
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron transfer in an acidophilic bacterium: interaction between a diheme cytochrome and a cupredoxin†
X.
Wang
a,
M.
Roger
b,
R.
Clément
a,
S.
Lecomte
c,
F.
Biaso
a,
L. A.
Abriata
 d,
P.
Mansuelle
e,
I.
Mazurenko
f,
M. T.
Giudici-Orticoni
a,
E.
Lojou
*a and
M.
Ilbert
*a
d,
P.
Mansuelle
e,
I.
Mazurenko
f,
M. T.
Giudici-Orticoni
a,
E.
Lojou
*a and
M.
Ilbert
*a
aAix Marseille Univ, CNRS, IMM, BIP, UMR 7281, 31 Chemin Aiguier, 13009 Marseille, France. E-mail: lojou@imm.cnrs.fr; milbert@imm.cnrs.fr
bSchool of Life Sciences, University of Dundee, Dundee, DD1 5EH, Scotland, UK
cInstitute for Chemistry and Biology of Membrane and Nano-objects, Allée Geoffroy St Hilaire, 33600 Pessac, France
dLaboratory for Biomolecular Modeling, École Polytechnique Fédérale de Lausanne and Swiss Institute of Bioinformatics, AAB014, Station 19, 1015 Lausanne, Switzerland
eAix Marseille Univ, CNRS, Institut de Microbiologie de la Méditerranée, FR 3479, Plate-forme Protéomique, Marseille Protéomique (MaP), B.P. 71, 13402 Marseille Cedex 20, France
fSchool of Biomedical Sciences, Leeds, LS2 9JT, UK
First published on 1st May 2018
Abstract
Acidithiobacillus ferrooxidans, a chemolithoautotrophic Gram-negative bacterium, has a remarkable ability to obtain energy from ferrous iron oxidation at pH 2. Several metalloproteins have been described as being involved in this respiratory chain coupling iron oxidation with oxygen reduction. However, their properties and physiological functions remain largely unknown, preventing a clear understanding of the global mechanism. In this work, we focus on two metalloproteins of this respiratory pathway, a diheme cytochrome c4 (Cyt c4) and a green copper protein (AcoP) of unknown function. We first demonstrate the formation of a complex between these two purified proteins, which allows homogeneous intermolecular electron-transfer in solution. We then mimic the physiological interaction between the two partners by replacing one at a time with electrodes displaying different chemical functionalities. From the electrochemical behavior of individual proteins, we show that, while electron transfer on AcoP requires weak electrostatic interaction, electron transfer on Cyt c4 tolerates different charge and hydrophobicity conditions, suggesting a pivotal role of this protein in the metabolic chain. The electrochemical study of the proteins incubated together demonstrates an intermolecular electron transfer involving the protein complex, in which AcoP is reduced through the high potential heme of Cyt c4. Modelling of the electrochemical signals at different scan rates allows us to estimate the rate constant of this intermolecular electron transfer in the range of a few s−1. Possible routes for electron transfer in the acidophilic bacterium are deduced.
Introduction
Biodiversity is an extraordinary source of microorganisms displaying unusual features such as resistance to high temperatures, high pressures, high salinity, extreme pHs, etc. Although these organisms were identified many years ago, the molecular factors that allow them to survive and grow under such extreme conditions are far from being completely understood. Extreme acidophiles grow optimally at an external pH of 3 or less. Their ability to survive such drastic environments has aroused a great deal of interest, from both a fundamental and an application point of view.1Acidithiobacillus ferrooxidans is one of the best studied model organisms and has been used for a long time in bioleaching mine processes and more recently in microbial fuel cells, thanks to its ability to gain energy through the oxidation of ferrous iron at pH as low as 2, thus allowing overcoming the issue of proton availability required to construct an efficient biocathode.2,3 It has been clearly shown that Fe2+ oxidation takes place outside the bacterial cell, while the electrons generated are driven into the periplasmic compartment via electron shuttling to the inner membrane where O2 reduction takes place.4Several metalloproteins involved in this metabolic chain have already been isolated (Scheme 1), and details on the components have been reported in comprehensive reviews.5,6 The primary electron acceptor, cytochrome Cyc2, is an outer-membrane monoheme c-type cytochrome. Then periplasmic proteins, including a blue copper protein, rusticyanin (Rus), and a diheme c-type cytochrome (Cyt c4), have been shown to transfer electrons to the terminal electron acceptor, cytochrome c oxidase (CcO), an inner-membrane protein belonging to the subgroup of heme-copper O2 reductases.7 All these proteins have in common a high value of their redox potentials,5 which fits with the redox potential of the Fe3+/Fe2+ redox couple at acidic pHs.8 A docking model suggests that Cyt c4 acts as a wire between Rus and CcO, with the heme of the highest potential (HemeH) in interaction with Rus, and the heme of the lowest potential (HemeL) in contact with CcO.9 Nevertheless, different models have been proposed to describe the full electron transfer (ET) pathway, and divergence exists concerning the sequential ET pathway between the proteins in the chain.10–12
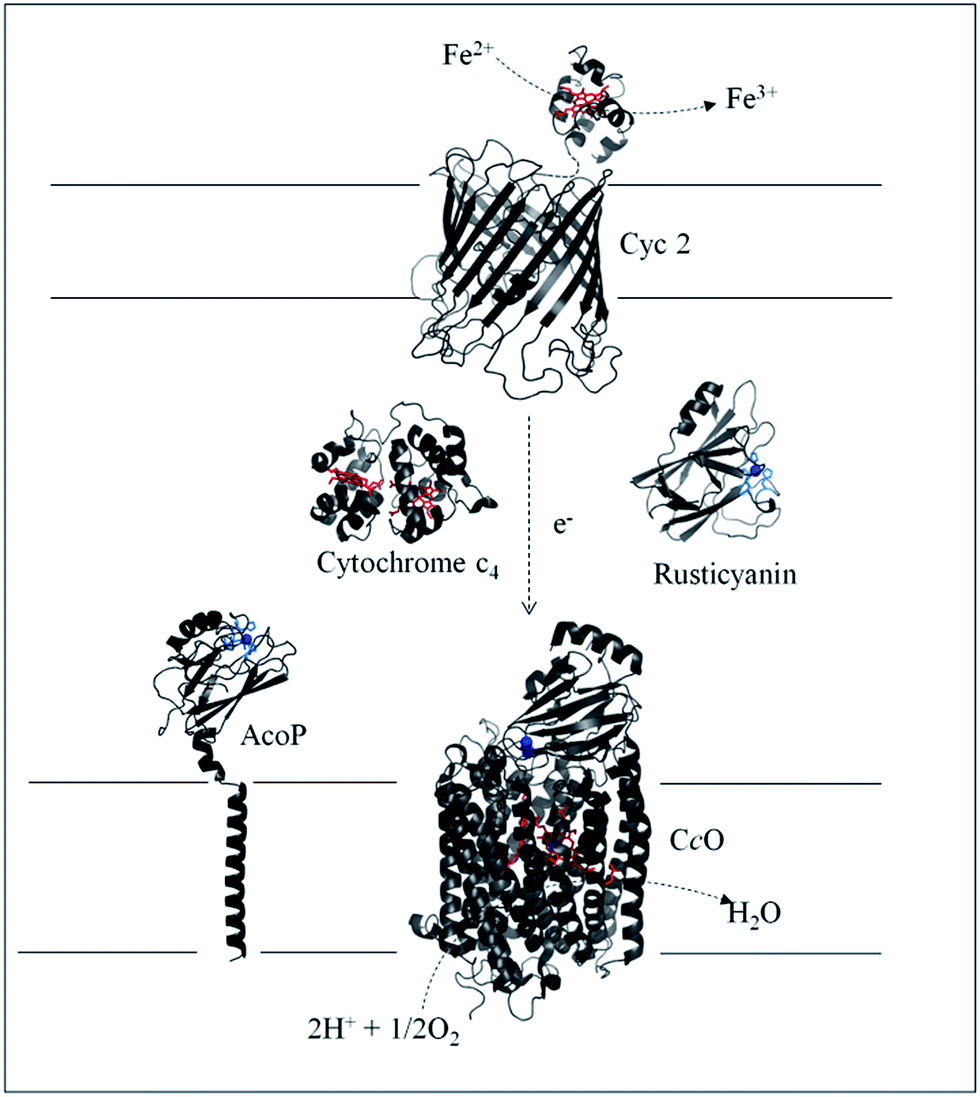 | ||
| Scheme 1 Iron respiratory chain of Acidithiobacillus ferrooxidans. Proteins known to be involved in the iron respiratory chain of A. ferrooxidans are depicted using Pymol Software. The structures of Cyt c4 (PDB: 1h1o) and Rus (PDB: 1RCY) are known. CcO of Paracoccus denitrificans is depicted (PDB: 3HB3) as an enzyme closely related to CcO of A. ferrooxidans. The structure of AcoP has been predicted using the Phyre 2 server and the transmembrane segment has been added. The structure of Cyc 2 was predicted.50 | ||
Another copper binding protein, AcoP short for “Acidophile Cytochrome c Oxidase Partner”, has been found to be directly involved in the respiratory chain.13 It has been shown that AcoP copurifies with CcO, an enzyme which, in other organisms, is well known to receive its electron from a soluble cytochrome c. Most studies have consequently assumed a direct ET between Cyt c4 and CcO in A. ferrooxidans, not taking into account AcoP.6,12 The presence of AcoP as an additional copper metalloprotein in tight interaction with CcO raises the question of its role in the respiratory chain. Its genetic organization confirms its involvement in the respiratory chain as the acoP gene is in an operon with the genes encoding the entire pathway.5 The presence of a copper site in AcoP may suggest a function beyond the stabilization effect on CcO activity in an acidic environment.13 Preliminary observations proposed a potential interaction between purified AcoP and Cyt c4.13 In addition, AcoP's intrinsic properties have recently been fully characterized and point out that it is a novel member of the cupredoxin family with a green copper centre, also called a type 1.5 copper centre, displaying the highest redox potential described to date for this subclass (+350 mV at pH 4.8 vs. Ag/AgCl).14,15 On the basis of its high redox potential and its cupredoxin fold, well suited for optimized ET, a role of AcoP as an electron shuttle might be envisioned, although it has not been demonstrated so far.
Electrochemical techniques are the methods of choice to study the thermodynamics and kinetics of ET processes involved in energy chains.16,17 ET between various types of electrodes, including carbon or gold surfaces, and small redox proteins such as heme,18–20 copper21 or iron–sulfur clusters22 containing proteins, was investigated early on to determine the key factors allowing the electron exchange at the interface. Intramolecular ET between several redox sites in the same protein has also been reported. In the cases where two redox sites are present, thanks to a specific orientation of one site over the other on the electrode, specific electrochemical signals have helped in the quantification of both the interfacial and the intramolecular ET.23,24 These preliminary studies have served the further quantification of the intermolecular ET within the transitory complex formed between an enzyme and its physiological partner. As examples among many others, second order rate constants between polyheme cytochrome c3 and hydrogenases,25,26 or nitrite reductases27 and azurin28 or cytochromes29 have been obtained by recording the current response to a potential step at electrochemical interfaces. Beyond this, all these studies opened the way for the study of direct catalysis using enzymes at electrochemical interfaces.30,31
In this paper, we focus on the interaction between AcoP and one of its putative partners in the ET chain, Cyt c4, two proteins operating in the pH range of 2.5–3 in the periplasmic compartment.5 To our knowledge, very few electrochemical studies have been reported on the interaction between two proteins interacting in an ET pathway even though not directly involved in an enzymatic process. One relevant but rare example is the early voltammetric study of the intermolecular ET between cytochrome c and cytochrome b5, two proteins shown to interact through a protein–protein ET complex.32 Thanks to the purification to the homogeneity of both AcoP and Cyt c4 from A. ferrooxidans, we now have the opportunity to investigate an additional ET chain by electrochemistry. In the work presented here, we first used biochemical techniques to demonstrate the occurrence of a complex between AcoP and Cyt c4 which allows ET in a homogeneous aqueous phase. To reveal the molecular determinants of a protein–protein interaction favourable to ET in the acidophilic chain of A. ferrooxidans, we then analysed the interfacial ET between each individual protein and an electrode, with the aim of mimicking partner–partner interaction. For this purpose, the surface chemistry of the electrode was modified and further modulated by pH and ionic strength. We finally took advantage of the knowledge of the electrode surface chemistry required for the interfacial ET on an individual protein, with the ultimate goal of analysing the consequence of the formation of a complex between AcoP and Cyt c4 on the electrochemical signal. We discuss the occurrence of intermolecular ET between the two proteins at the electrochemical interface. This ET process suggests new insights into the ET pathway allowing A. ferrooxidans to grow under acidic conditions.
Results and discussion
Complex formation and intermolecular electron transfer between Cyt c4 and AcoP in solution
To demonstrate the formation of a complex between AcoP and Cyt c4, we first purified both proteins to homogeneity as shown in Fig. 1. | ||
Fig. 1 AcoP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cyt c4 complex formation in solution from purified proteins. (A) Structure prediction and purified AcoP in solution or loaded on SDS-PAGE gel. The structure prediction of AcoP using the Phyre 2 server is in agreement with a cupredoxin fold mainly composed of β sheets previously observed by circular dichroism.14 The 4 residues identified as copper ligands15 are depicted in blue and are found in close proximity to the copper center (green sphere) which validate the model; (B) structure and purified Cyt c4 in solution or loaded on SDS-PAGE gel and blue stained. The structure (PDB: 1H1O) is shown in grey with the two hemes in magenta; (C) 5 μl of 100 μM Cyt c4, AcoP or AcoP:Cyt c4 1:1 complex were loaded on a “Modified-native” gel. An additional band labeled (*) is observed when the complex AcoP:Cyt c4 migrates on the gel. When mentioned the AcoP:Cyt c4 complex was incubated with 700 μM ascorbate before migration. :Cyt c4 complex formation in solution from purified proteins. (A) Structure prediction and purified AcoP in solution or loaded on SDS-PAGE gel. The structure prediction of AcoP using the Phyre 2 server is in agreement with a cupredoxin fold mainly composed of β sheets previously observed by circular dichroism.14 The 4 residues identified as copper ligands15 are depicted in blue and are found in close proximity to the copper center (green sphere) which validate the model; (B) structure and purified Cyt c4 in solution or loaded on SDS-PAGE gel and blue stained. The structure (PDB: 1H1O) is shown in grey with the two hemes in magenta; (C) 5 μl of 100 μM Cyt c4, AcoP or AcoP:Cyt c4 1:1 complex were loaded on a “Modified-native” gel. An additional band labeled (*) is observed when the complex AcoP:Cyt c4 migrates on the gel. When mentioned the AcoP:Cyt c4 complex was incubated with 700 μM ascorbate before migration. | ||
Both purified proteins were incubated for 12 hours, either separately or together, and subsequently loaded on a “modified-native” gel containing a small amount of SDS (0.0175% versus 0.1% in SDS-PAGE gel). As shown in Fig. 1C, in this experimental set-up, a clear additional band, of a higher molecular weight than the individual proteins, and labeled (*) could be visualized when AcoP and Cyt c4 were incubated together. This additional band was not seen using SDS-PAGE gel (ESI Fig. S1†). To confirm the presence of both proteins in the band labeled (*), N-terminal sequencing was performed. The results presented in ESI Fig. S2† unequivocally confirm the presence of AcoP and Cyt c4 and thus the existence of an observable AcoP/Cyt c4 complex. No complex formation occurred between AcoP and Cyt c4 in the presence of ascorbate, suggesting that it is dependent on the redox state of the proteins (Fig. 1C).
The involvement of AcoP in an electron pathway through Cyt c4 in solution was then evaluated. Purified Cyt c4 in its reduced form (Fig. 2A) was incubated with “as prep” AcoP in an equimolar amount. UV-vis spectroscopy clearly showed Cyt c4 oxidation in the presence of AcoP, revealed by the decrease of the β and γ bands of heme c groups at 523 and 552 nm, and a net change of the ratio between these two bands (Fig. 2A). For comparison, the spectrum of Cyt c4 reduced by ascorbate and oxidized by K2IrCl6 is shown in the Fig. 2A inset.
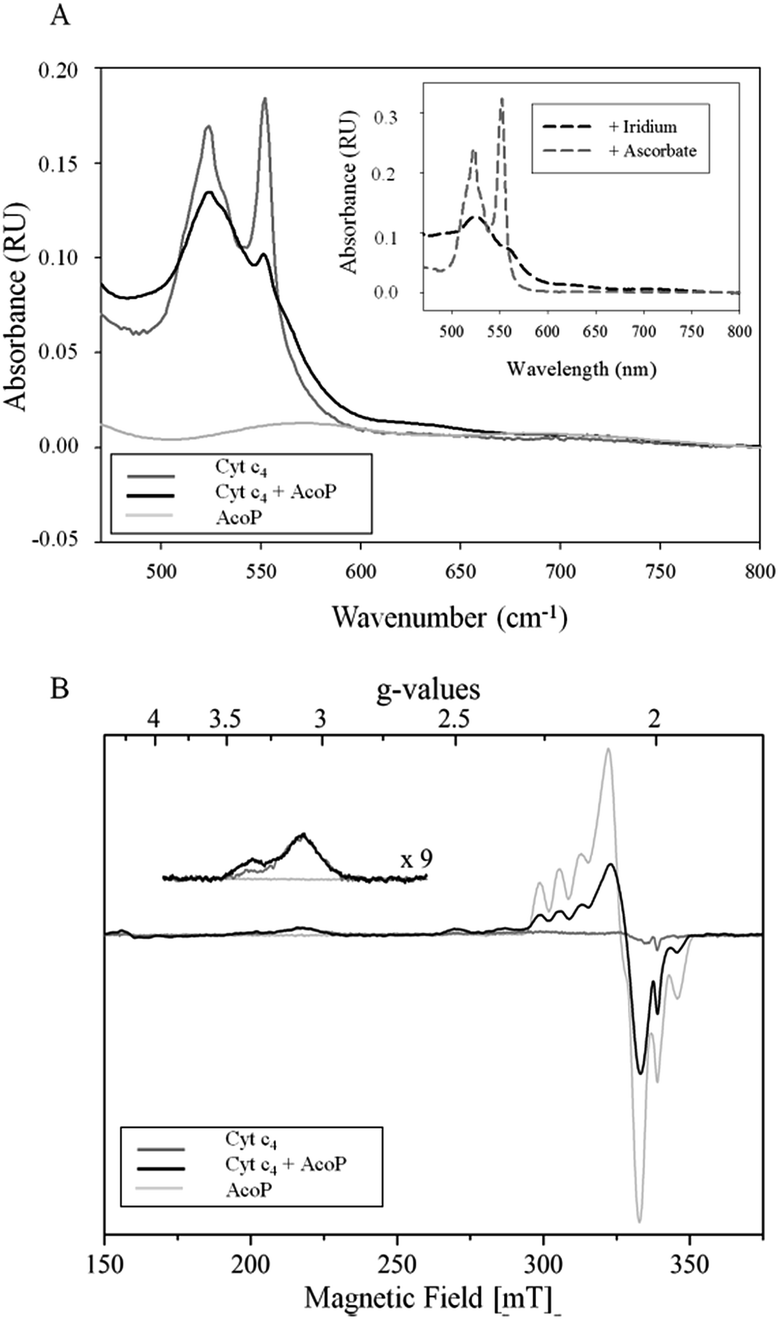 | ||
| Fig. 2 (A) UV-vis spectra of 15 μM AcoP “as prep” (light grey), 15 μM reduced Cyt c4 (dark grey), and AcoP/Cyt c4 1:1 mix (15 μM of each in the final mixture) (black) in 20 mM NaAC buffer, pH 4.8. Inset: typical spectra of oxidized Cyt c4 obtained after the addition of K2IrCl6 (black dashed line) or of reduced Cyt c4 obtained after the addition of ascorbate (grey dashed line); (B) EPR spectra of 100 μM AcoP “as prep” (light grey), 100 μM Cyt c4 untreated (dark grey) and AcoP/Cyt c4 1:1 mix (100 μM of each in the final mixture) (black) in 20 mM NaAc buffer, pH 4.8. Acquisition parameters: temperature = 15 K, microwave frequency = 9479 MHz, microwave power = 0.2 mW, and modulation amplitude = 2 mT. Inset: magnification of the gz-peak region of low-spin hemes (microwave power = 4 mW). | ||
Given the low absorption coefficient of AcoP at 570 nm (∼2500 M−1 cm−1 (ref. 15)) compared to that of Cyt c4 at a similar wavelength (ε552 nm = 46000 M−1 cm−1 (ref. 33)), the concomitant reduction of AcoP cannot be observed using this approach. To observe AcoP and Cyt c4 signatures independently, EPR measurements of frozen solutions of Cyt c4, AcoP and AcoP/Cyt c4 1:1 mix were performed (Fig. 2B). The Cyt c4 spectrum exhibits two positive peaks, at g = 3.39 and 3.13, corresponding to the gz-values of oxidized HemeH and HemeL, respectively.34 The broad signal around g = 2.3 can be attributed to the gy parts of heme signals. As expected for low-spin ferric heme signals with a maximum g value higher than 3, the gx contributions are too broad to be detected.35 The AcoP spectrum was characteristic of a rhombic Cu(II) centre with g-values equal to 2.193, 2.057 and 2.019, and copper hyperfine coupling constants of 66 × 10−4, 12 × 10−4 and 65 × 10−4 cm−1 respectively.14 The addition of one equivalent of Cyt c4 led to a significant decrease of the AcoP signal, suggesting the reduction of the Cu(II) site. In addition, the HemeH peak at g = 3.39 increases by a factor of 2.4 in intensity, which indicates an oxidation of the HemeH concomitant with the reduction of the AcoP copper site. Thus UV-vis and EPR spectroscopies both demonstrate that ET between AcoP and Cyt c4 occurs in solution in vitro, most probably through HemeH.
Interfacial electron transfer properties of AcoP
More insights into the conditions and kinetics of complex formation between AcoP and Cyt c4 were obtained by using electrochemical techniques. The additional interest of using electrochemistry was that the electrochemical interface could be easily tuned to give access to the key parameters allowing the relationship between interfacial ET and pH, ionic strength, etc., to be investigated. The data obtained by such an investigation could help to define the type of interaction involved in the interfacial ET between the electrode and each protein, and, beyond, between the two proteins if we consider the electrode as one of the partners. We first mimicked the AcoP-partner interaction by studying the electrochemical behavior of AcoP under different experimental conditions at two different electrodes, pyrolytic graphite (PG) and gold, the electrode mimicking its partner, Cyt c4.The role of potential electrostatic interactions was first evaluated by studying the electrochemical behavior of AcoP at a PG electrode. Three particular pHs, 3.5, 4.8 and 7, were chosen as they allowed both the global charge of the protein and the charge of the electrode to be tuned. The PG surface carries many carboxyl functions that induce a pKa close to 5.36 Modifying the electrode at these different pHs enabled positively (pH 3.5) or negatively charged (pH 7) electrodes to be created. AcoP has a theoretical pI of 7.2 and is thus globally positively charged at pH 3.5 and 4.8, and neutral at pH 7. Fig. 3 reports the voltammetric behavior of AcoP entrapped in the thin layer of the membrane electrode configuration at these 3 pHs in 20 mM NH4AC buffer before and after the addition of 200 mM NaCl, which was used to screen possible electrostatic interactions.37 The relationship between the mean potential and pH is given in ESI Fig. S3.†
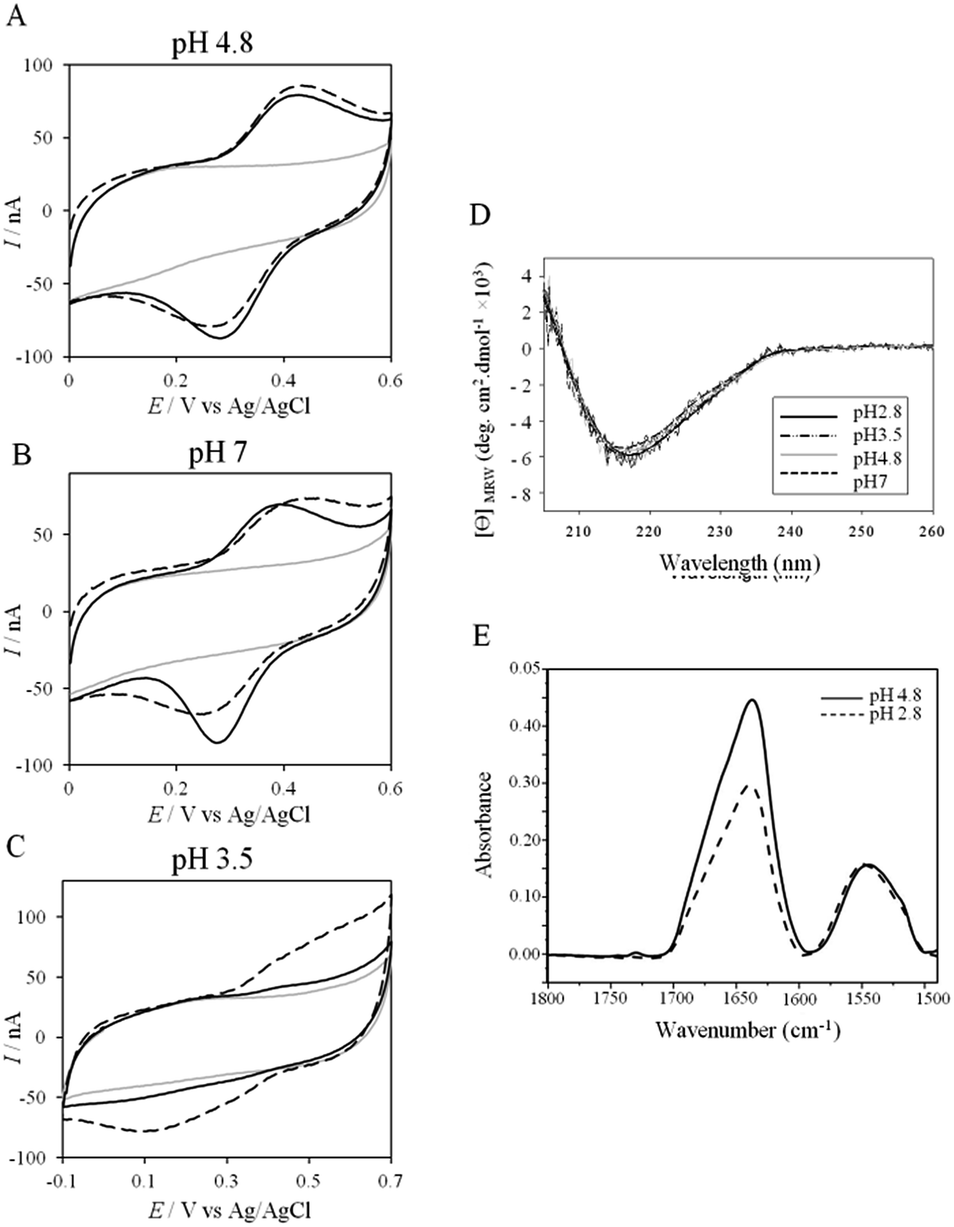 | ||
| Fig. 3 (A–C) CVs of 50 μM AcoP at a PG electrode in the membrane configuration at pH 4.8 (A), pH 7 (B), and pH 3.5 (C) in 20 mM NH4AC buffer, before (black solid lines) and after the addition of 200 mM NaCl (black dashed line). v = 20 mV s−1. Grey solid lines represent the CVs with no protein; (D) CD spectra of AcoP at different pHs in 10 mM NH4AC buffer at room temperature; (E) ATR-FTIR spectra of AcoP at 20 μM in 10 mM NH4AC at pH 4.8 (solid line) and pH 2.8 (dashed line). | ||
At pH 4.8, the one-electron redox wave characteristic of the Cu site in AcoP appears with an average potential of +350 ± 5 mV, in agreement with previous measurements either in solution using UV-vis titration14 or at a PG electrode.15 The difference between the cathodic and the anodic peaks, ΔEp, is close to 120 mV at 20 mV s−1, denoting a slow ET rate. The addition of up to 200 mM NaCl in the buffer did not induce a change either in the peak current or in ΔEp. When cyclic voltammetry (CV) was performed at pH 7, a redox wave at +340 ± 5 mV was observed displaying a ΔEp of 90 mV, which was not stable over consecutive cycles (ESI Fig. S3†). The addition of NaCl tended to increase ΔEp to 160 mV, but also induced the stability of the signal. Decreasing the pH to 3.5 or lower led to a drastic loss of the reversible redox wave, which hardly reappeared after NaCl addition. The transfer of the AcoP-based electrode from pH 3.5 back to pH 4.8 did not lead to the recovery of the redox signal.
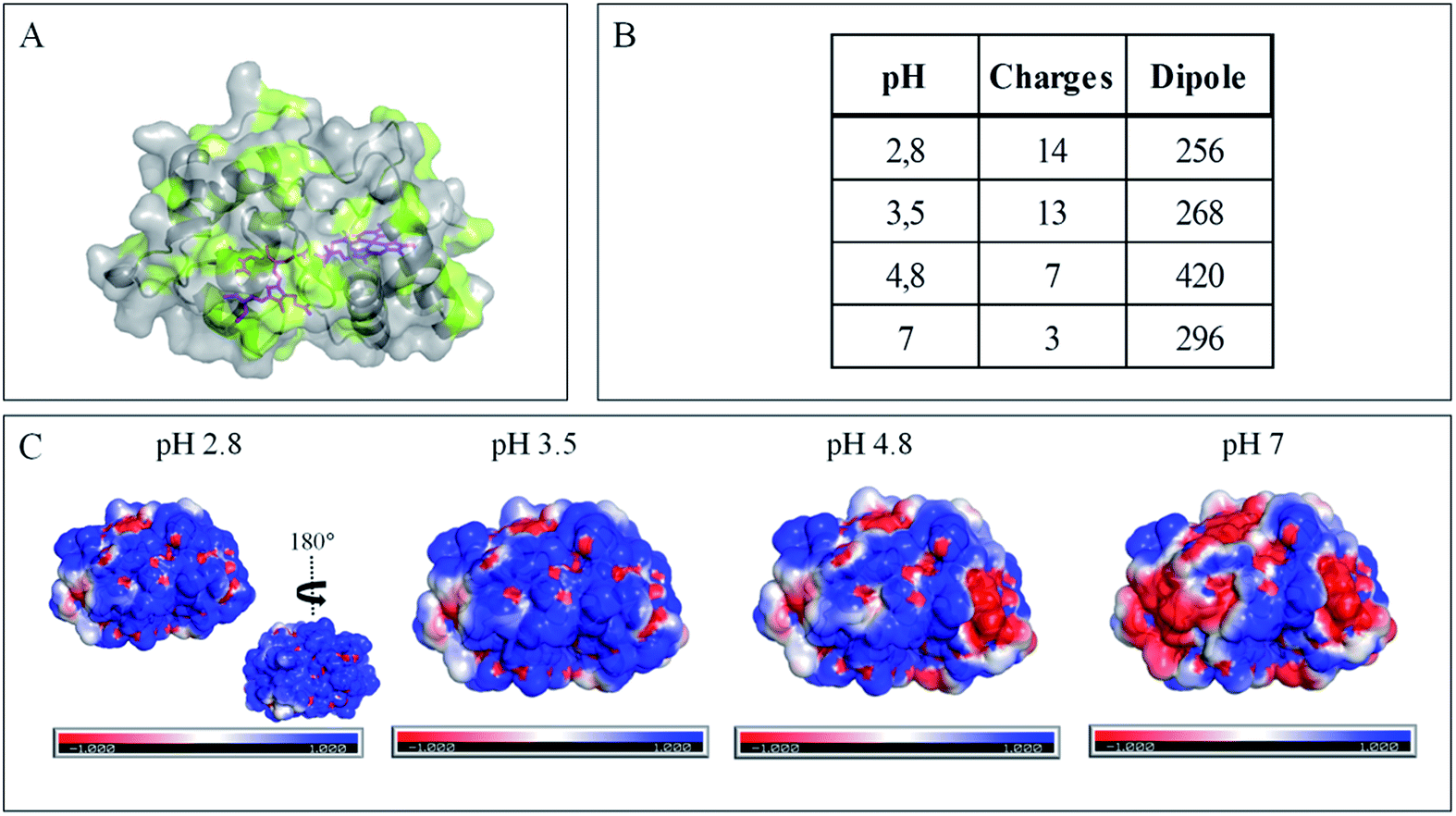
The evolution of the CV signal of AcoP as a function of buffer pH and ionic strength can be explained considering the distribution of protein charges at a given pH (Fig. 4), which induces dipole moments of 269, 216 and 145 Debye at pH 3.5, 4.8 and 7, respectively. Since we used a model of the AcoP structure, the exact dipole moments might be slightly different, but we believe that the trend is realistic. These dipole moments are 3–4 times lower than values obtained for proteins known to interact with the electrode surface through electrostatic interactions,38,39 suggesting that this type of interaction is not the main force driving the interaction of AcoP with the electrode. Accordingly, interfacial ET occurs only under conditions where electrostatic interactions are weak, such as pH 4.8, where AcoP is globally positively charged while PG is neutral, or pH 7, where the protein is globally neutral and the electrode is negatively charged. In the latter case, ET is stabilized by the presence of NaCl, which weakens electrostatic interactions.
 | ||
| Fig. 4 Impact of pH on exposed AcoP amino acids. (A) Hydrophobic surfaces (light green) depicted using Pymol software on the predicted structure of AcoP. (B) Dipole moment of AcoP and the number of charged atoms at different pHs using the Protein Dipole Moments server. (C) Electrostatic charges at the surface of AcoP as a function of pH is shown using Pymol with the following color code: positive charges in blue, negative charges in red, and neutral in white. | ||
The loss of the redox wave at low pHs was unexpected for an acidophilic protein and raises the question of AcoP structural stability on the PG electrode. UV-visible spectra had previously shown that copper geometry was not affected by changes in pH in the range from 3.5 to 7.4.14 The present work further demonstrates that even a pH as low as pH 2.8 does not induce any modification of the copper centre (ESI Fig. S4†). Circular Dichroïsm (CD) (Fig. 3D) and ATR FTIR (Fig. 3E) measurements of AcoP solutions at different pHs confirm that the decrease of pH has no effect on the secondary structure of AcoP. CD spectra can be superimposed in the pH range of 2.8–7, and no shift of the amide I band at around 1630 cm−1, characteristic of a structure rich in β-sheets, was observed in the ATR FTIR spectra.
UV-vis, CD and ATR-FTIR measurements underlined that AcoP was able to tolerate a broad range of pH conditions in solution, with no impact on its overall fold. These observations suggest that the absence of interfacial ET at low pH is most probably linked to the interaction with the electrode surface, which irreversibly impacts the native conformation of AcoP. Polarization modulation-infrared reflection-adsorption spectroscopy (PMIRRAS) on a Self-Assembled-Monolayer (SAM) on gold surfaces was used to study any conformational change of the protein upon immobilization on the electrode, especially at low pH. A 6-Mercaptohexanoic acid (MHA)-based SAM (Fig. 5A) was chosen because the induced surface chemistry and pKa (pKa of MHA as a SAM is close to 6 (ref. 18)) resemble those of PG. The carboxylic groups of the MHA-based SAM are observed at around 1735 cm−1. Both amide I and amide II bands appear on PMIRRAS spectra whatever the pH (Fig. 5A), demonstrating that AcoP is readily adsorbed on MHA-based SAMs at the two pHs under investigation. However, the position of the amide I band differs as a function of pH, shifting from 1655 cm−1 to 1670 cm−1 at pH 4.8 and 2.8, respectively. The typical band at 1655 cm−1 corresponds to the amide group involved in α-helices, while the band at 1670 cm−1 is assigned to the amide group implicated in turns. As described above, AcoP in solution is mainly structured in β-sheets, with a characteristic wavenumber of 1630 cm−1. The first conclusion is that adsorption on MHA-based SAMs induces either a modification of the protein secondary structure or a specific orientation of AcoP with the main contribution of the AcoP α-helix perpendicular to the surface according to PMIRRAS selection rules.40 At pH 4.8, because AcoP is still electroactive (Fig. 5B), strong denaturation is unlikely, but instead the maximum observed at 1655 cm−1 would reveal an orientation of the AcoP on the surface. At pH 2.8, the AcoP amide I band was shifted from 1655 cm−1 to 1670 cm−1. At this pH, the loss of electroactivity (Fig. 3) would be favorable for some modification of the secondary structure rather than variation in the orientation of AcoP. In conclusion, AcoP adsorption on hydrophilic surfaces at low pH affects its secondary structure, and hence interfacial ET.
 | ||
| Fig. 5 AcoP behavior on SAMs. (A) PMIRRAS spectrum of AcoP at 20 μM adsorbed for 1 h on MHA-Au in 10 mM NH4AC at pH 4.8 (solid line) and pH 2.8 (dashed line); (B) SWVs of AcoP adsorbed on MHA (solid line) or BT (dashed line) in 10 mM NH4AC at pH 4.8; (C) PMIRRAS spectra of AcoP at 20 μM in 10 mM NH4AC at pH 4.8 adsorbed for 1 h on MHA-Au (solid line) or BT-Au (dashed line). | ||
Native AcoP has a transmembrane segment and has been proposed to interact with the membrane bound CcO. Having ruled out a major role of electrostatic interactions, we proceeded to test whether hydrophobic interactions could rather drive interfacial ET and consequent complex formation. Fig. 4 underlines some hydrophobic patches on the surface of the protein which could be involved in the partner recognition. In a mimicking way, the putative recognition of AcoP with the electrode surface through hydrophobic interactions was studied on a hydrophobic butane thiol (BT)-based SAM on a gold electrode, on which fast ET on azurin was previously obtained.28 No SWV signal was observed with AcoP adsorbed on the BT-SAM (Fig. 5B). PMIRRAS was used to compare the conformation of the protein upon immobilization on the BT-SAM with that on MHA-based SAM surfaces (Fig. 5C). The spectra show that AcoP is also readily adsorbed on a BT-based SAM surface. Changes in the conformation of AcoP can be observed, with a loss of β sheets. The amide I position is shifted to 1670 cm−1, revealing the presence of a high level of turns in the structure of AcoP adsorbed on the hydrophobic surface. AcoP adsorption on a BT-based SAM electrode might induce a conformational change of the protein, explaining the absence of an electrochemical response on a hydrophobic surface.
The conclusions that can be drawn from electrochemistry and PMIRRAS are that hydrophobic surfaces, or strong electrostatic force between highly positively charged proteins at low pH and hydrophilic surfaces, irreversibly modify the AcoP structure and impair ET to the electrodes. This is in agreement with the fact that periplasmic proteins from acidophiles operate at pH lower than 3 and are highly positively charged (Fig. 4). Under such conditions, electrostatic interactions mostly involve the repulsion between positive charges impairing the ET. This would explain the requirement of weak electrostatic interactions between AcoP and the electrode for efficient ET. It cannot be excluded that hydrophobic interactions might drive the interaction between proteins in such an environment, but would involve small hydrophobic patches. A novel design of electrodes would be required to mimic such surfaces.
Interfacial ET properties of Cyt c4
To further evaluate the factors that can control protein–protein interaction in the metabolic chain of A. ferrooxidans, the electrochemical behavior of Cyt c4 was studied at the three pHs used previously, i.e. pH 3.5, 4.8 and 7, with a PG electrode acting as a partner (Fig. 6). At all these pHs, Cyt c4 is globally positively charged (the theoretical pI of Cyt c4 is 8) (Fig. 7). At pH 4.8 (Fig. 6A), two redox waves were observed, with mean potentials of +235 ± 5 mV and +125 ± 5 mV, corresponding to the high potential heme, HemeH, and the low potential heme, HemeL, respectively. These values are in good agreement with the previous ones determined at a gold electrode modified with bis(4-pyridyl)disulfide.9 The ratio of peak heights of HemeH and HemeL is close to 1, and ΔEp for both processes is around 30 mV at 20 mV s−1, showing that the ET rate is fast and equivalent for the two hemes. As with AcoP, the addition of NaCl did not change the profile of the CV. When the membrane was removed from the electrode, the redox waves for Cyt c4 persisted (Fig. 6D), showing the stable adsorption of some molecules. From the charge under the redox waves at different scan rates, a surface coverage of 20 ± 2 pmol cm−2 was calculated. At more basic and more acid pHs (Fig. 6B and C), the curves for the two redox processes for HemeH and HemeL were still well shaped. Notably at pH 7, the redox signal for Cyt c4 was not stable over time, even after the addition of NaCl (ESI Fig. S5†). Using CD and optical spectroscopy we did not see any critical modification of the Cyt c4 structure in solution in the pH range of 2.8–7 (Fig. 6E, ESI Fig. S6†). Cyt c4 loss of activity on the electrodes at pH 7 might have been due to electrostatic interaction with the negatively charged electrode surface which was far from physiological conditions for acidophilic organisms.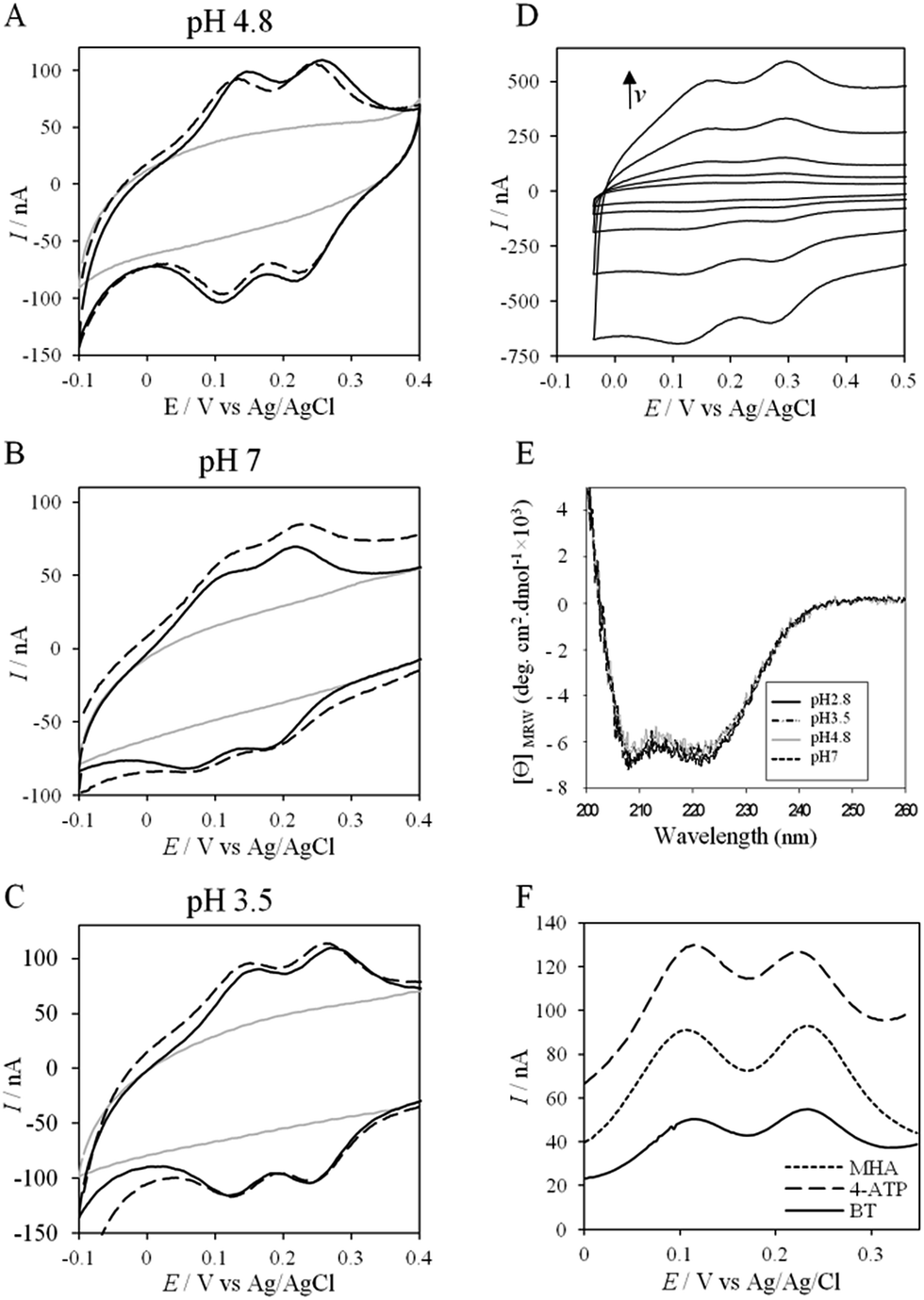 | ||
| Fig. 6 (A–C) CVs of 50 μM Cyt c4 at a PG electrode in the membrane configuration at pH 4.8 (A), pH 7 (B), and pH 3.5 (C) in 20 mM NH4AC buffer, before (black solid lines) and after the addition of 200 mM NaCl (black dashed line). v = 20 mV s−1. Grey solid lines represent the CVs with no protein; (D) CV of Cyt c4 adsorbed at the PG electrode at 5, 10, 20, 50 and 100 mV s−1; (E) CD spectra of Cyt c4 at different pHs in NH4AC 10 mM buffer at room temperature; (F) SWVs on MHA (short dashed line), 4-ATP (dashed line) and BT (solid line) in 20 mM NH4AC buffer pH 4.8. | ||
 | ||
| Fig. 7 Impact of pH on exposed Cyt c4 amino acids. (A) Hydrophobic surfaces (light green) depicted using Pymol software on the predicted structure of Cyt c4. (B) Dipole moment of Cyt c4 and the number of charged atoms at different pHs obtained using the Protein Dipole Moments Server. (C) Electrostatic charges at the surface of Cyt c4 as a function of pH is shown using Pymol with the following color code: positive charges in blue, negative charges in red, and neutral in white. | ||
Overall, the occurrence of a redox signature for both hemes, and their equal intensities whatever the pH, and hence whatever the charge of the PG electrode, suggest that Cyt c4 cannot be specifically oriented at the electrochemical interface. This is different from what was found for the Cyt c4 from Pseudomonas stutzeri, which had distinct positive and negative domains surrounding each heme at pH 7 (ESI Fig. S7†), properties that allowed its orientation on a negatively charged SAM electrode with one heme facing the electrode.23 The absence of such an orientation of Cyt c4 was further assessed by immobilizing the protein on SAM electrodes bearing different charges (Fig. 6F). Well-shaped SWV signals were obtained at negatively charged MHA and positively charged 4-aminothiol (4-ATP) SAM electrodes at pH 4.8, with redox potentials of 230 ± 5 mV and 115 ± 5 mV, corresponding to HemeH and HemeL. Cyt c4 from A. ferrooxidans displays a dipole moment ranging from 256 D to 420 D depending on the pH, half that of P. stutzeri Cyt c4 at pH 7. A comparison of their surface charges (ESI Fig. S7†) clarifies the non-orientation of A. ferrooxidans Cyt c4, as positive and negative charges are randomly exposed to the protein surface, in contrast with P. stutzeri Cyt c4. Interestingly, on a hydrophobic BT-based SAM, the redox waves relative to the two hemes also appeared at their expected redox potentials, demonstrating an efficient ET interaction between Cyt c4 and hydrophobic surfaces. This interaction can be easily understood by considering the large hydrophobic areas on the surface of the protein (Fig. 7).
This is also in agreement with molecular dynamic simulations that showed that the interaction between Cyt c4 and CoxB, the subunit of CcO transferring the electrons to the catalytic site, was stabilized by hydrophobic residues.12 Unlike AcoP, Cyt c4 thus appears as a versatile partner that should afford many types of interactions for ET with both hemes. In line with this conclusion, it was recently proposed that Cyt c4 from A. ferrooxidans would be involved in the anaerobic electron transfer chain coupling Fe3+ reduction to sulfur oxidation.41 In such a case, Cyt c4 would transfer electrons to other partners than CcO. Interestingly, versatility has also been demonstrated recently using the diheme cytochrome c550 from Thermus thermophilus.42 Whether it is a general property of diheme cytochromes is an interesting open question.
Electrochemical behavior of a mixture of AcoP and Cyt c4
Thanks to the knowledge of the electrochemical behavior of individual proteins, the kinetics of ET between AcoP and Cyt c4 was further studied using electrochemistry. Fig. 8 shows the CVs obtained at 20 mV s−1 at the PG electrode in the membrane configuration with a mixture of AcoP and Cyt c4 in a 1:1 ratio, incubated for 12 h at pH 4.8. The first CV cycle was run in the forward scan from −0.1 V to +0.6 V. The second cycle superimposed onto the first one (not shown). The signals for AcoP and Cyt c4 alone under the same experimental conditions are overlaid for comparison. Interesting features can be extracted from these CV experiments. Redox waves corresponding to HemeL in the mixture are similar to those found with Cyt c4 alone, both in terms of mean potential and peak height. Such a result demonstrates that the low potential HemeL is not affected by the presence of AcoP. The oxidative wave for HemeH is also conserved in the mixture compared to Cyt c4 alone. In contrast, no well-shaped redox signal at the expected potentials for AcoP can be observed in the CV of the mixture, suggesting that heterogeneous interfacial ET for AcoP is slowed down in the presence of Cyt c4. However, a reduction wave at a peak potential around +250 mV appears with a marked increase in the peak current compared to either HemeH of Cyt c4 or AcoP alone.
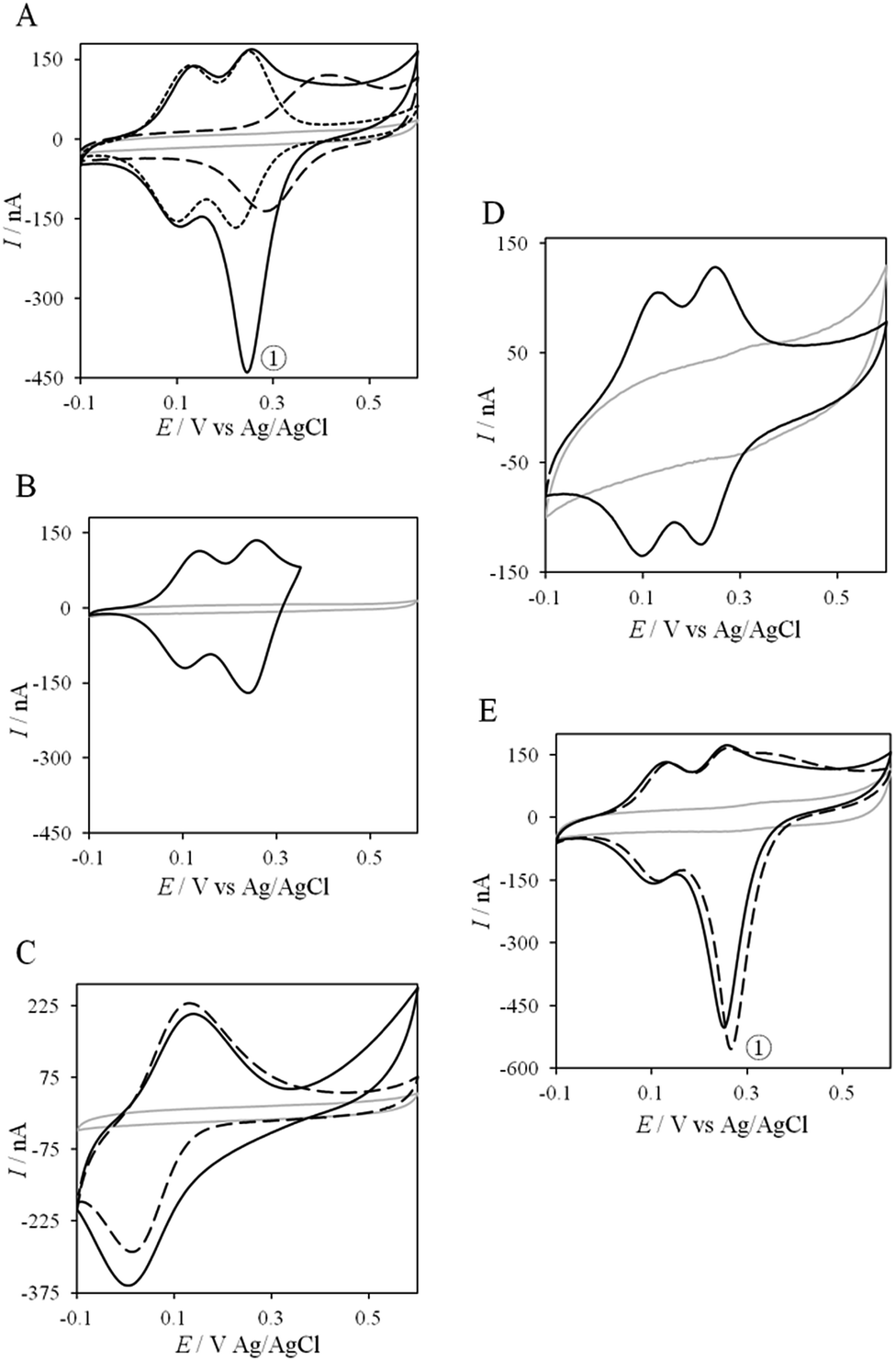 | ||
| Fig. 8 Electrochemical behavior of the AcoP:Cyt c4 complex at a PG electrode in the membrane configuration. (A) CV of 100 μM AcoP (dashed line), 100 μM Cyt c4 (short dashed line), and the 1:1 mixture of AcoP and Cyt c4 (100 μM of each in the final mixture) (solid line); (B) CV of the 1:1 mixture of AcoP and Cyt c4 in a restricted potential window; (C) CV of horse heart Cyt c (dashed line) and the 1:1 mixture of AcoP and Cyt c (solid line); (D) CV of the 1:1 mixture of AcoP H166A and Cyt c4; (E) CV of the 1:1 mixture of AcoP and Cyt c4 before (solid line) and after the addition of 400 mM NaCl (dashed line). Peak ① represents the intermolecular ET process. Grey solid lines represent the CVs with no protein. pH 4.8, 20 mM NH4AC, 20 mV s−1. | ||
The reduction wave at +250 mV, denoted peak ① in the following, strongly decreases when the forward scan of the CV is restricted to +350 mV, i.e., before the expected oxidation of AcoP (Fig. 8B). Peak ① thus involves the reduction of oxidized AcoP. Peak ① does not appear when the same experiment is performed with Horse heart Cyt c instead of Cyt c4 (Fig. 8C). This last result strongly suggests that peak ① is linked to a specific interaction between AcoP and Cyt c4. We repeated similar experiments with the mutant AcoPHis166A previously shown to have lost its redox properties, even though its secondary structure and copper content were unmodified (Fig. 8D).13 Peak ① was absent when AcoPHis166A was incubated with Cyt c4, underlining the importance of a redox active state of AcoP to observe the particular electrochemical behavior of the complex.
In a study focusing on the interaction between Cyt c and cytochrome b5 (Cyt b5), CVs of a mixture of these two redox proteins revealed a pre-peak before the reduction signal for Cyt b5. It was proposed that this reductive process was due to fast intermolecular ET between reduced Cyt b5 and oxidized Cyt c. Actually, the electrochemical interface was chemically modified so that Cyt c could be reduced at the interface only through the electrogenerated reduced form of Cyt b5.32 The situation is somewhat different in the case of AcoP and Cyt c4 because the interfacial ET for AcoP is hindered but not completely forbidden. Effectively, oxidation current is noticeable in Fig. 8A in the potential range for AcoP oxidation, which can be attributed to slow interfacial ET. Nevertheless, the typical CV signal we have observed with the protein mixture can also be attributed to intermolecular ET through the complex formed between Cyt c4 and AcoP, whose formation is not driven by electrostatic interactions. Indeed, the addition of 400 mM NaCl to the electrolyte did not drastically modify the CV (Fig. 8E). Peak ① is thus the result of intermolecular ET between electrogenerated reduced Cyt c4 and oxidized AcoP according to the processes illustrated in Scheme 2. Since we demonstrated that HemeL does not participate in the interaction with AcoP, its independent electrochemical processes are excluded from the ET model. To explain the full CV, we propose the following kinetic steps: (i) complex formation between the oxidized forms of the protein, (ii) heterogeneous reduction of Cyt c4 HemeH through favoured interaction of the complex with the PG electrode via the versatile adsorbed Cyt c4, (iii) reduction of AcoP through intermolecular ET from the reduced HemeH of Cyt c4 along peak ①, (iv) dissociation of the complex after the reduction of the proteins, (v) heterogeneous oxidation of Cyt c4, (vi) slow oxidation of AcoP at potentials above +350 mV, and (vii) reassociation of the two oxidized proteins through the complex.
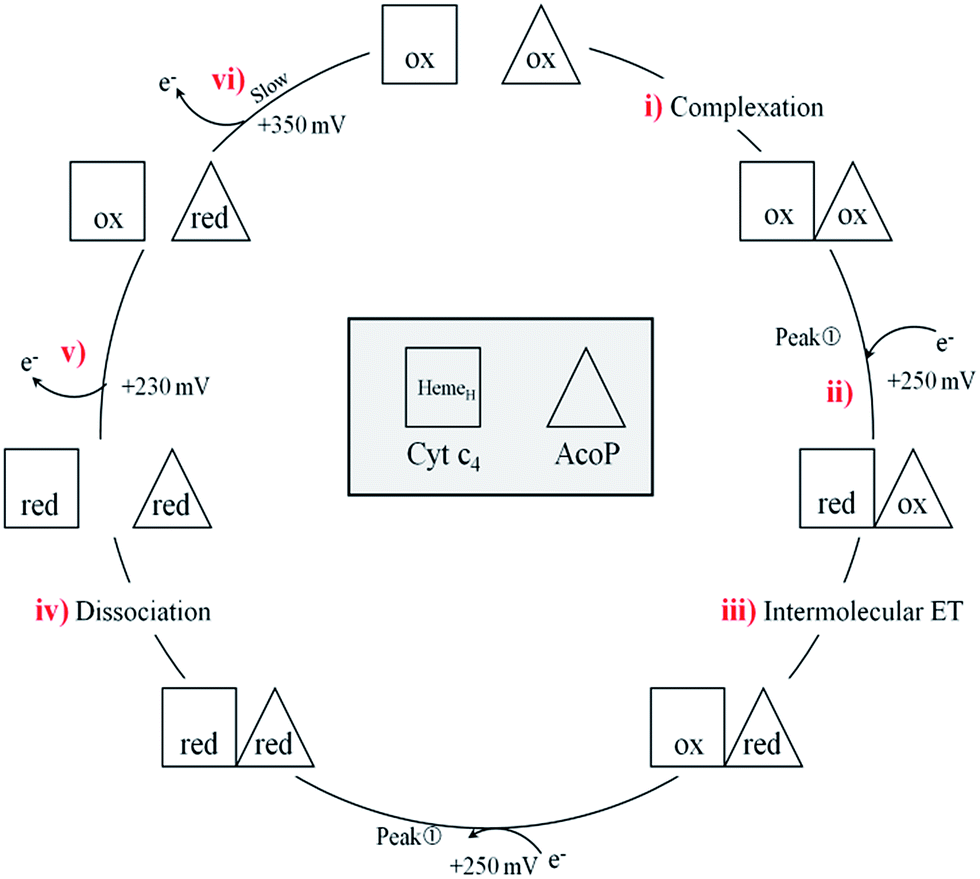 | ||
| Scheme 2 Electrochemical and chemical steps involved in the ET pathway from the oxidized form of the AcoP and HemeH Cyt c4 complex to the reduced form explaining the appearance of peak ① observed in Fig. 8. | ||
If the peculiar electrochemical behavior of the mixture of Cyt c4 and AcoP is linked to the formation of a complex that facilitates AcoP reduction, the sweep rate should affect the overall signal. To gain more insight into the redox processes involving the AcoP:Cyt c4 mixture, the CVs at 20 mV s−1 were compared to CVs obtained at 2 and 200 mV s−1. Typical CVs are shown in Fig. 9, where for each sweep rate, the signatures for AcoP and Cyt c4 alone and for the mixture are overlaid. At 2 mV s−1 (Fig. 9A), the CV of the complex is characterized by two redox waves assigned to Cyt c4, with a decrease in the peak height proportional to the sweep rate, as expected for thin layer electrochemistry. As the interfacial ET for AcoP alone is a slow process, the oxidative signal for AcoP at a low scan rate is now visible at the expected potential value. Nevertheless, peak ① is still observed, meaning that intermolecular ET is faster than the heterogeneous reduction of AcoP. The experiments described before were carried out at pH 4.8. In the context of an acidophilic chain, it should be more relevant to study the complex electrochemical behavior at a physiological pH. Thus, we recorded the CVs at pH 2.8, in the presence of the AcoP:Cyt c4 mixture at the PG membrane electrode. Peak ① linked to the reduction of AcoP via intermolecular ET through Cyt c4 was clearly observed (Fig. 9B). As the interfacial ET for AcoP alone is very slow at this low pH, AcoP reduction can only proceed through intermolecular ET via Cyt c4. This is an additional proof of the occurrence of an efficient ET complex between the two partners.
 | ||
| Fig. 9 Effect of the scan rate and pH on the electrochemical behavior of the AcoP:Cyt c4 complex at a PG electrode in the membrane configuration. CV at 2 mV s−1 of AcoP (long dashed line), Cyt c4 (short dashed line), and the 1:1 mixture of AcoP and Cyt c4 (solid line) at (A) pH 4.8 and (B) pH 2.8; (C) CVs as in (A) but at 200 mV s−1 and pH 4.8; grey solid lines represent the CVs with no protein in 20 mM NH4AC buffer. | ||
At 200 mV s−1, only the redox waves for Cyt c4 are observed (Fig. 9C). At this high speed, one or several kinetic processes may be limiting steps precluding the peak ① observation: slow AcoP oxidation, slow complex formation or slow intermolecular electron transfer within the complex. To verify the influence of each process and confirm complex formation, numerical modeling of the CVs as described in the ESI (Fig. S8†) was performed at 2, 20 and 200 mV s−1 as a function of the heterogeneous electron transfer constant of AcoP (k0), the kinetic rate constant of the complex formation (kas) and the constant of the intermolecular electron transfer within the complex (kinter). Fig. 10 illustrates one such simulation at 20 mV s−1.
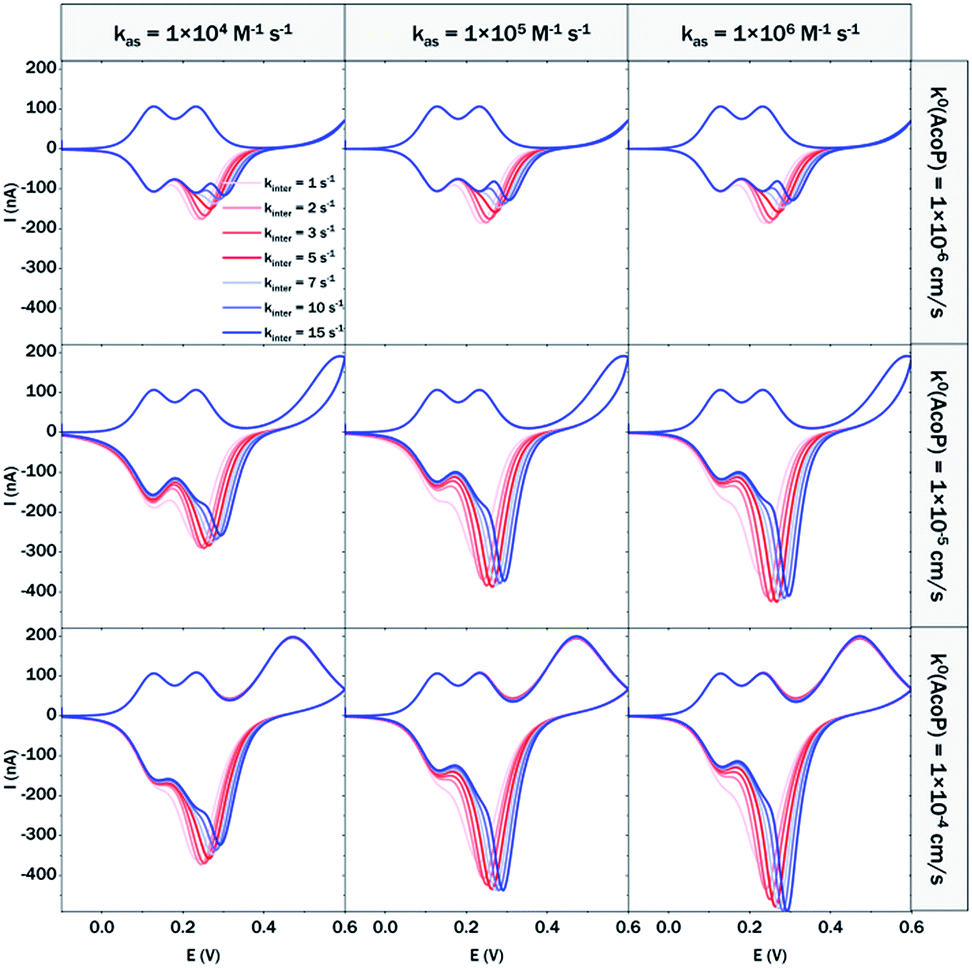 | ||
| Fig. 10 Modelling of the CVs showing the influence of three different constants, the heterogeneous electron transfer constant of AcoP (k0), the kinetic rate constant of the complex formation (kas) and the constant of the intermolecular electron transfer within the complex (kinter), on the shape of the simulated voltammograms at 20 mV s−1 (capacitive current is excluded). | ||
The shapes of the theoretical CV curves tend to match those of the experimental ones, confirming the intermolecular ET through a complex between adsorbed Cyt c4 and AcoP. The constant of the intermolecular electron transfer has the strongest influence on the potential of peak ① which only slightly varies as a function of the other two constants. This can be used for the estimation of this constant. Based on the simulation at the three different scan rates, and hypothesizing a fast complex formation which is thermodynamically favoured, the intermolecular electron transfer within the complex could be estimated to occur with a rate of 3–5 s−1. This value is smaller than other values estimated from photoinduced intracomplex ET between Cyt f and Cyt c6 from Chlamydomonas reinhardtii (9 × 102 s−1)43 or intracomplex ET obtained by laser excitation of ruthenium-labeled Cyt b5 and cyt c (4 × 105 s−1).44 Our intermolecular ET between AcoP and Cyt c4 is in the same range as the value reported for ET between Pseudomonas aeruginosa cyt c551 and Silene pratensis plastocyanin obtained by stopped-flow measurements (5.7 s−1).45 Two hypotheses may explain the slow intermolecular ET rate between AcoP and Cyt c4: (i) the adsorption of Cyt c4 at the electrode surface impairs the optimal ET complex; (ii) AcoP has additional functions other than accepting electrons from Cyt c4.
Experimental
Chemicals and biomaterials
(+)-Sodium L-ascorbate (ascorbate), sodium dithionite, potassium hexachloroiridate(IV) (K2IrCl6), CM sepharose fast flow resin (CMC resin), a HiLoad 26/10 SP Sepharose HP column (SP Sepharose column), a Mono S™ 5/50 GL column (MonoS column), ethylenediaminetetraacetic acid disodium salt dehydrate (EDTA), DNAse I from bovine pancreas, anti-protease (fast protease inhibiter tablets), n-dodecyl β-d-maltoside (DDM), sodium acetate (NaAC), ammonium acetate (NH4AC), 1-butanethiol (99%) (BT), 4-aminothiophenol (97%) (4-ATP), 6-mercaptohexanoic acid (90%) (MHA), 30% H2O2, NaOH, 98% H2SO4, absolute ethanol (EtOH), acetic acid (96%), and sodium chloride were bought from Sigma-Aldrich and used as received. All aqueous solutions were prepared with Milli Q (18.2 MΩ cm) water.Recombinant AcoP without its transmembrane segment was expressed and purified from Escherichia coli as described previously.14 AcoP protein concentration was determined by UV-vis absorbance spectroscopy, measuring the absorption intensity at 280 nm and using a sequence-derived theoretical molar extinction coefficient of 25440 M−1 cm−1. Purified AcoP was stored in 50 mM NaAC buffer at pH 4.8, containing 0.005% (w/v) DDM. When mentioned, AcoP was dialysed against 10 or 20 mM NH4AC buffers adjusted to different pH values, pH 2.8, pH 3.5, pH 4.8 and pH 7.
Cyt c4 was expressed as described previously in 100 l E. coli cultures9,34 using three separation steps on carboxymethyl cellulose, SP-Sepharose and monoS columns. The enriched fractions containing Cyt c4 in 80 mM NH4AC buffer at pH 4.8 were pooled and dialysed against 80 mM NH4AC buffer at pH 2.8, followed by another dialysis at pH 4.8. This pH variation allowed further elimination of E. coli acid unstable proteins. When mentioned, purified Cyt c4 was dialysed against 10 or 20 mM NH4AC buffers adjusted to different pH values, pH 2.8, pH 3.5, pH 4.8 and pH 7.
Cytochrome c (Cyt c) from an equine heart (≥95%, SDS-PAGE) was bought from Sigma-Aldrich. Cyt c was diluted in NH4AC 20 mM buffer at pH 4.8.
Protein modelling
For cupredoxins, being well known, several structures have been solved which serve as templates for modelling the globular soluble domain of AcoP. Here we used the Phyre2, Swiss-model and I-TASSER servers47 obtaining similar results. We note that, given the high homology with the templates, this model is expected to be of reasonable quality along most of the sequence.48,49 For Scheme 1, AcoP's TM helix was built manually by threading its sequence as an ideal helix. The model of Cyc2 in Scheme 1 is based on I-TASSER homology modelling guided by coevolution-based restraints as described in ref. 50. For Cyt c4 of Acidithiobacillus acidophilus (pdb: 1H1O) and Cyt c4 of Pseudomonas stutzeri, we used available X-ray structures (pdb: 1H1O and 1ETP, respectively).To study protein electrostatics, AcoP models and Cyt c4 structures were prepared with the PDB2PQR-Propka servers, using the Amber force field to assign electrostatic charges at the surface of the proteins and the Parse force fields to determine the dipole moments of proteins with PDB2PQR-Propka servers.51 Dipole moments were also determined from the pqr output files using the Protein Dipole Moments Server.52 Electrostatic charges at the surface of proteins and surface hydrophobicity were illustrated using PyMOL (The PyMOL Molecular Graphics System, Version 1.8 Schrodinger, LLC).
Polyacrylamide gels
Electrochemical measurements
Cyclic voltammetry (CV) and square wave voltammetry (SWV) were performed using an Autolab PGSTAT30 potentiostat analyzer controlled by Nova software (Eco Chemie). The electrochemical cell was equipped with three electrodes, a pyrolytic graphite (PG) or a gold electrode as the working electrode (S = 0.07 cm2), a platinum wire as an auxiliary electrode and an Ag/AgCl electrode as the reference electrode. All potentials are quoted vs. the Ag/AgCl reference electrode here. Potentials versus NHE can be obtained by adding 210 mV to the reported potentials. 20 mM NH4AC buffer was used as the electrolyte. All the electrochemical measurements were made at least in triplicate at 25 °C.The PG electrode surface was renewed by polishing with fine sand paper (P1200), and then briefly sonicated to remove free carbon particles. The membrane electrode configuration was used to entrap 2 μl of protein sample in a thin layer between the electrode and a dialysis membrane of suitable cutoff.53 The gold surfaces were cleaned with “piranha” solution (3H2SO4 98%:1H2O2 30%) for 4 min and rinsed extensively with water and later with ethanol. Self-assembled monolayers (SAMs) were formed by immersing the gold surfaces in 5 mM ethanol solutions of 4-ATP or MHA or BT for 18 hours. The surfaces were then cleaned with ethanol to remove all organic contaminants.
Spectroscopies
Circular Dichroism (CD) spectra of 15 μM proteins were recorded on a Jasco J-715 spectropolarimeter at 298 K in a 1 mm path length cell. Spectra were averaged from five scans and normalized for any variation in protein concentrations measured at 280 nm by optical spectroscopy using a Cary 50 Bio (Varian).UV-vis spectra were recorded on a Cary 50 Bio (Varian) spectrophotometer. Protein samples (AcoP or Cyt c4) were used at 15 μM, for a scan recorded from 200 nm to 1000 nm to follow metal center properties. When mentioned, oxidation or reduction of the samples was performed using an equimolar amount of protein and potassium hexachloroiridate(IV) (K2IrCl6) or ascorbate. Ascorbate can be removed for further analysis by using a desalting column (PD-10 column, GE Healthcare). Because iridium is a strong oxidizing agent that irreversibly modifies key residues, we used “as prep” AcoP which corresponds to ∼40% of the oxidized form.14
X-band electron paramagnetic resonance (EPR) spectra were recorded using a Bruker-Biospin EleXsys E500 spectrometer equipped with a standard rectangular Bruker EPR cavity (ER4102ST) connected to an Oxford Instruments helium flow cryostat (ESR900).
For polarization modulation-infrared reflection-adsorption spectroscopy (PM-IRRAS), the modified dried gold surface was placed at room temperature in the external beam of the FT-IR instrument on a Nicolet Nexus 870 FT-IR spectrometer (Madison, WI). The experimental set-up has been reported in a previous paper.54 The PMIRRAS spectra were recorded at 8 cm−1 resolution, with the co-addition of six hundred scans.
ATR-FTIR spectra were recorded on a Nicolet 6700 FT-IR spectrometer (Nicolet Instrument, Madison, WI) equipped with a liquid nitrogen cooled mercury–cadmium–telluride detector (Thermo Fisher Scientific, San Jose, CA), with a spectral resolution of 4 cm−1. Two hundred interferograms were co-added.
Conclusions
In this study, we demonstrated the formation of a complex between AcoP and Cyt c4, two proteins of an acidophilic bacterial respiratory chain. We used an electrode interface to mimic AcoP and Cyt c4 as putative redox partners in the energy chain. We found that electrostatic interactions do not drive efficient ET from the AcoP copper centre to the electrodes. In contrast, we showed that Cyt c4 is highly versatile, displaying ET at the two heme redox centres on varied types of surfaces, either positive, negative or hydrophobic. This suggests that Cyt c4 could be a hub that controls the rate of electrons towards different routes, depending on the metabolic or environmental states. We also demonstrated an intermolecular ET between AcoP and Cyt c4, allowing the reduction of AcoP through the high potential heme of Cyt c4 (Scheme 3). Thanks to modeling of the electrochemical signals, we were able to quantify the intermolecular ET rate within the complex. Although the results for the mixture suggest a likely directionality of ET from Cyt c4 to AcoP, the value of the intermolecular rate constant opens a new direction toward the physiological role of AcoP.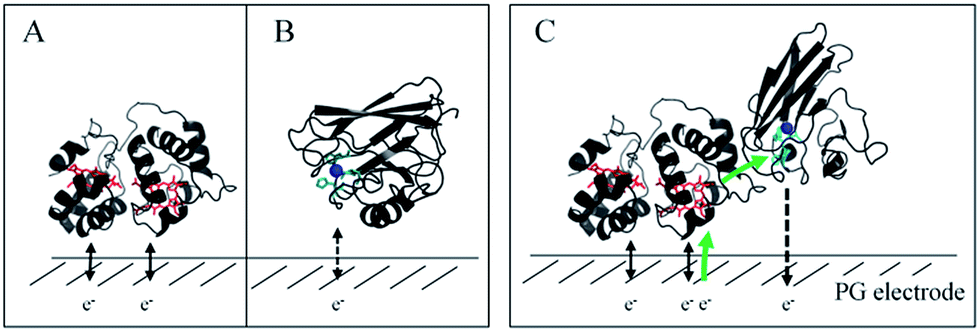 | ||
| Scheme 3 Proposed model of ET between redox proteins and the electrode surface observed on PG electrodes (see Fig. 8). Dashed lines represent PG electrodes. Structures of Cyt c4 or predicted AcoP are presented in grey ribbons with their respective redox centers, hemes (in red) and copper (in blue). (A) Interfacial ET between the electrode and the two heme centers, a reversible process, illustrated by black arrows; (B) slow interfacial ET between the AcoP copper center and the electrode shown by a dashed arrow; (C) additional intermolecular ET pathway within the AcoP:Cyt c4 complex depicted by a green arrow. This complex has been modelled with the HADDOCK web server46 by driving the docking with the constraint based on AcoP interacting with HemeH of Cyt c4. | ||
The electrochemical setup developed in this work allowed us to reconstitute on electrodes a portion of a respiratory chain and to quantify ET rates using small amounts of proteins and with the possibility to very easily tune the protein environment. We propose that this method could serve to get thermodynamic and kinetic data on other redox proteins, as well as between two interacting redox partners. Furthermore, for membrane proteins, the use of electrochemical interfaces appears more appropriate to mimic their physiological environment, in comparison with classical solution-state stopped-flow analysis.
Using the same strategy and by integrating the other partners of the respiratory chain, we should be able in the future to complete the understanding of the energy chain of A. ferrooxidans. For upstream Cyt c4, it was demonstrated that Rus was interacting with Cyt c4via HemeH.7 Rus and AcoP are both cupredoxins of high redox potential. The purpose served by AcoP is not clear as Rus could be sufficient to shuttle electrons in the periplasm. Their stoichiometry (Rus is highly abundant)55 and localization (membrane anchored for AcoP versus periplasm for Rus) might drive their specific function and explain their co-occurrence. It should be interesting to determine whether competition exists between the two cupredoxins in the respiratory chain, and under which conditions. Downstream, the question of the electron transfer to the terminal oxidase CcO should be addressed. In most known biological systems, Cyt c has been described as the electron donor to the CcO.56 However, in some biological systems, cupredoxins have been shown to replace Cyt c as electron donors to terminal enzymes depending on the metal occurrence in the bacterial environment.57 In the case of A. ferrooxidans, AcoP and Cyt c4 being under the control of the same promoter, and hence expressed under the same conditions, might both be able to transfer electrons to the CcO. We can hypothesize that AcoP may act as a second entry for electrons, while protecting the CcO copper center exposed to the acidic environment.11 Further studies are required to answer such questions.
For a long time A. ferrooxidans has attracted the interest of industries regarding its use in bioleaching processes. More recently, its advantage as a biocathodic catalyst in microbial fuel cells3 expanded the importance of its study, which emphasizes the need to better describe how this bacterium finds energy to grow using iron, a low energetic substrate. Even more interestingly, proteins in this ET chain share the common features of having a high redox potential. Such properties can be of great importance for ecologically friendly devices such as enzymatic fuel cells. These fuel cells are cathode-limited by the low oxygen affinity of currently used multicopper oxidases. CcO could be an alternative, but the most widely characterized CcOs display low redox potentials, a limitation that A. ferrooxidans CcO could overcome.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by ANR (ENZYMOR-ANR-16-CE05-0024) and Aix-Marseille University through X. Wang's funding. We acknowledge the national French EPR network (RENARD, IR3443) and the Aix-Marseille EPR facility. The authors thank P. Infossi, D. Byrne-Kodjabachian and Dr C. Gutierrez-Sanchez (BIP, IMM, Marseille) for technical help in enzyme purification and characterization. Marion Taris is acknowledged for her help in performing ATR-FTIR and PMIRRAS experiments. This work benefited from the facilities of the Platform of Fermentation of IMM and expertise from M. Bauzan, to whom the authors are very grateful. The authors would also like to thank Dr A. de Poulpiquet and Dr B. Gontero (BIP, Marseille) for fruitful discussion.References
- R. Quatrini and D. B. Johnson, Curr. Opin. Microbiol., 2018, 43, 139–147 CrossRef CAS PubMed.
- J. P. Cardenas, R. Quatrini and D. S. Holmes, Res. Microbiol., 2016, 167, 529–538 CrossRef CAS PubMed.
- N. Chabert, V. Bonnefoy and W. Achouak, Microb. Biotechnol., 2018, 11, 136–140 CrossRef CAS PubMed.
- S. J. Ferguson and W. J. Ingledew, Biochim. Biophys. Acta, 2008, 1777, 1471–1479 CrossRef CAS PubMed.
- M. Roger, C. Castelle, M. Guiral, P. Infossi, E. Lojou, M. T. Giudici-Orticoni and M. Ilbert, Biochem. Soc. Trans., 2012, 40, 1324–1329 CrossRef CAS PubMed.
- V. Bonnefoy and D. Holmes, Environ. Microbiol., 2012, 14, 1597–1611 CrossRef CAS PubMed.
- A. L. Ducluzeau, B. Schoepp-Cothenet, R. van Lis, F. Baymann, M. J. Russell and W. Nitschke, J. R. Soc., Interface, 2014, 11, 20140196 CrossRef PubMed.
- M. Ilbert and V. Bonnefoy, Biochim. Biophys. Acta, 2013, 1827, 161–175 CrossRef CAS PubMed.
- G. Malarte, G. Leroy, E. Lojou, C. Abergel, M. Bruschi and M. T. Giudici-Orticoni, Biochemistry, 2005, 44, 6471–6481 CrossRef CAS PubMed.
- C. Castelle, M. Guiral, G. Malarte, F. Ledgham, G. Leroy, M. Brugna and M. T. Giudici-Orticoni, J. Biol. Chem., 2008, 283, 25803–25811 CrossRef CAS PubMed.
- T.-F. Li, R. Painter, B. Ban and R. C. Blake, J. Biol. Chem., 2015, 290, 18293–18303 CrossRef CAS PubMed.
- M. C. Patra, S. K. Pradhan, S. N. Rath and J. Maharana, ISRN Biophys., 2013, 295718 Search PubMed.
- C. Castelle, M. Ilbert, P. Infossi, G. Leroy and M. T. Giudici-Orticoni, J. Biol. Chem., 2010, 285, 21519–21525 CrossRef CAS PubMed.
- M. Roger, F. Biaso, C. Castelle, M. Bauzan, F. Chaspoul, E. Lojou, G. Sciara, S. Caffari, M. T. Giudici-Orticoni and M. Ilbert, PLoS One, 2014, 9, e98941 Search PubMed.
- M. Roger, G. Sciara, F. Biaso, E. Lojou, X. Wang, M. Bauzan, M. T. Giudici-Orticoni, A. Vila and M. Ilbert, Biochim. Biophys. Acta, 2017, 1858, 351–359 CrossRef CAS PubMed.
- L. Jeuken, Adv. Biochem. Eng./Biotechnol., 2016, 158, 43–73 CrossRef PubMed.
- M. Roger, A. de Poulpiquet, A. Ciaccafava, M. Ilbert, M. Guiral, M. T. Giudici-Orticoni and E. Lojou, Anal. Bioanal. Chem., 2014, 406, 1011–1027 CrossRef CAS PubMed.
- B. Jin, G.-X. Wang, D. Millo, P. Hildenbrandt and X.-H. Xia, J. Phys. Chem. C, 2012, 116, 13038–13044 CAS.
- K. Bewley, K. Ellis, M. Firer-Sherwood and S. Elliott, Biochim. Biophys. Acta, 2013, 1827, 938–948 CrossRef CAS PubMed.
- D. Novak, M. Mojovic, A. Pavicevic, M. Zatloukalova, L. Hernychova, M. Bartosik and J. Vacek, Bioelectrochemistry, 2018, 119, 136–141 CrossRef CAS PubMed.
- P. Jensen, Q. Chi, J. Zhang and J. Ulstrup, J. Phys. Chem. C, 2009, 113, 13993–14000 CAS.
- P. Zanello, Coord. Chem. Rev., 2017, 335, 172–227 CrossRef CAS.
- Q. Chi, J. Zhang, T. Arslan, L. Borg, G. Pedersen, H. Christensen, R. Nazmudtinov and J. Ulstrup, J. Phys. Chem. B, 2010, 114, 5617–5624 CrossRef CAS PubMed.
- S. Casalini, M. Berto, A. Kovtun, A. Operamolla, G. Di Rocco, P. Facci, A. Liscio, G. Farinola, M. Borsari and C. Bortolotti, Electrochim. Acta, 2015, 178, 638–646 CrossRef CAS.
- A. Ciaccafava, M. Alberola, S. Hameury, P. Infossi, M. T. Giudici-Orticoni and E. Lojou, Electrochim. Acta, 2011, 56, 3359–3368 CrossRef CAS.
- Y. Sugimoto, Y. Kitazumi, O. Shirai, K. Nishikawa, Y. Higuchi, M. Yamamoto and K. Kano, Biochim. Biophys. Acta, 2017, 1865, 481–487 CrossRef CAS PubMed.
- R. Milton and S. Minteer, ChemPlusChem, 2017, 82, 513–521 CrossRef CAS.
- E. Lojou, F. Cutruzzola, M. Tegoni and P. Bianco, Electrochim. Acta, 2003, 48, 1055–1064 CrossRef CAS.
- H. Pedroso, C. Silveira, R. Almeida, A. Almeida, S. Besson, I. Moura, J. Moura and M. Almeida, Biochim. Biophys. Acta, 2016, 1857, 1412–1421 CrossRef CAS PubMed.
- L. Kertess, A. Adamska-Venkatesh, P. Rodriguez-Macia, O. Rudiger, W. Lubitz and T. Happe, Chem. Sci., 2017, 8, 8127–8137 RSC.
- F. A. Armstrong, R. M. Evans, S. V. Hexter, B. J. Murphy, M. M. Roessler and P. Wulff, Acc. Chem. Res., 2016, 49, 884–892 CrossRef CAS PubMed.
- R. Seetharaman, S. White and M. Rivera, Biochemistry, 1996, 35, 12455–12463 CrossRef CAS PubMed.
- M. T. Giudici-Orticoni, G. Leroy, W. Nitschke and M. Bruschi, Biochemistry, 2000, 39, 7205–7211 CrossRef CAS PubMed.
- C. Cavazza, M. T. Giudici-Orticoni, W. Nitschke, C. Appia, V. Bonnefoy and M. Bruschi, Eur. J. Biochem., 1996, 242, 308–314 CAS.
- F. A. Walker, Coord. Chem. Rev., 1999, 185–186, 471–534 CrossRef CAS.
- F. Armstrong, P. Cox, H. Hill, V. Lowe and B. Oliver, J. Electroanal. Chem., 1987, 217, 331–366 CrossRef CAS.
- Y. Sugimoto, Y. Kitazumi, O. Shirai, M. Yamamoto and K. Kano, J. Phys. Chem. B, 2016, 120, 3122–3128 CrossRef CAS PubMed.
- F. Oteri, M. Baaden, E. Lojou and S. Sacquin-Mora, J. Phys. Chem., 2014, 118, 13800–13811 CrossRef CAS PubMed.
- I. Mazurenko, K. Monsalve, J. Rouhana, P. Parent, C. Laffon, A. Le Goff, S. Szunerits, R. Boukherroub, M. T. Giudici-Orticoni, N. Mano and E. Lojou, ACS Appl. Mater. Interfaces, 2016, 8, 23074–23085 CAS.
- P. Olejnik, B. Palys, A. Kowalczyk and A. M. Nowicka, J. Phys. Chem. C, 2012, 116, 25911–25918 CAS.
- J. Kucera, E. Pakostova, J. Lochman, O. Janiczek and M. Mandl, Res. Microbiol., 2016, 167, 357–366 CrossRef CAS PubMed.
- F. Melin, B. Schoepp-Cothenet, S. Abdulkarim, M. R. Noor, T. Soulimane and P. Hellwig, Inorg. Chim. Acta, 2017, 468, 252–259 CrossRef CAS.
- T. Grove and N. Kostic, J. Am. Chem. Soc., 2003, 125, 10598–10607 CrossRef CAS PubMed.
- A. Willie, P. Stayton, S. Sligar, B. Durham and F. Millet, Biochemistry, 1992, 31, 7237–7242 CrossRef CAS PubMed.
- S. Takayama, K. Irie, H. Tai, T. Kawahara, S. Hirota, T. Takabe, L. Alcaraz, A. Donaire and Y. Ymamoto, J. Biol. Inorg Chem., 2009, 14, 821–828 CrossRef CAS PubMed.
- G. C. van Zundert and A. M. Bonvin, Methods Mol. Biol., 2014, 113, 163–179 Search PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS PubMed.
- L. A. Abriata, G. E. Tamo, B. Monasryrskyy, A. Kryshtafovych and M. Dal Peraro, Proteins, 2018, 97–112 CrossRef CAS PubMed.
- L. A. Abriata, L. N. Kinch, G. E. Tamo, B. Monasryrskyy, A. Kryshtafovych and M. Dal Peraro, Proteins, 2018, 16–26 CrossRef CAS PubMed.
- L. A. Abriata, bioRxiv, 2018, 262287, DOI:10.1101/262287.
- T. J. Dolinsky, J. E. Nielsen, J. A. McCammon and N. A. Baker, Nucleic Acids Res., 2004, 32, W665–W667 CrossRef CAS PubMed.
- C. E. Felder, J. Prilusky, I. Silman and J. L. Sussman, Nucleic Acids Res., 2007, 35, W512–W521 CrossRef PubMed.
- M. Correia dos Santos, P. Paes de Sousa, M. Simões Gonçalves, L. Krippahl, J. Moura, E. Lojou and P. Bianco, J. Electroanal. Chem., 2003, 541, 153–162 CrossRef CAS.
- C. Gutierrez-Sanchez, A. Ciaccafava, P.-Y. Blanchard, K. Monsalve, M. T. Giudici-Orticoni, S. Lecomte and E. Lojou, ACS Catal., 2016, 6, 5482–5492 CrossRef CAS.
- W. Nitschke and V. Bonnefoy, Energy acquisition in low pH environments, ed. R. Quatrini and D. B. Johnson, Caister Academic Press, UK, 2016, pp. 19–48 Search PubMed.
- S. Shimada, K. Shinsawa-Itoh, J. Baba, S. Aoe, A. Shimada, E. Yamashita, J. Kang, M. Tateno, S. Yoshikawa and T. Tsukihara, EMBO J., 2017, 36, 291–300 CrossRef CAS PubMed.
- S. Merchant and L. Bogorad, Mol. Cell. Biol., 1986, 6, 462–469 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: N-Terminal sequencing, distribution of charges and dipole moments of the proteins, and stability and modelling of the voltammetric signals. See DOI: 10.1039/c8sc01615a |
| This journal is © The Royal Society of Chemistry 2018 |