
Silver-catalyzed decarboxylative radical cascade cyclization toward benzimidazo[2,1-a]isoquinolin-6(5H)-ones†
Kai
Sun‡
 a,
Shi-Jun
Li‡
a,
Xiao-Lan
Chen
*ac,
Yan
Liu
a,
Xian-Qiang
Huang
b,
Dong-Hui
Wei
*a,
Ling-Bo
Qu
a,
Yu-Fen
Zhao
ac and
Bing
Yu
*a
a,
Shi-Jun
Li‡
a,
Xiao-Lan
Chen
*ac,
Yan
Liu
a,
Xian-Qiang
Huang
b,
Dong-Hui
Wei
*a,
Ling-Bo
Qu
a,
Yu-Fen
Zhao
ac and
Bing
Yu
*a
aCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, China. E-mail: chenxl@zzu.edu.cn; donghuiwei@zzu.edu.cn; bingyu@zzu.edu.cn
bSchool of Chemistry & Chemical Engineering, Liaocheng University, Liaocheng, Shandong 252059, China
cThe Key Laboratory for Chemical Biology of Fujian Province, Xiamen University, Xiamen 361005, China
First published on 4th February 2019
Abstract
A simple and efficient decarboxylative radical addition/cyclization strategy was developed, by which a wide range of benzimidazo[2,1-a]isoquinoline-6(5H)-ones were prepared in one-pot via reaction of functionalized 2-arylbenzoimidazoles and carboxylic acids in the presence of K2S2O8/AgNO3 under mild reaction conditions.
Structurally diverse polycycles containing benzimidazo-isoquinoline fused framework A, as shown in Scheme 1, compose an important part of privileged synthetic intermediates, biologically active molecules and functional materials.1Scheme 1 shows some important polyheterocycles that contain scaffold A. Among them, polyheterocycles B–E exhibit a wide variety of pharmaceutical activities, such as modulation of potassium ion flux, antitumor, etc.2 Compound F was reported to be a potential candidate for organic electronics.3 Nowadays, the development of efficient synthetic strategies for quickly accessing biologically and materially valuable benzimidazole-containing polycycles has been enthusiastically pursued and expected in the organic synthetic field.4
 | ||
| Scheme 1 Examples of benzimidazo-isoquinoline containing polyheterocycles. | ||
Most traditional synthetic methods for the construction of the above-mentioned benzimidazo-isoquinoline fused polycycles (Scheme 1) heavily relied on condensation.5 However, almost all of those classical methods suffer limitations such as not easily available starting materials, harsh reaction conditions and practical inconvenience. Very recently, Song and co-workers developed an elegant [4+2] annulative reaction for the construction of benzimidazole[2,1-a]isoquinolines from 2-arylimidazoles and α-diazoketoesters in the presence of a Cp*Rh(III) catalyst (Scheme 2a).2b However, this protocol involved the use of potentially explosive diazo compounds,6 and an expensive Rh catalyst, which might hinder its synthetic application. Over the past decade, the rapid development of radical cascade reactions for concise construction of structurally diverse polycycles has been witnessed.7 The remarkable merits of radical cascade reactions include atom/step economy, together with the reduction of work and time needed to carry them out.8 Over the past four decades, carboxylic acids, as commercially available and inexpensive compounds, have been widely used as alkyl radical precursors to construct C-heteroatom and C–C bonds via decarboxylation reactions.9 In particular, in the past ten years, many highly valuable decarboxylative cascade cyclization reactions have been successfully developed for accessing many different biologically and materially important heterocycles.10 As part of our continuing interest in the development of novel alkyl-radical-involved reactions,11 herein, we present an efficient silver-catalyzed decarboxylative cascade radical cyclization strategy, by which a large variety of benzimidazo[2,1-a]isoquinolins were prepared via reaction of functionalized 2-arylbenzoimidazoles with carboxylic acids in the presence of AgNO3/K2S2O8 in one pot under mild reaction conditions, as illustrated in Scheme 2b. To the best of our knowledge, this is the first example of constructing a large variety of biologically and materially valuable benzimidazo[2,1-a]isoquinolins via a silver-catalyzed decarboxylative cascade radical cyclization.
 | ||
| Scheme 2 Synthesis of benzimidazo-isoquinoline fused frameworks. | ||
We initiated our study by evaluating the model reaction of N-methacryloyl-2-phenylbenzoimidazole (1a) with pivalic acid (2a) under different reaction conditions, as summarized in Table S1 (ESI†). After an intensive experimentation, the optimal reaction conditions were thus established as follows: 1a (0.5 mmol), 2a (1 mmol), AgNO3 (15 mol%) and K2S2O8 (3 equiv.) in the mixed solvent of CH3CN/H2O (v/v = 1/1) at 80 °C for 8 h under a N2 atmosphere.
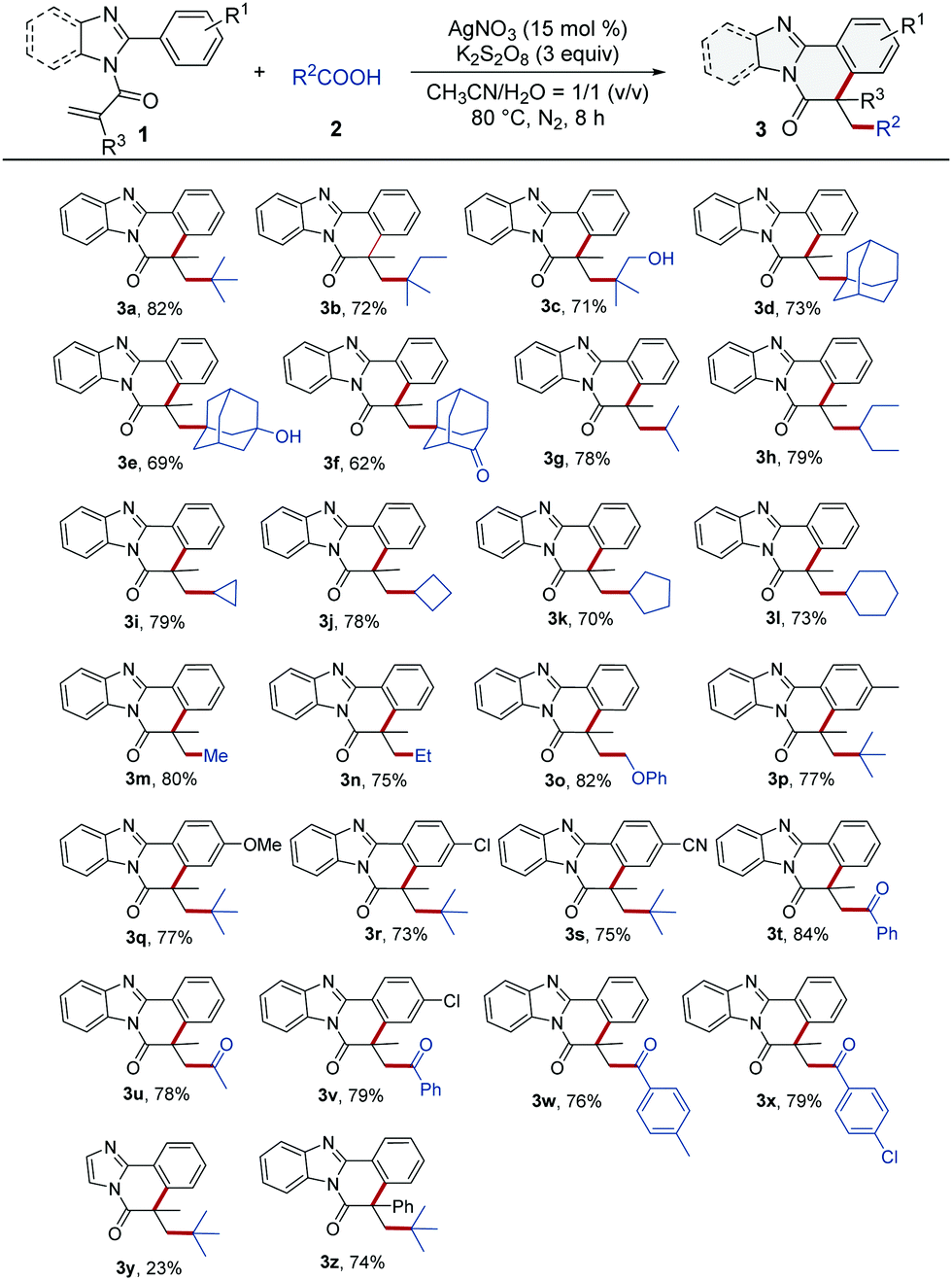
With the optimized reaction conditions established, we next explored the substrate scope of this Ag-catalyzed decarboxylative radical cyclization. As it can be seen in Table 1, many tertiary α-substituted aliphatic carboxylic acids reacted well with N-methacryloyl-2-phenyl-benzoimidazole (1a), giving the corresponding polycyclic products 3a–3f in good yields (62–82%). It is especially worth mentioning here that the pharmaceutically attractive12 adamantane motifs were successfully incorporated into the polycycles as meaningful functional groups of the benzimidazo-isoquinolines (3d–3f). Many secondary α-substituted carboxylic acids, including isobutyric acid, 2-ethylbutanoic acid, and even 3-, 4-, 5-, and 6-membered ring containing carboxylic acids were also efficiently converted into the corresponding products (3g–3l). Notably, three primary carboxylic acids, acetic acid, propionic acid and phenoxy acetic acid which were predicted to produce three unstable primary alkyl radicals via a decarboxylation process, were surprisingly feasible for this synthetic procedure, delivering the corresponding polycycles in 75–82% yields (3m–3o). It was also worth noticing that a number of functionally unstable groups such as the hydroxy group, ether group, and in particular the cyclopropyl group and cyclobutyl group contained in the starting carboxylic acids were well tolerated in these procedures, affording the corresponding products 3c, 3e, 3i, 3j, and 3o in good yields. Afterward, pivalic acid was used to react with various functionalized 2-arylbenzoimidazoles bearing electron-donating and electron-withdrawing groups like –Me, –OMe, –Cl, and –CN on the 2-phenyl ring, giving the products 3p–3s in good yields (73–77%), and no obvious electronic effects were observed in those cases (3p–3s). Encouraged by these results, we extended this decarboxylative addition/cyclization reaction to 2-oxopropanoic acid and 2-oxo-2-arylacetic acids. To our delight, the desired products were obtained in 76–79% yields (3t–3x). In addition, we carried out a reaction starting from N-methacryloyl-2-phenylimidazole and pivalic acid under the optimized reaction conditions, as illustrated in Table 1. However, we only get 23% yield of the corresponding product (3y). The low yield of 3y might be owing to the less favorable conformation of the related intermediate radical (for details, see the ESI†). Finally, we performed an experiment by reacting 2-phenyl-1-(2-phenylbenzimidazol-1-yl)prop-2-en-1-one (1z) with pivalic acid under the optimized reaction conditions. It was pleasing that 74% yield of 3z was obtained, indicating that the phenyl group directly attached to the vinyl group in 1z, basically didn’t interfere with the cyclization process toward the formation of the final product 3z.
| a Reaction conditions: 1 (0.5 mmol), 2 (1 mmol), AgNO3 (15 mol%), K2S2O8 (3 equiv.), CH3CN/H2O = 1/1 (v/v) (5 mL), 80 °C, 8 h. Isolated yields were given. |
|---|
|
To gain further insight into the reaction mechanism, control experiments were conducted (Scheme S1, ESI†). 4.0 equiv. of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) and 4.0 equiv. of BHT (2,6-di-tert-butyl-4-methylphenol) as radical scavengers were subjected to the reaction under standard conditions, respectively. As a result, the Ag-catalyzed decarboxylative cyclization reactions were remarkably suppressed, indicating that this cascade reaction may involve a radical process.
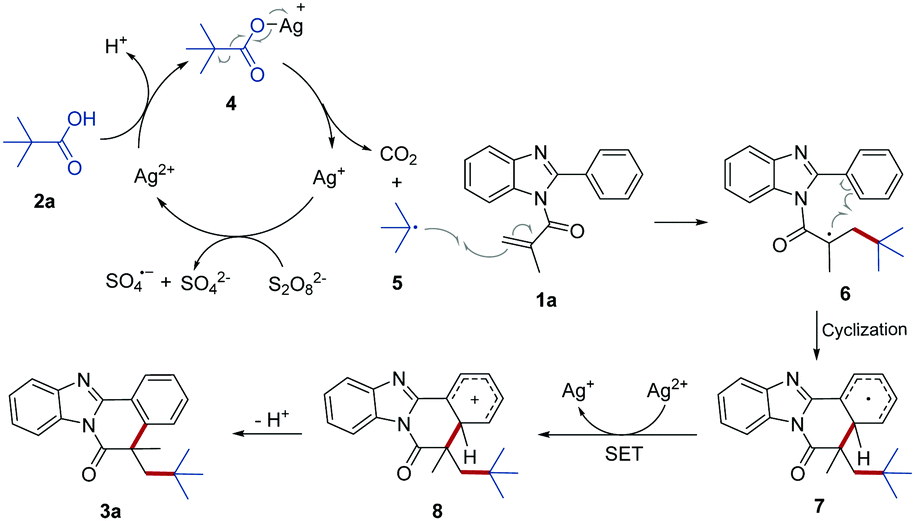
Based on the above experimental results and previous reports, we proposed a plausible mechanism of this sliver-catalyzed transformation as described in Scheme 3. Initially, Ag(I) was oxidized by persulfate (S2O82−) to generate the Ag(II) cation. Subsequently, the Ag(II) cation reacted with 2a to afford the silver carboxylate complex 4 with loss of a proton. Complex 4 then was homolytically cleaved to form t-butyl radical 5 with release of a CO2 molecule and regenerating the Ag(I) cation. Following that, the addition of reactive radical 5 to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of 1a delivered radical intermediate 6, which subsequently underwent an intramolecular cyclization to form intermediate 7. Intermediate 7 was further oxidized into the carbocation 8 by Ag(II) via an intermolecular single electron transfer (SET) process. Finally, rapid deprotonation of carbocation 8 regenerated the aromatic ring to give the final product 3.
C bond of 1a delivered radical intermediate 6, which subsequently underwent an intramolecular cyclization to form intermediate 7. Intermediate 7 was further oxidized into the carbocation 8 by Ag(II) via an intermolecular single electron transfer (SET) process. Finally, rapid deprotonation of carbocation 8 regenerated the aromatic ring to give the final product 3.
 | ||
| Scheme 3 Plausible mechanism of the decarboxylative cyclization. | ||
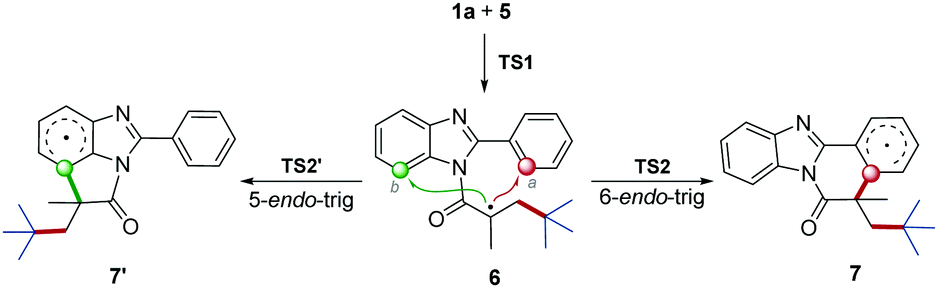
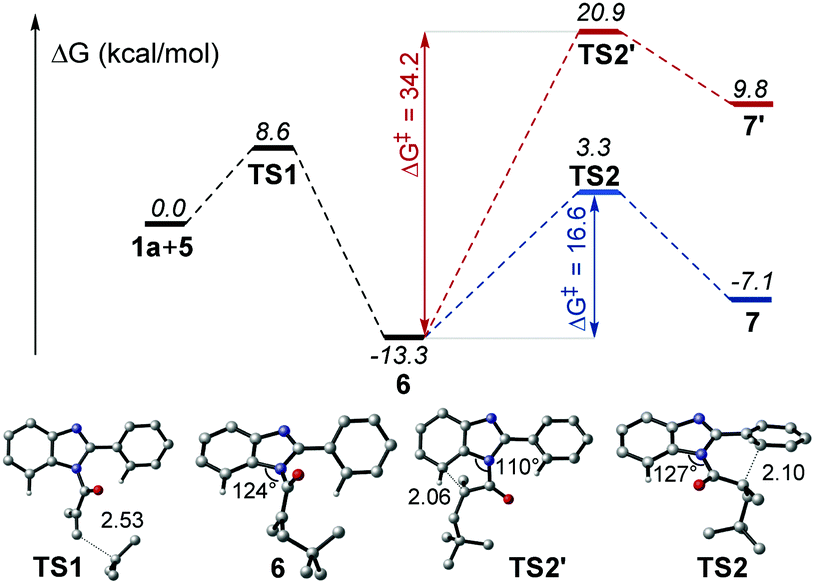
According to the proposed mechanism and previous reports,13 after the generation of radial intermediate 6, both the 6-endo-trig cyclization at the a-position and 5-endo-trig cyclization at the b-position, leading to the polycycle 7 and 7′ respectively, would be possible (Scheme 4). However, no 7′ product was obtained from our synthesis. To understand this regioselectivity, we have investigated the competing pathways for the structural transformations from 1a and 5 to the five- and six-membered ring intermediates 7 and 7′ by performing DFT calculations, which have been frequently carried out for the investigation of organic reaction mechanisms.14 As shown in Fig. 1, the two regioselective pathways would share the same mechanism for the addition of radical 5 to 1a to form intermediate 6via transition state TS1 with an energy barrier of 8.6 kcal mol−1, and then divorced. Starting from 6, the one pathway underwent six-membered ring transition state TS2 to generate 7, and the other pathway went through five-membered ring transition state TS2′ to form 7′. Obviously, the energy barrier associated with TS2 (16.6 kcal mol−1) is much lower than that associated with TS2′ (34.2 kcal mol−1), indicating that the six-membered ring product should be the main one in kinetics. Compared with the initial angle C–N–C (124°) in 6, it would be distorted much more in TS2′ (110°) than that (127°) in TS2, which demonstrates that the less distortion should be responsible for the lower energy of TS2. In addition, the relative Gibbs free energy of 7 would be much lower than that of 7′. Hence, the pathway associated with the six-membered product would be more energetically favourable in both kinetics and thermodynamics, which is in agreement with the experimental observation.
 | ||
| Scheme 4 The possible pathways of cyclization. | ||
 | ||
| Fig. 1 The Gibbs free energy profile for the competing reactions (distance in Å). | ||
In conclusion, we have disclosed a simple and efficient decarboxylative radical addition/cyclization strategy to construct the benzimidazo[2,1-a]isoquinolines from easily available 2-arylbenzimidazoles and carboxylic acids using inexpensive silver catalysis. A large number of carboxylic acids, including tertiary α-substituted tertiary, secondary and even primary carboxylic acids, as well as 2-oxocarboxylic acids were all suitable for being used as the alkyl radical precursors to initiate this radical addition/cyclization reaction. The wide reactant scope of carboxylic acids of this synthesis thus gave a big chance to incorporate many meaningful functional groups into the benzimidazo-quinolines. For example, the pharmaceutically attractive adamantane motifs were successfully introduced from the starting adamantane carboxylic acids into the biological and meaningful polycycle scaffold. Notably, several vulnerable functional groups contained in the starting carboxylic acids, such as the hydroxy group, ether group, and in particular the cyclopropyl group and cyclobutyl group, were well tolerated in this procedure, generating the corresponding polycycles in good yields. DFT calculations proved that the 6-endo-trig pathway is more energetically favourable in both kinetics and thermodynamics than the 5-endo-trig pathway. To the best of our knowledge, this is the first example of constructing a large variety of biologically and materially valuable benzimidazo[2,1-a]isoquinolin-6(5H)-ones via a silver-catalyzed decarboxylative cascade radical cyclization. Great advantages of this strategy include wide reactant scope, mild reaction conditions, experimental simplicity and easy workup. Further application of this strategy is currently ongoing in our laboratory.
We acknowledge the financial support from the National Natural Science Foundation of China (21501010), the 2017 Science and Technology Innovation Team in Henan Province (22120001), and the Major Scientific and Technological Projects in Henan Province (181100310500).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. V. Lygin and A. de Meijere, Angew. Chem., Int. Ed., 2010, 49, 9094–9124 CrossRef CAS PubMed; (b) F. Pan, C. Shu and L.-W. Ye, Org. Biomol. Chem., 2016, 14, 9456–9465 RSC; (c) B. Yu, B. Zou and C.-W. Hu, J. CO2 Util., 2018, 26, 314–322 CrossRef CAS; (d) B. Wu and N. Yoshikai, Org. Biomol. Chem., 2016, 14, 5402–5416 RSC; (e) X. X. Guo, D. W. Gu, Z. X. Wu and W. B. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed; (f) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395–3442 CrossRef CAS PubMed; (g) L.-Y. Xie, J. Qu, S. Peng, K.-J. Liu, Z. Wang, M.-H. Ding, Y. Wang, Z. Cao and W.-M. He, Green Chem., 2018, 20, 760–764 RSC; (h) M. H. Muhammad, X.-L. Chen, B. Yu, L.-B. Qu and Y.-F. Zhao, Pure Appl. Chem., 2019, 91, 33–41 CAS.
- (a) K. Alam, S. W. Hong, K. H. Oh and J. K. Park, Angew. Chem., Int. Ed., 2017, 56, 13387–13391 CrossRef CAS PubMed; (b) S. Mai, Y. Luo, X. Huang, Z. Shu, B. Li, Y. Lan and Q. Song, Chem. Commun., 2018, 54, 10240–10243 RSC.
- M. J. Taublaender, F. Glöcklhofer, M. Marchetti-Deschmann and M. Unterlass, Angew. Chem., Int. Ed., 2018, 57, 12270–12274 CrossRef CAS PubMed.
- (a) J. Yin, F. Zhou, L. Zhu, M. Yang, Y. Lan and J. You, Chem. Sci., 2018, 9, 5488–5493 RSC; (b) B. Liu, F. Hu and B.-F. Shi, Adv. Synth. Catal., 2014, 356, 2688–2696 CrossRef CAS; (c) Z. Zeng, H. Jin, K. Sekine, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 6935–6939 CrossRef CAS PubMed; (d) J. Cai, B. Wu, G. Rong, C. Zhang, L. Qiu and X. Xu, Org. Lett., 2018, 20, 2733–2736 CrossRef CAS PubMed; (e) C. C. McAtee, P. S. Riehl and C. S. Schindler, J. Am. Chem. Soc., 2017, 139, 2960–2963 CrossRef CAS PubMed; (f) J. Liu, Z. Xue, Z. Zeng, Y. Chen and G. Chen, Adv. Synth. Catal., 2016, 358, 3694–3699 CrossRef CAS.
- T. B. Nguyen, L. Ermolenko and A. Al-Mourabit, Chem. Commun., 2016, 52, 4914–4917 RSC.
- (a) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed; (b) Q.-Q. Cheng, Y. Deng, M. Lankelma and M. P. Doyle, Chem. Soc. Rev., 2017, 46, 5425–5443 RSC.
- (a) Y. Yang, B. Zhou, X. Zhu, G. Deng, Y. Liang and Y. Yang, Org. Lett., 2018, 20, 5402–5405 CrossRef CAS PubMed; (b) L. Lv, S. Lu, Y. Chen and Z. Li, Org. Chem. Front., 2017, 4, 2147–2152 RSC; (c) J. Li, W. J. Hao, P. Zhou, Y. L. Zhu, S. L. Wang, S. J. Tu and B. Jiang, RSC Adv., 2017, 7, 9693–9703 RSC; (d) Y. Yu, W. Yuan, H. Huang, Z. Cai, P. Liu and P. Sun, J. Org. Chem., 2018, 83, 1654–1660 CrossRef CAS PubMed; (e) M. P. Plesniak, H.-M. Huang and D. J. Procter, Nat. Rev. Chem., 2017, 1, 0077 CrossRef; (f) L. J. Sebren, J. J. Devery and C. R. J. Stephenson, ACS Catal., 2014, 4, 703–716 CrossRef CAS PubMed; (g) M.-H. Huang, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, Chem. Commun., 2018, 54, 10791–10811 RSC; (h) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed; (i) C. Jing, X. Chen, K. Sun, Y. Yang, T. Chen, Y. Liu, L. Qu, Y. Zhao and B. Yu, Org. Lett., 2019, 21, 486–489 CrossRef CAS PubMed; (j) K.-J. Liu, S. Jiang, L.-H. Lu, L.-L. Tang, S.-S. Tang, H.-S. Tang, Z. Tang, W.-M. He and X. Xu, Green Chem., 2018, 20, 3038–3043 RSC; (k) R. Li, X. Chen, S. Wei, K. Sun, L. Fan, Y. Liu, L. Qu, Y. Zhao and B. Yu, Adv. Synth. Catal., 2018, 360, 4807–4813 CrossRef.
- (a) M.-H. Huang, W.-J. Hao and B. Jiang, Chem. – Asian J., 2018, 13, 2958–2977 CrossRef CAS PubMed; (b) J. Sun, J.-K. Qiu, Y.-N. Wu, W.-J. Hao, C. Guo, G. Li, S.-J. Tu and B. Jiang, Org. Lett., 2017, 19, 754–757 CrossRef CAS PubMed; (c) F. Liu, J.-Y. Wang, P. Zhou, G. Li, W.-J. Hao, S.-J. Tu and B. Jiang, Angew. Chem., Int. Ed., 2017, 56, 15570–15574 CrossRef CAS PubMed; (d) Y. Liu, X.-L. Chen, F.-L. Zeng, K. Sun, C. Qu, L.-L. Fan, Z.-L. An, R. Li, C.-F. Jing, S.-K. Wei, L.-B. Qu, B. Yu, Y.-Q. Sun and Y.-F. Zhao, J. Org. Chem., 2018, 83, 11727–11735 CrossRef CAS PubMed; (e) W. Wei, J. Wen, D. Yang, M. Guo, Y. Wang, J. You and H. Wang, Chem. Commun., 2015, 51, 768–771 RSC; (f) W. Wei, H. Cui, D. Yang, H. Yue, C. He, Y. Zhang and H. Wang, Green Chem., 2017, 19, 5608–5613 RSC; (g) L.-Y. Xie, S. Peng, F. Liu, J.-Y. Yi, M. Wang, Z. Tang, X. Xu and W.-M. He, Adv. Synth. Catal., 2018, 360, 4259–4264 CrossRef CAS; (h) K. Sun, X.-L. Chen, S.-J. Li, D.-H. Wei, X.-C. Liu, Y.-L. Zhang, Y. Liu, L.-L. Fan, L.-B. Qu, B. Yu, K. Li, Y.-Q. Sun and Y.-F. Zhao, J. Org. Chem., 2018, 83, 14419–14430 CrossRef CAS PubMed; (i) G.-P. Yang, X. He, B. Yu and C.-W. Hu, Appl. Organomet. Chem., 2018, 32, e4532 CrossRef; (j) G.-P. Yang, S.-X. Shang, B. Yu and C.-W. Hu, Inorg. Chem. Front., 2018, 5, 2472–2477 RSC.
- (a) K. Xu, Z. M. Tan, H. N. Zhang, J. L. Liu, S. Zhang and Z. Q. Wang, Chem. Commun., 2017, 53, 10719–10722 RSC; (b) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (c) J. Dai, G. Wang, X. Xu and H. Xu, Chin. J. Org. Chem., 2013, 33, 2460–2468 CrossRef CAS; (d) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed; (e) Y. Jin and H. Fu, Asian J. Org. Chem., 2017, 6, 368–385 CrossRef CAS; (f) S. Zhang, Z. Tan, H. Zhang, J. Liu, W. Xu and K. Xu, Chem. Commun., 2017, 53, 11642–11645 RSC; (g) C.-F. Yang, J.-Y. Wang and S.-K. Tian, Chem. Commun., 2011, 47, 8343–8345 RSC; (h) W.-P. Mai, J.-T. Wang, L.-R. Yang, J.-W. Yuan, Y.-M. Xiao, P. Mao and L.-B. Qu, Org. Lett., 2013, 16, 204–207 CrossRef PubMed; (i) Z.-J. Liu, X. Lu, G. Wang, L. Li, W.-T. Jiang, Y.-D. Wang, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2016, 138, 9714–9719 CrossRef CAS PubMed; (j) G. Li, T. Wang, F. Fei, Y.-M. Su, Y. Li, Q. Lan and X.-S. Wang, Angew. Chem., Int. Ed., 2016, 55, 3491–3495 CrossRef CAS PubMed; (k) W. C. Yang, P. Dai, K. Luo, Y. G. Ji and L. Wu, Adv. Synth. Catal., 2017, 359, 2390–2395 CrossRef CAS; (l) J.-W. Yuan, L.-R. Yang, P. Mao and L.-B. Qu, Org. Chem. Front., 2017, 4, 545–554 RSC; (m) F. Gao, J.-T. Wang, L.-L. Liu, N. Ma, C. Yang, Y. Gao and W. Xia, Chem. Commun., 2017, 53, 8533–8536 RSC; (n) K. Sun, Z. Shi, Z. Liu, B. Luan, J. Zhu and Y. Xue, Org. Lett., 2018, 20, 6687–6690 CrossRef CAS PubMed.
- Q.-Z. Zheng and N. Jiao, Chem. Soc. Rev., 2016, 45, 4590–4627 RSC.
- (a) W.-M. Zhao, X.-L. Chen, J.-W. Yuan, L.-B. Qu, L.-K. Duan and Y.-F. Zhao, Chem. Commun., 2014, 50, 2018–2020 RSC; (b) H. Hu, X. Chen, K. Sun, J. Wang, Y. Liu, H. Liu, B. Yu, Y. Sun, L. Qu and Y. Zhao, Org. Chem. Front., 2018, 5, 2925–2929 RSC; (c) H. Hu, X. Chen, K. Sun, J. Wang, Y. Liu, H. Liu, L. Fan, B. Yu, Y. Sun, L. Qu and Y. Zhao, Org. Lett., 2018, 20, 6157–6160 CrossRef CAS PubMed.
- L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516–3604 CrossRef CAS PubMed.
- (a) W. Mai, J. Wang, L. Yang, J. Yuan, P. Mao, Y. Xiao and L. Qu, Chin. J. Org. Chem., 2014, 34, 1958–1965 CrossRef CAS; (b) Z. Li, Y. Zhang, L. Zhang and Z.-Q. Liu, Org. Lett., 2014, 16, 382–385 CrossRef CAS PubMed; (c) Z. Xu, C. Yan and Z.-Q. Liu, Org. Lett., 2014, 16, 5670–5673 CrossRef CAS PubMed.
- (a) N. Liu, Y.-F. Xie, C. Wang, S.-J. Li, D. Wei, M. Li and B. Dai, ACS Catal., 2018, 8, 9945–9957 CrossRef CAS; (b) Y. Wang, Y. Wang, X. Wang, X. Li, L.-B. Qu and D. Wei, Catal. Sci. Technol., 2018, 8, 4229–4240 RSC; (c) Y. Yang, C. Qu, X. Chen, K. Sun, L. Qu, W. Bi, H. Hu, R. Li, C. Jing, D. Wei, S. Wei, Y. Sun, H. Liu and Y. Zhao, Org. Lett., 2017, 19, 5864–5867 CrossRef CAS PubMed; (d) Y. Wang, Y. Qiao, D. Wei and M. Tang, Org. Chem. Front., 2017, 4, 1987–1998 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1869999. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc10243k |
| ‡ Kai Sun and Shi-Jun Li contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |