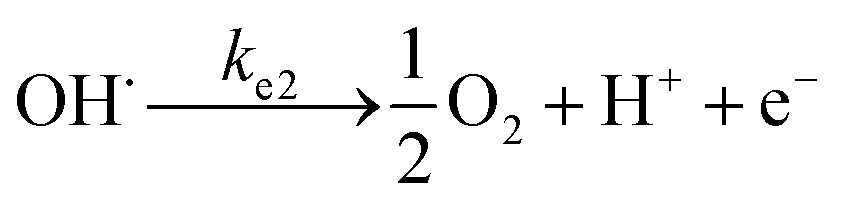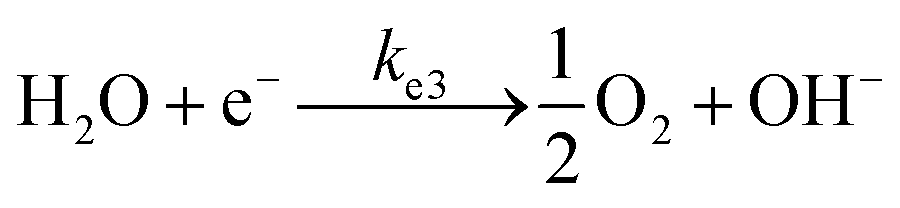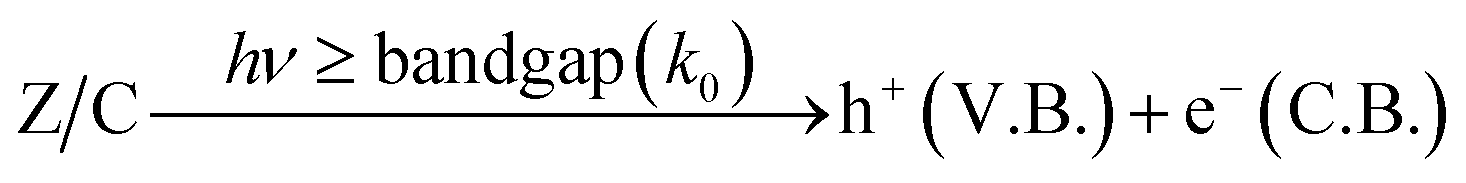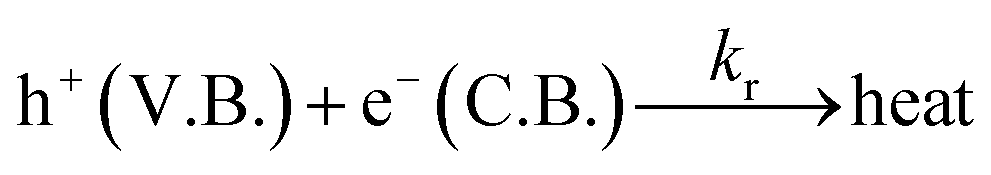DOI:
10.1039/C9NA00483A
(Paper)
Nanoscale Adv., 2019,
1, 3992-4008
Behavioral analysis of simultaneous photo-electro-catalytic degradation of antibiotic resistant E. coli and antibiotic via ZnO/CuI: a kinetic and mechanistic study†
Received
6th August 2019
, Accepted 18th August 2019
First published on 11th September 2019
Abstract
Visible light responsive semiconductor-based photocatalysis is known to be an efficient method for the disinfection of bacterial cells. Here, we address the issue of aqueous contamination by persistent pollutants such as antibiotics and antibiotic resistant bacteria (ARB) from an innovative angle. Simultaneous degradation of an antibiotic (chloramphenicol) and antibiotic resistant bacteria (chloramphenicol resistant E. coli) is performed to observe the effect of the presence of antibiotic in the reaction system when it is required for survival of the bacteria. A p–n junction-based ZnO/CuI composite is shown to demonstrate drastic enhancement in photocatalytic activity due to the inbuilt potential barrier suppressing charge carrier recombination. Moreover, an additional driving force for the suppression of recombination was provided by using a potential bias. Hydrothermally grown ZnO/CuI electrode films were characterized to assess optical, electrochemical, physicochemical and structural properties of the composite. Electrochemical impedance spectroscopy and diffuse reflectance spectroscopy were performed to obtain insights into the band bending, band edge potential, band gap and transmittance of the semiconductors. X-ray-based spectroscopic methods and zeta potential measurement demonstrated the surface properties and surface charges of the moieties in the reaction system, allowing us to deduce justifiable conclusions. A model based on the interaction of photogenerated radicals with the bacteria was developed and rate expressions were used to obtain the rate constants for the experimental results. Photoelectrocatalysis and photocatalysis followed first order rate kinetics; however, due to the unavailability of direct hole attack in photolysis, the electrolysis and electrocatalysis followed Langmuir–Hinshelwood kinetics. Bacterial disinfection was confirmed by K+ ion leaching and by structural changes in the membrane observed by FTIR of the cells after the reaction. We also addressed the issue of bacterial adhesion on the films restricting the mobility of radicals to interact with the bacteria, affecting the reusability of the catalyst films. The present work opens a wide avenue to discuss and address the improvement of the reusability of nanomaterial films for bacterial applications by controlling bacterial adhesion.
1. Introduction
Waterborne diseases are continuously increasing worldwide due to the ingestion of microbially contaminated water. The potential health consequences of contaminated water are huge. Therefore, its control is a primary concern and cannot be compromised.1 Antibiotics are used as chemical compounds, possessing antibacterial properties. However, excessive usage of antibiotics and their release from anthropogenic sources plays a major role in increasing the organic contamination content in water.2 In order to resolve these issues, semiconductor-based photocatalysis and photoelectrocatalysis processes have attracted immense attention in recent years. After the first report of photoelectrocatalysis using TiO2 coupled with a platinum electrode for water splitting,3 tremendous efforts have been made to develop a conducting substrate with an enhanced heterogeneous photocatalytic performance for pollutant degradation as well as for water splitting applications.4
The concentration of antibiotics is particularly high in hospital waste water (in μg mL−1), but lower in the case of municipal (μg mL−1) and surface water bodies (ng mL−1). The presence of microbes in environments with considerable concentrations of antibiotics brings about various transformations in the vital components of the microbial cell body,5 such as the development of proteins that can resist the effect of antibiotics. Therefore, disinfection of such microbes has become a topic of interest due to them becoming a persistent health threat. Antimicrobial resistance in pathogenic microorganisms gives rise to high morbidity and mortality. The increase in the concentration of such pathogens is exacerbated by the use of existing antibiotics for infection control. Therefore, this is a potential risk and has become a topic of primary concern.6,7
The photocatalytic activity of a semiconductor photocatalyst is positively related to the lifetime and diffusion length of the charge carriers. However, low mobility and accumulation of the carriers in the bands increase the rate of recombination in the semiconductor. Photoelectrocatalysis offers the advantage of reduced recombination by transporting the excited electrons from the conduction band to the counter electrode under an external potential. The external bias forces the electrons to move through the external circuit (anode), enabling the holes and electrons to actively participate in redox reactions. Reduced recombination of charge carriers eliminates a huge barrier limiting the usage of semiconductor photocatalysts and opens a new avenue of applications.
Catalysts in slurry form present numerous difficulties in commercial usage. Therefore, photocatalyst immobilization is a potential solution to promote the recovery of the catalyst to a great extent, facilitate catalyst separation and promote the effective reusability of pollutants. However, immobilization reduces the active surface area of the catalyst able to interact with the pollutant. Several studies have observed that surface area is not a dominating parameter responsible for higher photocatalytic activity. Growth of the catalyst on a substrate is one of the best ways to immobilize the catalyst for photoelectrocatalysis applications. The major difference between chemical coating processes and seed layer-based hydrothermal growth is that the former is conformal, while the latter is directional or anisotropic. This directional growth due to the seed layer provides a firm attachment with the substrate and efficient transportation of charge carriers. Conducting substrates such as FTO (fluorine-doped tin oxide) and ITO (indium-doped tin oxide) are widely used for photoelectrochemical applications. However, FTO is known for its better thermal stability and cost effectiveness than ITO. Therefore, Eswar et al. carried out a study to understand the effect of this substrate on the charge transfer and photoelectrocatalytic processes.8
Various attempts have been made to achieve the seed layer-based growth of ZnO with various morphologies for solar cell applications. However, there is no detailed kinetic study available of the disinfection of microorganisms using photoelectrocatalysis. Numerous studies have been performed to explore the effects of heterojunctions of ZnO/TiO2 formed by spin coating,9 ZnO/CdS/TiO2 using chemical bath deposition,10 and ZnO/vertically aligned MoS2 flakes on ITO using chemical vapor deposition11 for photocurrent measurements. Spray deposited ZnO/WO3 was reported for degradation of phthalic acid12 and ZnO array/Ag rods for degradation of rhodamine B.13 ZnO/Cu2O/CdS was immobilized on FTO for improved photon to current conversion efficiency14 and BiVO4/ZnO synthesised by liquid phase deposition followed by successive ionic layer adsorption and reaction (SILAR) was used for degradation of tetracycline.15 Elsewhere, core–shell C3N4/ZnO fabricated via a reflux method was demonstrated for degradation of phenols16 and Fe2O3/ZnO formed by liquid phase deposition was used for degradation of a variety of pollutants.17
In order to augment the visible light absorption range and to increase the charge carrier separation, a lower band gap semiconductor has to be incorporated. CuI has not been widely explored as a photocatalyst. However, the antibacterial feature of CuI has been demonstrated by Pramanik et al.18 To the best of our knowledge, only one study has been performed to understand the photocatalytic properties of CuI/Au composites for degradation of organic pollutants and ethanol oxidation.19 The band edge potentials of the conduction band and the valence band of CuI have also been mentioned in the literature. However, the band gap of a semiconductor can be tuned by the method of preparation and particle size of the particles.20
In the present study, an effort has been successfully made to synthesize a ZnO/CuI composite on an FTO substrate. A vertically aligned ZnO nanorods array has been grown on the FTO substrate using the hydrothermal approach followed by SILAR for growth of CuI. Optimization of the molar concentration of the grown species was achieved by comparing concentrations of 0.01, 0.025, 0.05 and 0.1 M. The optimized loading of ZnO was used to carry out a further detailed study on the photoelectrocatalytic simultaneous inactivation of an antibiotic resistant microorganism and degradation of the organic pollutant chloramphenicol. Inactivation of an antibiotic resistant strain of E. coli was compared with the simultaneous inactivation of a non-antibiotic resistant strain of E. coli. The synergistic effects of the ZnO/CuI composite and effect of incorporation of CuI were demonstrated in detail. A kinetic study was carried out based on a previous study and the data was fitted based on the model presented by Eswar et al.21 The photoelectrocatalytic mechanism was deduced with the support of various characterization techniques and scavenger studies. The problems of reusability, recyclability and photocorrosion have been addressed for the first time in detail specifically for the case of bacterial inactivation, as the attachment of bacteria on the catalyst covers the active sites of the catalyst and compromises the catalyst activity in the longer run.
2. Experimental
2.1 Materials and methods
2.1.1 Photocatalysts: preparation and characterization.
2.1.1.1 Zn2+ seed layer.
The Zn2+ seed layer was prepared using 0.01 M (0.087 g in 40 mL of ethanol) zinc acetate solution. First, FTO slides (3 × 4 cm, 1 cm spacing) were washed with deionized (DI) water, soap solution, acetone and isopropanol (IPA) for removal of dust particles and to degrease the slides. Then, the dried slides were dipped in the above-mentioned solution for 5 s (7 cycles).
2.1.1.2 ZnO nanorods.
A 30 mL solution of 0.01, 0.025, 0.5 and 0.1 M zinc nitrate and equimolar hexamine was prepared and transferred to a 50 mL Teflon reactor. The seed layer-coated slide was placed in the solution such that it covered the coated area. This Teflon-coated stainless steel reactor assembly was kept in the furnace at 90 °C for 6 h. A thick layer of grown ZnO was obtained after the reaction. This slide was washed with DI water and ethanol alternately until the pH of the DI water became ≈7. It was dried for 10 min and calcined at 300 °C for 2 h.
2.1.1.3 CuI and ZnO/CuI films.
CuI films were synthesised using SILAR. FTO slides (3 × 4 cm, 1 cm spacing) were washed with DI water, soap solution, acetone and IPA for removal of dust particles and to degrease the slides. A solution (A) of 0.1 M copper sulphate (1.2 g, 50 mL DI water) and 0.1 M (1.2 g, 50 mL DI water) sodium thiosulphate [cationic precursor] was prepared. Another solution (B) of 0.025 M (0.20 g, 50 mL DI water) potassium iodide [anionic precursor] was prepared.22 The slides were dipped alternately in solution A, DI water, solution B and DI water for 5 s and dried at 40 °C on a hot plate. This is considered as 1 cycle. The process was repeated for 10, 20, 30 and 40 cycles.
ZnO/CuI was also prepared by the SILAR method except using a ZnO-grown slide as substrate instead of bare FTO.
3. Results and discussion
3.1 Catalyst characterization
3.1.1 X-ray diffraction analyses.
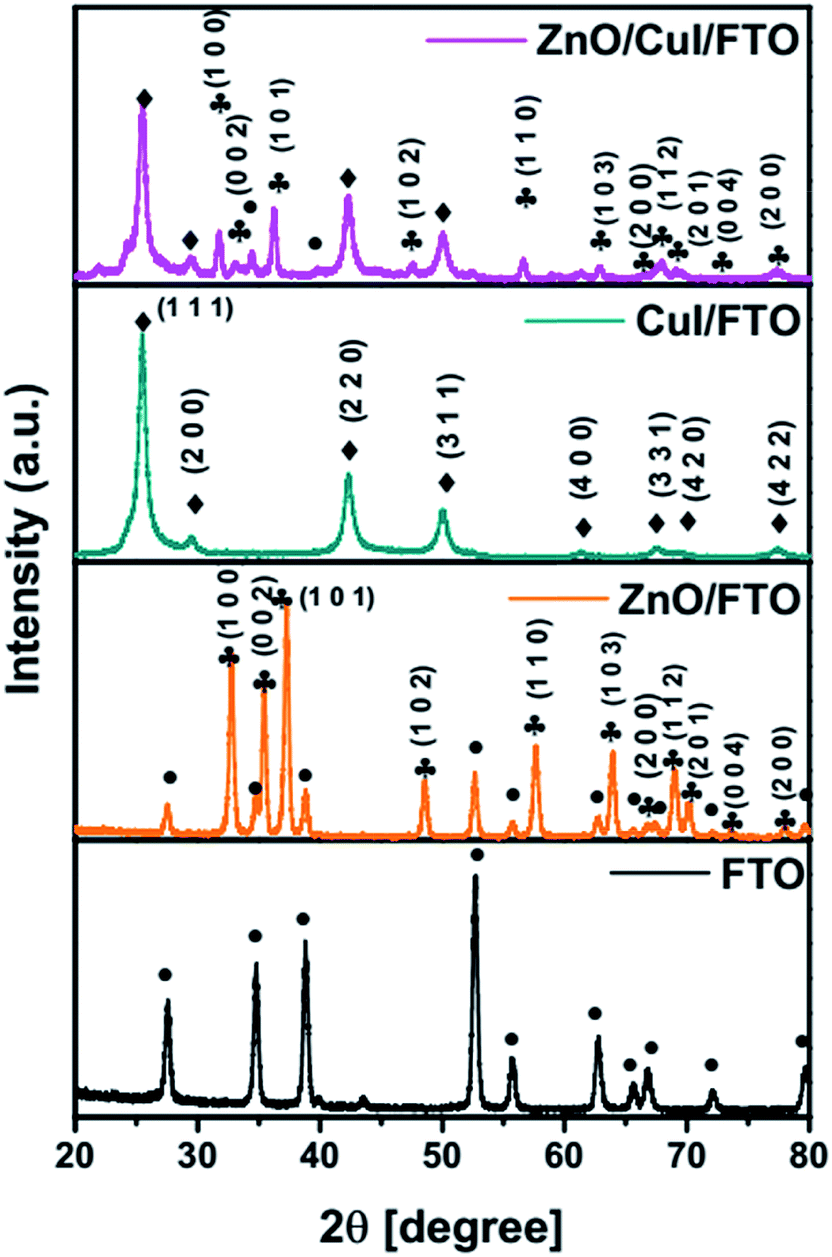
Diffraction patterns of the synthesised semiconductors ZnO, CuI and the composite ZnO/CuI were obtained at a scan rate of 1° min−1 with grazing angle of 3°. Because the catalysts were grown on a substrate i.e. FTO, the XRD pattern of FTO was recorded to identify the FTO peaks in the XRD patterns of the photocatalysts. Fig. 1 show the XRD patterns of FTO, ZnO/FTO, CuI/FTO and ZnO/CuI/FTO from bottom to the top of the figure, respectively. Major intense peaks of ZnO at 37.3°, 35.3° and 32.7° represent the planes (1 0 1), (0 0 2) and (1 0 0), respectively. Additionally, a number of FTO peaks were obtained at 27.5°, 34.6°, 38.8°, 52.6°, 55.7°, 62.7°, 72° and 79.6°. In the case of CuI, major intense peaks were obtained at 25.3° and 42.3° which correspond to the planes (1 1 1) and (2 2 0), respectively. The XRD patterns were verified with reference to the JCPDF database, indicating cards 00-001-0581 for CuI, 00-002-1340 for FTO and 00-001-1136 for ZnO. The composite showed the peaks of both ZnO and CuI. CuI is a cubic structure with the F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m space group, while ZnO has a hexagonal structure with the P63mc space group. The lattice parameters of CuI were calculated using Rietveld refinement as a = b = c = 6.0434 Å.
3m space group, while ZnO has a hexagonal structure with the P63mc space group. The lattice parameters of CuI were calculated using Rietveld refinement as a = b = c = 6.0434 Å.
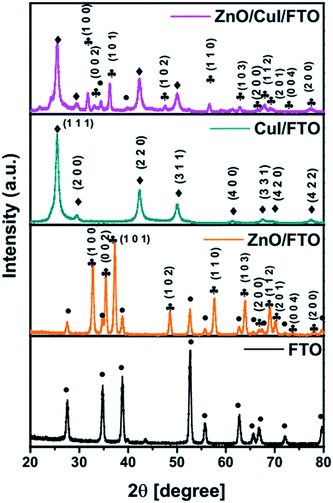 |
| | Fig. 1 XRD patterns of FTO, ZnO/FTO, CuI/FTO and ZnO/CuI/FTO. | |
3.1.2 Transmittance.
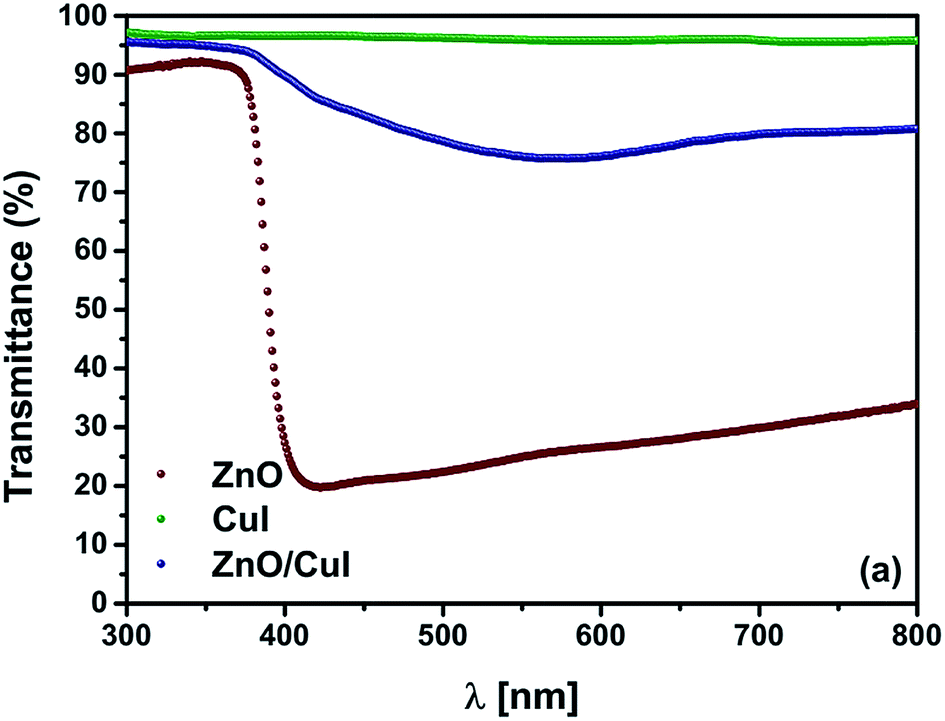
The optical properties of a semiconductor are the deciding factor in its selection for photocatalytic application. The transmittance spectrum of the solid film, the band gap of the semiconductors, and the type of band gap are all features of semiconductors that help in the prediction of their photocatalytic activity. Therefore, the transmittance spectra of the semiconductors and the composite were measured by a UV-vis spectrophotometer. Thereafter, calculation of the band gaps was done using Tauc plots based on information in the literature about indirect or direct band gaps. ZnO is well known as a direct band gap semiconductor.23–25 CuI has also been characterised as a direct band gap semiconductor.26 Tauc plots are plotted with [F(R)hν]1/n as ordinate and hν − Eg as abscissa (F(R) = [1–R]2/2R (Kubelka munk function), hν = photon energy, n = parameter denoting direct or indirect band gap 2 for indirect transition, 1/2 for direct transition). Hence, extrapolation of the line [F(R)hν]n = 0 to the abscissa gives the value of the band gap. Fig. 2(a) shows the transmittance spectra of ZnO, CuI and the composite ZnO/CuI. The ZnO films transmitted only 20–30% across the visible region whereas CuI transmitted around 95–97% across the UV and visible region. Therefore, incorporation of CuI into ZnO enhanced the transmittance of ZnO to 80% in the visible region. Fig. A.8(b) and (c) [shown in ESI†] show the Tauc plots of ZnO and CuI. The extrapolated values indicate the band gap of ZnO and CuI to be 3.13 and 2.86 eV, which is consistent with the literature.27,28 Various density functional theory (DFT) studies have revealed that ZnO is a direct band gap semiconductor29 while some studies have revealed that hexagonally structured nanowires, nanorods, nanobelts etc. have indirect band gaps.30 Therefore, the identification of the band gap of ZnO as direct or indirect is debatable. The fundamental way to calculate the band gap is from the absorption edge of the optical spectrum, which for ZnO is at 413 nm i.e. ≈3 eV. However, the Tauc plot for the case of a direct band gap (shown in the inset of Fig. A.8(c)†) implies a band gap of 3.23 eV, which contradicts the above-calculated value. Therefore, ZnO must possess an indirect band gap, with a value of 3.13 eV according to the corresponding Tauc plot. The extended tail in Fig. A.8(b)† is called the Urbach tail, and indicates the presence of defect levels between the valence and conduction bands, i.e. a forbidden gap.31
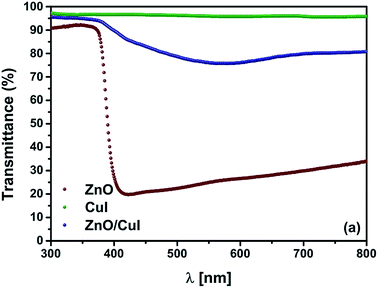 |
| | Fig. 2 (a) Transmittance spectra of ZnO, CuI and ZnO/CuI. | |
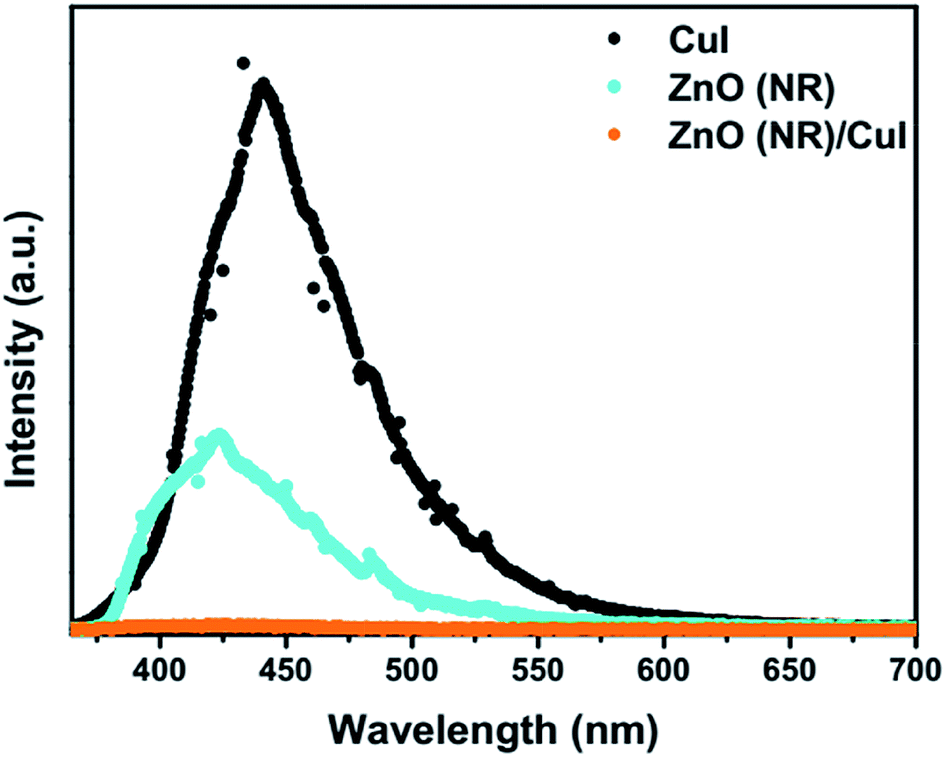
3.1.3 Photoluminescence.
Photoluminescence (PL) involves the emission of light as the result of absorption of photons. Therefore, to shed light on the emission, excitonic recombination, surface defects and band gap of the semiconductors, photoluminescence at room temperature was carried out for the composite ZnO/CuI and the semiconductors ZnO and CuI, as shown in Fig. 3(a). The high PL intensity in the case of CuI shows that it has a higher recombination rate than ZnO and ZnO/CuI. Suppression of recombination is evident in the case of the composite (Fig. 3(a)). Fig. A.9(b) and (c)† show the deconvoluted spectra, providing deeper information about the surface defects and the band gap. The first peak, at 437 nm for CuI and at 398 nm for ZnO, indicates the near band edge emission. Absorption of photons excites the electrons to the conduction band leaving behind holes in the valence band. Thereafter, the electrons and holes undergo momentum and energy relaxations to move to the conduction band minima and valence band maxima. The recombination between the charge carriers present at the band edges leads to the emission of photons of equal or lesser energy, which manifest as the PL energy of near band edge emission. The band gap of CuI and ZnO is 2.86 and 3.13 eV, respectively, calculated from the Tauc plots (Section 3.1.2). The PL energies of the first peaks are in good agreement with the band gaps obtained in Section 3.1.2. The literature suggests that the reason for the broadening of the peak is fluorescence due to the presence of excess iodine at the surface. Excitation with much higher energy than the band gap can liberate the iodine from CuI, resulting in the presence of iodine at the surface which generates some surface trapping sites at 0.2 eV above the valence band.32 The other deconvoluted peak at 465.7 nm is consistent with recombination between the electrons in the conduction band and holes at the hole trapping sites (the Urbach tail in Fig. A.8(b)† also confirms the existence of defect levels). ZnO possesses a hexagonal structure with Zn occupying half of the tetrahedral sites, with empty octahedral sites. Therefore, there are plenty of vacant sites available for incorporation of intrinsic defects (Zn interstitials). The possible donor defects in the ZnO lattice are Zn′/Zn′′ (doubly ionized defect), Zn×/Zn′ (singly ionized defect) and 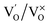 (single oxygen ion vacancy). The corresponding defect levels are in the range of 0.5–0.15 eV below the conduction band, as described elsewhere.33 The acceptor defects are composed of vacancies generated due to the
(single oxygen ion vacancy). The corresponding defect levels are in the range of 0.5–0.15 eV below the conduction band, as described elsewhere.33 The acceptor defects are composed of vacancies generated due to the 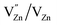 ,
, 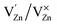 and
and 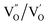 and lie in the range of 0.8 to 2.8 eV above the valence band.34 Therefore, the emission peaks at 421, 450 and 516 nm are transitions from the energy levels of defect sites (shallow traps/deep traps).35 It is evident from the X-ray photoelectron spectroscopy (XPS) O-1s spectrum (Section 3.1.5) that the concentration of oxygen vacancies in the composite is significant.
and lie in the range of 0.8 to 2.8 eV above the valence band.34 Therefore, the emission peaks at 421, 450 and 516 nm are transitions from the energy levels of defect sites (shallow traps/deep traps).35 It is evident from the X-ray photoelectron spectroscopy (XPS) O-1s spectrum (Section 3.1.5) that the concentration of oxygen vacancies in the composite is significant.
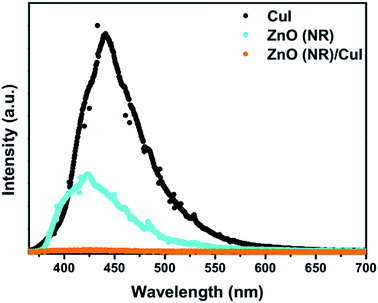 |
| | Fig. 3 Photoluminescence spectra of ZnO nanorods, CuI and ZnO/CuI. | |
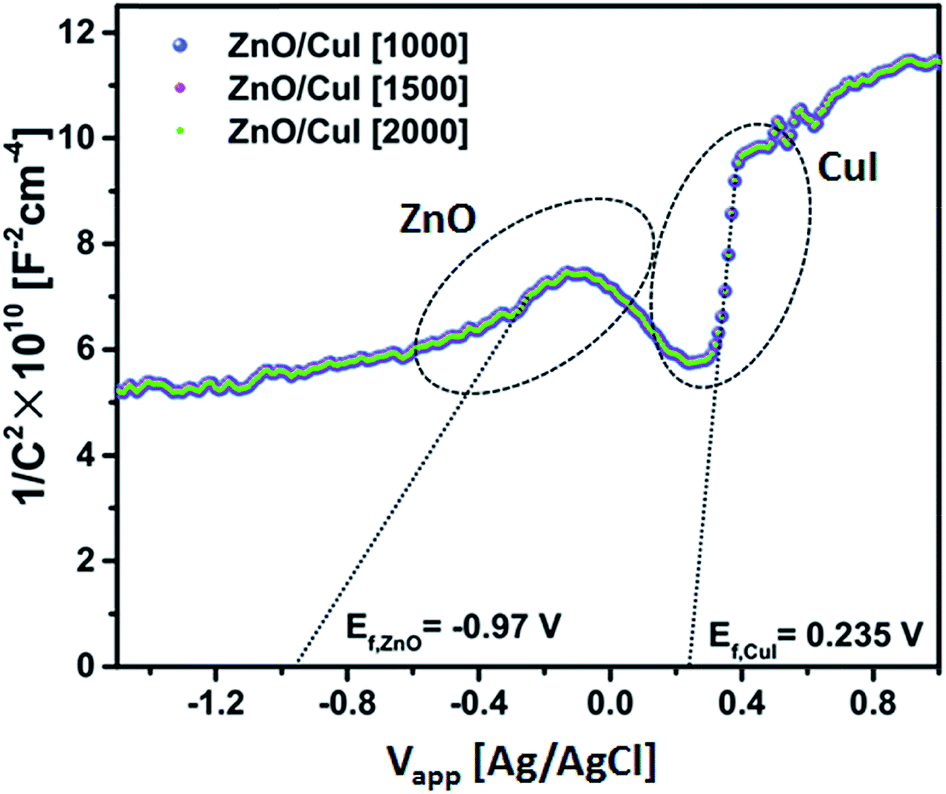
3.1.4 Band edge assessment (Mott–Schottky plot).
Electrochemical impedance measurements were done to assess the band edges of CuI. The extrapolated value of the abscissa at the inflation point of the curve gives the flat band potential (EFB) of the semiconductor. The flat band potential for CuI and ZnO is 0.235 V and −0.97 V, respectively, obtained from Fig. 4 at different frequencies (1000, 1500 and 2000 Hz). The flat band potential is the potential at which the bands are not polarized in the interfacial region in the vicinity of the electrolyte and do not exhibit any band bending due to the difference between the potential of the semiconductor and the redox potential of the electrolyte. For the Ag/AgCl electrode, conversion to the normal hydrogen scale can be obtained by adding 0.22 V to the Efp (flat band potential for the p-type semiconductor CuI). Hence, the valence band and conduction band are found to be at −7.815 V and −4.955 V.| | | ECB(NHE) = EFB(Ag/AgCl) + 0.22 | (1) |
| | | E(AVS) = −E(NHE) − 4.5 | (2) |
| | 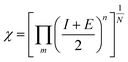 | (4) |
N = number of atoms; m = number of elements, n = no. of atoms of element.| | | ECB(NHE) = −0.97 + 0.22(EAg/AgCl) = −0.75 | (7) |
| | | ECB(AVS) = −ECB(NHE) − 4.5 = −3.75 V | (8) |
| | | EVB(AVS) = ECB(AVS) − Eg(3.13) = −6.88 V | (9) |
| | | ECB(NHE) = 0.235 + 0.22(EAg/AgCl) = −0.455 | (11) |
| | | ECB(AVS) = −ECB(NHE) − 4.5 = −4.955 V | (12) |
| | | EVB(AVS) = ECB(AVS) − Eg(2.86) = −7.815 V | (13) |
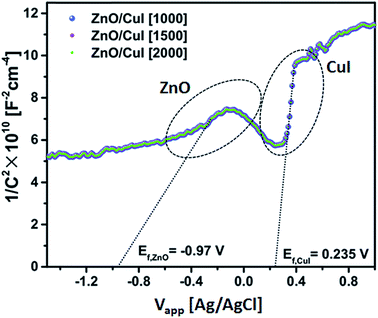 |
| | Fig. 4 Mott–Schottky plot of ZnO/CuI. | |
3.1.5 X-ray photoelectron spectroscopy.
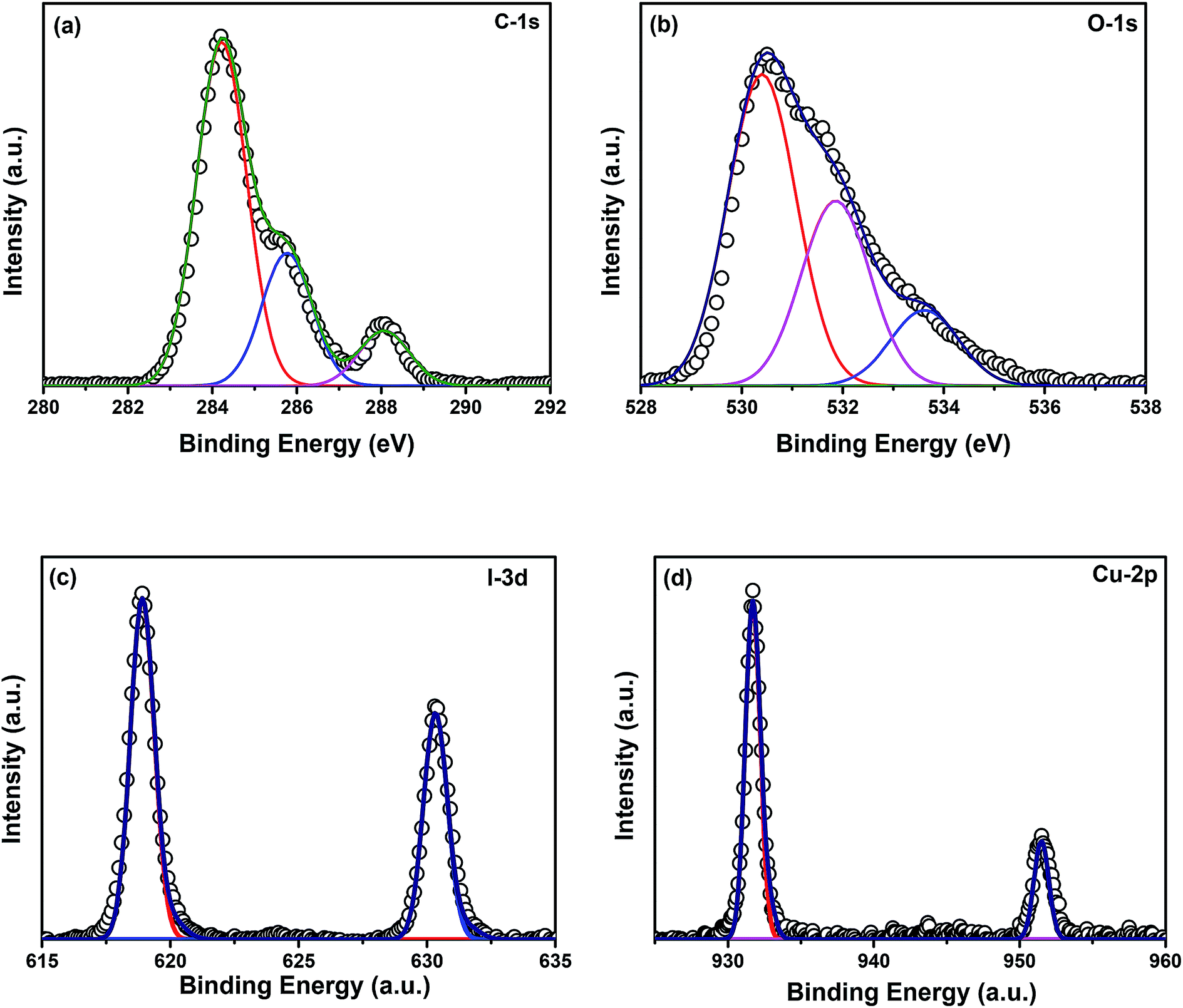
Elemental analyses of the composite were done by XPS. Fig. 5(a)–(d) show the XPS spectra of elements C, O, I, and Cu, respectively. The binding energy of carbon corresponds to 284.8 eV. No charge correction was required for the spectra. The peak at 284.8 eV represents the presence of adventitious carbon in the catalyst. The extended peaks of carbon may correspond to carbon bonded with the adsorbed hydroxyl groups at the catalyst surface. Fig. 5(b) represents the XPS spectrum of oxygen where the peak at 530 eV corresponds to the lattice oxygen in the form of Zn–O. The peak at 532 eV represents the presence of oxygen vacancies in the ZnO/CuI lattice and another extended peak at 534 eV shows the physisorption of hydroxyl ions at the catalyst surface. Fig. 5(c) shows the XPS spectrum of I-3d present in the lattice of ZnO/CuI, where the peak at ∼519 (618.8) eV reveals the presence of I-3d5/2.36 The spin orbit peak separation between 3d5/2 and 3d3/2 is observed to be 11.5 eV, which is consistent with the literature. Fig. 5(d) shows the XPS spectrum of Cu-2p. The Cu-2p3/2 and 2p1/2 peaks were observed at 931.6 and 951.35 eV, respectively. Therefore, the spin orbit separation is 19.75 eV which is consistent with the literature. However, the absence of any satellite peak around 940 eV clearly indicates the presence of Cu in the +1 oxidation state.36 Similarly, Fig. A.10(e)† presents the XPS spectrum of Zn-2p. In the case of ZnO, the Zn-2p3/2 peak appears at 1021.8 eV and the spin orbit peak separation between 2p3/2 and 2p1/2 is 23 eV. The surface amounts of Cu and Zn were calculated from the relative sensitivity factors (RSFs), found to be in a ratio of 3.29.
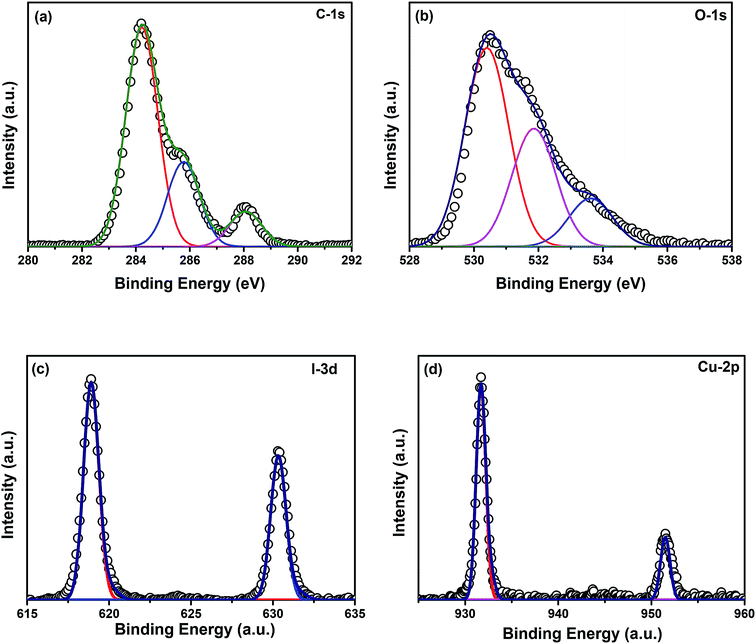 |
| | Fig. 5 XPS spectra of (a) C-1s, (b) O-1s, (c) I-3d, (d) Cu-2p. | |
3.1.6 Morphological and surface area analysis.
The morphological features of the substrate-grown photocatalysts were observed by scanning electron microscopy, as shown in Fig. 6(a)–(d). Fig. 6(a) indicates the horizontal view SEM image of ZnO grown using a concentration of 0.25 M on the surface of FTO. Well dispersed growth of nanorods can be seen and the inset shows the hexagonal faceted top of the nanorods. The growth mechanism of the nanorods is described in ESI and shown in Fig. A.11.† As the concentration of the growth solution is increased, the density of the rods increases. The increase in the concentration also enhances the homogeneous growth of the rods in the solution and at the surface of the top of the rods, as shown in Section 3.46. Fig. 6(b) shows the horizontal view of CuI coated using the SILAR method after 30 cycles. Fig. 6(c) presents the cross-sectional view of ZnO nanorods grown on FTO. The thickness of the film is observed to be ∼1.798 μm. Similarly, Fig. 6(d) shows the cross-sectional view of the composite ZnO/CuI. The thickness of the film obtained after the synthesis of the composite is ∼3.2 μm. The growth of ZnO on the substrate provides a surface roughness that enables easy adsorption of Cu and iodine ions to form CuI.
 |
| | Fig. 6 (a) SEM of ZnO nanorods (0.025 M), (b) CuI (30 cycles), (c) cross section of ZnO grown on FTO, (d) cross section of ZnO/CuI on FTO, (e) heterojunction of ZnO and CuI in the composite. | |
Fig. 6(e) shows the HRTEM image of the ZnO/CuI composite and the heterojunction of ZnO/CuI. This confirms the heterojunction formation in the composite, which facilitates the efficient charge carrier transport through the junction.
3.2 Photo-mediated processes
3.2.1 Photoelectrocatalysis.
In the case of photoelectrocatalysis, a conducting substrate is required, which acts as a working electrode. Several reports are available on the growth of photocatalysts on FTO (a conducting glass substrate).37,38 Growth of photocatalysts on this substrate enhances the charge dynamics, easy recovery and reusability. For commercial usage of photocatalysts, immobilization provides many benefits from an economical point of view.39,40
The experimental data obtained from photolysis, electrolysis, electrocatalysis, photocatalysis and photoelectrocatalysis were fitted with the model derived for bacteria in a previous study 8. The direct hole effect in the case of electrocatalysis was also taken into consideration. This implies that kh = 0 (kh = rate constant responsible for direct hole attack) for photolysis and electrolysis.
Therefore, the model contains the following equations.
For photolysis and electrolysis
| | 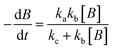 | (14) |
| | 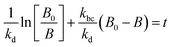 | (15) |
For electrocatalysis, photocatalysis and photoelectrocatalysis
| | 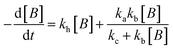 | (16) |
| | 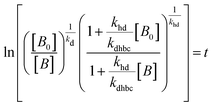 | (17) |
where,

,
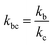
,
kd =
kh +
kabc,
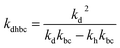
,
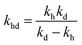
.
OriginPro 9 software was used to fit the data using this model. Non-linear implicit curve fitting was used to fit the data based on eqn (15) and linear fitting was used to fit the data based on eqn (17).
3.2.2 Photolysis and electrolysis.
The experiments in the presence of light and an external potential (1 V) were carried out with a lamp intensity of 57 W m−2.
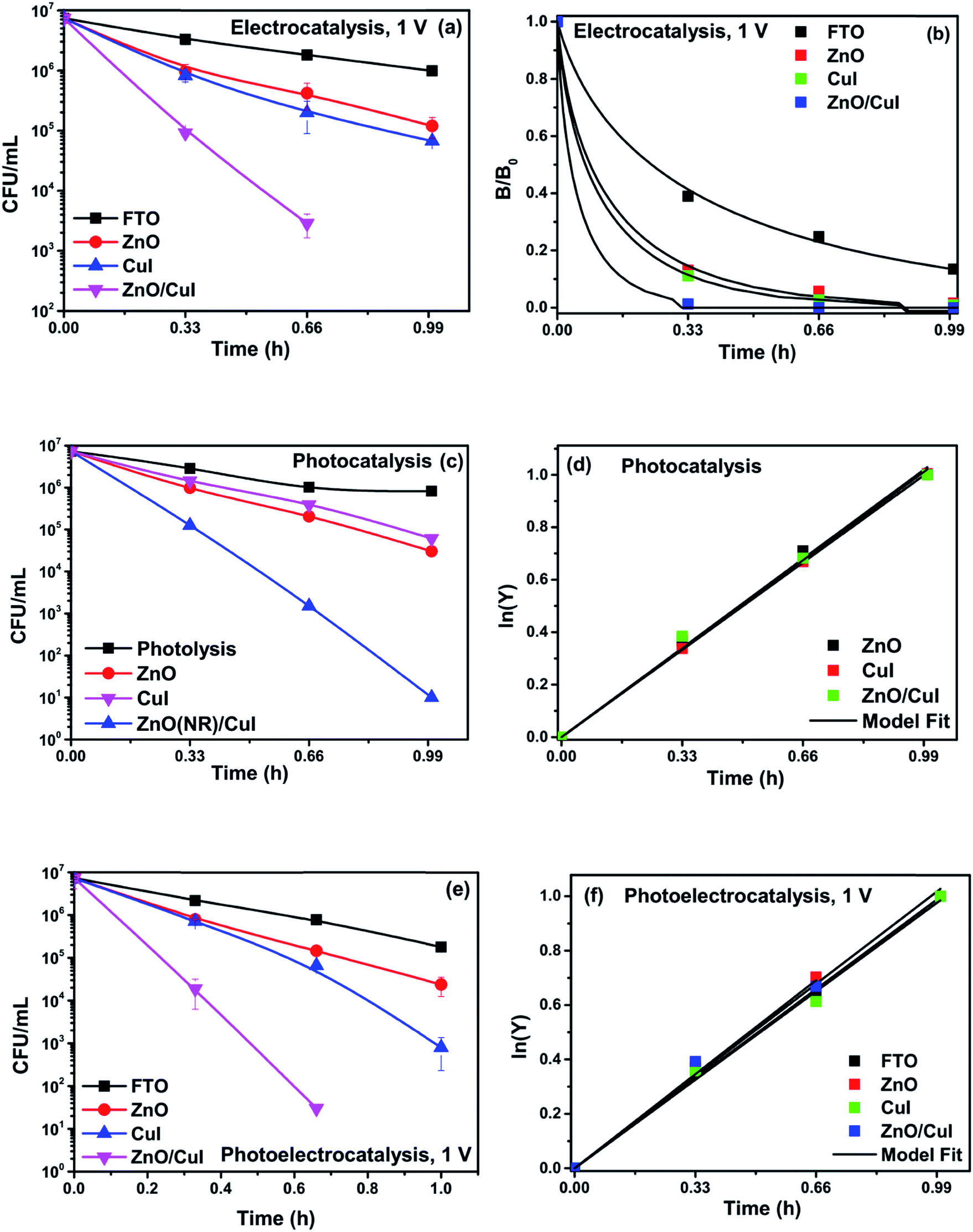
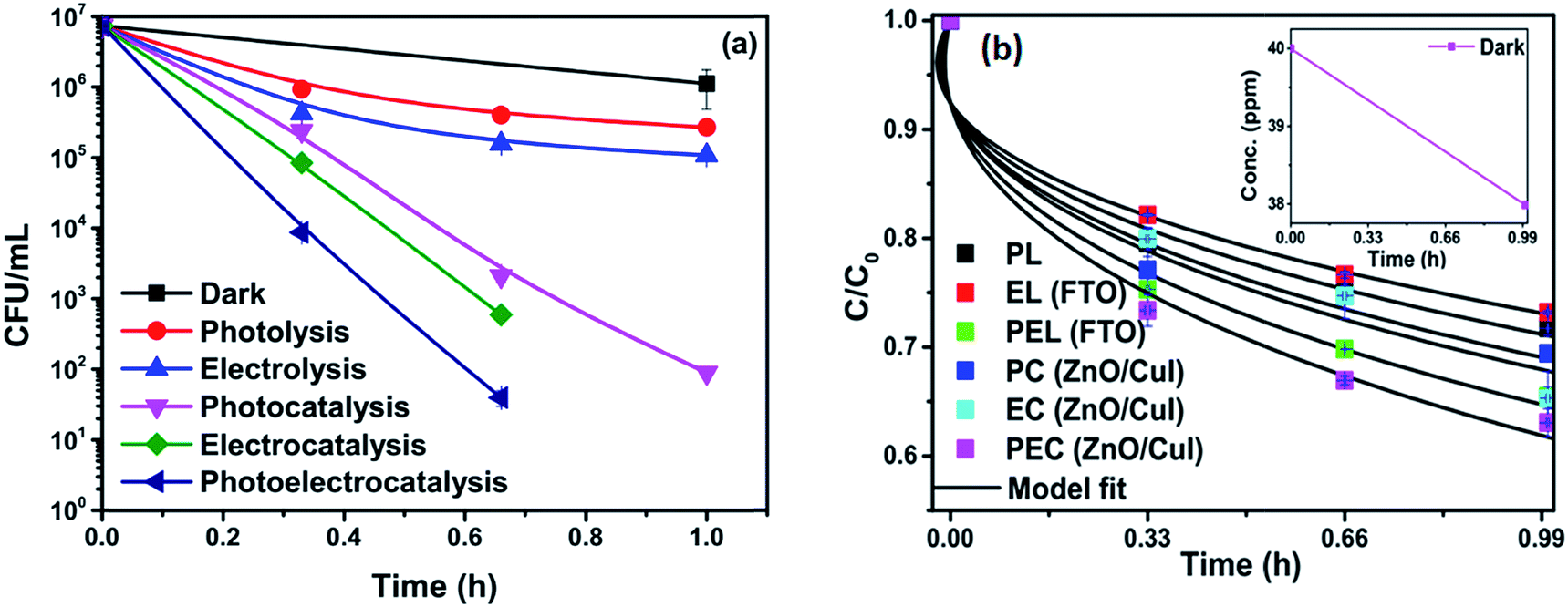
To analyse the effect of light and potential alone on the bacterial inactivation, photolysis and electrolysis experiments were carried out. Fig. 7 shows the photolytic inactivation of E. coli and the inset shows the model fit of the inactivation. A ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log-reduction in the bacterial concentration was observed when the bacterial suspension was exposed to light (metal halide lamp, in the absence of catalyst), as shown in Fig. 7. Similarly, upon applying 1 V potential, a 1log-reduction was observed. 1 V potential was applied for all the photoelectrocatalytic reactions as shown in Fig. 10(a). The kinetics of the inactivation reactions was plotted using the model derived previously,8 as shown in the inset of Fig. 7 and 10(b). The rate constants are tabulated in Table A.2.† The electrochemical reactions taking place in the reaction system are shown by eqn (18)–(24). Generation of hydroxyl radicals is responsible for the bactericidal effect during the electrolysis reaction.
log-reduction in the bacterial concentration was observed when the bacterial suspension was exposed to light (metal halide lamp, in the absence of catalyst), as shown in Fig. 7. Similarly, upon applying 1 V potential, a 1log-reduction was observed. 1 V potential was applied for all the photoelectrocatalytic reactions as shown in Fig. 10(a). The kinetics of the inactivation reactions was plotted using the model derived previously,8 as shown in the inset of Fig. 7 and 10(b). The rate constants are tabulated in Table A.2.† The electrochemical reactions taking place in the reaction system are shown by eqn (18)–(24). Generation of hydroxyl radicals is responsible for the bactericidal effect during the electrolysis reaction.
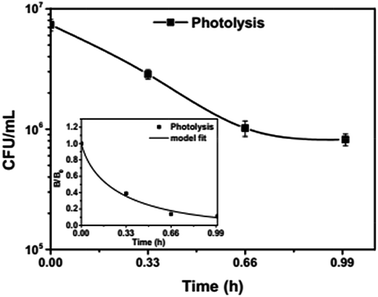 |
| | Fig. 7 Photolytic inactivation and kinetic profiles of bacteria (initial concentration ≈ 107 CFU mL−1), inset: model fit for photolysis. | |
Visible light is known to be lethal for bacterial species. The endogenous cellular photosensitizers present in the cell body such as cytochromes, flavins, porphyrins and NADH possess a wide absorption range. Therefore, bacterial cells absorb in the entire visible range. Nussbaum et al. demonstrated that at 630 nm, a phototoxic effect can be observed for E. coli.41 However, other reports suggest that the blue region of visible light i.e. 400–500 nm is lethal for bacteria.42 Therefore, it has been suggested that the presence of these endogenous photosensitizers leads to the generation of ROS (reactive oxygen species), which reduces the bacterial concentration over time.43 Therefore, photolysis was fitted with a model including the effect of radicals.
The absence of holes in both the processes, i.e. photolysis and electrolysis, excludes the mechanism of direct hole attack. Therefore, eqn (7) is used as the fitting function for the experimental data of these processes.
3.2.3 Catalyst loading.
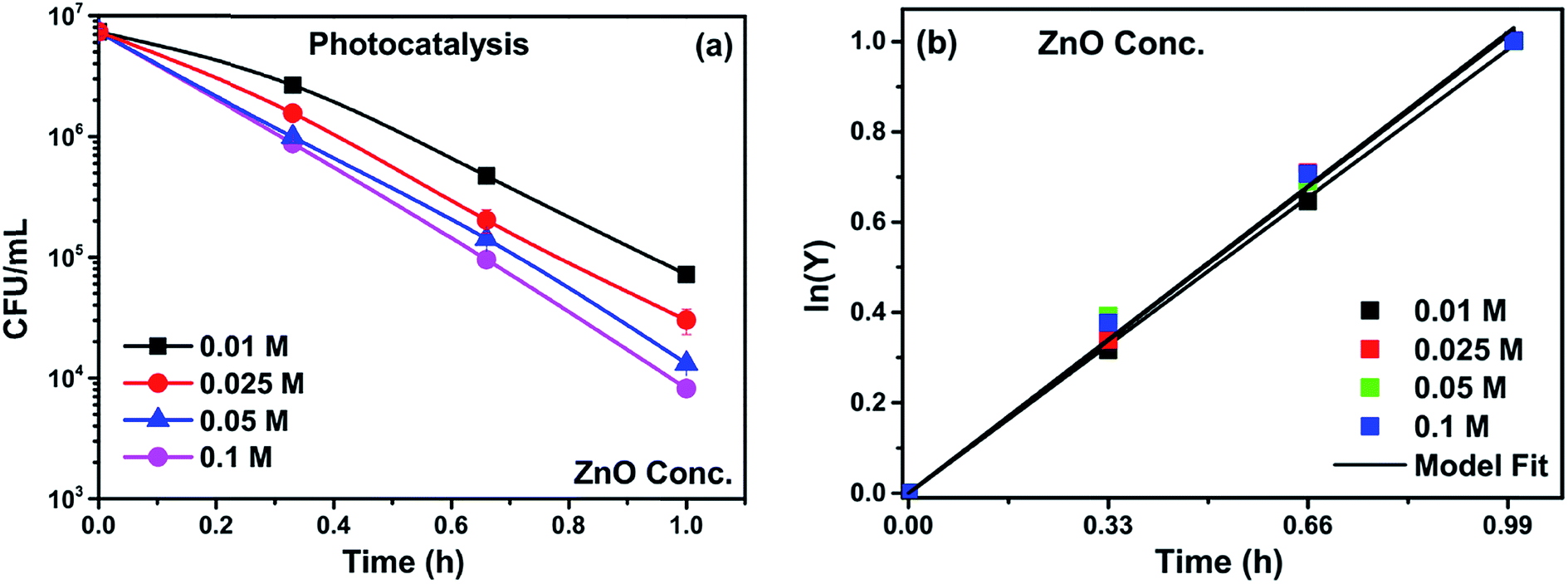
Catalyst loading was varied to optimize the loading concentration, keeping the seed layer concentration constant. At low concentrations of growth solutions, heterogeneous nucleation on the substrate dominates and results in the formation of vertical rods on the substrate (shown in Section 3.1.6). However, as the concentration of growth solution increases from 0.025 to 0.05 M, the rate of homogeneous nucleation also increases (as shown in Fig. 8). The structures on top of the electrode (flowers and rods of micron size) clearly indicate the increased rate of homogeneous nucleation independent of the substrate. When the electrodes with high catalyst concentrations are immersed in the system the homogeneously formed structures detach from the electrodes and suspend in the solution, which in turn increases the photocatalytic activity due to increased active surface area. However, this hinders the separation of the catalyst from the bacterial solution and the recovery of the catalyst. Therefore, for the current study, 0.025 M was considered the optimum loading for all subsequent composite syntheses. The inactivation plots for different concentrations are shown in Fig. 9(a). The inactivation follows first order rate kinetics consistent with the model used for the kinetic studies (eqn (9)).
 |
| | Fig. 8 SEM of electrodes with catalyst loading 0.05 M. | |
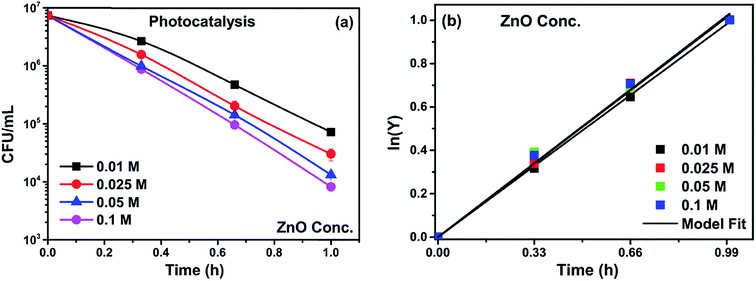 |
| | Fig. 9 (a) Photocatalytic inactivation and (b) kinetic profiles of bacteria (initial concentration ≈ 107 CFU mL−1) with catalyst loading 0.01 M, 0.025 M, 0.05 M and 0.1 M. | |
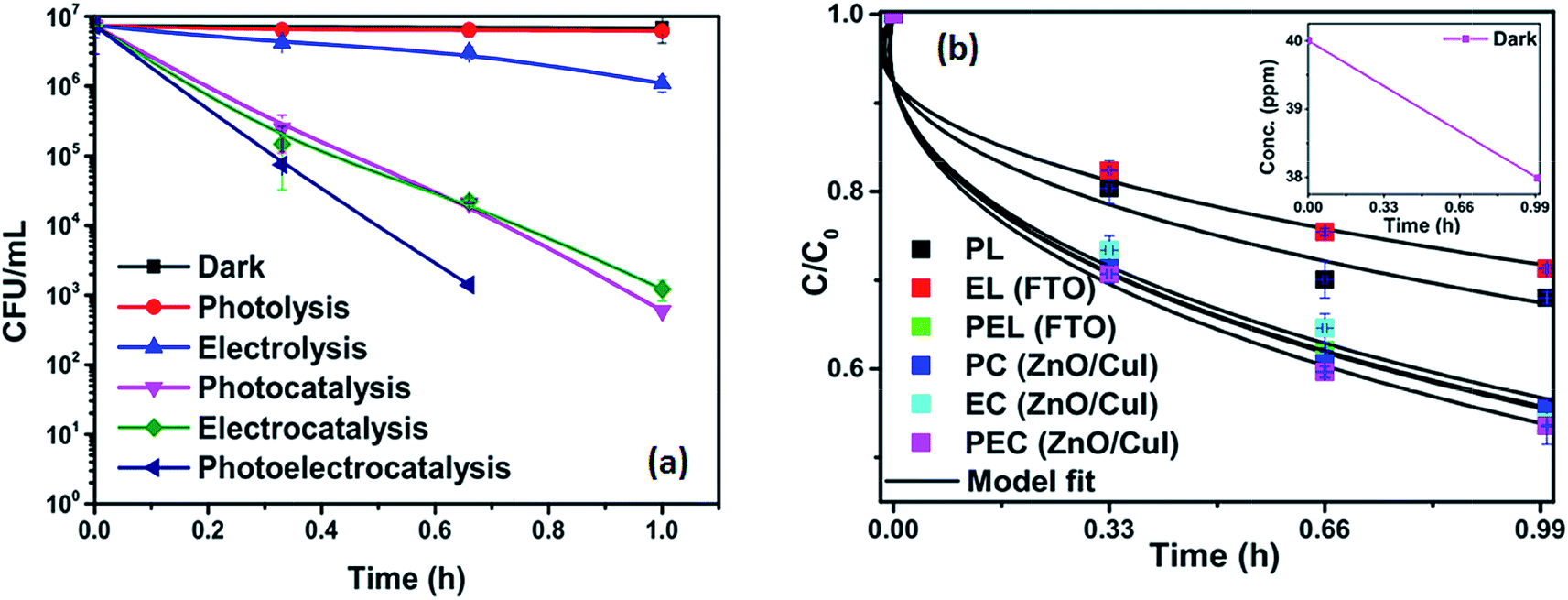
3.2.4 Effect of different catalysts for inactivation of E. coli.
Experiments were carried out using ZnO, CuI and the ZnO/CuI composite for the inactivation of E. coli. Fig. 10(a), (c) and (e) show the electrocatalytic, photocatalytic and photoelectrocatalytic (i.e. control experiments) inactivation of the bacterial cells and Fig. 10(b), (d) and (f) show the kinetic plots for electrocatalysis, photocatalysis and photoelectrocatalysis, respectively. Only a 1log-reduction in E. coli concentration was observed in the case of electrolysis, while for ZnO/CuI, photocatalysis and photoelectrocatalysis showed 5.8 and 5.56log-reductions in 1 h and 40 min, respectively. Electrocatalysis showed a 3.56log-reduction in 40 min. Moreover, electrocatalysis and photoelectrocatalysis achieved completely blank agar plates at the end of 1 h. Tremendous enhancement in the inactivation was seen when the bacterial solution was exposed in the presence of light. The increased rate of inactivation is due to the involvement of both superoxide and hydroxyl radicals as well as the holes in the redox reaction due to the appropriate oxidation and reduction potentials of water (solvent) (refer to eqn (13)–(16)), while in the case of electrocatalysis, only hydroxyl radicals take part in the reaction (refer to eqn (10)–(12)). Therefore, eqn (9) is used to fit the photocatalysis and photoelectrocatalysis data. However, eqn (7) is used to fit the electrocatalysis data due to the absence of holes during electrocatalysis.
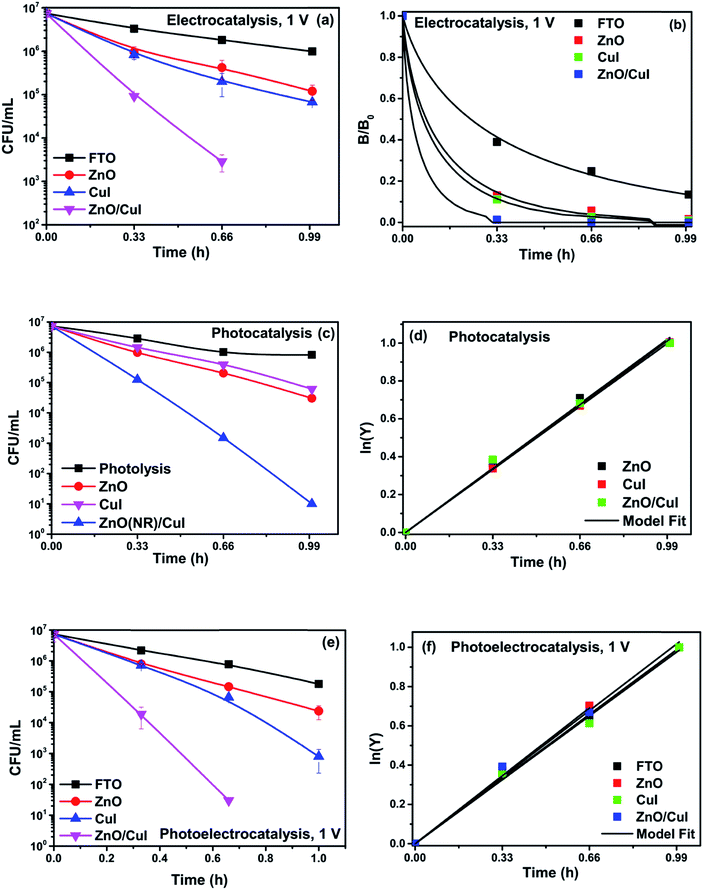 |
| | Fig. 10 Inactivation and kinetic plots of E. coli (initial concentration ≈ 107 CFU mL−1) with ZnO, CuI, ZnO/CuI composite: (a) & (b) electrocatalysis, (c) & (d) photocatalysis and (e) & (f) photoelectrocatalysis. | |
At the anode:
| |  | (18) |
| | 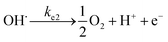 | (19) |
At the cathode:
| | 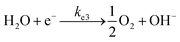 | (20) |
Photocatalysis:
| | 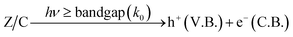 | (21) |
| | 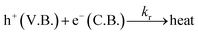 | (22) |
The conducting nature of transparent CuI films provide easy charge transport through the external circuit, therefore, the activity of CuI is better than ZnO in the case of electrocatalysis and photoelectrocatalysis.44
3.2.5 Effect of catalysts on chloramphenicol.
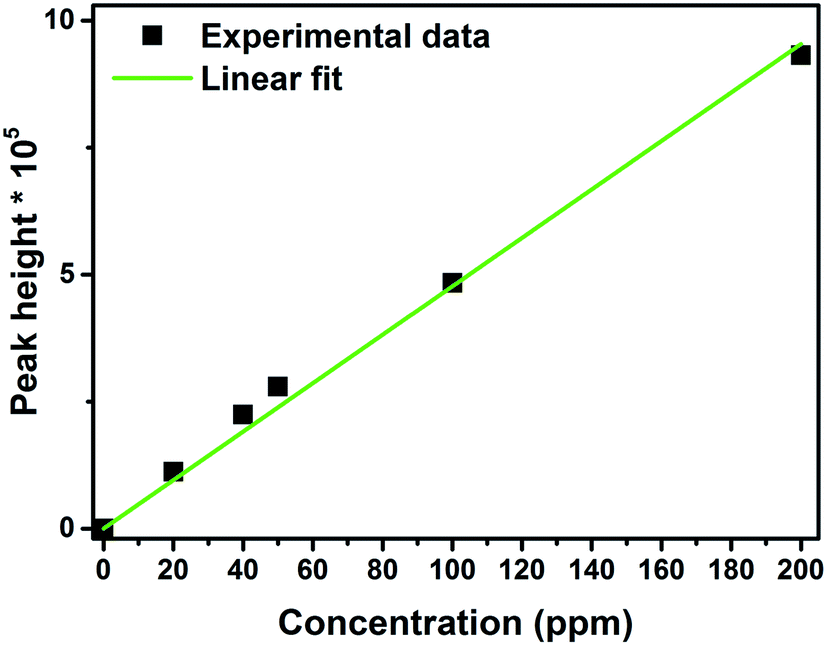
Photoelectrocatalytic degradation of chloramphenicol was carried out using ZnO, CuI and the composite ZnO/CuI. The chloramphenicol peak was observed at a retention time of 3.05 min. The calibration curve was plotted at different concentrations of chloramphenicol as shown in Fig. 11. All the experiments were carried out using 40 ppm solutions of chloramphenicol since the antibiotic resistant E. coli requires 34 ppm of chloramphenicol to survive in the natural state (MIC, minimum inhibitory concentration). Therefore, a concentration higher than 34 ppm was chosen for the present study.
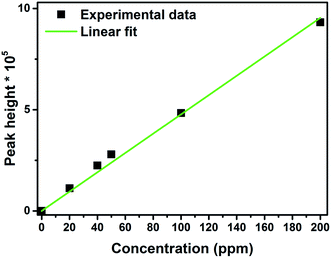 |
| | Fig. 11 Calibration curve for concentration of chloramphenicol using HPLC. | |
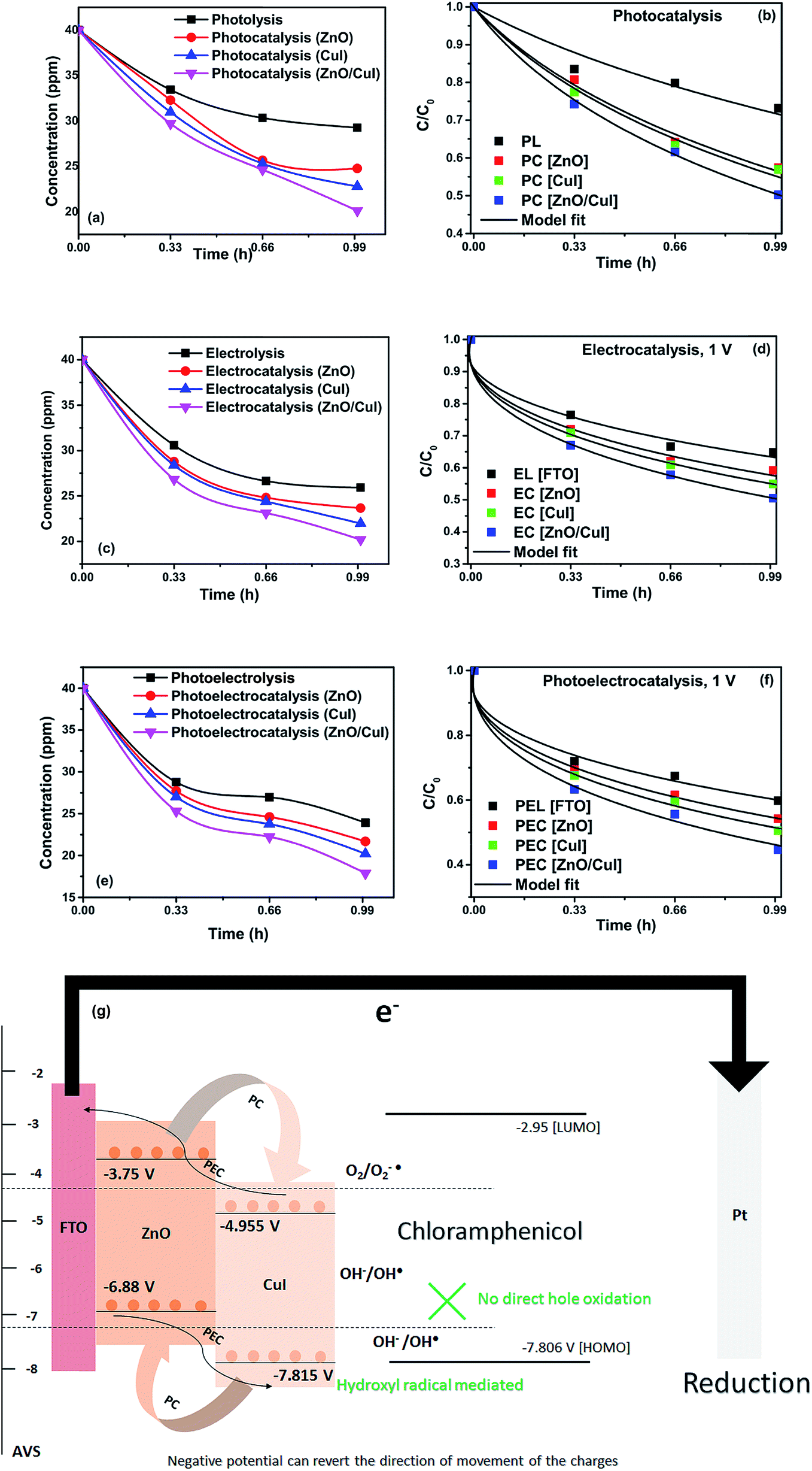
Photolysis, electrolysis and photoelectrolysis were performed as control experiments. Fig. 12(a), (c) and (e) show the degradation plots of chloramphenicol and Fig. 12(b), (d) and (f) show the kinetic plots of the degradation of chloramphenicol. The experimental data were fitted using a model explicitly designed for photoelectrocatalytic degradation of antibiotics, namely the model described earlier with some modification. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of chloramphenicol were taken from theoretical calculations described in the literature.45 The HOMO of chloramphenicol is very close to the valence band potential, which is energetically favourable for hydroxyl radical generation. However, the generation of hydroxyl radicals nullifies the effect of direct hole oxidation. Therefore, direct hole attack cannot be considered as a potential degradation mechanism, as shown schematically in Fig. 12(g). Thus, eqn (9) for photocatalysis and photoelectrocatalysis reduces to eqn (7). All the data for chloramphenicol degradation were fitted using eqn (7). The hydroxyl radicals and superoxide radicals generated via electrochemical and photocatalytic routes are responsible for the degradation of chloramphenicol. The rate constants of all the reactions are listed in Table A.3.† Photocatalytic, electrocatalytic and photoelectrocatalytic degradation using ZnO/CuI achieved 49.72%, 49.54% and 55.27% degradation, respectively. The difference in the degradation rates between the three processes is small due to the fact that the generation of hydroxyl radicals is prohibited in the case of photocatalysis, with superoxide radicals generated instead. It is suggested in the literature that HO2˙ and O2˙− are ineffective due to the high LUMO potential of chloramphenicol as shown in Fig. 12(g).46 However, electron transfer can directly cause structural change in chloramphenicol. Hydroxyl radicals generated via the electrochemical pathways described by eqn (10)–(12) are responsible for electrocatalysis. Photoelectrocatalytic degradation of chloramphenicol is induced by the electrons received through the counter electrode and hydroxyl radicals generated through the electrochemical pathway. However, the respective control experiments demonstrated only 26.86%, 35.21% and 40.21% degradation, respectively.
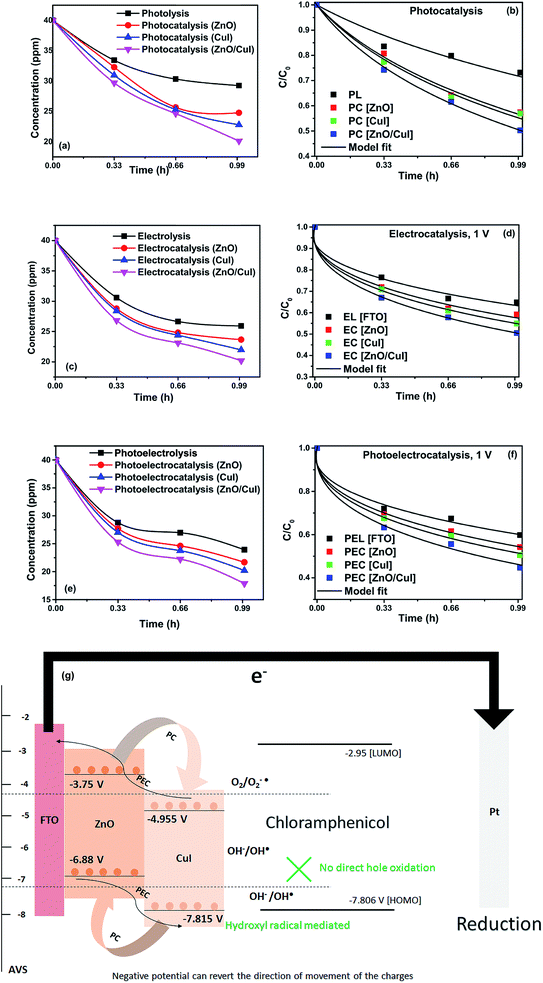 |
| | Fig. 12 Degradation and kinetic plots of chloramphenicol degradation with ZnO, CuI, ZnO/CuI composite (a) & (b) electrocatalysis, (c) & (d) photocatalysis and (e) & (f) photoelectrocatalysis. (g) Schematic of photoelectrocatalytic degradation of chloramphenicol. | |
3.2.6 Effect of catalyst on drug/chloramphenicol and bacteria together.
Non-antibiotic resistant bacteria are referred to as NARB and antibiotic resistant bacteria are referred to as ARB. The simultaneous inactivation of NARB and chloramphenicol is referred to as NARB + CHM and the simultaneous inactivation of ARB and chloramphenicol is referred to as ARB + CHM.
3.2.6.1 Effect on non-antibiotic resistant bacteria (NARB).
The effect of the catalysts for simultaneous degradation of bacteria and chloramphenicol was measured. Fig. 13(a) shows the inactivation profile of NARB in presence of 40 ppm chloramphenicol solution. Samples were taken at an interval of 20 min and plated without any delay to minimize the effect of the antibiotic action of chloramphenicol in the dark. The effect in the dark was the net reduction in bacterial concentration caused due to the presence of chloramphenicol. However, as the time of reaction progresses, the degradation of chloramphenicol also takes place (as shown in Fig. 13(b)). The simultaneous effect of chloramphenicol and catalyst enhances the rate of inactivation of NARB initially, therefore, the rates of inactivation are higher initially but reduced after 40 min due to the continuous degradation of chloramphenicol. Complete inactivation was observed for the electrocatalysis and photoelectrocatalysis. The concentration of chloramphenicol is reduced during the last 20 min, as evidenced by its lower effectiveness, due to the structural changes in chloramphenicol and competition between chloramphenicol and ROS for reaction with the bacteria. The probability of collision between radicals and dead bacterial cells increases as the time progresses, and the inactivation rate further decreases. In photolysis and electrolysis, due to the limited concentration of radicals, the kinetic rate of inactivation does not significantly decline. Therefore, all the processes are plotted using the model described earlier. The rate constants based on the model fitting are listed in Table A.4.† The fitting plots are shown in Fig. A.4 in ESI.†
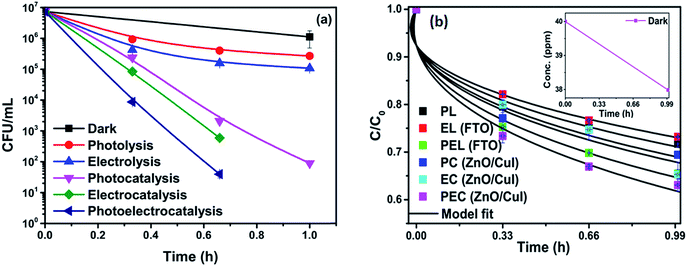 |
| | Fig. 13 (a) Simultaneous inactivation of NARB in NARB + CHM (40 ppm) and (b) kinetic plots of CHM in NARB + CHM. | |
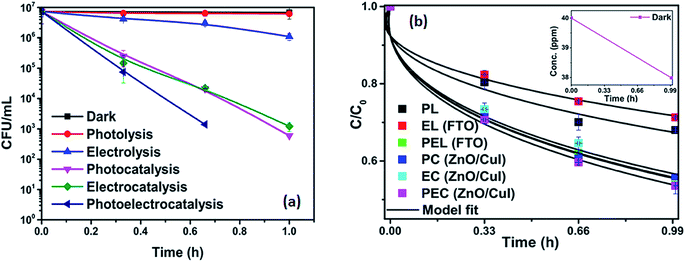 |
| | Fig. 14 (a) Simultaneous inactivation of ARB in ARB + CHM and (b) kinetic plots of CHM in ARB + CHM. | |
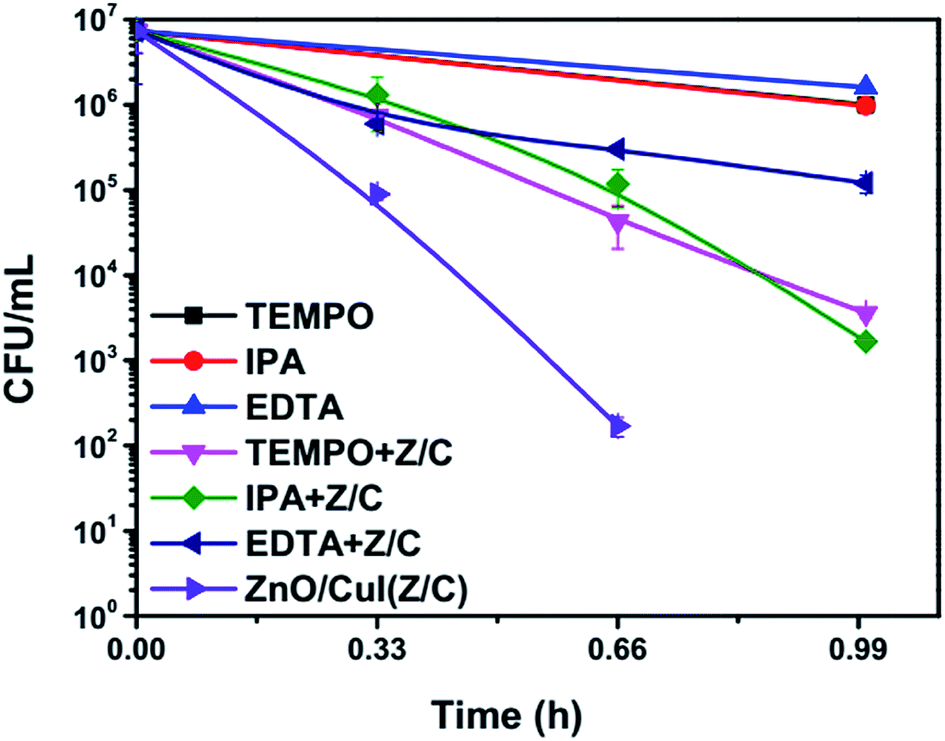
3.2.7 Effect of scavengers on the E. coli bacterial concentration.
Scavengers are chemical species used for trapping of different free radicals or moieties responsible for inactivation. This reveals information about the mechanism of the photocatalytic reaction. The scavengers used for the trapping of superoxide radicals, hydroxyl radicals and holes are TEMPO, IPA and EDTA, respectively.49–51 Control experiments were performed using 2 mmol L−1, 1 mmol L−1 and 1 mmol L−1 of TEMPO, IPA and EDTA, respectively. To determine the toxicity of the added chemicals at the concentrations used for the experiments, the photolytic inactivation reaction was also performed in the presence of the scavengers only. The plots shown below (Fig. 15) show that the scavengers alone demonstrate equivalent inactivation to that of photolysis. However, a significant reduction in the inactivation rate was observed in the case of EDTA, indicating the significant role of holes in the inactivation mechanism. Similarly, TEMPO also reduced the inactivation rate. Therefore, the results suggest that effective inactivation depends on the presence of superoxide radicals, which can be generated via splitting of H2O2 as shown in eqn (25) and (26). These results validate the mechanism proposed based on the band edge positions of the semiconductors. However, the photocatalytic mechanism proposed in Section 3.3 does not involve the direct generation of hydroxyl radicals due to the inappropriate location of the valence band edge of CuI. However, hydroxyl radical generation may take place by the electrochemical pathways illustrated in Section 3.2 (eqn (18)–(20)). Therefore, it can be said that the photoelectrocatalysis using ZnO/CuI uses all the possible active moieties (hydroxyl, superoxide radicals and holes), which augments the inactivation considerably.| | | H2O2 + OH˙ → H2O + HOO˙ | (25) |
| | | HOO˙ + OH˙ → H2O˙ + O2−˙ | (26) |
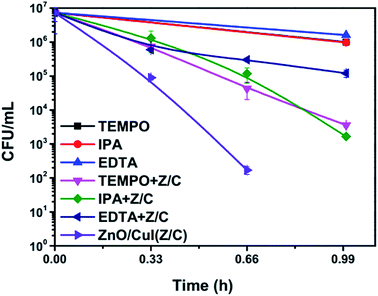 |
| | Fig. 15 Scavenger studies of photoelectrocatalytic inactivation of E. coli in presence of ZnO/CuI. | |
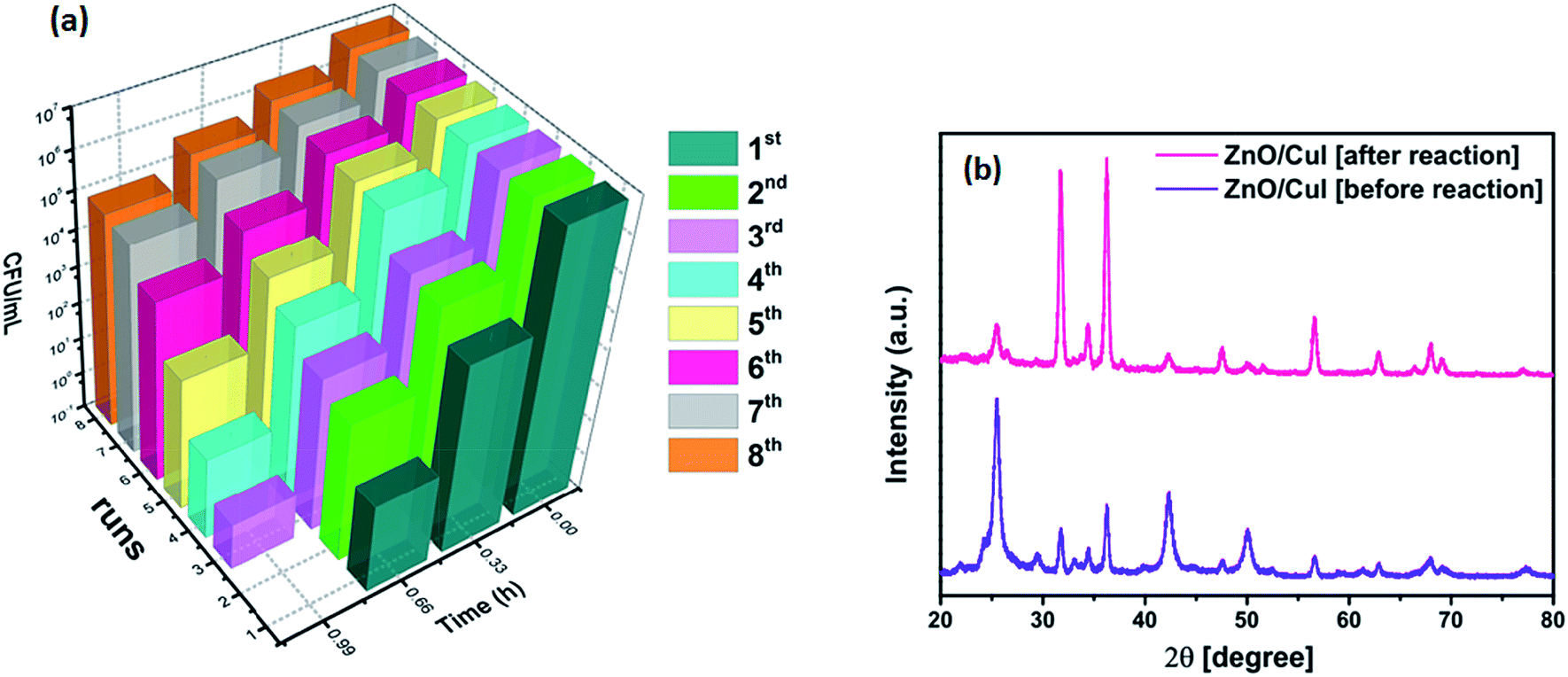
3.2.8 Reusability and photostability.
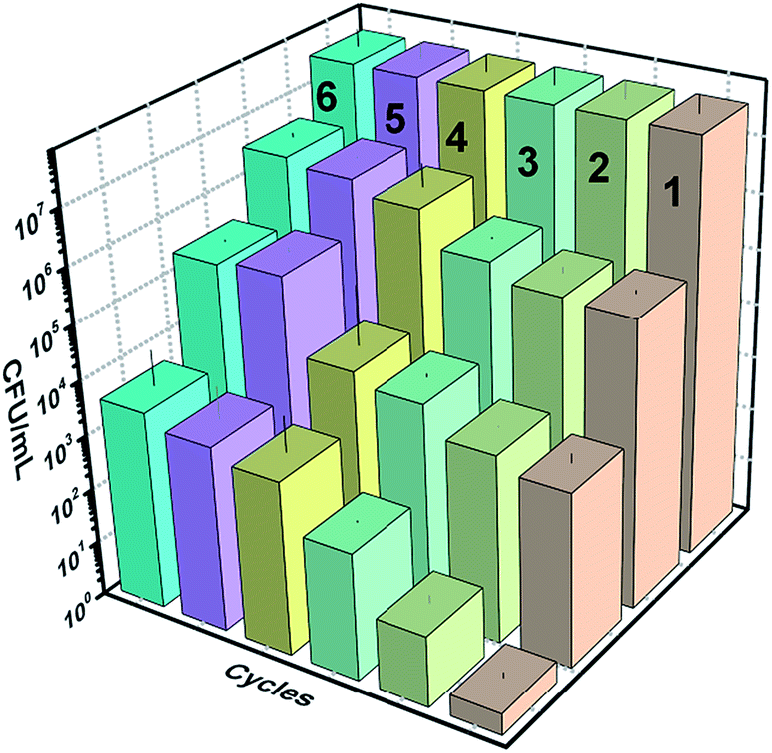
The reusability and photostability of a photocatalyst are the two properties that determine its commercial viability. Therefore, the immobilized catalyst was used for 8 cycles of reaction. The inactivation plots of E. coli for 8 cycles are shown in Fig. 16(a). The slides were washed and dried after each reaction. A reduction in the inactivation was observed after every cycle until the 5th cycle, after which it saturated, due to the reduced content of CuI after each reaction and coverage of the slide with dead bacterial cells (shown in Fig. A.2 in appendix 8†). The reduced content of CuI was confirmed by XRD (Fig. 16(b)). The crystal structure after reaction remained the same but a huge intensity difference was observed in the CuI peaks. The XPS spectra of the elements after the reaction are shown in Fig. A.1 in ESI.† The effect of powder catalysts and loading of CuI on inactivation of bacteria is also shown in Fig. A.6 and A.7, respectively, in ESI.†
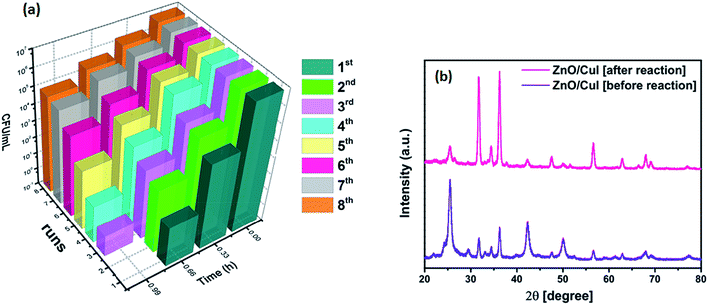 |
| | Fig. 16 (a) Reusability of the catalyst ZnO/CuI for 8 cycles, (b) XRD of the catalyst before and after 8 h of reaction. | |
3.3 Photoelectrocatalysis mechanism
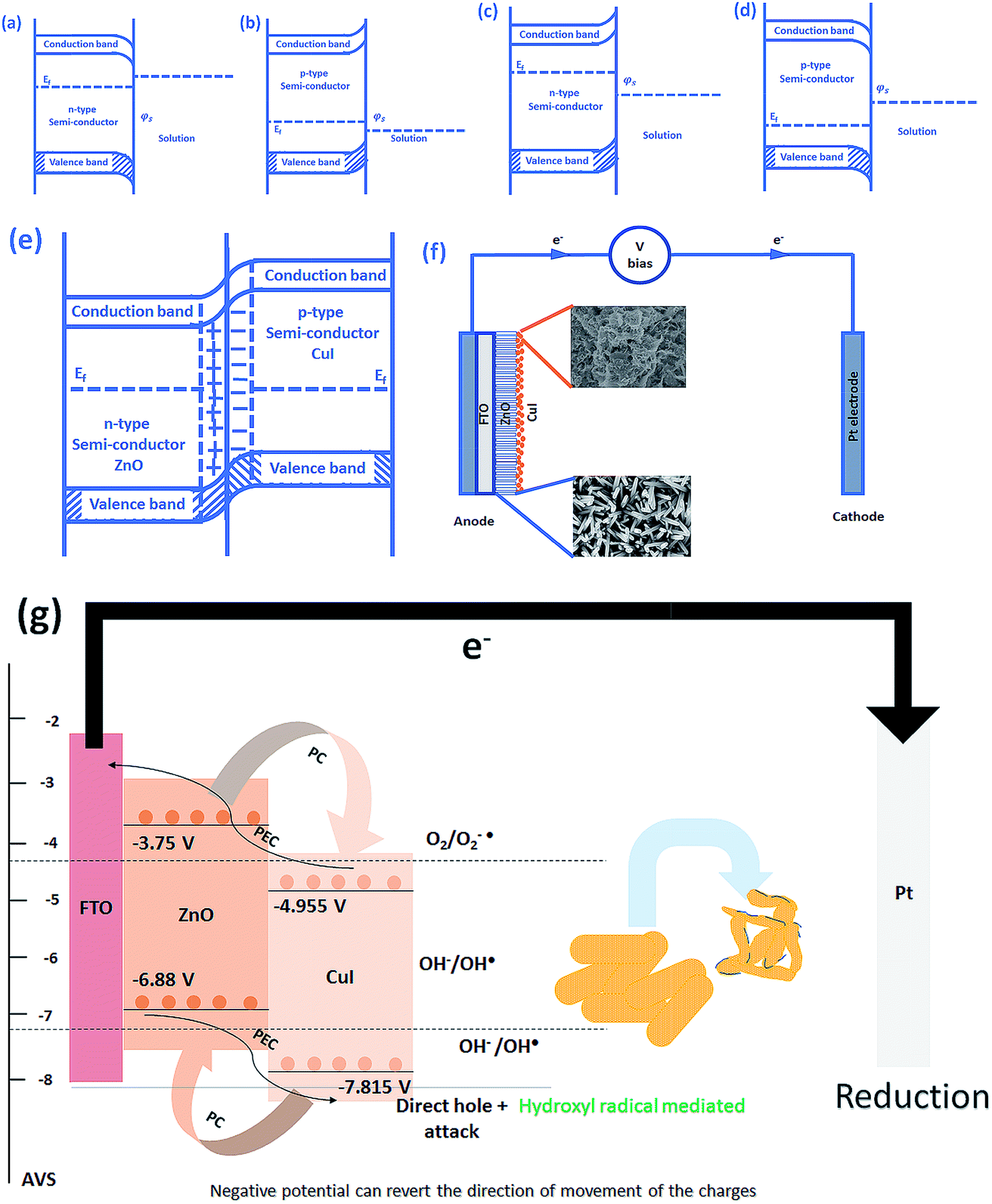
The schematic (Fig. 17) shown below represents the flow of electrons when a positive current is provided to the photoelectrochemical system. As described in the Experimental section, the system is a three-electrode system containing a working electrode of ZnO/CuI on the conducting substrate FTO, counter electrode of platinum and reference electrode of Ag/AgCl. When the working electrode (semiconductor composite) is exposed in the presence of irradiation, the absorption of a photon occurs. This leads to the excitation of electrons to the conduction band and creates vacancies i.e. holes in the valence band.
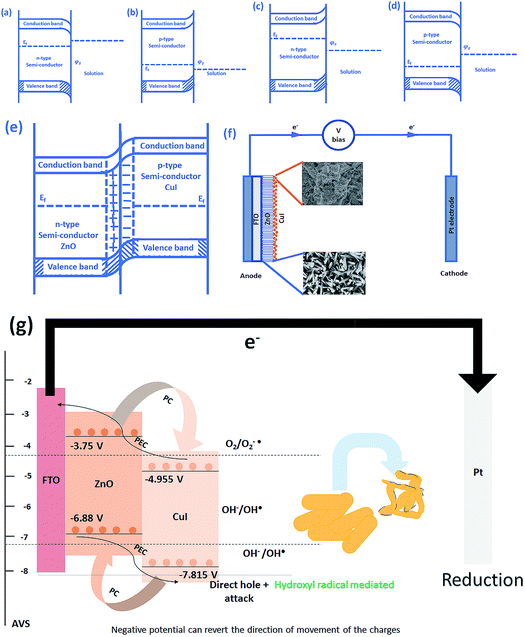 |
| | Fig. 17 Schematic of (a) downward bending in n-type semiconductor, (b) upward bending in p-type semiconductor, (c) upward bending in n-type semiconductor, (d) downward bending in p-type semiconductor, (e) band bending in p–n junction, (f) electron transport in a photoelectrocatalysis setup and (g) electron hole dynamics in the photoelectrocatalyst in case of bacteria (composite). | |
In the absence of any charge, these bands do not show any potential difference as a function of position in the semiconductor.52 However, when potential is applied, the energy (potential) of the bands changes with position in the semiconductor.53 Therefore, charge transfer occurs until the Fermi levels of the semiconductor and redox electrolyte equalize.54 A potential barrier is created at the surface of the catalyst exposed to the electrolyte/composite counterpart. If the potential at the surface, φS, in an n-type semiconductor is more positive than the flat band potential of the semiconductor (AVS scale), the bands tend to become more negative towards the surface and vice versa.54 Therefore, if the concentration of majority charge carriers at the surface is greater, downward bending occurs for an n-type semiconductor while upward band bending occurs for the p-type, and the space charge region is called an enrichment layer (shown in schematic Fig. 17(a) and (b)). Similarly, when the concentration of minority carriers is high at the surface, upward band bending occurs for n-type semiconductors and downward for p-type semiconductors, and the space charge region is called a depletion region (shown in schematic Fig. 17(c) and (d)).53 This band bending facilitates the charge transfer from the semiconductor to the solution until the Fermi levels equalize.
Due to the external potential applied in the system (positive current), the electrons move towards the Pt electrode i.e. from anode to cathode (in the case of an n-type semiconductor as photoanode). However, p-type semiconductors act as photocathodes, thus when applying positive potential the electrons move from the Pt electrode to the working electrodes.52 This condition in the case of a photocathode enhances the charge recombination due to the availability of electrons to recombine with the holes. Therefore, a negative potential must be applied in order to move the electrons from cathode to anode. ZnO/CuI forms a p–n junction.55 Due to the inbuilt potential barrier at the p–n junction the charges are separated and this facilitates the efficient charge transport of the carriers to the respective electrodes (shown in schematic Fig. 17(e)).56,57
For a p-type semiconductor, the Fermi level lies just above the valence band. Therefore, electron flow occurs towards the semiconductor in order to move the Fermi level upward and equalize the redox potential of the electrolyte. Thus, a gradient in potential is developed near the interface and facilitates the movement of electrons towards the interface and movement of holes away from the interface. However, when positive external potential is applied to a p-type semiconductor, its Fermi level moves downward, which reverses the band bending, providing holes at the semiconductor interface and causing the accumulation layer to be converted into a depletion layer. In our system, as a p-type semiconductor, CuI is in direct contact with the electrolyte, therefore, positive external bias is provided to utilize the holes at the interface for anodic (oxidation) reactions.
In the case of an n-type semiconductor, the Fermi level lies just below the conduction band. Therefore, the electrons move towards the solution to move the Fermi level downward. Thus, after equalization of the Fermi level with the redox potential of the electrolyte solution, the band bending occurs in the upward direction. Thus, holes accumulate at the interface for performing the electrochemical reaction. Electrons move away from the interface creating a depletion region at the interface. However, when negative external potential is applied, the Fermi level moves, and the bend bending is inverted. Thus, the depletion region is converted into an accumulation region, providing electrons at the interface for the cathodic reaction. Thus, in our system, i.e. n-type ZnO, positive external bias is provided to make use of the holes present at the interface to promote the anodic (oxidation) reaction and cathodic reaction at the counter electrode.
The band edge potentials of the semiconductors were calculated theoretically and experimentally.54 They are in good agreement with each other. The flat band potential of CuI was evaluated by Mott–Schottky plots. The band edge potentials are tabulated in Table 1. The charge transport is shown in the schematic Fig. 17(g). The charge dynamics was confirmed by the scavenger experiments discussed in Section 3.2.7. The electrons exceed the potential for generation of superoxide radicals while the transport of holes to the highest valence band orbital does not facilitate the formation of hydroxyl radicals. Therefore, due to the high oxidative power of holes, they directly attack the pollutant species and oxidize it. The direct hole attack was confirmed by using EDTA as a hole scavenger.51
Table 1 Band edge positions of photocatalysts
| Semiconductors |
Theoretical |
Mott–Schottky |
|
χ (Mulliken's electronegativity) |
V.B. (V) |
C.B. (V) |
V.B. (V) |
C.B. (V) |
| CuI |
5.49 |
−6.865 |
−4.005 |
−7.815 |
−4.955 |
| ZnO |
5.79 |
−7.39 |
−4.26 |
−6.88 |
−3.75 |
| Hydroxyl radical |
−4.4 |
| Superoxide radicals |
−7.3 |
3.4 Bacterial inactivation mechanism
3.4.1 K+ ion leaching.
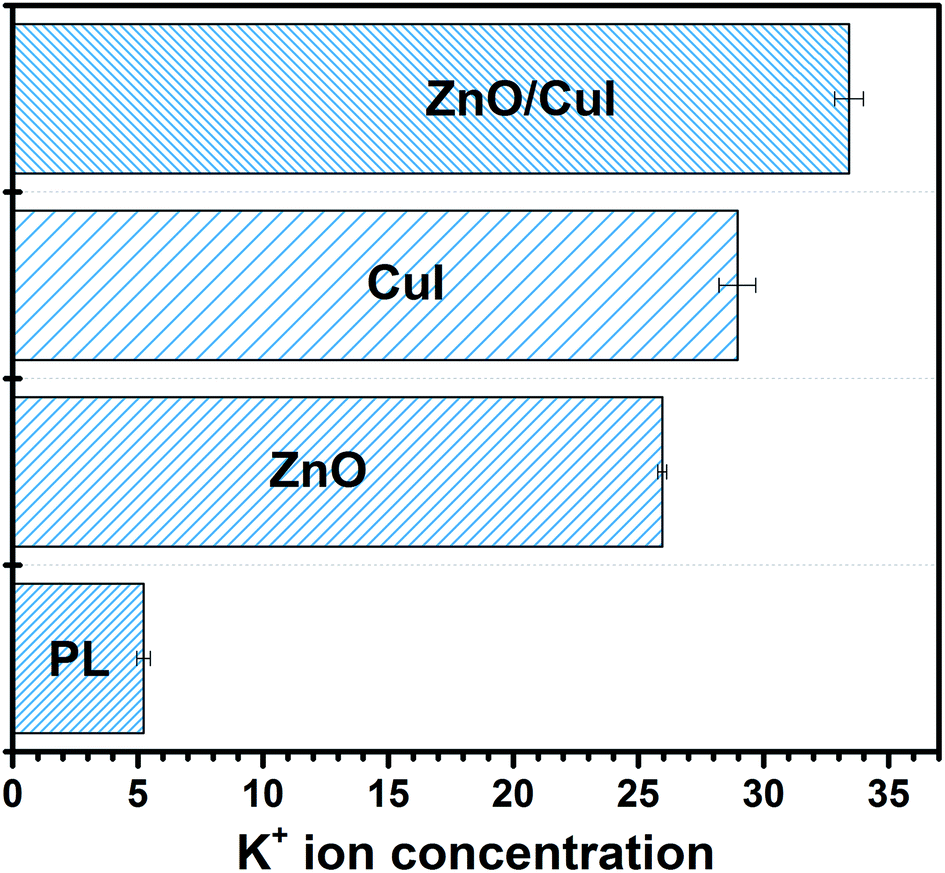
The presence of potassium ions in a bacterial cell body facilitates various tasks such as activation of intracellular enzymes, osmosis, and regulation of the pH in the cell body.58 Therefore, whenever there is any disturbance in the permeability of the cell membrane due to interaction with reactive oxygen species, K+ ions present in the cell body start leaching out into the reaction system. K+ leaching is indicative of the disruption of the membrane. Quantification of the potassium ions was carried out in order to understand the level of leaching in the semiconductors and the composites. ICP-MS analysis was performed to measure the concentration of K+ in the solution. Initially, the K+ concentration is zero, however, for photolysis and the use of ZnO, CuI and the composite ZnO/CuI, the concentration was found to be 5.22 ± 0.27, 25.95 ± 0.18, 28.95 ± 0.73 and 33.41 ± 0.58 ppb, respectively. This indicates that the loss in cell permeability is highest in the case of the composite due to a greater number of radicals. Several reports have measured the trend of leaching at different time intervals. Those studies observed that after a certain time period (30 min) the leached amount became steady (Fig. 18).59,60
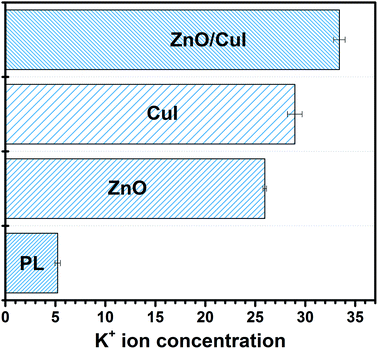 |
| | Fig. 18 K+ ion leaching for photocatalytic inhibition of bacteria by photolysis, ZnO, CuI, ZnO/CuI. | |
3.5 Effect of the washing using DMSO
Fig. 19 below shows the reusability plots after washing the catalyst slide using DMSO. Improvement in catalytic activity was observed, however, complete reusability was not achieved.
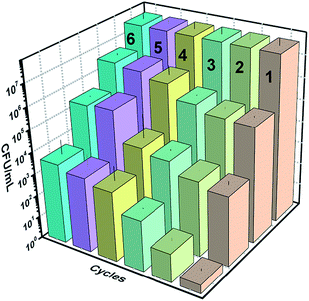 |
| | Fig. 19 Reusability plots after washing with DMSO. | |
4. Conclusions
A novel composite, immobilized ZnO/CuI on FTO, was successfully synthesised via the hydrothermal followed by SILAR approach. Better charge separation and extended visible light absorption were achieved. Morphologically, the dimensional anisotropy of the ZnO nanorods provides unidirectional transport of charge and an external bias of 1 V facilitates the suppression of recombination due to the movement of electrons through the external circuit to the counter electrode. The catalyst was used as photocatalyst, electrocatalyst and photoelectrocatalyst and demonstrated excellent activity for bacterial inactivation in all three cases. Simultaneous inactivation of ARB or NARB together with chloramphenicol was carried out to analyse the effect of different types of pollutants on each other. Increased inactivation of bacteria and reduced degradation of chloramphenicol was obtained for NARB with chloramphenicol in the system and vice versa for ARB in chloramphenicol. Bacterial inactivation was confirmed by K+ ion leaching, and the highest leaching was obtained for the composite (33.41 ppb) owing to its excellent photoactivity. FTIR confirmed the structural changes in the cell body after the reaction. Holes and superoxide radicals were identified as the responsible species for inactivation of bacterial cells, confirmed by scavenger studies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are thankful to Department of Science and Technology (DST) – India for the financial support, CeNSE for characterization facilities. Giridhar Madras thanks DST for the J. C. Bose fellowship. The authors thank Solid State and Structural Chemistry Unit, IISc for extending their Photoluminescence facilities. Authors want to thank Mr Sagar Nilawar for assistance in FTIR. Authors thank Ms Ratna Shree from Rahul Roy lab for providing the chloramphenicol resistant strain of E. coli.
References
- F. Edition, WHO Chron., 2011, 38, 104–108 Search PubMed.
- B. Ambrosetti, L. Campanella and R. Palmisano, J. Environ. Sci. Eng. A, 2015, 4, 273–281 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS.
- M. Frieri, K. Kumar and A. Boutin, Journal of Infection and Public Health, 2017, 10, 369–378 CrossRef.
- V. K. Sharma, N. Johnson, L. Cizmas, T. J. McDonald and H. Kim, Chemosphere, 2016, 150, 702–714 CrossRef CAS PubMed.
- S. B. Zaman, M. A. Hussain, R. Nye, V. Mehta, K. T. Mamun and N. Hossain, Cureus, 2017, 9, e1403 Search PubMed.
- N. K. R. Eswar, R. Gupta, P. C. Ramamurthy and G. Madras, Mol. Catal., 2018, 451, 161–169 CrossRef CAS.
- D. W. Kim, S. Lee, H. Suk Jung, J. Young Kim, H. Shin and K. S. Hong, Int. J. Hydrogen Energy, 2007, 32, 3137–3140 CrossRef CAS.
- J. Ouyang, M. Chang and X. Li, J. Mater. Sci., 2012, 47, 4187–4193 CrossRef CAS.
- T. N. Trung, D.-B. Seo, N. D. Quang, D. Kim and E.-T. Kim, Electrochim. Acta, 2018, 260, 150–156 CrossRef CAS.
- Y. Hunge, M. Mahadik, A. Moholkar and C. Bhosale, Appl. Surf. Sci., 2017, 420, 764–772 CrossRef CAS.
- S. Han, W. Qu, J. Xu, D. Wu, Z. Shi, Z. Wen, Y. Tian and X. Li, Phys. Status Solidi A, 2017, 214(9), 1700059 CrossRef.
- P.-Y. Kuang, X.-J. Zheng, J. Lin, X.-B. Huang, N. Li, X. Li and Z.-Q. Liu, ACS Omega, 2017, 2, 852–863 CrossRef CAS.
- J. Feng, L. Cheng, J. Zhang, O. K. Okoth and F. Chen, Ceram. Int., 2018, 44, 3672–3677 CrossRef CAS.
- J. Wang, Z. Yang, X. Gao, W. Yao, W. Wei, X. Chen, R. Zong and Y. Zhu, Appl. Catal., B, 2017, 217, 169–180 CrossRef CAS.
- L. Cheng, L. Liu, R. Li and J. Zhang, J. Electrochem. Soc., 2017, 164, H726–H733 CrossRef CAS.
- A. Pramanik, D. Laha, D. Bhattacharya, P. Pramanik and P. Karmakar, Colloids Surf., B, 2012, 96, 50–55 CrossRef CAS.
- M. Sun, C. Zhai, J. Hu, M. Zhu and J. Pan, J. Colloid Interface Sci., 2018, 511, 110–118 CrossRef CAS.
- M. Singh, M. Goyal and K. Devlal, Journal of Taibah University for Science, 2018, 12, 470–475 CrossRef.
- N. K. Eswar, R. Gupta, P. C. Ramamurthy and G. Madras, Mol. Catal., 2018, 451, 161–169 CrossRef CAS.
- B. N. Ezealigo, A. C. Nwanya, A. Simo, R. U. Osuji, R. Bucher, M. Maaza and F. Ezema, Arabian J. Chem., 2017, 0–11 Search PubMed.
- N. Kamarulzaman, M. F. Kasim and R. Rusdi, Nanoscale Res. Lett., 2015, 10, 346 CrossRef.
- P. Rodnyi and I. Khodyuk, Opt. Spectrosc., 2011, 111, 776–785 CrossRef CAS.
- M. Kahouli, A. Barhoumi, A. Bouzid, A. Al-Hajry and S. Guermazi, Superlattices Microstruct., 2015, 85, 7–23 CrossRef CAS.
- M. N. Amalina, Y. Azilawati, N. A. Rasheid and M. Rusop, Procedia Eng., 2013, 56, 731–736 CrossRef CAS.
- M. Nur Amalina, M. A. Azman, U. M. Noor and M. R. Mahmood, Adv. Mater. Res., 2012, 403–408, 451–455 Search PubMed.
- V. Srikant and D. R. Clarke, J. Appl. Phys., 1998, 83, 5447–5451 CrossRef CAS.
- C. Supatutkul, S. Pramchu, A. Jaroenjittichai and Y. Laosiritaworn, J. Phys.: Conf. Ser., 2017, 901, 012172 CrossRef.
- Y. Zhang, Y.-H. Wen, J.-C. Zheng and Z.-Z. Zhu, Appl. Phys. Lett., 2009, 94, 113114 CrossRef.
- E. Sheha, H. Khoder, T. S. Shanap, M. G. El-Shaarawy and M. K. El Mansy, Optik, 2012, 123, 1161–1166 CrossRef CAS.
- Y. Yang and Q. Gao, Langmuir, 2005, 21, 6866–6871 CrossRef CAS.
- L. Schmidt-Mende and J. L. MacManus-Driscoll, Mater. Today, 2007, 10, 40–48 CrossRef CAS.
- J. Han, P. Mantas and A. Senos, J. Eur. Ceram. Soc., 2002, 22, 49–59 CrossRef CAS.
- W. Zhang, X. Wu, H. Chen, J. Zhu and G. Huang, J. Appl. Phys., 2008, 103, 093718 CrossRef.
- M. Zi, J. Li, Z. Zhang, X. Wang, J. Han, X. Yang, Z. Qiu, H. Gong, Z. Ji and B. Cao, Phys. Status Solidi A, 2015, 212, 1466–1470 CrossRef CAS.
- Z. Qiu, T. Zeng, K. Ye, X. Yu, Y. Zhu and Y. Zhang, J. Electrochem. Soc., 2016, 163, H18–H23 CrossRef CAS.
- J. Wang, L. Pan, H. Meng, R. Han, Z. Huang and C. Zhang, Adv. Mater. Phys. Chem., 2016, 06(03), 16 Search PubMed.
- R. Ahumada-Lazo, L. M. Torres-Martínez, M. A. Ruíz-Gómez, O. E. Vega-Becerra and M. Z. Figueroa-Torres, Appl. Surf. Sci., 2014, 322, 35–40 CrossRef CAS.
- S. Sontakke, J. Modak and G. Madras, J. Adv. Oxid. Technol., 2011, 14, 86–92 CAS.
- E. L. Nussbaum, L. Lilge and T. Mazzulli, J. Clin. Laser Med. Surg., 2002, 20, 325–333 CrossRef.
- M. Eichler, R. Lavi, A. Shainberg and R. Lubart, Lasers Surg. Med., 2005, 37, 314–319 CrossRef.
- R. Lubart, A. Lipovski, Y. Nitzan and H. Friedmann, Laser Therapy, 2011, 20, 17–22 CrossRef CAS.
- K. Tennakone, G. R. R. A. Kumara, I. R. M. Kottegoda, V. P. S. Perera, G. M. L. P. Aponsu and K. G. U. Wijayantha, Sol. Energy Mater. Sol. Cells, 1998, 55, 283–289 CrossRef CAS.
- A. G. Al-Sehemi, A. Irfan, S. A. Alrumman and A. Hesham, Bull. Chem. Soc. Ethiop., 2016, 30, 307–316 CrossRef CAS.
- S. Kapoor and L. Varshney, J. Phys. Chem. A, 1997, 101, 7778–7782 CrossRef CAS.
- P. M. Hawkey, BMJ, 1998, 317, 657–660 CrossRef CAS.
- M. U. Rasheed, N. Thajuddin, P. Ahamed, Z. Teklemariam and K. Jamil, Rev. Inst. Med. Trop. Sao Paulo, 2014, 56, 341–346 CrossRef.
- N. Khadgi, A. R. Upreti and Y. Li, RSC Adv., 2017, 7, 27007–27016 RSC.
- K. Ouyang, K. Dai, S. L. Walker, Q. Huang, X. Yin and P. Cai, Sci. Rep., 2016, 6, 25702 CrossRef CAS.
- R. Gupta, B. Boruah, J. M. Modak and G. Madras, J. Photochem. Photobiol., A, 2019, 372, 108–121 CrossRef CAS.
-
J. O. M. Bockris and A. K. Reddy, Modern electrochemistry 2B: electrodics in chemistry, engineering, biology and environmental science, Springer Science & Business Media, 2001 Search PubMed.
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS.
- Y. Xu and M. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- N. Gupta, G. Alapatt, R. Singh, K. Poole and R. Podila, Int. J. Photoenergy, 2010, 2010, 13 Search PubMed.
- N. Gupta, G. Alapatt, R. Podila, R. Singh and K. Poole, Int. J. Photoenergy, 2009, 154059 Search PubMed.
- A. Samal, D. P. Das and G. Madras, Sci. Rep., 2018, 8, 2881 CrossRef.
- W. Epstein, Prog. Nucleic Acid Res. Mol. Biol., 2003, 75, 293–320 CAS.
- P. K. Samantaray, G. Madras and S. Bose, J. Membr. Sci., 2018, 548, 203–214 CrossRef CAS.
- C. Hu, J. Guo, J. Qu and X. Hu, Langmuir, 2007, 23, 4982–4987 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00483a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
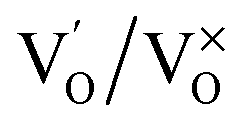 (single oxygen ion vacancy). The corresponding defect levels are in the range of 0.5–0.15 eV below the conduction band, as described elsewhere.33 The acceptor defects are composed of vacancies generated due to the
(single oxygen ion vacancy). The corresponding defect levels are in the range of 0.5–0.15 eV below the conduction band, as described elsewhere.33 The acceptor defects are composed of vacancies generated due to the 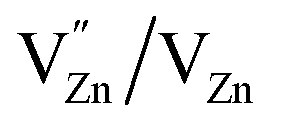 ,
, 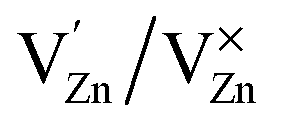 and
and  and lie in the range of 0.8 to 2.8 eV above the valence band.34 Therefore, the emission peaks at 421, 450 and 516 nm are transitions from the energy levels of defect sites (shallow traps/deep traps).35 It is evident from the X-ray photoelectron spectroscopy (XPS) O-1s spectrum (Section 3.1.5) that the concentration of oxygen vacancies in the composite is significant.
and lie in the range of 0.8 to 2.8 eV above the valence band.34 Therefore, the emission peaks at 421, 450 and 516 nm are transitions from the energy levels of defect sites (shallow traps/deep traps).35 It is evident from the X-ray photoelectron spectroscopy (XPS) O-1s spectrum (Section 3.1.5) that the concentration of oxygen vacancies in the composite is significant.
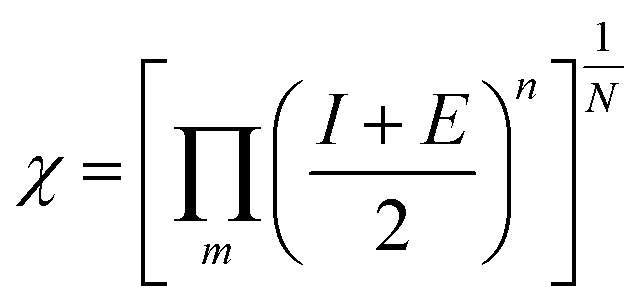

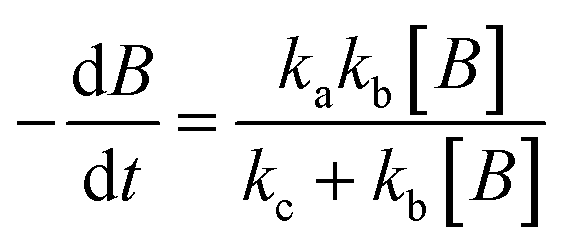
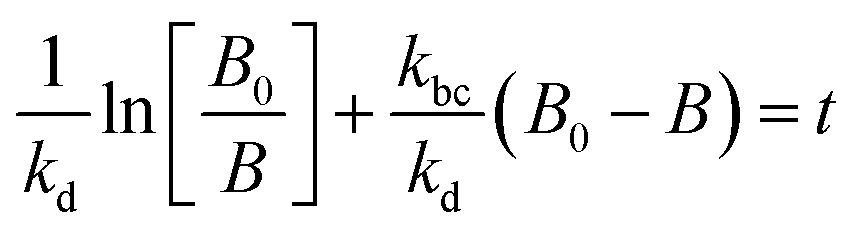
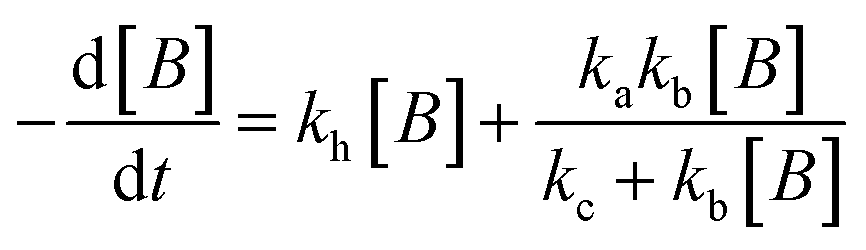
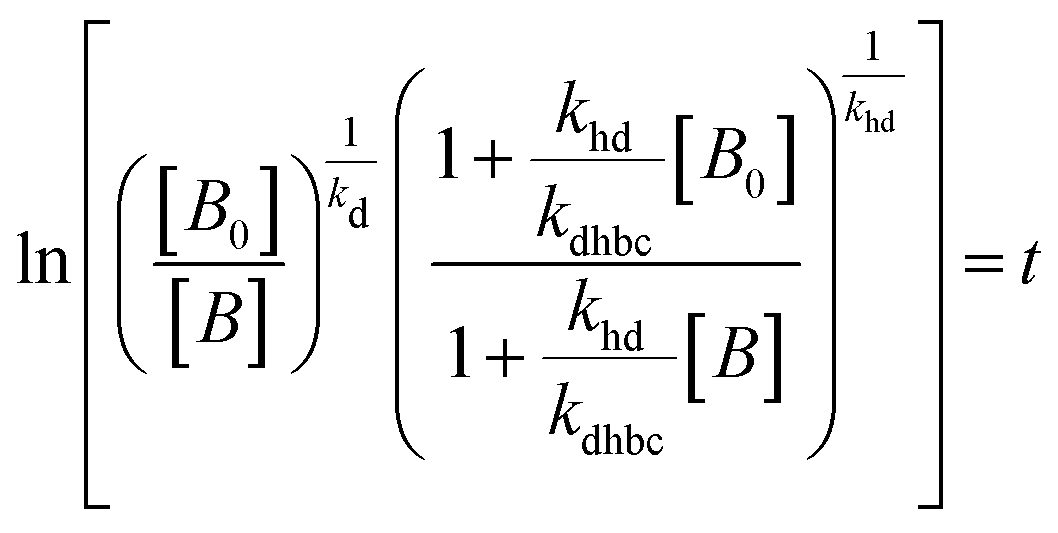
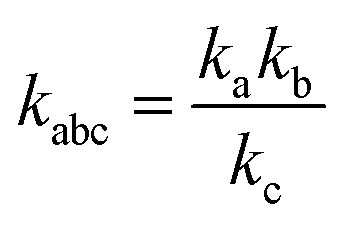 ,
,  , kd = kh + kabc,
, kd = kh + kabc, 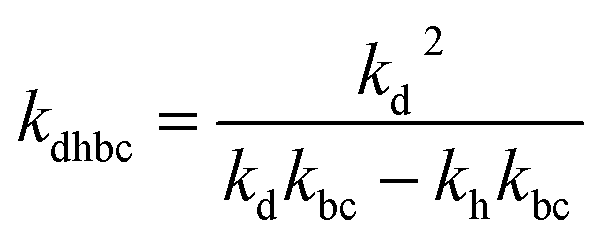 ,
, 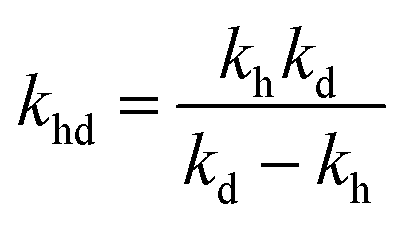 .
.