
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium-catalyzed asymmetric hydroamination and hydroindolation of keto-vinylidenecyclopropanes†
Song
Yang
a,
Quan-Zhe
Li
a,
Chen
Xu
a,
Qin
Xu
a and
Min
Shi
 *abc
*abc
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: mshi@sioc.ac.cn
bState Key Laboratory and Institute of Elemento-organic Chemistry, Nankai University, Tianjin 300071, P. R. China
cState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, University of Chinese Academy of Sciences, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 11th May 2018
Abstract
We reported a highly regio- and enantioselective hydroamination and hydroindolation of keto-vinylidenecyclopropanes via cationic Rh(I) catalysis in this context. The combination of various secondary amines and indoles with keto-vinylidenecyclopropanes afforded the corresponding hydrofunctionalization products in good to excellent yields with outstanding ee values under mild conditions. A new TMM–Rh model complex was proposed, providing an atom economical Rh-π-allyl precursor at the same time. Moreover, the resulting products could easily be transformed into more complex polyheterocycles upon further synthetic manipulation.
Introduction
Allylic substitutions1,2 and allylic oxidations3 are powerful synthetic tools for carbon–carbon and carbon–heteroatom bond formation and have a broad range of applications in the synthesis of biologically important molecules (Scheme 1a). Besides, recent studies show that even allylic alcohols can serve as allyl precursors.4,5 However, these methods suffer from drawbacks such as the required preinstallation of a leaving group or the use of stoichiometric amounts of oxidant, respectively. Thus, the development of new asymmetric carbon–carbon and carbon–heteroatom bond-forming reactions which fulfil the criteria of atom economy is of imminent importance to the evolution of chemical synthesis.6 In this respect, the atom economic pathway toward linear allylic products under Pd catalysis was pioneered by Trost and Yamamoto in the late 1990s and early 2000s utilizing mostly terminal allenes or internal Me-substituted alkynes.7 More examples using other metals were reported over the following years. Allenes,8,9 alkynes10,11 and conjugated dienes12,13 have been transformed into electrophilic metal-π-allyl intermediates using iridium, rhodium and other transition metal catalysts, which undergo nucleophilic attack to form hydrofunctionalization products (Scheme 1b). This strategy could be regarded as an atom-economic alternative to the traditional metal catalyzed allylic substitution and allylic oxidation.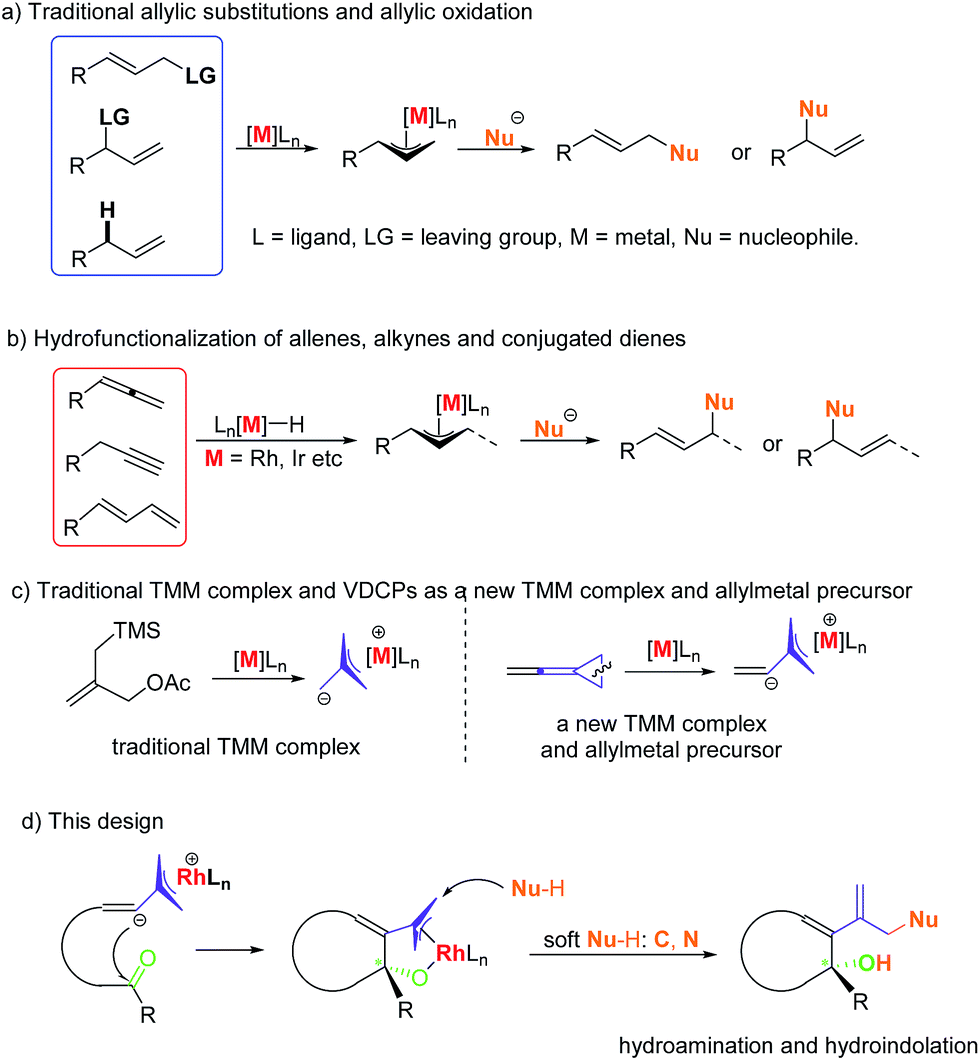 | ||
| Scheme 1 Previous work and this work. | ||
Vinylidenecyclopropanes (VDCPs), bearing an allene moiety connected to a highly strained cyclopropane ring, serve as fascinating building blocks in organic synthesis and have received great attention from organic chemists.14 Based on our ongoing investigation on metal-catalyzed transformations of VDCPs, we found that cationic Rh(I) complexes could insert into the weaker distal bond of the three-membered ring to give new trimethylenemethane (TMM) complexes of rhodium (Scheme 1c).15 However, unlike the traditional TMM metal complex, which has been used extensively in [3 + 2] cycloaddition reactions,16 this TMM–Rh species containing an inner olefinic moiety could react with an unsaturated functional group and then generate a new more reactive electrophilic Rh-π-allyl intermediate, which might be a productive electrophile for hydrofunctionalization (allylic substitution). Thus, we envisaged that VDCPs could be excellent candidates for the exploration of new reaction modes in the atom economic pathway of allylic substitution with “soft” carbon and heteroatom nucleophiles (those from conjugate acids with a pKa less than 25) (Scheme 1d, the design presented in this work).
Results and discussion
Experimental investigations
To test the feasibility of our hypothesis, we initially investigated the reaction of keto-VDCP 1a with indoline 2a as a coupling partner. Notably, indoline and indole moieties are ubiquitous structural elements of many natural compounds of biological interest.17 Therefore, the development of efficient processes for the functionalization of these compounds will facilitate access to pharmaceutically attractive molecules. To our delight, when [Rh(cod)Cl]2 and AgNTf2 were used as catalysts with (rac)-Binap as the ligand, hydroamination product rac-3aa could be successfully furnished in 75% yield in toluene at 90 °C (Table 1, entry 1). The structure of 3aa has been unequivocally determined by X-ray diffraction.18 Some other nucleophiles were also investigated in this transformation, such as alcohols, thiophenol, benzothiazole and diketones. Disappointedly, only when dibenzoylmethane was used as the nucleophile could the corresponding hydrofunctionalization product be obtained in moderate yield (see Table S1 in the ESI†). Interestingly, 35% yield of rac-3aa could also be obtained when [Ir(cod)Cl]2 was used to replace [Rh(cod)Cl]2. A subsequent survey of other coordinating anions indicated that the sterically bulky, more weakly coordinating BArF− anion (BArF− = B[(3,5-(CF3)2C6H3)]4−) was the best choice (entries 2–5).19 Different solvents were surveyed next. A better yield up to 92% was realized when 1,4-dioxane was employed (entries 6–8). Encouraged by these results, various chiral bisphosphine ligands were investigated next. An excellent yield (90%) and good enantioselectivity (up to −83% ee) could be realized by using (R)-Binap (L2) as the ligand. Higher ee values were afforded when (R)-Tol-Binap (L3) and (R)-H8-Binap (L4) were employed (entries 10 and 11). Unexpectedly, the use of either non-biaryl bisphosphine ligand L5 or monophosphine ligand L6 resulted in only trace amounts of the corresponding product being produced (entries 12 and 13). Gratifyingly, the best enantioselectivity (>99% ee) was achieved by employing (R)-SDP (L7) as a ligand (entry 14). For streamlining the operation, [Rh(cod)((R)-SDP)]BArF was prepared in advance and used as a catalyst, providing 3aa in 93% yield with >99% ee value (entry 15). As the catalytic activity of this [Rh(cod)((R)-SDP)]BArF catalyst was very high, the reaction could even be carried out in the presence of 2.5 mol% of the Rh catalyst without reduction of the product's yield and ee value (entry 16). In addition, lowering the reaction temperature did not give a better result (entry 17).|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | [Rh] | Additive | Ligand | Solvent | Yieldb [%] | eec [%] |
| a Reaction conditions: 1a (0.10 mmol), 2a (0.12 mmol), Rh catalyst (5 mol%), additive (10 mol%), ligand (10 mol%), and solvent (1.0 mL) for 4–12 h. b Isolated yield. c Determined by HPLC on a chiral stationary phase. d [Rh(cod)((R)-SDP)]BArF (2.5 mol%) was employed. e The reaction was conducted at 60 °C. Ts = 4-toluenesulfonyl, cod = cyclo-1,5-octadiene, and NaBArF = sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate. | ||||||
| 1 | [Rh(cod)Cl]2 | AgNTf2 | L1 | Toluene | 75 | — |
| 2 | [Rh(cod)Cl]2 | AgBF4 | L1 | Toluene | 58 | — |
| 3 | [Rh(cod)Cl]2 | AgOTf | L1 | Toluene | 62 | — |
| 4 | [Rh(cod)Cl]2 | AgOTs | L1 | Toluene | 73 | — |
| 5 | [Rh(cod)Cl]2 | NaBArF | L1 | Toluene | 85 | — |
| 6 | [Rh(cod)Cl]2 | NaBArF | L1 | Chlorobenzene | 78 | — |
| 7 | [Rh(cod)Cl]2 | NaBArF | L1 | 1,2-Dichloroethane | 56 | — |
| 8 | [Rh(cod)Cl]2 | NaBArF | L1 | Dioxane | 92 | — |
| 9 | [Rh(cod)Cl]2 | NaBArF | L2 | Dioxane | 90 | −83 |
| 10 | [Rh(cod)Cl]2 | NaBArF | L3 | Dioxane | 75 | −89 |
| 11 | [Rh(cod)Cl]2 | NaBArF | L4 | Dioxane | 77 | −93 |
| 12 | [Rh(cod)Cl]2 | NaBArF | L5 | Dioxane | Trace | — |
| 13 | [Rh(cod)Cl]2 | NaBArF | L6 | Dioxane | Trace | — |
| 14 | [Rh(cod)Cl]2 | NaBArF | L7 | Dioxane | 92 | >99 |
| 15 | [Rh(cod)((R)-SDP)]BArF | — | — | Dioxane | 93 | >99 |
| 16 | [Rh(cod)((R)-SDP)]BAr F | — | — | Dioxane | 91 | >99 |
| 17e | [Rh(cod)((R)-SDP)]BArF | — | — | Dioxane | 68 | >99 |
|
||||||

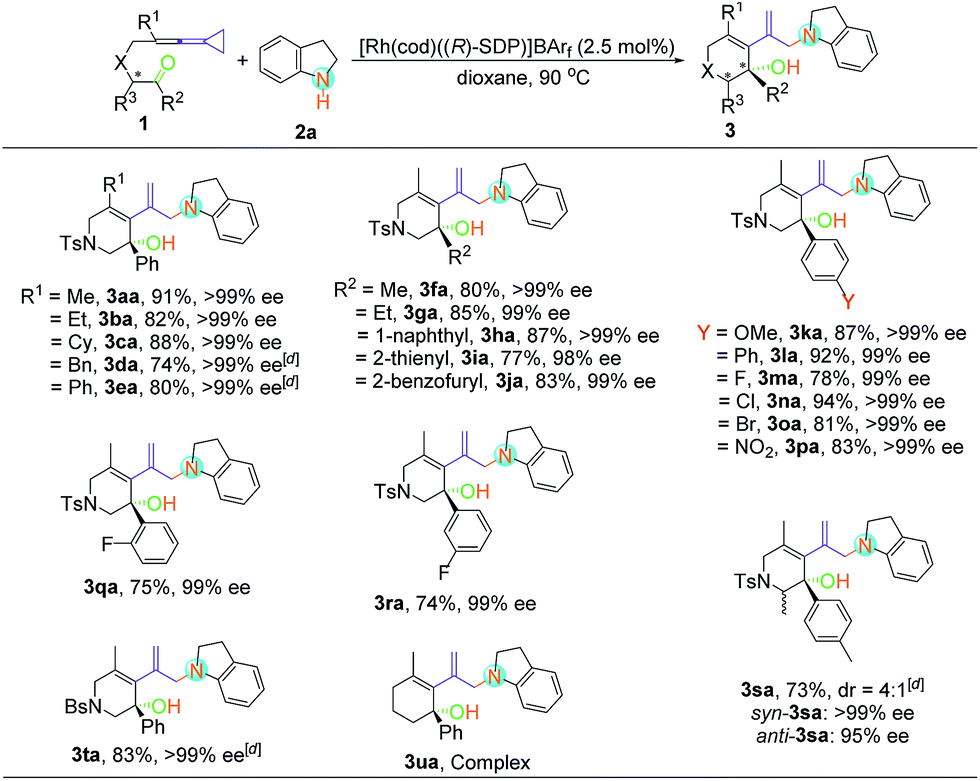
With the optimal reaction conditions in hand, the scope of this asymmetric hydroamination was then assessed through variation of the keto-VDCPs and secondary amines. We first examined the scope of keto-VDCP 1. As shown in Table 2, the substrate scope of this protocol was broad. For substrates 1b and 1c (R1 = primary or secondary alkyl groups), the desired products 3ba and 3ca were obtained in good to excellent yields (82% and 88%) with outstanding ee values (>99% ee). R1 could also be a benzyl or a phenyl group, giving the desired products 3da and 3ea in 74% and 80% yields along with >99% ee, respectively. We note that the optimized conditions should be modified with regard to the substituent groups or linkers in keto-VDCP 1. For substrates 1d, 1e, 1s and 1t, the reactions proceeded effectively to furnish the corresponding products (3da, 3ea, 3sa and 3ta) in good yields (73–83%) with excellent ee values (up to 99% ee) in the presence of [Rh(cod)Cl]2, AgNTf2 and (R)-SDP in toluene at 90 °C. It is noteworthy that the product 3sa contains a pair of diastereoisomers in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The relative configuration of syn-3sa was determined by nuclear Overhauser effect spectroscopy (see page S58 in the ESI†). The ketone moiety of keto-VDCP 1 was examined next. We found that R2 could be an aliphatic, naphthyl or heteroaromatic group, affording the corresponding products 3fa–3ja in good yields with excellent ee values. As for the substituents at the benzene ring, whether they were electron-rich or electron-poor, the reactions proceeded smoothly to produce the desired products 3ka–3pa in 78–94% yields with 99% ee values, even for strongly electron-withdrawing substituents such as the nitro group. Besides, no obvious reduction of yields and ee values was observed when different halogen atoms such as F, Cl or Br were introduced. Moreover, even when the halogen substituent was at different positions of the benzene ring, the desired products were produced in similar yields as in the cases of products 3ma, 3qa and 3ra. The use of NBs (4-bromobenzenesulfonyl amide) as a tether was also tolerated in this transformation, giving the corresponding product 3ta in 83% yield with >99% ee value. However, upon changing the linker to a carbon or an oxygen atom, or extending the carbon chain using a (CH2)2 or a (CH2)3 tether, only traces of expected product could be detected by thin-layer chromatography (TLC) monitoring (see Table S2 in the ESI†).
:1 ratio. The relative configuration of syn-3sa was determined by nuclear Overhauser effect spectroscopy (see page S58 in the ESI†). The ketone moiety of keto-VDCP 1 was examined next. We found that R2 could be an aliphatic, naphthyl or heteroaromatic group, affording the corresponding products 3fa–3ja in good yields with excellent ee values. As for the substituents at the benzene ring, whether they were electron-rich or electron-poor, the reactions proceeded smoothly to produce the desired products 3ka–3pa in 78–94% yields with 99% ee values, even for strongly electron-withdrawing substituents such as the nitro group. Besides, no obvious reduction of yields and ee values was observed when different halogen atoms such as F, Cl or Br were introduced. Moreover, even when the halogen substituent was at different positions of the benzene ring, the desired products were produced in similar yields as in the cases of products 3ma, 3qa and 3ra. The use of NBs (4-bromobenzenesulfonyl amide) as a tether was also tolerated in this transformation, giving the corresponding product 3ta in 83% yield with >99% ee value. However, upon changing the linker to a carbon or an oxygen atom, or extending the carbon chain using a (CH2)2 or a (CH2)3 tether, only traces of expected product could be detected by thin-layer chromatography (TLC) monitoring (see Table S2 in the ESI†).
| a Reactions were performed with keto-VDCP 1 (0.10 mmol), secondary amine 2a (0.12 mmol), and [Rh(cod)((R)-SDP)]BArF (2.5 mol%) in dioxane (1.0 mL) at 90 °C for 4–12 h. b Isolated yield. c Determined by HPLC on a chiral stationary phase. d [Rh(cod)Cl]2 (2.5 mol%), AgNTf2 (5.0 mol%), (R)-SDP (5.0 mol%), and toluene (1.0 mL) were used. |
|---|
|
With respect to secondary amine 2, various substituents at the indolines were firstly examined (Table 3). Substrates bearing chloro, bromo, methyl and nitryl groups at the different positions of indolines were smoothly transformed into the enantiomerically enriched hydroamination products with good yields (67–87%) and excellent enantioselectivities (97–99% ee). When 1,2,3,4-tetrahydroquinoline was used as the substrate, the desired product 3af could also be obtained with excellent yield and ee value. However, the yield of 3ag was slightly decreased when the N-heterocycle was extended to a 7-membered ring. As for non-cyclic secondary amines, the reactions proceeded smoothly to furnish the corresponding products 3ah–3aj in good to excellent yields with outstanding ee values. In addition, 3ak could be obtained with 2-anilinoethanol without the use of any protecting group. Disappointingly, none of the desired product was observed under the above conditions when dibenzylamine, diethylamine, morpholine, pyrrolidine and diphenylamine were employed (see Table S2 in the ESI†). It seems that the aryl group was essential because of its electronic nature.
| a Reactions were performed with keto-VDCP 1a (0.10 mmol), secondary amine 2 (0.12 mmol), and [Rh(cod)((R)-SDP)]BArF (2.5 mol%) in dioxane (1.0 mL) at 90 °C for 4–12 h. b Isolated yield. c Determined by HPLC on a chiral stationary phase. d [Rh(cod)Cl]2 (2.5 mol%), AgNTf2 (5.0 mol%), (R)-SDP (5.0 mol%), and toluene (1.0 mL) were used. |
|---|

|
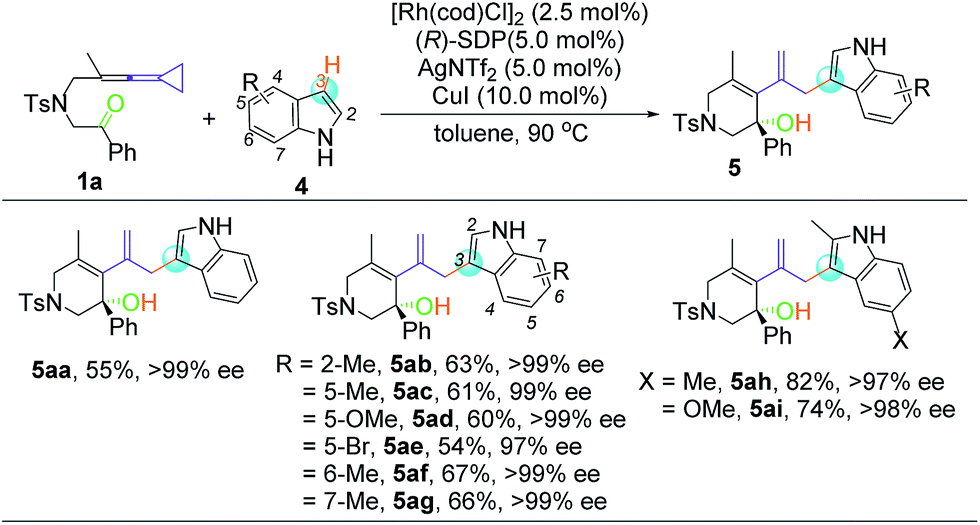
Despite the diverse reactivity of indoles, we observed selective bond formation at the 3-position upon coupling of keto-VDCP 1a and indoles to yield 5 as the only regioisomer (for details about optimization of the reaction conditions, please see Table S3 in the ESI†).20 The structure of 5aa has been unequivocally determined by X-ray diffraction.21 Then, we focused on developing an enantioselective cycloisomerization/cross coupling using indoles as the carbon nucleophile due to the importance of these heterocycles in natural and pharmaceutical products (Table 4). Efficient and selective indole−VDCP cross coupling occurs with a variety of indole substitution patterns. For example, a methyl group or methoxy group can be incorporated at the 2-, 5-, 6- and 7-positions of indole to afford the corresponding products in moderate yields, with up to 99% ee values (5ab, 5ac, 5af, and 5ag). In comparison, a lower yield and ee value are observed with 5-bromolindole (5ae, 57% yield and 97% ee value). Moreover, we also observed better reactivity with indoles containing two electron-donating groups (5ah and 5ai, up to 82% yield and 98% ee value).
The enantioselective N-substitution of indoles has rarely been explored due to the weak acidity of the N–H group, despite the fact that the products are privileged structural motifs in natural alkaloids and biologically active compounds.22 A one-pot protocol was thus developed, using Rh-catalyzed asymmetric hydroamination and subsequent oxidative dehydroaromatization of indoline 2, providing facile access to N1 allylic alkylation of indoles 6aa in 71% yield with >99% ee value and 6ab in 76% yield with >99% ee value (Scheme 2).
 | ||
| Scheme 2 One-pot asymmetric N1 allylic alkylation of indoles. | ||
Considering the easy-to-handle functional groups in products 3 and 6, further transformations of 3aa and 6aa were also examined (Scheme 3). The allyl-substituted product 7aa could be obtained in 91% yield from 3aa upon treatment with potassium carbonate and allyl bromide. The subsequent ruthenium-catalyzed intramolecular ring-closing olefin metathesis reaction of 7aa gave the bicyclic derivative 8aa in 71% yield with >99% ee. Its structure has been fully confirmed by NMR spectroscopic data including DEPT, COSY, HSQC and HMBC (see pages S112–S117 in the ESI†). Moreover, polycyclic indole 10aa could be obtained from propargyl-substituted product 9aa in the presence of [Au(tBuXPhos)(NCMe)][SbF6] (XPhos = 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl) (5 mol%) in 92% yield with 99% ee value. The absolute configuration of 10aa has been determined to be S by X-ray diffraction. The ORTEP drawing is shown in Scheme 3 and the CIF data are summarized in the ESI.†23
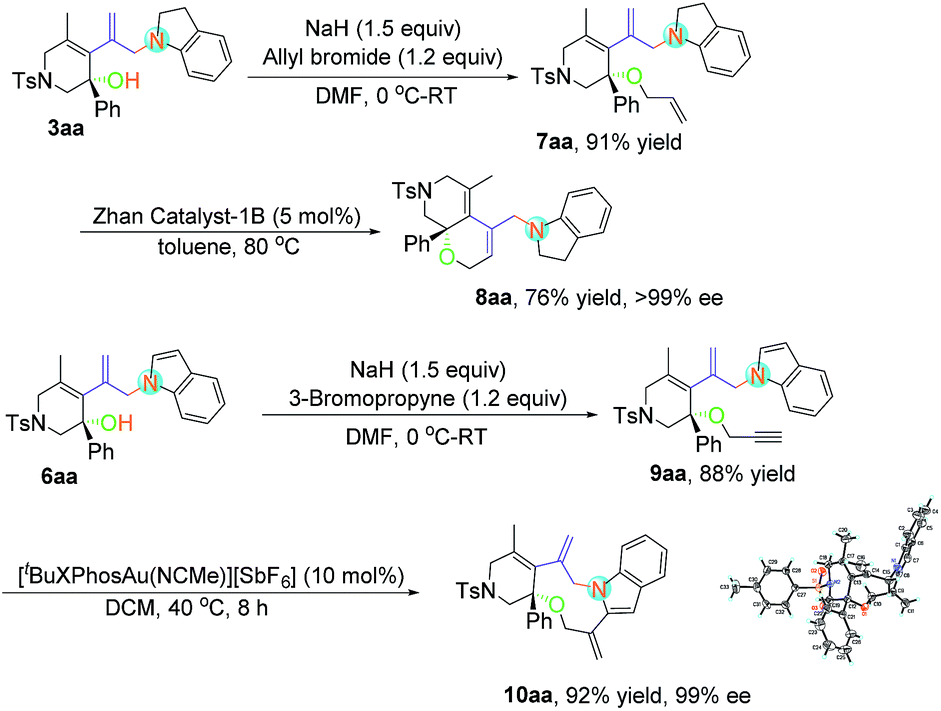 | ||
| Scheme 3 Derivatizations of the products 3aa and 6aa. | ||
Mechanistic proposal
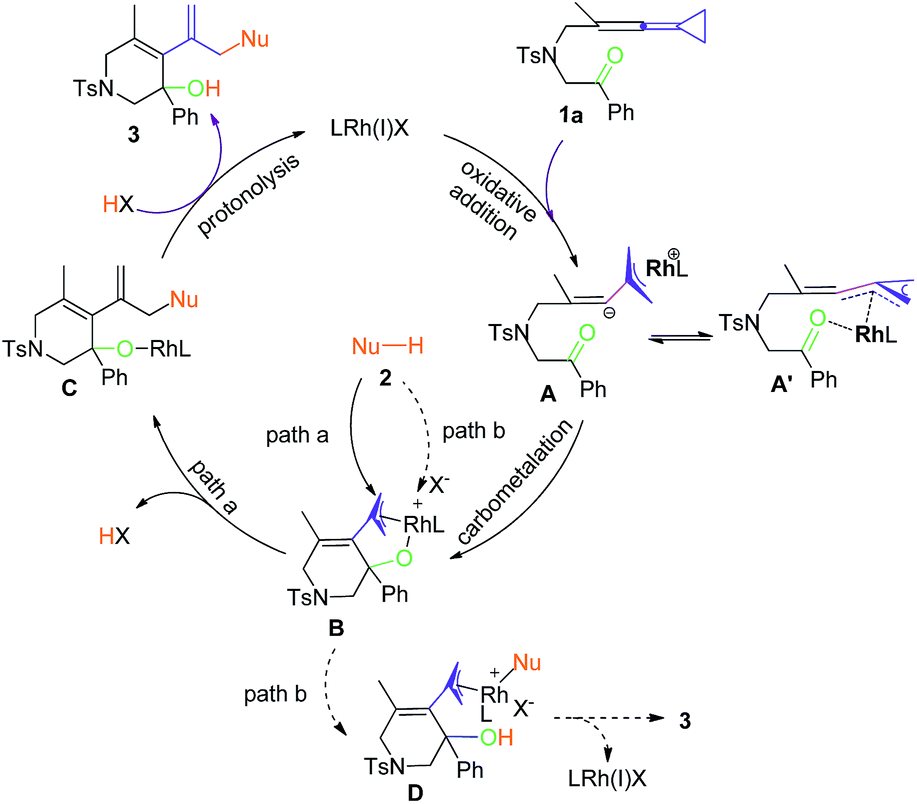 | ||
| Scheme 4 A plausible reaction mechanism. | ||
Conclusions
In conclusion, we have developed a novel Rh-catalyzed highly regio- and enantioselective hydrofunctionalization of keto-VDCPs with a wide range of soft nucleophiles. The combination of various secondary amines with keto-VDCPs could afford the hydroamination products in good to excellent yields with outstanding ee values. The highly enantioselective allylic alkylation at both of the C3 and N1 positions of indoles could be realized either by using indoles as nucleophiles directly or via a one-pot asymmetric hydroamination and subsequent oxidative dehydroaromatization of indolines. A new TMM–Rh model complex was proposed, which can act as a new atom economical Rh-π-allyl precursor at the same time. Moreover, the resulting multiple functionalized products could easily be transformed into more complex polyheterocycles under ruthenium or gold(I) catalysis. Further investigations to examine the mechanistic details more extensively and exploration of new methodologies based on this novel TMM–metal complex generated from functionalized VDCPs are currently underway in our laboratory.General procedure for the synthesis of product 3
A 10 mL dried tube was charged with keto-VDCP 1 (0.1 mmol, 1.0 equiv.) and [Rh(cod)(R-SDP)]BArF (2.5 mol%, 0.025 equiv.). The reaction tube was evacuated and backfilled with argon (repeated three times). Then, secondary amine 2 (0.12 mmol, 1.2 equiv.) and dioxane (1.0 mL) were added into the tube. The reaction mixture was stirred at 90 °C for 4–10 h. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (SiO2) to give the corresponding product 3.General procedure for the synthesis of product 5
A 10 mL dried tube was charged with Keto-VDCP 1a (0.1 mmol, 1.0 equiv.), [Rh(COD)Cl]2 (0.0025 mmol, 0.025 equiv.), (R)-SDP (0.005 mmol, 0.05 equiv.), AgNTf2 (0.005 mmol, 0.05 equiv.) and CuI (0.010 mmol, 0.10 equiv.). The reaction tube was evacuated and backfilled with argon (repeated three times). Then, indole 4 (0.12 mmol, 1.2 equiv.) and toluene (1.0 mL) were added into the tube. The reaction mixture was stirred at 90 °C for 4–10 h. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (SiO2) to give the corresponding product 5.Typical procedure for the preparation of compound 6aa
To a flame dried Schlenk tube were added compound 3aa (0.2 mmol), NaH (60% dispersion in mineral oil, 1.5 equiv.) and DMF (2.0 mL). The reaction mixture was stirred at 0 °C for 0.5 h before allyl bromide (1.2 equiv.) was added. The reaction mixture was stirred at 0 °C for another 4 h. Then, the reaction mixture was diluted with cold water and extracted with ether (4 mL × 3) and the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (SiO2) to give the corresponding product 7aa.Under an argon atmosphere, compound 7aa (0.1 mmo, 1.0 equiv.), Zhan-catalyst-1B (0.10 equiv.) and toluene (10 mL) were added into a Schlenk tube and then the mixture was heated at 80 °C for 12 h. Then, the solvent was removed under reduced pressure and the residue was purified by flash column chromatography (SiO2) to give the corresponding product 8aa.
Typical procedure for the preparation of compound 10aa
To a flame dried Schlenk tube were added compound 6aa (0.2 mmol), NaH (60% dispersion in mineral oil, 1.5 equiv.) and DMF (2.0 mL). The reaction mixture was stirred at 0 °C for 0.5 h before 3-bromopropyne bromide (1.2 equiv.) was added. The reaction mixture was stirred at 0 °C for another 4 h. Then, the reaction mixture was diluted with cold water and extracted with ether (4 mL × 3) and the combined organics were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (SiO2) to give the corresponding product 9aa.To a flame dried Schlenk tube were added 9aa (0.1 mmol, 1.0 equiv.), [Au(tBuXPhos)(NCMe)][SbF6] (10 mol%) and DCM (2.0 mL), and the resulting mixture was stirred at 10 °C for 8 h. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (SiO2) to give the corresponding product 10aa.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Basic Research Program of China [(973)-2015CB856603], the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDB20000000 and sioczz201808), the National Natural Science Foundation of China (20472096, 21372241, 21572052, 20672127, 21421091, 21372250, 21121062, 21302203, 21772037, 21772226, 20732008 and 21772037), and the Fundamental Research Funds for the Central Universities (222201717003).References
- For selected reviews on allylic substitutions, see (a) R. L. Grange, E. A. Clizbe and P. A. Evans, Synthesis, 2016, 48, 2911–2968 CrossRef; (b) S. Oliver and P. A. Evans, Synthesis, 2013, 45, 3179–3198 CrossRef; (c) Z. Lu and S.-M. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef PubMed; (d) B. M. Stoltz and J. T. Mohr, Chem. Asian J., 2007, 2, 1476–1491 CrossRef PubMed; (e) H. Miyabe and Y. Takemoto, Synlett, 2005, 2005, 1641–1655 Search PubMed; (f) G. Helmchen, J. Org. Chem., 1999, 576, 203–214 CrossRef; (g) B. M. Trost and D. L. V. Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef PubMed.
- For selected reviews on the applications of allylic substitutions in organic synthesis, see (a) J.-P. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539–2555 CrossRef PubMed; (b) A. D. Huters, E. D. Styduhar and N. K. Garg, Angew. Chem., Int. Ed., 2012, 51, 3758–3765 CrossRef PubMed; (c) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef PubMed.
- For selected examples of allylic C−H functionalization, see (a) G.-Y. Yin, Y.-C. Wu and G.-S. Liu, J. Am. Chem. Soc., 2010, 132, 11978–11987 CrossRef PubMed; (b) G.-S. Liu and S. S. Stahl, J. Am. Chem. Soc., 2007, 129, 6328–6335 CrossRef PubMed; (c) M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346–1347 CrossRef PubMed.
- For selected reviews on allylic substitutions via allylic alcohols, see (a) N. Butt, G.-Q. Yang and W.-B. Zhang, Chem. Rec., 2016, 16, 2683–2692 CrossRef PubMed; (b) N. A. Butt and W.-B. Zhang, Chem. Soc. Rev., 2015, 44, 7929–7967 RSC; (c) B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467–4483 RSC; (d) M. Bandini, Angew. Chem., Int. Ed., 2011, 50, 994–995 CrossRef PubMed; (e) K. J. Szabó, Synlett, 2006, 2006, 811–824 CrossRef; (f) Y. Tamaru, Eur. J. Org. Chem., 2005, 2005, 2647–2656 CrossRef; (g) J. Muzart, Tetrahedron, 2005, 61, 4179–4212 CrossRef.
- For selected examples on allylic substitutions via allylic alcohols, see (a) J.-Y. Jing, X.-H. Huo, J.-F. Shen, J.-K. Fu, Q.-H. Meng and W.-B. Zhang, Chem. Commun., 2017, 53, 5151–5154 RSC; (b) X.-H. Huo, G.-Q. Yang, D.-L. Liu, Y.-G. Liu, I. D. Gridnev and W.-B. Zhang, Angew. Chem., Int. Ed., 2014, 53, 6776–6780 CrossRef PubMed; (c) R. Shibuya, L. Lin, Y. Nakahara, K. Mashima and T. Ohshima, Angew. Chem., Int. Ed., 2014, 53, 4377–4381 CrossRef PubMed; (d) Y.-X. Li, Q.-Q. Xuan, L. Liu, D. Wang, Y.-J. Chen and C.-J. Li, J. Am. Chem. Soc., 2013, 135, 12536–12539 CrossRef PubMed; (e) K. Das, R. Shibuya, Y. Nakahara, N. Germain, T. Ohshima and K. Mashima, Angew. Chem., Int. Ed., 2012, 51, 150–154 CrossRef PubMed; (f) G.-X. Jiang and B. List, Angew. Chem., Int. Ed., 2011, 50, 9471–9474 CrossRef PubMed; (g) T. Ohshima, Y. Miyamoto, J. Ipposhi, Y. Nakahara, M. Utsunomiya and K. Mashima, J. Am. Chem. Soc., 2009, 131, 14317–14328 CrossRef PubMed; (h) C. Defieber, M. A. Ariger, P. Moriel and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 3139–3143 CrossRef PubMed; (i) F. Ozawa, H. Okamoto, S. Kawagishi, S. Yamamoto, T. Minami and M. Yoshifuji, J. Am. Chem. Soc., 2002, 124, 10968–10969 CrossRef PubMed.
- B. M. Trost, Science, 1991, 254, 1471–1477 Search PubMed.
- (a) N. T. Patil, N. K. Pahadi and Y. Yamamoto, Can. J. Chem., 2005, 83, 569–573 CrossRef; (b) N. T. Patil, N. K. Pahadi and Y. Yamamoto, Synthesis, 2004, 2004, 2186–2190 CrossRef; (c) N. T. Patil, F. Nawaz Khan and Y. Yamamoto, Tetrahedron Lett., 2004, 45, 8497–8499 CrossRef; (d) N. T. Patil, I. Kadota, A. Shibuya, Y. S. Gyoung and Y. Yamamoto, Adv. Synth. Catal., 2004, 346, 800–804 CrossRef; (e) I. Kadota, A. Shibuya, Y. S. Gyoung and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 10262–10263 CrossRef; (f) M. Al-Masum and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 3809–3810 CrossRef; (g) Y. Yamamoto, M. Al-Masum, N. Fujiwara and N. Asao, Tetrahedron Lett., 1995, 36, 2811–2814 CrossRef; (h) B. M. Trost and V. J. Gerusz, J. Am. Chem. Soc., 1995, 117, 5156–5157 CrossRef; (i) Y. Yamamoto, M. Al-Masum and N. Asao, J. Am. Chem. Soc., 1994, 116, 6019–6020 CrossRef; (j) B. M. Trost, W. Brieden and K. H. Baringhaus, Angew. Chem., Int. Ed., 1992, 31, 1335–1336 CrossRef.
- For examples of the use of allenes as electrophilic allylmetal precursors, see (a) D. Berthold and B. Breit, Org. Lett., 2018, 20, 598–601 CrossRef PubMed; (b) S. Parveen, C. Li, A. Hassan and B. Breit, Org. Lett., 2017, 19, 2326–2329 CrossRef PubMed; (c) H. Zhou, Y.-N. Wang, L. Zhang, M. Cai and S.-Z. Luo, J. Am. Chem. Soc., 2017, 139, 3631–3634 CrossRef PubMed; (d) Z. Liu and B. Breit, Angew. Chem., Int. Ed., 2016, 55, 8440–8443 CrossRef PubMed; (e) A. B. Pritzius and B. Breit, Angew. Chem., Int. Ed., 2015, 54, 3121–3125 CrossRef PubMed; (f) K. L. Butler, M. Tragni and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2012, 51, 5175–5178 CrossRef PubMed; (g) T. Kawamoto, S. Hirabayashi, X.-X. Guo, T. Nishimura and T. Hayashi, Chem. Commun., 2009, 0, 3528–3530 RSC; (h) R. L. LaLonde, B. D. Sherry, E. J. Kang and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 2452–2453 CrossRef PubMed; (i) J. S. Johnson and R. G. Bergman, J. Am. Chem. Soc., 2001, 123, 2923–2924 CrossRef PubMed.
- For examples of the use of allenes as nucleophilic allylmetal precursors, see (a) B. Sam, T. Luong and M. J. Krische, Angew. Chem., Int. Ed., 2015, 54, 5465–5469 CrossRef PubMed; (b) J. R. Zbieg, E. L. McInturff, J. C. Leung and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 1141–1144 CrossRef PubMed; (c) J. Moran, A. Preetz, R. A. Mesch and M. J. Krische, Nat. Chem., 2011, 3, 287–290 CrossRef PubMed; (d) S. B. Han, I. S. Kim, H. Han and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 6916–6917 CrossRef PubMed; (e) E. Skucas, J. R. Zbieg and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 5054–5055 CrossRef PubMed.
- For selected examples of the use of alkynes as electrophilic allylmetal precursors, see (a) J. Zheng and B. Breit, Org. Lett., 2018, 20, 1866–1870 CrossRef PubMed; (b) F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1029–1032 CrossRef PubMed; (c) F. A. Cruz, Y.-M. Zhu, Q. D. Tercenio, Z.-M. Shen and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 10641–10644 CrossRef PubMed; (d) Q.-A. Chen, Z.-W. Chen and V. M. Dong, J. Am. Chem. Soc., 2015, 137, 8392–8395 CrossRef PubMed; (e) A. Lumbroso, N. Abermil and B. Breit, Chem. Sci., 2012, 3, 789–793 RSC; (f) A. Lumbroso, P. Koschker, N. R. Vautravers and B. Breit, J. Am. Chem. Soc., 2011, 133, 2386–2389 CrossRef PubMed; (g) N. T. Patil, L. M. Lutete, H.-Y. Wu, N. K. Pahadi, I. D. Gridnev and Y. Yamamoto, J. Org. Chem., 2006, 71, 4270–4279 CrossRef PubMed; (h) G. B. Bajracharya, Z.-B. Huo and Y. Yamamoto, J. Org. Chem., 2005, 70, 4883–4886 CrossRef PubMed; (i) I. Kadota, L. M. Lutete, A. Shibuya and Y. Yamamoto, Tetrahedron Lett., 2001, 42, 6207–6210 CrossRef.
- For examples of the use of alkynes as nucleophilic allylmetal precursors, see (a) T. Liang, W. Zhang and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 16024–16027 CrossRef PubMed; (b) T. Liang, K. D. Nguyen, W.-D. Zhang and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 3161–3164 CrossRef PubMed; (c) B. Y. Park, K. D. Nguyen, M. R. Chaulagain, V. Komanduri and M. J. Krische, J. Am. Chem. Soc., 2014, 136, 11902–11905 CrossRef PubMed; (d) Y. Obora, S. Hatanaka and Y. Ishii, Org. Lett., 2009, 11, 3510–3513 CrossRef PubMed.
- For examples of the use of dienes as electrophilic allylmetal precursors, see (a) N. J. Adamson, K. C. E. Wilbur and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 2761–2764 CrossRef PubMed; (b) J. S. Marcum, C. C. Roberts, R. S. Manan, T. N. Cervarich and S. J. Meek, J. Am. Chem. Soc., 2017, 139, 15580–15583 CrossRef PubMed; (c) N. J. Adamson, E. Hull and S. J. Malcolmson, J. Am. Chem. Soc., 2017, 139, 7180–7183 CrossRef PubMed; (d) X.-H. Yang and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1774–1777 CrossRef PubMed; (e) X.-H. Yang, A. Lu and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 14049–14052 CrossRef PubMed; (f) M. J. Goldfogel and S. J. Meek, Chem. Sci., 2016, 7, 4079–4084 RSC; (g) Y. N. Timsina, R. K. Sharma and T. V. RajanBabu, Chem. Sci., 2015, 6, 3994–4008 RSC; (h) D. Banerjee, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 1630–1635 CrossRef PubMed; (i) L.-Y. Liao and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 10209–10211 CrossRef PubMed.
- For examples of the use of dienes as nucleophilic allylmetal precursors, see (a) K. D. Nguyen, D. Herkommer and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 14210–14213 CrossRef PubMed; (b) B. Y. Park, T. P. Montgomery, V. J. Garza and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 16320–16323 CrossRef PubMed; (c) A. Köpfer, B. Sam, B. Breit and M. J. Krische, Chem. Sci., 2013, 4, 1876–1880 RSC; (d) J. R. Zbieg, E. Yamaguchi, E. L. McInturff and M. J. Krische, Science, 2012, 336, 324–327 CrossRef PubMed; (e) E. L. McInturff, E. Yamaguchi and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 20628–20631 CrossRef PubMed.
- For some more recent papers related to VDCPs, please see (a) S. Yang, K.-H. Rui, X.-Y. Tang, Q. Xu and M. Shi, J. Am. Chem. Soc., 2017, 139, 5957–5964 CrossRef PubMed; (b) S. Yang, Q. Xu and M. Shi, Chem.–Eur. J., 2016, 22, 10387–10392 CrossRef PubMed; (c) S. Yang, W. Yuan, Q. Xu and M. Shi, Chem.–Eur. J., 2015, 21, 15964–15969 CrossRef PubMed; (d) D.-Y. Li, Y. Wei, I. Marek, X.-Y. Tang and M. Shi, Chem. Sci., 2015, 6, 5519–5525 RSC; (e) D.-H. Zhang, X.-Y. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913–924 CrossRef PubMed; (f) A. V. Stepakov, A. G. Larina, V. M. Boitsov, V. V. Gurzhiy, A. P. Molchanov and R. R. Kostikov, Tetrahedron Lett., 2014, 55, 2022–2026 CrossRef; (g) M.-Z. Miao, J. Cao, J.-J. Zhang, X. Huang and L.-L. Wu, J. Org. Chem., 2013, 78, 2687–2692 CrossRef PubMed; (h) B.-L. Lu and M. Shi, Angew. Chem., Int. Ed., 2011, 50, 12027–12031 CrossRef PubMed; (i) M. Shi, L.-X. Shao, J.-M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883–5913 CrossRef PubMed; (j) N. U. Zhanpeisov, K. Mizuno, M. Anpo and J. Leszczynski, Int. J. Quantum Chem., 2004, 96, 343–348 CrossRef; (k) H. Maeda, T. Hirai, A. Sugimoto and K. Mizuno, J. Org. Chem., 2003, 68, 7700–7706 CrossRef PubMed; (l) K. Mizuno, H. Maeda, H. Sugita, S. Nishioka, T. Hirai and A. Sugimoto, Org. Lett., 2001, 3, 581–584 CrossRef PubMed.
- (a) H. Y. Lee, Acc. Chem. Res., 2015, 48, 2308–2319 CrossRef PubMed; (b) P. Dowd, Acc. Chem. Res., 1972, 5, 242–248 CrossRef; (c) B. M. Trost, Angew. Chem., Int. Ed., 1986, 25, 1–20 CrossRef; (d) B. M. Trost and D. M. T. Chan, J. Am. Chem. Soc., 1979, 101, 6429–6432 CrossRef.
- For some selected papers related to TMM, please see (a) B. M. Trost and L. Debien, J. Am. Chem. Soc., 2015, 137, 11606–11609 CrossRef PubMed; (b) B. M. Trost, V. Ehmke, B. M. O'Keefe and D. A. Bringley, J. Am. Chem. Soc., 2014, 136, 8213–8216 CrossRef PubMed; (c) B. M. Trost, D. A. Bringley, T. Zhang and N. Cramer, J. Am. Chem. Soc., 2013, 135, 16720–16735 CrossRef PubMed; (d) B. M. Trost, T. M. Lam and M. A. Herbage, J. Am. Chem. Soc., 2013, 135, 2459–2461 CrossRef PubMed; (e) B. M. Trost and D. A. Bringley, Angew. Chem., Int. Ed., 2013, 52, 4466–4469 CrossRef PubMed; (f) E. E. Schultz and R. Sarpong, J. Am. Chem. Soc., 2013, 135, 4696–4699 CrossRef PubMed; (g) B. M. Trost and S. M. Silverman, J. Am. Chem. Soc., 2012, 134, 4941–4954 CrossRef PubMed; (h) B. M. Trost and T. M. Lam, J. Am. Chem. Soc., 2012, 134, 11319–11321 CrossRef PubMed; (i) B. M. Trost, S. M. Silverman and J. P. Stambuli, J. Am. Chem. Soc., 2011, 133, 19483–19497 CrossRef PubMed.
- (a) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef PubMed; (b) S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC; (c) M. Somei and F. Yamada, Nat. Prod. Rep., 2004, 21, 278–311 RSC; (d) D. J. Faulkner, Nat. Prod. Rep., 2002, 19, 1–49 Search PubMed; (e) J. Bosch and M. L. Bennasar, Synlett, 1995, 1995, 587–596 CrossRef.
- The crystal data of rac-3aa have been deposited at the CCDC with number 1538522.†.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef PubMed.
- For examples of the asymmetric C3 alkylation of indoles via allylic substitutions, see (a) S. Gao, Z.-J. Wu, X.-X. Fang, A.-J. Lin and H.-Q. Yao, Org. Lett., 2016, 18, 3906–3909 CrossRef PubMed; (b) B. Feng, X.-Y. Pu, Z.-C. Liu, W.-J. Xiao and J.-R. Chen, Org. Chem. Front., 2016, 3, 1246–1249 RSC; (c) Y.-L. Liu and H.-F. Du, Org. Lett., 2013, 15, 740–743 CrossRef PubMed; (d) Z.-Q. Liu, Z.-P. Cao and H.-F. Du, Org. Biomol. Chem., 2011, 9, 5369–5372 RSC; (e) T. Hoshi, K. Sasaki, S. Sato, Y. Ishii, T. Suzuki and H. Hagiwara, Org. Lett., 2011, 13, 932–935 CrossRef PubMed; (f) Z.-P. Cao, Y.-L. Liu, Z.-Q. Liu, X.-Q. Feng, M.-Y. Zhuang and H.-F. Du, Org. Lett., 2011, 13, 2164–2167 CrossRef PubMed; (g) K. Onitsuka, C. Kameyama and H. Sasai, Chem. Lett., 2009, 38, 444–445 CrossRef.
- The crystal data of rac-5aa have been deposited at the CCDC with number 1525845.†.
- For examples of the asymmetric N-substitution of indoles, see (a) K.-Y. Ye, Q. Cheng, C.-X. Zhuo, L. X. Dai and S.-L. You, Angew. Chem., Int. Ed., 2016, 55, 8113–8116 CrossRef PubMed; (b) W.-B. Liu, X. Zhang, L.-X. Dai and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 5183–5187 CrossRef PubMed; (c) B. M. Trost, M. Osipov and G.-B. Dong, J. Am. Chem. Soc., 2010, 132, 15800–15807 CrossRef PubMed; (d) L. Hong, W.-S. Sun, C.-X. Liu, L. Wang and R. Wang, Chem.–Eur. J., 2010, 16, 440–444 CrossRef PubMed; (e) Q. Cai, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2010, 49, 8666–8669 CrossRef PubMed; (f) L. M. Stanley and J. F. Hartwig, Angew. Chem., Int. Ed., 2009, 48, 7841–7844 CrossRef PubMed; (g) H.-L. Cui, X. Feng, J. Peng, J. Lei, K. Jiang and Y.-C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5737–5740 CrossRef PubMed; (h) M. Bandini, A. Eichholzer, M. Tragni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2008, 47, 3238–3241 CrossRef PubMed.
- The crystal data of 10aa have been deposited at the CCDC with number 1822276.†.
- (a) Y. Su, P. A. Inglesby and P. A. Evans, Angew. Chem., Int. Ed., 2018, 57, 673–677 CrossRef PubMed; (b) S. Mazumder, D. Shang, D. E. Negru, M. H. Baik and P. A. Evans, J. Am. Chem. Soc., 2012, 134, 20569–20572 CrossRef PubMed.
- (a) H. Matsushita and E. J. Negishi, J. Chem. Soc., Chem. Commun., 1982, 160–161 RSC; (b) B. M. Trost and T. R. Verhoeven, J. Org. Chem., 1976, 41, 3215–3216 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of the new compounds. CCDC 1538522, 1525845, and 1822276. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01595c |
| This journal is © The Royal Society of Chemistry 2018 |