
Electrochemical oxidative C–H/N–H cross-coupling for C–N bond formation with hydrogen evolution†
Yi
Yu‡
a,
Yong
Yuan‡
 b,
Huilin
Liu
a,
Min
He
a,
Mingzhu
Yang
a,
Pan
Liu
a,
Banying
Yu
a,
Xuanchi
Dong
a and
Aiwen
Lei
*ab
b,
Huilin
Liu
a,
Min
He
a,
Mingzhu
Yang
a,
Pan
Liu
a,
Banying
Yu
a,
Xuanchi
Dong
a and
Aiwen
Lei
*ab
aNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, P. R. China. E-mail: aiwenlei@whu.edu.cn
bCollege of Chemistry and Molecular Sciences, Institute for Advanced Studies (IAS), Wuhan University, Wuhan 430072, P. R. China
First published on 23rd January 2019
Abstract
Under metal catalyst-free and exogenous-oxidant-free conditions, a series of C-3 aminated imidazo[1,2-a]pyridines were synthesized by electrochemical intermolecular oxidative C–H/N–H cross-coupling. Furthermore, by using a catalytic amount of ferrocene as the mediator, electrochemical intramolecular oxidative C–H/N–H cross-coupling for the synthesis of 10H-benzo[4,5]imidazo[1,2-a]indole derivatives has also been accomplished.
An imidazo[1,2-a]pyridine core is a very important structural unit that widely occurs in many biologically active molecules,1 particularly in many commercially available drugs (such as alpidem, zolpidem, necopidem and saripidem).2 In order to obtain other new biologically active molecules, further modification or functionalization of imidazo[1,2-a]pyridines is highly necessary. Over the past few years, the modification of imidazo[1,2-a]pyridines has received much attention and studies have revealed that C-3 substituted imidazo[1,2-a]pyridines exhibit a better biological activity than imidazo[1,2-a]pyridines itself:3,4 for example, C-3 sulfenylated or aminated imidazo[1,2-a]pyridines. However, compared with extensive research on C-3 sulfenylation, the C-3 amination of imidazo[1,2-a]pyridines still remains underexplored,5 and the reported methods usually require stoichiometric oxidants, long reaction times, and/or high reaction temperature. Therefore, developing an alternative synthetic method to construct C-3 aminated imidazo[1,2-a]pyridines is desirable.
Electrochemical synthesis is an efficient and environmentally friendly synthetic method,6 which can realize C–X bond formation via electrochemical oxidative C–H/X–H cross-coupling with hydrogen evolution under metal catalyst-free and exogenous-oxidant-free conditions. In recent years, electrochemical oxidative C–H/X–H cross-coupling with hydrogen evolution has gained significant attention.7 However, until now, electrochemical oxidative C–H/N–H cross-coupling with hydrogen evolution for the synthesis of C-3 aminated imidazo[1,2-a]pyridines has not been reported. As a part of our recent research interest in the field of electrochemical oxidative C–X bond formation,8 we herein report an electrochemical oxidative C–H/N–H cross-coupling protocol for the synthesis of C-3 aminated imidazo[1,2-a]pyridines. Under metal catalyst-free and exogenous-oxidant-free conditions as well as using cheap and commercially available NaNO3 as the electrolyte, a series of significant C-3 aminated imidazo[1,2-a]pyridines were synthesized with hydrogen evolution (Scheme 1a). Furthermore, under electrochemical oxidative conditions, intramolecular oxidative C–H/N–H cross-coupling for the synthesis of 10H-benzo[4,5]imidazo[1,2-a]indole derivatives has also been accomplished (Scheme 1b).
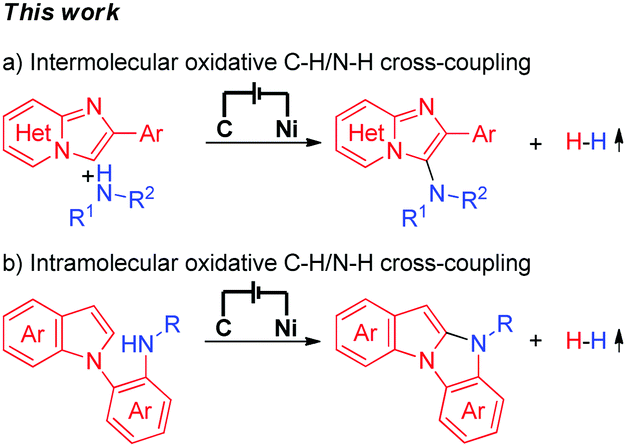 | ||
| Scheme 1 Electrochemical oxidative C–H/N–H cross-coupling for C–N bond formation with hydrogen evolution. | ||
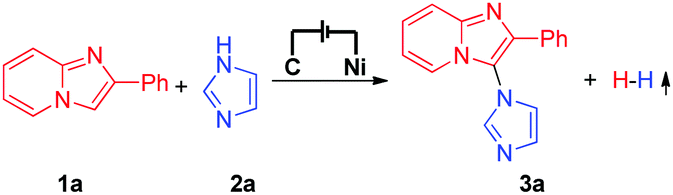
Our initial investigation started by using 2-phenylimidazo[1,2-a]pyridine (1a) and imidazole (2a) as the model substrates (Table 1). Interestingly, when the reaction was conducted at a constant current of 8 mA in MeCN/H2O/EtOH, the desired C–H/N–H cross-coupling product could be obtained in 78% 1H NMR yield (Table 1, entry 1). Control experiments revealed that the mixed solvent (MeCN/H2O/EtOH) and electricity were important for the reaction efficiency (Table 1, entries 2–5). Some organic electrolytes were then investigated, but all were less effective than NaNO3 (Table 1, entries 6 and 7). Either increasing the operating current to 16 mA or decreasing the operating current to 4 mA also led to a decreased reaction yield (Table 1, entries 8 and 9). Further investigation showed that the carbon rod anode and the nickel plate cathode were the best choice for the electrode materials (Table 1, entry 10).
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
| a Standard conditions: graphite rod anode, nickel plate cathode, constant current = 8 mA, 1a (0.3 mmol), 2a (2.0 equiv.), NaNO3 (0.3 mmol), MeCN (4.8 mL), H2O (1.0 mL), EtOH (1.2 mL), 50 °C, 5 h (5.0 F mol−1), undivided cell. b Yields were determined by 1H NMR using naphthalene as the internal standard. c Isolated yields. | ||
| 1 | Standard conditions | 78, 75c |
| 2 | No H2O (MeCN/EtOH = 5.8/1.2) | 66 |
| 3 | No EtOH (MeCN/H2O = 6/1) | 67 |
| 4 | No MeCN (H2O/EtOH = 1/6) | 40 |
| 5 | No constant current | 0 |
| 6 | n Bu4NBF4 instead of NaNO3 | 73 |
| 7 | n Bu4NPF6 instead of NaNO3 | 67 |
| 8 | 16 mA, 2.5 h | 65 |
| 9 | 4 mA, 10 h | 46 |
| 10 | C(+)|Fe(−) instead of C(+)|Ni(−) | 56 |
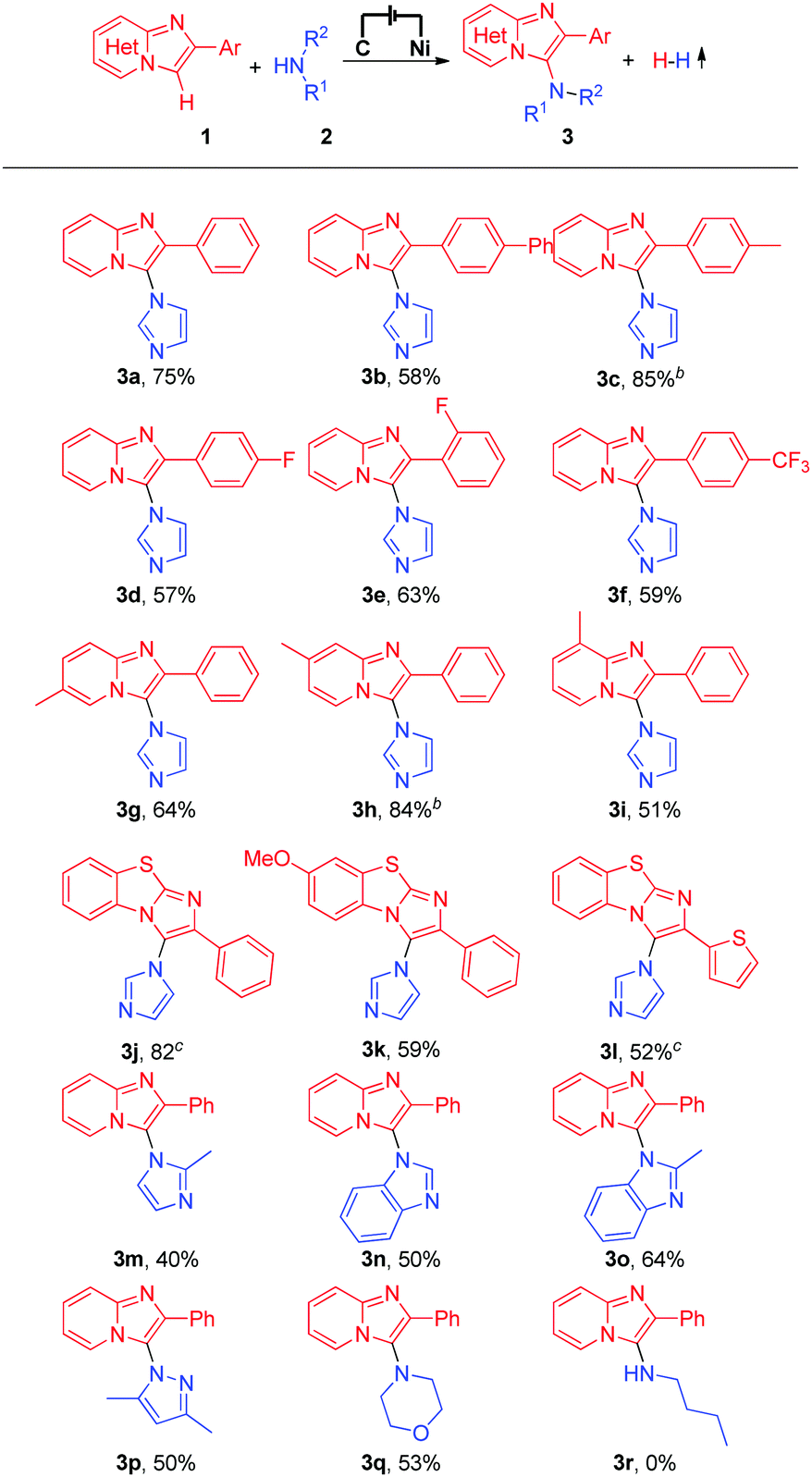
With the optimized reaction conditions in hand, we subsequently explored the substrate scope of this electrochemical oxidative C–H/N–H cross-coupling reaction with different imidazo[1,2-a]pyridines and N-nucleophiles (Table 2). First, the effect of different substituents on the 2-phenyl moiety of imidazo[1,2-a]pyridines was investigated. Imidazo[1,2-a]pyridine bearing an electron-neutral methyl substituent at the 2-phenyl moiety produced the C–H/N–H cross-coupling product in 75% yield (Table 2, 3a), while imidazo[1,2-a]pyridines with phenyl or electron-withdrawing −F and −CF3 substituents at the 2-phenyl moiety gave the corresponding products in moderate to good yields (Table 2, 3b to 3f). Moreover, substrates with a methyl group at the C-5, C-6, or C-7 position of 2-phenylimidazo[1,2-a]pyridines reacted smoothly with imidazole (2a) and afforded the C-3 aminated products in moderate to good yields (Table 2, 3g to 3i). It is worth noting that important benzo[d]-imidazo[2,1-b]thiazole derivatives were also compatible with the reaction conditions, generating the corresponding C–H/N–H cross-coupling products in moderate to high yields (Table 2, 3j to 3l). Different azoles and other N-nucleophiles were also applied as substrates in this electrochemical oxidative C–H/N–H cross-coupling reaction. Delightedly, 2-methyl-1H-imidazole, 1H-benzo[d]imidazole, 2-methyl-1H-benzo[d]imidazole, and even 3,5-dimethyl-1H-pyrazole and morpholine all were suitable substrates for this transformation (Table 2, 3m to 3q). However, when long-chain aliphatic amines, such as n-butylamine, were employed in the reaction, no desired product was detected (Table 2, 3r).
| a Standard conditions: graphite rod anode, nickel plate cathode, constant current = 8 mA, 4 (0.3 mmol), Cp2Fe (20 mol%), nBu4NBF4 (0.3 mmol), MeCN (3.5 mL), EtOH (3.5 mL), 70 °C, 4 h (4.0 F mol−1), undivided cell. |
|---|
|
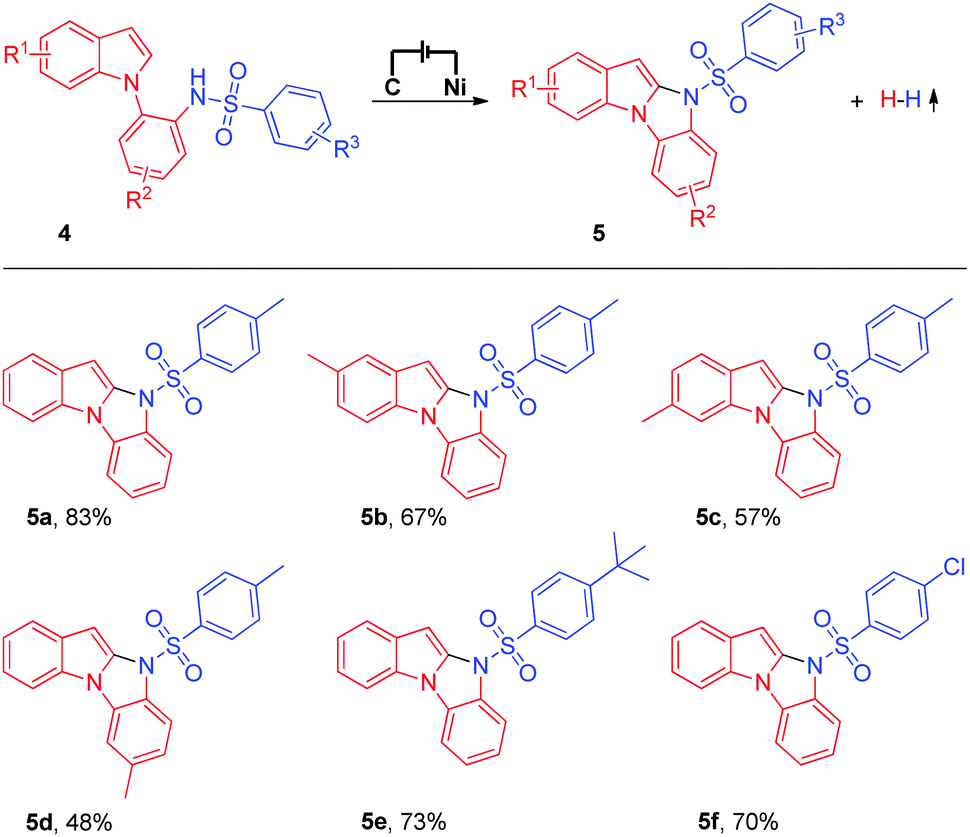
Having successfully established the feasibility of the electrochemical intermolecular oxidative C–H/N–H cross-coupling of imidazo[1,2-a]pyridines with N-nucleophiles, we became enthusiastic about the synthesis of significant 10H-benzo[4,5]imidazo[1,2-a]indole derivatives9 using the electrochemical intramolecular oxidative C–H/N–H cross-coupling strategy (Table 3). Unfortunately, under the current electrochemical conditions, the intramolecular oxidative C–H/N–H cross-coupling products could not be obtained. Next, we tried to modify the reaction conditions to obtain the target products (for details about the optimization of reaction conditions, see the ESI†). To our delight, when a catalytic amount of ferrocene (Cp2Fe) was added as the mediator, the intramolecular oxidative C–H/N–H cross-coupling reaction proceeded well and the corresponding 10H-benzo[4,5]imidazo[1,2-a]indole derivative could be isolated in 83% yield (Table 3, 5a). The substrate scope of the electrochemical intramolecular oxidative C–H/N–H cross-coupling reaction was then investigated. Various N-(2-(1H-indol-1-yl)phenyl)benzenesulfonamide derivatives were all suitable substrates in this transformation (Table 3, 5b to 5f).
To investigate the details of the mechanism for this electrochemical oxidative C–H/N–H cross-coupling reaction, several control experiments were carried out. First, cyclic voltammetry experiments on model substrates were carried out. 1a and 2a have very similar redox potentials. 1a could be oxidized when the oxidation potential exceeded 0.94 V, while 2a started to get oxidized from 0.93 V (for more details about the cyclic voltammetry experiments, see the ESI†). This result showed that 1a and 2a could be synchronously oxidized at the anode under our electrochemical conditions. Second, the model reaction and the electrolysis of 1a in the absence of 2a were investigated, respectively (Scheme 2). Homo-coupling products of 1a were detected in both reactions, while the capture product of A (see Scheme 3 for the structure of A) by H2O or EtOH was not detected. These results indicated that 1a could be converted to the corresponding radical under our electrochemical conditions and the pathway of radical cation intermediate A captured by a nucleophile might be ruled out.
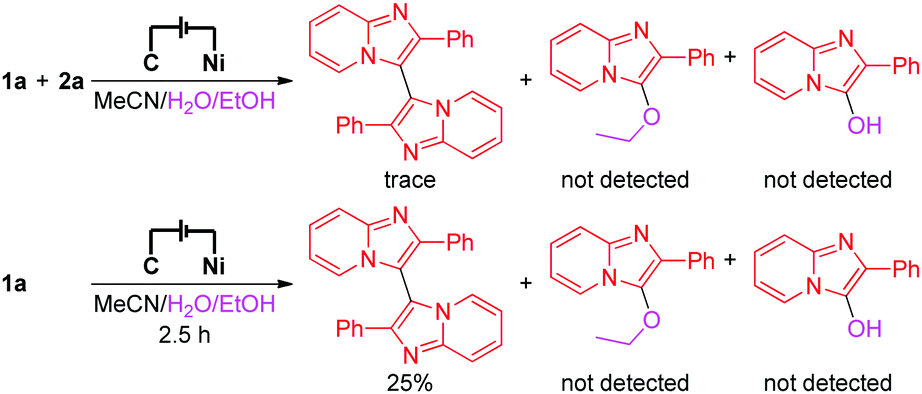 | ||
| Scheme 2 Control experiments. | ||
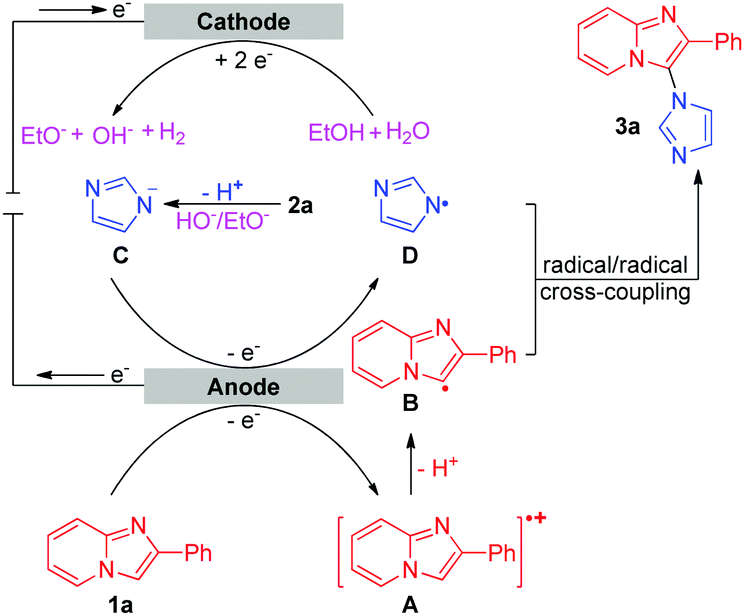 | ||
| Scheme 3 Proposed mechanism of the intermolecular oxidative C–H/N–H cross-coupling reaction. | ||
Based on the results of the above experiments and previous reports,5c,10 a possible mechanism for the intermolecular oxidative C–H/N–H cross-coupling reaction is shown in Scheme 3. The anodic oxidation and deprotonation of 1a produce radical B. The cathodic reduction of H2O or EtOH produces H2 and HO− or EtO−. The generated HO− or EtO− then deprotonates substrate 2a to afford its conjugate base C. C is further oxidized at the anode, forming the nitrogen radical D. Next, radical/radical cross-coupling affords the C–H/N–H cross-coupling product 3a. Note that the pathway of radical addition of nitrogen radical D to 1a could not be fully ruled out. In addition, for some alkyl amines, the corresponding nitrogen radicals are unstable, which may be the reason why 3r was not obtained. As for the intramolecular oxidative C–H/N–H cross-coupling reaction (Scheme 4), the reaction begins with the anodic oxidation of Cp2Fe to afford Cp2Fe+,11 and Cp2Fe+ then oxidizes the deprotonated 4 (A) to regenerate Cp2Fe and form nitrogen radical intermediate B. Sequential radical addition, single electron oxidation, and deprotonation finally afford the C–H/N–H cross-coupling product 5.
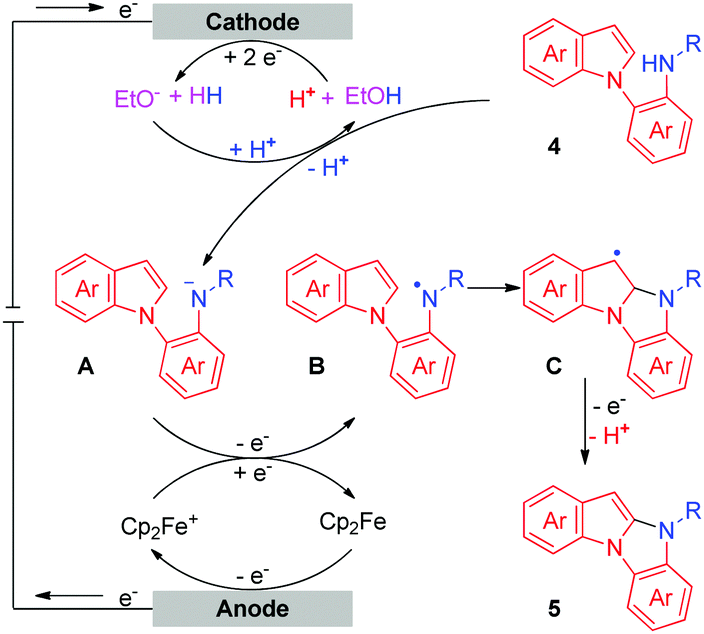 | ||
| Scheme 4 Proposed mechanism of the intramolecular oxidative C–H/N–H cross-coupling reaction. | ||
In summary, we have developed an electrochemical oxidative C–H/N–H cross-coupling reaction. Under metal catalyst-free and exogenous-oxidant-free conditions as well as using cheap and commercially available sodium nitrate as the electrolyte, a series of C-3 aminated imidazo[1,2-a]pyridines were synthesized with hydrogen evolution. Furthermore, by using a catalytic amount of ferrocene as the mediator, electrochemical intramolecular oxidative C–H/N–H cross-coupling for the synthesis of significant 10H-benzo[4,5]imidazo[1,2-a]indole derivatives has also been reported.
This work was supported by the National Natural Science Foundation of China (21520102003, 21702081, and 21702150), the Hubei Province Natural Science Foundation of China (2017CFA010), and the Jiangxi Provincial Education Department Foundation (GJJ160325). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. F. Byth, C. Geh, C. L. Forder, S. E. Oakes and A. P. Thomas, Mol. Cancer Ther., 2006, 5, 655 CrossRef CAS PubMed; (b) T. H. Al-Tel, R. A. Al-Qawasmeh and R. Zaarour, Eur. J. Med. Chem., 2011, 46, 1874–1881 CrossRef CAS PubMed.
- (a) Y. Rival, G. Grassy, A. Taudou and R. Ecalle, Eur. J. Med. Chem., 1991, 26, 13–18 CrossRef CAS; (b) K. Mizushige, T. Ueda, K. Yukiiri and H. Suzuki, Cardiovasc. Drug Rev., 2002, 20, 163–174 CrossRef CAS PubMed; (c) S. Boggs, V. I. Elitzin, K. Gudmundsson, M. T. Martin and M. J. Sharp, Org. Process Res. Dev., 2009, 13, 781–785 CrossRef CAS; (d) B. Du, A. Shan, X. Zhong, Y. Zhang, D. Chen and K. Cai, Am. J. Med. Sci., 2014, 347, 178–182 CrossRef PubMed.
- (a) H. Cao, H. Zhan, Y. Lin, X. Lin, Z. Du and H. Jiang, Org. Lett., 2012, 14, 1688–1691 CrossRef CAS PubMed; (b) H. Cao, S. Lei, N. Li, L. Chen, J. Liu, H. Cai, S. Qiu and J. Tan, Chem. Commun., 2015, 51, 1823–1825 RSC; (c) D. Yang, K. Yan, W. Wei, G. Li, S. Lu, C. Zhao, L. Tian and H. Wang, J. Org. Chem., 2015, 80, 11073–11079 CrossRef CAS PubMed; (d) S. Mitra, M. Ghosh, S. Mishra and A. Hajra, J. Org. Chem., 2015, 80, 8275–8281 CrossRef CAS PubMed; (e) P. Liu, Y. Gao, W. Gu, Z. Shen and P. Sun, J. Org. Chem., 2015, 80, 11559–11565 CrossRef CAS PubMed; (f) M.-A. Hiebel and S. Berteina-Raboin, Green Chem., 2015, 17, 937–944 RSC; (g) S. Lei, G. Chen, Y. Mai, L. Chen, H. Cai, J. Tan and H. Cao, Adv. Synth. Catal., 2016, 358, 67–73 CrossRef CAS; (h) P. Sun, D. Yang, W. Wei, M. Jiang, Z. Wang, L. Zhang, H. Zhang, Z. Zhang, Y. Wang and H. Wang, Green Chem., 2017, 19, 4785–4791 RSC; (i) R. Rahaman, S. Das and P. Barman, Green Chem., 2018, 20, 141–147 RSC.
- (a) L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305–312 CrossRef CAS; (b) Y. Uemura, S. Tanaka, S. Ida and T. Yuzuriha, J. Pharm. Pharmacol., 1993, 45, 1077–1081 CrossRef CAS PubMed; (c) G. A. Thompson, P. J. Whiting and K. A. Wafford, Behav. Pharmacol., 1995, 6, 116–117 CrossRef; (d) J. J. Kaminski and A. M. Doweyko, J. Med. Chem., 1997, 40, 427–436 CrossRef CAS PubMed; (e) K. C. Rupert, J. R. Henry, J. H. Dodd, S. A. Wadsworth, D. E. Cavender, G. C. Olini, B. Fahmy and J. J. Siekierka, Bioorg. Med. Chem. Lett., 2003, 13, 347–350 CrossRef CAS PubMed; (f) T. Meng, W. Wang, Z. Zhang, L. Ma, Y. Zhang, Z. Miao and J. Shen, Bioorg. Med. Chem., 2014, 22, 848–855 CrossRef CAS PubMed.
- (a) Y. Wang, B. Frett, N. McConnell and H.-Y. Li, Org. Biomol. Chem., 2015, 13, 2958–2964 RSC; (b) S. Samanta, C. Ravi, S. N. Rao, A. Joshi and S. Adimurthy, Org. Biomol. Chem., 2017, 15, 9590–9594 RSC; (c) H. Chen, H. Yi, Z. Tang, C. Bian, H. Zhang and A. Lei, Adv. Synth. Catal., 2018, 360, 3220–3227 CrossRef CAS; (d) K. Sun, S. Mu, Z. Liu, R. Feng, Y. Li, K. Pang and B. Zhang, Org. Biomol. Chem., 2018, 16, 6655–6658 RSC.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) S. Liang, K. Xu, C.-C. Zeng, H.-Y. Tian and B.-G. Sun, Adv. Synth. Catal., 2018, 360, 4266 CrossRef CAS; (c) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS; (d) J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed; (e) D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573–4591 CrossRef CAS PubMed; (f) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (g) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (h) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef CAS; (i) J.-i. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702–4730 CrossRef CAS PubMed; (j) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef CAS.
- (a) H. Gao, Z. Zha, Z. Zhang, H. Ma and Z. Wang, Chem. Commun., 2014, 50, 5034–5036 RSC; (b) A. Wiebe, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 14727–14731 CrossRef CAS PubMed; (c) J. Wu, Y. Zhou, Y. Zhou, C.-W. Chiang and A. Lei, ACS Catal., 2017, 7, 8320–8323 CrossRef CAS; (d) N. Fu, L. Li, Q. Yang and S. Luo, Org. Lett., 2017, 19, 2122–2125 CrossRef CAS PubMed; (e) N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS PubMed; (f) N. Sauermann, R. Mei and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5090–5094 CrossRef CAS PubMed; (g) A. Shao, N. Li, Y. Gao, J. Zhan, C.-W. Chiang and A. Lei, Chin. J. Chem., 2018, 36, 619–624 CrossRef CAS; (h) X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS; (i) Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494 CrossRef CAS PubMed; (j) S. Zhang, L. Li, H. Wang, Q. Li, W. Liu, K. Xu and C. Zeng, Org. Lett., 2018, 20, 252–255 CrossRef CAS PubMed; (k) Z.-J. Wu, S.-R. Li, H. Long and H.-C. Xu, Chem. Commun., 2018, 54, 4601–4604 RSC; (l) S. Zhang, L. Li, M. Xue, R. Zhang, K. Xu and C. Zeng, Org. Lett., 2018, 20, 3443–3446 CrossRef CAS PubMed.
- (a) P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009–3013 CrossRef CAS PubMed; (b) Y. Yuan, Y. Yu, J. Qiao, P. Liu, B. Yu, W. Zhang, H. Liu, M. He, Z. Huang and A. Lei, Chem. Commun., 2018, 54, 11471–11474 RSC; (c) Y. Yuan, Y. Chen, S. Tang, Z. Huang and A. Lei, Sci. Adv., 2018, 4, eaat5312 CrossRef PubMed; (d) Y. Yuan, Y. Cao, Y. Lin, Y. Li, Z. Huang and A. Lei, ACS Catal., 2018, 8, 10871–10875 CrossRef CAS; (e) X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195–4199 CrossRef CAS PubMed; (f) Y. Yuan, Y. Cao, J. Qiao, Y. Lin, X. Jiang, Y. Weng, S. Tang and A. Lei, Chin. J. Chem., 2019, 37, 49–52 CrossRef CAS; (g) S. Tang, S. Wang, Y. Liu, H. Cong and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 4737–4741 CrossRef CAS PubMed.
- (a) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS PubMed; (b) S. C. Teguh, N. Klonis, S. Duffy, L. Lucantoni, V. M. Avery, C. A. Hutton, J. B. Baell and L. Tilley, J. Med. Chem., 2013, 56, 6200–6215 CrossRef CAS PubMed; (c) L. Yin, Q. Hu, J. Emmerich, M. M.-C. Lo, E. Metzger, A. Ali and R. W. Hartmann, J. Med. Chem., 2014, 57, 5179–5189 CrossRef CAS PubMed.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- L. Zhu, P. Xiong, Z.-Y. Mao, Y.-H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 2226–2229 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc09899a |
| ‡ Yi Yu and Yong Yuan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |