
An overview on enhancing the stability of lead halide perovskite quantum dots and their applications in phosphor-converted LEDs
Yi
Wei
ab,
Ziyong
Cheng
*ab and
Jun
Lin
 *abc
*abc
aState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. E-mail: jlin@ciac.ac.cn; zycheng@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei, 230026, China
cSchool of Applied Physics and Materials, Wuyi University, Jiangmen, Guangdong 529020, P. R. China
First published on 22nd November 2018
Abstract
Beyond the unprecedented success achieved in photovoltaics (PVs), lead halide perovskites (LHPs) have shown great potential in other optoelectronic devices. Among them, nanometer-scale perovskite quantum dots (PQDs) with fascinating optical properties including high brightness, tunable emission wavelength, high color purity, and high defect tolerance have been regarded as promising alternative down-conversion materials in phosphor-converted light-emitting diodes (pc-LEDs) for lighting and next-generation of display technology. Despite the promising applications of perovskite materials in various fields, they have received strong criticism for the lack of stability. The poor stability has also attracted much attention. Within a few years, numerous strategies towards enhancing the stability have been developed. This review summarizes the mechanisms of intrinsic- and extrinsic-environment-induced decomposition of PQDs. Simultaneously, the strategies for improving the stability of PQDs are reviewed in detail, which can be classified into four types: (1) compositional engineering; (2) surface engineering; (3) matrix encapsulation; (4) device encapsulation. Finally, the challenges for applying PQDs in pc-LEDs are highlighted, and some possible solutions to improve the stability of PQDs together with suggestions for further improving the performance of pc-LEDs as well as the device lifetime are provided.
 Yi Wei | Yi Wei received his BS degree (2015) from Northeast Normal University (NENU), Republic of China. He is currently pursuing a PhD degree in chemistry at Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences, under the supervision of Prof. Ziyong Cheng and Prof. JunLin. His research focuses on improving the stability of lead halide perovskite quantum dots, and their applications in solid-state lightings and displays. |
 Ziyong Cheng | Ziyong Cheng earned his BS degree in materials engineering from Changchun University of Technology in 1994 and his PhD degree from the Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences, in 2006. Following postdoctoral studies at the Max-Planck Institute for Polymer Research (Mainz, Germany), he returned to CIAC (2008) to take up an associate professor position in inorganic chemistry. In 2013, he was promoted to a full professor. His research interests are nanostructured materials including perovskite quantum dots in photoelectric applications and polymer–inorganic nanocomposites for biomaterials related fields. |
 Jun Lin | Jun Lin was born in Changchun, China, in 1966. He received his BS and MS degrees in inorganic chemistry from Jilin University, China, in 1989 and 1992, respectively, and a PhD degree in inorganic chemistry from the Changchun Institute of Applied Chemistry in 1995. His postdoctoral studies were performed at the City University of Hong Kong (1996), Institute of New Materials (Germany, 1997), Virginia Commonwealth University (USA, 1998) and University of New Orleans (USA, 1999). He came back to China in 2000, and since then has been working as a professor at CIAC. His research interests include bulk and nanostructured luminescent materials and multifunctional composite materials together with their applications in the display, lighting, and biomedical fields. |
1. Introduction
Lead halide perovskites (LHPs) have been regarded as promising classes of materials for photovoltaics (PVs) and optoelectronic devices, owing to the unique characteristics, such as long charge carrier diffusion lengths, precise tunable bandgaps, high light-absorption coefficients, and high defect tolerance.1–10 Research on LHPs has been gaining increasingly intense interest over the past years. Since the first perovskite photovoltaics with a power conversion efficiency (PCE) of 3.9% came out in 2009, the new record is over 23% now.1,10 The great success of applying LHPs in PVs has triggered a number of corresponding research studies. Applications of LHPs in the fields including light-emitting diodes (LEDs),11–20 lasers,21–26 X-ray imaging,27–32 and photodetectors (PDs)33–38 have been massively investigated. Among them, LEDs in particular have attracted worldwide attention with a rapid rise in the electroluminescence (EL) efficiency of perovskite LEDs (PeLEDs).Perovskite nanomaterials could be divided into five main types by multiple dimensions:39,40 nanospheres41/nanocubes42 (both of them can be regarded as quantum dots), nanorods43/nanowires,44,45 nanoplates46/nanodisks,47 supercrystals,48 and polycrystalline films.49,50 Among them, polycrystalline films and colloidal quantum dots represent the most recent type of LHP materials with promising EL applications.51 However, due to the low film quality as well as low photoluminescence quantum yields (PL QYs) of LHP polycrystalline films, achieving high EL efficiency PeLEDs is somewhat difficult.12,15,52 The PL QYs of perovskite quantum dots (PQDs) solution are much higher.41,53–56 Unfortunately, the charge injection efficiency of PQDs is relatively low owing to the fact that the poor conductive surface ligands block the charge injection.42 The EQE was successfully improved via surface ligand density control, sufficient purification, etc.57–61 In comparison with the mature technology such as organic LEDs (OLEDs)62 and CdSe-based QD LEDs (QLEDs)63,64 that have optimized device structure, further studies are needed to boost the EL efficiency and brightness.
Although luminescent LHPs have led to rapid advancement in EL LEDs and presented the opportunity for lighting devices and the next-generation displays, phosphor-converted LEDs (pc-LEDs) remain in the mainstream at present due to their extraordinary luminous efficiency, high electro-optical conversion efficiency, long operation life, etc.65–70 The so-called pc-LEDs consist of inorganic phosphors as the down-conversion layer and a blue InGaN chip as the light source. Owing to the highly efficient blue InGaN chip which has been widely recognized as an optimal light emission source, utilizing appropriate color converters plays a pivotal role in the performance of pc-LEDs.
Recently, PQDs with almost every figure of merit, such as high brightness, tunable emission, high color purity, high light-absorption coefficient, high defect tolerance and facile fabrication, have shown promise as phosphors in pc-LEDs.56,71–85 Especially, PQDs present high color purity together with a narrow full width at half maximum (FWHM) ranging from 12 to 40 nm.54,71 The color gamut of pc-LEDs composed of PQDs could cover over 140% of the National Television System Committee (NTSC) standard, which is superior to commercial OLEDs and QLEDs.86–89 All these advantages suggest their potential as color-converters in pc-LEDs for solid-state lighting as well as future displays (Fig. 1).
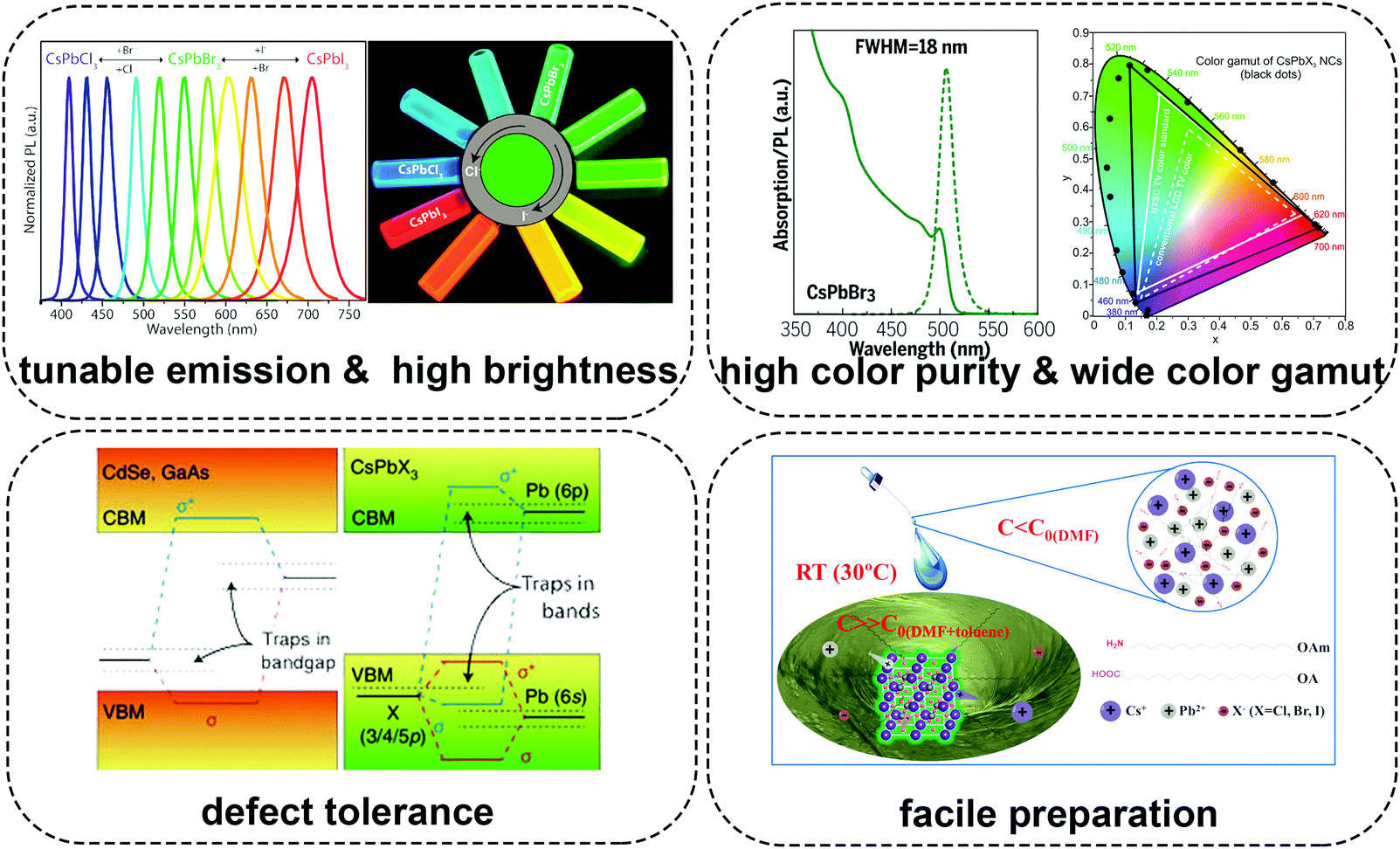 | ||
| Fig. 1 Schematic illustration describing the advantages of lead halide perovskite quantum dots in pc-LEDs. Figures in the section of tunable emission & high brightness are reprinted with permission from ref. 78. Copyright 2018, American Chemical Society. Figures in the section of high color purity are reprinted with permission from ref. 72. Copyright 2017, AAAS. Figures in the section of wide color gamut are reprinted with permission from ref. 71. Copyright 2015, American Chemical Society. Figures in the section of defect tolerance are reprinted with permission from ref. 73. Copyright 2018, Springer Nature. Figures in the section of facile preparation are reprinted with permission from ref. 55. Copyright 2016, Wiley-VCH. | ||
Despite the huge success achieved in various fields, the applications of perovskite materials are impeded by several issues related to their stability.90–96 (1) LHPs are highly sensitive to polar solvents due to their inherent ionic nature.71 They usually lose optical properties, surface ligands (for perovskite nanomaterials), and even structural integrity in polar organic solvents or water.97–99 Particularly, LHPs even decompose in a moist condition, which really impedes their practical applications.100 (2) Owing to the low formation energy, LHPs are vulnerable to environmental stress like light, oxygen, heat, etc.93,101–103 As the color-converter for practical pc-LED devices, they would function on a hot LED chip in ambient air. Environmental stress has become the most harmful factor that affects their performance. Therefore, environmental stability also has nearly become the primary concern for LHPs.94 (3) The photoluminescence (PL) of all kinds of luminescent materials would be inevitably affected upon heating, especially for LHPs.98 On the one hand, LHPs would be decomposed directly under exceedingly high temperature.104,105 On the other hand, thermal stress might accelerate the rates of oxidation and hydration, which amplify the oxygen- and moisture-induced PL quenching. (4) Fast ion-exchange takes place and brings in the severe PL emission shift when two LHPs with varied halide components are mixed.106–108 For example, the red and green PL shifts to yellow rapidly when green-emitting and red-emitting LHPs are mixed.109 It is worth noting that the ion-exchange occurs in gas, liquid, and solid phases, which seems to be irresistible.110–112 All these stability-related shortcomings mentioned above cause serious problems in material synthesis, storage, operation, and device fabrication. PQDs which represent one of the most recent types of LHP materials also suffer from the same problem.113,114 And not only that, PQDs reunite and lose colloidal stability as well as quantum efficiency under continuous illumination115 or during purification,97 which makes their application more challenging.
Stability is the common issue for all of the perovskite-related fields, and the poor stability limits their broad applications. Recently, the structural116 and irradiant96 effects on the optoelectronic properties and stability of perovskites have been reported. Nevertheless, most of the reviews have mainly focused on applications of LHPs in PV technologies.90,93,94 The ion-diffusion-induced optoelectronic device degradation and the strategies to improve the stability of perovskite films in EL type of LEDs have been reviewed recently.92 However, the degradation mechanisms for perovskite films in optoelectronic applications and PQDs in photonic applications are different. In this review, the origins of the instability of PQDs such as structural stability, interfacial stability, atmospheric stability (including the effects caused by light, oxygen and moisture), and thermal stability combined with the full understanding of degradation mechanisms under diverse conditions are summarized. Meanwhile, we review the recent studies on enhancing the stability of PQDs. According to the principle, these strategies can be classified into four types: (1) compositional engineering; (2) surface engineering; (3) matrix encapsulation; (4) device encapsulation. Their performance in pc-LEDs is also summarized. At last, based on the inspiring achievements in the issue, some possible solutions to improve the stability of PQDs and the device performance of pc-LEDs are highlighted, and the prospects for applying PQDs in pc-LEDs are presented.
2. Origins of the instability of PQDs
2.1. Instability derived from the crystal structure
Perovskite is a calcium titanium oxide mineral species consisting of calcium titanate, with the chemical formula of CaTiO3.117 The discovery of perovskite (CaTiO3) dates back to 1839 in the Ural Mountains of Russia by Gustav Rose. The mineral is named after the Russian mineralogist L. A. Perovski (1792–1856). Materials with a similar type of crystal structure as CaTiO3 are known as perovskites. And any compound denoted by the general chemical formula of ABX3 can be described as a perovskite. The previous research centered on compounds consisting of oxygen, tetravalent metal cations (such as Ge4+, Ti4+, Zr4+, etc.) and bivalent metal cations (e.g., Ca2+, Sr2+, Ba2+, etc.), which are known as perovskite oxides.118–120 Recent studies have focused on perovskites composed of halide anions (Cl−, Br−, or I−), bivalent metal cations (such as Pb2+, Sn2+, Cu2+, Ge2+, Eu2+, Ni2+, etc.), and monovalent inorganic metal cations or organic cations (e.g., Cs+, Rb+, CH3NH3+ (methylammonium, MA), CH(NH2)2+ (formamidinium, FA), etc.).40,121–124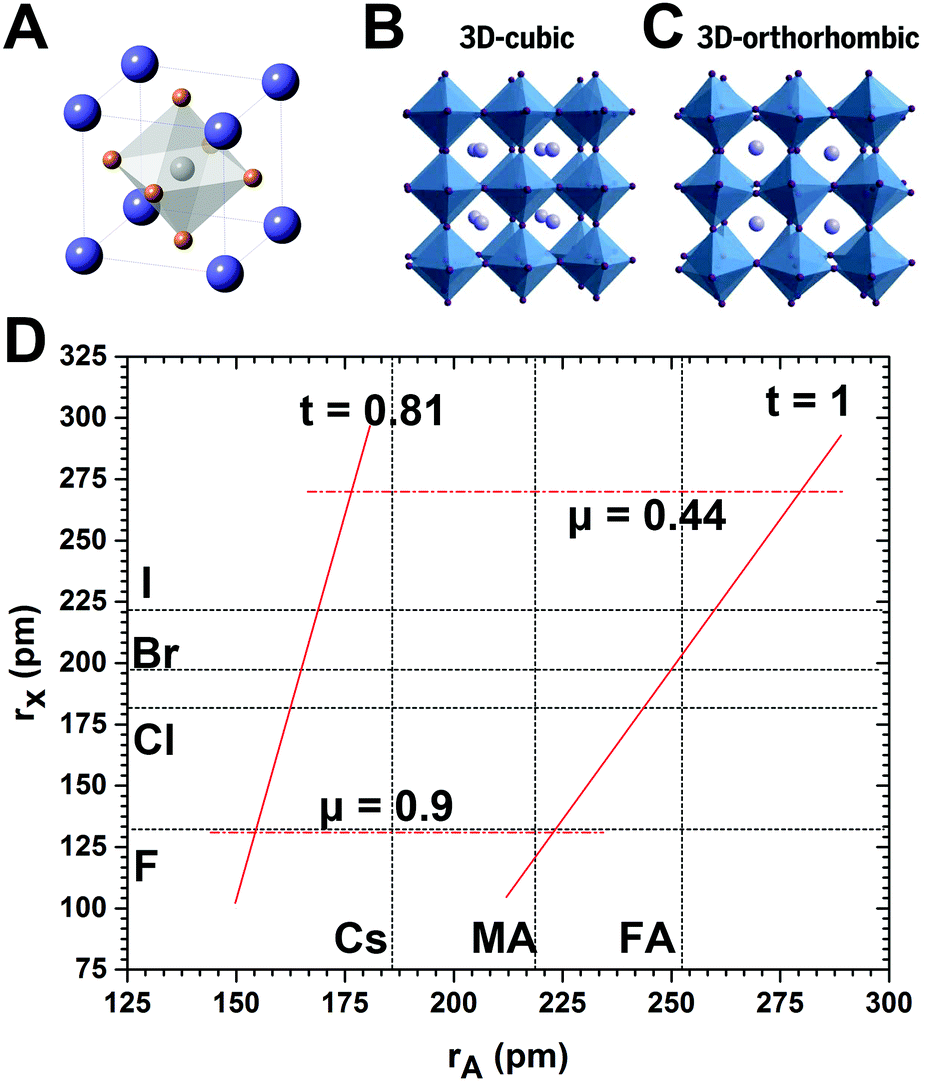 | ||
| Fig. 2 (A) Depiction of APbX3 perovskites with cubic structure, and crystal structures of lead halide perovskites with (B) cubic and (C) orthorhombic phase. (D) Formability of 3D lead halide perovskites as a function of A-site cation and halide anion radii. The solid and dashed lines mark the bounds of the tolerance and octahedral factors, respectively. (B and C) Reprinted with permission from ref. 72. Copyright 2017, AAAS. | ||
The A cations occupy the 12-fold coordinated sites surrounded by eight PbX6 octahedra. Therefore, the A cation is limited by the interstices of the 3D PbX6 framework. A large A cation cannot be accommodated into the interstice, while a small A cation filling in the interstice leads to collapse of the 3D perovskite structure. For an ideal cubic perovskite structure, A cation that should be accommodated into the framework and the ionic radii have the following geometrical relationship:
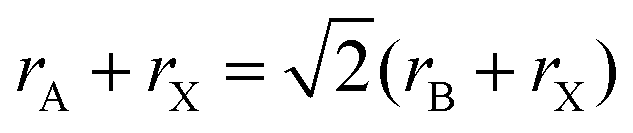 | (1) |
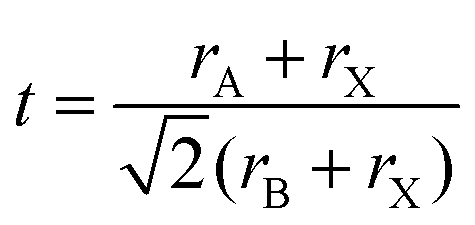 | (2) |
Although the formula has been proposed for nearly 100 years (in the early 1920s) by Goldschmidt, t is still widely accepted as a criterion for predicting the formability of 3D perovskite structures.136 Generally, most perovskites maintain 3D connectivity in the range of approximately 0.813 ≤ t ≤ 1.107.137 A recent study shows that the range of 0.9 ≤ t ≤ 1 is generally considered as a good fit for an ideal cubic perovskite structure, implying the likely formation of cubic structures.138 The range for the tolerance factor t between 0.71 and 0.9 implies the likelihood of an orthorhombic or rhombohedral structure owing to the distortion of the PbX6 octahedra. When the condition cannot be satisfied, the 3D octahedral framework would collapse. In detail, when t ≥ 1 or t ≤ 0.71, non-perovskite structures of CsNiBr3-type (1D hexagonal structures formed by the face-sharing of octahedra) or NH4CdCl3-type (1D orthorhombic structures formed by the edge-sharing of octahedra) with much large band gaps and poor electroconductivity are formed, respectively.72,139
The other semiempirical geometric parameter, octahedral factor, μ, can be used to predict the octahedral stability. The octahedral factor μ can be described as
| μ = rB/rX | (3) |
Typically, BX6 octahedra are stable in the range between 0.442 and 0.895. As shown in Fig. 2D, the combination of Goldschmidt tolerance factor t and octahedral factor μ provides a parameter space for 3D perovskite formability.140 It is easy to draw a conclusion that appropriate ionic radii of components are crucial for forming a stable cubic perovskite. Cubic structures of perovskites are usually stable at high temperature, and after cooling down to RT, tetragonal or orthorhombic phases with less symmetry are thermodynamically preferred.127 MA-based LHPs present the nearly ideal cubic perovskite structure, whereas Cs and FA ions are slightly small or large to perfectly accommodate A-sites.141 Both MAPbCl3 and MAPbBr3 have a cubic structure,142 and MAPbI3 perovskites have a tetragonal structure at RT.103,143 The bulk CsPbI3 perovskites have a 3D orthorhombic structure and rapidly transform into the wide-bandgap 1D orthorhombic phase at RT. FAPbI3 perovskites have a pseudocubic structure with relatively better stability, but still transform into the wide-bandgap 1D hexagonal phase in several months.113 CsPbBr3 perovskites are widely used in EL and color-conversion type LEDs, and initial research suggested the structure of CsPbBr3 nanocrystals (NCs) as cubic,71 but further studies revealed that both bulk CsPbBr3144 and CsPbBr3 NCs145 are actually orthorhombic.
Nanoscale perovskite materials are a little different from bulk perovskites. On the one hand, the high specific surface area might accelerate the decomposition.53 On the other hand, the nanoscale form of perovskites is helpful to stabilize the cubic phase due to the large contribution of surface energy.146 For instance, bulk CsPbI3 with cubic phase is only stable above 305 °C, and it will transform into a 3D orthorhombic phase with a larger band gap gradually and into a nonperovskite 1D orthorhombic structure in the end. CsPbI3 in nanodimension can maintain the cubic phase for months in ambient air after appropriate treatment.146 Recently, Pradhan et al.147 demonstrated that the reaction temperature greatly affected the phase-stability of CsPbI3 PQDs. The CsPbI3 PQDs obtained below 200 °C usually lost phase-stability. But increasing the reaction temperature up to 260 °C effectively stabilized the CsPbI3 PQDs, and the cubic to orthorhombic phase transformation can be restricted.
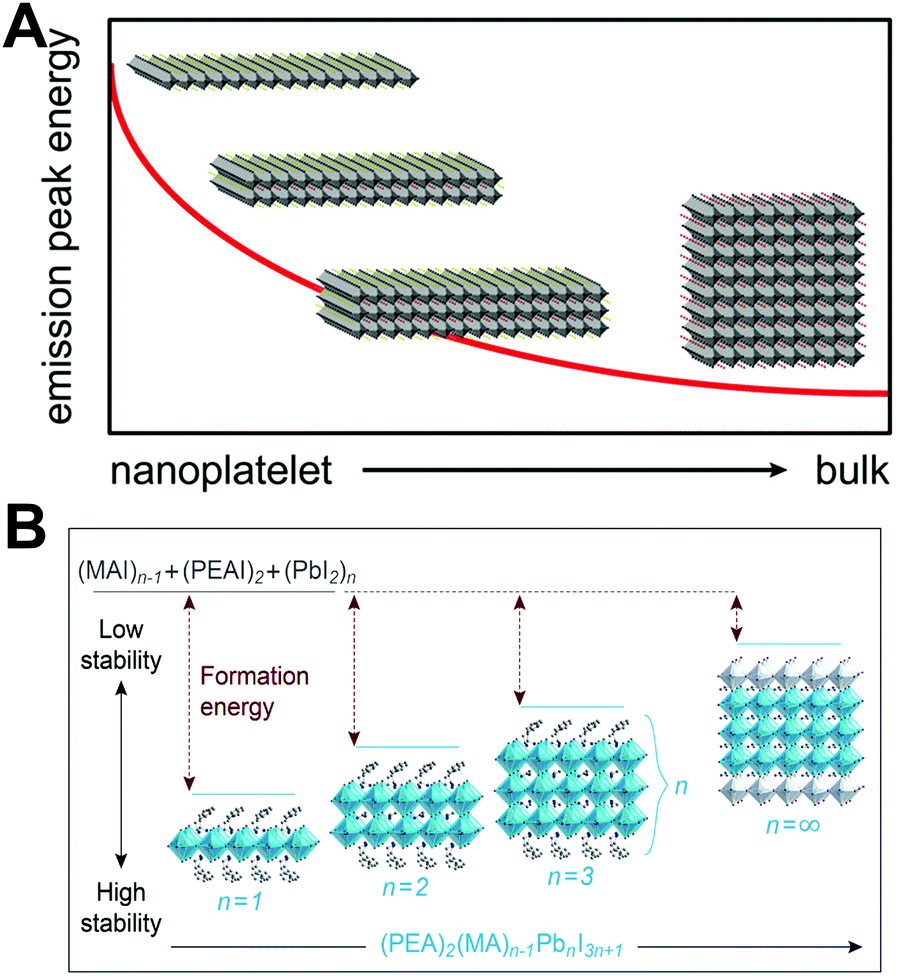 | ||
| Fig. 3 (A) Representation of the nanoplatelet thickness-dependent emission peak energy, where n = ∞ represents the 3D phase LHPs. Reprinted with permission from ref. 154. Copyright 2017, American Chemical Society. (B) Formation energies of (PEA)2(MA)n−1PbnI3n+1 perovskites with different n values: the higher formation energy implies better stability. Reprinted with permission from ref. 155. Copyright 2016, American Chemical Society. | ||
The Ruddlesden–Popper phase perovskites have better environmental stability than their 3D analogues, which is attributed to the relatively strong van der Waals interactions among long chain organic molecules resulting in an increased formation energy (Fig. 3B). Density functional theory (DFT) based calculations revealed that the energy of desorbing phenylethyl-ammonium iodide (PEAI) from the perovskite is 0.36 eV higher than that for methylammonium iodide (MAI). Hence, the PEA2PbI4 films show 1000 times slower decomposition than MAPbI3 films in ambient conditions.155
The long chain amines also result in poor electronic properties compared with 3D analogues.156 And the poor electronic properties of 2D perovskites impede their applications in solar cells and EL devices. Indeed, (quasi) 2D perovskites based solar cells suffer from a low PCE.157,158 However, the 2D–3D perovskite compounds fabricated by integrating an appropriate amount of long chain amines into 3D perovskite frameworks present high charge injection efficiency as well as high stability.159–165 Recently, a significant amount of effort has been devoted to 2D perovskites owing to their attractive characteristics, which are beneficial to develop high performance optoelectronic devices.166–174
2.2. Interface-induced instability
LHPs are ionic compounds, and their interactions with surface ligands also present ionic characteristics. Roo97et al. firstly elaborated the dynamical surface of CsPbBr3 PQDs by 1H solution nuclear magnetic resonance (NMR) spectroscopy. Briefly, surface ligands, oleic acid (OA) and oleylamine (OLA), are not tightly bound to the surface of CsPbBr3 PQDs. The dynamic surface is stabilized by oleylammonium bromide when OA as the only ligand for CsPbBr3 PQDs. While protonated OLA draws OA into the ligand shell, the dynamic surface is stabilized by oleylammonium bromide, oleylammonium oleate and OLA (Fig. 4A). The diffusion coefficient D was measured to further investigate the dynamic surface via Diffusion Ordered NMR Spectroscopy (DOSY). The parameter D is defined as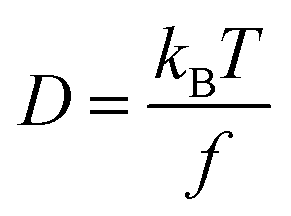 | (4) |
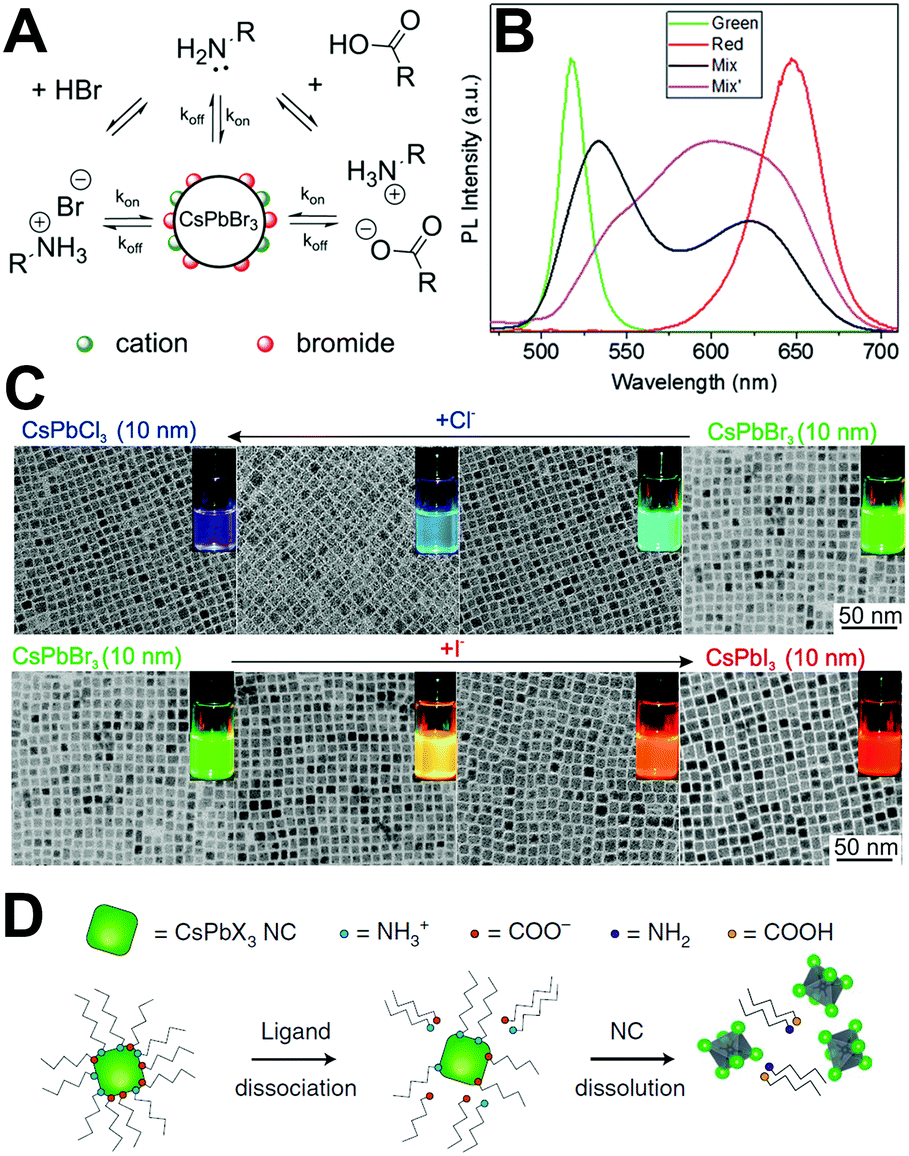 | ||
| Fig. 4 (A) Schematic representation of the dynamic surface of CsPbBr3 PQDs. Reprinted with permission from ref. 97. Copyright 2016, American Chemical Society. (B) PL spectra of green and red emissive PQDs mixed in the silicone resin. Reprinted with permission from ref. 107. Copyright 2016, The Royal Society of Chemistry. (C) Tuning the optical properties by treatment with various quantities of chloride or iodide anions. The figures show the evolution of TEM images and emission colors (under a UV lamp, λ = 365 nm) upon forming mixed-halide CsPb(Br/Cl)3 and CsPb(Br/I)3 to fully exchanged CsPbCl3 and CsPbI3. Reprinted with permission from ref. 106. Copyright 2015, American Chemical Society. (D) Schematic illustration that PQDs often lose their colloidal stability, or even structural integrity, due to the desorption of weakly bound ligands. Reprinted with permission from ref. 73. Copyright 2018, Springer Nature. | ||
2.3. Environmental stability
It is widely known that the LHPs suffer from rapid degradation when they are exposed to ambient air. Even when being carefully encapsulated in a resin sealant, perovskite materials are still sensitive to residual oxygen and moisture.175 An unconventional thorough encapsulation of the device is difficult and costly. Encapsulating them in an inert condition might help to prevent or mitigate degradation. However, as outdoor pc-LED devices, they must function under real-world atmospheric conditions and on a hot LED chip. Therefore, the environmental stability of LHPs has become the prior concern. Gaining insight into the degradation mechanisms including the degradation due to the effect of light, oxygen, and moisture on the material is crucial for the rational design of stable perovskite composites and for enhancing the duration of perovskite pc-LEDs.The light soaking effect is a common phenomenon and has attracted wide attention in PV technologies.179,180 Perovskites as the light-absorbing layer or light-emitting layer also present the light soaking effect.181–183 In the solar cells, the light soaking effect is associated with ion migration.184 In the light-emitting aspects, Scheblykin et al.185 found that the PL intensity as well as lifetime would be increased upon light irradiation in surface-deposited MAPbI3. The light-induced PL brightening is reversible enabled by switching off of the excitation light. It is interesting that the PL brightening was more pronounced in oxygen in contrast to nitrogen or vacuum, and a negligible difference was observed in ambient air or in pure oxygen conditions (Fig. 5A).
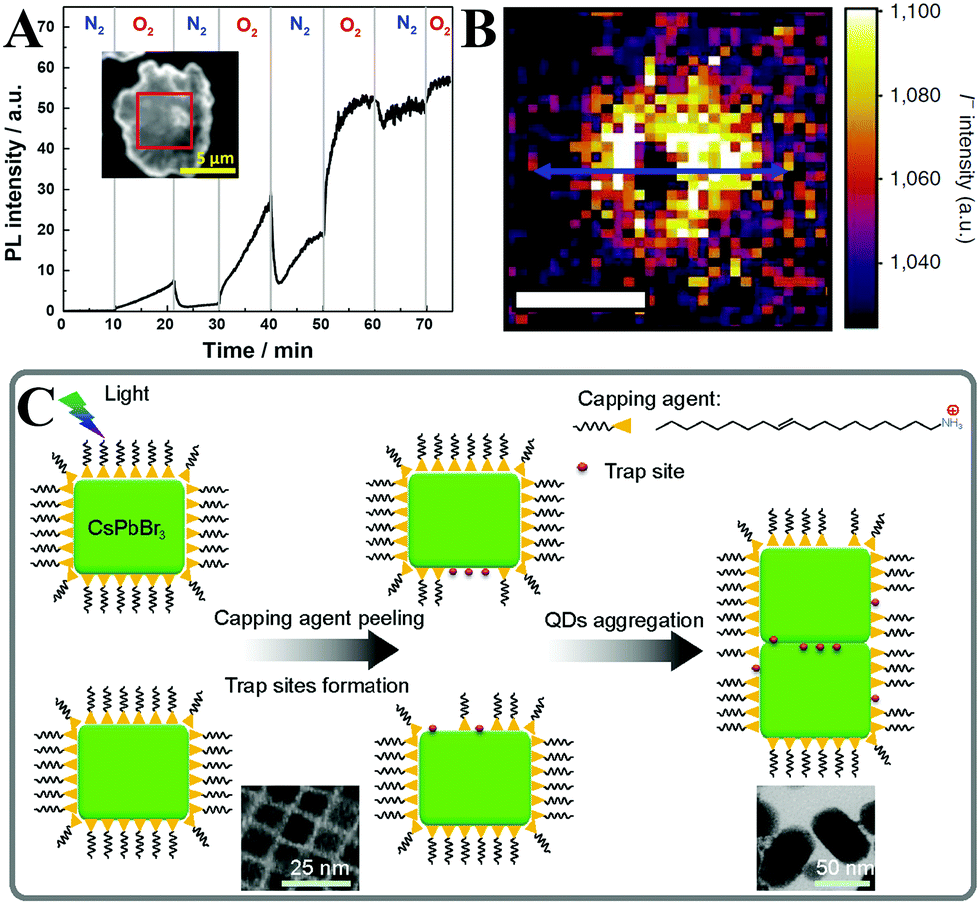 | ||
| Fig. 5 (A) The atmosphere effect on the PL enhancement of MAPbI3. The inset shows the micrograph of the sample and the selected region (red square) taken for the analysis. Reprinted with permission from ref. 185. Copyright 2015, The Royal Society of Chemistry. (B) ToF-SIMS image of the iodide (I−) distribution summed through the film depth, and the scale bar is 10 mm. Reprinted with permission from ref. 186. Copyright 2016, Springer Nature. (C) Schematic picture of the light-induced agglomeration of CsPbBr3 PQDs. The insets show the light-induced morphological evolution of CsPbBr3 PQDs. Reprinted with permission from ref. 189. Copyright 2016, Springer Nature. | ||
To further investigate the enhancement of both PL lifetime and intensity upon light irradiation, confocal fluorescence microscopy and time-of-flight secondary ion-mass spectrometry (ToF-SIMS) were carried out by Stranks et al.186 According to the ToF-SIMS image of the iodide distribution mapping and line scan, the regions irradiated by the laser show depleted levels of iodide (lower than the background iodide levels), whereas the adjacent regions show iodide-rich levels compared with the background iodide levels (Fig. 5B). The mechanisms of light-induced iodide redistribution resulting in PL brightening are proposed as follows: (1) before illumination, the trap density is high owing to the excess of iodide; (2) upon light irradiation, photo-generated electrons will fill the traps, which induces an electric field in MAPbI3 films. The electric field results in iodide migration from the irradiated region to the adjacent iodide vacancies; (3) under continuous illumination, the system attains a stabilized iodide concentration between the illuminated region and the adjacent dark spot. The PL emission reaches a steady level with orders of magnitude reduced trap density; (4) after removing the irradiation sources, some iodide ions are driven back from the dark region to the brightened region by concentration gradients. A similar phenomenon was also observed in MAPbI3−xBrx films.187
As for the perovskite nanomaterials, the PL intensity enhancement of MAPbBr3188 and CsPbBr377,115 PQDs was monitored by using a fluorescence spectrophotometer. Noticeably, such PL enhancement was only observed at the beginning of illumination by a precious in situ test, while the fluorescence intensity would decrease along with the prolonged illumination time even in a dry nitrogen atmosphere.115 Zheng et al. proposed a model (Fig. 5C) for light-induced regrowth based on the morphology evolution.189 After long-term light irradiation, the photo-generated carriers diffused to the surface of CsPbBr3 PQDs and were captured by ionic surface ligands. Some of the surface ligands diffused to the solvent. At the same time, the ligands in free state bound to the surface leading to aggregation of adjacent PQDs. As can be seen from the TEM images (inset of Fig. 5C), the CsPbBr3 PQDs with an incipient cubic shape aggregated into larger nanocrystals. The aggregation process brought in the increased trap states owing to the removal of the capping agent, leading to inferior optical performance.
Not only the inoxidizability of ground state LHPs, but also the PL was enhanced upon exposure to oxygen atmospheres, which has received extensive attention. Such “oxygen-boost” effect has also been found in traditional CdSe/CdS quantum dots191 and CdSe nanoplates.192 A full understanding of oxygen-induced PL brightening is helpful for optimizing the device performance and enhancing the short-term stability. A DFT modeling was conducted to reveal that oxygen molecules deactivated the interstitial iodide deep traps by forming moderately stable oxidized products in MAPbI3. The MAPbI3 forms emissive sub-band gap states upon illumination in an inert atmosphere, whereas the oxygen molecules (even in a small amount) could attenuate the trap density.193 Brovelli and coworkers194 carried out a spectro-electrochemical (SEC) experiment to investigate the selective carrier trapping and the influence of environmental oxygen on exciton recombination dynamics in CsPbBr3 PQDs. It was revealed that the photo-generated holes are captured by trapping states, which leads to severe PL quenching, whereas electron traps are nearly inconsequential to nonradiative decay. Therefore, suppression of hole traps results in brighter PL emission under oxidizing conditions.
In spite of the inoxidizability of ground state LHPs and the temporal “oxygen-boost” effects, the long-term oxidizability is completely different upon continuous light irradiation.195 The LHPs with photo-generated carriers are susceptible to oxygen molecules.196 The probable processes of photo-oxidation are as follows (Fig. 6):197 (1) oxygen molecules diffusing into the lattice and filling the vacancy; (2) photo-generated electrons in the conduction band (CB) and holes in the valance band (VB); (3) superoxide compound (O2−) formation from oxygen and CH3NH3PbI3; (4) decomposition to PbI2, H2O, I2 and CH3NH2. The proposed photo-oxidation reactions are
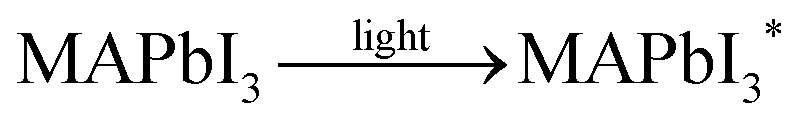 | (5) |
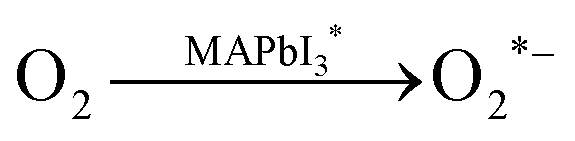 | (6) |
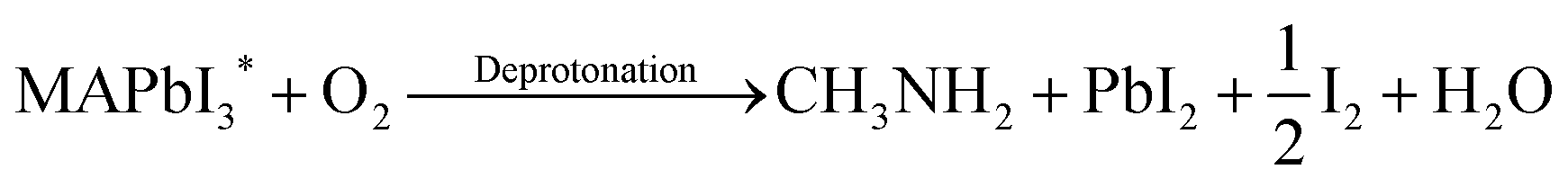 | (7) |
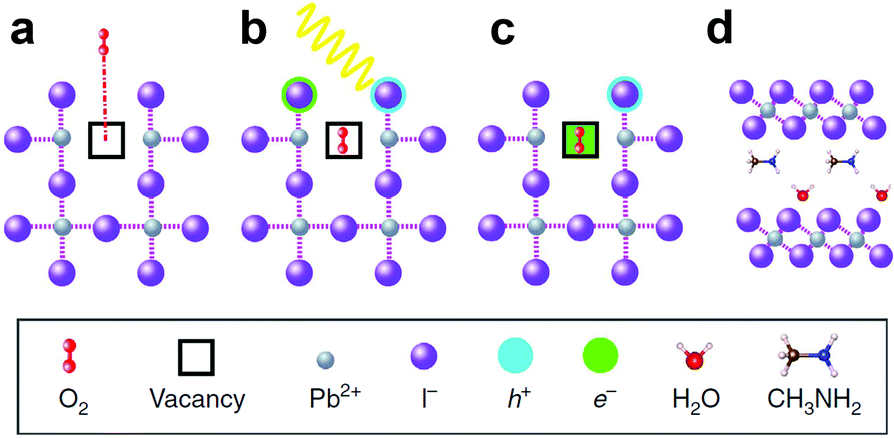 | ||
| Fig. 6 Schematic representation of the oxygen-induced decomposition. (a) Oxygen diffusion and incorporation into the lattice, (b) photoexcitation of CH3NH3PbI3 to create electrons and holes (c) superoxide formation from O2, and (d) reaction and degradation to PbI2, H2O, I2 and CH3NH2. Reprinted with permission from ref. 197. Copyright 2016, Springer Nature. | ||
The presence of decomposition products was verified by X-ray diffraction (XRD) and gas chromatography (GC), confirming the reliability of the photo-oxidation mechanism. As for the all-inorganic perovskites, A-sites composed of inorganic ions such as Cs+ and Rb+ show better chemical stability in comparison to the volatile organic component CH3NH3I. But unlike the specific photo-oxidation mechanism of hybrid MAPbI3, the process of photo-oxidation forming photo-active CsPbX3* and its reaction with oxygen molecules to form the final products is still ambiguous, except that some PbO was detected by the X-ray photoelectron spectroscopy (XPS) technique after photo-oxidation.115
Li et al.115 have systematically investigated CsPbBr3 PQDs’ photo-oxidation processes according to their morphology, structure, and PL evolutions. Their outstanding optical property can be retained in the first hour of illumination when they are encapsulated in a dry nitrogen atmosphere, whereas an apparent decay of PL intensity was observed upon continuous 450 nm pump illumination. The oxygen-induced fluorescence quenching can be divided into two categories. On the one hand, oxygen accelerates PQDs’ agglomeration and regrowth into large crystals with low PL QYs. On the other hand, oxygen molecules etch surface unstable nuclei, which leads to high density of surface defects.
Despite the disadvantage of photo-oxidation, this process is not as severe as we imagine. When they are incorporated in the full solar cell devices, most of the free electrons will be transferred from the CB to the electron acceptors and the free electron density is much lower than in the neat LHPs.198 When they are incorporated in the pc-LEDs, most excitons combined rapidly due to the direct bandgap in LHPs, the short PL lifetimes, and the high exciton binding energy, which means that the electron rich surface defects are less abundant.199 Experimentally, the device performance did decrease upon continuous light irradiation.115,189 But it is clear that photo-oxidation is not the predominant issue, the light-induced agglomeration of PQDs may be more serious in decreasing the performance of pc-LEDs. Therefore, it may be more effective to disperse PQDs preventing their agglomeration than constructing a prohibitively expensive oxygen barrier for improving the device lifetime of pc-LED devices.
Before the detailed decomposition mechanism is given, the material synthesis and device fabrication benefited from the water-assisted process should be discussed. The water-assisted resolvation and recrystallization of LHPs have been demonstrated to be a useful strategy for solar cell fabrication.202–204 Yang et al.205 systematically investigated the crystallinity, morphology, and carrier lifetime of MAPbBr3 films affected by postmoisture treatment. The maximum current efficiency of PeLEDs showed near 20-fold enhancement following the optimized postmoisture treatment in comparison with pristine MAPbBr3 films. Yin and coworkers206 developed a novel strategy for preparing highly luminescent CsPbX3 PQDs from non-emissive Cs4PbX6 nanocrystals through water-extraction of CsX. Additionally, the obtained CsPbX3 PQDs followed by the water-triggered transformation showed enhanced stability against moisture. Sahin et al.207 synthesized CsPbBr3 bundles utilizing the water-driven structural transition of CsPbBr3 nanowires (NWs). And such large bundles present better environmental stability. Rogach and coworkers208 synthesized shape-controlled and stable CsPbBr3 nanocrystals by introducing moderate levels of water into the reaction precursor. The crystallization environment was strongly affected by the additive water, which led to the formation of perovskite nanocrystals with varied morphology. Xu et al.209 firstly synthesized fluorescent MAPbX3 (X = Cl, Br) PQDs in an aqueous solution. The obtained PQDs were stable in ambient conditions and even in polar solvents, which was ascribed to the positively charged surface state of the PQDs and the proper synthetic ionic environment. Kim et al.210 reported a facile aqueous synthesis of rod-shaped luminescent APbX3 (A = Cs, MA) in acidic or basic media. Some Pb(OH)2 by-products formed on the surface of perovskites as confirmed by TEM and XRD measurements. Hence, the PL of non-ligand capped APbX3 nanorods can be retained even when being immersed in water over six months.
Benefited from the tremendous progress made in solar cells based on tri-iodide perovskites, the moisture-induced MAPbI3 decomposition mechanism has been elucidated by the experimental and theoretical study, which is given below:211
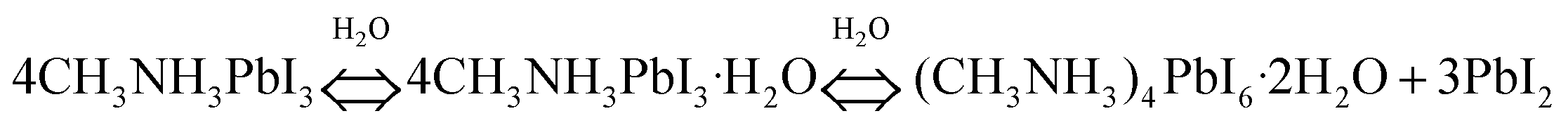 | (8) |
Only a small amount of the monohydrate phase primarily formed in the presence of moisture as confirmed by ellipsometry and XRD studies. The formation of the monohydrate compound has been proven to be reversible when placed in the dry conditions. Next, the dihydrate compound formed along with the hydration process (Fig. 7A). The phase separation caused by the formation of PbI2 results in accelerated hydration. The MA+ cations are not tightly bonded to the I− but interact with water in the dihydrate phase. Thereby, the dihydrate compounds are more likely to decompose into methylammonia (CH3NH2), HI, and PbI2. Walsh et al.212 proposed the decomposition pathway in the presence of water, as shown in Fig. 7B. Both CH3NH2 and HI byproducts are volatile and soluble in water. This pathway results in the formation of a PbI2 solid, which is in good agreement with the experimental observations.
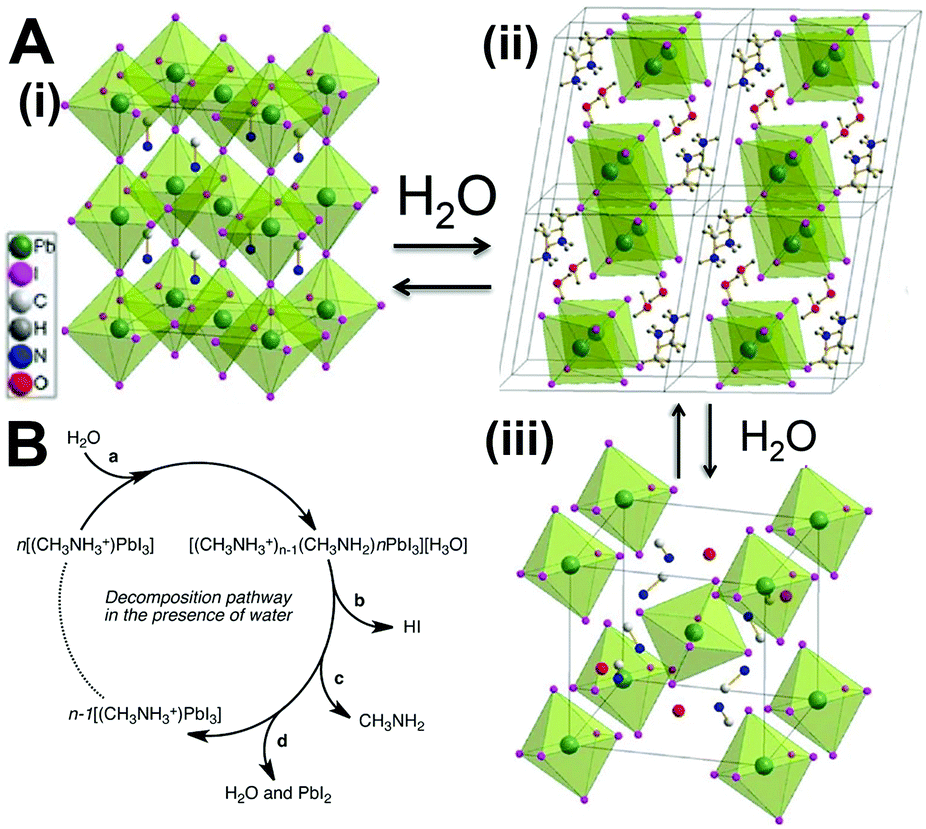 | ||
| Fig. 7 (A) The two hydrated structures of the MAPbI3 perovskite and the structural evolutions. Reprinted with permission from ref. 211. Copyright 2015, Wiley-VCH. (B) Possible decomposition pathway of MAPbI3 in the presence of water. Reprinted with permission from ref. 212. Copyright 2014, American Chemical Society. | ||
As for the all-inorganic perovskite, the pathways for moisture-induced degradation are not well-defined. But it is clear that the moisture-induced resolvation and recrystallization produce large crystal grains.115 Such large crystals might be beneficial to PV technologies but harmful to pc-LEDs because of loss of PL QYs.
The moisture-induced decomposition of perovskite materials results in a severe decline in device performance. And this process would be accelerated upon light irradiation by surface reconstruction.213,214 Functionalization of water-resisting layers can improve the humidity tolerance of perovskite films as well as passivate the surface defects, which is widely used in the PV and EL technologies.100,215 The situation is almost completely different for PQDs applied in pc-LEDs. The PQDs synthesized through the typical hot-injection method or the ligand-assisted reprecipitation (LARP) technique were capped by long-chain OA and OLA (for all-inorganic perovskites) or octylamine (for organic–inorganic hydride perovskites) molecules. In comparison with the bare perovskite films requiring extra passivation by hydrophobic ammonium cations, named the water-resisting layers, the OA molecules with longer carbon chains are more hydrophobic.
2.4. Thermal stability
Thermal degradation at high temperature is the most vital issue with respect to thermal stability. Thermogravimetric analysis (TGA) studies on hybrid LHPs show that the mass loss has two steps. The sublimation of HX and CH3NH2 is around 220 °C for MAPbBr3105 and 250 °C for MAPbI3.216 The second step begins at >500 °C owing to the sublimation of the PbX6 octahedra. This is consistent with the all-inorganic CsPbX3 that decomposes at >500 °C owing to the collapse of crystal structures.105 All the results suggest the high thermal stability of both organic–inorganic hybrid and all-inorganic perovskites. However, the oxygen- and moisture-induced decomposition would be accelerated and amplified at high temperature, and the combination of moisture and heat would lead to a more rapid decomposition.214 Moreover, for the reversible hydration/dehydration reaction, the monohydrate compounds easily decompose according to the following reaction:94,214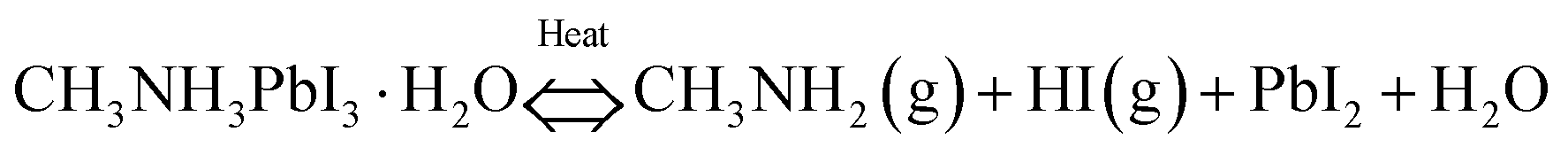 | (9) |
The formation of volatile HI and CH3NH2 leads to phase separation, which drastically accelerates the hydration.
Besides the decomposition under thermal stress severely affecting the PL property of LHPs, the thermal-induced PL quenching is universal and inevitable for all types of luminescent materials. Even the PL of commercial phosphor K2SiF6:Mn4+ suffers from thermal quenching of ∼25% upon heating from 293 to 453 K.217 Additionally, the core-only traditional CdSe quantum dots (QDs) show about 80% PL loss upon heating from 293 to 400 K.218 The optical property and stability can be enhanced by inorganic shell coating. Actually, the PL intensity of commercial CdSe/CdS/ZnS QDs still shows a decrease of about 30% of the initial value when heating from 293 to 373 K.218 The bulk MAPbBr3 perovskite shows severe PL quenching (nearly 100%) from 300 to 400 K, while the MAPbBr3 PQDs preserve an intensity of 30% in the same temperature range according to temperature dependent PL measurements.56 The neat CsPbBr3 PQDs prepared by the LARP technique show about 85% PL loss from 80 to 273 K,55 and another 80% (obtained from a similar protocol) from RT to 373 K.219 It should be noted that the thermal-induced PL quenching might dominatingly arise from the agglomeration of PQDs. Schaller et al.220 have systematically studied the temperature dependent fluorescence of CsPbBr3 PQDs by embedding the CsPbBr3 PQDs in a polymer matrix to minimize the aggregation-induced PL quenching. After diminishing the interactions of CsPbBr3 PQDs, the CsPbBr3–polymer composites show fluorescence loss of less than 10% when heating from 80 to 273 K, whereas the PL of neat CsPbBr3 PQDs reduced over 85% in the same condition. They proposed that the PL quenching of CsPbBr3–polymer composites corresponds to thermally activated halogen vacancies, supported by time-resolved and transient absorption (TA) measurements as well as DFT calculations. According to thermal cycling measurements (Fig. 8), the PL quenching of CsPbBr3 PQDs is largely reversible below 450 K. On the other hand, when CsPbBr3 PQDs are heated at higher temperature, they exhibit irreversible quenching. The irreversible loss is likely associated with the desorption and decomposition of organic ligands above 450 K. A similar PL resilience was also observed in the MAPbBr3 PQDs embedded in polymer hosts.75
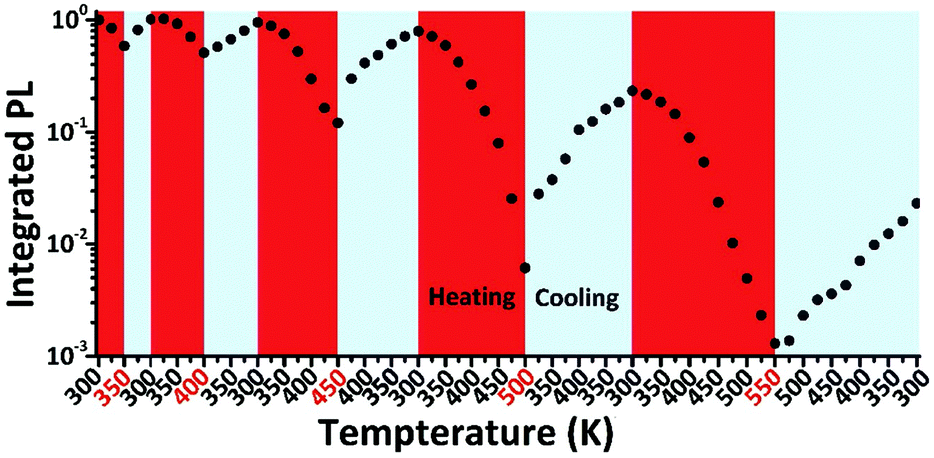 | ||
| Fig. 8 Thermal cycling measurements of CsPbBr3 PL. Reprinted with permission from ref. 220. Copyright 2017, Wiley-VCH. | ||
The reversible PL allows for device encapsulation at a relatively high temperature. It is easily seen that the PL persistence of PQDs at high temperatures is close to that of the core-only CdSe QDs, and the device performance may be affected at high operating temperatures. Some thermal barriers or appropriate heat sinks are highly expected for perovskite pc-LEDs at this stage. Maybe an inorganic shell protecting like a CdSe/CdS/ZnS core/shell structure holds great promise in further improving the thermal stability of PQDs.
As discussed above, light, oxygen, moisture, and heat, and their synergistic effects affect the stability of PQDs. Both photo-oxidation and moisture-induced decomposition take up a little proportion of the total perovskite materials in normal conditions. The chemical stability of PQDs is much higher than we imagine. But it does not mean that the encapsulations are no more necessary for PQDs. On the contrary, PQDs grow into large crystal grains with low PL QYs under moisture conditions and light irradiation. And such crystal-regrowth can be accelerated in the presence of oxygen. The formation of large perovskite crystals is the primary culprit for PL quenching rather than the material decomposition in normal conditions. Thermal-induced PL quenching also brings great difficulties in applying them for pc-LEDs. Although PQDs show high PL resilience, most PL quenched on the hot LED chip owing to their intrinsic poor PL thermal resistance. Additionally, it is well known that a chemical reaction can be accelerated at a higher temperature. The decomposition and crystal-regrowth can be accelerated at higher temperatures, which results in amplified PL quenching. Hence, designing thermally stable perovskite materials and better encapsulation to eliminate their agglomeration and regrowth are highly desired for pc-LED application.
3. Strategies toward improving the stability of PQDs
A great effort has been devoted to stabilize PQDs. Within several years, numerous strategies have been developed. Herein, we review the recent studies on enhancing the stability of PQDs (Fig. 9). It is worth noting that we mainly summarize the strategies for improving the stability of PQDs. Some of the methods used to stabilize perovskite films or perovskite crystal grains might be effective to improve the stability of PQDs, which are also included in this section.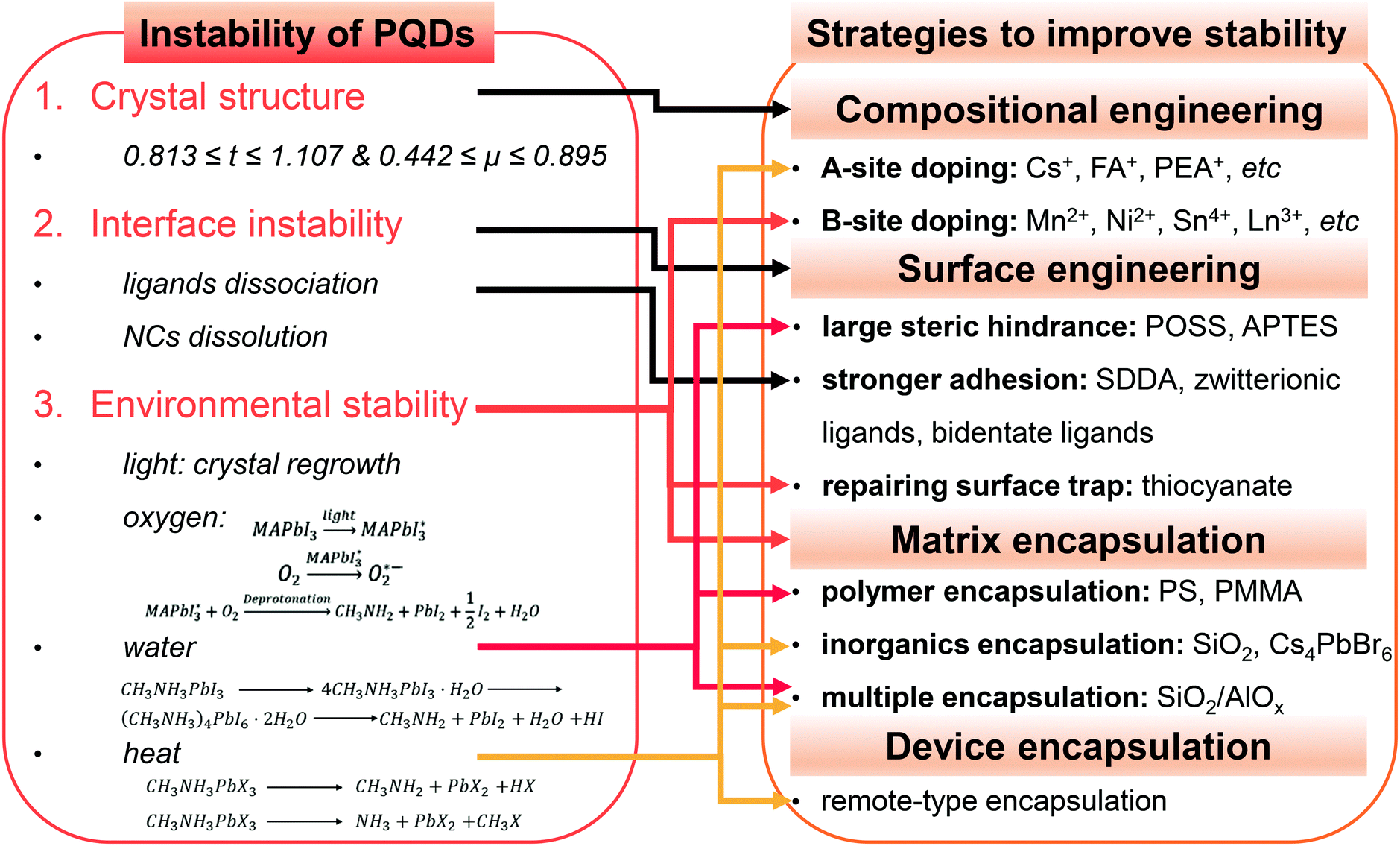 | ||
| Fig. 9 Schematic diagram summarizing the representative instability of PQDs and the corresponding solutions. | ||
3.1. Compositional engineering
All inorganic CsPbBr3 shows much higher thermal stability which could effectively suppress decomposition caused by the heat of LED chips, and its environmental stability is also superior to the hybrid perovskites.105 Due to the high chemical stability and high PL brightness (PL QYs up to 90% in solution) of CsPbBr3 PQDs, employing CsPbBr3 as a color converter in pc-LEDs has attracted great interest.
The red-emitting lead iodide perovskites are thermodynamically unstable compared with their bromide analogues. FAPbI34 and CsPbI371 with 3D perovskite structure crystallize into 1D hexagonal and 1D orthorhombic structures, respectively, because the FA+ ions are too large and the Cs+ ions are too small for 3D polymorphs. MAPbI3 PQDs with suitable ionic radii suffer from a rapid PL loss due to the poor chemical stability.104 This challenge which is faced when preparing stable red and near-infrared (NIR) nanoemitters, termed the “perovskite red wall”, could be addressed by a rational doping of FA+ into the CsPbI3 lattice.225 The as-designed FA0.1Cs0.9PbI3 PQDs show stable and bright red-emission with PL QYs over 70% because of the compensating inadaptable anion (Fig. 10A). Similarly, PeLEDs fabricated from the mixed cation FA0.8Cs0.2PbBr3 PQDs present significant enhancement of device performance that is most likely associated with the entropic stabilization.226 Etgar et al.227 synthesized RbxCs1−xPbX3 PQDs with tunable fluorescence and high PLQEs up to 60%. RbxCs1−xPbX3 PQDs retain the structural integrity and show a slight difference upon doping Rb+ cations into the lattice. A further stability test revealed that the mixed Rb/Cs bromide PQDs present a slight red-shift in absorption over months, but the chloride analogues show limited stability. Incorporation of K+ into CsPbBr3 PQDs by the postmodification method improves PL QYs from 65% to 83% (Fig. 10B).228 Similarly, the incorporation of K+ into CsPbCl3 PQDs brings in the improvement of PL QYs from 3.2% to 10.3%.229 Moreover, the introduction of K+ greatly enhanced the photostability and environmental stability. Especially, the deposited films composed of KxCs1−xPbBr3 PQDs maintain the initial brightness even after 153 h irradiation, whereas the PL intensity of pristine PQD films decreases to half of the initial value after 45 h. The incorporation of K+ leads to a K-rich phase at the interface, which is also demonstrated to be useful in the film applications. The “photo-brightening” phenomenon caused by halide migration has been discussed before. Surprisingly, the PL intensity of K-incorporated perovskite films is found to be stable to light exposure (Fig. 10C). The K-rich phase effectively inhibited the halide migration and suppressed the non-radiative decay.230 Multi A sites such as RbCsMAFA co-doping results in a high entropy system, which has been considered as an effective way to improve the device lifetime in the film applications.134,231 Nevertheless, the high entropy PQDs are rarely prepared and investigated, which might be attributed to self-purification effects of quantum dots.232
 | ||
| Fig. 10 (A) Optical absorption and PL spectra of FAPbI3 and FA0.1Cs0.9PbI3 PQDs before and after 6 months of storage. The insets are photographs of the FAPbI3 and FA0.1Cs0.9PbI3 PQDs’ colloidal solutions under daylight (upper image) and under UV lamp (λ = 365 nm; lower image) excitation. Reprinted with permission from ref. 225. Copyright 2017, American Chemical Society. (B) KxCs1−xPbBr3: (i) schematic of the addition of the K-oleate precursor into the as-prepared CsPbBr3 toluene solutions. (ii) PL emission spectra of the CsPbBr3 and K-modified CsPbBr3 PQDs. (iii) Optical images of the CsPbBr3 and K-modified CsPbBr3 PQD films as a function of the treatment time in the dark environment (50 °C, relative humidity 60%). Reprinted with permission from ref. 228. Copyright 2018, Wiley-VCH. (C) PLQE time course of perovskites illustrating that the K-doped perovskites are stable to light exposure. Reprinted with permission from ref. 230. Copyright 2018, Springer Nature. | ||
2D perovskites developed by introducing long chain amines such as oleylamine into perovskite structures have been introduced above. Owing to the relatively strong van der Waals interaction force, 2D perovskites show increased stability than their 3D polymorphs.155 Because the rich organic cations can be adapted into the perovskite lattice, abundant 2D perovskites have been synthesized so far. Among them, some 2D hybrid perovskites show broadband white light emission, which is related to the self-trapped excited states created by lattice deformation.233–242 These 2D LHPs can serve as a single-component, broadband white light emitter for pc-LEDs; however, the low PLQYs limit their applications in lighting devices. Zhang et al.243 synthesized highly luminescent 2D (PEA)2PbX4 (PEA = phenylethylamine, C8H9NH3) nanosheets (NSs). The obtained (PEA)2PbBr4 NSs shows the highest PLQY of 46.5% among the (PEA)2PbX4 family. The PL intensity of (PEA)2PbI4 NS solutions preserved a value of 53% under illumination for 60 minutes, while 22% of the original intensity was sustained for MAPbI3 QD solution. Formation of 2D–3D perovskite films by rationally introducing long chain amines leads to a decent device performance and long device lifetime for PV and EL applications, which has attracted intense attention.160–163 However, such research on multidimensional PQD structures has little been done so far. Mathews et al.244 prepared highly luminescent PQDs with an assumed 2D–3D core–shell structure. The core–shell PQDs show intriguing optical characteristics and enhanced environmental stability, which will be discussed in Section 3.3.2.
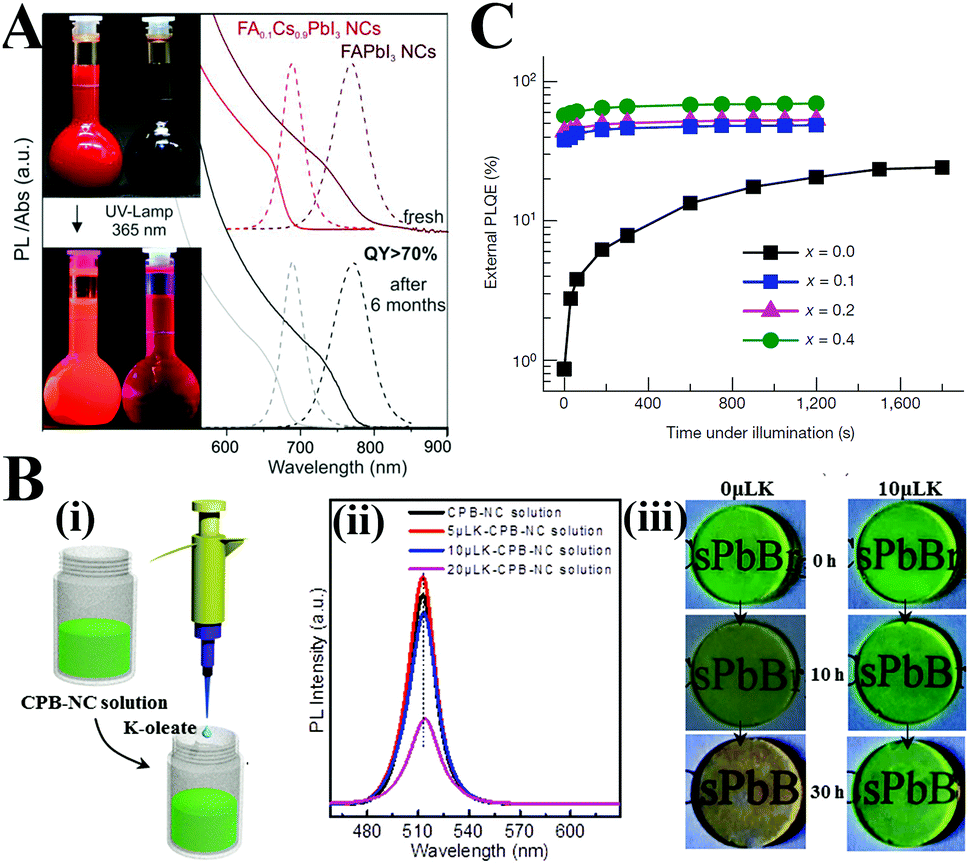
Among the numerous reports on this issue, doping Mn2+ into PQDs has received the most intention.102,249–262 Owing to the internal transition of the Mn2+ impurity from 4T1 to 6A1, the as-prepared CsPbCl3:Mn PQDs showed a second emission at the channel of ∼2.15 eV (Fig. 11A).249 The optimized Mn2+ doped CsPbCl3 PQDs showed an increased PL QY up to 27%, whereas the undoped PQDs had relatively low fluorescence brightness (PL QY < 5%). Zhang et al.251 prepared high Mn doped CsPbxMn1−xCl3 PQDs. The obtained PQDs retained the original tetragonal structure of the CsPbCl3 host even when 46% Pb2+ were replaced by Mn2+. The PL QYs of CsPbxMn1−xCl3 PQDs increased from 5 to 54% along with the increasing Mn substitution ratio. The crystal structures and the enhanced PL properties can be retained after being replaced under ambient air for over 3 months. Moreover, the bright orange-red light emitted by pc-LEDs fabricated from CsPb0.73Mn0.27Cl3 PQDs did not change even after continuous working for 200 h, which indicates the prolonged device lifetime of Mn-doped lead chloride PQDs. Chen and coworkers102 reported a Mn2+-doped strategy to improve the environmental stability and PL properties of PQDs. CsPbBr3:Mn PQDs preserved about 60% of the initial PL brightness upon exposure to ambient air for 120 days (Fig. 11B). In contrast, the PL of undoped CsPbBr3 QDs quenched within 30 days in ambient air. First-principle calculations corroborated that the enhanced stability and PL performance are ascribed to the increased formation energies by Mn2+ doping. Similarly, the α-CsPbI3 PQDs with metastable phase can be stabilized by incorporating Mn ions into the lattice.263 The highly luminescent CsPbxMn1−xI3 quantum dot films retain their initial α-phase and strong PL emission after one-month storage, whereas the luminescent film drop-cast from α-CsPbI3 PQDs completely degrade to nonemissive δ-CsPbI3 within 5 days (Fig. 11C). The calculations based on the DFT revealed that the enhanced robustness of α-CsPbI3 PQDs was associated with the increased Goldsmith tolerance factor and the cohesive energy through Mn2+ doping.
 | ||
| Fig. 11 (A) PL spectra of Mn-doped and undoped CsPbCl3 and CsPb(Cl/Br)3 PQDs. The insets are photographs of the sample under UV excitation. Reprinted with permission from ref. 249. Copyright 2016, American Chemical Society. (B) PL emission photographs of CsPbBr3:Mn QDs coated on the surface of a glass slide with different Mn2+ contents from 0 to 6.2 mol% taken under UV irradiation at the indicated time periods. (C) XRD patterns of CsPbI3 and CsPbxMn1−xI3 PQD films before and after 5/30 days of storage. (B and C) Reprinted with permission from ref. 102 and 263, respectively. Copyright 2017, American Chemical Society. | ||
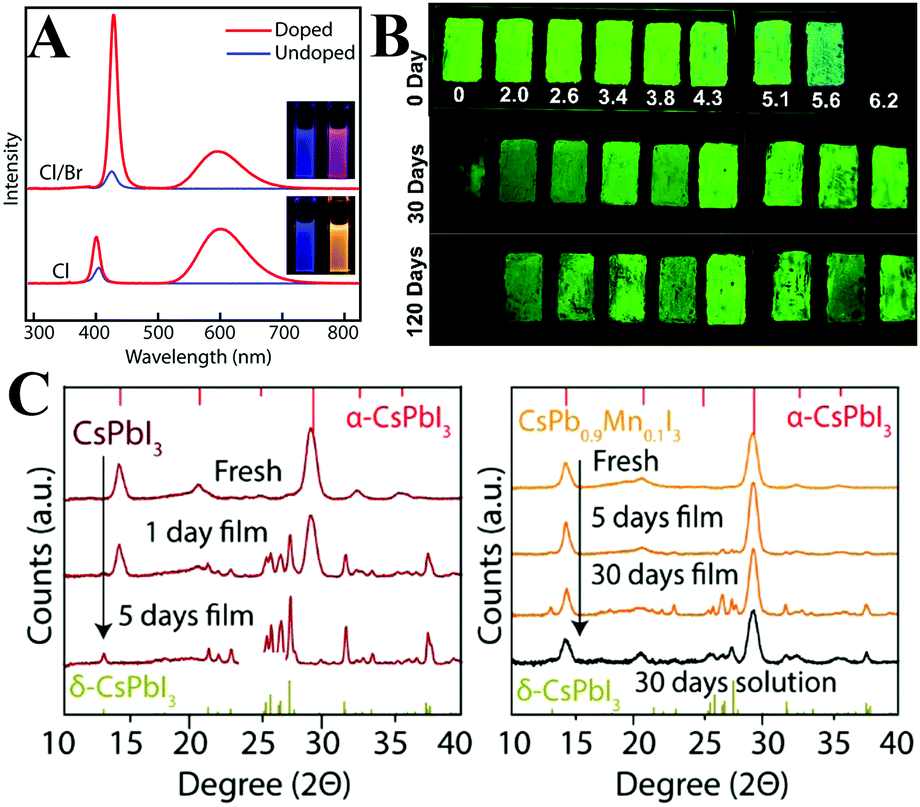
Böhm et al.264 synthesized lead-free CsSnX3 PQDs with tunable emission spanning from 442 nm to the NIR regions. Nevertheless, the bivalent Sn2+ ions are sensitive to oxygen and will be easily oxidized to Sn4+, which results in decreasing PL QYs to nondetectable values. Deng et al.265 fabricated CsSnBr3 hollow nanocages with better environmental stability by using stannous 2-ethylhexanoate as the tin source at a higher temperature (Fig. 12A). The absorbance spectra of CsSnBr3 nanocages show a few changes after 3 hour exposure to ambient air, whereas the CsSnBr3 nanocubes undergo a visible decomposition within 2 h. Surface modification with perfluorooctanoic acid (PFOA) can further improve the stability of CsSnBr3 nanocages. No obvious changes in the absorption spectra of PFOA treated CsSnBr3 nanocage films were observed after 16 h of storage in ambient air. The enhanced robustness might be attributed to the stronger interaction between Sn2+ and the electron-withdrawing F− in contrast to Br−. SnBr2266 and SnF2267 surface treatments were also demonstrated to be efficient strategies to improve the oxygen-stability of CsSnX3. Although these methods aim at protecting CsSnX3, the environmental stability of tin-based perovskites is still lower than that of lead-based perovskites. Deng and coworkers268 synthesized Cs2SnI6 NCs with variable shapes such as QDs, nanorods, nanowires, nanobelts and nanoplates by using SnI4 as precursor. The unoxidizable tetravalent tin ions impart ultrahigh stability to Cs2SnI6 NCs. It is worthy to note that the measured PL QYs of Sn-based perovskites are much lower (0.14% for CsSnBr3 nanocubes,264 2.1% for CsSnBr3 nanocages,265 0.48% for Cs2SnI6 QDs268) than those of Pb-based analogues. Overall, the optical properties of tin halide perovskites are far from the requirements of pc-LEDs.
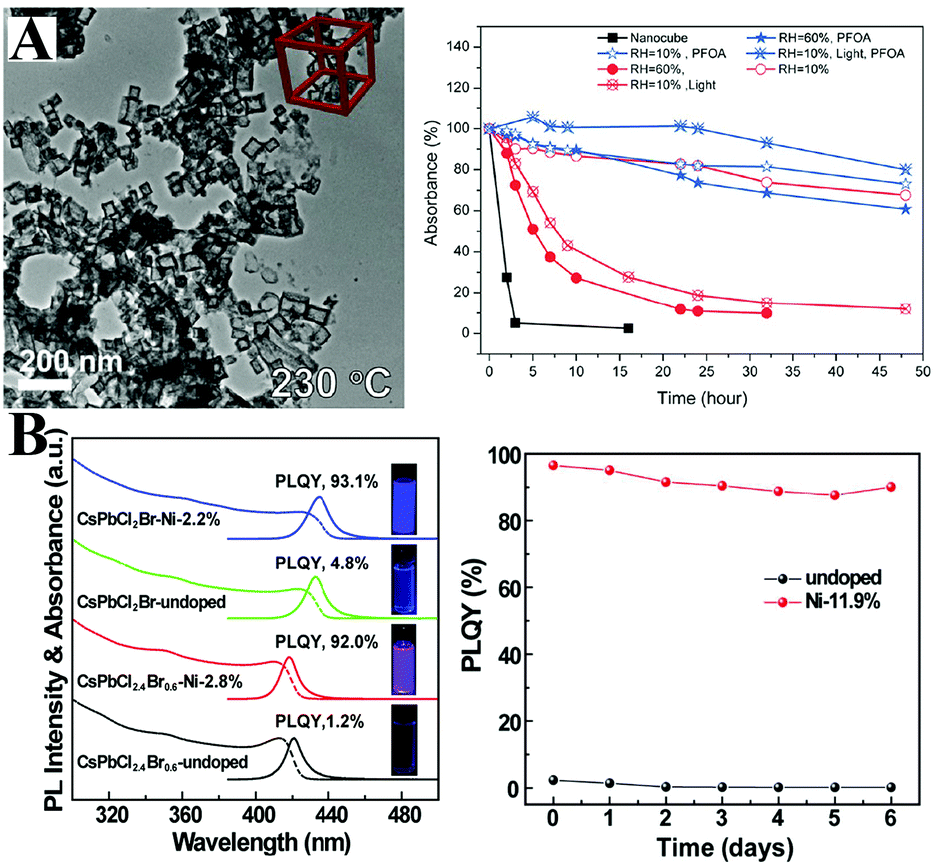 | ||
| Fig. 12 (A) TEM images of CsSnBr3 nanocages (left) and relative absorbance intensity at 620 nm as a function of time under various conditions (right). Reprinted with permission from ref. 265. Copyright 2017, American Chemical Society. (B) Absorption and PL spectra of undoped and doped CsPb(Cl/Br)3 PQDs. The inset shows the photographs of an QD solution under UV (365 nm) illumination. PL stability of pristine PQDs and Ni-doped PQDs (right). Reprinted with permission from ref. 274. Copyright 2017, American Chemical Society. | ||
Although a total substitution of Pb by Sn seems to be not reliable at the current stage, partial replacement may retain the outstanding optical properties of LHPs and reduce the toxicity of lead content.269 As expected, doping Sn into LHP films leads to a red-shift in absorption, which is attributed to the smaller bandgap of tin halide perovskites.270 However, doping Sn into CsPbBr3 NCs (synthesized at a low temperature) leads to an abnormal blueshift that is likely associated with the tilting of the PbBr6 octahedra.271,272 The PL QYs of the Sn(II)-doped NCs decreased due to the gradual oxidation of Sn2+. Liu and coworkers273 prepared CsPb1−xSnxBr3 PQDs at a higher temperature. The Sn2+ was oxidized to Sn4+ during synthesis and the tetravalent Sn ions doped into the CsPbBr3 hosts as confirmed by X-ray absorption near-edge spectroscopy (XANES). Hence, the CsPb1−xSnxBr3 PQDs with partial Sn4+ substitution present high environmental stability, unlike the Sn2+-substitution. Additionally, Sn(IV)-doping leads to increased PL QYs from 45% to 85%.
Despite the ultrahigh PL QYs of PQDs in the green and red region, the PLQYs of violet-emitting CsPbClxBr1−x PQDs are much lower. Recently, Sun et al.274 found that the poor PL properties of violet-emitting PQDs derive from halide vacancy-induced short-range disorder of the lattice. They prepared Ni2+-doped CsPbClxBr1−x:Ni PQDs with near-unity PL QYs in the violet spectral range (Fig. 12B). Ni-Doping led to improved local structural order of the lattice and deactivated structural defects. DFT calculations revealed that the defect formation energy increased by Ni-doping. Hence, highly violet emissive CsPbClxBr1−x:Ni PQDs show promise in violet-emitting perovskite-based devices. Meng et al.247 prepared stable blue emitters by doping Al3+ into CsPbBr3 PQDs. Due to the similar bond energy between Al–Br and Pb–Br, the Al3+ impurity can easily incorporate into the CsPbBr3 lattice.
Tang and coworkers275 firstly synthesized MA3Bi2Br9 PQDs with a PL QY of 12%. Through anion exchange reaction, the emission wavelength can be tuned from 360 to 540 nm. The PL QY of MA3Bi2Br9 PQDs can be further improved up to 54.1% by Cl-passivation.276 Beyond the improved PL, the Cl-treated MA3Bi2Br9 PQDs also show enhanced photostability as shown in Fig. 13A. They also synthesized blue emissive all-inorganic Cs3Bi2Br9 PQDs with a PL QY of 19.4% by utilizing ethanol as the antisolvent.277 The PL peaks can be tuned from 380 to 526 nm by anion exchange.278 Unlike the unstable CsPbI3, Cs3Bi2I9 PQDs manifest outstanding robustness in ambient air over months.279 Han et al.280 also fabricated all-inorganic Cs3Bi2X9 PQDs with the PL peak ranging from 400 to 560 nm. The obtained Cs3Bi2X9 PQDs retained the initial phase even after being stored in ambient air over 30 days, which indicates their high stability. Begum et al.281 reported CsPbBr3 NCs with Bi3+ substitution via an in situ doping approach. The PL QYs of CsPb1−xBixBr3 (0 ≤ x ≤ 2.1%) NCs decreased from 78% to 8% along with the increasing replacement by Bi3+ (Fig. 13B). Further research shows that substitution of Bi3+ remarkably increased the sub-band gap density of states, which also results in increased nonradiative recombination centers.282 Song and coworkers283 introduced Bi3+ and Mn2+ into the CsPbCl3 NCs to expand the emission spectra. The Bi3+/Mn2+ codoped NCs achieved single-component white emission by tuning the concentration of Bi3+ and Mn2+ under UV light excitation.
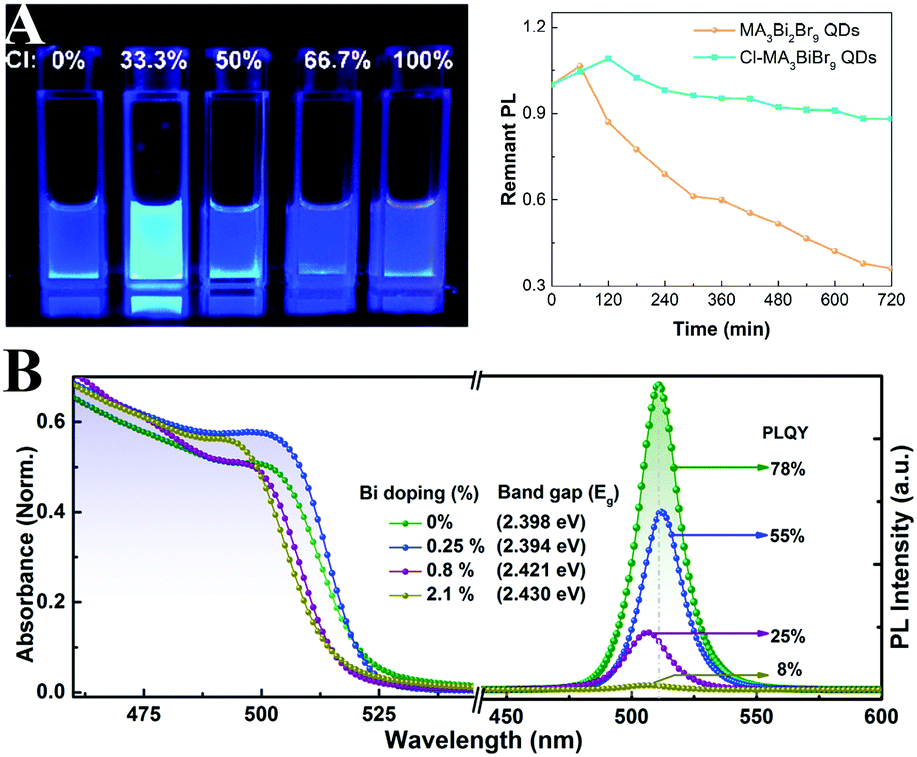 | ||
| Fig. 13 (A) Photographs of MA3Bi2Br9 QD solutions under 325 nm UV lamp excitation (left), and PL stability of pristine QDs and Cl-passivated QDs (right). Reprinted with permission. Copyright 2018 from ref. 276, American Chemical Society. (B) Absorption and PL spectra of CsPbBr3 with varied Bi-doping ratios. Reprinted with permission. Copyright 2016 from ref. 281, American Chemical Society. | ||
Antimony and bismuth belong to one family, and they exhibit similar properties in some respects. All-inorganic Cs3Sb2Br9 PQDs with high bright blue emission can be synthesized by the similar LARP method.284 The measured PL QY is 46%, which is superior to that of blue-emitting CsPbCl3 PQDs. Cs3Sb2Br9 PQDs retained 70% of original PL intensity after stored in air for 35 days (Fig. 14A). In addition, by halide exchange reactions, the PL emission of Cs3Sb2X9 PQDs can be tuned from 370 to 560 nm. The Sb3+-doped CsPb1−xSb2x/3I3 perovskite films were prepared by Chen and coworkers.285 The phase stability and film morphology are enhanced after Sb3+-doping. However, to the best of our knowledge, Sb3+-doped LHP nanomaterials have not been investigated yet. Sb-Doping may show promise in designing stable perovskite nanomaterials.
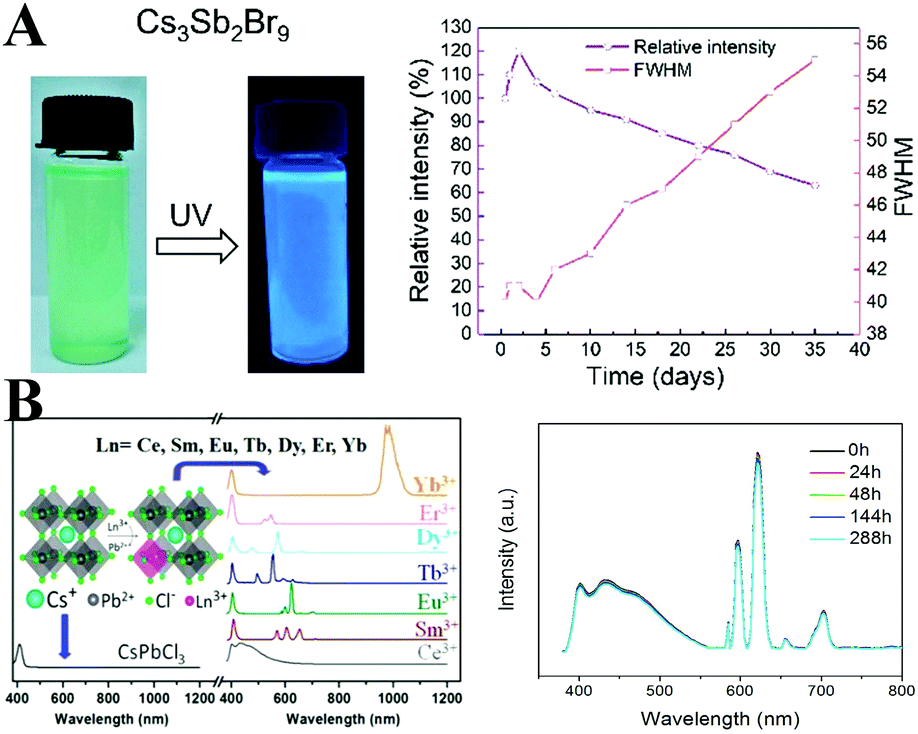 | ||
| Fig. 14 (A) Photographs of Cs3Sb2Br9 PQDs under UV lamp excitation (left), and environmental stability of Cs3Sb2Br9 PQDs (right). Reprinted with permission from ref. 284. Copyright 2018, American Chemical Society. (B) PL spectra of CsPbCl3 doped with different lanthanide ions (left) and the emission spectra of the white LED device fabricated from Ce3+ and Eu3+ ion codoped CsPbCl3 PQDs acquired at different working times at a bias of 3.0 V (right). Reprinted with permission from ref. 292. Copyright 2017, American Chemical Society. | ||
Recently, A2B(I)B′(III)X6-type double perovskites have attracted great attention, especially Cs2AgBiX6.286–289 Although the quantum efficiencies for Cs2AgBiX6 PQDs are rather low at the current stage, the environmental stability is much better than other lead-less halide perovskites. More research should be devoted to this issue.
Rare earth (RE) ion activated luminescent materials are widely used in lighting and display devices. Owing to the abundant energy level of the RE ion, the obtained RE-doped PQDs achieved multiple color emissions by energy transfer from the CsPbBr3 host to the RE impurities.290 McLeod et al.291 prepared Eu-doped MAPb1−xEuxBr3 PQDs by the LARP method. The Eu is demonstrated to be a suitable element for substituting Pb without the change of crystal structure. At high Eu-substitution, blue luminescence originating from Eu2+ was observed in MAPb1−xEuxBr3 PQDs, revealing their potential in optoelectronics. Song and coworkers292 incorporated diversified RE ions including Ce3+, Sm3+, Eu3+, Tb3+, Dy3+, Er3+, and Yb3+ into the wider band gap CsPbCl3 PQDs at a higher reaction synthetic temperature. The as-prepared CsPbCl3:RE PQDs exhibit unique RE emissions ranging from visible to NIR regions together with high PL QYs and high stability (Fig. 14B). Interestingly, the Yb3+-doped CsPbCl3 PQDs with a NIR emission centered at ∼1000 nm present a high PL QY of 170%, which is attributed to the quantum cutting of excitonic transition of CsPbCl3 hosts.293 The Yb3+/Er3+ co-doped CsPbCl3 PQDs emit 1533 nm NIR light. The co-doped PQDs present better stability than the undoped CsPbCl3 PQDs.294 Such materials can serve as optically active layers for silicon solar cells to improve the performance.295 The Ce3+ cation with a similar ionic radius and higher energy level located in the CB of the perovskite hosts has been regarded as a favorite dopant for CsPbBr3 PQDs. The CsPbBr3:Ce PQDs sustain the structural integrity of the perovskite and exhibit a PL enhancement from 41% to 89% owing to the modulation of PL kinetics by Ce3+-doping.296 The co-doping of Ce3+ and Eu3+ ions into CsPbCl3 hosts produces a cool white emission.292 And white light pc-LEDs fabricated by combining them on a 365 nm LED chip show a high luminous efficiency of 24 lm W−1 and a long device lifetime.
Overall, doping has been widely accepted as an effective strategy for tailoring and enhancing the optical performance and improving the stability of PQDs. The optical properties and the stability assessments of some promising PQDs are summarized in Table 1.
| PQDs | Synthetic method | Emission peak (nm) | FWHM (nm) | PL QYs (%) | Stability | Ref. |
|---|---|---|---|---|---|---|
| CsPbCl3 | LARP | 405 | 12 | 10 | 55 | |
| Ultrasonication | 10 | 8% (4 months, air) | 45 | |||
| Microwave irradiation | 410 | 14 | 7 | 80 | ||
| KxCs1−xPbCl3:Eu | Hot-injection (185 °C) | 408 | 12.1 | 31.2 | 229 | |
| CsPbCl3:Ni | Hot-injection (210 °C) | 407 | 96.5 | >90% (6 d, air) | 274 | |
| Cs2AgBiCl6 | LARP | 395 | 68 | 6.7 | 287 | |
| Cs3Bi2Br9 | LARP | 468 | 40 | 4.5 | 77% (400 min, UV) | 280 |
| MA3Bi2Br9 | LARP | 430 | 62 | 12 | 92% (25 h, UV) | 275 |
| Cl-MA3Bi2Br9 | LARP | 422 | 41 | 54.1 | 89% (12 h, UV) | 276 |
| Cs3Sb2Br9 | LARP | 410 | 41 | 46 | 70% (35 d, air) | 284 |
| 50% (108 h, UV) | ||||||
| CsPbBr1.5Cl1.5 | LARP | 455 | 16 | 37 | 55 | |
| CsPbBr3:Al | Hot-injection (150 °C) | 456 | 16 | 42 | 247 | |
| CsPb(Br/I)3:Al | Hot-injection (150 °C) | 536 | 40 | 247 | ||
| CsPbBr3 | LARP | 513 | 20 | 95 | 90% (30 d, air) | 55 |
| Ultrasonication | 92 | 89% (4 months, air) | 45 | |||
| Microwave irradiation | 517 | 17 | 90 | 80 | ||
| Hot-injection (160 °C) | 509 | 16 | ∼92 ± 2 | 322 | ||
| Hot-injection (180 °C) | 507 | 27 | 90 | 91% (30 d, air) | 55 | |
| MAPbBr3 | LARP | 505 | 21 | 70 | 56 | |
| Spray synthesis | 511 | 22 | ∼100 | 91% (2 months, air) | 81 | |
| FAPbBr3 | LARP | 530 | 22 | 75 | 38% (1 h, 100 °C) | 82 |
| Modified hot-injection (130 °C) | 530 | 22 | 85 | Retain PL after 2–3 cycles of purification | 224 | |
| (K/Cs)PbBr3 | Post-synthesis | 512 | 20 | 83 | 100% (153 h, blue light) | 228 |
| CsPb1−xSnxBr3 (0 ≤ x ≤ 0.1) | Post-synthesis | 479–512 | 62 | 272 | ||
| CsPbBr3:Sn(IV) | Hot-injection (180 °C) | 517 | 20 | 83 | 273 | |
| CsPbBr3:Ce | Hot-injection (185 °C) | 510 | 89 | 60% (30 d, air) | 296 | |
| CsPbBr3:Mn | Hot-injection (150 °C) | 514–517 | 20 | 90 | 60% (120 d, air) | 102 |
| CsPbCl3:Mn | Hot-injection (150 °C) | 589 | 27 | 249 | ||
| CsPb0.73Mn0.27Cl3 | Hot-injection (170 °C) | 580 | 54 | 40% (60 min, UV) | 251 | |
| (C4H9NH3)2PbBr4:Mn | Annealing at 125 °C | 600 | 37 | 80% (8 d, air) | 259 | |
| CsPbBr1.5I1.5 | LARP | 600 | 38 | 72 | 55 | |
| CsPbI3 | Ultrasonication | 90 | 0% (2 months, air) | 45 | ||
| Microwave irradiation | 691 | 35 | 70 | 80 | ||
| CsPbI3:Mn | Hot-injection (150 °C) | 680 | 40 | 82 ± 9 | Stable over a month | 263 |
| FA0.1Cs0.9PbI3 | Modified hot-injection (80 °C) | 685 | >70 | >95% for month | 225 | |
3.2. Surface engineering
The surface of semiconductors plays a significant role in carrier recombination processes, and the surface defects result in severe PL quenching.297 The surface defects are more crucial for colloidal QDs owing to the large surface-to-volume ratio.298 Although numerous theoretical299–301 and experimental302,303 research studies have revealed that the LHPs exhibit a high defect tolerance, they are not defect impervious. Moreover, the ill-passivated surfaces are highly susceptible to moisture.53 Therefore, it is essential to seek suitable surface ligands that not only enhance the PL performance but also improve the stability.Generally, PQDs are capped by long alkyl OA and OLA (octylamine for MAPbX3 PQDs) molecules. Jasieniak et al.304 proposed that the phase instability of CsPbI3 PQDs originates from the interaction between the ion-pairs of OA ligands and the surface of CsPbI3 PQDs. They stabilized CsPbI3 PQDs by replacing conventional OA with bis-(2,2,4-trimethylpentyl)phosphinic acid (TMPPA). The PL performance of OLA and TMPPA passivated CsPbI3 PQDs (denoted as CsPbI3–TMPPA) is similar to that of conventional OLA and OA capped CsPbI3 PQDs (denoted as CsPbI3–OA). Compared with CsPbI3–OA decomposing within 3 days, the PL intensity of CsPbI3–TMPPA nearly preserved the initial value upon storage over 20 days (Fig. 15A). Replacing OA with TMPPA effectively slows down the α-to-δ phase transformation of CsPbI3.
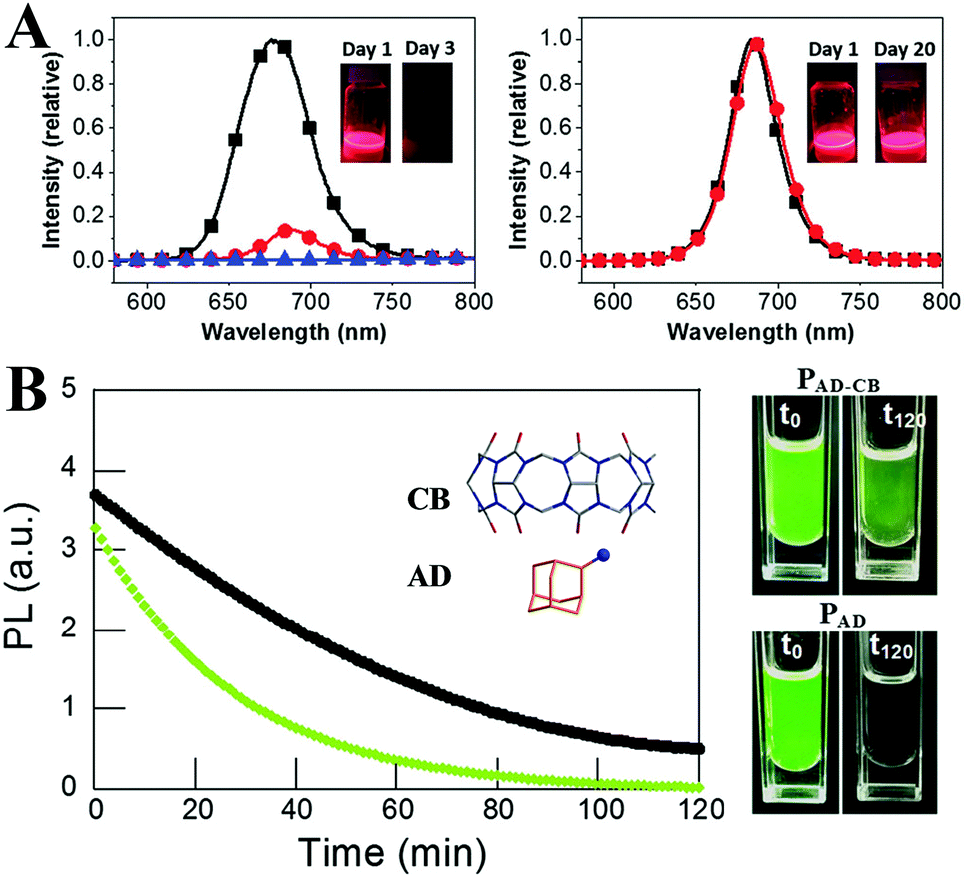 | ||
| Fig. 15 (A) PL spectra of CsPbI3–OA (left) and CsPbI3–TMPPA (right), respectively. The insets are the solutions of the respective PQDs under UV light at different times following synthesis. Reprinted with permission from ref. 304. Copyright 2017, The Royal Society of Chemistry. (B) PL of PAD–CB (black line) and PAD (green line) dispersed in toluene and in contact with water as a function of the irradiation time; the inset shows the molecular structures of CB and AD ligands. The right images are the colloidal dispersions immediately after the addition (left) of water and 120 min later (right). Reprinted with permission from ref. 308. Copyright 2016, Wiley-VCH. | ||
The molecules with large steric hindrance such as highly branched (3-aminopropyl)triethoxysilane (APTES),305,306 polyhedral silsesquioxane-[3-(2-aminoethyl)amino]propylheptaisobutyl substituted (NH2-POSS),305 1-tetradecylphosphonic acid (TDPA),307 2-adamantylammonium bromide (ADBr),308 trioctylphosphine oxide (TOPO),309 mercapto-β-cyclodextrin (SH-β-CD),310 poly(lactic acid) (PLA),311 and cage-like polyhedral oligomeric silsesquioxane (POSS)107 could bestow high protic solvent resistance to perovskites. From the stability test, such branched ligands could effectively prevent the protic solvents from penetrating into the PQDs’ surface, in contrast to the OA and OLA capped PQDs transforming into a non-luminescent state or decomposing directly in the same condition. Moreover, the light-induced agglomeration is impeded by the large steric hindrance, which ensures high photostability. For example, by using ADBr as the unique surface ligand, the PL QYs of MAPbBr3 quasi-spherical nanoparticles reach almost unity.308 The special ligands form cucurbit[7]-uril-adamantyl ammonium host–guest complexes upon illumination in humid conditions, which can avoid the photodarkening effect and improve the water stability (Fig. 15B). However, immoderate incorporation of branched ligands like APTES results in an ill-passivated surface.305
Ligand passivation always relates to the deactivated quenching defects. Trioctylphosphine (TOP),312–314 diphenylphosphinic acid (DPPA),315 di-dodecyl dimethyl ammonium bromide (DDAB),302,316 didodecyl dimethylammonium sulfide (SDDA),317 PEA,318 benzyl alcohol (BnOH),319 YCl3320 and ZnX2 salts321 can control the ligand binding motifs, and enhance the PL QYs and phase duration. For instance, Shen et al.312 synthesized CsPbI3 PQDs with PL QYs of near unity by using TOP-PbI2 as precursor (Fig. 16A). Time-resolved TA spectroscopy revealed negligible electron or hole trapping pathways for TOP-CsPbI3 PQDs, which implies that the TOP molecules well passivate the CsPbI3 surface. TOP-CsPbI3 samples retain ∼85% of the original PL intensity after being stored for 30 days. In comparison, the PL QYs of the non-TOP capped sample decreased from 86% to 60% within 1 month. The enhanced environmental stability is likely associated with the improved crystalline quality combined with less quenching defects in TOP-CsPbI3. Beyond the enhanced stability by TOP passivation, the PL intensity of aged CsPbI3 PQDs can recover to their initial value (Fig. 16B). This is attributed to the fact that some ions migrate to the surface of PQDs and repair the surface defects in the presence of TOP.313
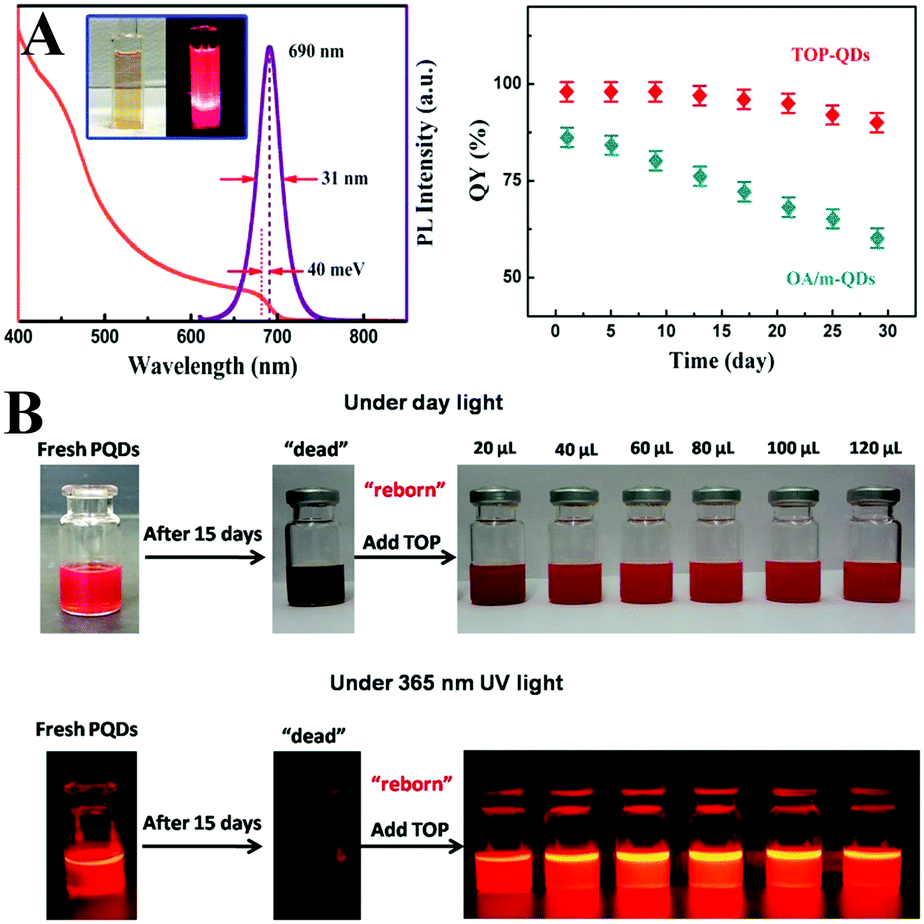 | ||
| Fig. 16 (A) UV-vis absorption and PL spectra of CsPbI3 PQDs. The inset shows the photos of the CsPbI3 solution under the room light (left) and UV lamp (λ = 365 nm) (right) excitation. The right images are the PL QYs of the OA/m- and TOP-CsPbI3 PQDs over 30 days of storage. Reprinted with permission from ref. 312. Copyright 2017, American Chemical Society. (B) Photos of fresh/aged PQDs and aged PQDs with different amounts of TOP under daylight (top) and UV light (bottom). Reprinted with permission from ref. 313. Copyright 2018, American Chemical Society. | ||
Post-synthetic treatment can also effectively eliminate the surface trap. Alivisatos et al.322 proposed that the surface trapping originates from the lead-rich surface of perovskites due to the lead-rich synthetic environments. Hence, they developed a postsynthetic thiocyanate surface treatment strategy to repair the lead-rich surface. As expected, the surface trap of both fresh (with PLQYs of ∼92%) and aged (with PLQYs of ∼63%) CsPbBr3 PQDs deactivated by thiocyanate treatment, and the treated samples showed near-unity PLQYs together with high stability (Fig. 17A). Samanta et al.323 reported a more general strategy to enhance the PL of CsPbX3 PQDs by treating them with tetrafluoroborate salts. The PL QYs of the green emissive CsPbBr3 and blue emissive CsPbBrxCl3−x could reach near unity. However, these two strategies seemed to make little success with CsPbBrxI3−x. CsPbBrxI3−x PQDs suffer from a faster degradation and PL quenching than their bromide counterparts.324 Therefore, a CsPbBr3 layer like CsPbI3@CsPbBr3 core–shell structures might protect CsPbI3. Ethanol or acetone could selectively etch the surface of iodine-containing CsPbBrxI3−x PQDs and result in a bromine-rich protective self-passivation layer. Consequently, the protective layer improves the stability of the CsPbBrxI3−x PQDs by three orders of magnitude (Fig. 17B). As for blue emissive perovskite NCs, Feldmann and coworkers325 found that dispersing CsPbBr3 nanoplates in PbBr2-ligand (OA and oleylamine) solutions can boost their PL QYs from 7% to 42%. The PbBr2-ligands could effectively repair the surface trap, which leads to PL enhancement. Moreover, the CsPbBr3 nanoplates show improved colloidal stability and photostability after PbBr2-ligands treatment (Fig. 17C).
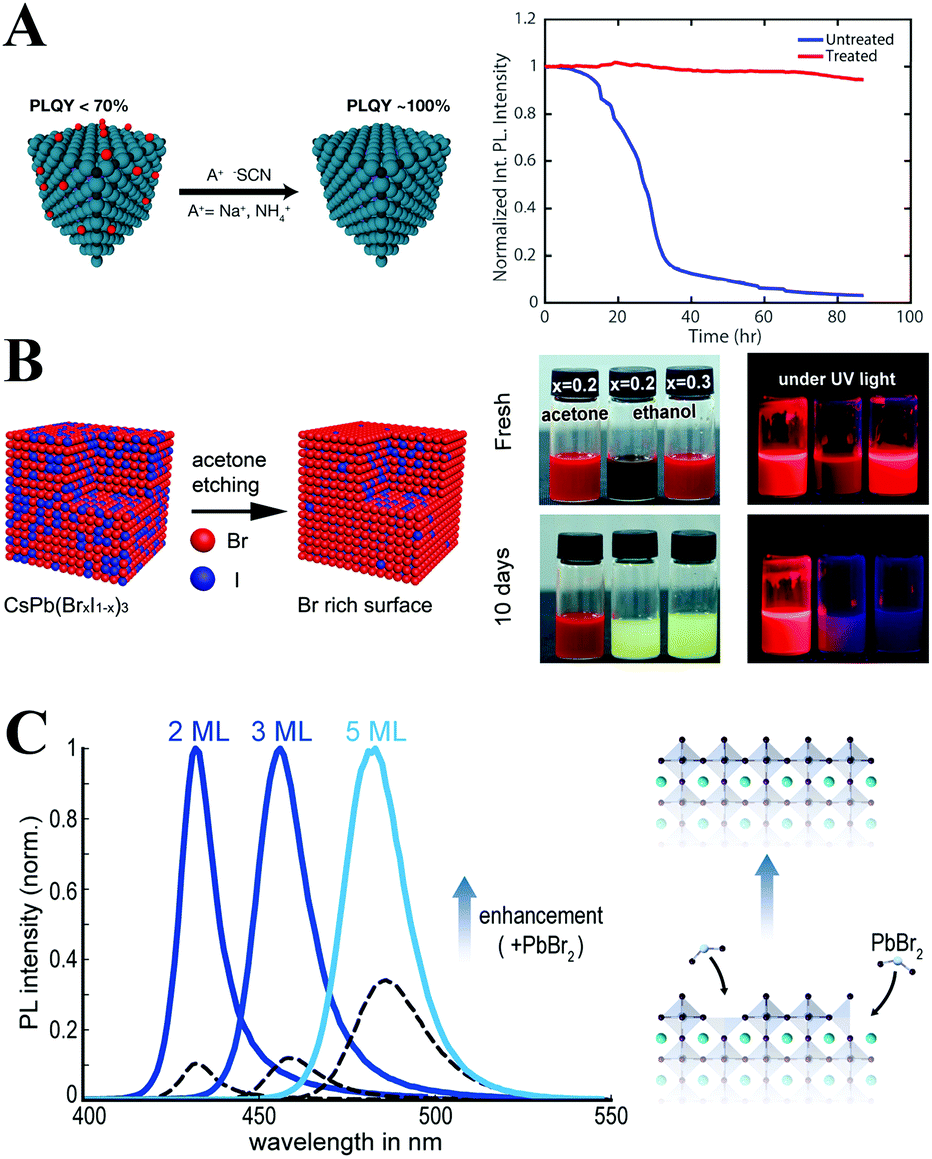 | ||
| Fig. 17 (A) Schematic of thiocyanate surface treatment on CsPbBr3 PQDs. Test of the optical stability of a dilute colloidal sample of treated and untreated particles under continuous illumination with ∼100 mW of 365 nm UV light (right). Reprinted with permission from ref. 322. Copyright 2017, American Chemical Society. (B) Schematic of acetone surface treatment on CsPb(Br/I)3 PQDs. Colloidal solution of CsPb(Brx/I1−x)3 (x = 0.1–0.3) samples in cyclohexane under daylight and UV light illumination. Reprinted with permission from ref. 324. Copyright 2017, The Royal Society of Chemistry. (C) PL spectra of initial CsPbBr3 nanoplates’ colloidal dispersions (black dashed lines) and enhanced dispersions (normalized, solid lines), and scheme for the repair process of surface defects initiated by a chemical post-treatment with a PbBr2 solution. Reprinted with permission from ref. 325. Copyright 2018, American Chemical Society. | ||
Zhong et al.326 proposed that the decomposition of MAPbI3 is attributed to the defective surface interacting with coordinated solvents and/or iodine vacancies. The noncoordinated acetonitrile (ACN) was chosen to dissolve the perovskite precursors. The obtained MAPbI3 PQDs manifest high air stability owing to the defect-less surface. Deng and coworkers327 made an X-type ligand to fix the surface defects of CsPbBr3 PQDs. The additive Pb2+ sites could fill the Cs+ terminated traps. The formation of PbBr2-terminated PQDs led to slight PL enhancement. Moreover, the compact ligand layer ensures stability in polar solvents.
The OA and OLA molecules are not tightly bound to the surface of PQDs but undergo fast exchange between their bound and free state. The dynamic surface might be the origin of the color lability. And the ligands are easily lost during purification, which leads to poor colloidal stability.97 Brutchey et al.328 firstly quantified the ligands binding to the PQDs. Acid- and amine-based ligands undergo exchange between their bound and free state, whereas the phosphonic acid ligands can tightly bind to the surface. Hence, employing phosphonic acid ligands can minimize the ligand loss. Besides phosphonic acid ligands, Sun and coworkers329 employed stearic acid (SA) and octadecylamine (ODA) with a similar long alkyl chain but a higher melting-point around 50–70 °C to cap FAPbBr3 PQDs. As a result, the ligand adsorption–desorption is eliminated in contrast to the PQDs capped by the liquid OA and OLA with a melting-point around 15–25 °C. The highly luminescent FAPbBr3 PQDs capped by solid ligands maintain 80% of the initial brightness in ambient air over 30 days, whereas the PL of liquid ligand capped PQDs decreases to 40% in the same condition (Fig. 18A). The improved stability can be attributed to the minimal dynamic-surface-induced ligand loss. Similarly, ligands with stronger adhesion can tightly bind to the surface of perovskites and avoid ligand loss during purification. In this respect, SDDA,317 long chain zwitterionic ligands,330 and bidentate ligands331 are employed to passivate the CsPbX3 PQDs. The ligands with strong adhesion allow for obtaining clean PQDs with high PL QYs above 90% following multi-purification.330 The SDDA capped samples show high PL after 34 hours of high-intensity pulsed laser irradiation.317 And the bidentate ligands can well-stabilize the cubic phase of CsPbI3 PQDs.331
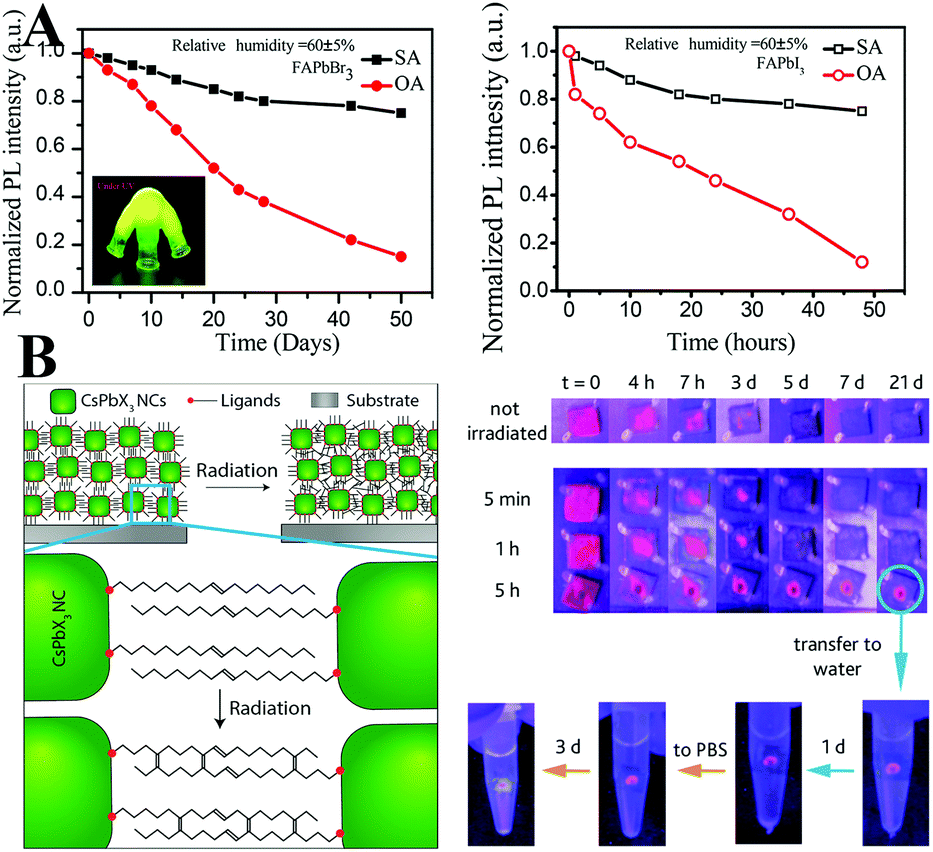 | ||
Fig. 18 (A) Change of PL intensity as a function of storage time of FAPbBr3 (left) and of FAPbI3 (right) PQDs. The inset of the left image is the photograph of SA capped FAPbBr3 solutions under UV light illumination. Reprinted with permission from ref. 329. Copyright 2017, American Chemical Society. (B) Scheme illustrating that intermolecular C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonding appears as a consequence of irradiation, linking adjacent NCs in the film (left), and water stability of pristine CsPbI3 QD films and irradiated films. Reprinted with permission from ref. 335. Copyright 2016, American Chemical Society. C bonding appears as a consequence of irradiation, linking adjacent NCs in the film (left), and water stability of pristine CsPbI3 QD films and irradiated films. Reprinted with permission from ref. 335. Copyright 2016, American Chemical Society. | ||
Crosslinking surface ligands could strengthen the interactions of PQDs and minimize ligand loss. Seferos and coworkers332 employed a thermally crosslinkable molecule 4-vinylbenzyl-dimethyloctadecylammonium chloride (V18) to passivate the MAPbBr3 PQDs. Tan and coworkers333 explored a trimethylaluminum (TMA) vapor crosslinking method and made CsPbX3 PQDs insoluble in solvents. Manna et al.334,335 deposited OA and OLA capped CsPbX3 PQDs on a silicon substrate. The intra- and intermolecular CC bonds form upon X-ray irradiation. The intermolecular CC bonds link the adjacent PQDs (Fig. 18B). Overall, based on the various crosslinking protocols, the obtained samples could retain high PL QYs in ambient air for a long period of time, because the ligand-crosslinking could minimize the ligand loss.
3.3. Matrix encapsulation
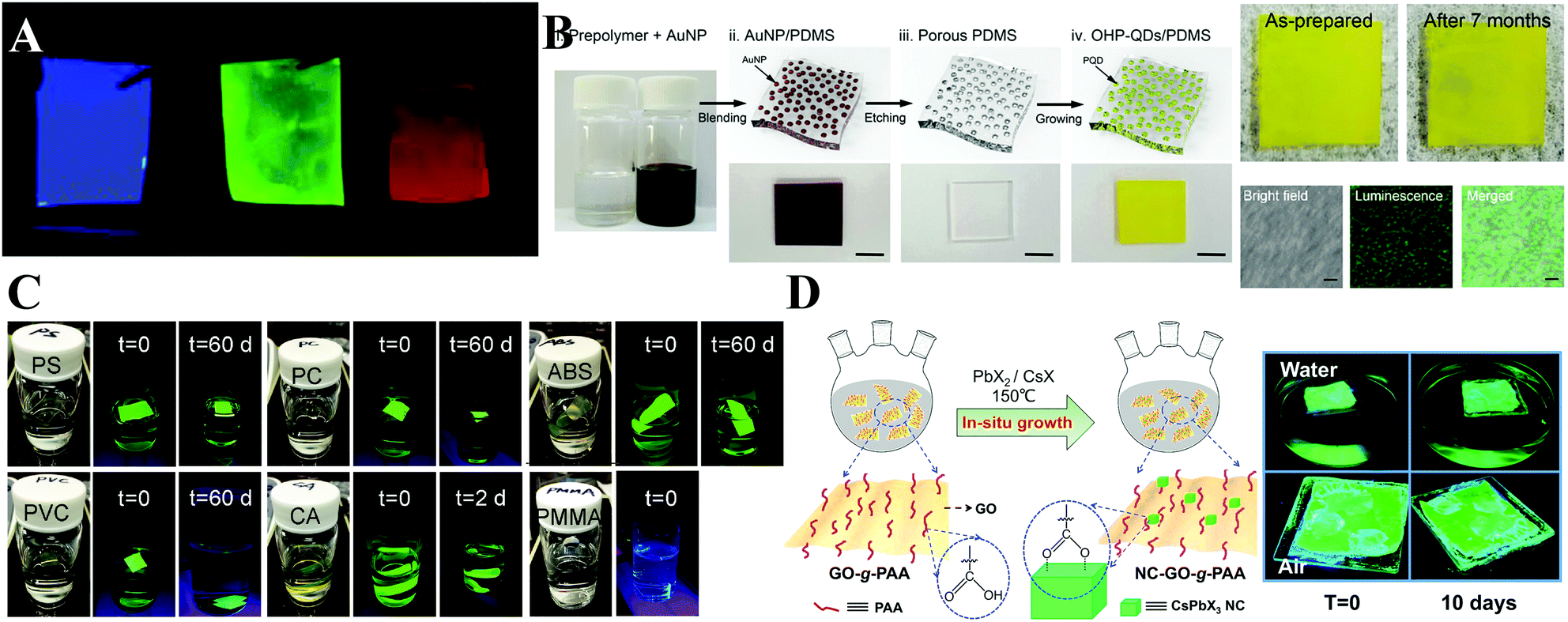
Among the various forms of perovskite–polymer composites, perovskite–polymer films have received a significant attention. On the one hand, numerous strategies have been developed for fabricating high-quality films. On the other hand, the luminescent films can be used as the backlight sources for LCDs. Sargent et al.99 cast highly luminescent CsPbBr3 quantum dot films by a centrifugal casting process without any matrix additives. Snaith et al.336 blended the as-prepared PQDs with polystyrene (PS) or polymethyl methacrylate (PMMA) beads, and then the mixture was cast onto a glass substrate and spin-coated to form a dry film (Fig. 19A). The polymer films can prevent anion exchange, which demonstrates their potential in pc-LEDs. Chen et al.337 employed a microfluidic spinning technique to embed CsPbBr3 PQDs into PMMA fiber films. Yoon and coworkers338 fabricated ethyl cellulose (EC) with CsPbBr3 QD films by coating the mixed EC and CsPbBr3 PQDs toluene solutions on a PET substrate via the doctor blade method. Kang et al.339 fabricated perovskite nanoparticle films by size exclusion lithography. Kim et al.340 blended polydimethylsiloxane (PDMS) and Au nanoparticles (NPs) in toluene and cast AuNP/PDMS films firstly. Then the Au NPs can be removed by aqua regia etching, resulting in porous PDMS films. The porous films can be employed as the template for growth of MAPbBr3 PQDs (Fig. 19B). The emission wavelength of MAPbBr3 PQDs can be tuned by controlling the size of Au NP templates. Kuo et al.341 encapsulated CsPbX3 PQDs in stretchable poly(styrene-butadiene-styrene) (SBS) fibers by an electrospinning strategy. The obtained fiber membranes can serve as water-proof multicolor converters for pc-LEDs. Chen and coworkers342 prepared uniform CsPbX3/PAN (polyacrylonitrile) nanofibers by an electrospinning technique. The hydrophobic PAN provides superior resistance towards humidity and water. Yang and coworkers343 synthesized CsPbX3/polymer fibers by in situ growth of PQDs in polymer fibers. PQDs with uniform size can be homogeneously encapsulated in the polymer fibers. Dong and coworkers75 dissolved the perovskite precursor in dimethyl formamide (DMF) solution and deposited the solution on as-prepared polymer films (PS, PMMA, polycarbonate (PC), acrylonitrile butadiene styrene (ABS), cellulose acetate (CA), and polyvinyl chloride (PVC) were used here). The polymer chains will swell when in contact with good solvents and take in the precursor solutions. After the baking process, the solvents are evaporated, and meanwhile the MAPbBr3 PQDs are formed in the polymer matrices. As a result, the MAPbBr3–PS, MAPbBr3–PC, MAPbBr3–ABS, MAPbBr3–PVC, and MAPbBr3–CA films retain high luminescence even when immersed in water over 60 days, whereas the luminescence of MAPbBr3–PMMA is quenched rapidly (Fig. 19C). The rapid PL loss is attributed to the low swelling ratio of PMMA in DMF solvent and thus the PQDs only form on the surface of PMMA films. Chen et al.344 prepared flexible CsPbBr3/ethylene vinyl acetate (EVA) composite films by crystallization of PQDs and dissolution of EVA in toluene. Their PL intensity did not change even after bending the films over 1000 cycles. Zhong et al.76 reported an in situ preparation of MAPbBr3/polyvinylidene fluoride (PVDF) composite films. Due to the strong interactions between the –NH3+ group in MAPbBr3 and the –CF2– groups in PVDF, the obtained MAPbBr3/PVDF films show increased PL QYs up to 94.6 ± 1%.
 | ||
| Fig. 19 (A) Images of the perovskite crystal/polymer composite films emitting blue ((OA/MA)PbCl3), green ((OA/MA)PbBr3), and red ((OA/MA)PbI3) light under UV lamp excitation. Reprinted with permission from ref. 336. Copyright 2015, American Chemical Society. (B) Schematic illustration of MAPbBr3-QDs/PDMS film synthesis. The accompanying photographs show the resulting films at each stage. Scale bar = 1 cm. On the right side are photographs of the MAPbBr3/PDMS films before and after 7 months of storage together with their bright field and luminescence images. Scale bar = 4 mm. Reprinted with permission from ref. 340. Copyright 2017, The Royal Society of Chemistry. (C) Photographs taken under white light or UV irradiation at the indicated time period. The composite film samples immersed in water are MAPbBr3–PS, MAPbBr3–PC, MAPbBr3–ABS, MAPbBr3–PVC, MAPbBr3–CA, and MAPbBr3–PMMA. Reprinted with permission from ref. 75. Copyright 2016, Wiley-VCH. (D) Illustration of the synthesis of the ternary NC–GO-g-PAA hybrid (left) and the photographs of glass slides coated with NC–GO-g-PAA nanorod films after soaking in water or exposure to air for 0 and 10 days. Reprinted with permission from ref. 346. Copyright 2017, American Chemical Society. | ||
When referring to the interactions between polymers and perovskites, poly(maleic anhydride-alt-1-octadecene) (PMA)345 was employed to tighten the ligand binding. The color converter composed of CsPbX3–PMA composites shows a prolonged lifetime in comparison with pristine PQDs. Liu and coworkers346 synthesized polyacrylic acid-grafted graphene oxide (GO-g-PAA), and the hybrid composites were used as ligands for the uptake of the perovskite precursors by coordinating PbBr2, forming a kinetically favorable nucleation point for in situ crystal growth (Fig. 19D). Note that such a carboxylate-group-rich polymer can minimize the ligand loss, which improves the colloidal stability of PQDs, but the environmental stability is still limited since the PQDs only lie on the polymer surface instead of being encapsulated. But the addition of the CsPbBr3–GO-g-PAA toluene solution into hexane can form self-assembled nanorod-shaped hierarchical superstructures. Thus, self-assembled nanorods provide favorable encapsulation of PQDs, which imparts superior environmental stability to PQDs. Quan et al.347 employed an amphiphilic block copolymer with a hydrophobic PS block and a coordinatable poly-2-vinylpyridine (P2VP) block as a confined nanoreactor for synthesizing CsPbBr3 PQDs. The PbBr2 precursors were uptaken by the P2VP block. After the addition of CsBr, a strong PL under UV excitation was observed, indicating the formation of CsPbBr3 PQDs in the nanoreactors. Liu and coworkers348 synthesized polystyrene-b-poly(ethyl oxide) (PS-b-PEO) grafted MAPbBr3 PQDs. After rehydrating the precipitate in water, the MAPbBr3 PQDs self-assembled into well-defined vesicular nanostructures, which suggests their great potential for cell imaging (Fig. 20A).
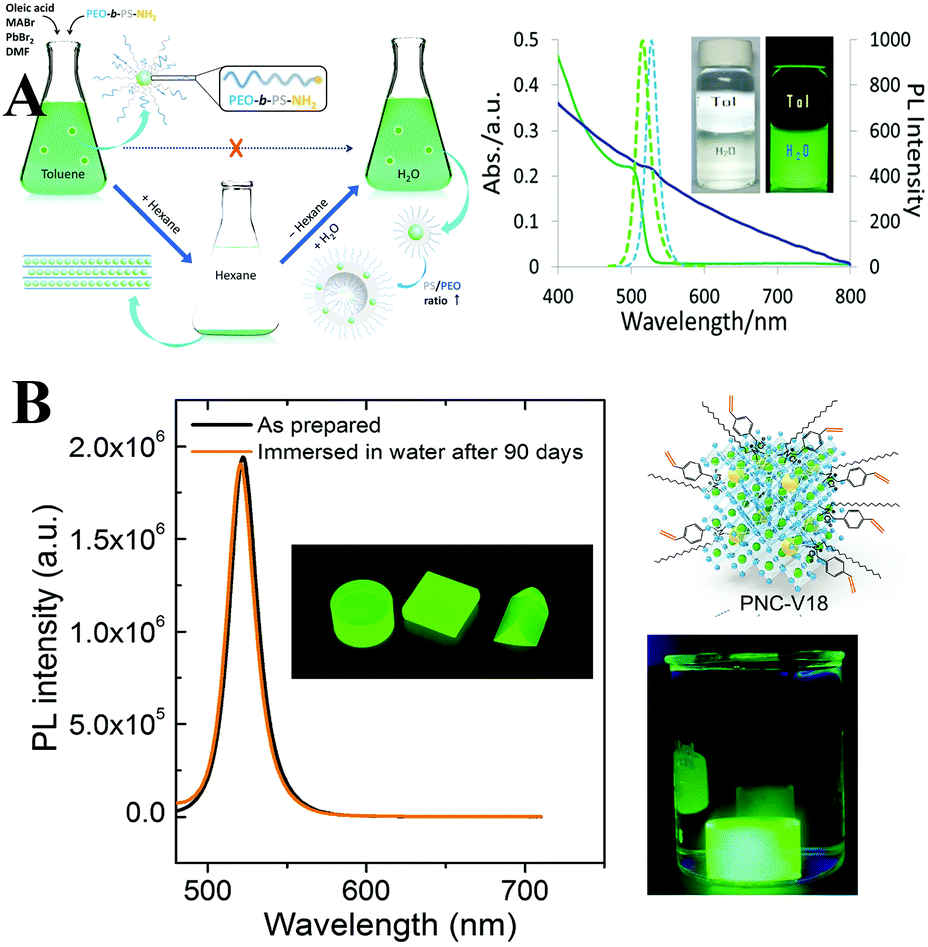 | ||
| Fig. 20 (A, i) Schematic illustration of the synthesis and toluene-to-water phase transfer of PEO-b-PS-NH2 grafted MAPbBr3 QDs. (ii) UV-Vis absorption (solid line) and PL emission spectra (dashed line) of PQD@PS106-b-PEO45 in toluene (green) and in water (blue). Inset: Photographs of an aqueous solution of PQD@PS58-b-PEO45 in a glass vial filled with toluene (left) and the same vial illuminated with a 365 nm UV lamp (right). Reprinted with permission from ref. 348. Copyright 2018, The Royal Society of Chemistry. (B) PL spectra of MAPbBr3–V18–polymer composites before and after being immersed in water for 90 d. The insets are the photo of bulk material under UV light excitation and the illustration of V18 modified MAPbBr3 PQDs, respectively. Reprinted with permission from ref. 332. Copyright 2017, Wiley-VCH. | ||
Carboxybenzene (CB) microcrystals with high transparencies, low toxicities, and low refractive index are regarded as a desired matrix for stabilizing perovskites. Chen et al.349 prepared highly stable macroscopic needle-shaped MAPbBr3–CB crystals through slow-cooling of the hot toluene solution containing dissolved CB and MAPbBr3 PQDs. Alivisatos et al.350 encapsulated PQDs in a macroscale polymer including PS, poly(lauryl methacrylate) (PLMA), and poly(styrene-ethylene-butylenestyrene) (SEBS) matrices. The further stability tests revealed that PQDs–PS is the most water resistant, and the photostability of PQDs–PLMA is the most prominent. Among the three kinds of perovskite–polymer composites, PQDs–SEBS shows high water and photo stability. Yu and coworkers351 prepared white-emitting composites by encapsulating CsPb(Br/I)3 PQDs with anthracene. The anthracene can work as a blue-emitting component upon UV light excitation, and simultaneously, it can protect PQDs against environmental-induced decomposition. The polymerizable V18 ligands make it possible to incorporate the PQDs into a polymer matrix by copolymerization.332 A PQDs–polymer bulk composite was obtained by blending 2,2-azobisisobutyronitrile (AIBN) as the radical initiator with monomers in V18–PQDs solutions. A negligible difference in PL spectra was observed when the bulk composites were immersed in water over 90 days, which demonstrated the ultrahigh stability towards superior water resistance (Fig. 20B). Kovalenko et al.71 conducted photo-induced polymerization by employing bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide (Irgacure 819) as the photoinitiator and the obtained CsPbX3 NCs–PMMA polymer monoliths exhibited high luminescence.
Tüysüz et al.352 regarded CsPbI3 PQDs as a photocatalyst that could produce radicals under light irradiation and polymerize monomers. 2,2′,5′,2′′-ter-3,4-Ethylenedioxythiophene (TerEDOT) was polymerized over CsPbI3 PQDs with the help of 1,4-benzoquinone as the electron acceptor upon visible light irradiation. As a result, CsPbI3 PQDs are encapsulated in the poly(3,4-ethylenedioxythiophene) (PEDOT) networks, and simultaneously the initial cubic structure is retained. Recently, Tan et al.353 used CsPbBr3 PQDs as the photoinitiator in the polymerization of vinyl monomers. Upon white light illumination, polymer chains grow on the surface of PQDs directly, which results in the individually encapsulated PQDs@polymer nanoparticles with monodisperse core–shell structure (Fig. 21A).
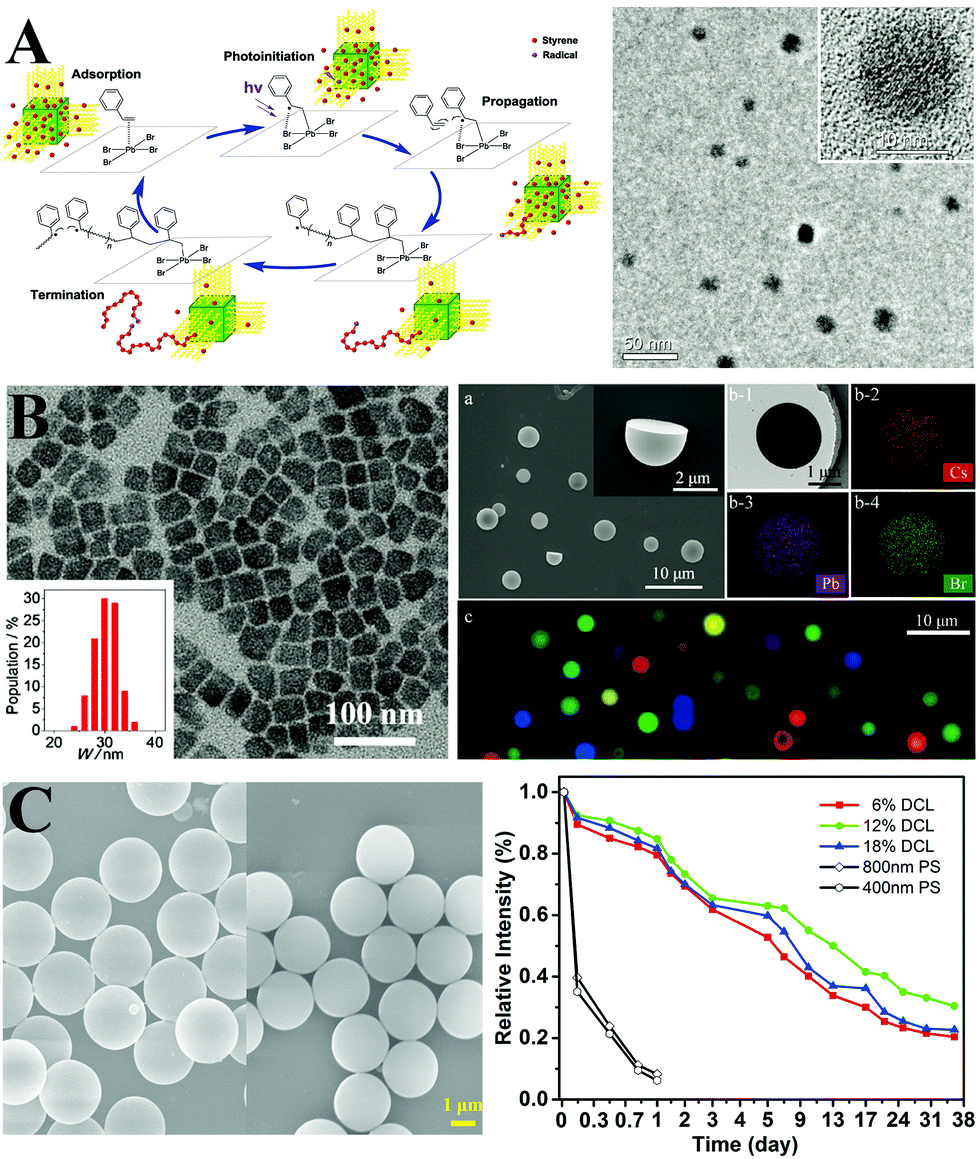 | ||
| Fig. 21 (A) Proposed reaction mechanism (left) of the perovskite photoactivated polymerization of styrene and TEM and HRTEM images (right) of perovskite–polystyrene nanocomposites. Reprinted with permission from ref. 353. Copyright 2018, Wiley-VCH. (B) TEM images of PVP-capped PQDs (left), and (right) SEM images of CsPbX3 NCs@MHSs, elemental mapping of single CsPbBr3 NCs@MHSs, and the fluorescence image of a mixture of neat and mixed-halide NC-tagged beads emitting single-color signals at 429, 455, 490, 512, 543, 581, and 642 nm. (C) SEM image of pure polymer spheres (left) with a size of 3 μm and PQDs@PS (right) composites. And PL spectra of CsPbBr3 NCs packed into PS spheres with different degrees of crosslinking and different diameters. (B and C) Reprinted with permission from ref. 354 and 356. Copyright 2017, Wiley-VCH. | ||
Aside from the perovskite–polymer films and monoliths, perovskite–polymer nanocomposites also contribute to the enhanced stability owing to the compact encapsulation. Moreover, the monodisperse nanocomposites offer the possibility for cell imaging which is not feasible with bulk materials. In this respect, Fu and co-workers354 developed a room-temperature solution self-assembly approach to synthesize polyvinylpyrrolidone (PVP)-capped colloidal CsPbX3 nanocrystals (NCs) and embedded the NCs in polystyrene microhemispheres (MHSs) by assembling PS chains on NCs. The obtained luminescent NCs@MHSs with diameters of 4 ± 1 μm exhibit high water stability and nontoxicity, and can be employed as probes for cell imaging (Fig. 21B). Li and coworkers355 prepared monodisperse CsPbBr3@PS microspheres by electrospraying methods. PS encapsulation leads to the improved stability against anion exchange and water. Lin and coworkers356 synthesized PQDs@PS composite beads via swelling the crosslinked beads in PQDs toluene solution and then shrinking in hexane. Further studies revealed that most PQDs might lie on the periphery of PS beads, and the small size (400 and 800 nm) of PS beads results in insufficient protection and limited water stability. The relatively large size (about 3 μm) of PS beads significantly enhanced the stability, especially, the PS beads with a hydrophobic surface well resisted aqueous, acid, and alkali solutions (Fig. 21C). The PQDs@PS retains a relatively high PL efficacy in such harsh environments, which can be applied as a cellular labelling agent and down-converter for pc-LEDs.
Silica coating. Silica is chemically stable and transparent in the whole visible region that does not bring in changes in the optical properties of luminescent materials but can protect materials against moisture-induced damage. Silica-coating has been widely used in the lanthanide-doped upconversion nanoparticles,357 traditional QDs,358 magnetic nanoparticles,359etc. However, one primary problem with the silica coating for PQDs is that in a typical silica coating process via Stöber360 or reverse microemulsion methods,361 one inevitably uses tetraethyl orthosilicate (TEOS) as the precursor which decomposes in a strong alkaline alcohol solution. The coating of silica layers on PQDs is nearly impossible since the perovskite decomposes under such conditions before silica formation. There are three alternative methods available to overcome this problem: (1) using a pre-synthesized porous silica material; (2) selecting other precursors as silica sources; (3) altering the synthetic conditions for silica formation.
Liu et al.109 firstly blended pre-synthesized CsPbBr3 PQDs with silica spheres with a pore size of 12–15 nm. After integrating PQDs into mesoporous silica, the obtained composites show enhanced thermal stability and photostability (Fig. 22A). Kovalenko et al.362 infiltrated CsPbBr3 precursor solutions into mesoporous silica, and removal of solvents by baking resulted in the formation of PQDs in the silica pores. Yamauchi et al.363 prepared MAPbBrxIx−3 PQDs inside the mesoporous silica. Quantum confinement was observed by tuning the size of the QDs by altering the pore size of the silica templates (Fig. 22B). They further used gyroidal mesoporous silica364 and mesoporous silica films365 as templates for MAPbX3 growth. The 3D gyroidal template promotes the formation of monodispersed larger structures such as nanowires or nanorods due to the easier escaping of solvents. On the other hand, by utilizing mesoporous silica films as the template, CsPbBr3 PQDs with 3.1 nm of average size formed within the pores. Owing to the strong quantum confinement effects, the composite films show blue-shift in PL emission together with increased PL QYs in comparison with the bulk materials.
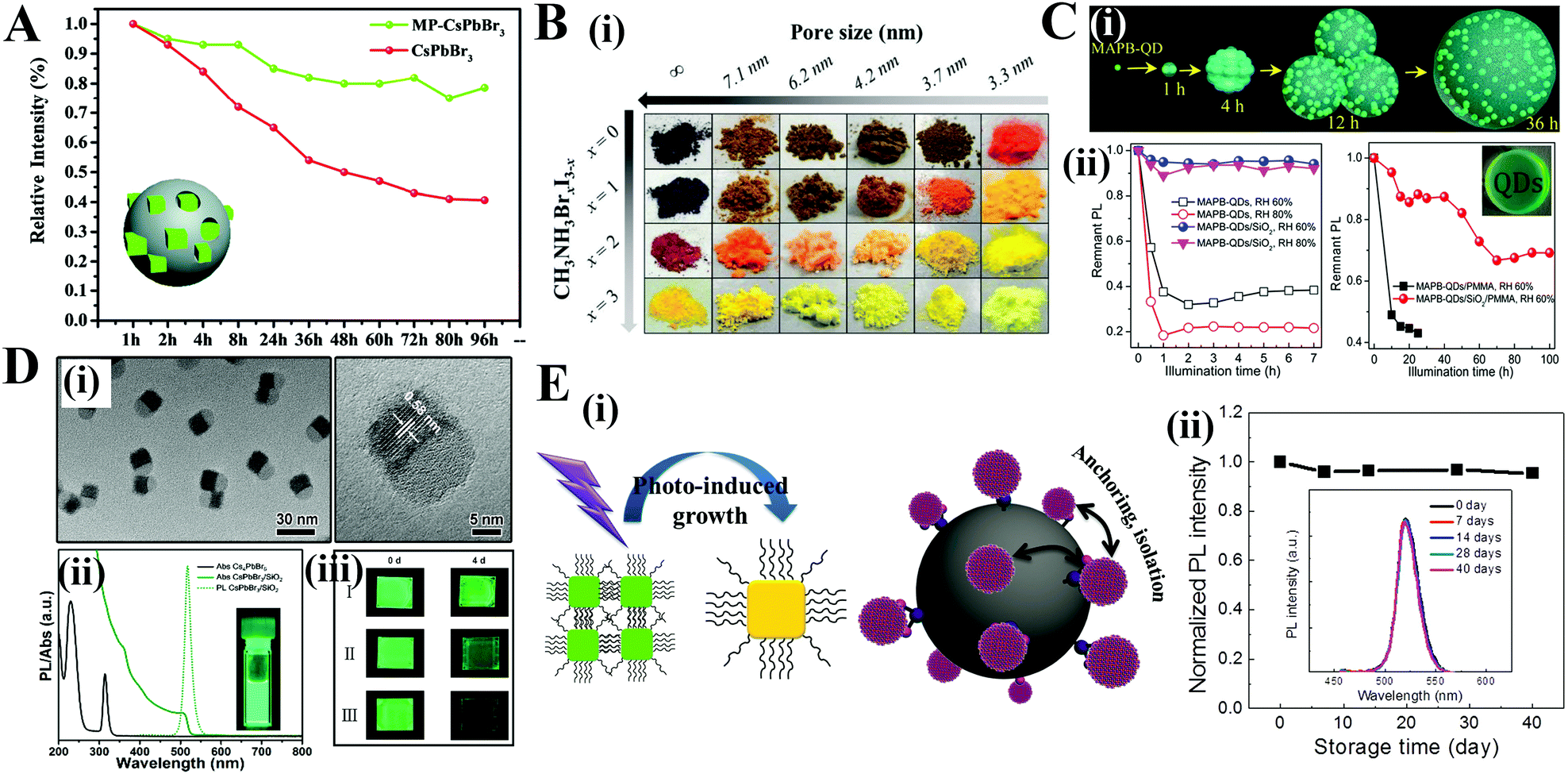 | ||
| Fig. 22 (A) Change of PL intensity as a function of UV illumination time of CsPbBr3–SiO2 and CsPbBr3. The inset is the illustration of mesoporous silica spheres embedded with CsPbBr3 PQDs. Reprinted with permission from ref. 109. Copyright 2016, Wiley-VCH. (B) Photographs of the mesoporous silica powders after impregnation and crystallization. Reprinted with permission from ref. 363. Copyright 2016, American Chemical Society. (C) MAPbBr3–SiO2: (i) schematic of the MAPbBr3–SiO2 nanocomposites’ formation process with increasing reaction time. (ii) Photostability of the MAPbBr3 (left) and MAPbBr3–SiO2 powders and their films with different RH values (right) under 470 nm LED light illumination. Inset: Optical image of the MAPbBr3–SiO2–PMMA film. Reprinted with permission from ref. 188. Copyright 2016, American Chemical Society. (D) CsPbBr3/SiO2 Janus: (i) TEM image of the obtained CsPbBr3/SiO2 Janus NCs. (ii) Absorption and PL spectra (λexc = 380 nm) of pristine Cs4PbBr6 NCs (black solid line) and CsPbBr3/SiO2 NCs. The inset photograph shows a strong green PL emission under UV light (λ = 365 nm). Reprinted with permission from ref. 77. Copyright 2017, American Chemical Society. (E) CsPbBr3 QDs/A-SiO2 composites: (i) schematic illustration of photo-induced regrowth of PQDs (left) and the isolation effect of PQDs on silica sphere surface (right). (ii) Material stability after storage without any protection. Reprinted with permission from ref. 370. Copyright 2017, Wiley-VCH. | ||
Li et al.188 employed tetramethyl orthosilicate (TMOS) as the silica precursor, which is more sensitive to water and easily hydrolyzed in a moisture condition than TEOS. The silica barriers formed on the MAPbBr3 PQDs in the waterless toluene (with a H2O content of 0.0184%), protecting MAPbBr3 PQDs against moisture-induced degradation (Fig. 22C). Yin and coworkers77 prepared isolated CsPbX3/SiO2 Janus nanoparticles via a water-triggered transformation process combined with a sol–gel protocol (Fig. 22D). The PL intensity of PQDs declined to 85% upon continuous illumination, whereas the CsPbX3/SiO2 Janus nanoparticles preserve the value of 98%. Jang et al.366 employed perhydropolysilazane (PHPS) as a silica precursor. The silica formed on the CsPbX3 PQDs by curing in a moisture condition. The formation of silica barriers provides good thermal stability and chemical stability for PQDs. Leng and coworkers367 constructed an amorphous silica layer by inserting TEOS into freshly synthesized CsPbBr3 PQDs. The silica layer formed on the PQDs rapidly under high temperature. The obtained CsPbBr3–SiO2 composites present enhanced photostability, nonblinking optical properties, and improved PL performance. Recently, monodisperse CsPbBr3@SiO2 core–shell nanoparticles have been synthesized by manipulating synthetic conditions such as temperature, precursors, and pH.368,369 Compared with the large-sized aggregates, the ultrastable CsPbBr3@SiO2 nanoparticles show potential in high-quality film applications, cell imaging, etc.
Zeng and coworkers370 reported that, in addition to silica coating, anchoring PQDs on silica surface can also improve the stability. The anchoring-induced isolation prevents the PQDs contacting each other, which limits the possibility of crystal regrowth (Fig. 22E). The obtained PQDs–SiO2 composites show excellent stability against light irradiation and air damage. After 40 d storage in ambient air, the PL intensity did not change as revealed by fluorescence spectrophotometric detection.
When referring to the anchoring effects, some reports comment that the nanoparticle anchoring on PQDs also contributes to the stability. Zhou et al.371 synthesized CsPbX3/ZnS heterostructures by anchoring ZnS nanoparticles on the PQDs’ surface. ZnS anchoring results in rich electronic properties as confirmed by DFT calculations, which enhanced the chemical stability. Kamat et al.372 prepared a Au–CsPbBr3 hybrid nanosystem by anchoring Au nanoparticles on CsPbBr3. An excess of Au anchoring leads to ePL quenching, whereas an appropriate Au-anchoring can improve the brightness of PQDs.373
Porous materials coating. As can be seen from the silica coating, many efforts have been devoted into integrating PQDs into as-prepared mesoporous SiO2. Direct growth of SiO2 protecting layers on PQDs is somewhat challenging, because the optical performance would be deteriorated due to the harsh synthetic environment and time-consuming formation process for the protective layer. And a fast formation of the protective layer leads to strong phase separation between the perovskite and the protective layer. Accordingly, introducing PQDs into pre-synthesized porous materials is relatively attainable.
The idea of embedding perovskites into mesoporous Al2O3374 and TiO2375 to enhance their stability has been investigated in the PV technology. Ye et al.376 infused Cs+ into zeolites to substitute Na+ in the pore by ion-exchange reaction. Then, Cs+ cations react with PbBr2, and form PQDs inside the zeolite pores (Fig. 23A). Hierarchical CaF2 matrices also can be used to encapsulate PQDs.377 The outstanding PL performance of pristine PQDs such as high brightness, high color purity, and quantum confinement effects is well preserved. Additionally, the co-doping of Ce3+/Tb3+ into CaF2 exhibits strong green light upon UV excitation. Thus, the luminescent CaF2 matrices not only protect PQDs against environment-induced decomposition but also enrich their emitting colors (Fig. 23B). Hou and coworkers378 utilized magnesium silicate hollow porous spheres to encapsulate MAPbX3 PQDs. The strategy was demonstrated to be useful for multicolor displays, for which the obtained composites show tunable emission and enhanced thermal- and photo-stability.
 | ||
| Fig. 23 (A) CsPbX3–zeolite-Y: (i) schematic illustration of the two-step synthesis of CsPbX3–zeolite-Y composites. Spontaneous decomposition of CsPb(Br0.5I0.5)3 evidenced by an emission blueshift over the course of multiple days for QDs embedded in zeolite-Y (ii) and bare QDs (iii). Reprinted with permission from ref. 376. Copyright 2017, Wiley-VCH. (B) CsPbX3–CaF2: (i) PL spectra of as-prepared CaF2:Ce/Tb-CsPbCl1.5Br1.5 (top) and CaF2:Ce/Tb-CsPbBr1.5I1.5 (bottom) excited at 306 nm. (ii) Photostability test of CaF2–CsPbBr3 composites and pristine CsPbBr3 PQDs. Reprinted with permission from ref. 377. Copyright 2018, Wiley-VCH. (C) MAPbX3@MOF: (A) schematic illustrations of the reversible on/off switching of the luminescence signal. (i) As-printed Pb-MOF on a commercial parchment paper. (ii) MABr treatment leads to the formation of luminescent MAPbBr3@Pb-MOF. (iii) The luminescence quenched after MeOH treatment. (iv) The luminescence can be recovered after MABr treatment. The left images are the illustration of MAPbBr3 formed in the Pb-MOF matrix and their optical images under sunlight and UV lamp illumination. Reprinted with permission from ref. 380. Copyright 2017, Springer Nature. | ||
As one of the famous porous material families, metal–organic frameworks (MOFs) with tunable pore size hold great potential for encapsulation of perovskites. Zhang and coworkers379 firstly employed oriented MOF films as template for the growth of MAPbI2X PQDs. The ultrasmall PQDs formed on introducing perovskite precursors into MOF pores. Further studies revealed that the PQDs in MOF films are environmentally stable and insensitive to air exposure. Li et al.380 synthesized invisible and printable Pb-MOF. By MAX-triggering, luminescent MAPbX3 formed in the Pb-MOF matrix. Interestingly, the PL of the MAPbX3@MOF is reversible by environmental stimuli in response to encryption and decryption processes, which indicates that MAPbX3@MOF composites can act as a smart luminescent system for application regarding confidential information (Fig. 23C).
Salt coating. Encapsulation in a dense inorganic ionic salt matrix also provides a robust surrounding for PQDs, which could greatly enhance the stability. Zhong et al.381 proposed a one-step reprecipitation process for fabrication of MAPbBr3/NaNO3 nanocomposites. The PL intensity of pristine PQDs dropped to a value of 10% after 14 h UV excitation, whereas the MAPbBr3/NaNO3 composites preserved 80% of the initial value. Gaponik et al.112 incorporated CsPbBr3 PQDs into different KX salts under a high pressure (2.2 GPa) and showed that the CsPbX3/KX pellets with bright emission can be tuned covering the whole visible spectra. Li and coworkers382 conducted an ion exchange of CsPbCl3 PQDs to prepare CsPbBr3@NH4Br composites. The rich-bromide environments led to a sufficiently passivated surface of CsPbBr3 PQDs, resulting in the decreased surface trapping as well as enhanced stability towards water resistance (Fig. 24A). Upon water contact with the CsPbBr3@NH4Br composites, the NH4Br framework firstly dissolved in water and then released NH4+ and Br− ions, which cap the PQDs and make the perovskite isolated from water.
 | ||
| Fig. 24 Water resistance test of (A) pristine CsPbBr3, CsPbBr3@NH4Br, and (B) pristine CsPbBr3 QD films, and CsPbBr3/AlOx. (C) CsPbBr3/TiO2: (i) optical images of the fresh CsPbBr3 PQDs in toluene, dried CsPbBr3 powder in Milli-Q water, CsPbBr3/TiOx composite in water, and CsPbBr3/TiO2 NCs in water (top) and those samples in water with different extended time under ambient conditions (bottom); (ii) the relative PL intensity of CsPbBr3 NCs (without precipitation), dried CsPbBr3 powder, CsPbBr3/TiOx powder, and CsPbBr3/TiO2 powder after immersing in water; (iii) the relative PL intensity of CsPbBr3/TiO2 NCs after immersing in water (0–12 weeks), the inset shows a TEM image of CsPbBr3/TiO2 NCs after immersing in water for 12 weeks. (A) Reprinted with permission from ref. 382. Copyright 2017, The Royal Society of Chemistry. (B and C) Reprinted with permission from ref. 383 and 384. Copyright 2017, Wiley-VCH. | ||
Oxide coating. Most inorganic oxides are highly robust. Oxides have been regarded as a practical alternative for coating PQDs. Buonsanti et al.383 prepared CsPbBr3/AlOx composites via an atomic layer deposition (ALD) technique. By virtue of the low-temperature and fast ALD process, the amorphous alumina (AlOx) matrix grew on the perovskite directly, which forms a tight protection for CsPbBr3 PQDs. The obtained CsPbBr3/AlOx composites show exceptional stability towards air, high-energy light irradiation, and thermal and water treatment (Fig. 24B). Zheng et al.384 coated the TiO2 shell on CsPbBr3 PQDs by calcining the titanium precursor with CsPbBr3 PQDs at 300 °C. The monodispersed CsPbBr3/TiO2 nanocomposites preserve the initial characteristic including size, structure, and PL intensity even being immersed in water over three months (Fig. 24C).
Core–shell structure. Despite the fact that various strategies such as halide-rich surfaces, passivated ligands, polymer frameworks, and inorganic matrices have been developed as the protective shells for encapsulating PQDs, an ideal core–shell PQD structure similar to the monodisperse CdSe@ZnS architecture is still highly expected. On the one hand, the compact shell seals the PQDs and prevents oxygen and water molecules from permeating into the perovskite cores, which ensures stability. On the other hand, the shell passivates the surface and reduces the nonradiative recombination at the surface defects. Moreover, such a thin shell minimizes the nonluminescent protective materials, which maximizes the PL performance.
Despite the fact that monodisperse perovskite–polymer core–shell structural nanocomposites (CsPbBr3@PS,353 CsPbBr3@PS-b-P2VP347) at a single-particle level have been achieved, the thermal stability is still limited. The monodisperse CsPbBr3/TiO2 nanocomposites seem to provide perfect protection, but most optical property is sacrificed during TiO2 shell formation (calcination at 300 °C). Mathews et al.244 prepared core–shell type MA-octylammonium lead bromide perovskite nanoparticles. Although it is difficult to obtain the direct evidence of the presence of the 2D perovskite shell, the improved stability implies the successful shell formation (Fig. 25A). Tian et al.385 synthesized CsPbBr3@amorphous CsPbBrx (denoted as CsPbBr3@A-CsPbBrx) core–shell-type QDs (Fig. 25B). After A-CsPbBrx coating, the QDs show strong blue emission with PL QYs above 80%. Simultaneously, the A-CsPbBrx shell protects the CsPbBr3 core against photooxidation. Recently, Li et al.386 reported a controllable phase transformation from CsPbBr3 to the CsPbBr3/Rb4PbBr6 core/shell structure by postsynthetic rubidium oleate treatment of CsPbBr3 PQDs. The robust Rb4PbBr6 shells render ultrahigh stability to CsPbBr3 cores. The PL intensity of CsPbBr3/Rb4PbBr6 nanocrystals remains above 90% of the original emission after 42 h of light illumination, which is superior to that of the commercial CdSe/CdS QDs (Fig. 25C).
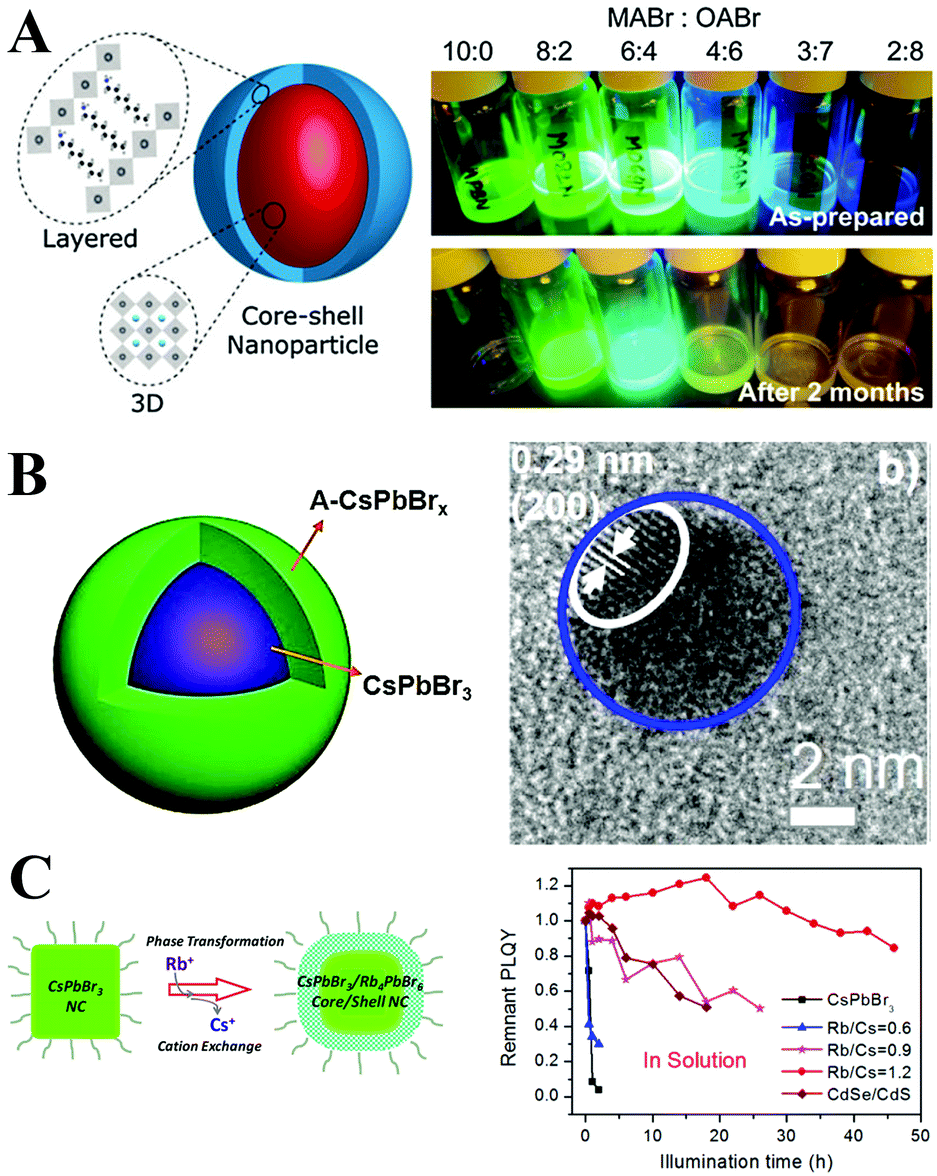 | ||
| Fig. 25 (A) Schematic showing the core–shell type layer growth of octylammonium lead bromide nanomaterials over MAPbBr3 NPs (left) and photographic images of mixed organolead bromide perovskite NPs with different MABr–OABr ratios in toluene solution under UV exposure over different time intervals (right). Reprinted with permission from ref. 244. Copyright 2016, The Royal Society of Chemistry. (B) Diagrammatic sketch of CsPbBr3@A-CsPbBrx QDs (left) and their corresponding HRTEM (right) figures. Reprinted with permission from ref. 385. Copyright 2017, American Chemical Society. (C) Diagrammatic sketch of CsPbBr3@Rb4PbBr6 NCs (left) and their photostability (right). Reprinted with permission from ref. 386. Copyright 2018, American Chemical Society. | ||
Endotaxial systems. Inspired by the successful synthesis of endotaxial systems,387 endotaxy with the same chemical elements but in a different crystal structure could cover the PQDs and prevent their agglomeration. Cs4PbBr6 and CsPb2Br5 composed of the same chemical elements as the CsPbBr3 perovskite could provide endotaxy to CsPbBr3. Among them, the tetragonal phase CsPb2Br5 with a sandwich structure consists of Cs+ and (Pb2Br5)− layers, which is quite different from the 3D structure of CsPbBr3 perovskites.23,388 Nevertheless, their optical properties are similar: narrow bandwidth, high bright green fluorescence, tunable bandgap, etc. The fabrication of binary CsPbBr3–CsPb2Br5 perovskites has attracted extensive interest.389–392 The presence of CsPb2Br5 can increase the PL lifetime of CsPbBr3 by decreasing nonradiative energy transfer to the trap states.389
As for the Cs4PbBr6, the PbBr64− octahedra are completely decoupled in all dimensions, which is often referred as the zero-dimensional (0D) perovskite.393–395 The rhombic prism hexabromide Cs4PbBr6 with individual PbBr64− clusters has a wide band gap of 3.95 eV.393 Additionally, the 0D perovskites present much better stability than the 3D perovskites. It is known that the 3D CsPbI3 rapidly degrades into a 1D orthorhombic structure within several days, while Cs4PbI6 maintains the original hexagonal phase under ambient conditions over one month without any sign of variation.393 Hence, the 0D Cs4PbX6 can be regarded as the best host for perovskite endotaxial systems. Interestingly, obtaining pure phase Cs4PbBr6 is always difficult since it usually coexists with CsPbBr3. And at an early stage, a stable green light of Cs4PbBr6 under UV excitation was observed. Therefore, the 0D Cs4PbBr6 has been regarded as a highly efficient, highly stable luminescent phosphor and superior to its 3D polymorphs. Recent studies revealed that the green PL in Cs4PbBr6 is attributed to the presence of a small amount of CsPbBr3 NCs, but the content of CsPbBr3 was too low to be detected by the XRD technique.396
Sargent et al.397 developed a theoretical model showing that the lattice of cubic-phase CsPbBr3 PQDs can well match the hexagonal-phase Cs4PbBr6 matrix. They further synthesized the CsPbBr3–Cs4PbBr6 endotaxial system as confirmed by XRD and high resolution transmission electron microscopy (HRTEM) studies. The bulk CsPbBr3–Cs4PbBr6 composites have reached PL QYs of 90% owing to the improved passivation and well dispersibility in the Cs4PbBr6 matrix (Fig. 26A). Further research revealed that embedding CsPbBr3 PQDs into the Cs4PbBr6 matrix could effectively enhance the oscillator strength, which may help radiative decay which competes with the Auger process and improve the performance of PeLED devices.398 Sun and coworkers399 demonstrated that the ligand-free CsPbBr3–Cs4PbBr6 composites ensure thermal stability and high temperature operation, which enable temperature-insensitive two-photon lasers. Zhong et al.400 synthesized centimeter-sized CsPbBr3–Cs4PbBr6 composites via an HBr-assisted slow cooling strategy. The obtained composites show PL QYs as high as 97%, and can be used as high-efficiency phosphors in wide color gamut pc-LEDs. Wang et al.401 prepared CsPbBr3–Cs4PbBr6 nanocrystals with PL QYs of 83%. The CsPbBr3 PQDs were homogeneously embedded in the Cs4PbBr6 matrix, which ensures the PL property and stability. Li et al.402 further optimized the synthesis and achieved the preparation of CsPbX3/Cs4PbX6 nanocrystals with core–shell structure in a single particle level. Sun et al.403 developed a scalable and high-yield recipe for the synthesis of Cs4PbBr6/CsPbBr3 composites with PLQYs of 95% (Fig. 26B). Overall, CsPbX3–Cs4PbX6 endotaxial systems hold great promise for photonic applications due to the superior optical property and stability.
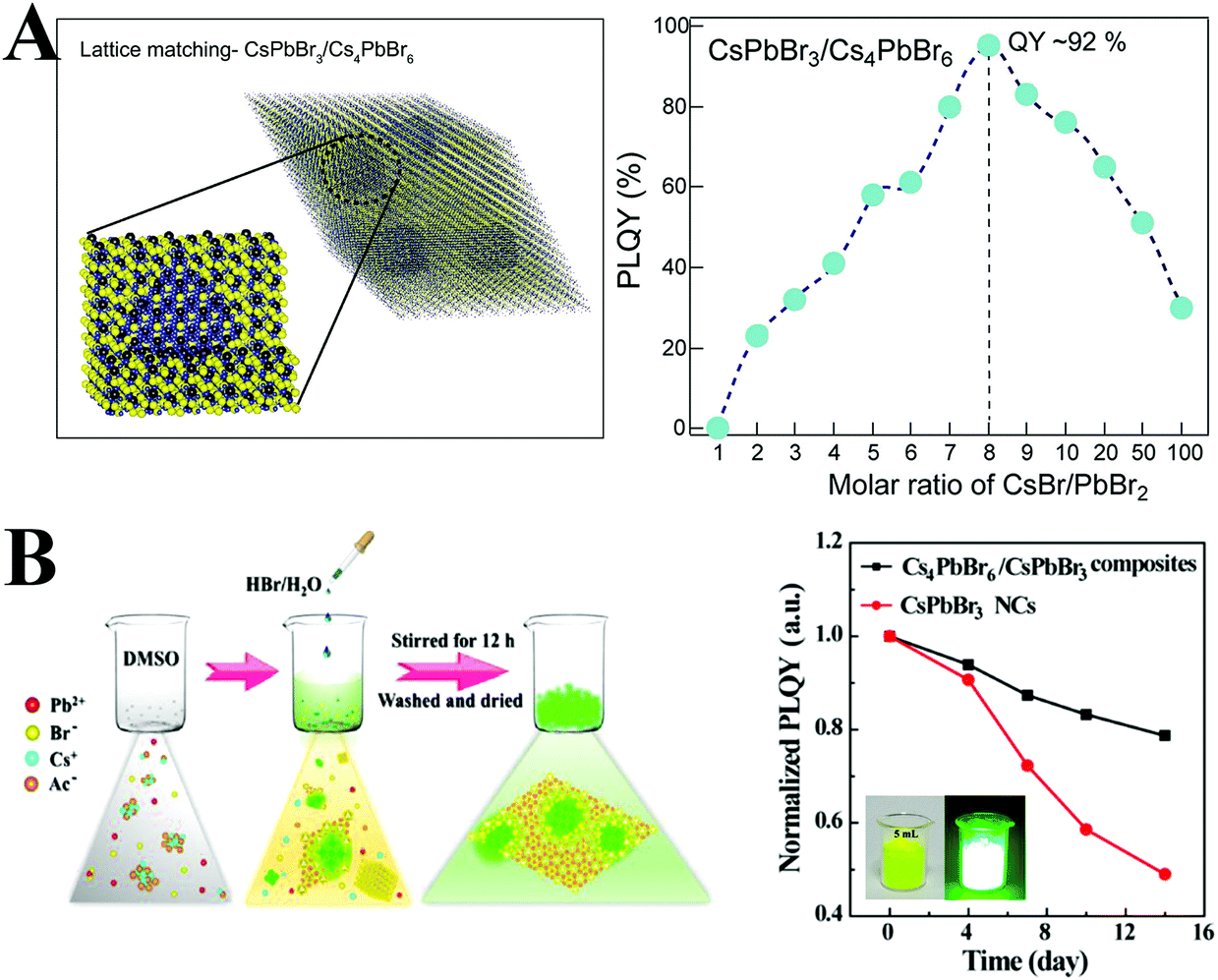 | ||
| Fig. 26 (A) Theoretical model for the material with various cubic perovskite inclusions in each rhombic prism matrix (left), and PLQYs of materials resulting from different ratios of CsBr and PbBr2 (right). Reprinted with permission from ref. 397. Copyright 2017, Wiley-VCH. (B) Schematic illustration of the room-temperature self-assembly process for the product of the Cs4PbBr6/CsPbBr3 composites (left), and PLQYs of Cs4PbBr6/CsPbBr3 composites and CsPbBr3 NCs exposed to air for different times (right). Reprinted with permission from ref. 403. Copyright 2018, American Chemical Society. | ||
Multi-step treatment. The stability of PQDs can be further enhanced by applying multiple protections. Liu et al.404 reported a three-step treatment of CsPbBr3 PQDs to improve the stability including (1) SDDA passivation, (2) mesoporous silica integration, and (3) PMMA coating. The final products present a high PLQY of 63% as well as high thermal and water resistance. Li et al.405 synthesized CsPbBr3-silica/alumina monoliths by employing compact SiO2 and Al2O3 layers to coat CsPbBr3 PQDs. Incorporation of silica and alumina layers greatly decreases the pinhole of films and minimizes water and oxygen permeability (Fig. 27A). Zeng and coworkers406 proposed a double-protection for CsPbBr3 PQDs by Cs4PbBr6 crystalline structures and SiO2 microspheres. The CsPbBr3@Cs4PbBr6@SiO2 composites show reversible fluorescence response between 30 and 150 °C. The thermal-modulated PL on/off function indicates their potential in thermal anti-counterfeiting. Recently, Xia et al.407 found that the surface defective layer of CsPbX3 PQDs can be dissolved in water by postsynthetic water treatment. Therefore, after separating the supernatant CsPbX3 PQDs hexane solution from water, their PL QYs are drastically improved. A small fraction of water still dissolved in the hexane solution, hence, they further used TMOS to run out of the residual water and form silica shell on the PQDs (Fig. 27B).
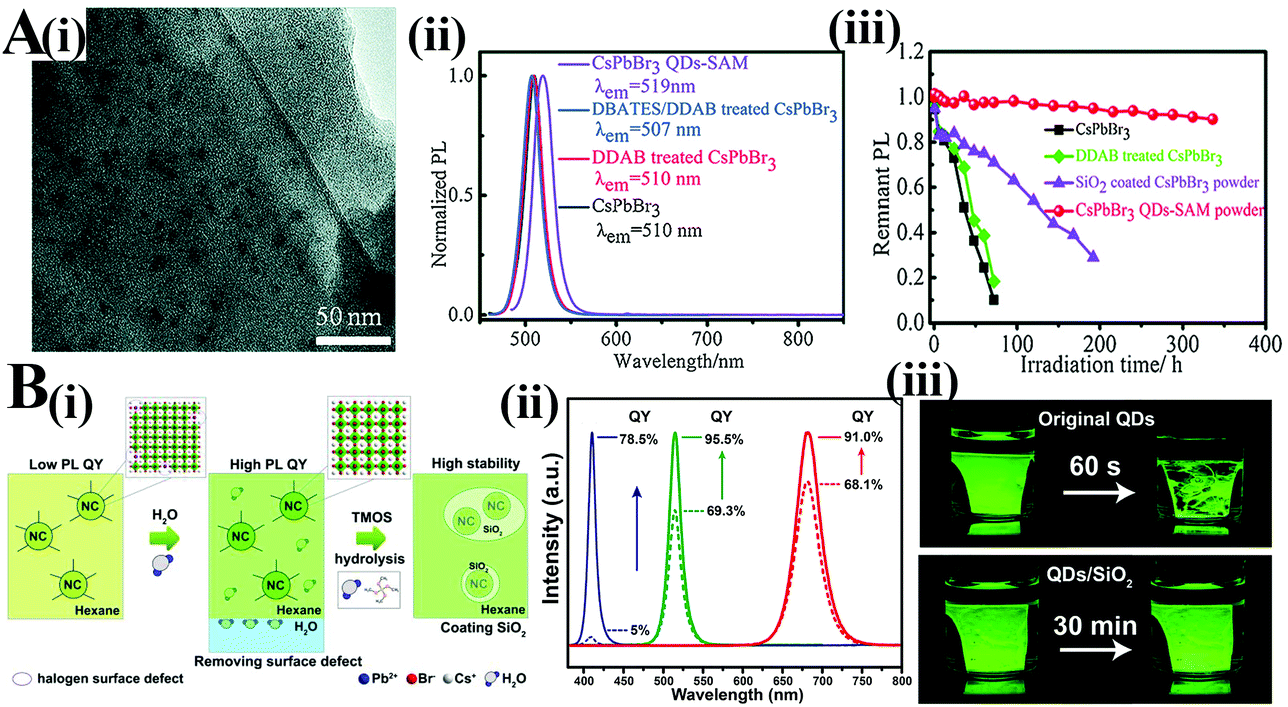 | ||
| Fig. 27 (A) CsPbBr3 QDs-SAM: (i) The TEM image of CsPbBr3 QDs-SAM. (ii) Evolution of PL spectra of CsPbBr3 PQDs after adding DDAB and DBATES. (iii) Photostability of the CsPbBr3 QDs-SAM powder, SiO2 coated CsPbBr3 PQDs powder, DDAB treated CsPbBr3 PQDs, and bare PQDs under illumination with a 470 nm LED light. Reprinted with permission from ref. 405. Copyright 2017, Wiley-VCH. (B, i) Illustration of the water treatment and silica encapsulation process of CsPbBr3 PQDs. (ii) PL emission spectra of the untreated (dashed line) and water treated (solid line) CsPbCl3 (blue), CsPbBr3 (green), and CsPbI3 (red) PQDs. (iii) Optical images of pristine CsPbBr3 and CsPbBr3/SiO2 films under UV illumination (λ = 365 nm) after 30 min of soaking in water. Reprinted with permission from ref. 407. Copyright 2018, American Chemical Society. | ||
Single-step treatment. A multi-step protection process can further improve the stability of PQDs. However, some stabilization processes can also damage luminescent PQDs. As a result, the stability is enhanced but the PL efficiency is sacrificed after tedious multi-step treatments. It is necessary to highlight some studies of applying protection materials as a function of both surface passivation and encapsulation by a single-step treatment. The synergetic effects could maintain the optical property and endow PQDs with high stability, indicating the superiority than a single protection process.
Zhang et al.74 used APTES to passivate the surface of PQDs, and simultaneously, the ligand layer formed a cross-linked silica layer over the CsPbX3 PQDs (Fig. 28A). The single protection material enables highly stable and efficient perovskite composites by passivating the surface and covering PQDs from contacting the environment directly. Combined with XPS and 1H NMR results, it is reasonable to speculate that the Pb(II) on the surface of QDs covalently coordinated with the amine group of ATPES molecules.306 González-Pedro and coworkers408 found that the PL of PQDs can be recovered after postsynthetic treatment with APTES. Further investigations revealed that the recovered PL originated from the ligands preferentially attached to the PQD surface through hydrogen bonds formed between silanol groups and bromide dangling bonds. Liu and coworkers409 employed reactive methacrylic acid (MAA) to passivate the CsPbX3 surface. UV-induced polymerization was conducted based on the free double bonds on the perovskite surface with POSS-appended methacrylate (MA-POSS), methyl methacrylate (MMA) monomer, and AIBN initiator. The copolymerization products can serve as luminescent inks and water-resistant films for pc-LEDs (Fig. 28B). Do et al.410 employed a novel inorganic polymer matrix, a dual-silicon nitride and silicon oxide nitride polymer, polysilazane (PSZ), to passivate the surface of PQDs and encapsulate CsPbBr3. The bifunctional inorganic PSZ layer can improve the thermal stability and maintain the PL efficiency of PQDs.
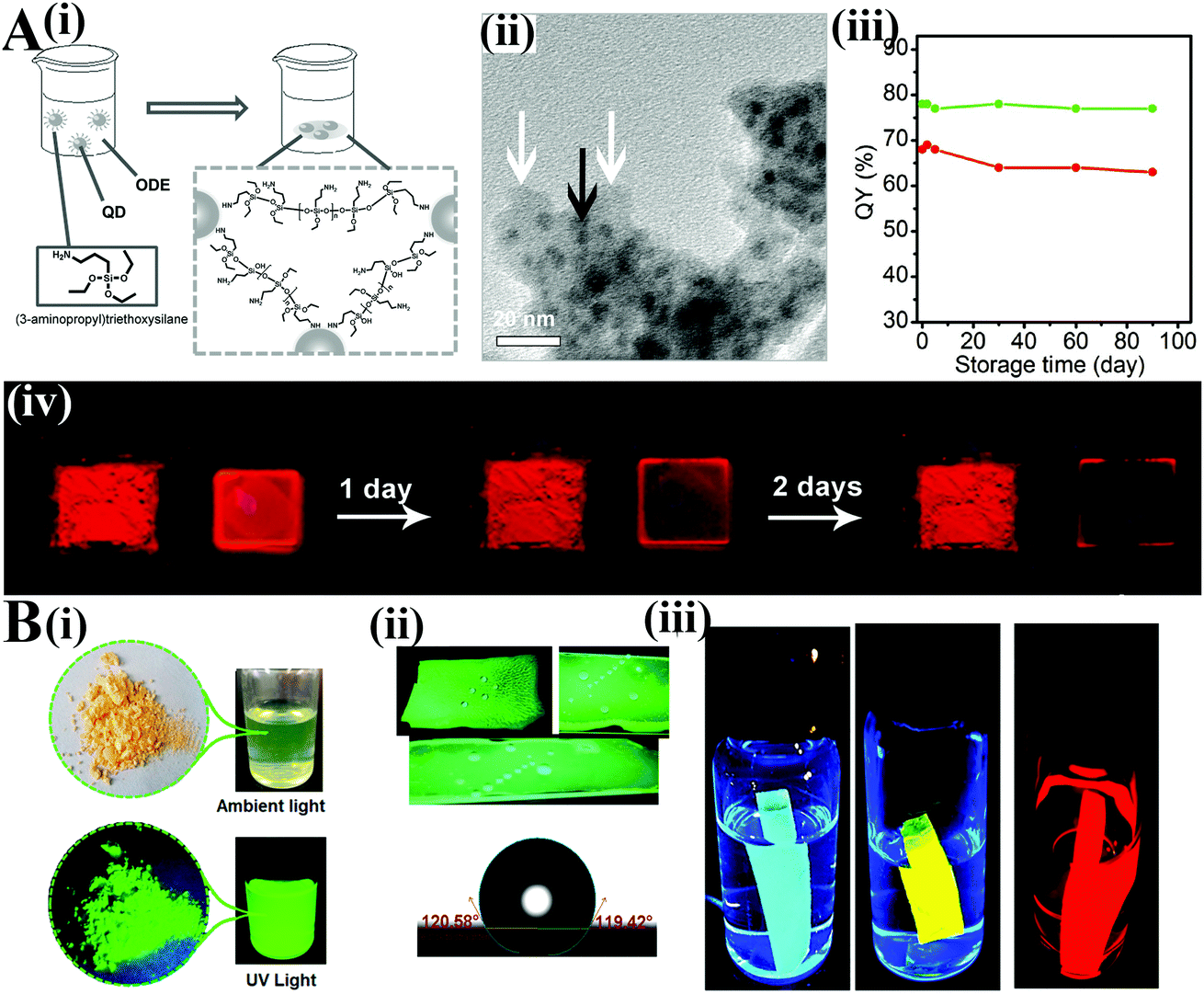 | ||
| Fig. 28 (A) QD/silica composites: (i) schematic illustration of formation of QD/silica composites. (ii) TEM images of CsPbBr3/silica. (iii) The PL QY stability of CsPbBr3/silica composites. (iv) The photographs of red QD/silica powder and the red OLA–QD film under UV light. Reprinted with permission from ref. 74. Copyright 2016, Wiley-VCH. (B) Characterization of PMPOPNC: (i) as-prepared powder of PMPOPNC under ambient light and UV light, respectively. (ii) Photos showing the hydrophobicity of the PMPOPNC film. (iii) Photographs of films of PMPOPNC–CsPb(Br/Cl)3, PMPOPNC–CsPb(Br/I)3, and PMPOPNC–CsPbI3 taken under UV irradiation after immersion in water for 60 days. Reprinted with permission from ref. 409. Copyright 2018, American Chemical Society. | ||
3.4. Device encapsulation
LED device encapsulation is a process of combining the InGaN chip and other packaging materials including phosphors and encapsulants to fabricate the final devices.66,70 Device encapsulation not only plays a crucial role in mechanical protection but also greatly affects the device performance.411As described above, thermal-induced PL quenching may arise from agglomeration and ligand loss. Homogeneously dispersing PQDs into some kinds of matrices can mitigate agglomeration. After separating PQDs from each other, the PL quenching is largely reversible below 450 K owing to the undamaged ligands, as confirmed by thermal cycling tests.220 The recoverable PL allows for high temperature processing such as the thermal curing of silicone resin. However, most thermal curing of silicone resin occurs at ∼420 K, which is close to the ligand loss temperature.
To minimize the thermal silicone curing-induced PL loss, some polymer matrices such as PMMA56 with high transmittance as well as a lower curing temperature (about 350 K) or UV curable optical adhesives76,175 hold great promise for perovskite-based device encapsulation. Employing transparent ceramics or glass ceramics to integrate phosphors, i.e. phosphor-in-glass (PiG), can be an effective strategy for device encapsulation.414 PiG could provide excellent heat and moisture resistance for phosphors. Recently, stable PQDs-glass emitters were prepared by the melt-growth PiG technique.415–419 The surrounded amorphous phosphate or borosilicate glass matrices greatly enhanced the stability towards water and oxygen exposure.
The fast ion-exchange takes place between two LHPs with varied halide components, which leads to the severe emission shift. Mixing green-emitting and red-emitting PQDs directly in the soft resin gel bring in the strong yellow-emitting PQDs.109 To block the anion exchange, changing surface ligands or embedding PQDs in matrices is necessary. Additionally, individually cast thin films with different color emitting PQDs could also effectively block the ion exchange.336
Reabsorption is a common issue for all kinds of QDs. The narrow bandwidth leads to a low color rendering index (CRI) value; hence encapsulating various color-emitting QDs is imperative to improve the CRI value. However, owing to the relatively small Stokes shift (20–80 meV for CsPbBr3420), these different color-emitting QDs suffer from severe reabsorption. Owing to the absorption spectra of red-emitting QDs covering the whole PL emission spectra of green-emitting QDs, the emitted green light would be reabsorbed by red-emitting QDs, which decreases the device performance. A multi-layer encapsulation technique was carried out to overcome the reabsorption. The red-emitting QDs/encapsulant were cast on the LED chips firstly, followed by the casting of green-emitting QDs/encapsulant. Thus the device performance can be significantly improved.164,400
Hydrophobic OA and OLA capped PQDs can homogeneously disperse in silicone resin due to the similar surface property, but PQDs with hydrophilic ligands such as PVP–PQDs cannot well disperse in silicone resin.413 A similar agglomeration was observed when embedding TOP capped QDs into the PMMA matrix owing to the poor compatibility, which accelerates the PL quenching.421 Thereby, selecting an appropriate encapsulant is important for improving device performance and duration.
A typical device architecture of pc-LED is shown in Fig. 29A. As can be seen from prototype pc-LEDs, the heat is easily transferred from the chip to the color converter, and accumulated in the converter layer due to the low thermal conductivity as well as the poor heat dissipation of the encapsulant–air interface. In this respect, a remote-type device was constructed to mitigate the thermal-induced PL quenching. Wang and coworkers422 prepared the red CsPbBrI2/PMMA film and stocked on a Y3Al5O12:Ce (YAG)-based PiG. The obtained bi-layered color converters were coupled with the blue chips to construct the remote-type white LEDs (wLEDs) (Fig. 29B). As expected, the luminescence intensity of PQDs increased along with the driven current increasing from 20 mA to 300 mA, which demonstrates the minimum thermal quenching by remote encapsulation (Fig. 29C and D). Zhang et al.74 synthesized small sized PQDs and embedded them in the silica matrix. The high temperature spectra show that the PQDs/silica composites lost nearly all emission upon heating to 350 K due to the easily reuniting small PQDs. They adopted a remote phosphor configuration to separate them from the hot LED chips. The remote-type wLEDs show preferable stability. The PL intensity presented a slight change after the wLED functioned for 10 h.
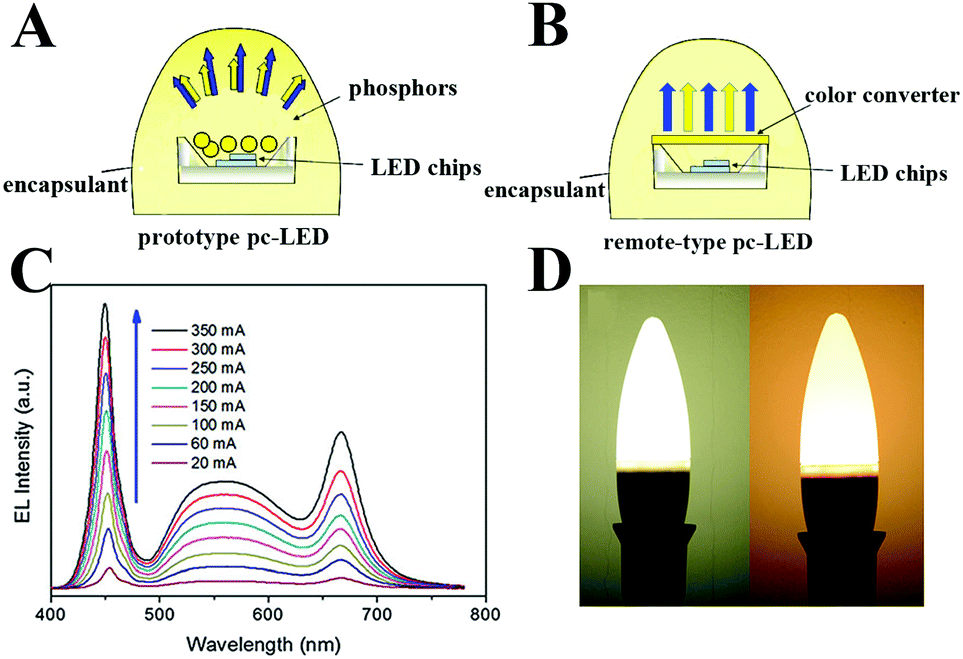 | ||
| Fig. 29 Schematic structure of prototype (A) and remote-type (B) pc-LEDs. (C) EL spectra of CsPbBrI2 PQD-modified pc-LEDs under various operational currents. (D) Photographs of pc-LEDs with and without the CsPbBrI2 QD film. (C and D) Reprinted with permission from ref. 422. Copyright 2016, The Royal Society of Chemistry. | ||
4. Application in pc-LEDs
Pc-LEDs are promising solid-state light sources and widely used in the fields of lighting and backlight down-converters for liquid crystal displays (LCDs) owing to their low cost, high electro-optical conversion efficiency, and environmentally friendly characteristics.66,68,69 The LED chip is a crucial component that can convert electricity into near UV or visible light. Owing to the highly reliable and low-cost InGaN chip, the LED chip has been widely recognized as an optimal light excitation source. Hence, utilizing an appropriate phosphor to convert the chip light greatly determines the performance of pc-LEDs.Luminous efficacy (LE) is a quantitative measure of the ability of how well a light source produces visible light, which has been considered to be the foremost index for pc-LEDs. The luminous flux reflects the varying sensitivity of human eyes to different wavelengths of light, which is different from radiant flux, the measure of the total power of electromagnetic radiation (including infrared, ultraviolet, and visible light). The luminous efficacy is defined as
 | (10) |
The CRI refers to the ability of a light source to reveal the colors of various objects faithfully in comparison with an ideal or natural light source. The color of objects is more faithful under a high CRI light source illumination. The CRI is associated with the spectrum of the light source. The CRI of sunlight is defined as 100, and the CRI of an incandescent lamp is close to 100, whereas the CRI of most pc-LEDs is around 80.423 The CRI and LE are two independent parameters to evaluate the device performance, but a high CRI usually brings in a low LE.424
Correlated color temperature (CCT) is the temperature of an ideal black-body radiator that radiates light of a color comparable to that of the light source. As the temperature rises, the black body radiates from reddish, orange, white, and bluish white light. Therefore, the color of a black body can be simply described by absolute temperature. By analogy, the color of nearly Planckian light sources including a tungsten lamp and sunlight can be judged by their temperature. But for the light source spectra that are not Planckian, such as pc-LEDs, color temperature is not a well-defined attribute; the concept of CCT was developed to map such light sources. CCT over 5000 K is called cool white light, while lower CCT is called warm white light. In general, a warm white light can promote relaxation, which is more useful for lighting applications.
Optimizing the LE, CRI, and CCT is crucial for promoting their applications in lighting and displays. Incorporating an appropriate phosphor can greatly adjust these parameters of pc-LEDs. Fig. 30 demonstrates the evolution of general pc-LEDs for five generations. Most types of commercial LEDs consist of blue chips and yellow-emitting YAG:Ce3+ phosphors, which are also named as the first generation of white-LEDs. However, excitation of the YAG:Ce3+ phosphor produces a cold white light with CCT above 5000 K and CRI below 80. To obtain warmer white light, red-emitting nitride phosphors with high thermal stability were explored. As a result, a warm white light with higher CRI can be obtained but the LE of the LED is slightly sacrificed. To meet the requirements for high-quality display applications, narrow-band K2SiF6:Mn4+ (KSF) was developed. KSF-based devices show wide color gamut in contrast to earlier generations of pc-LEDs. Recently, the Illuminating Engineering Society of North America proposed a new measurement system, a color fidelity index (CFI, Rf) and a color gamut index (CGI, Rg) as technical memorandum TM-30-2015 to accurately assess the color rendition and the color performances of fourth-generation pc-LEDs. However, it is found that trichromatic (blue, green, and red) warm white-light LEDs with a Ra score exceeding 90 are hard to reach a preferable Rf value over 90.86 High-quality pc-LEDs should be able to produce white colors in the range of warm to cool. Recently, color-reproducible and healthy white-light LEDs were developed for next-generation lighting.87 These smart white-light LEDs show tunable capability for the circadian effect, which is used for controlling melatonin secretion and more suitable for bio- and medical-related applications. The spectrum of QDs containing white-light LEDs should match daily variations in sunlight under a natural circadian rhythm.
 | ||
| Fig. 30 Schematic diagram of the development approaches for the first through fifth generations for general WLED lighting devices. Reproduced with permission from ref. 87. Copyright 2017, Springer Nature. | ||
Overall, PQDs can be employed as promising phosphors in pc-LEDs. But the poor stability limits their application. To enhance the stability of PQDs, the strategies including compositional engineering, surface engineering, matrix encapsulation and device encapsulation enable stable perovskite-based color converters for pc-LEDs. The reported improved stability and device performance are summarized in Table 2.
| Composites | PL QYs | Stability | Device structure | Luminous efficacy and device lifetime | Other index | Ref. |
|---|---|---|---|---|---|---|
| MAPbBr3 | 70% | Blue chip/KSF (silicone)/MAPbBr3 (PMMA) | 48 lm W−1 | 130% NTSC | 56 | |
| (4.9 mA) | (0.33, 0.27) | |||||
| CsPbCl3:Ce,Eu | 40% (35 h, UV) | UV chip (365 nm)/CsPbCl3:Ce, Eu (PMMA) | 24 lm W−1 | (0.32, 0.26) | 292 | |
| Unchanged (working over 288 h) | ||||||
| MAPbBr3 | 72% | 85% (120 h, UV) | Blue chip (455 nm)/MAPbBr3@POSS, KSF (silicone resin) | 38 lm W−1 | (0.30, 0.30) | 83 |
| CsPbX3@POSS | 62% (green) | Stable in water over 10 weeks | Blue chip (455 nm)/CsPbBr3@POSS, CsPbBr1.2I1.8 (silicone resin) | 14.1 lm W−1 | (0.349, 0.383) | 107 |
| 45% (red) | ||||||
| MAPbX3/SiO2 | 56% (blue) | 96% (72 h, air) | Blue chip (447 nm)/CsPbBr3/SiO2 CsPbI1.5Br3/SiO2 (PS) | 54 lm W−1 | (0.31, 0.34) | 306 |
| 95% (green) | 66% (30 min, 60 °C) | CCT: 6581 K | ||||
| 70% (red) | CRI: 85 | |||||
| 121% NTSC | ||||||
| CsPbBr3–TDPA | 68% | 80% (300 min, water) | Blue chip (453 nm)/CsPbBr3–TDPA, KSF (silicone resin) | 63 lm W−1 | (0.31, 0.29) | 307 |
| (20 mA) | CCT: 7072 K CRI: 83 | |||||
| 57 lm W−1 | ||||||
| (Working for 15 h) | ||||||
| CsPbX3–PLA | 33–90% | 80% (50h, working on LED) | Blue chip/CsPbBr3–PLA (PMMA)/(Ba, Ca, Sr)3SiO5:Eu | 62.93 lm W−1 | (0.38, 0.37) | 311 |
| (20 mA) | CCT: 4000 | |||||
| 50 lm W−1 | ||||||
| (Working for 50 h) | ||||||
| CsPbX3/PMMA | 45% | 75% (3 d, 30 °C air with 70% humidity) | Blue chip (460 nm)/CsPbX3/PMMA, CdSe QDs (silicone resin) | (0.34, 0.33) | 337 | |
| CRI: 89.2 | ||||||
| CsPbBr3/EC | 37.2% | 95% (150 h, air) | Blue chip/CsPbBr3/EC, Sr2Si5N8:Eu2+ (silicone) | 65.78 lm W−1 | CCT: 7540 K | 338 |
| (20 mA) | CRI: 67.93 | |||||
| CsPbX3@SBS | 11% (blue) | 100% (10 min, water) | Blue chip (450 nm)/CsPb(Br/I)3@SBS/CsPbBr3@SBS | 9 lm W−1 | 105% NTSC | 341 |
| 23% (green) | (10 mA) | |||||
| 12.2% (red) | ||||||
| MAPbBr3@PS | 48% | 95% (60 d, water) | Blue chip (450 nm)/red QDs@PS/MAPbBr3@PS (remote-type) | 95% rec. 2020 | 75 | |
| 65% (5 h, 100 °C) | ||||||
| CsPbX3/PS | 48% (green) | 70% (192 h, water) | Blue chip (410 nm)/CdSe QDs (ethoxyline resin)/CsPbBr3/PS fiber | (0.34, 0.33) | 343 | |
| 50% (2 h, 80 °C) | CCT: 5376 K | |||||
| CsPbBr3/EVA | 40.5% | 100% (192 h, air) | Blue chip (460 nm)/CsPbBr3/EVA/(Sr,Ca)AlSiN3:Eu2+ | 37.7 lm W−1 | CRI: 74.7 | 344 |
| ∼100% (720 h, water) | (20 mA) | CCT: 2347 K | ||||
| MAPbX3/PVDF | 38.4% (blue) 95% (green) 11% (red) | 92% (400 h, UV) | Blue chip/KSF (adhesive)/MAPbBr3/PVDF | 109 lm W−1 | 121% NTSC | 76 |
| (20 mA) | (0.272, 0.278) | |||||
| CsPbX3–CB | 80% (30 min, UV) | Blue chip/CsPbBr3–CB, CsPb(Br0.4I0.6)3–CB | (0.41, 0.37) | 349 | ||
| CsPb(Br/I)3@anthracene | 41.9% | 50% (100 °C) | UV chip (385 nm)/CsPb(Br0.6I0.4)3@anthracene (PMMA) | (0.35, 0.30) | 351 | |
| CsPbX3–PS | 44% (green) | 82% (24 h, water) | Blue chip/CsPbBr1.2I1.8–PS/CsPbBr3–PS | 353 | ||
| CsPbX3@PS | 68% (green) | 60% (16 d, UV) | Blue chip (450 nm)/CsPbBr3@SiO2, CsPb(Br0.4I0.6)3 (silicone resin) | (0.31, 0.30) | 356 | |
| 30% (30 d, water) | ||||||
| CsPbBr3–SiO2 | ∼100% (cooling from 100 °C to RT) | Blue chip (450 nm)/CsPbBr3–SiO2, CsPb(Br0.4I0.6)3 (silicone resin) | 30 lm W−1 | 113% NTSC | 109 | |
| 80% (96 h, UV) | (0.24, 0.28) | |||||
| CsPbBr3/SiO2 | 80% | 98% (10 h, UV) | Blue chip/CdSe NCs (PMMA)/CsPbBr3/SiO2 (PMMA) | 56 lm W−1 | 138% NTSC | 77 |
| (5 mA) | (0.30, 0.32) | |||||
| Unchanged after working for 1 h (1 mA) | ||||||
| CsPbBr3–SiO2 | 71.8% | 63.5% (80 °C) | Blue chip (450 nm)/CsPbBr3–SiO2, CsPbBr1.2I1.8 (silicone resin) | 35.4 lm W−1 | 126% NTSC | 366 |
| (20 mA) | (0.33, 0.36) CCT: 5623 K | |||||
| CsPbX3/SiO2 | 85% (green) | 95% (90 d, air) | Blue chip (458 nm)/CsPbBr3/SiO2, red QDs (PMMA) (remote-type) | 61.2 lm W−1 | 120% NTSC | 74 |
| 88% (red) | Stable (5 d, UV) | (20 mA) | (0.33, 0.33) | |||
| Little variation after working for 10 h | ||||||
| CsPbX3/SiO2 | 11.2% (blue) | 93% (30 d, air) | Blue chip (441 nm)/CsPbBr3/SiO2, CsPb(Br0.3/I0.7)3/SiO2 (PMMA) | 35.32 lm W−1 | (0.30, 0.31) | 369 |
| 82% (green) | Little variation after working for 40 h | |||||
| 73% (red) | ||||||
| CsPbX3–zeolites | 50% (80 °C) | Blue chip (460 nm)/CsPb(Br0.4I0.6)3, CsPbBr3–zeolites (silicone resin) | 2 lm W−1 | 127% NTSC (0.33, 0.36) | 376 | |
| (20 mA) | CCT: 5623 K | |||||
| CsPbX3–CaF2 | 50% (blue) | 56% (80 h, UV) | Blue chip (460 nm)/KSF (silicone resin)/CsPbBr3–CaF2 (PMMA) | 62.7 lm W−1 | 122% NTSC (0.33, 0.30) | 377 |
| 82% (green) | 60% (2 d, air with 100% humidity) | (20 mA) | ||||
| 66% (red) | 56.3 lm W−1 | |||||
| (Working for 8 h) | ||||||
| MAPbBr3/NaNO3 | 42% | 80% (14 h, UV) | Blue chip (450 nm)/KSF, MAPbBr3/NaNO3 (silicone resin) | 22.39 lm W−1 | 127% NTSC (0.28, 0.32) | 381 |
| 30% (300 min, 100 °C) | (20 mA) | CRI: 86.6 | ||||
| ∼11 lm W−1 | ||||||
| (Working for 4 h, 20 mA) | ||||||
| CsPbBr3@NH4Br | 64.21% | 40% (3.5 h, water) | Blue chip/CsPbBr3@NH4Br, KSF (silicone resin) | 90.6% rec. 2020 | 382 | |
| 96% (cooling from 100 °C to RT) | (0.331, 0.331) | |||||
| CsPbBr3/Cs4PbBr6 | 97% | Blue chip/KSF (silicone resin)/CsPbBr3/Cs4PbBr6 | 151 lm W−1 | 95% rec. 2020 | 400 | |
| (20 mA) | ||||||
| CsPbBr3/Cs4PbBr6 | 95% | 93% (80 min, blue light irradiation) | CsPbBr3/Cs4PbBr6, KSF | 73.8 lm W−1) (5 mA) | 403 | |
| 80% (14 d, air) | ||||||
| CsPbBr3–SDDA/SiO2/PMMA | 63% | ∼100% (cooling from 100 °C to RT) | Blue chip (458 nm)/CsPbBr3–SDDA/SiO2/PMMA, KSF (silicone resin) | 102% NTSC (0.271, 0.232) | 404 | |
| CsPbBr3/SiO2–Al2O3 | 90% | 90% (34 h, air) | Blue chip (455 nm)/CsPbBr3, CsPbBr3/SiO2–Al2O3 (PDMS) | 80.77 | 405 | |
| 90% (300 h, UV) | ||||||
| 70% (1 h, 100 °C) | ||||||
| CsPbX3–PMPOPNC | 67% (blue) | 95% (60 d, water) | Blue chip (460 nm)/red YAG (silicone resin)/CsPbX3–PMPOPNC (silicone resin) | 26.3 | (0.35, 0.32) | 409 |
| 85% (green) | >80% (120 °C) | (20 mA) | CCT: 4325 K | |||
| 69% (red) | 81% (156 h, UV) | CRI: 70.5 | ||||
| CsPbBr3/PSZ | 81.7% | >60% (70 h, UV) | Blue chip (455 nm)/CsPbBr3/PSZ, KSF (PMMA) | 138.6 | 111% NTSC | 410 |
| ∼100% (60 d, air) | (60 mA) | (0.31, 0.33) | ||||
| 52% (100 °C) | CCT: 6762 K | |||||
| CsPbBr3–phospho–silicate glass | 63% | 85% (5 d, UV) | Blue chip (460 nm)/CsPbBr3 glass/CaAlSiN3:Eu2+ (epoxy resin) | 50.5 | CCT: 3674 K | 415 |
| (20 mA) | CRI: 83.4 | |||||
| CsPbBrI2 | 65% | Blue chip (460 nm)/YAG:Ce PiG/CsPbBrI2 (PMMA) (remote-type) | 58 (20 mA) | (0.33, 0.32) | 422 | |
| CCT: 5907 K | ||||||
| CRI: 90 | ||||||
5. Conclusion and perspective
Lead halide perovskite quantum dots with intriguing optoelectronic properties have shown potential in a wide range of optoelectronic and photonic applications. Applying PQDs as phosphors for pc-LEDs to obtain excellent luminous efficacy and wide color gamut is very promising for next-generation high-quality lighting and vivid color display applications. At present, the poor stability is the major obstacle. There has been tremendous progress in the past few years in understanding the degradation mechanisms and enhancing the stability of PQDs.In this review, we summarized the degradation mechanism of perovskites in different conditions and tried to correlate the exterior-environment-induced factors with pc-LED device working mechanisms. Overall, ground-state PQDs are chemically stable, and only a small amount of the perovskite hydrated in normal conditions. The fluorescence loss is mainly ascribed to the agglomeration together with the increased surface trap. A better understanding of PL quenching promotes the development of various strategies for enhancing the stability of PQDs. According to the principle of these strategies, we can categorize them into four types.
The ionic radius plays a crucial role in the structural stability of perovskites. A large A cation cannot be accommodated into the interstice of the PbX6 framework, whereas a small A cation filling in the interstice will lead to the collapse of the 3D perovskite structure. Selecting appropriate or rationally mixing different A ions effectively stabilizes the 3D perovskite structure. Additionally, plenty of studies have shown that B-site doping can not only adjust the optoelectronic property but can also contribute to their stability. Although substitution of Pb2+ with some metal ions is possible to yield new deep defect states, an appropriate doping leads to improved optical performance and stability. Manipulating the perovskite composition can enhance the intrinsic stability of perovskites without sacrificing the photonic and electronic characteristics. At this stage, a full understanding of structural reorganization through component engineering at the atom level is highly expected.
The ill-passivated surface not only leads to the carriers’ nonradiative recombination through surface localized states but is also susceptible to water and oxygen molecules. Moreover, conventional OA and OLA ligands are easily lost during purification. Hence, incorporation of suitable surface ligands to replace OA/OLA can increase the PL and stability of PQDs. In this respect, using branched molecules with large steric hindrance or strengthening the interactions between the surface and ligands has made a huge success.
Matrix encapsulation is the most widely used strategy to enhance the perovskite stability. Particularly, a compact protective layer covering PQDs could endow them with superior water resistance. Some polymer matrices with carboxyl or amine groups can passivate their surface and result in enhanced PL performances, but their thermal stability is still limited. Most inorganics are dense and thermally stable, but a controllable formation of an inorganic layer to cover PQDs is difficult because of the high synthetic temperature and relatively complicated synthetic conditions. Hence, more efforts should be devoted in this direction.
Device encapsulation is a process of integrating luminescent powders with LED chips. In addition to the powders, employing free-standing perovskite films is a more convenient strategy. Additional deposition of encapsulant over perovskite–polymer films can further improve the duration. Furthermore, adopting a suitable encapsulant can homogeneously disperse PQDs and prevent the agglomeration-induced PL quenching. Meanwhile, encapsulants form a mechanical protection, which mitigates the damage by oxygen and moisture. The most difficult issue in this regard is the limited thermal stability of PQD-based phosphors. Although some methods are developed such as the PiG technique or a remote-type device architecture can relieve the thermal-induced degradation, the thermal stability should be further improved.
In summary, the poor stability of PQDs has aroused a wide concern, and simultaneously, many strategies have been developed to overcome this problem in the past few years. As a result, the stability of PQDs and the corresponding device lifetime of pc-LEDs have been improved; however, they are still far away from the commercial requirements. Further stabilizing perovskites and preventing their decomposition are needed. Among the various protection methods, the outstanding photoluminescence property can be retained through compositional or surface engineering routes, which show promise in the applications of photonic as well as optoelectronic devices. However, the electroconductivity is sacrificed after compact polymer or inorganics coating. Thus, the obtained composites are no longer suitable for optoelectronic devices. Although immense progress has been made in enhancing the stability against water and oxygen damage, their thermal stability is still limited. This situation is quiet like the core-only CdSe QDs. Therefore, a compact and monodispersed shell shielding that ensures thermal stability and simultaneously electroconductivity is more favorable. Our review on understanding of the origins of instability and the corresponding strategies for enhancing the stability is helpful to design stable perovskite light emitters. We hope that this review could encourage more researchers to devote additional efforts to address the stability of PQDs. And we are convinced that a highly luminescent and stable color-converter together with a high optical performance and long device lifetime pc-LED will come out in the near future.
List of acronyms and abbreviations
| ABS | Acrylonitrile butadiene styrene |
| ACN | Acetonitrile |
| ADBr | 2-Adamantylammonium bromide |
| AIBN | 2,2-Azobisisobutyronitrile |
| ALD | Atomic layer deposition |
| APTES | (3-Aminopropyl)triethoxysilane |
| BnOH | Benzyl alcohol |
| CA | Cellulose acetate |
| CB | Conduction band |
| CBB | Carboxybenzene |
| CCT | Correlated color temperature |
| CRI | Color rendering index |
| DDAB | Di-dodecyl dimethyl ammonium bromide |
| DFT | Density functional theory |
| DMF | Dimethyl formamide |
| DOSY | Diffusion Ordered NMR Spectroscopy |
| DPPA | Diphenylphosphinic acid |
| EC | Ethyl cellulose |
| EL | Electroluminescence |
| EQE | External quantum efficiency |
| EVA | Ethylene vinyl acetate |
| FA | Formamidinium, CH(NH2)2+ |
| FWHM | Full width at half maximum |
| GC | Gas chromatography |
| GO-g-PAA | Polyacrylic acid-grafted graphene oxide |
| HRTEM | High resolution transmission electron microscopy |
| Irgacure 819 | Bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide |
| LARP | Ligand-assisted reprecipitation |
| LCDs | Liquid crystal displays |
| LE | Luminous efficacy |
| LEDs | Light-emitting diodes |
| LHPs | Lead halide perovskites |
| MA | Methylammonium, CH3NH3+ |
| MAA | Methacrylic acid |
| MMA | Methyl methacrylate |
| MA-POSS | POSS-appended methacrylate |
| MOF | Metal–organic framework |
| NCs | Nanocrystals |
| NH2-POSS | Polyhedral silsesquioxane-[3-(2-aminoethyl)amino]propylheptaisobutyl substituted |
| NIR | Near-infrared |
| NMR | Nuclear magnetic resonance |
| NPs | Nanoparticles |
| NSs | Nanosheets |
| NTSC | National Television System Committee |
| OA | Oleic acid |
| OLA | Oleylamine |
| OLEDs | Organic LEDs |
| P2VP | Poly-2-vinylpyridine |
| PC | Polycarbonate |
| PCE | Power conversion efficiency |
| pc-LEDs | Phosphor-converted light-emitting diodes |
| PDs | Photodetectors |
| PDMS | Polydimethylsiloxane |
| PeLEDs | Perovskite LEDs |
| PEA | Phenylethylamine, C8H9NH3 |
| PEDOT | Poly(3,4-ethylenedioxythiophene) |
| PiG | Phosphor-in-glass |
| PL | Photoluminescence |
| PLA | Poly(lactic acid) |
| PLMA | Poly(lauryl methacrylate) |
| PL QYs | Photoluminescence quantum yields |
| PMA | Poly(maleic anhydride-alt-1-octadecene) |
| PMMA | Polymethyl methacrylate |
| POSS | Polyhedral oligomeric silsesquioxane |
| PQDs | Perovskite quantum dots |
| PS | Polystyrene |
| PS-b-PEO | Polystyrene-b-poly(ethyl oxide) |
| PSZ | Polysilazane |
| PVs | Photovoltaics |
| PVC | Polyvinyl chloride |
| PVDF | Polyvinylidene fluoride |
| PVP | Polyvinylpyrrolidone |
| QLEDs | QD LEDs |
| RE | Rare earth |
| RT | Room temperature |
| SBS | Poly(styrene-butadiene-styrene) |
| SDDA | Didodecyl dimethylammonium sulfide |
| SEBS | Poly(styrene-ethylene-butylenestyrene) |
| SEC | Spectro-electrochemical |
| SH-β-CD | Mercapto-β-cyclodextrin |
| TA | Transient absorption |
| TDPA | 1-Tetradecylphosphonic acid |
| TerEDOT | 2,2′,5′,2′′-Ter-3,4-ethylenedioxythiophene |
| TEOS | Tetraethyl orthosilicate |
| TGA | Thermogravimetric analysis |
| TMA | Trimethylaluminum |
| TMOS | Tetramethyl orthosilicate |
| TMPPA | Bis-(2,2,4-trimethylpentyl)phosphinic acid |
| ToF-SIMS | Time-of-flight secondary ion-mass spectrometry |
| TOP | Trioctylphosphine |
| TOPO | Trioctylphosphine oxide |
| V18 | 4-Vinylbenzyl-dimethyloctadecylammonium chloride |
| VB | Valance band |
| wLEDs | White LEDs |
| XANES | X-ray absorption near-edge spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
| YAG | Y3Al5O12 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (NSFC 51772288, 51472231, 51720105015, and 51750110511), Science and Technology Development Planning Project of Jilin Province (20170101187JC and 20170414003GH), Jiangmen Innovative Research Team Program (2017), Major program of basic research and applied research of Guangdong Province (2017KZDXM083), the Key Research Program of Frontier Sciences, CAS (Grant No. YZDY-SSW-JSC018), and the CAS-Croucher Funding Scheme for Joint Laboratories (CAS18204).References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395 CrossRef CAS PubMed.
- D. W. de Quilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683–686 CrossRef CAS PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312–316 CrossRef CAS PubMed.
- F. Ye, H. Chen, F. Xie, W. Tang, M. Yin, J. He, E. Bi, Y. Wang, X. Yang and L. Han, Energy Environ. Sci., 2016, 9, 2295–2301 RSC.
- N. Li, Z. Zhu, C.-C. Chueh, H. Liu, B. Peng, A. Petrone, X. Li, L. Wang and A. K. Y. Jen, Adv. Energy Mater., 2017, 7, 1601307 CrossRef.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- T. M. Brenner, D. A. Egger, L. Kronik, G. Hodes and D. Cahen, Nat. Rev. Mater., 2016, 1, 15007 CrossRef CAS.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
- Z.-K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS PubMed.
- H. Cho, S.-H. Jeong, M.-H. Park, Y.-H. Kim, C. Wolf, C.-L. Lee, J. H. Heo, A. Sadhanala, N. Myoung and S. Yoo, Science, 2015, 350, 1222–1225 CrossRef CAS PubMed.
- M. Yuan, L. N. Quan, R. Comin, G. Walters, R. Sabatini, O. Voznyy, S. Hoogland, Y. Zhao, E. M. Beauregard and P. Kanjanaboos, Nat. Nanotechnol., 2016, 11, 872 CrossRef CAS PubMed.
- N. Wang, L. Cheng, R. Ge, S. Zhang, Y. Miao, W. Zou, C. Yi, Y. Sun, Y. Cao, R. Yang, Y. Wei, Q. Guo, Y. Ke, M. Yu, Y. Jin, Y. Liu, Q. Ding, D. Di, L. Yang, G. Xing, H. Tian, C. Jin, F. Cao, R. H. Friend, J. Wang and W. Huang, Nat. Photonics, 2016, 10, 699–704 CrossRef CAS.
- Y. Ling, Y. Tian, X. Wang, J. C. Wang, J. M. Knox, F. Perez-Orive, Y. Du, L. Tan, K. Hanson, B. Ma and H. Gao, Adv. Mater., 2016, 28, 8983–8989 CrossRef CAS PubMed.
- L. Zhang, X. Yang, Q. Jiang, P. Wang, Z. Yin, X. Zhang, H. Tan, Y. M. Yang, M. Wei, B. R. Sutherland, E. H. Sargent and J. You, Nat. Commun., 2017, 8, 15640 CrossRef CAS PubMed.
- W. Zou, R. Li, S. Zhang, Y. Liu, N. Wang, Y. Cao, Y. Miao, M. Xu, Q. Guo, D. Di, L. Zhang, C. Yi, F. Gao, R. H. Friend, J. Wang and W. Huang, Nat. Commun., 2018, 9, 608 CrossRef PubMed.
- J. Xing, Y. Zhao, M. Askerka, L. N. Quan, X. Gong, W. Zhao, J. Zhao, H. Tan, G. Long, L. Gao, Z. Yang, O. Voznyy, J. Tang, Z.-H. Lu, Q. Xiong and E. H. Sargent, Nat. Commun., 2018, 9, 3541 CrossRef PubMed.
- L. N. Quan, F. P. García de Arquer, R. P. Sabatini and E. H. Sargent, Adv. Mater., 2018, 30, 1801996 CrossRef PubMed.
- M. Bidikoudi, E. Fresta and R. D. Costa, Chem. Commun., 2018, 54, 8150–8169 RSC.
- H. Zhu, Y. Fu, F. Meng, X. Wu, Z. Gong, Q. Ding, M. V. Gustafsson, M. T. Trinh, S. Jin and X. Zhu, Nat. Mater., 2015, 14, 636–642 CrossRef CAS PubMed.
- S. A. Veldhuis, P. P. Boix, N. Yantara, M. Li, T. C. Sum, N. Mathews and S. G. Mhaisalkar, Adv. Mater., 2016, 28, 6804–6834 CrossRef CAS PubMed.
- X. Tang, Z. Hu, W. Yuan, W. Hu, H. Shao, D. Han, J. Zheng, J. Hao, Z. Zang, J. Du, Y. Leng, L. Fang and M. Zhou, Adv. Opt. Mater., 2017, 5, 1600788 CrossRef.
- Y. Wang, X. Li, V. Nalla, H. Zeng and H. Sun, Adv. Funct. Mater., 2017, 27, 1605088 CrossRef.
- W. Zheng, P. Huang, Z. Gong, D. Tu, J. Xu, Q. Zou, R. Li, W. You, J.-C. G. Bünzli and X. Chen, Nat. Commun., 2018, 9, 3462 CrossRef PubMed.
- L. Jiang, R. Liu, R. Su, Y. Yu, H. Xu, Y. Wei, Z.-K. Zhou and X. Wang, Nanoscale, 2018, 10, 13565–13571 RSC.
- Y. C. Kim, K. H. Kim, D.-Y. Son, D.-N. Jeong, J.-Y. Seo, Y. S. Choi, I. T. Han, S. Y. Lee and N.-G. Park, Nature, 2017, 550, 87 CrossRef CAS PubMed.
- W. Wei, Y. Zhang, Q. Xu, H. Wei, Y. Fang, Q. Wang, Y. Deng, T. Li, A. Gruverman, L. Cao and J. Huang, Nat. Photonics, 2017, 11, 315 CrossRef CAS.
- S. Shrestha, R. Fischer, G. J. Matt, P. Feldner, T. Michel, A. Osvet, I. Levchuk, B. Merle, S. Golkar, H. Chen, S. F. Tedde, O. Schmidt, R. Hock, M. Rührig, M. Göken, W. Heiss, G. Anton and C. J. Brabec, Nat. Photonics, 2017, 11, 436 CrossRef CAS.
- Q. Chen, J. Wu, X. Ou, B. Huang, J. Almutlaq, A. A. Zhumekenov, X. Guan, S. Han, L. Liang, Z. Yi, J. Li, X. Xie, Y. Wang, Y. Li, D. Fan, D. B. L. Teh, A. H. All, O. F. Mohammed, O. M. Bakr, T. Wu, M. Bettinelli, H. Yang, W. Huang and X. Liu, Nature, 2018, 561, 88–93 CrossRef CAS PubMed.
- P. Büchele, M. Richter, S. F. Tedde, G. J. Matt, G. N. Ankah, R. Fischer, M. Biele, W. Metzger, S. Lilliu, O. Bikondoa, J. E. Macdonald, C. J. Brabec, T. Kraus, U. Lemmer and O. Schmidt, Nat. Photonics, 2015, 9, 843–848 CrossRef.
- J. H. Heo, D. H. Shin, J. K. Park, D. H. Kim, S. J. Lee and S. H. Im, Adv. Mater., 2018, 30, 1801743 CrossRef PubMed.
- L. Dou, Y. M. Yang, J. You, Z. Hong, W.-H. Chang, G. Li and Y. Yang, Nat. Commun., 2014, 5, 5404 CrossRef CAS PubMed.
- M. I. Saidaminov, V. Adinolfi, R. Comin, A. L. Abdelhady, W. Peng, I. Dursun, M. Yuan, S. Hoogland, E. H. Sargent and O. M. Bakr, Nat. Commun., 2015, 6, 8724 CrossRef CAS PubMed.
- Y. Guo, C. Liu, H. Tanaka and E. Nakamura, J. Phys. Chem. Lett., 2015, 6, 535–539 CrossRef CAS PubMed.
- W. Deng, L. Huang, X. Xu, X. Zhang, X. Jin, S.-T. Lee and J. Jie, Nano Lett., 2017, 17, 2482–2489 CrossRef CAS PubMed.
- H. Wang and D. H. Kim, Chem. Soc. Rev., 2017, 46, 5204–5236 RSC.
- Y. Dong, Y. Zou, J. Song, X. Song and H. Zeng, J. Mater. Chem. C, 2017, 5, 11369–11394 RSC.
- L. Polavarapu, B. Nickel, J. Feldmann and A. S. Urban, Adv. Energy Mater., 2017, 7, 1700267 CrossRef.
- S.-T. Ha, R. Su, J. Xing, Q. Zhang and Q. Xiong, Chem. Sci., 2017, 8, 2522–2536 RSC.
- Z. Shi, Y. Li, Y. Zhang, Y. Chen, X. Li, D. Wu, T. Xu, C. Shan and G. Du, Nano Lett., 2017, 17, 313–321 CrossRef CAS PubMed.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- A. B. Wong, M. Lai, S. W. Eaton, Y. Yu, E. Lin, L. Dou, A. Fu and P. Yang, Nano Lett., 2015, 15, 5519–5524 CrossRef CAS PubMed.
- Y. Liu, M. Guo, S. Dong, X. Jiao, T. Wang and D. Chen, J. Mater. Chem. C, 2018, 6, 7797–7802 RSC.
- Y. Tong, B. J. Bohn, E. Bladt, K. Wang, P. Müller-Buschbaum, S. Bals, A. S. Urban, L. Polavarapu and J. Feldmann, Angew. Chem., Int. Ed., 2017, 56, 13887–13892 CrossRef CAS PubMed.
- Y. Ling, Z. Yuan, Y. Tian, X. Wang, J. C. Wang, Y. Xin, K. Hanson, B. Ma and H. Gao, Adv. Mater., 2016, 28, 305–311 CrossRef CAS PubMed.
- Q. Liao, K. Hu, H. Zhang, X. Wang, J. Yao and H. Fu, Adv. Mater., 2015, 27, 3405–3410 CrossRef CAS PubMed.
- Y. Tong, E.-P. Yao, A. Manzi, E. Bladt, K. Wang, M. Döblinger, S. Bals, P. Müller-Buschbaum, A. S. Urban, L. Polavarapu and J. Feldmann, Adv. Mater., 2018, 30, 1801117 CrossRef PubMed.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang, C. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef CAS PubMed.
- Y. Cao, N. Wang, H. Tian, J. Guo, Y. Wei, H. Chen, Y. Miao, W. Zou, K. Pan, Y. He, H. Cao, Y. Ke, M. Xu, Y. Wang, M. Yang, K. Du, Z. Fu, D. Kong, D. Dai, Y. Jin, G. Li, H. Li, Q. Peng, J. Wang and W. Huang, Nature, 2018, 562, 249–253 CrossRef PubMed.
- Q. Shan, J. Song, Y. Zou, J. Li, L. Xu, J. Xue, Y. Dong, B. Han, J. Chen and H. Zeng, Small, 2017, 13, 1701770 CrossRef PubMed.
- C. Wu, Y. Zou, T. Wu, M. Ban, V. Pecunia, Y. Han, Q. Liu, T. Song, S. Duhm and B. Sun, Adv. Funct. Mater., 2017, 27, 1700338 CrossRef.
- H.-C. Wang, Z. Bao, H.-Y. Tsai, A.-C. Tang and R.-S. Liu, Small, 2018, 14, 1702433 CrossRef PubMed.
- A. Swarnkar, R. Chulliyil, V. K. Ravi, M. Irfanullah, A. Chowdhury and A. Nag, Angew. Chem., 2015, 127, 15644–15648 CrossRef.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- F. Zhang, H. Zhong, C. Chen, X.-g. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- J. Li, L. Xu, T. Wang, J. Song, J. Chen, J. Xue, Y. Dong, B. Cai, Q. Shan, B. Han and H. Zeng, Adv. Mater., 2017, 29, 1603885 CrossRef PubMed.
- D. Han, M. Imran, M. Zhang, S. Chang, X.-g. Wu, X. Zhang, J. Tang, M. Wang, S. Ali, X. Li, G. Yu, J. Han, L. Wang, B. Zou and H. Zhong, ACS Nano, 2018, 12, 8808–8816 CrossRef CAS PubMed.
- J. Song, J. Li, L. Xu, J. Li, F. Zhang, B. Han, Q. Shan and H. Zeng, Adv. Mater., 2018, 30, 1800764 CrossRef PubMed.
- K. Hoshi, T. Chiba, J. Sato, Y. Hayashi, Y. Takahashi, H. Ebe, S. Ohisa and J. Kido, ACS Appl. Mater. Interfaces, 2018, 10, 24607–24612 CrossRef CAS PubMed.
- T. Chiba, K. Hoshi, Y.-J. Pu, Y. Takeda, Y. Hayashi, S. Ohisa, S. Kawata and J. Kido, ACS Appl. Mater. Interfaces, 2017, 9, 18054–18060 CrossRef CAS PubMed.
- B. Geffroy, P. Le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- W. K. Bae, J. Kwak, J. W. Park, K. Char, C. Lee and S. Lee, Adv. Mater., 2009, 21, 1690–1694 CrossRef CAS.
- X. Dai, Z. Zhang, Y. Jin, Y. Niu, H. Cao, X. Liang, L. Chen, J. Wang and X. Peng, Nature, 2014, 515, 96–99 CrossRef CAS PubMed.
- P. Pust, V. Weiler, C. Hecht, A. Tücks, A. S. Wochnik, A.-K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891–896 CrossRef CAS PubMed.
- G. Li, Y. Tian, Y. Zhao and J. Lin, Chem. Soc. Rev., 2015, 44, 8688–8713 RSC.
- L. Wang, R.-J. Xie, T. Suehiro, T. Takeda and N. Hirosaki, Chem. Rev., 2018, 118, 1951–2009 CrossRef CAS PubMed.
- Z. Xia and A. Meijerink, Chem. Soc. Rev., 2017, 46, 275–299 RSC.
- N. C. George, K. A. Denault and R. Seshadri, Annu. Rev. Mater. Res., 2013, 43, 481–501 CrossRef CAS.
- C. C. Lin and R.-S. Liu, J. Phys. Chem. Lett., 2011, 2, 1268–1277 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- C. Sun, Y. Zhang, C. Ruan, C. Yin, X. Wang, Y. Wang and W. W. Yu, Adv. Mater., 2016, 28, 10088–10094 CrossRef CAS PubMed.
- Y. Wang, J. He, H. Chen, J. Chen, R. Zhu, P. Ma, A. Towers, Y. Lin, A. J. Gesquiere, S. T. Wu and Y. Dong, Adv. Mater., 2016, 28, 10710–10717 CrossRef CAS PubMed.
- Q. Zhou, Z. Bai, W. g. Lu, Y. Wang, B. Zou and H. Zhong, Adv. Mater., 2016, 28, 9163–9168 CrossRef CAS PubMed.
- H. Hu, L. Wu, Y. Tan, Q. Zhong, M. Chen, Y. Qiu, D. Yang, B. Sun, Q. Zhang and Y. Yin, J. Am. Chem. Soc., 2017, 140, 406–412 CrossRef PubMed.
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656–2664 CrossRef CAS PubMed.
- Y. Tong, E. Bladt, M. F. Aygüler, A. Manzi, K. Z. Milowska, V. A. Hintermayr, P. Docampo, S. Bals, A. S. Urban, L. Polavarapu and J. Feldmann, Angew. Chem., Int. Ed., 2016, 55, 13887–13892 CrossRef CAS PubMed.
- H. Liu, Z. Wu, H. Gao, J. Shao, H. Zou, D. Yao, Y. Liu, H. Zhang and B. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 42919–42927 CrossRef CAS PubMed.
- S. W. Dai, B. W. Hsu, C. Y. Chen, C. A. Lee, H. Y. Liu, H. F. Wang, Y. C. Huang, T. L. Wu, A. Manikandan, R. M. Ho, C.-S. Tsao, C.-H. Cheng, Y.-L. Chueh and H.-W. Lin, Adv. Mater., 2018, 30, 1705532 CrossRef PubMed.
- D. N. Minh, J. Kim, J. Hyon, J. H. Sim, H. H. Sowlih, C. Seo, J. Nam, S. Eom, S. Suk, S. Lee, E. Kim and Y. Kang, Chem. Mater., 2017, 29, 5713–5719 CrossRef CAS.
- H. Huang, Q. Xue, B. Chen, Y. Xiong, J. Schneider, C. Zhi, H. Zhong and A. L. Rogach, Angew. Chem., Int. Ed., 2017, 56, 9571–9576 CrossRef CAS PubMed.
- S. Chang, Z. Bai and H. Zhong, Adv. Opt. Mater., 2018, 6, 1800380 CrossRef.
- T. Guner and M. M. Demir, Phys. Status Solidi A, 2018, 215, 1800120 CrossRef.
- H. C. Yoon, H. Kang, S. Lee, J. H. Oh, H. Yang and Y. R. Do, ACS Appl. Mater. Interfaces, 2016, 8, 18189–18200 CrossRef CAS PubMed.
- H. C. Yoon, J. H. Oh, S. Lee, J. B. Park and Y. R. Do, Sci. Rep., 2017, 7, 2808 CrossRef PubMed.
- X. Dai, Y. Deng, X. Peng and Y. Jin, Adv. Mater., 2017, 29, 1607022 CrossRef PubMed.
- Y. E. Panfil, M. Oded and U. Banin, Angew. Chem., Int. Ed., 2018, 57, 4274–4295 CrossRef CAS PubMed.
- N. H. Tiep, Z. Ku and H. J. Fan, Adv. Energy Mater., 2016, 6, 1501420 CrossRef.
- J. You, L. Meng, T.-B. Song, T.-F. Guo, Y. M. Yang, W.-H. Chang, Z. Hong, H. Chen, H. Zhou, Q. Chen, Y. Liu, N. D. Marco and Y. Yang, Nat. Nanotechnol., 2016, 11, 75–81 CrossRef CAS PubMed.
- H. Cho, Y. H. Kim, C. Wolf, H. D. Lee and T. W. Lee, Adv. Mater., 2018, 30, 1704587 CrossRef PubMed.
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 RSC.
- T. Leijtens, G. E. Eperon, N. K. Noel, S. N. Habisreutinger, A. Petrozza and H. J. Snaith, Adv. Energy Mater., 2015, 5, 1500963 CrossRef.
- L. Gomez, C. de Weerd, J. L. Hueso and T. Gregorkiewicz, Nanoscale, 2017, 9, 631–636 RSC.
- F. Lang, O. Shargaieva, V. V. Brus, H. C. Neitzert, J. Rappich and N. H. Nickel, Adv. Mater., 2018, 30, 1702905 CrossRef PubMed.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- X. Li, F. Cao, D. Yu, J. Chen, Z. Sun, Y. Shen, Y. Zhu, L. Wang, Y. Wei, Y. Wu and H. Zeng, Small, 2017, 13, 1603996 CrossRef PubMed.
- Y. Kim, E. Yassitepe, O. Voznyy, R. Comin, G. Walters, X. Gong, P. Kanjanaboos, A. F. Nogueira and E. H. Sargent, ACS Appl. Mater. Interfaces, 2015, 7, 25007–25013 CrossRef CAS PubMed.
- S. Yang, Y. Wang, P. Liu, Y.-B. Cheng, H. J. Zhao and H. G. Yang, Nat. Energy, 2016, 1, 15016 CrossRef CAS.
- R. Roesch, T. Faber, E. von Hauff, T. M. Brown, M. Lira-Cantu and H. Hoppe, Adv. Energy Mater., 2015, 5, 1501924 Search PubMed.
- S. Zou, Y. Liu, J. Li, C. Liu, R. Feng, F. Jiang, Y. Li, J. Song, H. Zeng, M. Hong and X. Chen, J. Am. Chem. Soc., 2017, 139, 11443–11450 CrossRef CAS PubMed.
- G. P. Nagabhushana, R. Shivaramaiah and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7717–7721 CrossRef CAS PubMed.
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 RSC.
- M. Kulbak, S. Gupta, N. Kedem, I. Levine, T. Bendikov, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2015, 7, 167–172 CrossRef PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- H. Huang, B. Chen, Z. Wang, T. F. Hung, A. S. Susha, H. Zhong and A. L. Rogach, Chem. Sci., 2016, 7, 5699–5703 RSC.
- V. K. Ravi, R. A. Scheidt, A. Nag, M. Kuno and P. V. Kamat, ACS Energy Lett., 2018, 3, 1049–1055 CrossRef CAS.
- H. C. Wang, S. Y. Lin, A. C. Tang, B. P. Singh, H. C. Tong, C. Y. Chen, Y. C. Lee, T. L. Tsai and R. S. Liu, Angew. Chem., Int. Ed., 2016, 55, 7924–7929 CrossRef CAS PubMed.
- Q. A. Akkerman, V. D’Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- X. Chen, H. Hu, Z. Xia, W. Gao, W. Gou, Y. Qu and Y. Ma, J. Mater. Chem. C, 2017, 5, 309–313 RSC.
- C. Guhrenz, A. Benad, C. Ziegler, D. Haubold, N. Gaponik and A. Eychmüller, Chem. Mater., 2016, 28, 9033–9040 CrossRef CAS.
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS PubMed.
- D. Yang, X. Li and H. Zeng, Adv. Mater. Interfaces, 2018, 5, 1701662 CrossRef.
- S. Huang, Z. Li, B. Wang, N. Zhu, C. Zhang, L. Kong, Q. Zhang, A. Shan and L. Li, ACS Appl. Mater. Interfaces, 2017, 9, 7249–7258 CrossRef CAS PubMed.
- K. Chen, S. Schünemann, S. Song and H. Tüysüz, Chem. Soc. Rev., 2018, 47, 7045–7077 RSC.
- Z. Cheng and J. Lin, CrystEngComm, 2010, 12, 2646–2662 RSC.
- K. Huang, R. S. Tichy and J. B. Goodenough, J. Am. Ceram. Soc., 1998, 81, 2565–2575 CrossRef CAS.
- D. Lebeugle, D. Colson, A. Forget and M. Viret, Appl. Phys. Lett., 2007, 91, 022907 CrossRef.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- J. S. Manser, J. A. Christians and P. V. Kamat, Chem. Rev., 2016, 116, 12956–13008 CrossRef CAS PubMed.
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC.
- Z. Shi, J. Guo, Y. Chen, Q. Li, Y. Pan, H. Zhang, Y. Xia and W. Huang, Adv. Mater., 2017, 29, 1605005 CrossRef PubMed.
- X. Guo and C. Burda, Coord. Chem. Rev., 2016, 320, 53–65 CrossRef.
- C. C. Stoumpos and M. G. Kanatzidis, Acc. Chem. Res., 2015, 48, 2791–2802 CrossRef CAS PubMed.
- E. Salje, Philos. Trans. R. Soc., A, 1989, 328, 409–416 CrossRef CAS.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- K. Aleksandrov, Ferroelectrics, 1976, 14, 801–805 CrossRef CAS.
- H. D. Megaw, Crystal structures: a working approach, Saunders Limited, 1973 Search PubMed.
- W. J. Yin, T. Shi and Y. Yan, Adv. Mater., 2014, 26, 4653–4658 CrossRef CAS PubMed.
- C. Quarti, E. Mosconi, J. M. Ball, V. D'Innocenzo, C. Tao, S. Pathak, H. J. Snaith, A. Petrozza and F. De Angelis, Energy Environ. Sci., 2016, 9, 155–163 RSC.
- S. Colella, E. Mosconi, P. Fedeli, A. Listorti, F. Gazza, F. Orlandi, P. Ferro, T. Besagni, A. Rizzo, G. Calestani, G. Gigli, F. D. Angelis and R. Mosca, Chem. Mater., 2013, 25, 4613–4618 CrossRef CAS.
- D. P. Mcmeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Graetzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- V. Goldschmidt, Akad. Oslo I. Mat-Nat. K1, 1926, 8, 112–117 Search PubMed.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2015, 6, 3430–3433 RSC.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2014, 5, 4712–4715 RSC.
- M.-G. Ju, J. Dai, L. Ma and X. C. Zeng, J. Am. Chem. Soc., 2017, 139, 8038–8043 CrossRef CAS PubMed.
- W. Travis, E. Glover, H. Bronstein, D. Scanlon and R. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- C. Li, X. Lu, W. Ding, L. Feng, Y. Gao and Z. Guo, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 702–707 CrossRef CAS PubMed.
- M. R. Filip, G. E. Eperon, H. J. Snaith and F. Giustino, Nat. Commun., 2014, 5, 5757 CrossRef CAS PubMed.
- A. Sadhanala, S. Ahmad, B. Zhao, N. Giesbrecht, P. M. Pearce, F. Deschler, R. L. Z. Hoye, K. C. Gödel, T. Bein, P. Docampo, S. E. Dutton, M. F. L. De Volder and R. H. Friend, Nano Lett., 2015, 15, 6095–6101 CrossRef CAS PubMed.
- J.-P. Correa-Baena, A. Abate, M. Saliba, W. Tress, T. J. Jacobsson, M. Grätzel and A. Hagfeldt, Energy Environ. Sci., 2017, 10, 710–727 RSC.
- C. C. Stoumpos, C. D. Malliakas, J. A. Peters, Z. Liu, M. Sebastian, J. Im, T. C. Chasapis, A. C. Wibowo, D. Y. Chung and A. J. Freeman, Cryst. Growth Des., 2013, 13, 2722–2727 CrossRef CAS.
- F. Bertolotti, L. Protesescu, M. V. Kovalenko, S. Yakunin, A. Cervellino, S. J. Billinge, M. W. Terban, J. S. Pedersen, N. Masciocchi and A. Guagliardi, ACS Nano, 2017, 11, 3819–3831 CrossRef CAS PubMed.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed.
- A. Dutta, S. K. Dutta, S. Das Adhikari and N. Pradhan, Angew. Chem., Int. Ed., 2018, 57, 9083–9087 CrossRef CAS PubMed.
- L. Pedesseau, D. Sapori, B. Traore, R. Robles, H.-H. Fang, M. A. Loi, H. Tsai, W. Nie, J.-C. Blancon, A. Neukirch, S. Tretiak, A. D. Mohite, C. Katan, J. Even and M. Kepenekian, ACS Nano, 2016, 10, 9776–9786 CrossRef CAS PubMed.
- Z. Yuan, Y. Shu, Y. Xin and B. Ma, Chem. Commun., 2016, 52, 3887–3890 RSC.
- J. Jagielski, S. Kumar, W.-Y. Yu and C.-J. Shih, J. Mater. Chem. C, 2017, 5, 5610–5627 RSC.
- K. Tanaka, T. Takahashi, T. Ban, T. Kondo, K. Uchida and N. Miura, Solid State Commun., 2003, 127, 619–623 CrossRef CAS.
- S. Kumar, J. Jagielski, S. Yakunin, P. Rice, Y.-C. Chiu, M. Wang, G. Nedelcu, Y. Kim, S. Lin, E. J. Santos, M. V. Kovalenko and C.-J. Shih, ACS Nano, 2016, 10, 9720–9729 CrossRef CAS PubMed.
- Z. Wu, C. Ji, Z. Sun, S. Wang, S. Zhao, W. Zhang, L. Li and J. Luo, J. Mater. Chem. C, 2018, 6, 1171–1175 RSC.
- M. C. Weidman, A. J. Goodman and W. A. Tisdale, Chem. Mater., 2017, 29, 5019–5030 CrossRef CAS.
- L. N. Quan, M. Yuan, R. Comin, O. Voznyy, E. M. Beauregard, S. Hoogland, A. Buin, A. R. Kirmani, K. Zhao, A. Amassian, D. H. Kim and E. H. Sargent, J. Am. Chem. Soc., 2016, 138, 2649–2655 CrossRef CAS PubMed.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53, 11232–11235 CrossRef CAS PubMed.
- D. H. Cao, C. C. Stoumpos, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 7843–7850 CrossRef CAS PubMed.
- C. R. Kagan, D. B. Mitzi and C. D. Dimitrakopoulos, Science, 1999, 286, 945–947 CrossRef CAS PubMed.
- G. Grancini, C. Roldán-Carmona, I. Zimmermann, E. Mosconi, X. Lee, D. Martineau, S. Narbey, F. Oswald, F. De Angelis and M. Graetzel, Nat. Commun., 2017, 8, 15684 CrossRef CAS PubMed.
- C. Ma, C. Leng, Y. Ji, X. Wei, K. Sun, L. Tang, J. Yang, W. Luo, C. Li and Y. Deng, Nanoscale, 2016, 8, 18309–18314 RSC.
- G. Li, T. Zhang, N. Guo, F. Xu, X. Qian and Y. Zhao, Angew. Chem., Int. Ed., 2016, 55, 13460–13464 CrossRef CAS PubMed.
- Z. Wang, Q. Lin, F. P. Chmiel, N. Sakai, L. M. Herz and H. J. Snaith, Nat. Energy, 2017, 2, 17135 CrossRef CAS.
- F. Li, Y. Pei, F. Xiao, T. Zeng, Z. Yang, J. Xu, J. Sun, B. Peng and M. Liu, Nanoscale, 2018, 10, 6318–6322 RSC.
- T. Zhang, L. Xie, L. Chen, N. Guo, G. Li, Z. Tian, B. Mao and Y. Zhao, Adv. Funct. Mater., 2017, 27, 1603568 CrossRef.
- T. Ye, A. Bruno, G. Han, T. M. Koh, J. Li, N. F. Jamaludin, C. Soci, S. G. Mhaisalkar and W. L. Leong, Adv. Funct. Mater., 2018, 28, 1801654 CrossRef.
- Y. Hu, T. Qiu, F. Bai, W. Ruan and S. Zhang, Adv. Energy Mater., 2018, 8, 1703620 CrossRef.
- Y. Tian, C. Zhou, M. Worku, X. Wang, Y. Ling, H. Gao, Y. Zhou, Y. Miao, J. Guan and B. Ma, Adv. Mater., 2018, 30, 1707093 CrossRef PubMed.
- Y. Chen, Y. Sun, J. Peng, J. Tang, K. Zheng and Z. Liang, Adv. Mater., 2018, 30, 1703487 CrossRef PubMed.
- K. Zheng, Y. Chen, Y. Sun, J. Chen, P. Chábera, R. Schaller, M. J. Al-Marri, S. E. Canton, Z. Liang and T. Pullerits, J. Mater. Chem. A, 2018, 6, 6244–6250 RSC.
- H. Zhang, Q. Liao, Y. Wu, Z. Zhang, Q. Gao, P. Liu, M. Li, J. Yao and H. Fu, Adv. Mater., 2018, 30, 1706186 CrossRef PubMed.
- J. Li, Q. Yu, Y. He, C. C. Stoumpos, G. Niu, G. G. Trimarchi, H. Guo, G. Dong, D. Wang, L. Wang and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 11085–11090 CrossRef CAS PubMed.
- E. Shi, Y. Gao, B. P. Finkenauer, Akriti, A. H. Coffey and L. Dou, Chem. Soc. Rev., 2018, 47, 6046–6072 RSC.
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2018, 3, 54–62 CrossRef CAS.
- L. Dou, A. B. Wong, Y. Yu, M. Lai, N. Kornienko, S. W. Eaton, A. Fu, C. G. Bischak, J. Ma, T. Ding, N. S. Ginsberg, L.-W. Wang, A. P. Alivisatos and P. Yang, Science, 2015, 349, 1518–1521 CrossRef CAS PubMed.
- J. Ren, X. Dong, G. Zhang, T. Li and Y. Wang, New J. Chem., 2017, 41, 13961–13967 RSC.
- F. S. Zu, P. Amsalem, I. Salzmann, R. B. Wang, M. Ralaiarisoa, S. Kowarik, S. Duhm and N. Koch, Adv. Opt. Mater., 2017, 5, 1700139 CrossRef.
- H. Tsai, R. Asadpour, J.-C. Blancon, C. C. Stoumpos, O. Durand, J. W. Strzalka, B. Chen, R. Verduzco, P. M. Ajayan, S. Tretiak, J. Even, M. A. Alam, M. G. Kanatzidis, W. Nie and A. D. Mohite, Science, 2018, 360, 67–70 CrossRef CAS PubMed.
- A. Ummadisingu, L. Steier, J.-Y. Seo, T. Matsui, A. Abate, W. Tress and M. Grätzel, Nature, 2017, 545, 208–212 CrossRef CAS PubMed.
- G. F. Samu, C. Janáky and P. V. Kamat, ACS Energy Lett., 2017, 2, 1860–1861 CrossRef CAS.
- T. Zhang, S. H. Cheung, X. Meng, L. Zhu, Y. Bai, C. H. Y. Ho, S. Xiao, Q. Xue, S. K. So and S. Yang, J. Phys. Chem. Lett., 2017, 8, 5069–5076 CrossRef CAS PubMed.
- W. Nie, J.-C. Blancon, A. J. Neukirch, K. Appavoo, H. Tsai, M. Chhowalla, M. A. Alam, M. Y. Sfeir, C. Katan and J. Even, Nat. Commun., 2016, 7, 11574 CrossRef CAS PubMed.
- C. Zhao, B. Chen, X. Qiao, L. Luan, K. Lu and B. Hu, Adv. Energy Mater., 2015, 5, 1500279 CrossRef.
- J. F. Galisteo-López, M. Anaya, M. Calvo and H. Míguez, J. Phys. Chem. Lett., 2015, 6, 2200–2205 CrossRef PubMed.
- H. J. Snaith, A. Abate, J. M. Ball, G. E. Eperon, T. Leijtens, N. K. Noel, S. D. Stranks, J. T.-W. Wang, K. Wojciechowski and W. Zhang, J. Phys. Chem. Lett., 2014, 5, 1511–1515 CrossRef CAS PubMed.
- Y. Tian, M. Peter, E. Unger, M. Abdellah, K. Zheng, T. Pullerits, A. Yartsev, V. Sundström and I. G. Scheblykin, Phys. Chem. Chem. Phys., 2015, 17, 24978–24987 RSC.
- W. Zhang, V. M. Burlakov, D. J. Graham, T. Leijtens, A. Osherov, V. Bulović, H. J. Snaith, D. S. Ginger and S. D. Stranks, Nat. Commun., 2016, 7, 11683 CrossRef PubMed.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- S. Huang, Z. Li, L. Kong, N. Zhu, A. Shan and L. Li, J. Am. Chem. Soc., 2016, 138, 5749–5752 CrossRef CAS PubMed.
- J. Chen, D. Liu, M. J. Al-Marri, L. Nuuttila, H. Lehtivuori and K. Zheng, Sci. China Mater., 2016, 59, 719–727 CrossRef CAS.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum and J. E. Moser, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- M. Lorenzon, V. Pinchetti, F. Bruni, W. K. Bae, F. Meinardi, V. I. Klimov and S. Brovelli, Nano Lett., 2017, 17, 1071–1081 CrossRef CAS PubMed.
- M. Lorenzon, S. Christodoulou, G. Vaccaro, J. Pedrini, F. Meinardi, I. Moreels and S. Brovelli, Nat. Commun., 2015, 6, 6434 CrossRef CAS PubMed.
- D. Meggiolaro, E. Mosconi and F. De Angelis, ACS Energy Lett., 2017, 2, 2794–2798 CrossRef CAS.
- M. Lorenzon, L. Sortino, Q. Akkerman, S. Accornero, J. Pedrini, M. Prato, V. Pinchetti, F. Meinardi, L. Manna and S. Brovelli, Nano Lett., 2017, 17, 3844–3853 CrossRef CAS PubMed.
- M. S. Abdou, F. P. Orfino, Y. Son and S. Holdcroft, J. Am. Chem. Soc., 1997, 119, 4518–4524 CrossRef CAS.
- H. Kautsky, Trans. Faraday Soc., 1939, 35, 216–219 RSC.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- V. Sarritzu, N. Sestu, D. Marongiu, X. Chang, Q. Wang, S. Masi, S. Colella, A. Rizzo, A. Gocalinska, E. Pelucchi, M. L. Mercuri, F. Quochi, M. Saba, A. Mura and G. Bongiovanni, Adv. Opt. Mater., 2018, 6, 1701254 CrossRef.
- J. A. Christians, P. A. Miranda Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530–1538 CrossRef CAS PubMed.
- E. Mosconi, J. M. Azpiroz and F. De Angelis, Chem. Mater., 2015, 27, 4885–4892 CrossRef CAS.
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-b. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- X. Gong, M. Li, X. B. Shi, H. Ma, Z. K. Wang and L. S. Liao, Adv. Funct. Mater., 2015, 25, 6671–6678 CrossRef CAS.
- C. Müller, T. Glaser, M. Plogmeyer, M. Sendner, S. Döring, A. A. Bakulin, C. Brzuska, R. Scheer, M. S. Pshenichnikov, W. Kowalsky, A. Pucci and R. Lovrincic, Chem. Mater., 2015, 27, 7835–7841 CrossRef.
- H. Wang, X. Li, M. Yuan and X. Yang, Small, 2018, 14, 1703410 CrossRef PubMed.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS PubMed.
- B. Akbali, G. Topcu, T. Guner, M. Ozcan, M. M. Demir and H. Sahin, Phys. Rev. Mater., 2018, 2, 034601 CrossRef.
- X. Zhang, X. Bai, H. Wu, X. Zhang, C. Sun, Y. Zhang, W. Zhang, W. Zheng, W. W. Yu and A. L. Rogach, Angew. Chem., 2018, 130, 3395–3400 CrossRef.
- C. Geng, S. Xu, H. Zhong, A. L. Rogach and W. Bi, Angew. Chem., Int. Ed., 2018, 57, 9650–9654 CrossRef CAS PubMed.
- A. Jana and K. S. Kim, ACS Energy Lett., 2018, 3, 2120–2126 CrossRef CAS.
- A. M. A. Leguy, Y. Hu, M. Campoy-Quiles, M. I. Alonso, O. J. Weber, P. Azarhoosh, M. van Schilfgaarde, M. T. Weller, T. Bein, J. Nelson, P. Docampo and P. R. F. Barnes, Chem. Mater., 2015, 27, 3397–3407 CrossRef CAS.
- J. M. Frost, K. T. Butler, F. Brivio, C. H. Hendon, M. Van Schilfgaarde and A. Walsh, Nano Lett., 2014, 14, 2584–2590 CrossRef CAS PubMed.
- L. Zhang, M.-G. Ju and W. Liang, Phys. Chem. Chem. Phys., 2016, 18, 23174–23183 RSC.
- S. N. Habisreutinger, T. Leijtens, G. E. Eperon, S. D. Stranks, R. J. Nicholas and H. J. Snaith, Nano Lett., 2014, 14, 5561–5568 CrossRef CAS PubMed.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 17102 CrossRef CAS.
- A. Dualeh, P. Gao, S. I. Seok, M. K. Nazeeruddin and M. Grätzel, Chem. Mater., 2014, 26, 6160–6164 CrossRef CAS.
- Q. Shao, L. Wang, L. Song, Y. Dong, C. Liang, J. He and J. Jiang, J. Alloys Compd., 2017, 695, 221–226 CrossRef CAS.
- Y. Zhao, C. Riemersma, F. Pietra, R. Koole, C. de Mello Donegá and A. Meijerink, ACS Nano, 2012, 6, 9058–9067 CrossRef CAS PubMed.
- S. Wei, Y. Yang, X. Kang, L. Wang, L. Huang and D. Pan, Chem. Commun., 2016, 52, 7265–7268 RSC.
- B. T. Diroll, G. Nedelcu, M. V. Kovalenko and R. D. Schaller, Adv. Funct. Mater., 2017, 27, 1606075 CrossRef.
- Q. Jiang, M. Chen, J. Li, M. Wang, X. Zeng, T. Besara, J. Lu, Y. Xin, X. Shan, B. Pan, C. Wang, S. Lin, T. Siegrist, Q. Xiao and Z. Yu, ACS Nano, 2017, 11, 1073–1079 CrossRef CAS PubMed.
- Y. Zhou, J. Chen, O. M. Bakr and H.-T. Sun, Chem. Mater., 2018, 30, 6589–6613 CrossRef CAS.
- F. C. Hanusch, E. Wiesenmayer, E. Mankel, A. Binek, P. Angloher, C. Fraunhofer, N. Giesbrecht, J. M. Feckl, W. Jaegermann, D. Johrendt, T. Bein and P. Docampo, J. Phys. Chem. Lett., 2014, 5, 2791–2795 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Bertolotti, N. Masciocchi, A. Guagliardi and M. V. Kovalenko, J. Am. Chem. Soc., 2016, 138, 14202–14205 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, S. Kumar, J. Bär, F. Bertolotti, N. Masciocchi, A. Guagliardi, M. Grotevent, I. Shorubalko, M. I. Bodnarchuk, C.-J. Shih and M. V. Kovalenko, ACS Nano, 2017, 11, 3119–3134 CrossRef CAS PubMed.
- X. Zhang, H. Liu, W. Wang, J. Zhang, B. Xu, K. L. Karen, Y. Zheng, S. Liu, S. Chen, K. Wang and X. W. Sun, Adv. Mater., 2017, 29, 1606405 CrossRef PubMed.
- D. Amgar, T. Binyamin, V. Uvarov and L. Etgar, Nanoscale, 2018, 10, 6060–6068 RSC.
- S. Huang, B. Wang, Q. Zhang, Z. Li, A. Shan and L. Li, Adv. Opt. Mater., 2018, 6, 1701106 CrossRef.
- Y. Liu, G. Pan, R. Wang, H. Shao, H. Wang, W. Xu, H. Cui and H. Song, Nanoscale, 2018, 10, 14067–14072 RSC.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. Cacovich, C. Stavrakas, B. Philippe, J. M. Richter, M. Alsari, E. P. Booker, E. M. Hutter, A. J. Pearson, S. Lilliu, T. J. Savenije, H. Rensmo, G. Divitini, C. Ducati, R. H. Friend and S. D. Stranks, Nature, 2018, 555, 497–501 CrossRef CAS PubMed.
- B. Philippe, M. Saliba, J.-P. Correa-Baena, U. B. Cappel, S.-H. Turren-Cruz, M. Grätzel, A. Hagfeldt and H. k. Rensmo, Chem. Mater., 2017, 29, 3589–3596 CrossRef CAS.
- G. M. Dalpian and J. R. Chelikowsky, Phys. Rev. Lett., 2006, 96, 226802 CrossRef PubMed.
- D. Cortecchia, S. Neutzner, A. R. Srimath Kandada, E. Mosconi, D. Meggiolaro, F. De Angelis, C. Soci and A. Petrozza, J. Am. Chem. Soc., 2017, 139, 39–42 CrossRef CAS PubMed.
- C. Quarti, N. Marchal and D. Beljonne, J. Phys. Chem. Lett., 2018, 9, 3416–3424 CrossRef CAS PubMed.
- S. Yang, Z. Lin, J. Wang, Y. Chen, Z. Liu, E. Yang, J. Zhang and Q. Ling, ACS Appl. Mater. Interfaces, 2018, 10, 15980–15987 CrossRef CAS PubMed.
- A. García-Fernández, J. M. Bermúdez-García, S. Castro-García, A. L. Llamas-Saiz, R. Artiaga, J. J. López-Beceiro, M. Sánchez-Andújar and M. A. Señarís-Rodríguez, Inorg. Chem., 2018, 57, 3215–3222 CrossRef PubMed.
- I. Neogi, A. Bruno, D. Bahulayan, T. W. Goh, B. Ghosh, R. Ganguly, D. Cortecchia, T. C. Sum, C. Soci, N. Mathews and S. G. Mhaisalkar, ChemSusChem, 2017, 10, 3765–3772 CrossRef CAS PubMed.
- L. Mao, Y. Wu, C. C. Stoumpos, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 5210–5215 CrossRef CAS PubMed.
- S. Krishnamurthy, R. Naphade, W. J. Mir, S. Gosavi, S. Chakraborty, R. Vaidhyanathan and S. Ogale, Adv. Opt. Mater., 2018, 6, 1800751 CrossRef.
- S. Wang, Y. Yao, J. Kong, S. Zhao, Z. Sun, Z. Wu, L. Li and J. Luo, Chem. Commun., 2018, 54, 4053–4056 RSC.
- Z. Zhuang, C. Peng, G. Zhang, H. Yang, J. Yin and H. Fei, Angew. Chem., 2017, 129, 14603–14608 CrossRef.
- H. Hu, S. Morris, X. Qiao, D. Zhao, T. Salim, B. Chen, E. Chia and Y.-M. Lam, J. Mater. Chem. C, 2018, 6, 10301–10307 RSC.
- S. Yang, W. Niu, A. L. Wang, Z. Fan, B. Chen, C. Tan, Q. Lu and H. Zhang, Angew. Chem., Int. Ed., 2017, 56, 4252–4255 CrossRef CAS PubMed.
- S. Bhaumik, S. A. Veldhuis, Y. F. Ng, M. Li, S. K. Muduli, T. C. Sum, B. Damodaran, S. Mhaisalkar and N. Mathews, Chem. Commun., 2016, 52, 7118–7121 RSC.
- H. Zhu, M. T. Trinh, J. Wang, Y. Fu, P. P. Joshi, K. Miyata, S. Jin and X. Y. Zhu, Adv. Mater., 2017, 29, 1603072 CrossRef PubMed.
- A. Swarnkar, W. J. Mir and A. Nag, ACS Energy Lett., 2018, 3, 286–289 CrossRef CAS.
- M. Liu, G. Zhong, Y. Yin, J. Miao, K. Li, C. Wang, X. Xu, C. Shen and H. Meng, Adv. Sci., 2017, 4, 1700335 CrossRef PubMed.
- F. Yang, D. Hirotani, G. Kapil, M. A. Kamarudin, C. H. Ng, Y. Zhang, Q. Shen and S. Hayase, Angew. Chem., Int. Ed., 2018, 57 Search PubMed.
- W. Liu, Q. Lin, H. Li, K. Wu, I. n. Robel, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2016, 138, 14954–14961 CrossRef CAS PubMed.
- D. Parobek, B. J. Roman, Y. Dong, H. Jin, E. Lee, M. Sheldon and D. H. Son, Nano Lett., 2016, 16, 7376–7380 CrossRef CAS PubMed.
- H. Liu, Z. Wu, J. Shao, D. Yao, H. Gao, Y. Liu, W. Yu, H. Zhang and B. Yang, ACS Nano, 2017, 11, 2239–2247 CrossRef CAS PubMed.
- F. Li, Z. Xia, Y. Gong, L. Gu and Q. Liu, J. Mater. Chem. C, 2017, 5, 9281–9287 RSC.
- P. K. Santra and P. V. Kamat, J. Am. Chem. Soc., 2012, 134, 2508–2511 CrossRef CAS PubMed.
- W. J. Mir, M. Jagadeeswararao, S. Das and A. Nag, ACS Energy Lett., 2017, 2, 537–543 CrossRef CAS.
- A. K. Guria, S. K. Dutta, S. D. Adhikari and N. Pradhan, ACS Energy Lett., 2017, 2, 1014–1021 CrossRef CAS.
- K. Xu, C. C. Lin, X. Xie and A. Meijerink, Chem. Mater., 2017, 29, 4265–4272 CrossRef CAS PubMed.
- G. Huang, C. Wang, S. Xu, S. Zong, J. Lu, Z. Wang, C. Lu and Y. Cui, Adv. Mater., 2017, 29, 1700095 CrossRef PubMed.
- S. Das Adhikari, S. K. Dutta, A. Dutta, A. K. Guria and N. Pradhan, Angew. Chem., 2017, 129, 8872–8876 CrossRef.
- A. Biswas, R. Bakthavatsalam and J. Kundu, Chem. Mater., 2017, 29, 7816–7825 CrossRef CAS.
- P. Arunkumar, K. H. Gil, S. Won, S. Unithrattil, Y. H. Kim, H. J. Kim and W. B. Im, J. Phys. Chem. Lett., 2017, 8, 4161–4166 CrossRef CAS PubMed.
- F. Li, Z. Xia, C. Pan, Y. Gong, L. Gu, Q. Liu and J. Z. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 11739–11746 CrossRef CAS PubMed.
- G. Fang, D. Chen, S. Zhou, X. Chen, L. Lei, J. Zhong and Z. Ji, J. Mater. Chem. C, 2018, 6, 5908–5915 RSC.
- Q. A. Akkerman, D. Meggiolaro, Z. Dang, F. De Angelis and L. Manna, ACS Energy Lett., 2017, 2, 2183–2186 CrossRef CAS PubMed.
- T. C. Jellicoe, J. M. Richter, H. F. Glass, M. Tabachnyk, R. Brady, S. n. E. Dutton, A. Rao, R. H. Friend, D. Credgington, N. C. Greenham and M. L. Böhm, J. Am. Chem. Soc., 2016, 138, 2941–2944 CrossRef CAS PubMed.
- A. Wang, Y. Guo, F. Muhammad and Z. Deng, Chem. Mater., 2017, 29, 6493–6501 CrossRef CAS.
- D. Moghe, L. Wang, C. J. Traverse, A. Redoute, M. Sponseller, P. R. Brown, V. Bulović and R. R. Lunt, Nano Energy, 2016, 28, 469–474 CrossRef CAS.
- G. Xing, M. H. Kumar, W. K. Chong, X. Liu, Y. Cai, H. Ding, M. Asta, M. Grätzel, S. Mhaisalkar, N. Mathews and T. C. Sum, Adv. Mater., 2016, 28, 8191–8196 CrossRef CAS PubMed.
- A. Wang, X. Yan, M. Zhang, S. Sun, M. Yang, W. Shen, X. Pan, P. Wang and Z. Deng, Chem. Mater., 2016, 28, 8132–8140 CrossRef CAS.
- M. Li, X. Zhang, K. Matras-Postolek, H.-S. Chen and P. Yang, J. Mater. Chem. C, 2018, 6, 5506–5513 RSC.
- F. Liu, C. Ding, Y. Zhang, T. S. Ripolles, T. Kamisaka, T. Toyoda, S. Hayase, T. Minemoto, K. Yoshino, S. Dai, M. Yanagida, H. Noguchi and Q. Shen, J. Am. Chem. Soc., 2017, 139, 16708–16719 CrossRef CAS PubMed.
- X. Zhang, W. Cao, W. Wang, B. Xu, S. Liu, H. Dai, S. Chen, K. Wang and X. W. Sun, Nano Energy, 2016, 30, 511–516 CrossRef CAS.
- W. Van der Stam, J. J. Geuchies, T. Altantzis, K. H. Van Den Bos, J. D. Meeldijk, S. Van Aert, S. Bals, D. Vanmaekelbergh and C. de Mello Donega, J. Am. Chem. Soc., 2017, 139, 4087–4097 CrossRef CAS PubMed.
- H. C. Wang, W. Wang, A. C. Tang, H. Y. Tsai, Z. Bao, T. Ihara, N. Yarita, H. Tahara, Y. Kanemitsu, S. Chen and R. S. Liu, Angew. Chem., 2017, 129, 13838–13842 CrossRef.
- Z.-J. Yong, S.-Q. Guo, J.-P. Ma, J. Zhang, Z.-Y. Li, Y.-M. Chen, B.-B. Zhang, Y. Zhou, J. Shu, J.-L. Gu, L.-R. Zheng, O. M. Bakr and H.-T. Sun, J. Am. Chem. Soc., 2018, 140, 9942–9951 CrossRef CAS PubMed.
- M. Leng, Z. Chen, Y. Yang, Z. Li, K. Zeng, K. Li, G. Niu, Y. He, Q. Zhou and J. Tang, Angew. Chem., Int. Ed., 2016, 55, 15012–15016 CrossRef CAS PubMed.
- M. Leng, Y. Yang, Z. Chen, W. Gao, J. Zhang, G. Niu, D. Li, H. Song, J. Zhang, S. Jin and J. Tang, Nano Lett., 2018, 18, 6076–6083 CrossRef CAS PubMed.
- M. Leng, Y. Yang, K. Zeng, Z. Chen, Z. Tan, S. Li, J. Li, B. Xu, D. Li, M. P. Hautzinger, Y. Fu, T. Zhai, L. Xu, G. Niu, S. Jin and J. Tang, Adv. Funct. Mater., 2018, 28, 1704446 CrossRef.
- Y. Lou, M. Fang, J. Chen and Y. Zhao, Chem. Commun., 2018, 54, 3779–3782 RSC.
- R. D. Nelson, K. Santra, Y. Wang, A. Hadi, J. W. Petrich and M. G. Panthani, Chem. Commun., 2018, 54, 3640–3643 RSC.
- B. Yang, J. Chen, F. Hong, X. Mao, K. Zheng, S. Yang, Y. Li, T. Pullerits, W. Deng and K. Han, Angew. Chem., 2017, 129, 12645–12649 CrossRef.
- R. Begum, M. R. Parida, A. L. Abdelhady, B. Murali, N. M. Alyami, G. H. Ahmed, M. N. Hedhili, O. M. Bakr and O. F. Mohammed, J. Am. Chem. Soc., 2016, 139, 731–737 CrossRef PubMed.
- P. K. Nayak, M. Sendner, B. Wenger, Z. Wang, K. Sharma, A. J. Ramadan, R. Lovrincic, A. Pucci, P. K. Madhu and H. J. Snaith, J. Am. Chem. Soc., 2018, 140, 574–577 CrossRef CAS PubMed.
- H. Shao, X. Bai, H. Cui, G. Pan, P. Jing, S. Qu, J. Zhu, Y. Zhai, B. Dong and H. Song, Nanoscale, 2018, 10, 1023–1029 RSC.
- J. Zhang, Y. Yang, H. Deng, U. Farooq, X. Yang, J. Khan, J. Tang and H. Song, ACS Nano, 2017, 11, 9294–9302 CrossRef CAS PubMed.
- S. Xiang, W. Li, Y. Wei, J. Liu, H. Liu, L. Zhu and H. Chen, Nanoscale, 2018, 10, 9996–10004 RSC.
- S. E. Creutz, E. N. Crites, M. C. De Siena and D. R. Gamelin, Nano Lett., 2018, 18, 1118–1123 CrossRef CAS PubMed.
- B. Yang, J. Chen, S. Yang, F. Hong, L. Sun, P. Han, T. Pullerits, W. Deng and K. Han, Angew. Chem., 2018, 130, 5457–5461 CrossRef.
- L. Zhou, Y. F. Xu, B. X. Chen, D. B. Kuang and C. Y. Su, Small, 2018, 14, 1703762 CrossRef PubMed.
- J. Sun, J. Yang, J. I. Lee, J. H. Cho and M. S. Kang, J. Phys. Chem. Lett., 2018, 9, 1573–1583 CrossRef CAS PubMed.
- Q. Hu, Z. Li, Z. Tan, H. Song, C. Ge, G. Niu, J. Han and J. Tang, Adv. Opt. Mater., 2018, 6, 1700864 CrossRef.
- L. Liu, J. Li and J. A. McLeod, Nanoscale, 2018, 10, 11452–11459 RSC.
- G. Pan, X. Bai, D. Yang, X. Chen, P. Jing, S. Qu, L. Zhang, D. Zhou, J. Zhu, W. Xu, B. Dong and H. Song, Nano Lett., 2017, 17, 8005–8011 CrossRef CAS PubMed.
- T. J. Milstein, D. M. Kroupa and D. R. Gamelin, Nano Lett., 2018, 18, 3792–3799 CrossRef CAS PubMed.
- X. Zhang, Y. Zhang, X. Zhang, W. Yin, Y. Wang, H. Wang, M. Lu, Z. Li, Z. Gu and W. Yu, J. Mater. Chem. C, 2018, 6, 10101–10105 RSC.
- D. Zhou, D. Liu, G. Pan, X. Chen, D. Li, W. Xu, X. Bai and H. Song, Adv. Mater., 2017, 29, 1704149 CrossRef PubMed.
- J.-S. Yao, J. Ge, B.-N. Han, K.-H. Wang, H.-B. Yao, H.-L. Yu, J.-H. Li, B.-S. Zhu, J.-Z. Song, C. Chen, Q. Zhang, H.-B. Zeng, Y. Luo and S.-H. Yu, J. Am. Chem. Soc., 2018, 140, 3626–3634 CrossRef CAS PubMed.
- L. Qu and X. Peng, J. Am. Chem. Soc., 2002, 124, 2049–2055 CrossRef CAS PubMed.
- C. Pu and X. Peng, J. Am. Chem. Soc., 2016, 138, 8134–8142 CrossRef CAS PubMed.
- J. Kang and L.-W. Wang, J. Phys. Chem. Lett., 2017, 8, 489–493 CrossRef CAS PubMed.
- Y. Liu, H. Xiao and W. A. Goddard III, Nano Lett., 2016, 16, 3335–3340 CrossRef CAS PubMed.
- S. ten Brinck and I. Infante, ACS Energy Lett., 2016, 1, 1266–1272 CrossRef CAS.
- J. Pan, L. N. Quan, Y. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. Yuan, L. Sinatra, N. M. Alyami, J. Liu, E. Yassitepe, Z. Yang, O. Voznyy, R. Comin, M. N. Hedhili, O. F. Mohammed, Z. H. Lu, D. H. Kim, E. H. Sargent and O. M. Bakr, Adv. Mater., 2016, 28, 8718–8725 CrossRef CAS PubMed.
- N. K. Noel, A. Abate, S. D. Stranks, E. S. Parrott, V. M. Burlakov, A. Goriely and H. J. Snaith, ACS Nano, 2014, 8, 9815–9821 CrossRef CAS PubMed.
- C. Wang, A. S. Chesman and J. J. Jasieniak, Chem. Commun., 2017, 53, 232–235 RSC.
- B. Luo, Y. C. Pu, S. A. Lindley, Y. Yang, L. Lu, Y. Li, X. Li and J. Z. Zhang, Angew. Chem., 2016, 128, 9010–9014 CrossRef.
- Z. Liu, Y. Zhang, Y. Fan, Z. Chen, Z. Tang, J. Zhao, Y. Lv, J. Lin, X. Guo, J. Zhang and X. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 13053–13061 CrossRef CAS PubMed.
- T. Xuan, X. Yang, S. Lou, J. Huang, Y. Liu, J. Yu, H. Li, K.-L. Wong, C. Wang and J. Wang, Nanoscale, 2017, 9, 15286–15290 RSC.
- S. Gonzalez-Carrero, L. Francés-Soriano, M. González-Béjar, S. Agouram, R. E. Galian and J. Pérez-Prieto, Small, 2016, 12, 5245–5250 CrossRef CAS PubMed.
- L. Wu, Q. Zhong, D. Yang, M. Chen, H. Hu, Q. Pan, H. Liu, M. Cao, Y. Xu, B. Sun and Q. Zhang, Langmuir, 2017, 33, 12689–12696 CrossRef CAS PubMed.
- Q.-L. Li, W.-X. Lu, N. Wan and S.-N. Ding, Chem. Commun., 2016, 52, 12342–12345 RSC.
- L. Rao, Y. Tang, C. Yan, J. Li, G. Zhong, K. Tang, B. Yu, Z. Li and J. Z. Zhang, J. Mater. Chem. C, 2018, 6, 5375–5383 RSC.
- F. Liu, Y. Zhang, C. Ding, S. Kobayashi, T. Izuishi, N. Nakazawa, T. Toyoda, T. Ohta, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, ACS Nano, 2017, 11, 10373–10383 CrossRef CAS PubMed.
- H. Wang, N. Sui, X. Bai, Y. Zhang, Q. Rice, F. J. Seo, Q. Zhang, V. L. Colvin and W. W. Yu, J. Phys. Chem. Lett., 2018, 9, 4166–4173 CrossRef CAS PubMed.
- C. Lu, H. Li, K. Kolodziejski, C. Dun, W. Huang, D. Carroll and S. M. Geyer, Nano Res., 2018, 11, 762–768 CrossRef CAS.
- K. P. Mubiayi, N. Moloto and M. J. Moloto, CrystEngComm, 2018, 20, 5275–5280 RSC.
- H. Wu, Y. Zhang, M. Lu, X. Zhang, C. Sun, T. Zhang, V. L. Colvin and W. Y. William, Nanoscale, 2018, 10, 4173–4178 RSC.
- J. Pan, S. P. Sarmah, B. Murali, I. Dursun, W. Peng, M. R. Parida, J. Liu, L. Sinatra, N. Alyami, C. Zhao, E. Alarousu, T. K. Ng, B. S. Ooi, O. M. Bakr and O. F. Mohammed, J. Phys. Chem. Lett., 2015, 6, 5027–5033 CrossRef CAS PubMed.
- G. Li, J. Huang, H. Zhu, Y. Li, J.-X. Tang and Y. Jiang, Chem. Mater., 2018, 30, 6099–6107 CrossRef CAS.
- S. A. Veldhuis, Y. K. E. Tay, A. Bruno, S. S. Dintakurti, S. Bhaumik, S. K. Muduli, M. Li, N. Mathews, T. C. Sum and S. G. Mhaisalkar, Nano Lett., 2017, 17, 7424–7432 CrossRef CAS PubMed.
- G. H. Ahmed, J. K. El-Demellawi, J. Yin, J. Pan, D. B. Velusamy, M. N. Hedhili, E. Alarousu, O. M. Bakr, H. N. Alshareef and O. F. Mohammed, ACS Energy Lett., 2018, 2301–2307 CrossRef CAS.
- J. Y. Woo, Y. Kim, J. Bae, T. G. Kim, J. W. Kim, D. C. Lee and S. Jeong, Chem. Mater., 2017, 29, 7088–7092 CrossRef CAS.
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566–6569 CrossRef CAS PubMed.
- T. Ahmed, S. Seth and A. Samanta, Chem. Mater., 2018, 30, 3633–3637 CrossRef CAS.
- Q. Jing, M. Zhang, X. Huang, X. Ren, P. Wang and Z. Lu, Nanoscale, 2017, 9, 7391–7396 RSC.
- B. J. Bohn, Y. Tong, M. Gramlich, M. L. Lai, M. Döblinger, K. Wang, R. L. Z. Hoye, P. Müller-Buschbaum, S. D. Stranks, A. S. Urban, L. Polavarapu and J. Feldmann, Nano Lett., 2018, 18, 5231–5238 CrossRef CAS PubMed.
- F. Zhang, S. Huang, P. Wang, X. Chen, S. Zhao, Y. Dong and H. Zhong, Chem. Mater., 2017, 29, 3793–3799 CrossRef CAS.
- L. Ruan, W. Shen, A. Wang, Q. Zhou, H. Zhang and Z. Deng, Nanoscale, 2017, 9, 7252–7259 RSC.
- S. R. Smock, T. J. Williams and R. L. Brutchey, Angew. Chem., 2018, 130, 11885–11889 CrossRef.
- Q. Li, H. Li, H. Shen, F. Wang, F. Zhao, F. Li, X. Zhang, D. Li, X. Jin and W. Sun, ACS Photonics, 2017, 4, 2504–2512 CrossRef CAS.
- F. Krieg, S. T. Ochsenbein, S. Yakunin, S. ten Brinck, P. Aellen, A. Süess, B. Clerc, D. Guggisberg, O. Nazarenko, Y. Shynkarenko, S. Kumar, C.-J. Shih, I. Infante and M. V. Kovalenko, ACS Energy Lett., 2018, 3, 641–646 CrossRef CAS PubMed.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili, A.-H. Emwas, O. F. Mohammed, Z. Ning and O. M. Bakr, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS PubMed.
- H. Sun, Z. Yang, M. Wei, W. Sun, X. Li, S. Ye, Y. Zhao, H. Tan, E. L. Kynaston, T. B. Schon, H. Yan, Z.-H. Lu, G. A. Ozin, E. H. Sargent and D. S. Seferos, Adv. Mater., 2017, 29, 1701153 CrossRef PubMed.
- G. Li, F. W. R. Rivarola, N. J. Davis, S. Bai, T. C. Jellicoe, F. de la Peña, S. Hou, C. Ducati, F. Gao, R. H. Friend, N. C. Greenham and Z.-K. Tan, Adv. Mater., 2016, 28, 3528–3534 CrossRef CAS PubMed.
- F. Palazon, F. Di Stasio, Q. A. Akkerman, R. Krahne, M. Prato and L. Manna, Chem. Mater., 2016, 28, 2902–2906 CrossRef CAS PubMed.
- F. Palazon, Q. A. Akkerman, M. Prato and L. Manna, ACS Nano, 2015, 10, 1224–1230 CrossRef PubMed.
- S. Pathak, N. Sakai, F. Wisnivesky Rocca Rivarola, S. D. Stranks, J. Liu, G. E. Eperon, C. Ducati, K. Wojciechowski, J. T. Griffiths, A. A. Haghighirad, A. Pellaroque, R. H. Friend and H. J. Snaith, Chem. Mater., 2015, 27, 8066–8075 CrossRef CAS.
- K. Ma, X.-Y. Du, Y.-W. Zhang and S. Chen, J. Mater. Chem. C, 2017, 5, 9398–9404 RSC.
- Y. H. Song, J. S. Yoo, B. K. Kang, S. H. Choi, E. K. Ji, H. S. Jung and D. H. Yoon, Nanoscale, 2016, 8, 19523–19526 RSC.
- D. N. Minh, S. Eom, L. A. T. Nguyen, J. Kim, J. H. Sim, C. Seo, J. Nam, S. Lee, S. Suk, J. Kim and Y. Kang, Adv. Mater., 2018, 30, 1802555 CrossRef PubMed.
- W. Cha, H.-J. Kim, S. Lee and J. Kim, J. Mater. Chem. C, 2017, 5, 6667–6671 RSC.
- C. C. Lin, D.-H. Jiang, C.-C. Kuo, C.-J. Cho, Y.-H. Tsai, T. Satoh and C. Su, ACS Appl. Mater. Interfaces, 2018, 10, 2210–2215 CrossRef CAS PubMed.
- P.-C. Tsai, J.-Y. Chen, E. Ercan, C.-C. Chueh, S.-H. Tung and W.-C. Chen, Small, 2018, 14, 1704379 CrossRef PubMed.
- H. Liao, S. Guo, S. Cao, L. Wang, F. Gao, Z. Yang, J. Zheng and W. Yang, Adv. Opt. Mater., 2018, 6, 1800346 CrossRef.
- Y. Li, Y. Lv, Z. Guo, L. Dong, J. Zheng, C. Chai, N. Chen, Y. Lu and C. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 15888–15894 CrossRef CAS PubMed.
- M. Meyns, M. Perálvarez, A. Heuer-Jungemann, W. Hertog, M. Ibáñez, R. Nafria, A. Genc, J. Arbiol, M. V. Kovalenko and J. Carreras, ACS Appl. Mater. Interfaces, 2016, 8, 19579–19586 CrossRef CAS PubMed.
- A. Pan, M. J. Jurow, F. Qiu, J. Yang, B. Ren, J. J. Urban, L. He and Y. Liu, Nano Lett., 2017, 17, 6759–6765 CrossRef CAS PubMed.
- S. Hou, Y. Guo, Y. Tang and Q. Quan, ACS Appl. Mater. Interfaces, 2017, 9, 18417–18422 CrossRef CAS PubMed.
- S. Yang, F. Zhang, J. Tai, Y. Li, Y. Yang, H. Wang, J. Zhang, Z. Xie, B. Xu, H. Zhong, K. Liu and B. Yang, Nanoscale, 2018, 10, 5820–5826 RSC.
- W. Xu, Z. Cai, F. Li, J. Dong, Y. Wang, Y. Jiang and X. Chen, Nano Res., 2017, 10, 2692–2698 CrossRef CAS.
- S. N. Raja, Y. Bekenstein, M. A. Koc, S. Fischer, D. Zhang, L. Lin, R. O. Ritchie, P. Yang and A. P. Alivisatos, ACS Appl. Mater. Interfaces, 2016, 8, 35523–35533 CrossRef CAS PubMed.
- X. Shen, C. Sun, X. Bai, X. Zhang, Y. Wang, Y. Wang, H. Song and W. W. Yu, ACS Appl. Mater. Interfaces, 2018, 10, 16768–16775 CrossRef CAS PubMed.
- K. Chen, X. Deng, G. Dodekatos and H. Tüysüz, J. Am. Chem. Soc., 2017, 139, 12267–12273 CrossRef CAS PubMed.
- Y. C. Wong, J. De Andrew Ng and Z. K. Tan, Adv. Mater., 2018, 30, 1800774 CrossRef PubMed.
- H. Zhang, X. Wang, Q. Liao, Z. Xu, H. Li, L. Zheng and H. Fu, Adv. Funct. Mater., 2017, 27, 1604382 CrossRef.
- X. Yang, T. Xu, Y. Zhu, J. Cai, K. Gu, J. Zhu, Y. Wang, J. Shen and C. Li, J. Mater. Chem. C, 2018, 6, 7971–7975 RSC.
- Y. Wei, X. Deng, Z. Xie, X. Cai, S. Liang, P. a. Ma, Z. Hou, Z. Cheng and J. Lin, Adv. Funct. Mater., 2017, 27, 1703535 CrossRef.
- J.-N. Liu, W.-B. Bu and J.-L. Shi, Acc. Chem. Res., 2015, 48, 1797–1805 CrossRef CAS PubMed.
- D. Gerion, F. Pinaud, S. C. Williams, W. J. Parak, D. Zanchet, S. Weiss and A. P. Alivisatos, J. Phys. Chem. B, 2001, 105, 8861–8871 CrossRef CAS.
- D. K. Yi, S. T. Selvan, S. S. Lee, G. C. Papaefthymiou, D. Kundaliya and J. Y. Ying, J. Am. Chem. Soc., 2005, 127, 4990–4991 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- J. Ziegler, S. Xu, E. Kucur, F. Meister, M. Batentschuk, F. Gindele and T. Nann, Adv. Mater., 2008, 20, 4068–4073 CrossRef CAS.
- D. N. Dirin, L. Protesescu, D. Trummer, I. V. Kochetygov, S. Yakunin, F. Krumeich, N. P. Stadie and M. V. Kovalenko, Nano Lett., 2016, 16, 5866–5874 CrossRef CAS PubMed.
- V. Malgras, S. Tominaka, J. W. Ryan, J. Henzie, T. Takei, K. Ohara and Y. Yamauchi, J. Am. Chem. Soc., 2016, 138, 13874–13881 CrossRef CAS PubMed.
- V. Malgras, J. Henzie, T. Takei and Y. Yamauchi, Chem. Commun., 2017, 53, 2359–2362 RSC.
- V. Malgras, J. Henzie, T. Takei and Y. Yamauchi, Angew. Chem., 2018, 130, 9019–9023 CrossRef.
- D. H. Park, J. S. Han, W. Kim and H. S. Jang, Dyes Pigm., 2018, 149, 246–252 CrossRef CAS.
- Z. Hu, Z. Liu, Y. Bian, S. Li, X. Tang, J. Du, Z. Zang, M. Zhou, W. Hu, Y. Tian and Y. Leng, Adv. Opt. Mater., 2018, 6, 1700997 CrossRef.
- Q. Zhong, M. Cao, H. Hu, D. Yang, M. Chen, P. Li, L. Wu and Q. Zhang, ACS Nano, 2018, 12, 8579–8587 CrossRef CAS PubMed.
- N. Ding, D. Zhou, X. Sun, W. Xu, H. Xu, G. Pan, D. Li, S. Zhang, B. Dong and H. Song, Nanotechnology, 2018, 29, 345703 CrossRef PubMed.
- X. Li, Y. Wang, H. Sun and H. Zeng, Adv. Mater., 2017, 29, 1701185 CrossRef PubMed.
- W. Chen, J. Hao, W. Hu, Z. Zang, X. Tang, L. Fang, T. Niu and M. Zhou, Small, 2017, 13, 1604085 CrossRef PubMed.
- S. K. Balakrishnan and P. V. Kamat, ACS Energy Lett., 2016, 2, 88–93 CrossRef.
- B. J. Roman, J. Otto, C. Galik, R. Downing and M. Sheldon, Nano Lett., 2017, 17, 5561–5566 CrossRef CAS PubMed.
- S. Guarnera, A. Abate, W. Zhang, J. M. Foster, G. Richardson, A. Petrozza and H. J. Snaith, J. Phys. Chem. Lett., 2015, 6, 432–437 CrossRef CAS PubMed.
- T. Leijtens, B. Lauber, G. E. Eperon, S. D. Stranks and H. J. Snaith, J. Phys. Chem. Lett., 2014, 5, 1096–1102 CrossRef CAS PubMed.
- J. Y. Sun, F. T. Rabouw, X. F. Yang, X. Y. Huang, X. P. Jing, S. Ye and Q. Y. Zhang, Adv. Funct. Mater., 2017, 27, 1704371 CrossRef.
- Y. Wei, H. Xiao, Z. Xie, S. Liang, S. Liang, X. Cai, S. Huang, A. A. Al Kheraif, H. S. Jang, Z. Cheng and J. Lin, Adv. Opt. Mater., 2018, 6, 1701343 CrossRef.
- Z. Zhao, Z. Wu, J. Cheng, L. Jing and Y. Hou, J. Phys. Chem. C, 2018, 122, 16887–16893 CrossRef CAS.
- Z. Chen, Z.-G. Gu, W.-Q. Fu, F. Wang and J. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 28737–28742 CrossRef CAS PubMed.
- C. Zhang, B. Wang, W. Li, S. Huang, L. Kong, Z. Li and L. Li, Nat. Commun., 2017, 8, 1138 CrossRef PubMed.
- G. Yang, Q. Fan, B. Chen, Q. Zhou and H. Zhong, J. Mater. Chem. C, 2016, 4, 11387–11391 RSC.
- S. Lou, T. Xuan, C. Yu, M. Cao, C. Xia, J. Wang and H. Li, J. Mater. Chem. C, 2017, 5, 7431–7435 RSC.
- A. Loiudice, S. Saris, E. Oveisi, D. T. Alexander and R. Buonsanti, Angew. Chem., Int. Ed., 2017, 56, 10696–10701 CrossRef CAS PubMed.
- Z. J. Li, E. Hofman, J. Li, A. H. Davis, C. H. Tung, L. Z. Wu and W. Zheng, Adv. Funct. Mater., 2018, 28, 1704288 CrossRef.
- S. Wang, C. Bi, J. Yuan, L. Zhang and J. Tian, ACS Energy Lett., 2018, 3, 245–251 CrossRef CAS.
- B. Wang, C. Zhang, S. Huang, Z. Li, L. Kong, L. Jin, J. Wang, K. Wu and L. Li, ACS Appl. Mater. Interfaces, 2018, 10, 23303–23310 CrossRef CAS PubMed.
- Z. Ning, X. Gong, R. Comin, G. Walters, F. Fan, O. Voznyy, E. Yassitepe, A. Buin, S. Hoogland and E. H. Sargent, Nature, 2015, 523, 324 CrossRef CAS PubMed.
- K.-H. Wang, L. Wu, L. Li, H.-B. Yao, H.-S. Qian and S.-H. Yu, Angew. Chem., Int. Ed., 2016, 55, 8328–8332 CrossRef CAS PubMed.
- X. Zhang, B. Xu, J. Zhang, Y. Gao, Y. Zheng, K. Wang and X. W. Sun, Adv. Funct. Mater., 2016, 26, 4595–4600 CrossRef CAS.
- X. Zhang, Z. Jin, J. Zhang, D. Bai, H. Bian, K. Wang, J. Sun, Q. Wang and S. F. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 7145–7154 CrossRef CAS PubMed.
- W. Shen, L. Ruan, Z. Shen and Z. Deng, Chem. Commun., 2018, 54, 2804–2807 RSC.
- P. Song, B. Qiao, D. Song, Z. Liang, D. Gao, J. Cao, Z. Shen, Z. Xu and S. Zhao, J. Alloys Compd., 2018, 767, 98–105 CrossRef CAS.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- Z. Liu, Y. Bekenstein, X. Ye, S. C. Nguyen, J. Swabeck, D. Zhang, S.-T. Lee, P. Yang, W. Ma and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 5309–5312 CrossRef CAS PubMed.
- Y. Zhang, M. I. Saidaminov, I. Dursun, H. Yang, B. Murali, E. Alarousu, E. Yengel, B. A. Alshankiti, O. M. Bakr and O. F. Mohammed, J. Phys. Chem. Lett., 2017, 8, 961–965 CrossRef CAS PubMed.
- Q. A. Akkerman, A. L. Abdelhady and L. Manna, J. Phys. Chem. Lett., 2018, 9, 2326–2337 CrossRef CAS PubMed.
- L. N. Quan, R. Quintero-Bermudez, O. Voznyy, G. Walters, A. Jain, J. Z. Fan, X. Zheng, Z. Yang and E. H. Sargent, Adv. Mater., 2017, 29, 1605945 CrossRef PubMed.
- J. Xu, W. Huang, P. Li, D. R. Onken, C. Dun, Y. Guo, K. B. Ucer, C. Lu, H. Wang, S. M. Geyer, R. T. Williams and D. L. Carroll, Adv. Mater., 2017, 29, 1703703 CrossRef PubMed.
- Y. Wang, D. Yu, Z. Wang, X. Li, X. Chen, V. Nalla, H. Zeng and H. Sun, Small, 2017, 13, 1701587 CrossRef PubMed.
- X. Chen, F. Zhang, Y. Ge, L. Shi, S. Huang, J. Tang, Z. Lv, L. Zhang, B. Zou and H. Zhong, Adv. Funct. Mater., 2018, 28, 1706567 CrossRef.
- T. Xuan, S. Lou, J. Huang, L. Cao, X. Yang, H. Li and J. Wang, Nanoscale, 2018, 10, 9840–9844 RSC.
- C. Jia, H. Li, X. Meng and H. Li, Chem. Commun., 2018, 54, 6300–6303 RSC.
- Y.-M. Chen, Y. Zhou, Q. Zhao, J.-Y. Zhang, J.-P. Ma, T.-T. Xuan, S.-Q. Guo, Z.-J. Yong, J. Wang, Y. Kuroiwa, C. Moriyoshi and H.-T. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 15905–15912 CrossRef CAS PubMed.
- X. Zhang, H.-C. Wang, A.-C. Tang, S.-Y. Lin, H.-C. Tong, C.-Y. Chen, Y.-C. Lee, T.-L. Tsai and R.-S. Liu, Chem. Mater., 2016, 28, 8493–8497 CrossRef CAS.
- Z. Li, L. Kong, S. Huang and L. Li, Angew. Chem., 2017, 129, 8246–8250 CrossRef.
- L. Xu, J. Chen, J. Song, J. Li, J. Xue, Y. Dong, B. Cai, Q. Shan, B. Han and H. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 26556–26564 CrossRef CAS PubMed.
- Y. Liu, F. Li, Q. Liu and Z. Xia, Chem. Mater., 2018, 30, 6922–6929 CrossRef CAS.
- V. González-Pedro, S. A. Veldhuis, R. Begum, M. J. Bañuls, A. Bruno, N. Mathews, S. Mhaisalkar and Á. Maquieira, ACS Energy Lett., 2018, 3, 1409–1414 CrossRef.
- A. Pan, J. Wang, M. J. Jurow, M. Jia, Y. Liu, Y. Wu, Y. Zhang, L. He and Y. Liu, Chem. Mater., 2018, 30, 2771–2780 CrossRef CAS.
- H. C. Yoon, S. Lee, J. K. Song, H. Yang and Y. R. Do, ACS Appl. Mater. Interfaces, 2018, 10, 11756–11767 CrossRef CAS PubMed.
- S. Liu and X. Luo, LED packaging for lighting applications: design, manufacturing, and testing, John Wiley & Sons, 2011 Search PubMed.
- M. Bahadur, A. W. Norris, A. Zarisfi, J. S. Alger and C. C. Windiate, Sixth International Conference on Solid State Lighting, 2006.
- J. Hai, H. Li, Y. Zhao, F. Chen, Y. Peng and B. Wang, Chem. Commun., 2017, 53, 5400–5403 RSC.
- R. Zhang, H. Lin, Y. Yu, D. Chen, J. Xu and Y. Wang, Laser Photonics Rev., 2014, 8, 158–164 CrossRef CAS.
- X. Di, Z. Hu, J. Jiang, M. He, L. Zhou, W. Xiang and X. Liang, Chem. Commun., 2017, 53, 11068–11071 RSC.
- S. Liu, Y. Luo, M. He, X. Liang and W. Xiang, J. Eur. Ceram. Soc., 2017, 38, 1998–2004 CrossRef.
- B. Ai, C. Liu, J. Wang, J. Xie, J. Han and X. Zhao, J. Am. Ceram. Soc., 2016, 99, 2875–2877 CrossRef CAS.
- B. Ai, C. Liu, Z. Deng, J. Wang, J. Han and X. Zhao, Phys. Chem. Chem. Phys., 2017, 19, 17349–17355 RSC.
- R. Yuan, L. Shen, C. Shen, J. Liu, L. Zhou, W. Xiang and X. Liang, Chem. Commun., 2018, 54, 3395–3398 RSC.
- M. C. Brennan, J. E. Herr, T. S. Nguyen-Beck, J. Zinna, S. Draguta, S. Rouvimov, J. Parkhill and M. Kuno, J. Am. Chem. Soc., 2017, 139, 12201–12208 CrossRef CAS PubMed.
- N. Reitinger, A. Hohenau, S. Köstler, J. R. Krenn and A. Leitner, Phys. Status Solidi A, 2011, 208, 710–714 CrossRef CAS.
- J. Zhou, F. Huang, H. Lin, Z. Lin, J. Xu and Y. Wang, J. Mater. Chem. C, 2016, 4, 7601–7606 RSC.
- Y. Ohno, Fourth International Conference on Solid State Lighting, 2004.
- J. Zhang, R. Hu, X. Yu, B. Xie and X. Luo, Opt. Laser Technol., 2017, 88, 161–165 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |