
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProton-coupled multi-electron transfer and its relevance for artificial photosynthesis and photoredox catalysis
Andrea
Pannwitz†
 and
Oliver S.
Wenger
*
and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: oliver.wenger@unibas.ch
First published on 27th February 2019
Abstract
The conversion of CO2, H2O, or N2 to energy-rich compounds such as CH3OH, H2 or NH3 requires the properly orchestrated transfer of multiple electrons and protons. Artificial photosynthetic systems therefore must be able to synchronize the rapid primary photoinduced transfer of single electrons to the slower catalytic (multi-electron) turnover of substrates, and this generates a need for temporary accumulation and storage of redox equivalents. This is a very difficult task, particularly in absence of sacrificial reagents. Toward this end, proton-coupled multi-electron transfer (PCMET) driven by light is now receiving increased attention. This invited Feature article considers recent pertinent studies of donor–sensitizer–acceptor compounds and inorganic–organic hybrid systems, as well as some recent photoredox catalysis studies of proton-coupled multi-electron reductions. Key principles for successful light-driven accumulation and storage of redox equivalents are discussed, and the relevance of PCMET for the formation of solar fuels and for photoredox catalysis is emphasized.
1. Introduction
Basic research on artificial photosynthesis addresses diverse fundamental challenges ranging from photoinduced electron transfer to the development of suitable catalysts for the activation of small inert molecules. The conversion of CO2 to HCOOH or CH3OH is especially challenging as it relies on the transfer of multiple electrons and protons,1–3 and the same is true for water splitting or N2 fixation.4,5 In photoredox catalysis applied to synthetic organic chemistry, multi-electron transfer coupled to proton transfer can potentially yield products that are inaccessible via classic single-electron transfer (SET) reaction pathways. Furthermore, the transfer of multiple charges often operates at lower energy demand, as observed for example in the proton-coupled electrochemical reduction of CO2.1 It is therefore desirable to understand the basic principles of proton-coupled multi-electron transfer (PCMET).6 The specific case of a one proton and two electrons transfer resembles a net hydride transfer. Some tungsten-based metal hydrides have promising reactivities via proton-coupled electron transfer in the electronic ground state,7–9 and some iridium-based metal hydrides show immense potential in their photoexcited state under photoelectrochemical and photochemically reducing conditions for the production of H2 and NADH analogs.10–12 Generally, the reaction products of a hydride transfer can be very different from those resulting from PCMET,13,14 and we hope that selective formal and real hydride transfers, as well as other charge accumulated chemical conversions will be part of the common repertoire of light-driven reactions in the close future. This Feature article focuses on recent progress in the emerging field of PCMET, with particular focus on light-driven reactions of this type.The primary photochemical reactions in oxygenic photosynthesis are photoinduced long-range electron transfer and proton-coupled electron transfer (PCET). These reactions take place within the confined structure of the transmembrane protein super complex photosystem II in the chloroplasts.15 Upon absorption of four photons, the oxygen-evolving complex buried in this protein super complex accumulates four redox equivalents and oxidizes two water molecules to oxygen by liberating four protons in a series of PCET steps.16,17 Fundamental studies of photoinduced electron transfer often rely on donor–sensitizer–acceptor (D–S–A) compounds, in which electron–hole pairs comprised of oxidized donor (D+) and reduced acceptor (A−) are formed after photoexcitation of the sensitizer (Fig. 1a).18 It is tempting to think that light absorption and photoinduced electron transfer simply has to occur multiple times in such D–S–A compounds to result in the formation of D4+ and A4− (Fig. 1b), which then undergo multi-electron chemistry with suitable substrates, for example the splitting of water into O2 and H2. As noted before, it is not that simple in purely molecular systems.19,20 Aside from the need for suitable oxidation and reduction catalysts, the accumulation of multiple oxidation and reduction equivalents is very difficult, because many counter-productive processes can occur upon repeated excitation of a photosystem, as discussed in detail below.19,20 However, accumulation and temporary storage of redox equivalents seems indispensable, because the oxidation and reduction processes leading to conversion of energy-poor into more energy-rich compounds are unlikely to operate at identical rates. Moreover, the timescale of photoinduced electron transfer and single PCET steps is usually orders of magnitude shorter than the timescale of catalytic turnovers, and the available flux of photons can be comparatively low.21
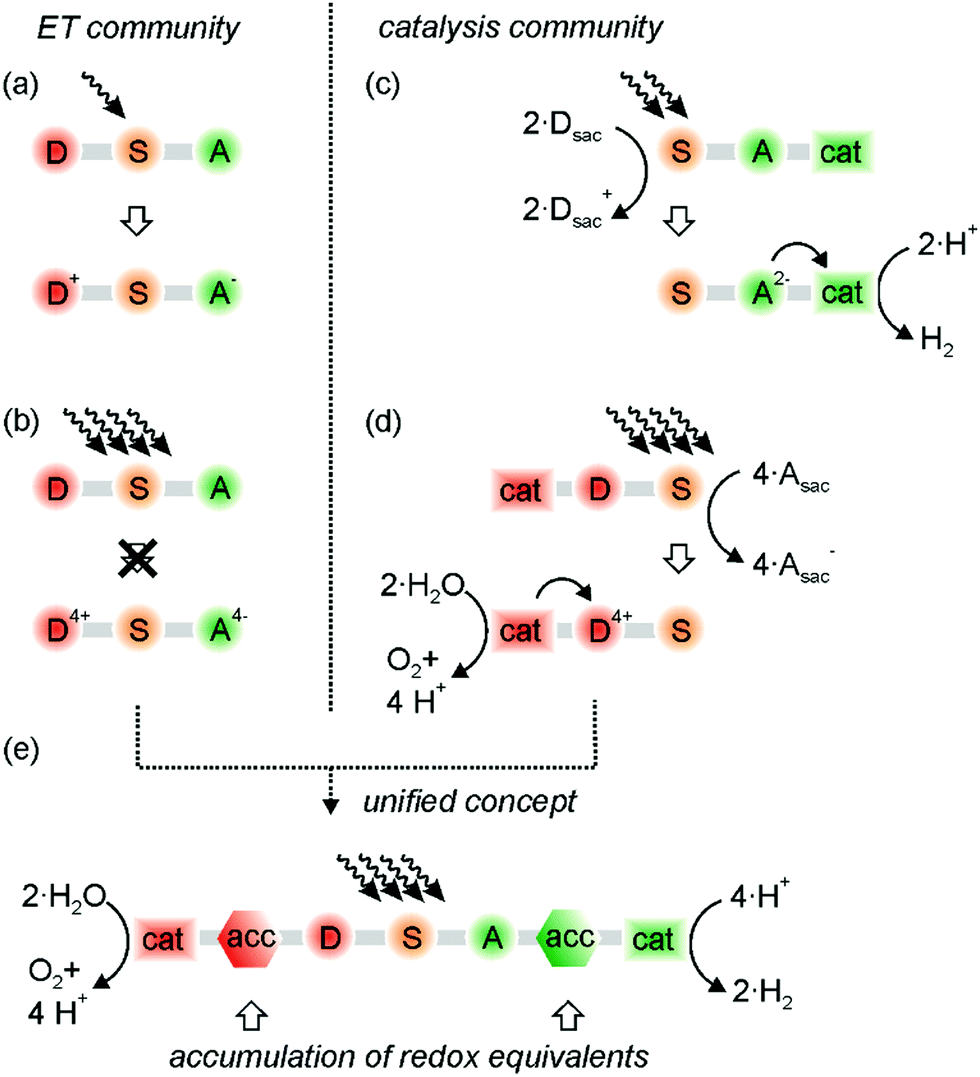 | ||
| Fig. 1 Some key concepts explored with donor–sensitizer–acceptor (D–S–A) compounds: (a) formation of a classic charge-separated state; (b) unrealistic scenario of multi-photon excitation and accumulation of multiple holes and electrons; (c) light-driven electron accumulation using sacrificial donors (Dsac) and proton reduction to H2 with a suitable catalyst; (d) light-driven hole accumulation using sacrificial acceptors (Asac) and water oxidation with a suitable catalyst: (e) water splitting without sacrificial reagents relying on temporary accumulation and storage of redox equivalents. | ||
The catalysis community has so far largely circumvented the fundamental challenges outlined above by focusing on half-reactions either in electrochemical settings, or by using sacrificial reagents (Fig. 1c and d). For example, the photochemical reduction of CO2 to various products has been achieved with sacrificial electron donors,1–3,22 and the catalyzed oxidation of water is now possible with spectacular turnover rates when using sacrificial oxidants.4,23 Interestingly, these oxidants are very often chemical oxidants, and not even photochemically produced ones.23,24 Sacrificial reagents decompose in the course of their oxidation or reduction, rendering undesired reverse reactions ineffective. However, they are energy-rich substances such as triethylamine, NADH analogs, or peroxydisulfate,25 and their use will not permit sustainable light-to-chemical energy conversion.
Proton-coupled multi-electron transfer (PCMET) connects the two research areas shown on the left- and right-hand sides of Fig. 1, because it can enable the accumulation and temporary storage of redox equivalents in similar manner as in bacterial photosynthesis. In our view, this is the missing link between the communities investigating the fast photoinduced electron transfer processes on the one hand and the (slower) catalysis of small-molecule activation by multi-electron transfer on the other hand (Fig. 1e). As we will emphasize in this Feature article, electron transfer reactions that are coupled to proton transfer seem essential, and the accumulation of oxidative and reductive equivalents seems superior over the accumulation of charges in the form of holes and electrons. Once the light-induced separation and the temporary storage of accumulated redox equivalents will become possible in an efficient manner, concepts as illustrated in Fig. 1e will finally become realizable, and oxidation and reduction half-reactions can be connected, eliminating the need for sacrificial reagents. Ultimately, artificial photosynthesis will of course have to rely on very robust accumulator and catalyst materials in order to become practical, but molecular systems remain indispensable for fundamental investigations.
Herein we first consider some light-induced SET processes in donor–sensitizer–acceptor compounds and discuss how the coupling to proton transfer can be beneficial for the temporary separation of (single) redox equivalents and, in the future, for the accumulation of multiple redox equivalents. Then we introduce some of the key challenges associated with photoinduced multi-electron transfer, before discussing recent case studies of PCMET and multi-electron photocatalysis in purely molecular compounds, as well as in some inorganic–organic hybrid systems involving nanoparticles or quantum dots. Lastly, we give some conclusions and an outlook.
2. From separating electrons and holes to separating redox equivalents: importance of photoinduced proton-coupled electron transfer
Traditional studies of donor–acceptor compounds only deal with the light-induced transfer of a single electron from the donor to the acceptor, which usually leads to short-lived electron–hole pairs (Fig. 1a). Porphyrin–quinone dyads are typical examples, and they resemble in their function the role of the P680/QA couple in bacterial photosynthesis:26 following energy transfer from the antenna system to P680, that chlorophyll unit donates an electron to the quinone called QA (purple arrows in Fig. 2a). Subsequently, onward electron transfer from quinone A to quinone B occurs, and the latter is protonated to its semiquinone form (upper red arrow in Fig. 2a). On the oxidative side, P680+ is reduced back to its neutral form by the nearby tyrosine Z (YZ) residue, and the latter is deprotonated to yield a tyrosyl radical (lower red arrow in Fig. 2a).15,17,27 The overall reaction up to this point corresponds to long-range electron transfer from YZ to QB, coupled to deprotonation of the former and protonation of the latter, yielding YZ˙ and QBH. Somewhat surprisingly, this overall reaction sequence had not been emulated in artificial donor–acceptor compounds until very recently,28 even though PCET in artificial YZ mimics has received much attention.29–43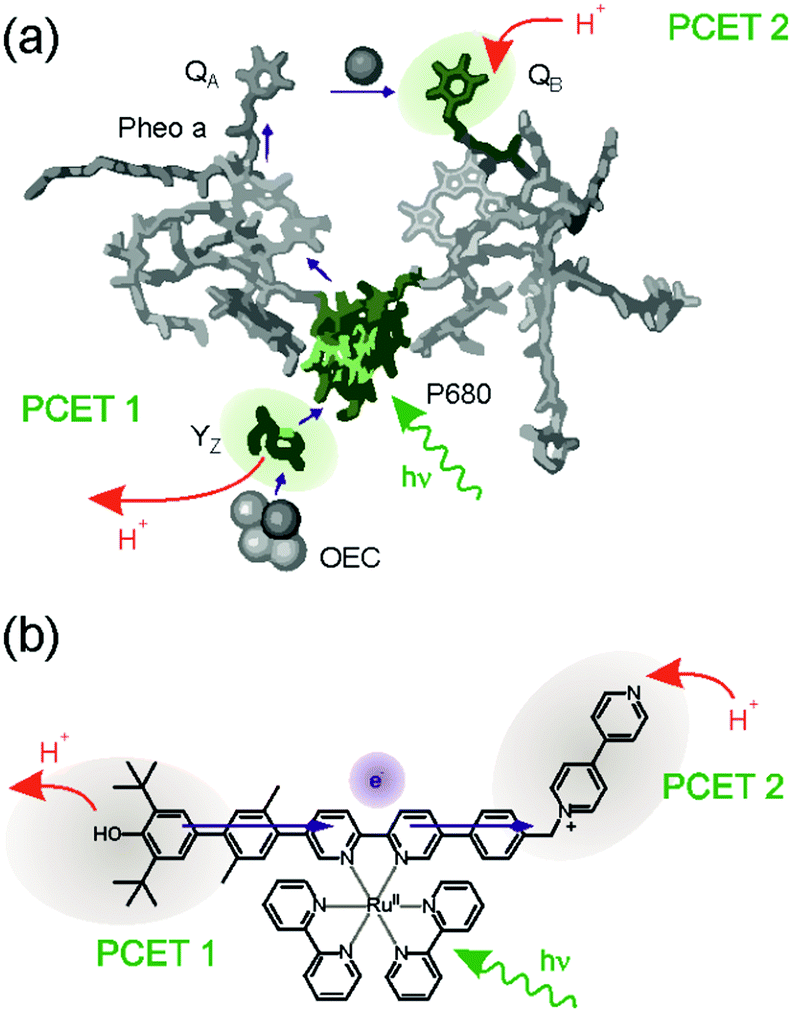 | ||
| Fig. 2 (a) Electron transfer (purple arrows) and proton transfer (red arrows) events occurring in photosystem II after excitation of P680. (b) Molecular triad emulating the function of the tyrosine Z, P680, and QB units of photosystem II. PCET occurs both at the donor and acceptor units, leading to oxidized and deprotonated donor, as well as reduced and protonated acceptor upon photoexcitation. Adapted with permission from ref. 28 Copyright 2017 American Chemical Society. | ||
The donor–sensitizer–acceptor compound in Fig. 2b contains all the necessary components to emulate the global reaction sequence seen for photosystem II in Fig. 2a. The central Ru(bpy)32+ (bpy = 2,2′-bipyridine) complex acts as the photosensitizer similar to P680 in the natural system, the phenolic unit emulates the function of YZ, and the N-methylbipyridinium (MQ+) moiety is a combined proton–electron acceptor that functions analogously to QB.28 Visible light triggers concerted proton–electron transfer (CPET) between the phenol and the excited Ru(bpy)32+ complex whereby the phenolic proton is released to the solvent, a mixture between pyridine and pyridinium. The one-electron reduced sensitizer rapidly passes the additional electron on to the MQ+ acceptor, which is then protonated by pyridinium. The net result is a photoproduct storing ca. 1.2 eV in the form of an oxidized and deprotonated donor (a neutral phenoxyl radical) combined with a reduced and protonated acceptor (MQH+). Conceptually, this is different from the classic case of a charge-separated state in the form of an electron–hole pair (Fig. 1a), because the donor and acceptor charges are unchanged with respect to the initial (ground) state. This is interesting for multi-electron transfer, because the build-up of positive charge on the donor unit as well as the build-up of negative charge at the acceptor is avoided by PCET. The accumulation of multiple reduction equivalents (comprised of an equal number of protons and electrons) is more favorable than the mere accumulation of several electrons, and likewise the accumulation of multiple oxidation equivalents (i.e., the release of an equal number of protons and electrons) is energetically less demanding. The overall photoinduced reaction in the molecular triad of Fig. 2b involves the transfer of one electron and two protons. Related reactions have been observed in so-called proton-relays,44 and were recently termed E2PT.45,46 The photogenerated radical pair state in the triad from Fig. 2b has a lifetime of 1.9 μs in de-aerated pyridine–pyridinium buffer at room temperature, and this is very similar to the typical lifetimes of classic charge-separated states in comparable molecular triads under similar conditions.47–51 Thus, the fact that PCET is involved at both the donor and the acceptor unit has little influence on the kinetic stability of the photoproduct, but it could make accumulation of redox equivalents after secondary photoexcitation more feasible as noted above. However, even when the build-up of charges is avoided, many challenges remain for productive multi-electron transfer, as discussed in the following.
3. Some fundamental challenges in photoinduced multi-electron transfer
Once a charge-separated state (such as that in the lower part of Fig. 1a) or a radical pair photoproduct (for example that resulting from the triad in Fig. 2b) has been formed, further excitation is usually necessary to accumulate charges or redox equivalents. Then, several hurdles must be overcome.19,20,52 For example, when taking the charge-separated state in Fig. 1a as the simplest example, one immediately notices that the oxidized donor (D+) is a strong acceptor because it lacks an electron. Analogously, the reduced acceptor (A−) has an excess electron and consequently is a potent donor. Thus, when exciting the sensitizer in the D+–S–A− primary photoproduct, oxidative quenching by D+ (Fig. 3a) or reductive quenching by A− (Fig. 3b) are usually the thermodynamically much preferred reactions, both ultimately leading to charge-recombination.52 To make things worse, D+ and A− are usually strongly colored radicals, and when they absorb the secondary excitation light directly, their oxidizing/reducing power is further amplified,53,54 and charge-recombination is bound to occur. This has been shown in recent two-pulse (pump–pump–probe) studies.55,56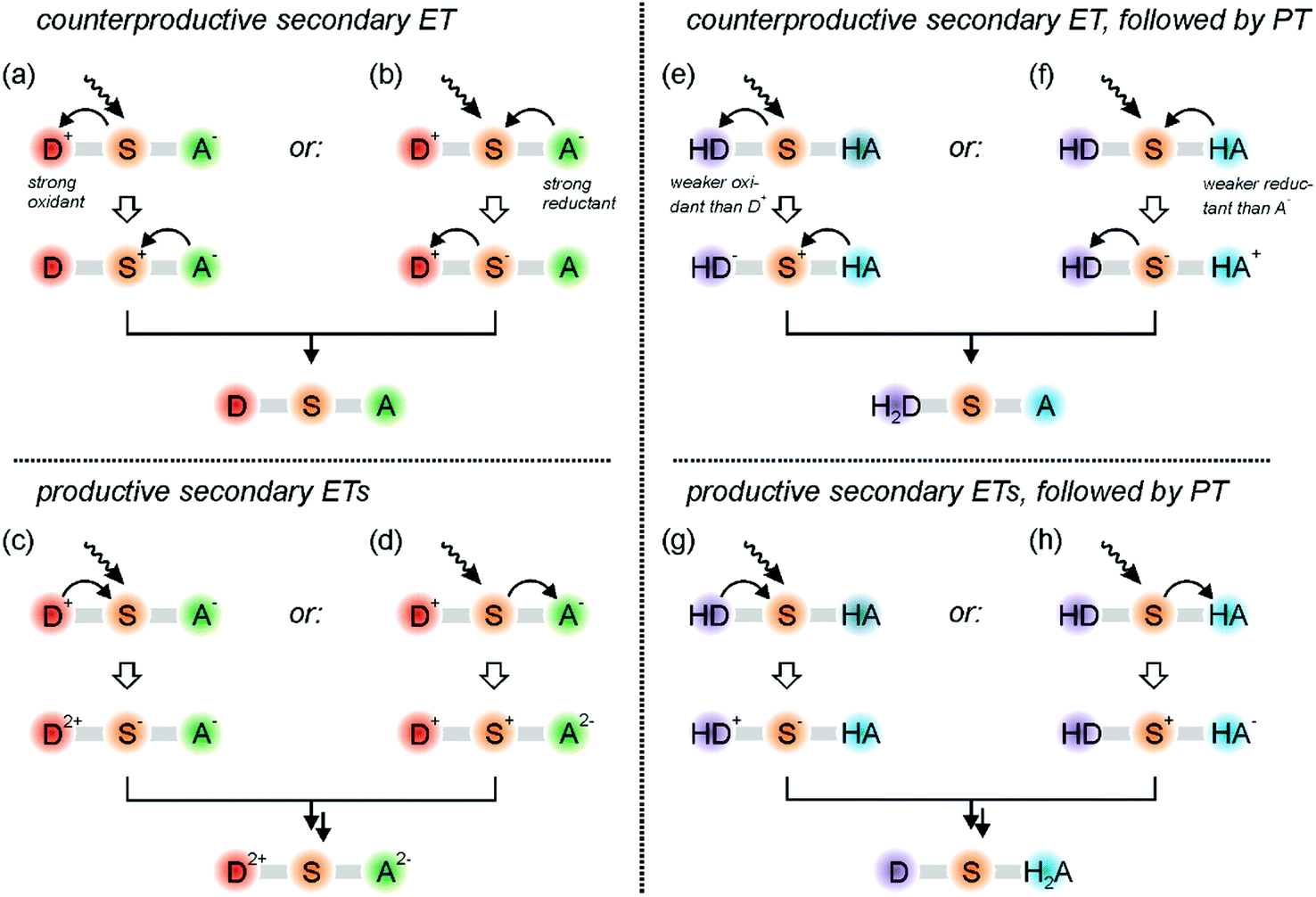 | ||
| Fig. 3 Secondary excitation of donor–sensitizer–acceptor (D–S–A) compounds after absorption of a first photon and formation of either a single electron–hole pair (a–d), or the separation of a single oxidation equivalent from a single reduction equivalent (e–h). On the left-hand side only electron transfer reactions in a D–S–A compound occur, whilst the reactions on the right-hand side involve PCET processes in a H2D–S–A triad comprised of a (doubly) deprotonatable donor and a (twofold) protonatable acceptor. | ||
Productive secondary electron transfers, either further oxidation of D+ to D2+ by the excited sensitizer (Fig. 3c) or further reduction of A− to A2− (Fig. 3d) have much less driving-force than the counter-productive reactions in Fig. 3a and b. If they nevertheless do occur, further charge-shift reactions (for example electron transfer from S− to A−; Fig. 3c) are in competition with the thermodynamically more favorable recombination reaction leading back to the D+–S–A− state. Thus, the formation of the target D2+–S–A2− photoproduct faces very significant thermodynamic and kinetic challenges, and consequently it is not so surprising that this has not been observed for purely molecular donor–sensitizer–acceptor compounds until now.
However, a few years ago the groups of Hammarström and Odobel reported on a molecular system attached to TiO2 in which the nanoparticles act as a two-electron acceptor.57,58 Upon consecutive excitation of a Ru(bpy)32+ sensitizer, two electrons are transferred from an oligo-triarylamine donor to the semiconductor. The large density of acceptor states in the latter greatly facilitates the uptake of two electrons. This is a significant benefit compared to most molecular acceptors and makes the overall two-electron transfer more readily possible. Nevertheless, given a quantum yield of ca. 0.3 for both primary and secondary charge-separation in that system, the overall quantum yield for the formation of the final two-electron charge-separated state (D2+–S–A2−) is limited to 0.09.57,58 Related work with TiO2 as an acceptor has been reported recently by Ardo.59 Furthermore, the inverse situation was realized with a semiconductor quantum dot (CdS) acting as a multi-electron donor whilst an electrostatically bound molecular viologen-derivative served as a two-electron acceptor.60 There are now a handful of cases in which charge-accumulation in purely molecular systems has been achieved without sacrificial reagents.20,61–63 However, for these systems it was crucial to have multiple donors for the accumulation of electrons on a single acceptor, or conversely, to have multiple acceptors for the accumulation of holes on a single donor.62–67
Fig. 3a–d summarizes the current struggles in the field.19,20,68 For solving these challenges in the future, PCET must be considered. When combining a two-electron deprotonatable donor (H2D) with an acceptor (A) that is protonatable upon reduction, some advantages emerge. In that case, absorption of a first photon by a sensitizer can lead to a primary photoproduct comprised of oxidized and singly deprotonated donor (HD) along with reduced and singly protonated acceptor (HA), which is in analogy to what has been observed for the triad in Fig. 2b. When now the sensitizer of this primary photoproduct state is excited again (Fig. 3e), then counter-productive electron transfer to the oxidized donor is still possible, but HD is a significantly weaker oxidant than D+. In other words, undesired reverse electron transfer becomes thermodynamically less favorable, and the desired accumulative electron transfer involving HD becomes thermodynamically less unfavorable (Fig. 3g). The same logic applies to the reductive side: energy-wasting reverse electron transfer from HA to photoexcited S is thermodynamically less favorable (Fig. 3f), and therefore the energy-storing onward electron transfer to produce HA− (or H2A in case of PCET) is less unfavorable (Fig. 3h). Thus, as long as the involved electron transfer (or PCET) processes occur in the so-called Marcus normal region, donor deprotonation and acceptor protonation is beneficial for the light-driven accumulation of redox equivalents. There can also be advantages regarding the lifetimes of the photoproducts as discussed in the next section.
4. PCMET in molecular donor–bridge–acceptor compounds
Along with the group of Hamm, we recently synthesized and explored a molecular pentad comprised of a central anthraquinone (AQ) acceptor flanked by two Ru(bpy)32+ sensitizers and two peripheral triarylamine (TAA) donors (Fig. 4a).67,69 Unlike triethylamine or related sacrificial donors, the triarylamine units are reversible single-electron donors, and hence this molecular pentad is useful for exploration of reversible multi-electron transfer and charge accumulation. With the experimental setup used in our investigations, both Ru(bpy)32+ units of a given pentad can be promoted to their photoactive 3MLCT states within the same laser pulse. This leads to intramolecular electron transfer from the TAA donors to the central acceptor unit within 65 ps, forming the quinone dianion (AQ2−) in neat CH3CN at room temperature. The pertinent AQ2− infrared absorption bands exhibit a quadratic dependence on the laser excitation power, as expected for a two-photon absorption process in the low power regime. Under optimized conditions 15% of all excited pentads undergo double electron transfer to form AQ2− whilst the remaining 85% exhibit only single electron transfer to result in AQ−. Once formed, the AQ2− photoproduct is kinetically remarkably stable, exhibiting a lifetime of 870 ns in de-aerated CH3CN at room temperature. This long-lived photoproduct stores ca. 3.5 eV of energy, whilst common donor–sensitizer–acceptor triads typically store only 1.4–2.0 eV.47,49,51,70 There were only two prior studies of charge-separated states in which two electrons are reversibly accumulated on a single acceptor of a purely molecular system, and in both cases the lifetime of that state was shorter than 10 ns.64,65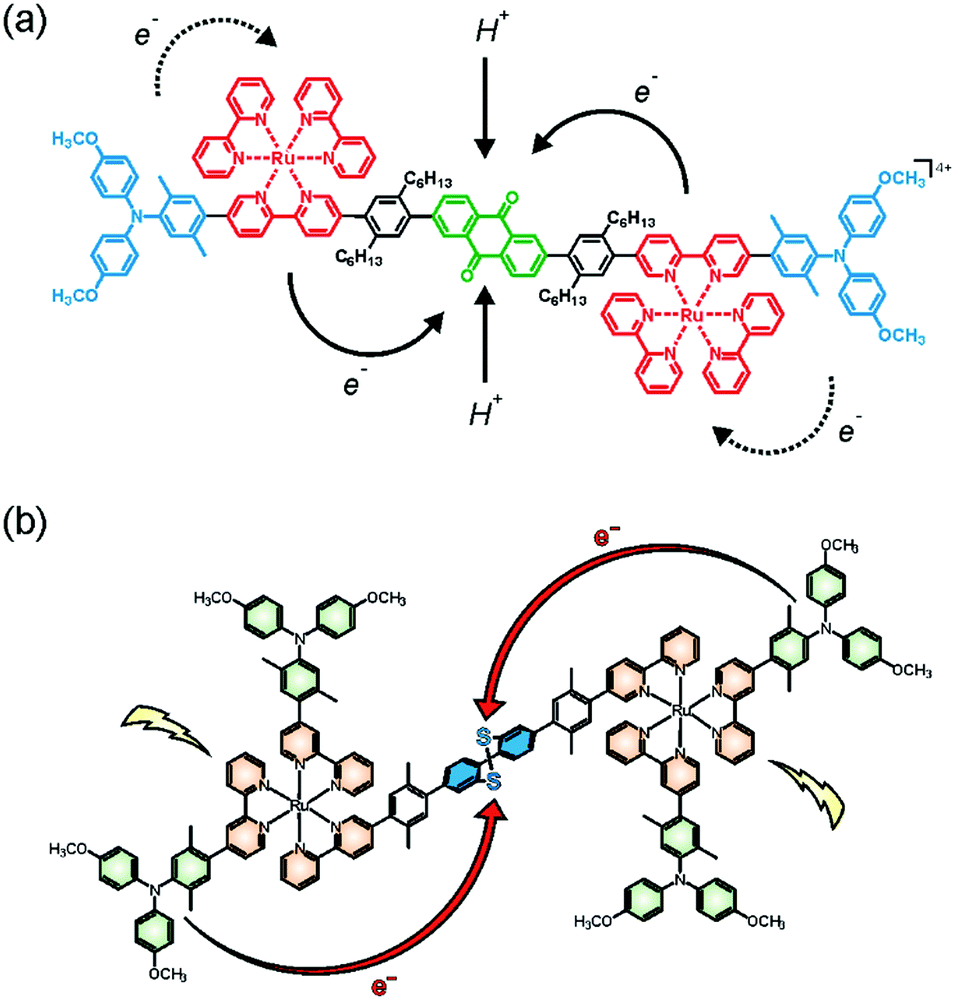 | ||
| Fig. 4 (a) Molecular pentad for reversible accumulation of two electrons and two protons on an anthraquinone acceptor;67,69 (b) electron accumulation on a dibenzo[1,2]dithiin acceptor exhibiting redox potential inversion.71,72 Copyright 2017 & 2018 American Chemical Society. | ||
In presence of 0.2 M p-toluenesulfonic acid (TsOH), AQ2− is protonated and the hydroquinone AQH2 forms, leading to an overall photoproduct comprised of two TAA+ units and one AQH2 with a lifetime of 4.7 μs in CH3CN at room temperature.69 Given the large energetic stabilization resulting from protonation of AQ2− (ca. 1.5 eV), the lifetime prolongation from 870 ns to 4.7 μs is relatively modest and could point to an important inverted driving-force effect for the AQ2− state storing 3.5 eV. Transient infrared spectroscopy shows that AQH2 forms via a stepwise electron transfer, proton transfer mechanism, with the AQ2− intermediate clearly visible after 65 ps whilst the first proton transfer step occurs with a time constant of 3 ns. By contrast, the intramolecular re-oxidation of AQH2 by the two TAA+ units is a proton-coupled reaction with an H/D kinetic isotope effect (KIE) of 1.4 (involving TsO− as base). However, there is no evidence for the formation of a semiquinone (AQH) intermediate and the starting materials (TAA, AQ) are re-instated directly, suggesting that oxidation of AQH2 occurs via a concerted two-electron, two-proton coupled reaction.69
In a related electrochemical study, Meyer and coworkers explored the two-electron reduction of tetramethylquinone by a riboflavin derivative bound to an oxide surface.73 This reaction occurs via protonation of the quinone and a rate-limiting hydride transfer.
The compound in Fig. 4b is comprised of four TAA donors, two Ru(bpy)32+ sensitizers, and one dibenzo[1,2]dithiin (PhSSPh) acceptor and functions as a molecular pentad similar to the compound in Fig. 4a.71 The key difference is that the PhSSPh acceptor exhibits very strong redox potential inversion, i.e., the second reduction to form to the dianion is far easier (ca. 1.3 V) than the first reduction of the neutral species to the monoanion due to structural changes associated with the second reduction.74 It was hoped that this would facilitate the observation of a two-electron reduction photoproduct, which is normally formed in small quantity compared to products resulting from single electron transfer.71 Excitation of the Ru(bpy)32+ units at 532 nm with ns laser pulses leads to the desired dithiolate photoproduct (PhS−PhS−) with a quantum yield of 0.5% in neat CH3CN, and reverse electron transfer to the two TAA+ units occurs with a time constant of 66 ns at room temperature. In presence of 0.2 M TsOH, the dithiol (PhSHPhSH) photoproduct accumulates under steady-state irradiation with a 455 nm LED and then remains stable on a minute timescale. The PCET mechanisms were not investigated in detail and it is not clear to what extent concerted and stepwise processes are involved in the overall reversible two-electron, two-proton-coupled reaction. However, it is very obvious that PCET entails an enormous lifetime prolongation for the two-electron reduced photoproduct.
The compound in Fig. 4b, as well as a simpler variant containing no peripheral triarylamine units, are able to catalyze the reduction of aliphatic disulfides to thiols in presence of excess sacrificial donors.72 Mechanistic studies reveal that this occurs via a thiolate–disulfide interchange reaction involving the two-electron reduced form of the dibenzo[1,2]dithiin unit. This provides the proof-of-concept that multielectron photoredox chemistry with organic substrates is possible after initial charge accumulation on integrated donor–sensitizer–acceptor compounds.
The concept of redox potential inversion seems generally promising for the light-driven accumulation of redox equivalents. The acceptor present in the compound of Fig. 4b had been investigated previously by Benniston and Harriman (Fig. 5a),75 whilst Glass and Evans explored a related 4,4′-bpy compound (Fig. 5c) as well as its methyl viologen analogue (Fig. 5b).74 More recently, Cattaneo, Siewert, and Meyer developed a 2,2′-bpy version of that same disulfide switch (Fig. 5d), for which it was found that water triggers redox potential inversion due to protonation of the radical monoanion at its basic N-atom.76 It will be interesting to see how that bpy ligand behaves electrochemically and photochemically when coordinated to a metal center.
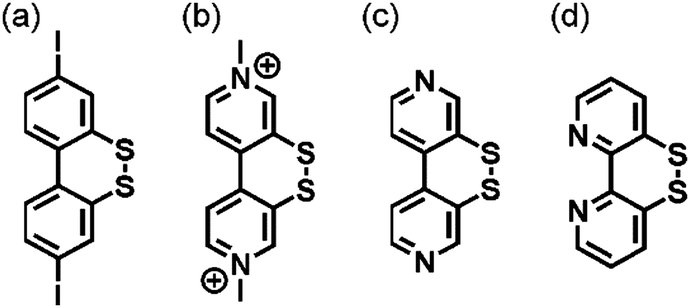 | ||
| Fig. 5 Compounds exhibiting redox potential inversion.74–76 | ||
5. PCMET in photoredox catalysis
In photoredox catalysis, the photoinitiated transfer of a single electron to a substrate is often applied to generate radical intermediates that are coupled to a reaction partner to give access to products of added value,77,78 and photoinduced PCET in the initial charge transfer can be very beneficial in this context.79 In the course of our investigations of photoinduced multi-electron transfer, we became interested in the reductive amination by photoredox catalysis. We found that a combination of ascorbate (AscH−) and mercaptopropionic acid (MPA) is well suited for the light-driven reductive amination of a range of aliphatic aldehydes and ketones with anilines and aliphatic amines.80 After reductive quenching of 3MLCT-excited Ru(bpy)32+, single electron transfer (SET) to iminium cations (formed via condensation of the substrates) produces highly reactive α-amino alkyl radicals as key intermediates (rectangular box in Fig. 6a). A cyclopropyl-based ketone substrate was used as a radical clock to gain information on the kinetics of the subsequent hydrogen atom transfer (HAT) yielding the final product. As long as only AscH− is present, the cyclopropyl-ring opens more rapidly than HAT can occur, leading to 2-pentanone (bottom right corner of Fig. 6a). When a mixture of MPA and AscH− is used, the ring-retention reductive amination product is instead obtained, indicating that MPA acts as a rapid (>107 M−1 s−1) HAT donor to the α-amino alkyl radical intermediate. However, MPA alone does not lead to any product accumulation since HAT between MPA and the α-amino alkyl radical is reversible due to similar bond dissociation energies (BDEs) associated with the relevant S–H and C–H bonds (Fig. 6b). The reaction between AscH− and the α-amino alkyl radical is exergonic by ca. 16 kcal mol−1 (Fig. 6b) but comparatively slow (Fig. 6a) because AscH− is an anion and the α-amino alkyl radical is an electron-rich, nucleophilic intermediate.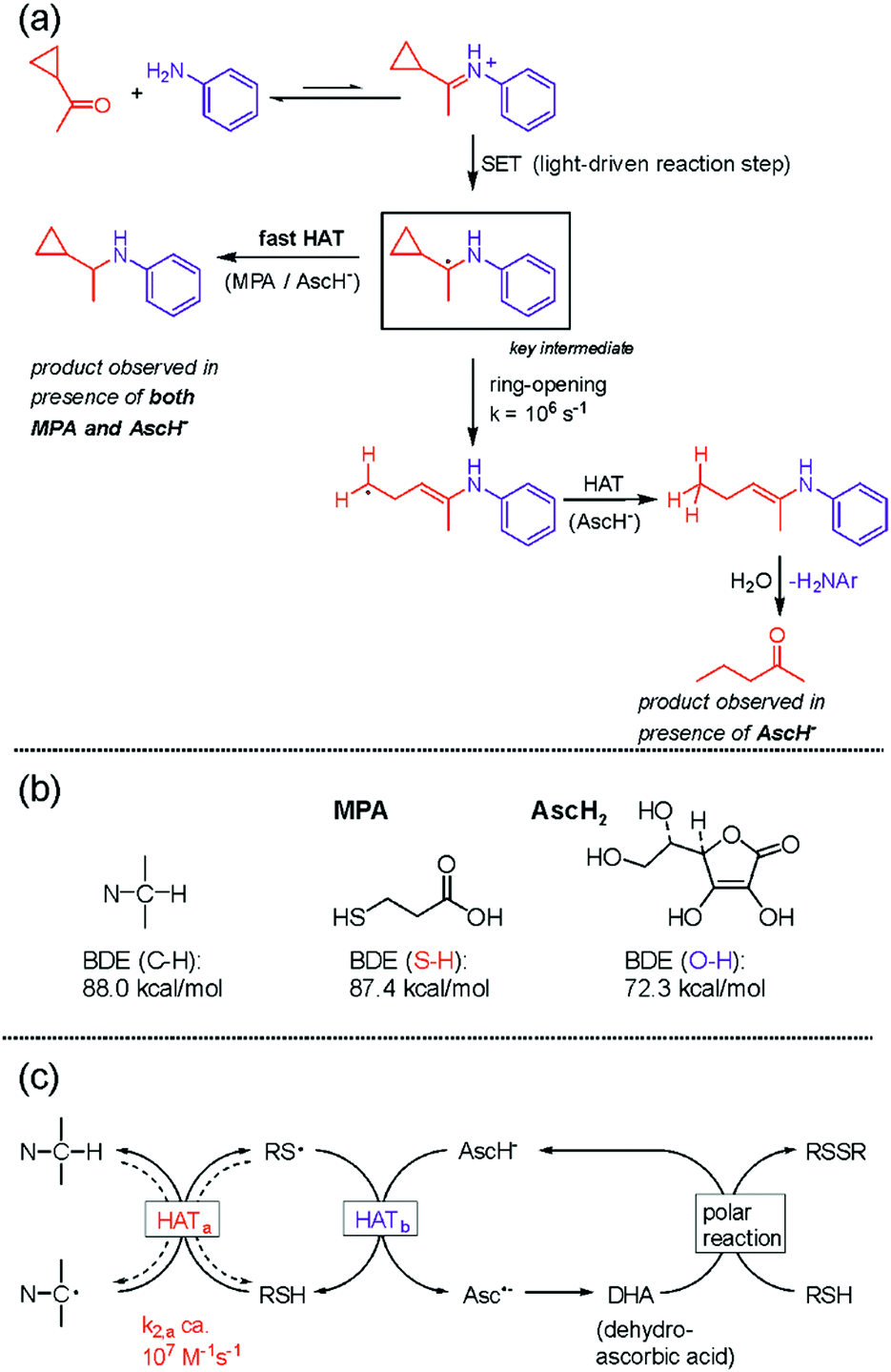 | ||
| Fig. 6 Reductive amination by photoredox catalysis:80 (a) radical clock experiment used to estimate the kinetics of hydrogen atom transfer (HAT); (b) relevant bond dissociation energies (BDEs); (c) overall mechanism involving polarity-matched HAT. | ||
When both MPA and AscH− are present, initial rapid HAT between MPA and the α-amino alkyl radical produces an electrophilic thiyl radical (HATa in Fig. 6c) that can be intercepted by AscH− (HATb in Fig. 6c) before the undesired reverse reaction occurs. This concept of polarity-matching had long been known,81 but came only recently into the focus of photoredox studies.82 The dehydroascorbic acid (DHA) oxidation product is then regenerated to AscH− by MPA, yielding the disulfide of MPA as a terminal oxidation product and making AscH− a co-catalyst of the overall reaction. Thus, the overall process of reductive amination by photoredox catalysis involves an initial electron transfer followed by HAT, which in sum corresponds to a proton-coupled two-electron transfer reaction. Following Mayer's recent notion that PCET and HAT are part of a continuum of related reaction types,83 it seems justified to classify this reductive amination method as a PCMET process. A key difference to the PCMET studies discussed in the prior section is that only the initial electron transfer is photoinduced and the subsequent HAT is a thermal process, whilst in the compounds from Fig. 4 every reduction step requires photoexcitation. Meanwhile, another photoredox method for reductive amination has also been reported.84 The concept of two-electron reduction by sequential photoinduced electron transfer and HAT has been further exploited for the enantioselective synthesis of amines by combined photoredox and enzymatic catalysis.85
Weiss and coworkers discovered that nitrobenzene can be reduced to aniline by photocatalysis with MPA-capped CdS quantum dots in a sequence of 6 consecutive PCET steps (Fig. 7).86 MPA serves as a source of electrons and protons, and methanol solvent also acts as an electron donor. Excitation with 405 nm light produces excitons which are reductively quenched by surface-bound MPA, leading to one-electron reduced quantum dots that deliver one electron at a time to the substrate. Nitrosobenzene is more readily reduced than nitrobenzene hence former is not observed, but the phenylhydroxylamine intermediate and the aniline product are both detectable by GC-MS. It was estimated that ca. 80 molecules of nitrobenzene, nitrosobenzene, phenylhydroxylamine, or aniline can bind to a given quantum dot, and it seems plausible that all of the 6 reduction steps are “static” PCET reactions from quantum dots to pre-adsorbed molecules which do not desorb from the surface until the final product is formed. Acidic conditions are beneficial for avoiding catalyst poisoning by aniline, and moreover, protons are then readily available for PCET. Due to uncertainties in the reduction potential for the quantum dots it is not possible to know the absolute values of the driving-forces for the individual reaction steps. However, mechanistic studies suggest that electron transfer, and not proton transfer, is the rate-limiting step for each reduction.
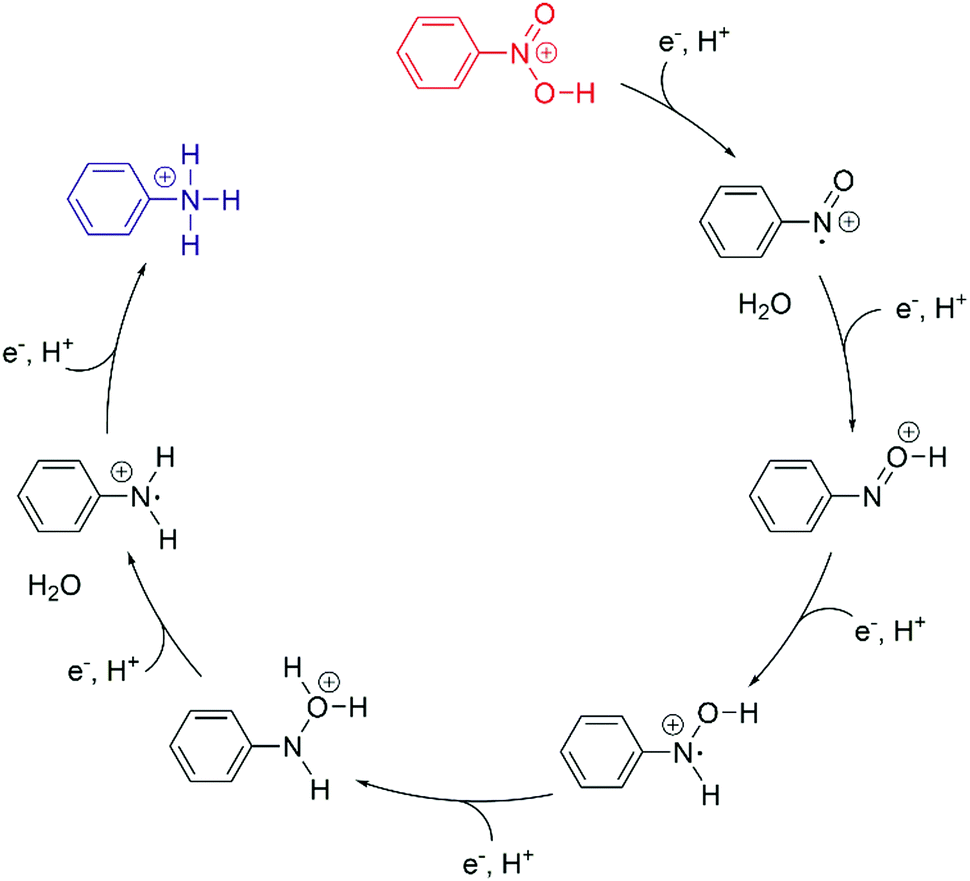 | ||
| Fig. 7 Photocatalytic conversion of nitrobenzene to aniline in six consecutive PCET steps using MPA-capped CdS quantum dots as sensitizers and MPA/methanol as electron source.86 The overall reaction was performed under acidic conditions and proceeds through nitrosobenzene and phenylhydroxylamine intermediates. | ||
The quantum dot based catalytic system is very robust and exhibited no detectable decrease in activity even after the transfer of 4.5 × 106 electrons per quantum dot. Turnover rates of 23 electrons donated per quantum dot and second were obtained, and 2.6 aniline molecules could be produced per quantum dot and second. However, with a photon flux of 4.6 × 1017 photons cm−2 s−1 there is no charge accumulation in the quantum dots.
In a recent spectacular case of PCMET involving a light-driven reaction step, dinitrogen splitting was achieved using a rhenium complex with a pincer ligand that can act as a 2 e−/2 H+ reservoir via interconversion between imine and amine forms.87 Benzamide and benzonitrile can be cleanly formed from N2 in overall 61% yield in a three-step cycle. The cycle starts with electrochemical N2 activation leading to the dinuclear N2-bridged complex, continues with photochemical splitting of the latter into nitride complexes, and ends with thermal nitrogen transfer from the nitride complexes to the benzoyl chloride substrate.
6. PCMET in nanocrystals and photocatalysis with some inorganic–organic hybrid systems
When ZnO or TiO2 nanoparticles in toluene are irradiated with UV light, they can undergo a photoinduced redox reaction with residual ethanol remaining from their synthesis and then remain relatively stable in their intensely blue colored reduced forms. Mayer and coworkers demonstrated that the description as ZnO/e− and TiO2/e− is incomplete because the reduction reaction is proton-coupled, i.e., these oxide nanoparticles are in fact PCET reagents.88,89 Addition of the stable 2,4,6-tri-tert-butylphenoxyl (tBu3ArO˙) or TEMPO radicals to suspensions of photoirradiated ZnO and TiO2 nanoparticles yields tBu3ArOH and TEMPOH, demonstrating that the reduced oxide particles are combined proton–electron donors. The likely reaction mechanism is concerted proton–electron transfer, because an initial outer-sphere electron transfer (as commonly proposed for interfacial redox reactions) would have to form tBu3PhO− or TEMPO− in a thermodynamically very unfavorable step. Titration experiments revealed that the average number of reduction equivalents is 4 for ZnO nanoparticles of 3.9 nm diameter after 30 minutes of irradiation, whereas for TiO2 nanoparticles of 3 nm diameter the average number is 45 after 1 hour. This latter value corresponds to reduction of 10% of all Ti4+ ions to Ti3+, whilst in ZnO the added electrons occupy more delocalized, conduction band-like orbitals. The active protons presumably originate from the particle synthesis and from the photochemical charging itself. Residual ethanol from the synthesis is oxidized to acetaldehyde, and this liberates protons. Moreover, surface hydroxide groups are likely to be present. Conceptually, the PCET chemistry observed for these metal oxides is similar to the accumulation of both e− and Li+ upon charging of a lithium battery anode.90In follow-up studies Mayer, Gamelin, and coworkers explored the chemical reduction of ZnO nanocrystals in aprotic solvents in which the proton/electron stoichiometry can be controlled precisely.91,92 By adding CoCp*2 (Cp* = η5-pentamethylcyclopentadienyl) or CrCp*2 in combination with acid to toluene/THF suspensions of ZnO nanocrystals, the number of electrons per nanocrystal (〈ne−〉) was determined as a function of driving-force (Fig. 8a). The linear correlation between the added numbers of electrons and protons indicates a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 e−/H+ process. Depending on the size of the nanocrystal, a different maximum number of electrons (〈ne−〉max) can be stored per nanocrystal. For instance, nanocrystals with an average size of 1.7 nm store up to 8 electrons, whereas nanocrystals with an average radius of 3.5 nm saturate at about 120 electrons. 〈ne−〉max varies systematically with the volume of the nanocrystals, and for CoCp2* a nearly constant maximum carrier density of (4.4 ± 1.0) × 1020 cm−3 for particle sizes between 1.5 and 5 nm was found. This corresponds to approximately one additional electron for every 100 Zn2+ ions. Even though CrCp*2 is ca. 400 mV less reducing than CoCp*2, the latter only transfers ca. 3 times more electrons than CrCp*2. However, the effective reduction potential of the nanocrystals is not constant, and hence the higher reducing power of CoCp*2 does not lead to a charge carrier density that is orders of magnitude higher, but rather it leads to the introduction of higher energy electrons. The charge balancing protons possibly intercalate into the lattice of the nanocrystals rather than merely attaching to their surface, as the very large difference in electron addition between H+ and CoCp*2+ cation suggests. CoCp*2 alone adds up to 40 electrons to nanocrystals of 4.9 nm radius, but when protons balance the charge, this reductant adds over 200 electrons to nanocrystals of that size.
:1 e−/H+ process. Depending on the size of the nanocrystal, a different maximum number of electrons (〈ne−〉max) can be stored per nanocrystal. For instance, nanocrystals with an average size of 1.7 nm store up to 8 electrons, whereas nanocrystals with an average radius of 3.5 nm saturate at about 120 electrons. 〈ne−〉max varies systematically with the volume of the nanocrystals, and for CoCp2* a nearly constant maximum carrier density of (4.4 ± 1.0) × 1020 cm−3 for particle sizes between 1.5 and 5 nm was found. This corresponds to approximately one additional electron for every 100 Zn2+ ions. Even though CrCp*2 is ca. 400 mV less reducing than CoCp*2, the latter only transfers ca. 3 times more electrons than CrCp*2. However, the effective reduction potential of the nanocrystals is not constant, and hence the higher reducing power of CoCp*2 does not lead to a charge carrier density that is orders of magnitude higher, but rather it leads to the introduction of higher energy electrons. The charge balancing protons possibly intercalate into the lattice of the nanocrystals rather than merely attaching to their surface, as the very large difference in electron addition between H+ and CoCp*2+ cation suggests. CoCp*2 alone adds up to 40 electrons to nanocrystals of 4.9 nm radius, but when protons balance the charge, this reductant adds over 200 electrons to nanocrystals of that size.
 | ||
| Fig. 8 (a) Chemical reduction of ZnO nanoparticles with CoCp*2 in presence of acid;91 (b) the assembly of Pt nanoparticles with CdS/CdSe quantum dots via polyacrylate linkers leads to a system that is able to produce H2 upon irradiation with visible light.93 | ||
Aside from these fundamental studies of PCET with oxide materials, nanoparticles continue to be of interest for the photochemical H2 production. Wu and coworkers explored self-assembled frameworks between ligand-free CdSe/CdS quantum dots (r ≈ 2.0 nm) and Pt nanoparticles (r ≈ 3.5 nm) that are decorated with polyacrylate ligands.93 The latter have pendant carboxylate groups that coordinate the Cd2+ ions of the quantum dots with binding constants of ∼106 M−1 in water, leading to a network in which multiple photosensitizers (CdSe/CdS) are connected per Pt proton reduction catalyst (Fig. 8b). This enables interparticle electron transfer on a timescale of ca. 65 ps after excitation of the CdSe/CdS quantum dots. Continuous irradiation at 450 nm over 8 hours yields a total TON of more than 1.64 × 107 mol of H2 per mol of Pt and a TOF of 570 s−1 with triethylamine (Et3N) as sacrificial reagent. Only trace amounts of H2 were formed in a reference experiment in which MPA-capped CdSe/CdS quantum dots were used, highlighting the advantage of the covalent framework for electron accumulation on Pt and H2 production.
Cronin, Symes, and coworkers demonstrated that the [P2W18O62]6− polyoxoanion can reversibly accept 18 electrons and protons in an electrochemical flow system.94 When exposed to Pt/C, an initial rate of 3.5 mmol of H2 per hour per mg of Pt was formed. This polyoxoanion can therefore be used in electrolytic cells for the on-demand liberation of H2. Alternatively, it can be employed as an electrolyte in redox flow batteries, where it enabled an energy density of up to 225 W h l−1.94
The photocatalytic H2 evolution on Pt nanoparticles using molecular photosensitizers, notably Ru(bpy)32+ derivatives, continues to be of interest. In this context, recent work on dendrimer-like systems by Sakai and coworkers seems noteworthy.95,96 By using bpy ligands with covalently attached methyl viologen acceptors and sacrificial reagents, these researchers achieved multi-electron storage, and in presence of Pt colloids catalytic H2 evolution was observed. Conceptually this is related to earlier work on electron accumulation with sacrificial reagents by MacDonnell, Campagna, and various other research groups.97–105
7. Conclusions and outlook
The basic principles of single PCET events become increasingly well understood,83,106 and the interest in photoinduced reactions involving the transfer of multiple electrons and protons is currently growing. In PCET reactions, concerted mechanisms avoid the formation of charged high-energy intermediates which would be formed in stepwise electron transfer–proton transfer (or opposite order) processes.30,52,107 In multi-electron transfer reactions, protonation of reduction products and deprotonation of oxidation products permits the accumulation of multiple redox equivalents without the build-up of charge.28,69 Nature follows this principle for example in the oxygen-evolving Mn4Ca cluster or in plastoquinone,15,21 and recently explored artificial model systems emulate this behavior.28,69,71,72 In most cases of reversible redox reactions explored to date, only one electron per absorbed quantum of light can be transferred, sometimes coupled to proton transfer. Consequently, the light-driven accumulation of redox equivalents is usually a multi-step process requiring the sequential absorption of several photons, and each individual electron transfer step can be coupled to proton transfer in a different manner. In other words, many combinations of concerted proton–electron transfers and consecutive electron, proton transfer reaction sequences are possible. Mechanistic investigations as detailed as those performed in the past for single PCET events are therefore very difficult for PCMET. Pump–pump–probe experiments can provide valuable mechanistic insight into light-induced PCMET, but such experiments tend to be significantly more challenging than ordinary transient absorption studies.55,57,64,108Proton-coupling of multi-electron transfer reactions not only helps to avoid the build-up of charges when accumulating multiple redox equivalents, but in some cases it can even provide a distinct thermodynamic advantage by making a secondary redox process more energetically favorable than a primary redox reaction. For instance, under sufficiently acidic conditions, semiquinone is more readily reduced than p-benzoquinone, and consequently the two-electron reduction of p-benzoquinone to hydroquinone is facilitated.109 In non-proton-coupled redox reactions, every additional redox step is usually thermodynamically less favored than the preceding step, except in the somewhat special cases of redox potential inversion.71,72,74–76,110
As noted in the introduction, temporary storage of redox equivalents is needed to synchronize the fast primary photoinduced electron transfer events with the slower multi-electron turnovers in catalytic reaction centers, but there is also the important issue of photon flux. Two-electron transfer is difficult to induce upon single excitation.111 The need for the consecutive absorption of multiple photons leads to a situation in which all relevant intermediate photoproducts must have a sufficiently long lifetime to survive until the photon needed to perform the next redox step reaches the system. Proton-coupled reactions lead to less reactive intermediates, increasing the chances for productive secondary reactions before energy-wasting reverse reactions occur.28,69 The use of antenna systems could compensate for low photon fluxes.68,112
The fundamental studies performed over the past few years on donor–bridge–acceptor compounds provided some insight into reversible multi-electron transfer reactions, and in the mid- to long-term, this could help eliminate the need for sacrificial reagents in research on artificial photosynthetic systems. Spatial separation of oxidation and reduction events then becomes an important issue.113,114
Sacrificial reagents are less problematic for multi-electron reductions (or oxidations) of organic substrates via photoredox catalysis, at least when the products are sufficiently valuable and when the side products resulting from the decomposition of the sacrificial reagent are readily separable. Until recently, two-electron reductions via photoredox catalysis were comparatively rare, because most photoredox catalysis operates on an SET basis generating reactive radical intermediates that must be intercepted rapidly. This has been exploited more frequently for coupling rather than reduction reactions, also in cases in which PCET was involved in the photoredox catalysis.79 Until now, multi-electron reductions via photoredox catalysis mostly rely on an initial photoinduced and a secondary thermal step,80,85 for example SET followed by subsequent thermal HAT. This is fundamentally different from the cases discussed in Sections 4 and 6 in which all individual electron transfer (or PCET) elementary steps are in fact photoinduced.
In photochemistry, there is always a kinetic challenge, because chemical reactions must be initiated before electronically excited states deactivate. For photoinduced multi-electron transfer, the situation is worse because usually multiple photons must be absorbed sequentially in order to drive multiple subsequent electron transfer reactions, and each additional electron transfer step tends to become more challenging than the preceding step. The studies discussed herein demonstrate that when occurring in proton-coupled fashion, photoinduced multi-electron transfer can proceed more readily. PCMET seems vital for the production of solar fuels and it offers attractive opportunities for photoredox catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Swiss National Science Foundation through grant number 200021_178760 is gratefully acknowledged. O. S. W. thanks all his co-workers for their contributions to the research performed by his group cited in this article. Their names appear in the references. Special thanks go to Peter Hamm and Margherita Orazietti (University of Zurich) for a fruitful collaboration.Notes and references
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- C. D. Windle and R. N. Perutz, Coord. Chem. Rev., 2012, 256, 2562–2570 CrossRef CAS.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS.
- X. Sala, S. Maji, R. Bofill, J. Garcia-Anton, L. Escriche and A. Llobet, Acc. Chem. Res., 2014, 47, 504–516 CrossRef CAS PubMed.
- J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802–6827 CrossRef CAS PubMed.
- M. T. M. Koper, Chem. Sci., 2013, 4, 2710–2723 RSC.
- M. Bourrez, R. Steinmetz, S. Ott, F. Gloaguen and L. Hammarström, Nat. Chem., 2015, 7, 140–145 CrossRef CAS PubMed.
- T. F. Liu, M. Y. Guo, A. Orthaber, R. Lomoth, M. Lundberg, S. Ott and L. Hammarström, Nat. Chem., 2018, 10, 881–887 CrossRef CAS PubMed.
- T. Huang, E. S. Rountree, A. P. Traywick, M. Bayoumi and J. L. Dempsey, J. Am. Chem. Soc., 2018, 140, 14655–14669 CrossRef CAS PubMed.
- M. B. Chambers, D. A. Kurtz, C. L. Pitman, M. K. Brennaman and A. J. M. Miller, J. Am. Chem. Soc., 2016, 138, 13509–13512 CrossRef CAS PubMed.
- C. L. Pitman and A. J. M. Miller, ACS Catal., 2014, 4, 2727–2733 CrossRef CAS.
- S. M. Barrett, C. L. Pitman, A. G. Walden and A. J. M. Miller, J. Am. Chem. Soc., 2014, 136, 14718–14721 CrossRef CAS PubMed.
- X. Guo, H. Zipse and H. Mayr, J. Am. Chem. Soc., 2014, 136, 13863–13873 CrossRef CAS PubMed.
- R. N. Perutz and B. Procacci, Chem. Rev., 2016, 116, 8506–8544 CrossRef CAS PubMed.
- G. Renger and T. Renger, Photosynth. Res., 2008, 98, 53–80 CrossRef CAS PubMed.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
- T. J. Meyer, M. H. V. Huynh and H. H. Thorp, Angew. Chem., Int. Ed., 2007, 46, 5284–5304 CrossRef CAS PubMed.
- P. F. Barbara, T. J. Meyer and M. A. Ratner, J. Phys. Chem., 1996, 100, 13148–13168 CrossRef CAS.
- L. Hammarström, Acc. Chem. Res., 2015, 48, 840–850 CrossRef PubMed.
- Y. Pellegrin and F. Odobel, Coord. Chem. Rev., 2011, 255, 2578–2593 CrossRef CAS.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Wiley-Blackwell, 2nd edn, 2014 Search PubMed.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- L. L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. C. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247–2252 RSC.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- B. A. Barry and G. T. Babcock, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7099–7103 CrossRef CAS.
- A. Pannwitz and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 13308–13311 CrossRef CAS PubMed.
- A. Magnuson, H. Berglund, P. Korall, L. Hammarström, B. Åkermark, S. Styring and L. C. Sun, J. Am. Chem. Soc., 1997, 119, 10720–10725 CrossRef CAS.
- S. Y. Reece and D. G. Nocera, Annu. Rev. Biochem., 2009, 78, 673–699 CrossRef CAS PubMed.
- A. Pannwitz and O. S. Wenger, Dalton Trans., 2019 10.1039/c8dt04373f.
- T. Irebo, M.-T. Zhang, T. F. Markle, A. M. Scott and L. Hammarström, J. Am. Chem. Soc., 2012, 134, 16247–16254 CrossRef CAS PubMed.
- M. Sjödin, S. Styring, B. Åkermark, L. C. Sun and L. Hammarström, J. Am. Chem. Soc., 2000, 122, 3932–3936 CrossRef.
- M.-T. Zhang, T. Irebo, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2011, 133, 13224–13227 CrossRef CAS PubMed.
- J. Bonin, C. Costentin, C. Louault, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2011, 133, 6668–6674 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2006, 128, 4552–4553 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2007, 129, 5870–5879 CrossRef CAS PubMed.
- M. A. Bowring, L. R. Bradshaw, G. A. Parada, T. P. Pollock, R. J. Fernandez-Teran, S. S. Kolmar, B. Q. Mercado, C. W. Schlenker, D. R. Gamelin and J. M. Mayer, J. Am. Chem. Soc., 2018, 140, 7449–7452 CrossRef CAS PubMed.
- J. Chen, M. Kuss-Petermann and O. S. Wenger, Chem. – Eur. J., 2014, 20, 4098–4104 CrossRef CAS PubMed.
- M. Kuss-Petermann, H. Wolf, D. Stalke and O. S. Wenger, J. Am. Chem. Soc., 2012, 134, 12844–12854 CrossRef CAS PubMed.
- J. C. Lennox, D. A. Kurtz, T. Huang and J. L. Dempsey, ACS Energy Lett., 2017, 2, 1246–1256 CrossRef.
- J. C. Lennox and J. L. Dempsey, J. Phys. Chem. B, 2017, 121, 10530–10542 CrossRef CAS PubMed.
- M. Natali, A. Amati, N. Demitri and E. Iengo, Chem. Commun., 2018, 54, 6148–6152 RSC.
- J. Bonin, C. Costentin, M. Robert, J. M. Savéant and C. Tard, Acc. Chem. Res., 2012, 45, 372–381 CrossRef CAS PubMed.
- M. T. Huynh, S. J. Mora, M. Villalba, M. E. Tejeda-Ferrari, P. A. Liddell, B. R. Cherry, A. L. Teillout, C. W. Machan, C. P. Kubiak, D. Gust, T. A. Moore, S. Hammes-Schiffer and A. L. Moore, ACS Cent. Sci., 2017, 3, 372–380 CrossRef CAS PubMed.
- G. Chararalambidis, S. Das, A. Trapali, A. Quaranta, M. Orio, Z. Halime, P. Fertey, R. Guillot, A. Coutsolelos, W. Leibl, A. Aukauloo and M. Sircoglou, Angew. Chem., Int. Ed., 2018, 57, 9013–9017 CrossRef CAS PubMed.
- S. Neumann and O. S. Wenger, Inorg. Chem., 2019, 58, 855–860 CrossRef CAS PubMed.
- M. Kuss-Petermann and O. S. Wenger, J. Am. Chem. Soc., 2016, 138, 1349–1358 CrossRef CAS PubMed.
- J. Hankache and O. S. Wenger, Chem. Commun., 2011, 47, 10145–10147 RSC.
- M. Kuss-Petermann and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 815–819 CrossRef CAS PubMed.
- B. Geiss and C. Lambert, Chem. Commun., 2009, 1670–1672 RSC.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjö, S. Styring, V. Sundström and L. Hammarström, Acc. Chem. Res., 2009, 42, 1899–1909 CrossRef CAS PubMed.
- M. Fujitsuka, S. S. Kim, C. Lu, S. Tojo and T. Majima, J. Phys. Chem. B, 2015, 119, 7275–7282 CrossRef CAS PubMed.
- J. A. Christensen, B. T. Phelan, S. Chaudhuri, A. Acharya, V. S. Batista and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 5290–5299 CrossRef CAS PubMed.
- M. Kuss-Petermann and O. S. Wenger, Helv. Chim. Acta, 2017, 100, e1600283 CrossRef.
- M. H. Ha-Thi, V. T. Pham, T. Pino, V. Maslova, A. Quaranta, C. Lefumeux, W. Leibl and A. Aukauloo, Photochem. Photobiol. Sci., 2018, 17, 903–909 RSC.
- S. Karlsson, J. Boixel, Y. Pellegrin, E. Blart, H. C. Becker, F. Odobel and L. Hammarström, J. Am. Chem. Soc., 2010, 132, 17977–17979 CrossRef CAS PubMed.
- S. Karlsson, J. Boixel, Y. Pellegrin, E. Blart, H. C. Becker, F. Odobel and L. Hammarström, Faraday Discuss., 2012, 155, 233–252 RSC.
- H. Y. Chen and S. Ardo, Nat. Chem., 2018, 10, 17–23 CrossRef CAS PubMed.
- R. M. Young, S. C. Jensen, K. Edme, Y. L. Wu, M. D. Krzyaniak, N. A. Vermeulen, E. J. Dale, J. F. Stoddart, E. A. Weiss, M. R. Wasielewski and D. T. Co, J. Am. Chem. Soc., 2016, 138, 6163–6170 CrossRef CAS PubMed.
- A. G. Bonn and O. S. Wenger, Chimia, 2015, 69, 17–21 CrossRef CAS PubMed.
- S. Mendes Marinho, M.-H. Ha-Thi, V.-T. Pham, A. Quaranta, T. Pino, C. Lefumeux, T. Chamaillé, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2017, 56, 15936–15940 CrossRef CAS PubMed.
- T. T. Tran, M. H. Ha-Thi, T. Pino, A. Quaranta, C. Lefumeux, W. Leibl and A. Aukauloo, J. Phys. Chem. Lett., 2018, 9, 1086–1091 CrossRef CAS PubMed.
- M. P. O'Neil, M. P. Niemczyk, W. A. Svec, D. Gosztola, G. L. Gaines and M. R. Wasielewski, Science, 1992, 257, 63–65 CrossRef PubMed.
- H. Imahori, M. Hasegawa, S. Taniguchi, M. Aoki, T. Okada and Y. Sakata, Chem. Lett., 1998, 721–722 CrossRef CAS.
- T. H. Ghaddar, J. F. Wishart, D. W. Thompson, J. K. Whitesell and M. A. Fox, J. Am. Chem. Soc., 2002, 124, 8285–8289 CrossRef CAS PubMed.
- M. Orazietti, M. Kuss-Petermann, P. Hamm and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 9407–9410 CrossRef CAS PubMed.
- L. Favereau, A. Makhal, Y. Pellegrin, E. Blart, J. Petersson, E. Goransson, L. Hammarström and F. Odobel, J. Am. Chem. Soc., 2016, 138, 3752–3760 CrossRef CAS PubMed.
- M. Kuss-Petermann, M. Orazietti, M. Neuburger, P. Hamm and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 5225–5232 CrossRef CAS PubMed.
- G. N. Lim, C. O. Obondi and F. D'Souza, Angew. Chem., Int. Ed., 2016, 55, 11517–11521 CrossRef CAS PubMed.
- J. Nomrowski and O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 5343–5346 CrossRef CAS PubMed.
- J. Nomrowski, X. W. Guo and O. S. Wenger, Chem. – Eur. J., 2018, 24, 14084–14087 CrossRef CAS PubMed.
- N. Song, C. J. Dares, M. V. Sheridan and T. J. Meyer, J. Phys. Chem. C, 2016, 120, 23984–23988 CrossRef CAS.
- G. B. Hall, R. Kottani, G. A. N. Felton, T. Yamamoto, D. H. Evans, R. S. Glass and D. L. Lichtenberger, J. Am. Chem. Soc., 2014, 136, 4012–4018 CrossRef CAS PubMed.
- A. C. Benniston, J. Hagon, X. Y. He, S. J. Yang and R. W. Harrington, Org. Lett., 2012, 14, 506–509 CrossRef CAS PubMed.
- M. Cattaneo, C. E. Schiewer, A. Schober, S. Dechert, I. Siewert and F. Meyer, Chem. – Eur. J., 2018, 24, 4864–4870 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed.
- X. Guo and O. S. Wenger, Angew. Chem., Int. Ed., 2018, 57, 2469–2473 CrossRef CAS PubMed.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182–1187 CrossRef CAS PubMed.
- J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, Acc. Chem. Res., 2018, 51, 2391–2399 CrossRef CAS PubMed.
- R. Alam and G. A. Molander, Org. Lett., 2018, 20, 2680–2684 CrossRef CAS PubMed.
- X. Guo, Y. Okamoto, M. R. Schreier, T. R. Ward and O. S. Wenger, Chem. Sci., 2018, 9, 5052–5056 RSC.
- S. C. Jensen, S. B. Homan and E. A. Weiss, J. Am. Chem. Soc., 2016, 138, 1591–1600 CrossRef CAS PubMed.
- F. Schendzielorz, M. Finger, J. Abbenseth, C. Würtele, V. Krewald and S. Schneider, Angew. Chem., Int. Ed., 2019, 58, 830–834 CrossRef CAS PubMed.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Science, 2012, 336, 1298–1301 CrossRef CAS PubMed.
- A. M. Schimpf, C. E. Gunthardt, J. D. Rinehart, J. M. Mayer and D. R. Gamelin, J. Am. Chem. Soc., 2013, 135, 16569–16577 CrossRef CAS PubMed.
- C. N. Valdez, M. F. Delley and J. M. Mayer, J. Am. Chem. Soc., 2018, 140, 8924–8933 CrossRef CAS PubMed.
- C. N. Valdez, A. M. Schimpf, D. R. Gamelin and J. M. Mayer, J. Am. Chem. Soc., 2016, 138, 1377–1385 CrossRef CAS PubMed.
- C. N. Valdez, M. Braten, A. Soria, D. R. Gamelin and J. M. Mayer, J. Am. Chem. Soc., 2013, 135, 8492–8495 CrossRef CAS PubMed.
- X. B. Li, Y. J. Gao, Y. Wang, F. Zhan, X. Y. Zhang, Q. Y. Kong, N. J. Zhao, Q. Guo, H. L. Wu, Z. J. Li, Y. Tao, J. P. Zhang, B. Chen, C. H. Tung and L. Z. Wu, J. Am. Chem. Soc., 2017, 139, 4789–4796 CrossRef CAS PubMed.
- J. J. Chen, M. D. Symes and L. Cronin, Nat. Chem., 2018, 10, 1042–1047 CrossRef CAS PubMed.
- K. Yamamoto, A. Call and K. Sakai, Chem. – Eur. J., 2018, 24, 16620–16629 CrossRef CAS PubMed.
- K. Kitamoto, M. Ogawa, G. Ajayakumar, S. Masaoka, H. B. Kraatz and K. Sakai, Inorg. Chem. Front., 2016, 3, 671–680 RSC.
- R. Konduri, N. R. de Tacconi, K. Rajeshwar and F. M. MacDonnell, J. Am. Chem. Soc., 2004, 126, 11621–11629 CrossRef CAS PubMed.
- R. Konduri, H. W. Ye, F. M. MacDonnell, S. Serroni, S. Campagna and K. Rajeshwar, Angew. Chem., Int. Ed., 2002, 41, 3185–3187 CrossRef CAS PubMed.
- M. Skaisgirski, X. Guo and O. S. Wenger, Inorg. Chem., 2017, 56, 2432–2439 CrossRef CAS PubMed.
- J. F. Lefebvre, J. Schindler, P. Traber, Y. Zhang, S. Kupfer, S. Grafe, I. Baussanne, M. Demeunynck, J. M. Mouesca, S. Gambarelli, V. Artero, B. Dietzek and M. Chavarot-Kerlidou, Chem. Sci., 2018, 9, 4152–4159 RSC.
- B. Matt, J. Fize, J. Moussa, H. Amouri, A. Pereira, V. Artero, G. Izzet and A. Proust, Energy Environ. Sci., 2013, 6, 1504–1508 RSC.
- G. Knör, A. Vogler, S. Roffia, F. Paolucci and V. Balzani, Chem. Commun., 1996, 1643–1644 RSC.
- D. Polyansky, D. Cabelli, J. T. Muckerman, E. Fujita, T. Koizumi, T. Fukushima, T. Wada and K. Tanaka, Angew. Chem., Int. Ed., 2007, 46, 4169–4172 CrossRef CAS PubMed.
- G. F. Manbeck and K. J. Brewer, Coord. Chem. Rev., 2013, 257, 1660–1675 CrossRef CAS.
- A. G. Bonn and O. S. Wenger, Phys. Chem. Chem. Phys., 2015, 17, 24001–24010 RSC.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- J. M. Mayer, Annu. Rev. Phys. Chem., 2004, 55, 363–390 CrossRef CAS PubMed.
- C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS PubMed.
- M. Quan, D. Sanchez, M. F. Wasylkiw and D. K. Smith, J. Am. Chem. Soc., 2007, 129, 12847–12856 CrossRef CAS PubMed.
- S. Lachmanová, G. Dupeyre, J. Tarábek, P. Ochsenbein, C. Perruchot, I. Ciofini, M. Hromadová, L. Pospisil and P. P. Lainé, J. Am. Chem. Soc., 2015, 137, 11349–11364 CrossRef PubMed.
- H. Kim, B. Keller, R. Ho-Wu, N. Abeyasinghe, R. J. Vazquez, T. Goodson and P. M. Zimmerman, J. Am. Chem. Soc., 2018, 140, 7760–7763 CrossRef CAS PubMed.
- M. O. Senge, A. A. Ryan, K. A. Letchford, S. A. MacGowan and T. Mielke, Symmetry, 2014, 6, 781–843 CrossRef CAS.
- G. Steinberg-Yfrach, P. A. Liddell, S. C. Hung, A. L. Moore, D. Gust and T. A. Moore, Nature, 1997, 385, 239–241 CrossRef CAS.
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Roger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS PubMed.
Footnote |
| † Current address: Leiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2019 |