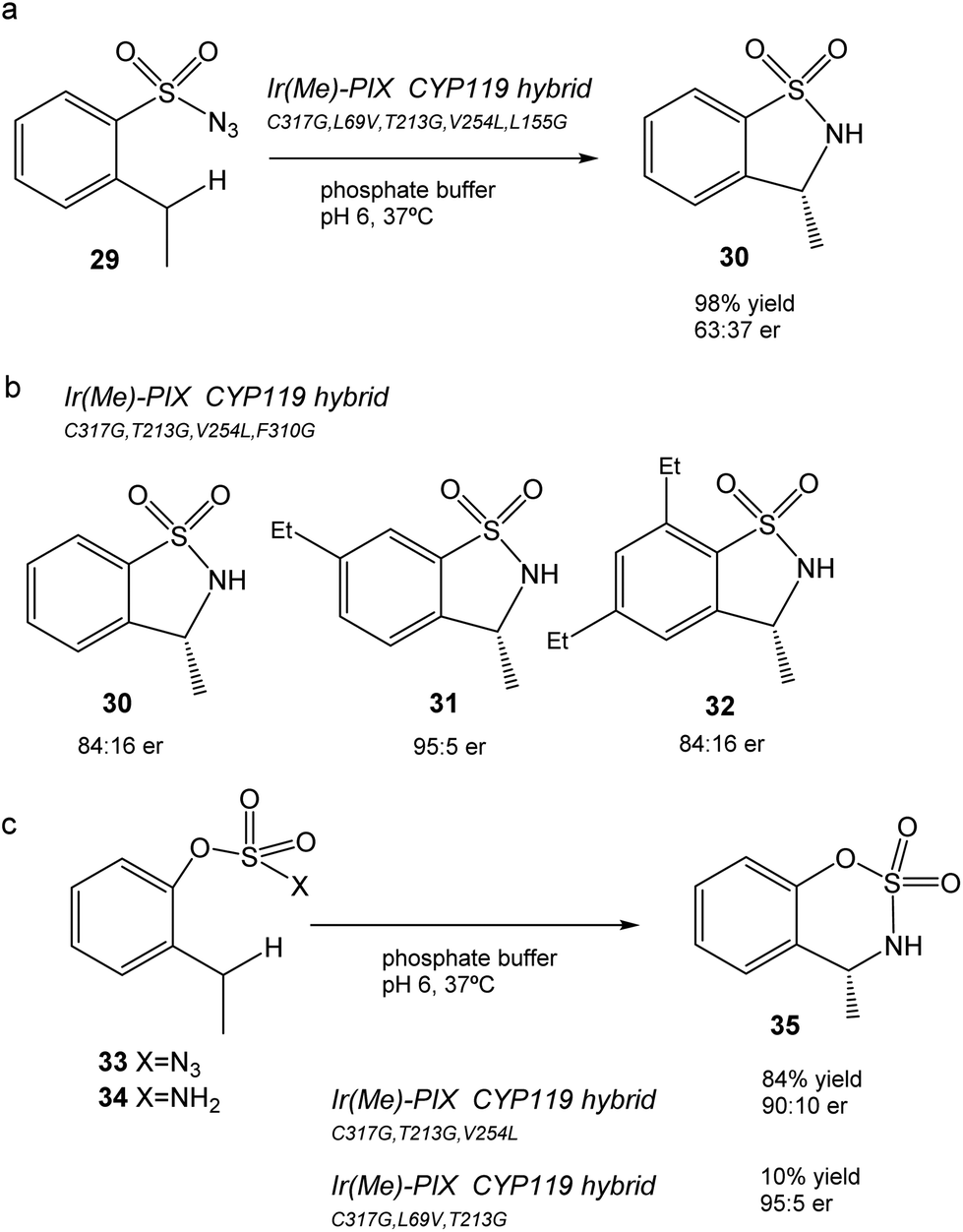 | ||
| Fig. 12 Chemoselective C–H bond amination to the formation of benzo-fused sulfamates using different artificial Ir(Me)-PIX CYP119 variant enzymes. (a) Reaction of sulfonyl azide 29. (b) Different sulphonamides synthesized (30–32) by using different artificial enzyme variants as catalysts. (c) C–H amination of aryloxysulfonyl azides 33–34. | ||
Initially, the previous artificial enzyme Ir(Me)-PIX CYP119, with the CYP119 variant containing five mutations (C317G, L69V, T213G, V254L, and L155G), showed excellent catalytic performance in the C–H reaction to transform sulfonyl azide 29 in the sulfonamide 30 with a yield of 98% with an enantiomeric ratio of 63:37 (Fig. 12a).43
Different variants of CYP119 were created, in which 3 to 5 amino acids were modified from those mutated at the active sites by means of molecular biology techniques.44 These were used to create new Ir-artificial enzymes. By changing the mutations in the variant, this artificial metalloenzyme showed excellent results in the C–H activation, for example with 29 improving the enantiomeric ratio (84:16) or using different substrates that produce different sulfonamides 31–32, particularly 31 in almost quantitative yields with excellent enantioselectivity (95:5 er) and chemoselectivity (20:1) (Fig. 12b).43
In addition, the preparation of Ir-CYP119 artificial enzymes, where the protein contained three mutations, showed excellent results in the enantioselective C–H amination of aryloxysulfonyl azides 33–34, especially in 33 (with azide instead of an amino group), where >80% yield of product 35 was obtained with a high enantiomeric ratio (90:10) (Fig. 12c). This result represents a challenging C–H amination reaction, because no known enzyme catalyzes the transformation of compound 33 to 35, and forms the product with low yield and enantioselectivity with rhodium catalysts.
Myoglobins are another type of hemoprotein used as a starting point for the creation of artificial metalloenzymes through this reconstitution strategy.42
In this case, the Hartwig group applied a similar strategy discussed earlier in a previous section to create a number of novel artificial enzymes by adding abiological metal complexes such as the metal–porphyrin complexes of Ru, Rh, Ir, Co, Cu, Mn and Ag to apo-PIX-proteins.42 These artificial [M]-myoglobins, which contain various metals and axial ligands, were tested in the C–H functionalization reaction of diazoester 36 to form chiral dihydrobenzofuran 37 (Fig. 13). All native myoglobins (containing iron complex) were completely inactive in the reaction, whereas the new artificial myoglobins containing different metal complexes (Co(Cl), Rh or Ir(Cl), among others) were active catalysts in this C–H activation process.
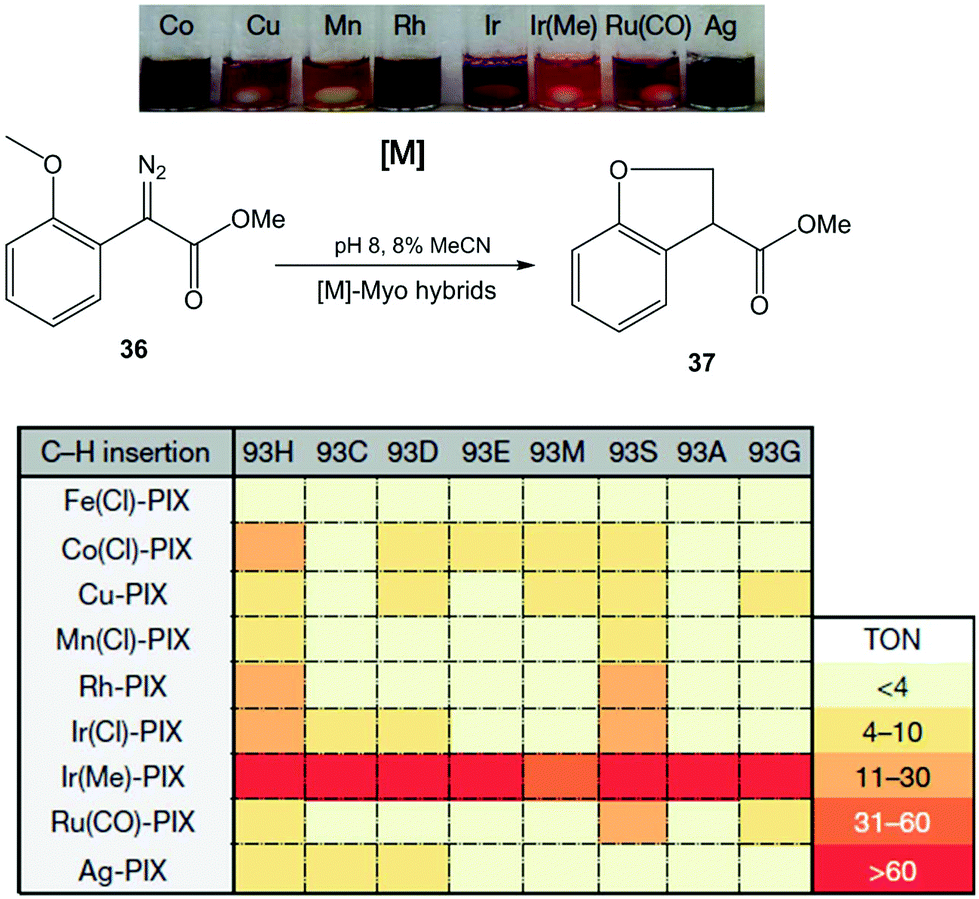 | ||
| Fig. 13 Evaluation of the artificial Myo catalysts for the insertion of carbenes into C–H bonds. TON: turnover number. The figure is adapted from ref. 42. | ||
Differences were also observed with the same metal complex introduced in Myo-H93X, each with a different mutation at the axial ligand position (H93X) (Fig. 13). However, the best catalyst for C–H functionalization was the variant containing Ir(Me)-PIX synthesised incorporating both a non-native metal (Ir) and an axial ligand (–CH3) that cannot be incorporated by standard mutagenesis techniques. In this case, good results were obtained in almost all cases, independently of the mutation at position 93 (50% yield without any mutation) (Fig. 13).
The directed evolution strategy was also used in the enzyme to find the optimal Ir(Me)PIX-Myo artificial enzymes able to perform C–H activation on a variety of substrates.42
After four rounds and more than 20 mutants, the variant containing mutations in 93G, 64L, 43L and 99F (Fig. 14) was the optimal Ir-PIX-Myo enzyme that catalyzed the production of 38 with excellent enantioselectivity, with an enantiomeric ratio of 98:2 (TON of 92)42 instead of the non-selective results using the starting non-evolved enzyme (Fig. 14), demonstrating that the combination of chemical and molecular biological strategies allows the expansion and scope of the most difficult C–H functionalization of these new artificial enzymes.
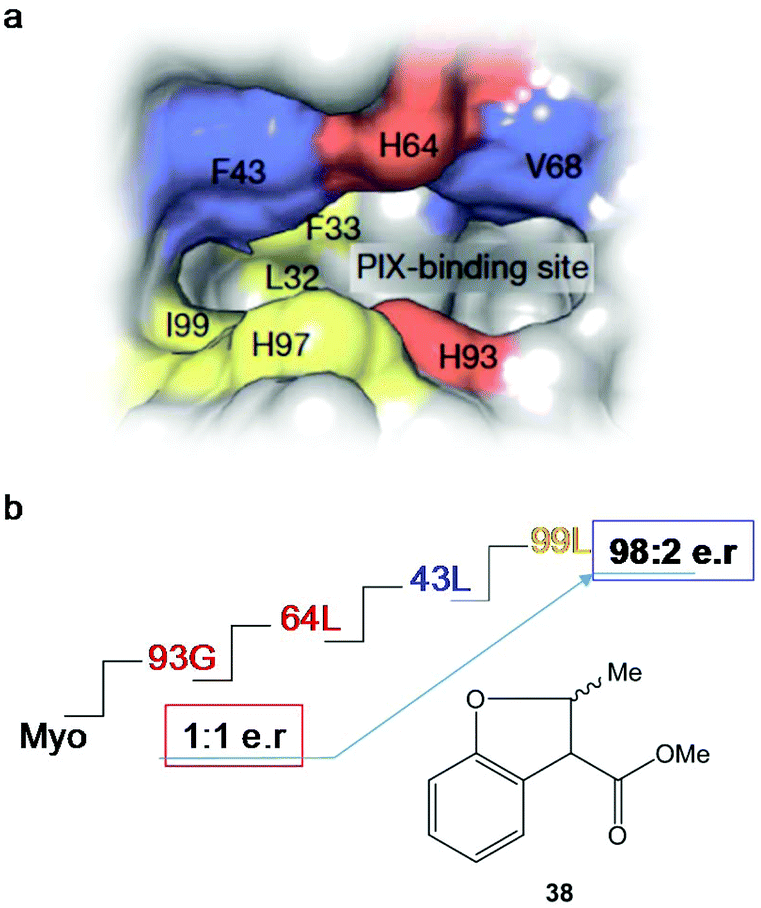 | ||
| Fig. 14 Strategy using directed evolution on myoglobin to produce new enantioselective Ir-Myo-mutants. (a) Image of the active site and its surrounding produced in Chimera from PDB 1MBN. (b) Directed evolution (4-rounds) to produce the best final mutant to obtain the most enantioselective artificial enzyme in the synthesis of 38. The figure is adapted from ref. 42. | ||
5. Conclusions
C–H bond activation is one of the main challenges faced in organic chemistry. In this review, we have compiled recent advances in the design of biocatalysts for C–H bond functionalization, using combinations of proteins/enzymes and metals or organometallic complexes for the creation of new artificial metalloenzymes. This is an important area, but as the chemistry of C–H activation under mild conditions is still in its infancy, new research studies are still necessary to improve this technology for industrial applications.The use of genetic tools, especially directed evolution, has allowed the transformation of natural enzymes with specific activity to synthetic enzymes, many of them not existing in Nature. Direct insertion of organometallic complexes into the specific regions of a protein has proved to be a solid technique for performing C–H activation reactions as well as photocatalysis.
In addition, the search for new alternatives in the development of these types of reactions with biohybrids is a safe bet. One of the examples in which biocatalysis has a wide field to explore is the use of Earth-abundant metals such as Fe, Cu, Co or Mn as catalysts in C–H reactions. This can be achieved in some cases by inserting a metallic complex into a protein structure, but research is still needed to expand this field.
The development of methodologies that allow the generation of alternative active sites in specific enzymes, generating enzymatic and metallic activities, has been the key for the success of biocatalysis. Future advances in molecular genetics, protein engineering, bioinformatics or nanotechnology will pave the way for further research. The development of artificial metalloenzymes that combine multiple catalytic activities in a single structure will reduce the steps necessary to obtain a final product. This is really interesting from an economic point of view, due to its possible impact on the chemical or pharmaceutical industries, for example.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Government, the Spanish National Research Council (CSIC), the Ministry of Education, Youth and Sports of the Community of Madrid and the European Social Fund (PEJD-2017PRE/SAL-3762). The authors thank the European Cooperation in Science and Technology (COST) program under CA15106 grant (CHAOS: CH Activation in Organic Synthesis).References
- J. C. Lewis, Beyond the Second Coordination Sphere: Engineering Dirhodium Artificial Metalloenzymes To Enable Protein Control of Transition Metal Catalysis, Acc. Chem. Res., 2019, 52, 576–584 CrossRef CAS.
- U. Markel, D. F. Sauer, J. Schiffels, J. Okuda and U. Schwaneberg, Towards the Evolution of Artificial Metalloenzymes-A Protein Engineer's Perspective, Angew. Chem., Int. Ed., 2019, 58, 4454–4464 CrossRef CAS PubMed.
- G. Roelfes, LmrR: A Privileged Scaffold for Artificial Metalloenzymes, Acc. Chem. Res., 2019, 52, 545–556 CrossRef CAS.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Protein Design: Toward Functional Metalloenzymes, Chem. Rev., 2014, 114, 3495–3578 CrossRef CAS PubMed.
- J. C. Lewis, Artificial Metalloenzymes and Metallopeptide Catalysts for Organic Synthesis, ACS Catal., 2013, 3, 2954–2975 CrossRef CAS.
- A. D. Liang, J. Serrano-Plana, R. L. Peterson and T. R. Ward, Artificial Metalloenzymes Based on the Biotin–Streptavidin Technology: Enzymatic Cascades and Directed Evolution, Acc. Chem. Res., 2019, 52, 585–595 CrossRef CAS.
- K. M. Lancaster, Revving up an Artificial Metalloenzyme, Science, 2018, 361, 1071–1072 CrossRef CAS PubMed.
- F. Nastri, M. Chino, O. Maglio, A. Bhagi-Damodaran, Y. Lu and A. Lombardi, Design and Engineering of Artificial Oxygen-Activating Metalloenzymes, Chem. Soc. Rev., 2016, 45, 5020–5054 RSC.
- Artificial Metalloenzymes and MetalloDNAzymes in Catalysis, ed. M. Diéguez, J.-E., Bäckvall and O., Pàmies, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018 Search PubMed.
- R. K. Zhang, X. Huang and F. H. Arnold, Selective C–H Bond Functionalization with Engineered Heme Proteins: New Tools to Generate Complexity, Curr. Opin. Chem. Biol., 2019, 49, 67–75 CrossRef CAS.
- M. Dürrenberger and T. R. Ward, Recent Achievements in the Design and Engineering of Artificial Metalloenzymes, Curr. Opin. Chem. Biol., 2014, 19, 99–106 CrossRef PubMed.
- M. Filice, O. Romero, A. Aires, J. M. Guisan, A. Rumbero and J. M. Palomo, Preparation of an Immobilized Lipase-Palladium Artificial Metalloenzyme as Catalyst in the Heck Reaction: Role of the Solid Phase, Adv. Synth. Catal., 2015, 357, 2687–2696 CrossRef CAS.
- D. F. Sauer, T. Himiyama, K. Tachikawa, K. Fukumoto, A. Onoda, E. Mizohata, T. Inoue, M. Bocola, U. Schwaneberg and T. Hayashi, et al., A Highly Active Biohybrid Catalyst for Olefin Metathesis in Water: Impact of a Hydrophobic Cavity in a β-Barrel Protein, ACS Catal., 2015, 5, 7519–7522 CrossRef CAS.
- A. Chatterjee, H. Mallin, J. Klehr, J. Vallapurackal, A. D. Finke, L. Vera, M. Marsh and T. R. Ward, An Enantioselective Artificial Suzukiase Based on the Biotin–streptavidin Technology, Chem. Sci., 2016, 7, 673–677 RSC.
- P. Srivastava, H. Yang, K. Ellis-Guardiola and J. C. Lewis, Engineering a Dirhodium Artificial Metalloenzyme for Selective Olefin Cyclopropanation, Nat. Commun., 2015, 6, 7789 CrossRef CAS.
- K. Oohora, Y. Kihira, E. Mizohata, T. Inoue and T. Hayashi, C(Sp 3)–H Bond Hydroxylation Catalyzed by Myoglobin Reconstituted with Manganese Porphycene, J. Am. Chem. Soc., 2013, 135, 17282–17285 CrossRef CAS.
- C. L. Davies, E. L. Dux and A. K. Duhme-Klair, Supramolecular Interactions between Functional Metal Complexes and Proteins, Dalton Trans., 2009, 46, 10141 RSC.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Palladium-Catalyzed Cross-Coupling Reactions in Total Synthesis, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS.
- C. H. Jun, Transition Metal-Catalyzed Carbon–carbon Bond Activation, Chem. Soc. Rev., 2004, 33, 610–618 RSC.
- K. Chen and F. H. Arnold, Tuning the Activity of an Enzyme for Unusual Environments: Sequential Random Mutagenesis of Subtilisin E for Catalysis in Dimethylformamide, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5618–5622 CrossRef CAS PubMed.
- F. H. Arnold, Directed Evolution: Bringing New Chemistry to Life, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS.
- C. K. Prier, R. K. Zhang, A. R. Buller, S. Brinkmann-Chen and F. H. Arnold, Enantioselective, Intermolecular Benzylic C-H Amination Catalysed by an Engineered Iron-Haem Enzyme, Nat. Chem., 2017, 9, 629–634 CrossRef CAS.
- X.-G. Zhou, X.-Q. Yu, J.-S. Huang and C.-M. Che, Asymmetric amidation of saturated C–H bonds catalysed by chiral ruthenium and manganese porphyrins, Chem. Commun., 1999, 23, 2377–2378 RSC.
- R. K. Zhang, K. Chen, X. Huang, L. Wohlschlager, H. Renata and F. H. Arnold, Enzymatic assembly of carbon–carbon bonds via iron-catalysed sp3 C–H functionalization, Nature, 2019, 565, 67–194 CrossRef CAS.
- E. Moore and R. Fasan, Effect of proximal ligand substitutions on the carbene and nitrene transferase activity of myoglobin, Tetrahedron, 2019, 75, 2357–2363 CrossRef CAS.
- A. Tinoco, Y. Wei, J. P. Bacik, D. Carminati, E. Moore, N. Ando, Y. Zhang and R. Fasan, Origin of High Stereocontrol in Olefin Cyclopropanation Catalyzed by an Engineered Carbene Transferase, ACS Catal., 2019, 9, 1514–1524 CrossRef CAS.
- A. Chandgude and R. Fasan, Highly Diastereo- and Enantioselective Synthesis of Nitrile-Substituted Cyclopropanes by Myoglobin-Mediated Carbene Transfer Catalysis, Angew. Chem., Int. Ed., 2018, 57, 15852–15856 CrossRef CAS.
- D. Vargas, A. Tinoco, V. Tyagi and R. Fasan, Myoglobin-catalyzed C—H functionalization of unprotected indoles, Angew. Chem., Int. Ed., 2018, 57, 9911–9915 CrossRef CAS PubMed.
- J. B. Chen and Y. X. Jia, Recent progress in transition-metal-catalyzed enantioselective indole functionalizations, Org. Biomol. Chem., 2017, 15, 3550–3567 RSC.
- A. J. Kochanowska-Karamyan and M. T. Hamann, Marine Indole Alkaloids: Potential New Drug Leads for the Control of Depression and Anxiety, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS.
- X. Gao, B. Wu, Z. Yan and Y. G. Zhou, Copper-catalyzed enantioselective C–H functionalization of indoles with an axially chiral bipyridine ligand, Org. Biomol. Chem., 2016, 14, 8237–8240 RSC.
- S. A. Loskot, D. K. Romney, F. H. Arnold and B. M. Stoltz, Enantioselective Total Synthesis of Nigelladine A via Late-Stage C-H Oxidation Enabled by an Engineered P450 Enzyme, J. Am. Chem. Soc., 2017, 139, 10196–10199 CrossRef CAS.
- K. Oohora, H. Meichin, Y. Kihira, H. Sugimoto, Y. Shiro and T. Hayashi, Manganese(V) Porphycene Complex Responsible for Inert C−H Bond Hydroxylation in a Myoglobin Matrix, J. Am. Chem. Soc., 2017, 139, 18460–18463 CrossRef CAS.
- I. Cho, Z. Jia and F. H. Arnold, Genetically Tunable Enzymatic C - H Amidation for Lactam Synthesis, Science, 2019, 364, 575–578 CrossRef CAS.
- T. Heinisch and T. R. Ward, Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Challenges and Opportunities, Acc. Chem. Res., 2016, 49, 1711–1721 CrossRef CAS.
- T. Heinisch, F. Schwizer, B. Garabedian, E. Csibra, M. Jeschek, J. Vallapurackal, V. B. Pinheiro, P. Marlière, S. Panke and T. R. Ward, E. Coli Surface Display of Streptavidin for Directed Evolution of an Allylic Deallylase, Chem. Sci., 2018, 9, 5383–5388 RSC.
- M. Szponarski, F. Schwizer, T. R. Ward and K. Gademann, On-Cell Catalysis by Surface Engineering of Live Cells with an Artificial Metalloenzyme, Commun. Chem., 2018, 1, 84 CrossRef.
- T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Biotinylated Rh(III) Complexes in Engineered Streptavidin for Accelerated Asymmetric C-H Activation, Science, 2012, 338, 500–503 CrossRef CAS.
- I. S. Hassan, A. N. Ta, M. W. Danneman, N. Semakul, M. Burns, C. H. Basch, V. N. Dippon, B. R. McNaughton and T. Rovis, Asymmetric δ-Lactam Synthesis with a Monomeric Streptavidin Artificial Metalloenzyme, J. Am. Chem. Soc., 2019, 141, 4815–4819 CrossRef CAS.
- H. Halan, A. Colbert, T. T. Nguyen, M. Kato and L. Cheruzel, Correlating the para-Substituent Effects on Ru(II)-Polypyridine Photophysical Properties and on the Corresponding Hybrid P450 BM3 Enzymes Photocatalytic Activity, Inorg. Chem., 2017, 56, 6558–6564 CrossRef.
- M. Kato, D. Nguyen, M. Gonzalez, A. Cortez, S. E. Mullen and L. E. Cheruzel, Regio- and stereoselective hydroxylation of 10-undecenoic acid with a light-driven P450 BM3 biocatalyst yielding a valuable synthon for natural product synthesis, Bioorg. Med. Chem., 2014, 22, 5687–5691 CrossRef CAS.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Abiological Catalysis by Artificial Haem Proteins Containing Noble Metals in Place of Iron, Nature, 2016, 534, 534–537 CrossRef CAS.
- P. Dydio, H. M. Key, H. Hayashi, D. S. Clark and J. F. Hartwig, Chemoselective, Enzymatic C-H Bond Amination Catalyzed by a Cytochrome P450 Containing an Ir(Me)-PIX Cofactor, J. Am. Chem. Soc., 2017, 139, 1750–1753 CrossRef CAS.
- P. Dydio, H. M. Key, A. Nazarenko, J. Y.-E. Rha, V. Seyedkazemi, D. S. Clark and J. F. Hartwig, An artificial metalloenzyme with the kinetics of native enzymes, Science, 2016, 354, 102 CrossRef CAS.
- K. H. Chan, X. Guan, V. K. Y. Lo and C.-M. Che, Elevated catalytic activity of ruthenium(II)-porphyrin-catalyzed carbene/nitrene transfer and insertion reactions with N-heterocyclic carbene ligands, Angew. Chem., Int. Ed., 2014, 53, 2982 CrossRef CAS.
- K. Oohora, A. Onoda and T. Hayashi, Hemoproteins Reconstituted with Artificial Metal Complexes as Biohybrid Catalysts, Acc. Chem. Res., 2019, 52, 945–954 CrossRef CAS.
- P. R. Ortiz de Montellano, Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes, Chem. Rev., 2010, 110, 932–948 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |