
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrene hydrogel for promoting direct bioelectrochemistry: ATP-independent electroenzymatic reduction of N2†
David P.
Hickey
a,
Koun
Lim
a,
Rong
Cai
a,
Ashlea R.
Patterson
a,
Mengwei
Yuan
a,
Selmihan
Sahin
ab,
Sofiene
Abdellaoui
a and
Shelley D.
Minteer
 *a
*a
aDepartment of Chemistry, University of Utah, 315 S 1400 E Room 2020, Salt Lake City, Utah 84112, USA. E-mail: minteer@chem.utah.edu
bDepartment of Chemistry, Faculty of Arts and Sciences, Suleyman Demirel University, Cunur, Isparta 32260, Turkey
First published on 14th May 2018
Abstract
Enzymatic bioelectrocatalysis often requires an artificial redox mediator to observe significant electron transfer rates. The use of such mediators can add a substantial overpotential and obfuscate the protein's native kinetics, which limits the voltage of a biofuel cell and alters the analytical performance of biosensors. Herein, we describe a material for facilitating direct electrochemical communication with redox proteins based on a novel pyrene-modified linear poly(ethyleneimine). This method was applied for promoting direct bioelectrocatalytic reduction of O2 by laccase and, by immobilizing the catalytic subunit of nitrogenase (MoFe protein), to demonstrate the ATP-independent direct electroenzymatic reduction of N2 to NH3.
Introduction
Oxidoreductases makeup a class of proteins that facilitate electron transfer reactions and play a critical role in virtually all metabolic pathways. As a result of their biological importance and use in bioelectrochemical devices, there has been a broad, sustained effort to develop strategies for interfacing redox enzymes to an electrode surface.1–15 The primary challenge associated with ‘wiring’ proteins to an electrochemical interface is the large polypeptide shell that often acts as an insulator between the redox-active cofactor and an electrode surface.According to Marcus theory, the terminal redox species of an electron transport chain needs to be within 14 Å of the electrode to enable electron transfer at a rate that is fast relative to the enzymatic reaction.16,17 An approach commonly employed to minimize this distance involves the use of a unique docking motif to immobilize the protein in a desired orientation; however, such strategies are often accomplished at the cost of exceptionally high enzyme loading due to denaturation of the protein during the immobilization/orientation process.18 This led us to consider the possibility of designing polymer materials with a focus on preserving residual protein activity rather than protein orientation at the electrode interface; thereby, preserving sufficient active protein to enable direct electron transfer (DET) through random orientation alone. The primary benefit to this conceptual approach is that it does not require a specific enzymatic binding motif, and therefore enables a “plug and play” template to study the direct electrochemistry for a broad range of redox proteins.
Cross-linked hydrogels are commonly used to immobilize biological catalysts onto a variety of electrode surfaces and can stabilize proteins through electrostatic interactions; however, their tendency to form a large porous network disfavours the spatial positioning of protein near the electrode surface necessary for direct electron transfer (DET). Conversely, pyrene has long been reported to adhere to carbon surfaces through non-covalent pi–pi interactions.19 While many reports have utilized pyrene in an attempt to anchor proteins directly to a carbon electrode,19–22 we hypothesized that pyrene could be employed to bind a hydrogel onto a carbon surface, thereby creating a matrix to entrap and stabilize a redox enzyme at the electrode interface (Fig. 1).
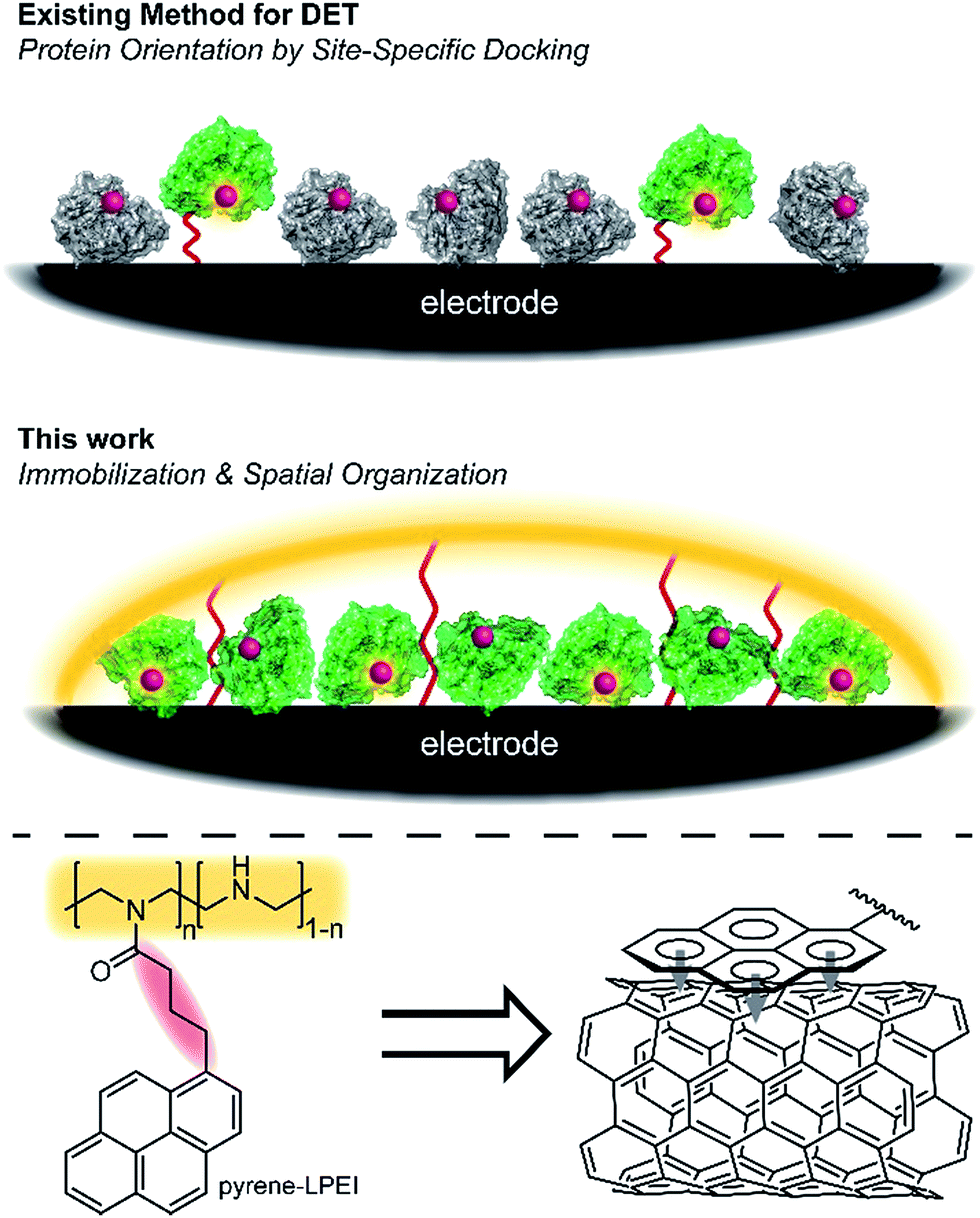 | ||
| Fig. 1 Scheme depicting the docking approach as compared to the hydrogel immobilization approach described here. Active redox protein is depicted in green, while inactive/denatured redox protein is depicted as grey. | ||
Herein, we describe a broadly applicable approach for direct electrochemical communication with redox-active proteins based on linear poly(ethylenimine) (LPEI) that has been covalently modified with pyrene moieties. The newly prepared pyrene-modified LPEI (pyrene-LPEI) can be used to immobilize a wide variety of oxidoreductases by cross-linking in the presence multiwalled carbon nanotubes (MWCNTs) at a carbon electrode to enable direct bioelectrocatalysis. We aim to demonstrate the robustness of our approach by employing pyrene-LPEI to facilitate DET in two example redox proteins, laccase and nitrogenase, for the electroenzymatic reduction of O2 at exceptionally low overpotentials, and the bioelectrosynthesis of NH3 from N2, respectively.
Results and discussion
To initiate our investigation, we prepared pyrene-LPEI by reacting an excess of 1-pyrenebutyric acid N-hydroxysuccinimide ester with high molecular weight LPEI overnight at room temperature followed by a thorough purification process to remove any unreacted pyrenebutyric acid NHS (a complete description of synthetic procedures and polymer characterizations is provided in the ESI†). The resulting polymer was determined by 1H-NMR to be substituted with pyrene pendant moieties on 22% of the backbone amines, and readily soluble in water up 10 mg mL−1. Similar to pyrene, pyrene-LPEI exhibits two strong fluorescence bands at 475 and 375 nm that correspond to a self-pi-stacked excimer and ‘free’ pyrene, where the ratio of intensity for these peaks skews towards free pyrene at low concentrations (Fig. S8a†).‡ To determine the ability of pyrene-LPEI to pi-stack onto MWCNTs, a Toray carbon paper electrode coated in MWCNT–COOH was incubated in a fluorescence cuvette for 10 minutes. The resulting solution exhibited significant quenching of the excimer fluorescence band in the presence of both bare Toray paper and MWCNT-coated Toray paper (Fig. S8b and c†). This is consistent with static quenching caused by the formation of pi–pi stacking complexes between pyrene and various carbon surfaces.23With polymer in hand, we sought to determine the feasibility of utilizing a cross-linked pyrene-modified hydrogel to immobilize and stabilize redox proteins at a carbon electrode surface. Several previous reports have demonstrated the ability of laccase from Trametes versicolor to undergo DET in the electroenzymatic reduction of O2.24–26 Laccase contains a single Cu atom near the protein surface (Cu–I) responsible for transferring electrons from an electron donor to a three-Cu cluster in the protein interior that catalyses the reduction of O2 to water at 0.615 ± 0.007 V vs. SCE (Fig. 2), and therefore direct bioelectrocatalysis of laccase depends on a sufficient quantity of the enzyme being oriented so the Cu–I site is within ∼14 Å of the electrode surface.27 Based on the ubiquity of laccase throughout the bioelectrocatalysis literature and its robust and well-understood electron transfer mechanism, we employed it as a benchmark for preliminary investigations focused on optimizing the newly prepared pyrene-LPEI.
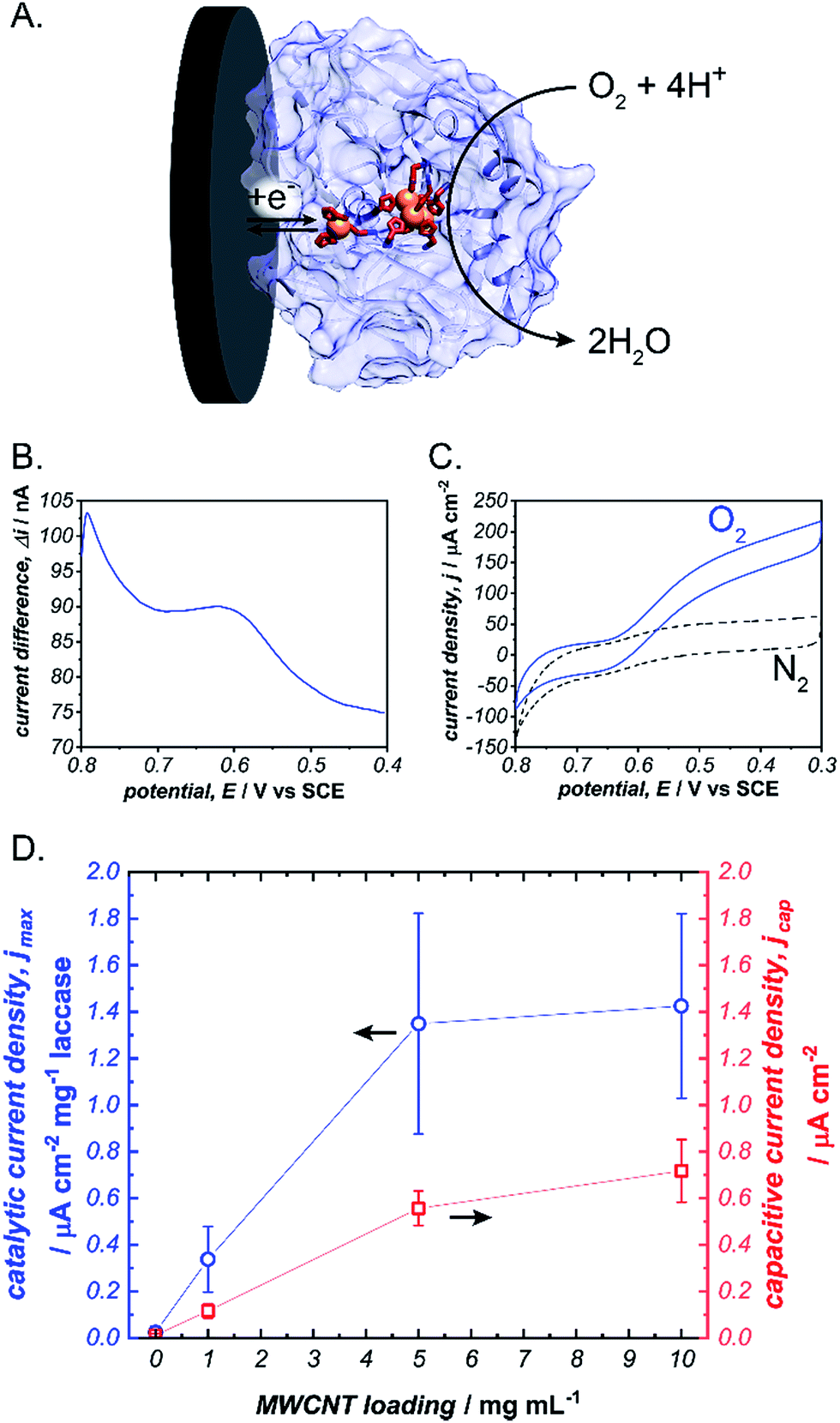 | ||
Fig. 2 (A) Scheme depicting the electrochemical reduction of O2 to H2O by laccase (blue). Square wave voltammogram for the Cu–I site of laccase immobilized in a cross-linked pyrene-LPEI film (B) and cyclic voltammograms of the same film under a bubbling of nitrogen ( ) or oxygen ( ) or oxygen ( ) (C). A plot depicting the background-subtracted catalytic current density (jmax) ( ) (C). A plot depicting the background-subtracted catalytic current density (jmax) ( ) and capacitive current density ( ) and capacitive current density ( ) for pyrene-LPEI/laccase films in the presence of O2 as a function of carbon nanotube loading (D). CVs were performed using 100 mM citrate buffer at pH 4.5, 25 °C and a scan rate of 5 mV s−1. ) for pyrene-LPEI/laccase films in the presence of O2 as a function of carbon nanotube loading (D). CVs were performed using 100 mM citrate buffer at pH 4.5, 25 °C and a scan rate of 5 mV s−1. | ||
Laccase/pyrene-LPEI bioelectrode films were prepared by cross-linking pyrene-LPEI in the presence of laccase with ethyleneglycol diglycidyl ether (EGDGE) and coating the resulting solution onto 0.25 cm2 Toray paper electrodes so that each film contained 89 μg of enzyme per electrode with varying concentrations of MWCNTs. Bioelectrocatalytic activity of laccase-modified electrodes was determined using cyclic voltammetry under bubbling of O2 gas (Fig. 2). The resulting films generated maximum catalytic current densities (jmax) of 40 ± 10, 390 ± 80, 1230 ± 280, and 1880 ± 80 μA cm−2 mg−1 laccase when containing 0, 1, 5 and 10 mg mL−1 of carboxylated MWCNTs (MWCNT–COOH). As a point of comparison, a commonly employed method for bioelectrocatalytic reduction of O2 by laccase utilizes anthracene-modified MWCNTs (An-MWCNTs) to orient the Cu–I centre towards the electrode surface and produces a limiting catalytic current density of 45 μA cm−2 mg−1 laccase.28
A proposed cause for low catalytic current per enzyme observed in the previously published An-MWCNT docking method comes from decomposition of the enzyme during the electrode preparation process.28 Therefore, we utilized a spectroscopic assay in which 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) is oxidized by laccase in the presence of O2, to determine residual specific activity of the enzyme immobilized in pyrene-LPEI vs. docked to An-MWCNTs in a film of tetrabutylammonium bromide modified Nafion© (TBAB/Nafion). For this assay, laccase was either immobilized onto the side of a cuvette with pyrene-LPEI or TBAB/Nafion with An-MWCNTs so that the films were submerged into assay solution but out of the beam path. By comparing the specific activity for each set of conditions with that of laccase dissolved in an aqueous solution, we found that for films of An-MWCNTs in TBAB/Nafion only 0.3 ± 0.1% of laccase remained active, while 15 ± 1% of laccase remained active in pyrene-LPEI films (full UV-Vis characterization of laccase is provided in the ESI†). This suggests that a sufficient amount of residually active redox protein at the electrode surface may result in a statistical distribution of protein that is properly oriented through random chance.
Cross-linked films of LPEI have previously been utilized to facilitate DET with redox proteins by enabling favourable electrostatic interactions between the polymer, enzyme, and electrode surface. To determine the extent that electrostatics of this type facilitate DET for pyrene-LPEI films, we performed a control experiment in which laccase was immobilized using unmodified LPEI in the presence of MWCNT–COOHs. Electrochemical analysis of the resulting films exhibited negligible bioelectrocatalytic responses; therefore, suggesting that electrostatic interactions alone cannot account for the electrochemical results obtained using pyrene-LPEI. A more complete description of these controls and the corresponding results are provided in the ESI.†
It should be noted that, while there is a clear correlation between jmax and concentration of CNTs, this comes at the cost of increased capacitance that can obscure finer electroenzymatic features in a CV. Nevertheless, our initial results demonstrate that pyrene-LPEI films can immobilize a redox enzyme in the presence of CNTs to enhance bioelectrocatalytic turnover rates, but also suggest that the polymer may be used without CNT additives to study mechanistic features of more complex oxidoreductases. With this in mind, we turned our attention to a Mo-dependent nitrogenase that has been shown to catalyse the in vivo reduction of N2 to ammonia.
Making up 78% of the Earth's atmosphere, N2 is among the most abundant raw materials on the planet and is consequently an ideal substrate for the production of ammonia-based fertilizers. However, its gaseous nature and the exceptional strength of a N–N triple bond make N2 a kinetic and thermodynamic sink, and as a result, the current process for converting N2 to ammonia (the Haber–Bosch process) consumes >1% of global energy produced per year.29 A promising alternative to the Haber–Bosch process employs a Mo-dependent nitrogenase protein complex to catalyse the electrochemical reduction of N2 to ammonia.30
Specifically, Mo-dependent nitrogenase is part of a bi-enzyme cascade that consists of a reducing protein (Fe protein) and a catalytic protein (MoFe). The MoFe protein contains a catalytic [7Fe–9S–Mo–C-homocitrate] (FeMoco) and a [8Fe–7S] cluster (P-cluster) that acts as an electron transfer bridge between the Fe protein and FeMoco.31,32 Mo-dependent nitrogenase is thought to operate by a redox-gated activation mechanism whereby the Fe protein binds transiently to the MoFe protein and where electron transfer is coupled to the hydrolysis of ATP to enable N2 reduction at the FeMoco by following equation:
| N2 + 8H+ + 8e− + 16ATP → 2NH3 + H2 + 16ADP + 16Pi |
To achieve sustainable substrate reduction by nitrogenase without the need for ATP, there is considerable interest in delivering electrons directly to the MoFe protein. However, success is commonly limited to N2H4, H+, NO2−, HCN and N3− substrates at very low rates or by photochemical methods.33–36 We recently reported utilizing cobaltocene as a redox mediator to “wire” the P-cluster to an electrode surface and effectively regenerate the MoFe protein electrochemically.37 While this strategy was effective for electroenzymatically reducing NO2− and N3−, the large overpotential of cobaltocene resulted in excessive electrochemical proton reduction to H2, which acts as an inhibitor to the MoFe protein and consequently prevented the electrochemical reduction of N2. Therefore, we hypothesized that pyrene-LPEI could be used to directly interface the P-cluster to minimize the required overpotential, limit electrochemical proton reduction and thereby enable electroenzymatic N2 reduction using only the MoFe protein.
To test this, we immobilized the MoFe protein in pyrene-LPEI onto Toray paper electrodes using the same procedure that was developed for laccase immobilization without MWCNTs. A combination of cyclic voltammetry and square wave voltammetry was used to identify a predominant redox feature at −0.51 ± 0.01 V vs. SCE. Based on previously suggested theoretical potentials, this peak was assigned to the P-cluster. As a control, the same experiment was performed with denatured MoFe, which resulted in shifted potential for the apparent P-cluster to −0.45 ± 0.01 V vs. SCE (Fig. 3). This suggests that the reversible redox peak is evidence of direct electrochemical communication with the non-denatured MoFe protein.
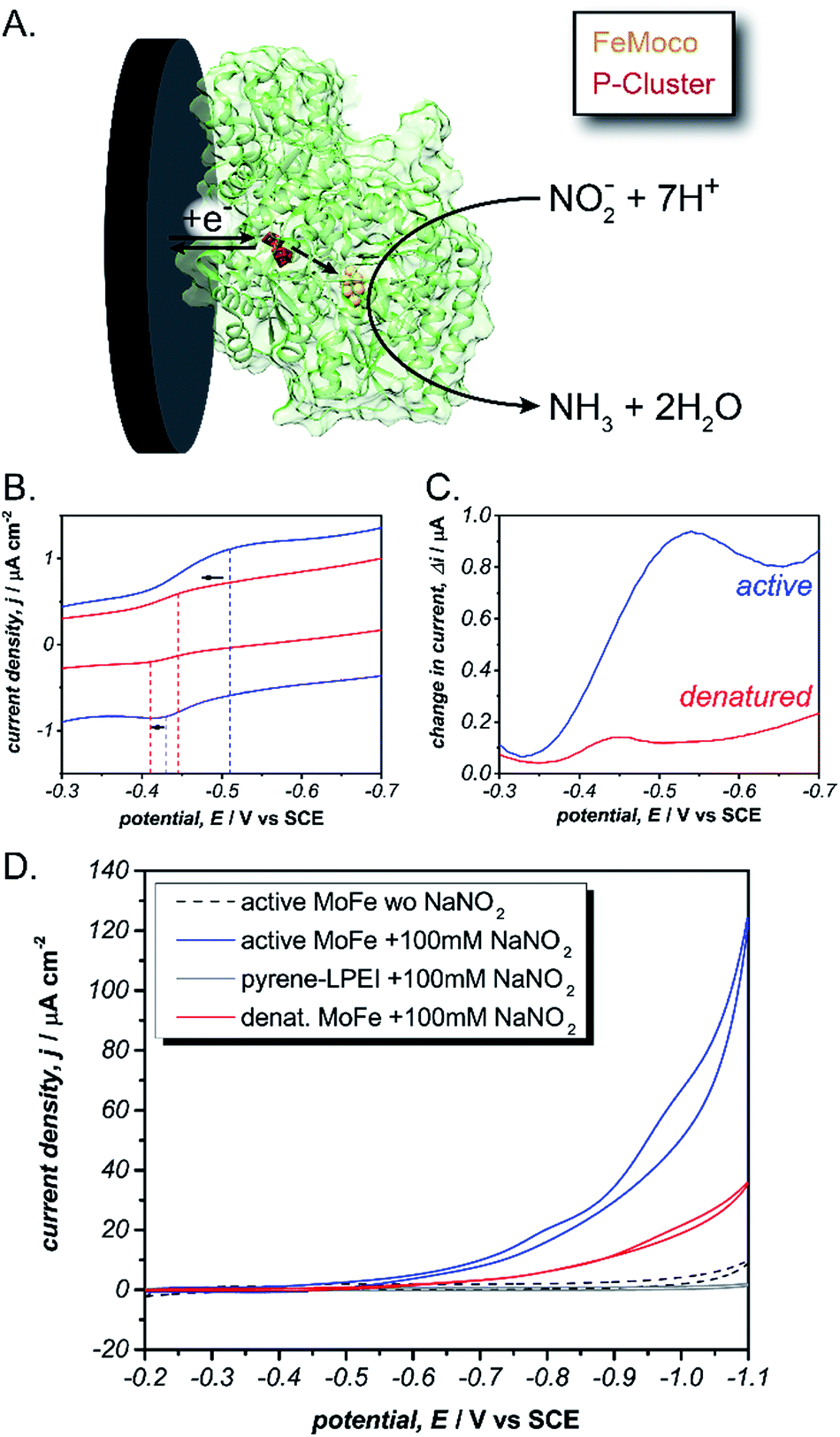 | ||
Fig. 3 (A) Scheme depicting the proposed direct electron transfer pathway for reduction of nitrite by MoFe protein (green). Cyclic voltammogram (B) and square wave voltammogram (C) for pyrene-LPEI films containing active ( ) or denatured ( ) or denatured ( ) MoFe protein. (D) Cyclic voltammogram of pyrene-LPEI films in containing active MoFe in the absence of nitrite ( ) MoFe protein. (D) Cyclic voltammogram of pyrene-LPEI films in containing active MoFe in the absence of nitrite ( ) or films containing active MoFe ( ) or films containing active MoFe ( ), denatured MoFe ( ), denatured MoFe ( ) and without MoFe ( ) and without MoFe ( ) in the presence of 100 mM NaNO2. CVs were performed using 100 mM MOPS buffer at pH 7.0, 25 °C and a scan rate of 5 mV s−1. ) in the presence of 100 mM NaNO2. CVs were performed using 100 mM MOPS buffer at pH 7.0, 25 °C and a scan rate of 5 mV s−1. | ||
For further evidence of direct electron transfer, we aimed to determine the ability of immobilized MoFe protein to exhibit direct bioelectrocatalysis. While N2 is the native substrate for nitrogenase, its gaseous nature makes it difficult to utilize in activity assays and previous Fe protein-decoupled studies have employed NO2− as a water soluble nitrogenase substrate.32 Consequently, our initial electrochemical activity experiments for the MoFe protein were performed using aqueous NaNO2. Cyclic voltammograms of the active MoFe protein immobilized in pyrene-LPEI exhibits a dramatically increased current density in the presence of 100 mM NaNO2 when compared to either the denatured MoFe protein or the pyrene-LPEI film alone (Fig. 3). It should be noted that the control employing denatured MoFe protein exhibits a small increase in current upon addition of NO2−; however, this is consistent with previous observations that the denatured P-cluster maintains residual activity towards nitrite. Nevertheless, these results suggest that the MoFe protein is both active inside the pyrene-LPEI film and is capable of direct bioelectrocatalysis.
Next, we aimed to explore the ability of pyrene-LPEI/MoFe protein films to reduce N2via direct bioelectrocatalysis. Due to the high sensitivity of the in vitro MoFe protein to O2, extreme care was taken to ensure that the electrochemical cell remained completely anaerobic. Bioelectrochemical activity of pyrene-LPEI/MoFe protein to N2 was measured by cyclic voltammetry under Ar and increasing time of bubbling in ultra high purity N2 (Fig. 4A). The resulting voltammogram demonstrates an increasing current density with additional bubbling of N2, which suggests that this is the first observation of bioelectrochemical N2 reduction in the absence of ATP. To confirm this result, we performed constant potential bulk electrolysis using electrodes coated with pyrene-LPEI containing active MoFe protein, denatured MoFe protein, BSA, and no protein in a sealed cell after bubbling ultra high purity N2 for 20 minutes. Products of the resulting electrolyses were analysed by a fluorometric assay to confirm and quantify the production of NH3. An aliquot of each electrolysis solution was combined with o-phthalaldehyde, which forms a fluorescent complex with NH3.25,32 Product analysis demonstrated that the bioelectrosynthetic reduction of N2 produced 180 ± 30 nmol of NH3 (1.1 ± 0.2 μmol mg−1 MoFe) compared to 44 ± 9 nmol NH3 under Ar, and 10 ± 9 nmol NH3 produced under N2 using pyrene-LPEI without the MoFe protein (Fig. 4B).
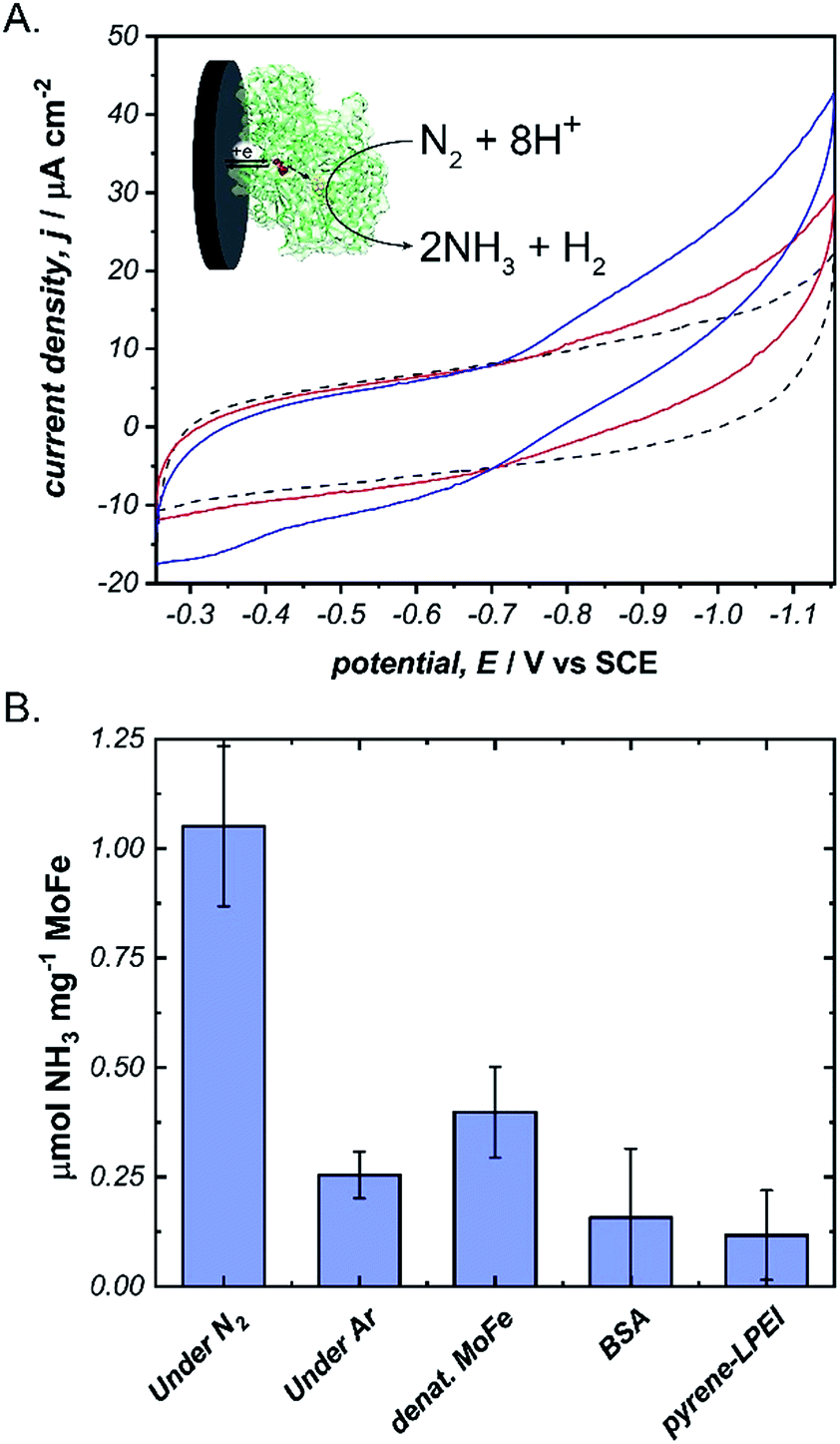 | ||
Fig. 4 (A) Cyclic voltammograms of pyrene-LPEI films containing MoFe protein and 5 mg mL−1 MWCNT–COOH under Ar ( ), and after 5 min ( ), and after 5 min ( ), or 10 min ( ), or 10 min ( ) of bubbling ultra high purity N2. (B) Production of NH3 using N2 as the only substrate after constant potential bulk electrolysis of pyrene-LPEI/MoFe films for 8 h. CVs were performed using 100 mM MOPS buffer at pH 7.0, 25 °C and a scan rate of 5 mV s−1. Bulk electrolysis was performed using 100 mM MOPS buffer pH 7.0 at −1.1 V vs. SCE and 25 °C. Error bars represent one standard deviation, where n = 3. ) of bubbling ultra high purity N2. (B) Production of NH3 using N2 as the only substrate after constant potential bulk electrolysis of pyrene-LPEI/MoFe films for 8 h. CVs were performed using 100 mM MOPS buffer at pH 7.0, 25 °C and a scan rate of 5 mV s−1. Bulk electrolysis was performed using 100 mM MOPS buffer pH 7.0 at −1.1 V vs. SCE and 25 °C. Error bars represent one standard deviation, where n = 3. | ||
Given the success of this method for studying direct bioelectrochemistry of laccase and nitrogenase, we sought to investigate the extent of applicability to a small sample of additional redox proteins containing a relatively diverse set of electrochemical potentials and redox cofactors. The resulting pyrene-LPEI/protein combinations were studied by CV and SWV to confirm the electrochemical potential of each observed cofactor. The results are summarized in Table 1 and representative CVs are displayed in Fig. 5.
| Enzyme | Cofactor | pH | Redox potential, E/V vs. SCE | ||
|---|---|---|---|---|---|
| This work | Literature | Ref. | |||
| Laccase | Cu-l | 4.5 | −0.615 ± 0.007 | −0.556 | 25 |
| Cytochrome C | Heme | 7.0 | −0.200 ± 0.006 | −0.174 | 38 |
| MoFe protein (nitrogenase) | P-Cluster [8Fe–7S] | 7.0 | −0.512 ± 0.008 | −0.544 | 39 |
| Ferredoxin | [2Fe–2S] | 7.0 | −0.583 ± 0.003 | −0.609 | 38 |
| Flavodoxin | FMN | 7.0 | −0.727 ± 0.008 | −0.688 | 40 |
| Free FMN | FMN | 7.0 | −0.437 ± 0.003 | −0.462 | 40 |
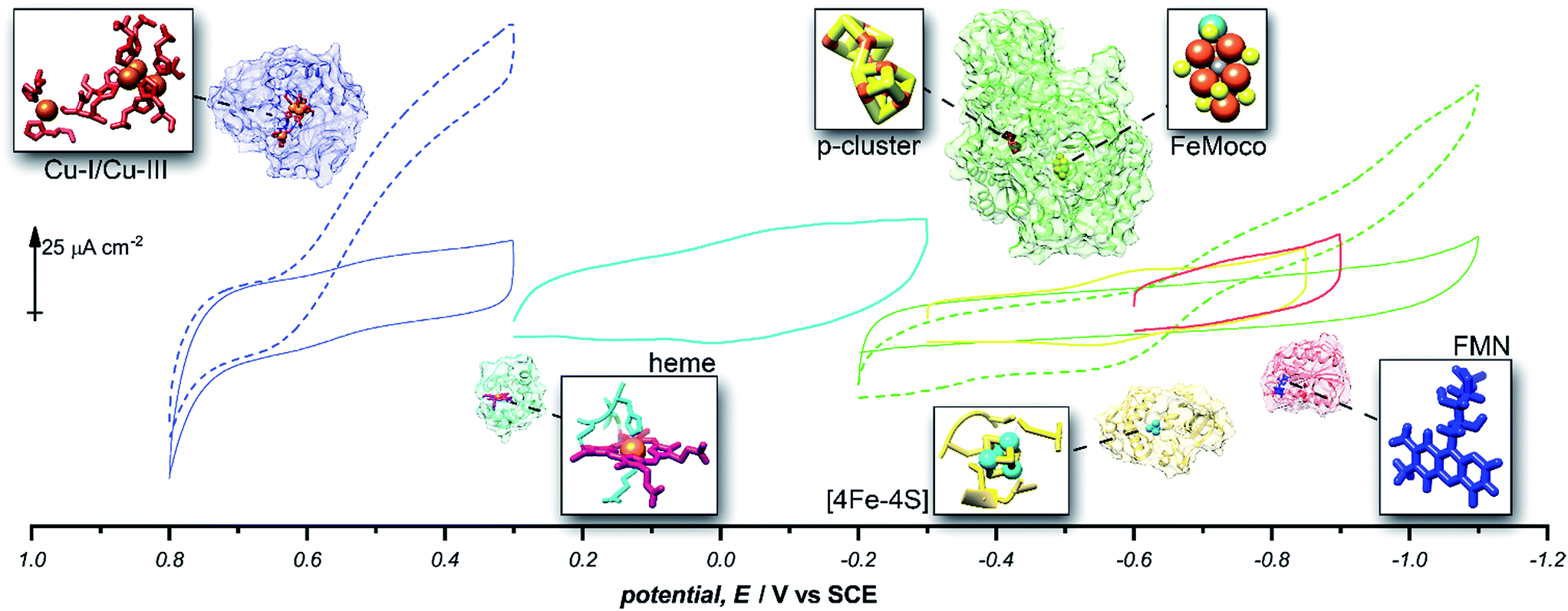 | ||
Fig. 5 Representative cyclic voltammograms of pyrene-LPEI films containing 5 mg mL−1 MWCNT–COOH and laccase under nitrogen ( ), laccase under O2 ( ), laccase under O2 ( ), cytochrome C ( ), cytochrome C ( ), ferredoxin ( ), ferredoxin ( ), flavodoxin ( ), flavodoxin ( ), MoFe protein under argon ( ), MoFe protein under argon ( ), and MoFe protein under N2 ( ), and MoFe protein under N2 ( ). All pyrene-LPEI/protein films were prepared using a single procedure (ESI†) without optimization for individual redox proteins. CVs were performed using 100 mM citrate (pH 4.5, for laccase), MOPS (pH 7.0 for MoFe protein), or phosphate (pH 7.0) at 25 °C and a scan rate of 5 mV s−1. ). All pyrene-LPEI/protein films were prepared using a single procedure (ESI†) without optimization for individual redox proteins. CVs were performed using 100 mM citrate (pH 4.5, for laccase), MOPS (pH 7.0 for MoFe protein), or phosphate (pH 7.0) at 25 °C and a scan rate of 5 mV s−1. | ||
Conclusions
In conclusion, we report a robust method based on a novel pyrene-modified LPEI material for creating a direct bioelectrochemical interface between a series of redox proteins and a carbon electrode without the need for specific orientation. Using this method, we were able to demonstrate direct electron transfer to MoFe protein of the nitrogenase complex and subsequent direct bioelectrochemical synthesis of NH3 from N2 without the need for ATP nor an artificial redox mediator. Future work will employ this method to study the direct electrochemical kinetics for the P-cluster and FeMoco of MoFe protein independently to provide a better understanding of the role each cofactor plays in the overall enzymatic mechanism of N2 reduction by nitrogenase.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Army Research Office MURI, the USDA NIFA program, and the Department of Energy SBIR with Fulcrum Bioscience for funding.Notes and references
- M. Cooney, V. Svoboda, C. Lau, G. Martin and S. D. Minteer, Energy Environ. Sci., 2008, 1, 320–337 Search PubMed.
- Y. Degani and A. Heller, J. Am. Chem. Soc., 1989, 111, 2357–2358 CrossRef.
- Y. Fu, P. Li, L. Bu, T. Wang, Q. Xie, X. Xu, L. Lei, C. Zou and S. Yao, J. Phys. Chem. C, 2010, 114, 1472–1480 Search PubMed.
- J. W. Gallaway and S. A. Calabrese Barton, J. Am. Chem. Soc., 2008, 130, 8527–8536 CrossRef PubMed.
- B. A. Gregg and A. Heller, Anal. Chem., 1990, 62, 258–263 CrossRef PubMed.
- T. L. Klotzbach, M. Watt, Y. Ansari and S. D. Minteer, J. Membr. Sci., 2008, 311, 81–88 CrossRef.
- C. G. Koopal, B. de Ruiter and R. J. Nolte, J. Chem. Soc., Chem. Commun., 1991, 1691–1692 RSC.
- S. A. Merchant, T. O. Tran, M. T. Meredith, T. C. Cline, D. T. Glatzhofer and D. W. Schmidtke, Langmuir, 2009, 25, 7736–7742 CrossRef PubMed.
- B. C. Thompson, O. Winther-Jensen, J. Vongsvivut, B. Winther-Jensen and D. R. MacFarlane, Macromol. Rapid Commun., 2010, 31, 1293–1297 CrossRef PubMed.
- B. F. Yon-Hin, M. Smolander, T. Crompton and C. R. Lowe, Anal. Chem., 1993, 65, 2067–2071 CrossRef.
- I. Willner, E. Katz, A. Riklin and R. Kasher, J. Am. Chem. Soc., 1992, 114, 10965–10966 CrossRef.
- I. Willner, N. Lapidot, A. Riklin, R. Kasher, E. Zahavy and E. Katz, J. Am. Chem. Soc., 1994, 116, 1428–1441 CrossRef.
- L. D. Mazzi, M. T. Sharon, K. Eugenii and W. Itamar, Angew. Chem., Int. Ed. Engl., 1995, 34, 1604–1606 CrossRef.
- A. Narvaez, E. Dominguez, I. Katakis, E. Katz, K. T. Ranjit, I. Ben-Dov and I. Willner, J. Electroanal. Chem., 1997, 430, 227–233 CrossRef.
- P. Fernando, K. Eugenii, H. S. Vered and W. Itamar, Chem. - Eur. J., 1998, 4, 1068–1073 CrossRef.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef.
- C. C. Moser, J. M. Keske, K. Warncke, R. S. Farid and P. L. Dutton, Nature, 1992, 355, 796 CrossRef PubMed.
- C. Léger and P. Bertrand, Chem. Rev., 2008, 108, 2379–2438 CrossRef PubMed.
- F. Giroud and S. D. Minteer, Electrochem. Commun., 2013, 34, 157–160 CrossRef.
- N. Lalaoui, K. Elouarzaki, A. Le Goff, M. Holzinger and S. Cosnier, Chem. Commun., 2013, 49, 9281–9283 RSC.
- I. Mazurenko, K. Monsalve, P. Infossi, M.-T. Giudici-Orticoni, F. Topin, N. Mano and E. Lojou, Energy Environ. Sci., 2017, 10, 1966–1982 Search PubMed.
- C. Gutiérrez-Sánchez, W. Jia, Y. Beyl, M. Pita, W. Schuhmann, A. L. De Lacey and L. Stoica, Electrochim. Acta, 2012, 82, 218–223 CrossRef.
- L. Liu, T. Wang, J. Li, Z.-X. Guo, L. Dai, D. Zhang and D. Zhu, Chem. Phys. Lett., 2003, 367, 747–752 CrossRef.
- D. Ivnitski and P. Atanassov, Electroanalysis, 2007, 19, 2307–2313 CrossRef.
- M. S. Thorum, C. A. Anderson, J. J. Hatch, A. S. Campbell, N. M. Marshall, S. C. Zimmerman, Y. Lu and A. A. Gewirth, J. Phys. Chem. Lett., 2010, 1, 2251–2254 CrossRef PubMed.
- G. Gupta, V. Rajendran and P. Atanassov, Electroanalysis, 2004, 16, 1182–1185 CrossRef.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef PubMed.
- M. T. Meredith, M. Minson, D. Hickey, K. Artyushkova, D. T. Glatzhofer and S. D. Minteer, ACS Catal., 2011, 1, 1683–1690 CrossRef.
- B. E. Smith, Science, 2002, 297, 1654–1655 CrossRef PubMed.
- R. D. Milton, R. Cai, S. Abdellaoui, D. Leech, A. L. De Lacey, M. Pita and S. D. Minteer, Angew. Chem., Int. Ed., 2017, 56, 2680–2683 CrossRef PubMed.
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3012 CrossRef PubMed.
- I. Dance, Chem. Commun., 2013, 49, 10893–10907 RSC.
- K. Danyal, B. S. Inglet, K. A. Vincent, B. M. Barney, B. M. Hoffman, F. A. Armstrong, D. R. Dean and L. C. Seefeldt, J. Am. Chem. Soc., 2010, 132, 13197–13199 CrossRef PubMed.
- L. E. Roth and F. A. Tezcan, J. Am. Chem. Soc., 2012, 134, 8416–8419 CrossRef PubMed.
- K. Danyal, A. J. Rasmussen, S. M. Keable, B. S. Inglet, S. Shaw, O. A. Zadvornyy, S. Duval, D. R. Dean, S. Raugei and J. W. Peters, Biochemistry, 2015, 54, 2456–2462 CrossRef PubMed.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters and L. C. Seefeldt, Science, 2016, 352, 448–450 CrossRef PubMed.
- R. D. Milton, S. Abdellaoui, N. Khadka, D. R. Dean, D. Leech, L. C. Seefeldt and S. D. Minteer, Energy Environ. Sci., 2016, 9, 2550–2554 Search PubMed.
- C. Xiang, Y. Zou, L.-X. Sun and F. Xu, Electrochem. Commun., 2008, 10, 38–41 CrossRef.
- R. Y. Igarashi and L. C. Seefeldt, in Catalysts for Nitrogen Fixation: Nitrogenases, Relevant Chemical Models and Commercial Processes, ed. B. E. Smith, R. L. Richards and W. E. Newton, Springer, Netherlands, 2004, ch. 5, vol. 1, pp. 97–140 Search PubMed.
- H. A. Heering and W. R. Hagen, J. Electroanal. Chem., 1996, 404, 249–260 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01638k |
| ‡ It should be noted that ‘free’ pyrene in this context is still covalently bound to the LPEI backbone but is not complexed to any other polymer-bound pyrene. |
| This journal is © The Royal Society of Chemistry 2018 |