
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sequence-activated AND logic dual-channel fluorescent probe for tracking programmable drug release†
Chenxu
Yan
a,
Zhiqian
Guo
 *a,
Yajing
Liu
b,
Ping
Shi
b,
He
Tian
a and
Wei-Hong
Zhu
*a
*a,
Yajing
Liu
b,
Ping
Shi
b,
He
Tian
a and
Wei-Hong
Zhu
*a
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, Shanghai Key Laboratory of Functional Materials Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai, 200237, China. E-mail: whzhu@ecust.edu.cn; guozq@ecust.edu.cn
bState Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai, 200237, China
First published on 22nd June 2018
Abstract
The translation of biomarker sensing into programmable diagnostics or therapeutic applications in vivo is greatly challenging, especially for eliminating the ‘false positive’ signals from OR logic gates. Herein we present a sense-of-logic dual-channel nanoprobe, operating through a sequence-activated AND logic gate by responding ultra-sensitively to pH changes and being subsequently triggered with biothiol for the controllable release of anti-cancer drugs. Specifically, programmable drug release is conducted in a multistage tumor microenvironment (acidic endocytic organelles followed by abnormal glutathione-overexpressing cell cytosol), which is synchronous with dual-channel near-infrared (NIR) fluorescence output. This approach represents the merging of sensing and release, including logically enabled molecular design, biomarker sensing, and controllable drug release. Impressively, the sequential AND logic feature within an unprecedented framework provides feedback on the diversity and complexity of biological milieu, along with remarkably enhancing the tumor therapeutic efficiency via its precise targeting ability and programmable drug release.
Introduction
Smart nanoprobes capable of intracellular information processing provide exceptional opportunities for biomarker sensing, molecular imaging,1–10 and drug delivery.11–20 To date, it is still a tremendous challenge to differentiate concurrent biomarkers, such as dysregulated pH, matrix metalloproteinase activity and aberrant biothiol levels. Current multi-stimulus-responsive probes are predominantly operated by “OR” logic gates in response to each stimulus.21–24 However, nonspecific activation in complex biological milieu always leads to “false positive” signals with difficulty in accurate recognition.25,26 Inspired by the Boolean logic idea,27–30 we envision that the incorporation of sequence-activated fluorescence responses from biomarkers might afford precise behavior in vivo, then produce specific output information for intelligent recognition. A small number of AND logic-based prodrugs have been reported, but they still lack the ability to couple the sequence-dependent response to concurrent biomarkers with specificity.31,32 In diagnostics or therapeutic applications, a sequential AND logic fluorescent nanoprobe that enables the translation of biomarker sensing into a programmable response in vivo has not yet been reported.Herein we present a proof-of-concept study of a sequence-activated AND logic dual-channel near-infrared (NIR) fluorescent probe P(Cy-S-CPT), which functions as a programmable sensor and then releases anticancer drugs. To demonstrate the feasibility of our strategy (Fig. 1), two tumor-associated biomarkers, low cellular pH and overexpressed glutathione (GSH),33–35 served as the model stimuli. The smart nanoprobe P(Cy-S-CPT) is composed of two functional components: an ionizable tertiary amine-containing diblock copolymer which renders an ultra-sensitive response to small pH differences between acidic tumor cells and blood,36–39 and a dual-channel NIR fluorescence component Cy-S-CPT for tracking the biothiol-triggered prodrug release in vivo. In Cy-S-CPT, the cyanine (Cy) derivative served as the dual-channel near-infrared fluorophore and the disulfide-bridged anticancer prodrug camptothecin (-S-CPT) served as the activatable unit. As expected, the nanoprobe becomes activatable and detectable with pH changes, resulting in the subsequent overexpression of GSH coupled with acidic pH, leading to another NIR readout channel (Fig. 1). Specifically, only when the pH and biothiol stimuli are sequence-dependently triggered is the drug release conducted in a multistage tumor microenvironment (acidic endocytic organelles followed by abnormal GSH-overexpressing cell cytosol), which is synchronous with in vivo dual-channel NIR fluorescence output. Notably, this strategy would protect prodrugs from destructive environments and undesirable interactions, and would ensure the initiation of designated drug release when needed.
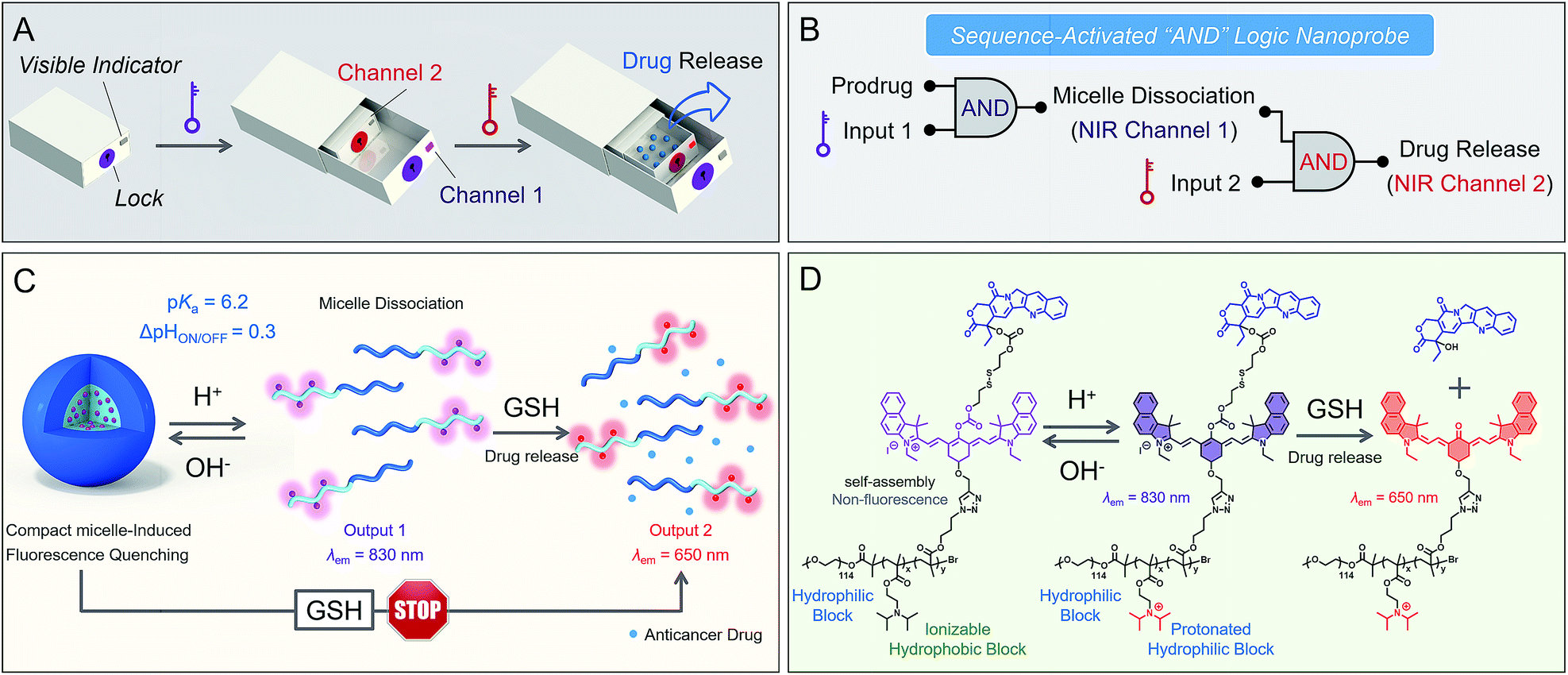 | ||
| Fig. 1 Sequence-activated AND logic dual-channel NIR fluorescent probe. (A and B) Logic circuit diagram of the sequence-activated AND logic nanoprobe for real-time tracking and programmable drug release via a dual NIR channel. (C and D) Schematic design of the sequence-activated AND logic dual-channel NIR theranostic nanoprodrug. At pH > pKa, the probe self-assembles as a compact micelle, leading to fluorescence quenching at 830 nm. At pH < pKa, the probe is in a micelle-dissociated state with a dramatic increase in fluorescence at 830 nm. Subsequently, biothiol-driven dual-channel NIR fluorescence (a remarkable shift from 830 to 650 nm) occurs for tracking the programmable responses in synergy with active camptothecin (CPT) release. | ||
Results and discussion
Activatable dual-channel NIR fluorescence signals
Firstly, we focus on whether P(Cy-S-CPT) could sense subtle pH differences between the acidic endocytic organelles of tumor cells (5.0–6.0) and blood (7.4). Notably, P(Cy-S-CPT) displays a sharp and reversible pH transition (pKa = 6.3, ΔpHON/OFF = 0.3, Fig. 2A and S1 in the ESI†). At pH 7.4, P(Cy-S-CPT) forms a compact self-assembled micelle with a spherical morphology, which completely silences the fluorophores40 (Fig. 2A and B). Moreover, the aggregation-caused NIR decrease of the monomer Cy-S-CPT confirms the micellization-induced quenching effect (Fig. S2†). As expected, when the pH is decreased to 6.0 below the probe’s transition, the protonation of the hydrophobic block makes the original compact micelle disassemble, and then leads to a dramatic NIR fluorescence enhancement at 830 nm (Fig. 2A and D). Thus, P(Cy-S-CPT) could be established as an ultra-sensitive pH-activatable NIR nanoprobe, which remains silent in the blood circulation but performs selective fluorescence activation in acidic tumor microenvironments.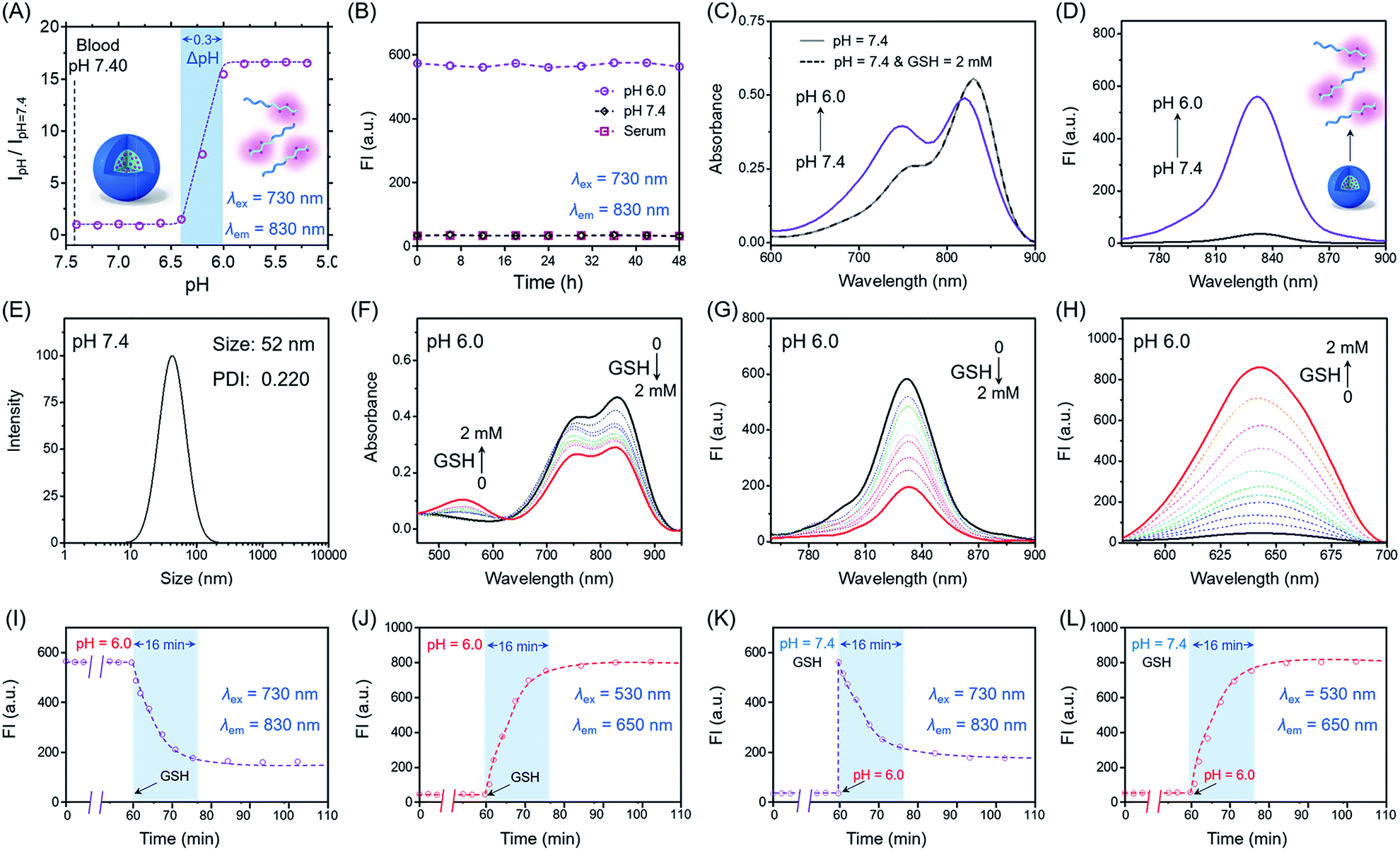 | ||
| Fig. 2 Self-assembly and dual-channel response of the NIR fluorescent probe. (A) Fluorescence intensity as a function of pH for P(Cy-S-CPT). At a high pH (for example, 7.4), P(Cy-S-CPT) remains silent. At a pH below its transition (pH = 6.3), the nanoprobe can be activated as a result of micelle dissociation. For P(Cy-S-CPT), the pH response is extremely sharp (ΔpHON/OFF = 0.3). (B) The P(Cy-S-CPT) nanoprobe remains stable in PBS and human serum over 48 h at 37 °C. Absorption (C) and emission spectra (D) with λex = 730 nm of P(Cy-S-CPT) in aqueous solution at pH 7.4 (with and without GSH) and 6.0. (E) Size distribution of P(Cy-S-CPT) in aqueous solution at pH 7.4. The absorption spectra (F) and fluorescence emission spectra with (G) λex = 730 nm and (H) λex = 530 nm of P(Cy-S-CPT) in the presence of GSH (each point was recorded after exposure to GSH for 15 min at 37 °C) in aqueous solution at pH 6.0. (I–L) Time dependence of fluorescence intensity of P(Cy-S-CPT) (I and J) in aqueous solution at pH 6.0, with GSH added at 1 h and (K and L) in the presence of GSH, adjusting the pH from 7.4 to 6.0 at 1 h at 830 and 650 nm. | ||
Subsequently, biothiol-driven dual-channel NIR fluorescence (a remarkable shift from 830 to 650 nm) was investigated for tracking the programmable responses in synergy with active CPT release. P(Cy-S-CPT) was initially at pH 6.0 (below its transition) in aqueous solution, wherein the micelle disassembled with remarkable NIR emission at 830 nm. Upon the addition of GSH (2 mM), P(Cy-S-CPT) containing disulfide linkages displayed an obvious wavelength-shifting change in both the absorption and emission spectra,41,42 along with a color change from green to purple-red. The absorption peak at 730 nm decreased sharply, and a new band centered at 530 nm was observed, along with a distinct isosbestic point at 635 nm (Fig. 2F). Concomitantly, a remarkable hypsochromic shift was also observed in the emission spectra. That is, an NIR fluorescence band at 830 nm was decreased, accompanied by a sharp increase at 650 nm (Fig. 2G and H). It took within 16 min to reach the reaction equilibrium at 37 °C. In this case, the dual-channel NIR fluorescence responses at pH 6.0 make it possible to simultaneously track where, when, and how the prodrug is activated and released in vivo (Fig. 1).
Sequence-activated AND logic behaviors
Unprecedentedly, P(Cy-S-CPT) shows quite different GSH responses at pH levels above its transition (pH > 6.3). At pH 7.4, we found that P(Cy-S-CPT) produced neither colorimetric nor fluorescence spectral changes upon the addition of GSH (Fig. 2C, K and L). This could be ascribed to the preferentially self-assembled compact micelles of P(Cy-S-CPT) at pH 7.4, which thereby reduced the permeation of GSH into the hydrophobic core. Consequently, the release of active camptothecin (CPT) is effectively hampered. Also, if GSH is absent, P(Cy-S-CPT) as a prodrug remains in an intact state regardless of its micelle self-assembly or disassembly (Fig. 2I and J). All TEM images and HPLC analyses provided solid evidence for the aforementioned activation process (Fig. S3 and S4†).As such, an AND logic gate was built up so that the output active CPT is produced only if H+ and GSH are both present as dual inputs. In particular, a programmable fluorescence response was carried out by the two sequence-dependent inputs (Fig. 1A and B). Specifically, output 1 is fluorescence at 830 nm (NIR Channel 1) which indicates whether the nanoprobe becomes activated and converts to a secondary form (disassembly); subsequently coupled to the second input of GSH, the disassembled nanoprobe exhibits another readout fluorescence at 650 nm (NIR Channel 2) as output 2 and the concomitant release of active CPT (Fig. 1B and C). Taken all together, the sequential AND logic gate is successfully constructed, providing a promising strategy for selective sensing and targeted drug release.
To characterize the nanostructure of the self-assembled nanoprobe at pH 7.4, a solution of P(Cy-S-CPT) was obtained by dialysis. We observed that the assemblies had an average size of 52 nm measured by dynamic laser scattering (DLS) with a polydispersion index (PDI) of 0.22 (Fig. 2E and S3†). Importantly, these well-defined compact nanoassemblies of P(Cy-S-CPT) maintain their initial good stability over 48 h in human serum at 37 °C (Fig. 2B). To gain further insight into their self-assembled behavior, the critical micelle concentration (CMC) of P(Cy-S-CPT) was also measured (Fig. S5†). Notably, the CMC value of P(Cy-S-CPT) is calculated to be only about 0.85 μg mL−1. This remarkably low CMC value further confirms the compact and stable nanostructure of P(Cy-S-CPT).
Prior to investigating the programmable responses in living systems, the specificity of sequence-activated P(Cy-S-CPT) for H+ and GSH was evaluated with potential competitive species including amino acids, enzymes, serum markers, metabolic substances and various tissue homogenates (including the heart, liver, spleen, lung and kidney of nude mice). As expected, at a pH above its transition, P(Cy-S-CPT), which forms a compact micellar structure, did not show any fluorescence response to any of the species (Fig. S6 and S7†). By comparison, only H+ elicited a dramatic increase in the fluorescence intensity at 830 nm, suggesting a pH-induced dissociation of the original compact micelle. Furthermore, only upon the addition of H+ and then GSH did P(Cy-S-CPT) exhibit an obvious enhancement at another new fluorescence channel of 650 nm, while the propagation of fluorescence at the 830 nm channel became quenched. Thus, this selective dual-channel fluorescence response promises the preservation of prodrugs from nonspecific activation, while being able to initiate the designated drug release when H+ and GSH are sequence-dependently triggered.
Real-time tracking of each step that leads to drug release in living cells
As is well known, cancer cells possess acidic endocytic organelles and abnormal GSH-overexpressing cell cytosol. To establish the sequential AND logic behavior in a multistaged tumor environment, A549 cancer cells were incubated with P(Cy-S-CPT). As expected, the fluorescent dots at 830 nm were firstly observed (Fig. 3C), indicating that this nanoprobe was activated and became disassembled inside the endosomes/lysosomes with lower pH values. Concurrently, there was observed abundant 650 nm fluorescence in the cytoplasm, indicative of the probe’s cleavage by GSH after disassembling. In contrast, after pretreating A549 cells with NEM (GSH inhibitor), the fluorescence of P(Cy-S-CPT) at 830 nm was still observed while there was no fluorescence at 650 nm (Fig. 3A). Moreover, after pretreating A549 cells with NaHCO3, the two fluorescence channels of P(Cy-S-CPT) at 830 and 650 nm were not observed (Fig. 3B). In conjunction with the in vitro results, it can be concluded that we have successfully developed a sequence-activated AND logic probe for the selective intracellular sensing of a multistage environment (Fig. 3G).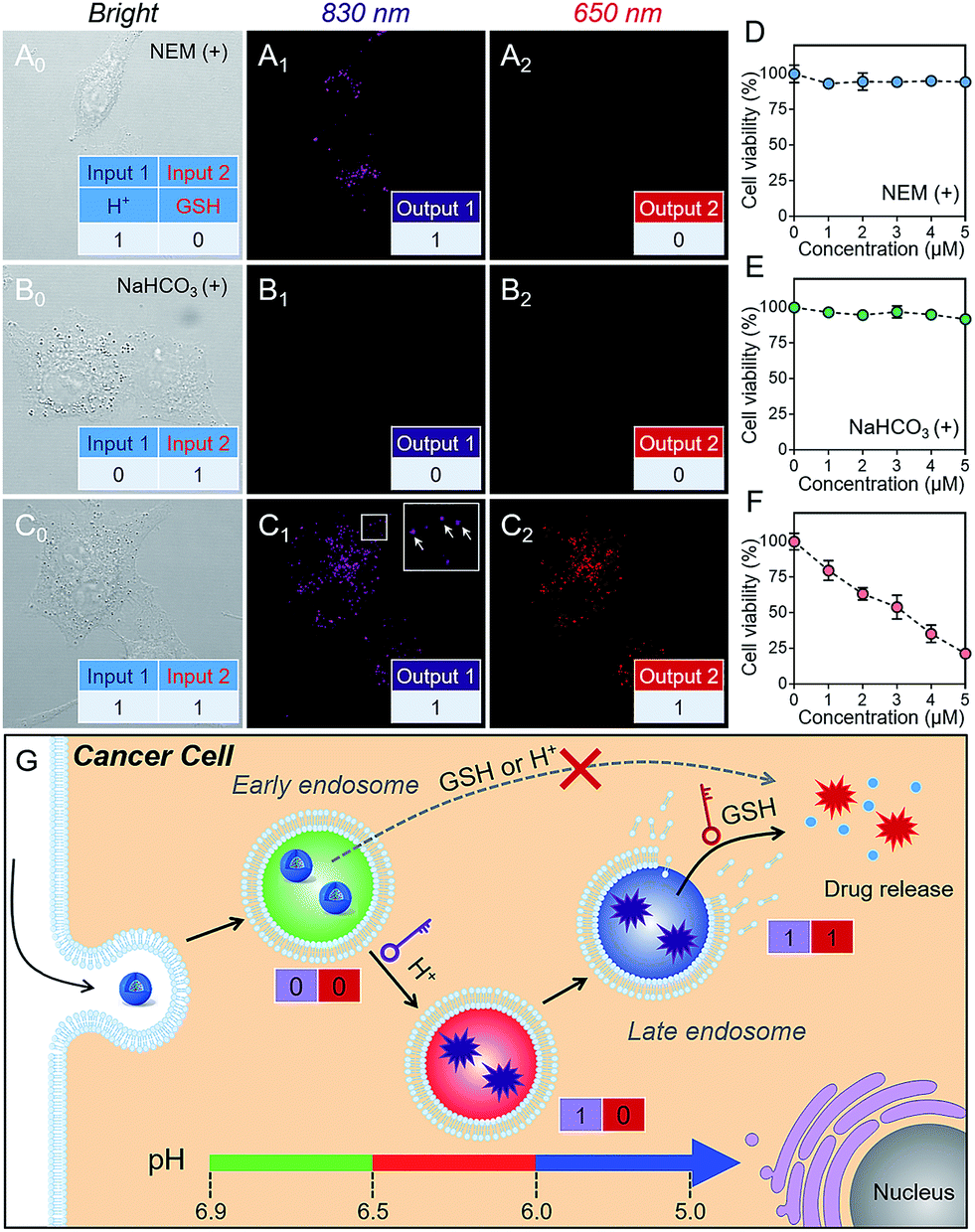 | ||
| Fig. 3 Dual-channel fluorescence tracking of the programmable drug release in vitro. A549 cancer cells were incubated with NEM & P(Cy-SCPT) (A), NaHCO3 & P(Cy-SCPT) (B) and P(Cy-SCPT) (C) at 37 °C for 1 h. Note: Channel 1: the violet signal (λex = 730 nm, λem = 830 nm) indicates the fluorescence from H+-triggered disassembling; Channel 2: the red signal (λex = 530 nm, λem = 650 nm) indicates prodrug activation by GSH. A549 cells & NEM (D), A549 cells & NaHCO3 (E) and A549 cells (F) were incubated with various concentrations of P(Cy-SCPT) for 24 h. The cell viability was determined using a MTT assay. (G) Schematic design of sequence-activated AND logic dual-channel NIR nanotheranostics. | ||
To further validate the biocompatibility, the cytotoxicity of P(Cy-S-CPT) against A549 cancer cells and normal cell lines was investigated by using a standard MTT assay. As expected, the nanoprobe P(Cy-S-CPT) exhibited a much higher cytotoxicity against cancer cells and no cytotoxicity against normal cells (Fig. S8†). Meanwhile, when A549 cancer cells were preincubated with NaHCO3 or NEM (Fig. 3D–F), P(Cy-S-CPT) exhibited no cytotoxic effects. Taken together, all of these results validated that toxicity results from the H+ and GSH sequential-driven release of active CPT through induced cell apoptosis (Fig. 3G), which is synchronous with dual-channel NIR fluorescence imaging. Clearly, this nanoprobe is selectively intracellularly activated in cancer cells, resulting in programmable drug release and minimizing the side-effects on normal cells.
In vivo dual-channel imaging of the programmable drug release in tumors
The above promising results inspired us to further explore the feasibility of P(Cy-S-CPT) in living mice. To date, almost all reported drug delivery systems suffer from only one readout channel, which could produce “false positive” information from the programmable behavior of theranostics in vivo. In the prodrug P(Cy-S-CPT), the remarkable shift from the dual-channel response made it perfect to simultaneously track each step of the CPT release. After the intravenous injection of A549 xenograft tumor-bearing mice, in vivo and ex vivo fluorescence bioimaging was recorded. In this way, the in vivo behavior of the small molecule Cy-S-CPT (as a control) was monitored by a dual read-out fluorescence channel (yellow-red represented at 830 nm; rainbow represented at 650 nm). As shown in Fig. 4A and B, only a small accumulated amount of Cy-S-CPT was observed at the tumor, but there were strong fluorescence signals from the liver in the dual read-out channel, suggestive of a continuous CPT release that was triggered by nonspecific activation and liver metabolism function towards the small molecular prodrug. In contrast, a specific targeted tumor accumulation of P(Cy-S-CPT) was visualized through both the 830 and 650 nm fluorescence channels (Fig. 4C and D). This could be attributed to the synergistic effect of the nanoprobe P(Cy-S-CPT): passive targeting from the EPR effect and activatable targeting (sequence-dependent pH and GSH triggering in a multistage tumor environment).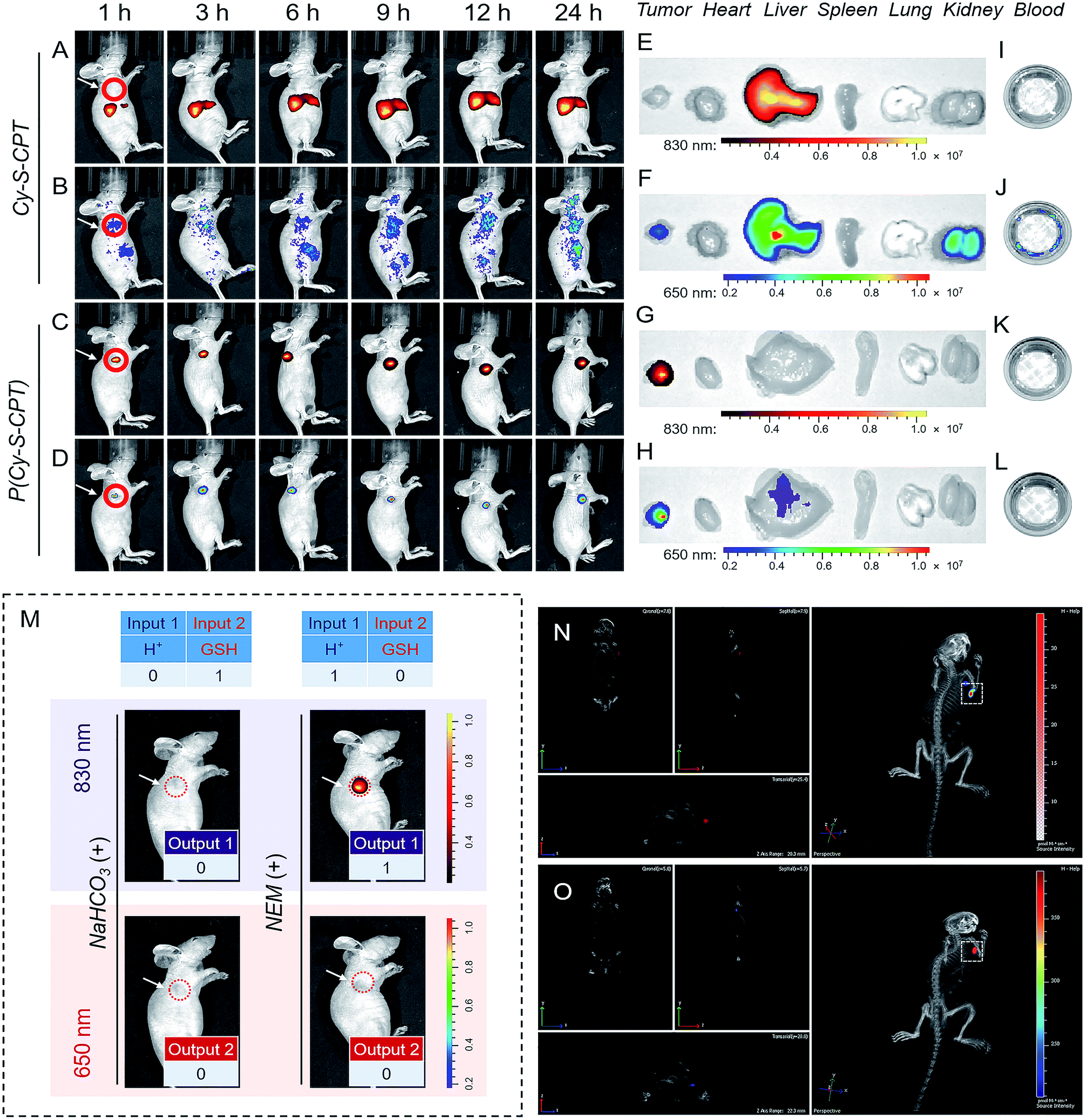 | ||
| Fig. 4 Dual-channel fluorescence tracking of the programmable drug release in vivo. In vivo dual-channel NIR-fluorescence imaging of A549 xenograft tumor-bearing mice at various times (1, 3, 6, 9, 12 and 24 h) after the intravenous injection of Cy-S-CPT (A and B) and P(Cy-S-CPT) (C and D) administered at a CPT-equivalent dose of 0.1 mg kg−1. The tumor site is circled in red. Ex vivo NIR-fluorescence imaging of the excised organs (heart, liver, spleen, lung, kidney and tumor) and blood at 24 h after the intravenous injection of Cy-S-CPT (E, F, I and J) and P(Cy-S-CPT) (G, H, K and L). (M) NaHCO3 or NEM was intratumorally injected 2 h before the intravenous injection of P(Cy-S-CPT). Note: fluorescence signals at 830 nm (yellow-red scale: A, C, E, G, I and K) and 650 nm (rainbow scale: B, D, F, H, J and L). (N and O) Three-dimensional in vivo imaging after the intravenous injection of P(Cy-S-CPT) for 24 h. Note: NIR fluorescence signals at 830 nm (N, white-red scale) and 650 nm (O, rainbow scale). | ||
After 24 h of intravenous injection, the tumors and major organs of the mice were collected to investigate the biodistribution of Cy-S-CPT and P(Cy-S-CPT), respectively. As shown in Fig. 4E and F, the dual-channel fluorescence of Cy-S-CPT revealed that it is mainly distributed in the liver region, suggestive of a continuous CPT release triggered by liver metabolism function. Comparatively, the ex vivo fluorescence images of the excised tumors with P(Cy-S-CPT) confirmed a high specific accumulation and then activation (Fig. 4G and H). Accordingly, there was much weaker fluorescence in the liver and no fluorescence in the other organs (heart, spleen, lung and kidney). Notably, there was no fluorescence signal in the blood, indicating that P(Cy-S-CPT) undergoes no destruction with a high stability in the circulatory system. Furthermore, for H+ and GSH inhibition control, NaHCO3 or NEM was intratumorally injected 2 h before P(Cy-S-CPT) administration. As shown in Fig. 4M, sequential AND logic behavior in vivo was achieved in tumor-bearing mice. These programmable dual-channel NIR fluorescence responses are consistent with the cell imaging and MTT assay. All of these results show that P(Cy-S-CPT) possesses striking characteristics of having well-targeting properties, and sustained and precisely controllable programmable drug release.
Real-time three-dimensional imaging is a powerful tool for accurate disease diagnostics, especially for suspicious lesions, with high spatiotemporal precision. The preferable characteristics of P(Cy-S-CPT) with an 830 nm fluorescence channel (activated by H+) shifting to a 650 nm fluorescence channel (activated by H+ and then GSH) make it suitable for performing real-time three-dimensional bioimaging. After a tail vein injection of P(Cy-S-CPT) into the mice, real-time, high-resolution three-dimensional fluorescence images were obtained with an IVIS Spectrum CT imaging system. As shown in Fig. 4N and O and Video 1 and 2 (in the ESI†), P(Cy-S-CPT) exhibited strong 830 and 650 nm fluorescence signals in the tumor, thus achieving real-time tracking from different perspectives, showing when the nanoprobe is disassembled and how the active drugs are released in vivo.
To evaluate the efficient tumor accumulation and specific programmable drug release, the in vivo antitumor activity of P(Cy-S-CPT) was systematically studied. Specifically, CPT, Cy-S-CPT and P(Cy-S-CPT) were intravenously injected into A549 tumor-bearing mice at a 10 mg CPT kg−1 dose, and the tumor growth was continuously monitored. As shown in Fig. 5A, in the mice treated with P(Cy-S-CPT), the tumor volume was significantly reduced compared with the control group (p < 0.001). More impressively, the tumor in the P(Cy-S-CPT) group was largely eradicated with an inhibition of 93.6% without there being a noticeable influence on body weight (Fig. 5B, C and D). Obviously, P(Cy-S-CPT) can distinctly reduce the side effects on normal tissues and can promote therapeutic efficiency via accurate recognition and tumor specific programmable drug release.
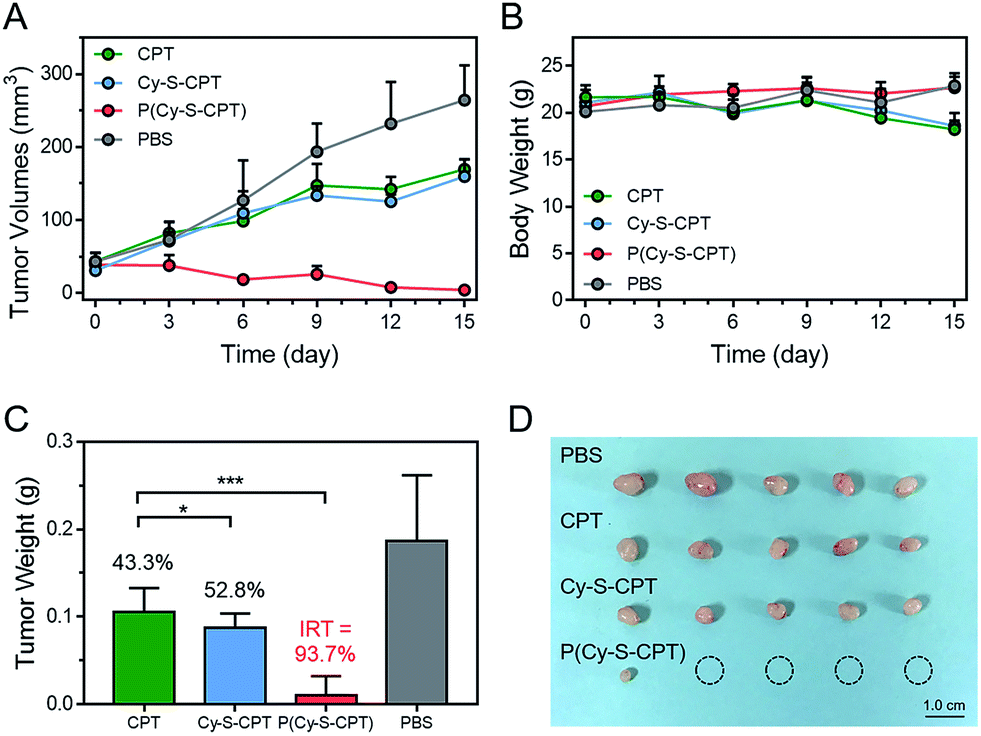 | ||
| Fig. 5 In vivo antitumor activities of the sequence-activated AND logic nanoprodrug against the A549 tumor xenograft antitumor activities of PBS, CPT, Cy-S-CPT, and P(Cy-S-CPT) administered at a CPT-equivalent dose of 10 mg kg−1 every 3 days against A549 xenograft tumors via intravenous injections. The results are summarized as the tumor volumes of the mice bearing A549 tumors exposed to various treatments (A), body weight changes (B), the tumor weights of each group of the mice at the end of the experiment and the inhibition rates of tumor growth (IRT) (*p < 0.1; ***p < 0.001) (C) and an image of the tumors (D). | ||
Conclusions
In summary, we developed an unprecedented sequence-activated AND logic dual-channel NIR fluorescence probe P(Cy-S-CPT) that can sense a multistage tumor environment prior to the release of an active drug. The sense-of-logic dual-channel nanoprobe can stay silent in the blood circulatory system, and is activated synchronously with in vivo dual-channel NIR fluorescence output in response to sequence-dependent stimuli such as ultra-small pH changes (ΔpHON/OFF = 0.3) and overexpressed biothiols. The perfect integration of functional sensing and drug release is a breakthrough in the real-time tracking of each step that leads to drug release in vivo, along with three-dimensional bioimaging from the dual-channel NIR fluorescence feedback. Impressively, P(Cy-S-CPT) exhibits excellent multistage tumor targeting ability, as well as a significant enhancement in antitumor activity in vivo, nearly eradicating the tumor. The strategy protects the prodrug from destructive environments and undesirable interactions, while also being able to initiate designated drug release when needed. This sense-of-logic nanoprobe P(Cy-S-CPT) provides a prototype for the development of in vivo intelligent biosensing probes for a precise programmable drug-delivery system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSFC/China (21788102, 21421004, 21636002 and 21622602), the National Key Research and Development Program (2017YFC0906900, 2016YFA02003), Oriental Scholarship, Scientific Committee of Shanghai (14ZR1409700 and 15XD1501400), Shanghai Pujiang Program (13PJD010), the Fok Ying Tong Education Foundation (142014), the Fundamental Research Funds for the Central Universities (WK1013002), and Programme of Introducing Talents of Discipline to Universities (B16017). This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and was approved by the Institutional Animal Care and Use Committee of the National Tissue Engineering Center (Shanghai, China).Notes and references
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- T. Gunnlaugsson, Nat. Chem., 2016, 8, 6–7 CrossRef PubMed.
- C. Y. Yu, H. Xu, S. Ji, R. T. Kwok, J. W. Lam, X. Li, S. Krishnan, D. Ding and B. Z. Tang, Adv. Mater., 2017, 29, 1606167 CrossRef PubMed.
- X. Li, C. Y. Kim, S. Lee, D. Lee, H. M. Chung, G. Kim, S. H. Heo, C. Kim, K. S. Hong and J. Yoon, J. Am. Chem. Soc., 2017, 139, 10880–10886 CrossRef PubMed.
- H. S. Jung, J. Han, H. Shi, S. Koo, H. Singh, H. J. Kim, J. L. Sessler, J. Y. Lee, J. H. Kim and J. S. Kim, J. Am. Chem. Soc., 2017, 139, 7595–7602 CrossRef PubMed.
- J. Peng, A. Samanta, X. Zeng, S. Han, L. Wang, D. Su, D. T. Loong, N. Y. Kang, S. J. Park, A. H. All, W. Jiang, L. Yuan, X. Liu and Y. T. Chang, Angew. Chem., Int. Ed., 2017, 56, 4165–4169 CrossRef PubMed.
- Y. Yuan, C. J. Zhang, R. T. K. Kwok, D. Mao, B. Z. Tang and B. Liu, Chem. Sci., 2017, 8, 2723–2728 RSC.
- L. He, X. Yang, K. Xu, X. Kong and W. Lin, Chem. Sci., 2017, 8, 6257–6265 RSC.
- F. Li, J. Lu, X. Kong, T. Hyeon and D. Ling, Adv. Mater., 2017, 29, 1605897 CrossRef PubMed.
- L. K. Truman, S. Comby and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2012, 51, 9624–9627 CrossRef PubMed.
- M. H. Lee, E. J. Kim, H. Lee, H. M. Kim, M. J. Chang, S. Y. Park, K. S. Hong, J. S. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 16380–16387 CrossRef PubMed.
- Y. Yuan, R. T. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546–2554 CrossRef PubMed.
- M. Ye, Y. Han, J. Tang, Y. Piao, X. Liu, Z. Zhou, J. Gao, J. Rao and Y. Shen, Adv. Mater., 2017, 29, 1702342 CrossRef PubMed.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef PubMed.
- X. Wu, Y. J. Tan, H. T. Toh, L. H. Nguyen, S. H. Kho, S. Y. Chew, H. S. Yoon and X. W. Liu, Chem. Sci., 2017, 8, 3980–3988 RSC.
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef PubMed.
- S. Wang, J. Lin, Z. Wang, Z. Zhou, R. Bai, N. Lu, Y. Liu, X. Fu, O. Jacobson, W. Fan, J. Qu, S. Chen, T. Wang, P. Huang and X. Chen, Adv. Mater., 2017, 29, 1701013 CrossRef PubMed.
- X. Xu, P. E. Saw, W. Tao, Y. Li, X. Ji, S. Bhasin, Y. Liu, D. Ayyash, J. Rasmussen, M. Huo, J. Shi and O. C. Farokhzad, Adv. Mater., 2017, 29, 1700141 CrossRef PubMed.
- J. Li, A. Dirisala, Z. Ge, Y. Wang, W. Yin, W. Ke, K. Toh, J. Xie, Y. Matsumoto, Y. Anraku, K. Osada and K. Kataoka, Angew. Chem., Int. Ed., 2017, 56, 14025–14030 CrossRef PubMed.
- J. Li, Y. Li, Y. Wang, W. Ke, W. Chen, W. Wang and Z. Ge, Nano Lett., 2017, 17, 6983–6990 CrossRef PubMed.
- X. Zhao, C. X. Yang, L. G. Chen and X. P. Yan, Nat. Commun., 2017, 8, 14998 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef PubMed.
- T. Ma, Y. Hou, J. Zeng, C. Liu, P. Zhang, L. Jing, D. Shangguan and M. Gao, J. Am. Chem. Soc., 2018, 140, 211–218 CrossRef PubMed.
- G. Yang, L. Xu, Y. Chao, J. Xu, X. Sun, Y. Wu, R. Peng and Z. Liu, Nat. Commun., 2017, 8, 902 CrossRef PubMed.
- A. Zakharchenko, N. Guz, A. M. Laradji, E. Katz and S. Minko, Nature Catalysis, 2017, 1, 73–81 CrossRef.
- Y. Li, S. Sun, L. Fan, S. Hu, Y. Huang, K. Zhang, Z. Nie and S. Yao, Angew. Chem., Int. Ed., 2017, 56, 14888–14892 CrossRef PubMed.
- J. Andréasson and U. Pischel, Chem. Soc. Rev., 2015, 44, 1053–1069 RSC.
- S. Erbas-Cakmak, S. Kolemen, A. C. Sedgwick, T. Gunnlaugsson, T. D. James, J. Yoon and E. U. Akkaya, Chem. Soc. Rev., 2018, 47, 2228–2248 RSC.
- A. P. de Silva, I. M. Dixon, H. Q. N. Gunaratne, T. Gunnlaugsson, P. R. S. Maxwell and T. E. Rice, J. Am. Chem. Soc., 1999, 121, 1393–1394 CrossRef.
- A. P. de Silva and S. Uchiyama, Nat. Nanotechnol., 2007, 2, 399–410 CrossRef PubMed.
- J. Guo, J. Zhuang, F. Wang, K. R. Raghupathi and S. Thayumanavan, J. Am. Chem. Soc., 2014, 136, 2220–2223 CrossRef PubMed.
- T. Soboleva, H. J. Esquer, A. D. Benninghoff and L. M. Berreau, J. Am. Chem. Soc., 2017, 139, 9435–9438 CrossRef PubMed.
- M. H. Lee, A. Sharma, M. J. Chang, J. Lee, S. Son, J. L. Sessler, C. Kang and J. S. Kim, Chem. Soc. Rev., 2018, 47, 28–52 RSC.
- Z. Liu, X. Zhou, Y. Miao, Y. Hu, N. Kwon, X. Wu and J. Yoon, Angew. Chem., Int. Ed., 2017, 56, 5812–5816 CrossRef PubMed.
- J. Liu, Y. Q. Sun, Y. Huo, H. Zhang, L. Wang, P. Zhang, D. Song, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 574–577 CrossRef PubMed.
- J. T. Hou, W. X. Ren, K. Li, J. Seo, A. Sharma, X. Q. Yu and J. S. Kim, Chem. Soc. Rev., 2017, 46, 2076–2090 RSC.
- Y. Wang, K. Zhou, G. Huang, C. Hensley, X. Huang, X. Ma, T. Zhao, B. D. Sumer, R. J. DeBerardinis and J. Gao, Nat. Mater., 2014, 13, 204–212 CrossRef PubMed.
- Z. Wang, M. Luo, C. Mao, Q. Wei, T. Zhao, Y. Li, G. Huang and J. Gao, Angew. Chem., Int. Ed., 2017, 56, 1319–1323 CrossRef PubMed.
- J. Chen, J. Ding, Y. Wang, J. Cheng, S. Ji, X. Zhuang and X. Chen, Adv. Mater., 2017, 29, 1701170 CrossRef PubMed.
- K. Zhou, H. Liu, S. Zhang, X. Huang, Y. Wang, G. Huang, B. D. Sumer and J. Gao, J. Am. Chem. Soc., 2012, 134, 7803–7811 CrossRef PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef PubMed.
- M. Z. Ye, X. H. Wang, J. B. Tang, Z. Q. Guo, Y. Q. Shen, H. Tian and W. H. Zhu, Chem. Sci., 2016, 7, 4958–4965 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02079e |
| This journal is © The Royal Society of Chemistry 2018 |