
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct and indirect hyperpolarisation of amines using parahydrogen†
Wissam
Iali
,
Peter J.
Rayner
 ,
Adel
Alshehri
,
A. Jonathan.
Holmes
,
Amy J.
Ruddlesden
and
Simon B.
Duckett
*
,
Adel
Alshehri
,
A. Jonathan.
Holmes
,
Amy J.
Ruddlesden
and
Simon B.
Duckett
*
Centre for Hyperpolarisation in Magnetic Resonance (CHyM), Department of Chemistry, University of York, Heslington, YO10 5DD, UK. E-mail: simon.duckett@york.ac.uk
First published on 9th March 2018
Abstract
Nuclear Magnetic Resonance (NMR) and Magnetic Resonance Imaging (MRI) are two widely used techniques for the study of molecules and materials. Hyperpolarisation methods, such as Signal Amplification By Reversible Exchange (SABRE), turn typically weak magnetic resonance responses into strong signals. In this article we detail how it is possible to hyperpolarise the 1H, 13C and 15N nuclei of a range of amines. This involved showing how primary amines form stable but labile complexes of the type [Ir(H)2(IMes)(amine)3]Cl that allow parahydrogen to relay its latent polarisation into the amine. By optimising the temperature and parahydrogen pressure a 1000-fold per proton NH signal gain for deuterated benzylamine is achieved at 9.4 T. Additionally, we show that sterically hindered and electron poor amines that bind poorly to iridium can be hyperpolarised by either employing a co-ligand for complex stabilisation, or harnessing the fact that it is possible to exchange hyperpolarised protons between amines in a mixture, through the recently reported SABRE-RELAY method. These chemical refinements have significant potential to extend the classes of agent that can be hyperpolarised by readily accessible parahydrogen.
Introduction
Hyperpolarisation methods are used to overcome the inherent insensitivity of Nuclear Magnetic Resonance (NMR) spectroscopy and Magnetic Resonance Imaging (MRI) where their use may lead to dramatic time and cost savings. One such hyperpolarisation method, Parahydrogen Induced Polarisation (PHIP),1 produces the required non-Boltzmann nuclear spin distribution by the incorporation of parahydrogen (p-H2), an example of a nuclear singlet, into a suitable substrate molecule. This effect was shown to yield an enhanced NMR signal in 1987 (ref. 2) and has been the subject of intense investigation.1,3–6 A drawback of PHIP though, is the requirement for chemical change, caused by p-H2 addition to an unsaturated centre such as an alkene. However, recently a p-H2 technique that does not change the chemical identity of the sensitised molecule, called Signal Amplification By Reversible Exchange (SABRE), was reported.7,8 In this process, p-H2 is not directly incorporated into the substrate. Instead, polarisation is transferred via the J-coupling network that exists within a metal complex that co-locates p-H2 derived hydride ligands and a weakly bound substrate (ligand).9–11 Ligand exchange with excess unbound substrate and p-H2 enables the build-up of a pool of polarised substrate molecules in solution in a catalytic fashion as shown in Scheme 1.12 The SABRE polarisation of 1H nuclei typically utilises a 4JHH coupling between the catalysts hydride and substrate ligand protons. Tessari et al. have quantified these small spin–spin couplings to be ≈1.2 Hz.13 Alternatively, stronger 2JHN couplings have now been used to achieve 15N polarisation transfer at micro-Tesla fields in a variant known as SABRE-SHEATH (SABRE-in shield enables alignment transfer to heteronuclei).14,15 Intra-molecular spin–spin coupling networks within the substrate subsequently enables transfer to remote spins which do not exhibit direct coupling to the hydride ligands.16 | ||
| Scheme 1 Route to SABRE hyperpolarisation of an amine, NH2R. | ||
One of the most effective precatalysts for this process is [IrCl(COD)(IMes)] (1) [where IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene, COD = cis,cis-1,5-cyclooctadiene] which, after reaction with H2 and an excess of substrate, typically forms [Ir(H)2(IMes)(substrate)3]Cl in protic solvents such as methanol.17 Neutral active catalysts of the type [Ir(H)2(Cl)(IMes)(substrate)2] have also been reported to achieve similar results.18 These metal based polarisation transfer catalysts have been shown to act on a range of substrates that contain multiple bonds to nitrogen, such as nicotinamide,19,20 isoniazid,21,22 metronidazole,23 pyrazole,24 imines,25 diazirines26 and nitriles,27 and lead to polarised 1H, 13C, 15N, 19F, 29Si, 31P, and 129Sn nuclei that yield substantially enhanced NMR responses in just a few seconds.19,28–33 In fact, 1H polarisations of 50% have been reported, while for 15N, values of over 20% have been achieved.19,23
While SABRE-induced polarisation can also be achieved using in-field rf. transfer methods,34–37 whose efficiency varies with pulse sequence,37–39 spontaneous polarisation transfer occurs readily at low-field and it is this method we employ here. Moreover, as predicted,9 it has also been established that SABRE can be used to produce hyperpolarised singlet states40 with long-lifetimes through transfer in ultra-low field, or after the implementation of rf. transfer.41–46 Hence the diversity of applications found for this approach is growing and it clearly reflects not only a successful medium to test hyperpolarisation concepts but a potential route to transform the analytical potential of NMR.47–50
In this article, we introduce a new class of substrate into the SABRE repertoire, the amine. This is achieved by the formation of iridium–amine complexes of type [Ir(H)2(IMes)(RNH2)3]Cl (2, Scheme 1), whose kinetic behaviour is determined. Whilst the synthesis and use of iridium–amine complexes has been reported for catalytic transformations such as hydrogenation,51–53 we use them here for polarisation transfer catalysis. We have recently shown a limited number of amines are amenable to SABRE.54 Here, we start by detailing the hyperpolarisation of ammonia and benzylamine (BnNH2) and its associated optimisation to achieve large NMR signal enhancements. We then show how hyperpolarisation can be achieved in a range of primary amines. Upon changing to sterically bulky primary amines, secondary amines or aromatic amines, we show that an active SABRE catalyst does not form upon reaction with 1. However, we exemplify co-ligand and relayed polarisation transfer protocols to overcome this limitation and hence expand further the range of amines amenable to polarisation by p-H2.
Results and discussion
Direct 1H hyperpolarisation of ammonia and BnNH2 by SABRE
Our objective was to investigate the efficiency of the SABRE polarization of amines and ammonia and to determine their ligand exchange dynamics. A 5 mM solution of 1 in dry dichloromethane-d2 containing an ≈6-fold excess of NH3 relative to 1 at 298 K was therefore prepared. The aprotic solvent ensures that we maintain the necessary J-coupling network in [Ir(H)2(IMes)(NH3)3]Cl (2-NH3) during the study, as rapid 2H exchange results to form ND3 in deuterated protic solvents. This complex yields a hydride signal at δ −23.8, alongside a broad response at δ 0.47 for free NH3. The corresponding equatorial and axial NH3 ligand 1H NMR signals of 2-NH3 appear at δ 2.19 and 2.88 respectively. 2D 1H–15N HMQC measurements were subsequently used to locate the corresponding 15N signals for these ligands at daxial −47.8 and dequ −35.5. Full characterisation data for 2-NH3 is available in the ESI.†54 EXSY methods were then used to probe NH3 and H2 loss in 2-NH3. At 298 K, the associated rate constant for NH3 loss proved to be 1.64 s−1 while that of H2 loss is 0.32 s−1. For comparison, the dissociation rate for pyridine in [Ir(H)2(IMes)(py)3]Cl is 13.2 s−1 and suggests a higher stability for 2-NH3 which agrees with the greater basicity of NH3 relative to pyridine.55As 2-NH3 undergoes both NH3 and H2 loss in solution, we sought to prove that it underwent SABRE catalysis. Thus, a 3 bar pressure of p-H2 was introduced at 298 K and polarisation transfer was conducted at 60 G. A 1H NMR spectrum at 9.4 T was then recorded which showed a 154-fold signal enhancement per proton for the free NH3 response while the corresponding equatorial ligand signal, at δ 2.19, showed a 77-fold enhanced response (Fig. 1). Hence 2-NH3 acts as a SABRE catalyst as it produces a hyperpolarised free ammonia response. In the presence of water, the observed signal enhancement of the protons in free NH3 decreased to 40-fold per proton, matching that now observed for the equatorially bound NH3 ligand This drop is reflected in the signal at δ 1.88, for what is a H2O response, exhibiting a 75-fold signal gain per proton due to concomitant proton exchange; the ratio of 2-NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:NH3 in this sample was 1:5:17.5. Under these conditions, the T1 value for free NH3 in the presence of the active SABRE catalyst was measured by inversion recovery to be 5.5 s.
:H2O:NH3 in this sample was 1:5:17.5. Under these conditions, the T1 value for free NH3 in the presence of the active SABRE catalyst was measured by inversion recovery to be 5.5 s.
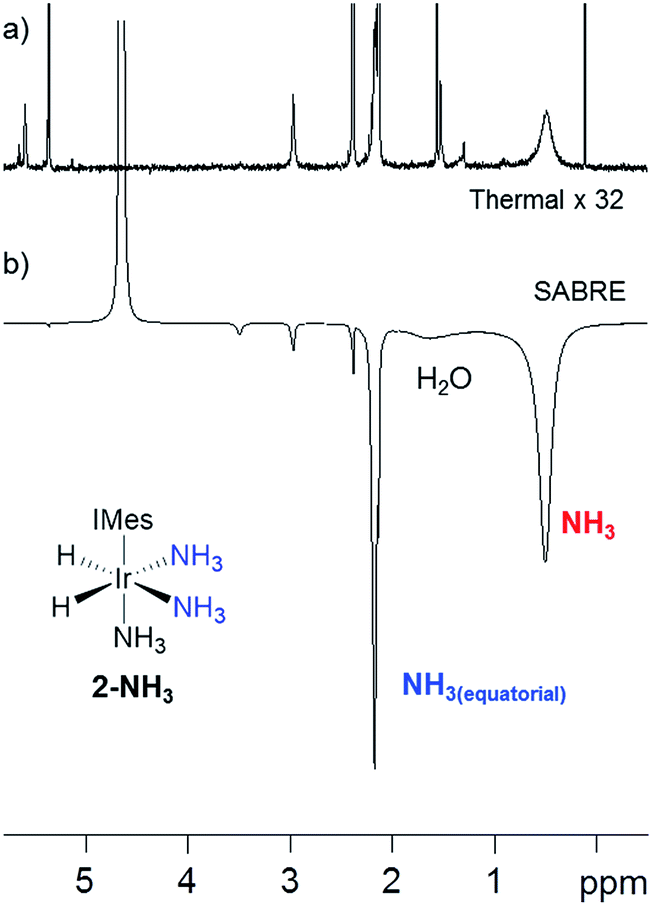 | ||
Fig. 1 (a) The thermally polarised 1H NMR SABRE spectrum (× 32 vertical expansion) recorded of 2-NH3 (formed by reaction of 1 with NH3 and H2) in dichloromethane-d2 at 298 K. (b) The corresponding SABRE polarised 9.4 T 1H NMR spectrum after transfer under p-H2 at 60 G. The hyperpolarised responses of free  and and  of 2-NH3 and residual H2O are indicated. of 2-NH3 and residual H2O are indicated. | ||
The SABRE-induced hyperpolarisation of benzylamine (BnNH2) was also investigated. A sample containing 1 (5 mM) and BnNH2 (10 eq.) in dichloromethane-d2 solution was exposed to 3 bar of H2. The immediate formation of [Ir(H)2(IMes)(BnNH2)3]Cl (2-BnNH2) was observed. It gives a characteristic hydride resonance in the 1H NMR spectrum at δ −23.97. Full characterisation data for this product is available in the ESI.† Interestingly, the 1H NMR spectrum of 2-BnNH2 showed that the BnNH2 ligand that lies trans to hydride, yields inequivalent responses for its NH2 protons at δ 4.92 and 2.30, and CH2 protons at δ 3.60 and 3.18. This is due to hindered rotation around the Ir–N bond which results in an up/down distinction for the resonances of the equatorial ligand. In contrast, the axial ligand yields single responses which are equivalent at δ 4.24 (NH2) and δ 3.83 (CH2) due to free rotation on the NMR timescale about the Ir–N bond. The corresponding EXSY-derived rate constant for equatorial BnNH2 loss from 2-BnNH2 was 3.33 s−1 while the rate of H2 loss was 2.83 s−1 at 298 K. Hence the rate of BnNH2 loss is higher than that of NH3 loss in 2-NH3. This difference is due to NH3 forming a stronger Ir–N bond as reflected in their relative pKb values and suggests that it might perform better under SABRE that NH3.
This was examined by p-H2-based polarisation transfer at 60 G which resulted in hyperpolarised free BnNH2 in solution. The signal enhancements were quantified to be 72-(NH2), 56-(CH2) and 194-fold (Ph) per proton as shown in Fig. 2a. However, by using d7-BnNH2 instead we were able to focus the SABRE polarisation into the two amino protons alone and this led to an improved signal enhancement of 916-fold per proton (Fig. 2b).
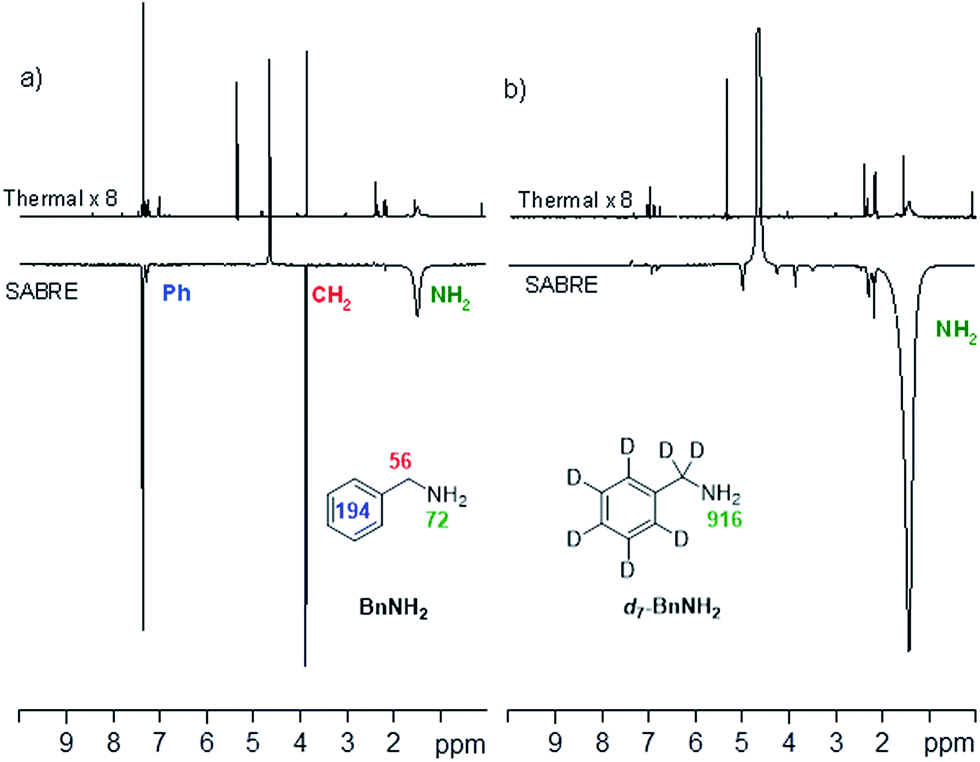 | ||
| Fig. 2 (a) 1H NMR spectra of BnNH2, thermally polarised, top, and hyperpolarised, bottom. (b) 1H NMR spectra for d7-BnNH2, thermally polarised, top, and hyperpolarised, bottom. | ||
In order to investigate the T1 contribution to this effect we determined values for BnNH2 and d7-BnNH2 at 9.4 T. BnNH2 proved to have effective T1 values of 1.1 s (NH2) and 4.7 s (CH2) respectively while its 2H-labelled variant exhibited a similar 1.1 s T1 value for the amino group in the presence of the active catalyst. Hence, the improved NH signal gain seen with d7-BnNH2 is due to a reduction in spin dilution which leads to more efficient SABRE transfer. The relaxation rates for BnNH2 and d7-BnNH2 are both slower in the absence of the active SABRE catalyst in agreement with earlier reports that the catalyst plays a role in reducing relaxation times due to reversible binding. Consequently, BnNH2 now shows T1 values of 9.0 s (NH2) and 11.0 s (CH2), whereas d7-BnNH2 has a T1 value of 10.1 s for its NH2 group.
Effect of catalyst to substrate ratio on SABRE polarisation
Previous studies have shown that the SABRE effect is dependent upon the catalyst to substrate ratio as a consequence of kinetic and relaxation effects.19,55 Therefore, we studied the effect of changing the ratio of BnNH2 relative to 1 from 4-fold to 20-fold in a series of further experiments, undertaking the associated SABRE transfer studies at 60 G and 298 K. It was found that similar total polarisation levels result within experimental error during these experiments (see ESI†). Hence, we conclude that the observed signal enhancements under these conditions are essentially independent of ligand excess which suggests that slow exchange and fast relaxation within the catalyst restrict the maximum polarisation level.Effect of p-H2 pressure on SABRE polarisation of BnNH2
As SABRE derives its polarisation from p-H2, it could be the limiting reagent in this catalytic process and therefore affect the observed substrate polarisation level.19 Up until this point, we have been utilising 3 bar pressure of p-H2 which reflects an ca. 6-fold excess when compared to the 50 mM substrate present in a 5 mm NMR tube. A sample containing 1 (5 mM), BnNH2 (50 mM, 10 eq.) in dichloromethane-d2 solution was therefore prepared and exposed to between 2 and 4 bar of p-H2. The resulting signal gains, after polarisation transfer at 60 G, are shown in Fig. S14 (see ESI†) and a strong dependence on p-H2 pressure is seen. This is consistent with the fact that H2 exchange takes place after ligand dissociation and the remaining equatorially bound BnNH2 ligand will experience a higher level of latent p-H2 polarisation (see Scheme 1). When d7-BnNH2 is examined with 4 bar of p-H2, the NH signal gain increases to 1079-fold per proton from the 916-fold signal gain achieved with 3 bar.Effect of temperature on SABRE polarisation of BnNH2
The temperature at which SABRE is conducted is also known to affect the efficiency of the polarisation transfer due to changes in the lifetime of the SABRE-active catalyst. We found here that cooling a dichloromethane-d2 solution containing 1, BnNH2 and 3 bar p-H2 to 288 K results in a reduction in the level of signal enhancement when compared to 298 K data (Fig. S15, ESI†). Conversely 308 K gave an improved response with the overall polarisation level increasing by ∼40%. This fits with the observed rate constant for BnNH2 dissociation increasing to 9.85 s−1 from the 3.33 s−1 value at 298 K. We therefore conclude the retained polarisation level in BnNH2 is improved by the faster rate of substrate dissociation and shorter catalyst lifetime. For NH3, a 251-fold 1H signal gain per NH proton is observed at 308 K when compared to the 154-fold value at 298 K. This is consistent with the increase in the NH3 dissociation rate constant to 10.42 s−1 at 308 K when compared to 1.64 s−1 at 298 K.SABRE transfer to 13C and 15N
SABRE-induced hyperpolarisation of 13C was also observed for BnNH2. Whilst polarisation transfer into the ortho phenyl carbon was readily observed using a standard 13C acquisition sequence after polarisation transfer 60 G under 4 bar p-H2, the other 13C resonances had poor signal-to-noise ratios. We overcame this by using a 1H–13C refocussed INEPT experiment that gave rise to a spectrum showing all 5 carbon environments after polarisation transfer at 60 G. We utilised long-range J-H–C-couplings to transfer this polarisation. 13C signal gains of up to 65-fold were achieved using this method (Fig. 3a). We further note that there is a very strong polarisation transfer field dependence on the BnNH213C signal intensities which is consistent with earlier reports on pyridine.30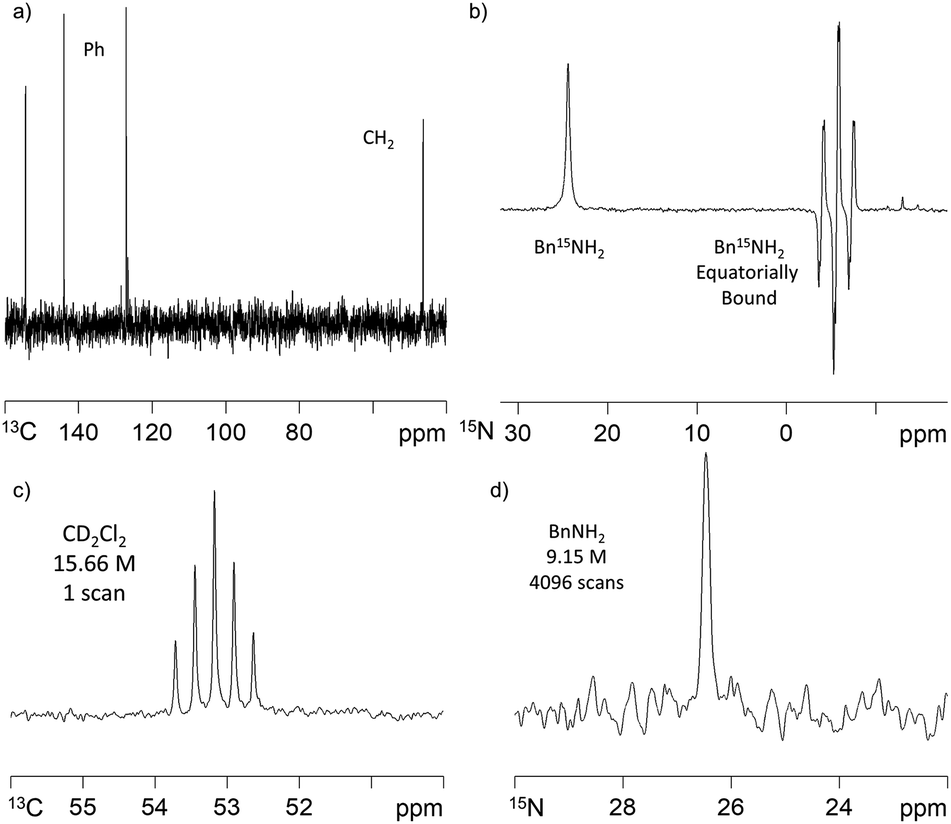 | ||
| Fig. 3 (a) 1H–13C refocussed INEPT NMR spectrum of hyperpolarised BnNH2 (35 mM) achieved via2-BnNH2 (5 mM) under SABRE in dichloromethane-d2 solution after transfer at 60 G and 308 K; (b) 15N NMR spectrum of 15N-labelled Bn15NH2 (35 mM) after SABRE transfer via2-BnNH2 (5 mM) at 60 G and 308 K which gives rise to hyperpolarised resonances for free (δ 24.42) and equatorially bound (δ −5.59) substrate; (c) single scan thermally polarised 13C NMR spectrum in CD2Cl2 (15.66 M) and (d) 4096 scan thermally polarised 15N NMR spectrum of BnNH2 (9.15 M). | ||
When Bn15NH2 is used instead of BnNH2, the detection of a hyperpolarised 15N response is readily evident as shown in Fig. 3b. The 15N signal gain for the free material in solution proved to be ∼880-fold after polarisation transfer at 60 G and 308 K. The equatorially bound 15N resonance at δ −5.59, is 4 times larger than the free amine signal. As the ratio of free amine to equatorially bound Bn15NH2 in solution is actually 7:2, the rate of Bn15NH2 loss must be relatively slow, even at 308 K. Under this 60 G condition, polarisation transfer is likely to occur via the 3JHH coupling between the Bn15NH2 and the hydride ligands. To investigate the effect of using a 2JHN coupling we repeated this measurement after polarization transfer within a μ-metal shield (ca. 350-fold shielding). Under these SABRE-SHEATH type conditions,14,15 an ∼800-fold 15N-signal gain was observed and further optimisation may therefore be needed to maximise this response. The corresponding 1H signal gains with this 15N labelled material after transfer at 60 G were now 33-(NH2), 34-(CH2) and 52-fold (Ph). These compare to the analogous values of 72-, 56- and 192-fold respectively with Bn14NH2. Interestingly, the 1H polarisation levels therefore decrease with 15N addition and we propose that this is an example of spin dilution.
Expanding the substrate range
In order to test the generality of amine polarisation via SABRE, we prepared a series of samples containing 1 (5 mM) and 10 eq. of the substrates shown in Fig. 4 in dichloromethane-d2 solution. These substrates include a number of primary amines and each is successfully hyperpolarised after transfer at 60 G upon reaction with 1 and p-H2. In fact, SABRE polarisation of phenylethylamine (PEA) and phenylpropylamine (PPA) results in strong signal enhancements and transfer is found to proceed across the corresponding C2 and C3 carbon chains into their phenyl rings. For PEA we found that the NH21H signal gain is actually increased to 108-fold per proton compared to the 72-fold BnNH2 value, and that the CH2CH2 bridge gave 50-fold (NC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2) and 45-fold (C2) enhancements per proton. The 5-proton containing phenyl group gave a 92-fold gain per proton.
2) and 45-fold (C2) enhancements per proton. The 5-proton containing phenyl group gave a 92-fold gain per proton.
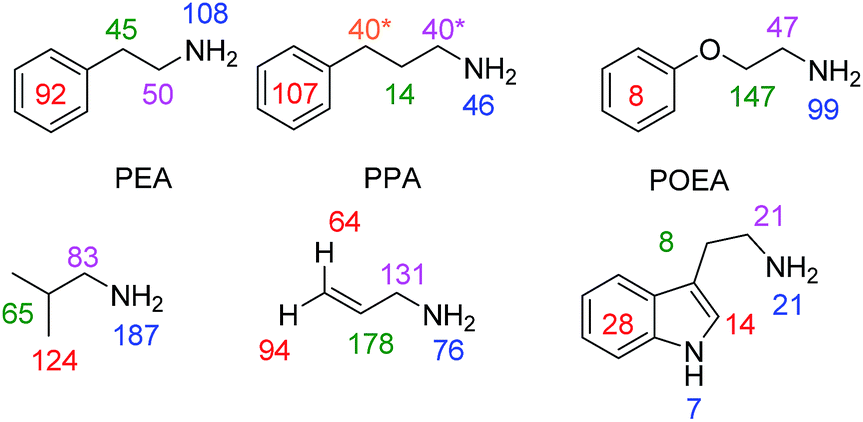 | ||
| Fig. 4 Amine substrates polarised by SABRE using precatalyst 1 in dichloromethane-d2 solution. Per proton signal gains are given for the indicated 1H sites (* average across two sites due to peak overlap) observed at 9.4 T. Corresponding 1H NMR spectra for thermally polarised and SABRE polarised experiments are given in the ESI.† | ||
Spin-isolation of the phenyl group, by introducing an ether linkage, as in phenoxyethylamine (POEA) resulted in signal enhancements of 99-(N2), 47-(NC2), 147-(C2O) and as expected, just 8-fold (Ph) per proton for our test sample. We therefore conclude that polarisation transfer across the oxygen linker is inefficient at 60 G and a stronger aliphatic proton response results. The amines isobutylamine, allylamine and tryptamine were also studied as shown in Fig. 4. In all cases, the formation of [Ir(H)2(IMes)(amine)3]Cl was indicated (see ESI†) and polarisation transfer results.
When secondary amines, such as dibenzylamine, were examined, no evidence for the formation of an active SABRE catalyst was observed. A similar result was observed for sterically hindered primary amines, such as isopropylamine and aromatic amines, such as aniline. Sterically demanding substrates, such as 2,6-lutidine, have been previously shown to be unable to be polarised using SABRE.56 A full list of the amines probed in this study is available in the ESI.† We therefore postulate that sterically demanding or electron deficient amines fail to activate and form the necessary [Ir(H)2(IMes)(amine)3]Cl SABRE catalyst.
This problem could be overcome for aniline by the addition of the co-ligand 1-methyl 1,2,3-triazole (mtz) or CH3CN. For the corresponding sample containing 1 (5 mM), aniline (10 eq.) and mtz (3 eq.) in dichloromethane-d2 we achieved signal enhancements of 51-fold for the NH2 group and 17-fold for the phenyl group, per proton. These signal gains are summarised in Fig. 5. When CH3CN (8 eq.) is used instead of mtz, the polarisation levels increase to 306-(NH2) and 193-fold (Ph) per proton. The active complex in this SABRE process was characterised as [Ir(H)2(IMes)(aniline)2(CH3CN)]Cl and yields a distinctive hydride resonance at δ −24.78 (see ESI†). Utilisation of such a co-substrate strategy was however unsuccessful for the secondary amines as detailed in the ESI.†
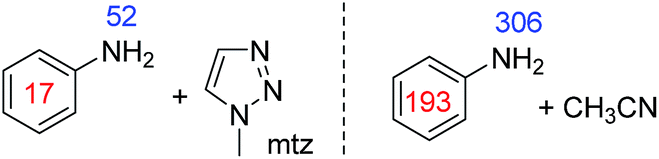 | ||
| Fig. 5 1H NMR signal gains per proton observed for the indicated aniline resonances when hyperpolarised by SABRE in the presence of the described co-ligand at 9.4 T. | ||
Indirect hyperpolarisation of amines by SABRE-RELAY
As expected, substrate binding to the metal centre is needed for polarisation transfer to occur. We hypothesised that these amines might also be hyperpolarised indirectly. In this scenario, hyperpolarisation of a primary amine or ammonia is achieved and subsequent proton exchange, which may be mediated by residual water, allows for a polarised proton to be shuttled into the non-SABRE-active amine. Subsequent intra-substrate polarisation transfer then relays the signal gain more widely in this agent (Scheme 2).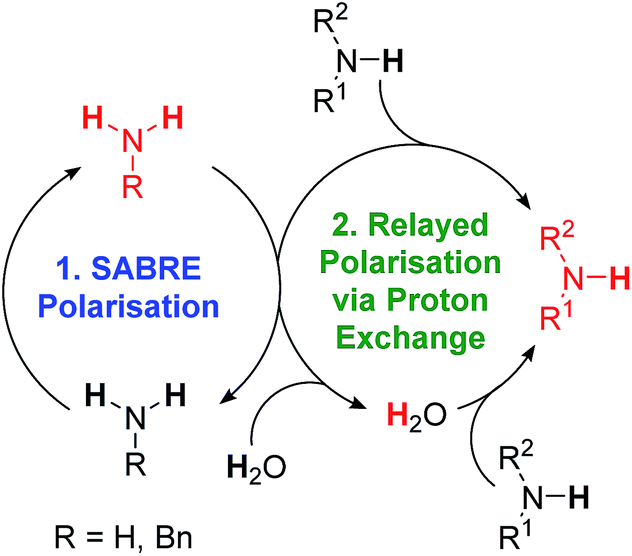 | ||
| Scheme 2 SABRE-RELAY polarisation of amines. (1) SABRE polarisation of an intermediary transfer agent, in this case a primary amine or ammonia. (2) Polarisation is then relayed into the target amine via proton exchange, either directly or via residual water present in the sample. | ||
In order to test this hypothesis, a series of samples containing 1 (5 mM), target amine (10 eq.) and NH3 (3–5 eq.) were prepared in dichloromethane-d2 solution. 2-NH3 formed in all cases as confirmed by the presence of a hydride resonance in the corresponding 1H NMR spectra at δ −23.8. Polarisation transfer was then conducted at 60 G, and the resulting signal gains that were observed at 9.4 T are presented in Fig. 6.
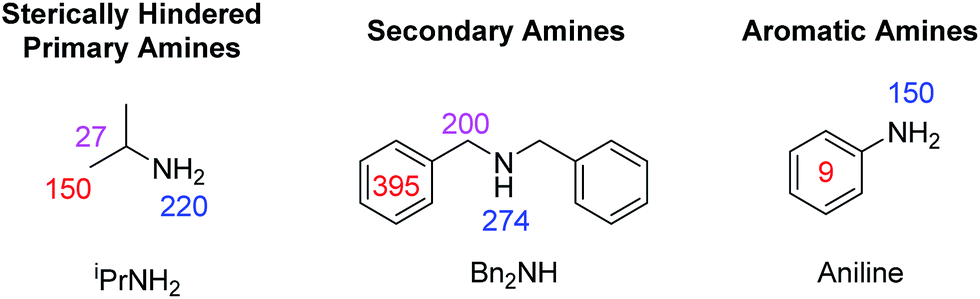 | ||
| Fig. 6 1H NMR signal gains observed per proton for the indicated amine resonances when hyperpolarised by SABRE-RELAY using 2-NH3 at 9.4 T. | ||
For isopropylamine (iPrNH2), the SABRE-RELAY polarised NH2 signal showed a 220-fold signal gain while 27- and 150-fold enhancements were seen for the CH and CH3 resonances respectively. This reflects a breakthrough as iPrNH2 was unable to be directly polarised by SABRE due to its steric bulk preventing adequate binding. Dibenzylamine (Bn2NH) was also successfully polarised using this method, and yields 1H signal gains of 274-(NH), 200-(CH2) and 395-fold (Ph) per proton. Additionally, a 13C spectrum can be acquired in a single scan on these materials after polarisation transfer at 60 G such that a 475-fold signal gain for the ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2 resonance is observed. Full NMR spectra are available in the ESI.† Furthermore, the aromatic amine, aniline, now exhibits a 150-fold NH2 proton signal enhancement and a 9-fold signal gain for the phenyl ring under analogous conditions. We note that these signal gains are lower than those seen when CH3CN is used as a co-ligand to achieve direct SABRE transfer as detailed in Fig. 5. We suggest that this difference in behaviour arises because a 60 G polarisation transfer field is no-longer optimal for intra-molecular polarisation transfer after proton exchange. This is clearly is not the case for transfer via directly bound aniline and the complexes scalar coupling network which is in fact commonly maximised for 1H transfer at 60 G.
H2 resonance is observed. Full NMR spectra are available in the ESI.† Furthermore, the aromatic amine, aniline, now exhibits a 150-fold NH2 proton signal enhancement and a 9-fold signal gain for the phenyl ring under analogous conditions. We note that these signal gains are lower than those seen when CH3CN is used as a co-ligand to achieve direct SABRE transfer as detailed in Fig. 5. We suggest that this difference in behaviour arises because a 60 G polarisation transfer field is no-longer optimal for intra-molecular polarisation transfer after proton exchange. This is clearly is not the case for transfer via directly bound aniline and the complexes scalar coupling network which is in fact commonly maximised for 1H transfer at 60 G.
From these results we can conclude that the SABRE-RELAY effect is able to polarise sterically hindered primary amines, secondary amines and aromatic amines that are not themselves accessible to SABRE. Thus, the scope of amine polarisation is vastly increased.
Conclusions
In summary, we have shown here how SABRE can be used to hyperpolarise a series of primary amines. This substrate extension opens up the SABRE approach to operate with a much wider range of analytes than was previously thought possible, as we extend beyond the original aromatic N-heterocycles, imines and nitriles. Activity is achieved by the formation of a series of complexes of the form [Ir(H)2(IMes)(amine)3]Cl. Relaxation studies, in conjunction with ligand dissociation rate measurements were used to demonstrate that the high relative stability of these complexes acts to limit the degree of SABRE signal gain. This hypothesis is consistent with the fact that increasing the p-H2 pressure or reaction temperature leads to improved signal gains. Therefore, significant catalyst optimisation will be important if very high levels of hyperpolarisation are to be achieved by this route in the future.Nonetheless, in the case of BnNH2, 1H NMR signal enhancement values of ∼100-fold per NH proton were achieved for benzylamine using [IrCl(COD)(IMes)]. Consequently, when d7-benzylamine was used, the resulting focusing of the hyperpolarisation into the NH2 resonance resulted in a 900-fold signal enhancement per proton at 9.4 T with a p-H2 pressure of 3 bar. This value reduced to 33-fold for Bn15NH2 after transfer at 60 G. Hence, we predict that further improvements can be made through a more detailed study of the effect of isotopic labelling.18,19,57 We have also demonstrated transfer to 13C and 15N with diagnostic NMR spectra being collected at a 35 mM concentration in a single scan. We predict that application of high-field SABRE transfer techniques,34–37,39 such as the LIGHT-SABRE38 approach, might subsequently enable this process to work inside the magnet, but note that a rigorous study of the effect the polarisation transfer field plays on the resulting signal enhancement levels is justified.
In the course of these studies we found that sterically hindered primary amines, secondary amines and aromatic amines were unable to form an active SABRE catalyst of the type [Ir(H)2(IMes)(amine)3]Cl. This meant that direct polarisation transfer via such a complex was not possible. We found for aniline that the addition of a co-ligand such as CH3CN overcame this problem via the formation of [Ir(H)2(IMes)(aniline)2(CH3CN)]Cl such that signal enhancements of up to 306-fold per NH proton could be achieved.
An indirect route was described to overcome this limitation more generally, such that hindered primary amines, secondary amines and aromatic amines can be hyperpolarised by SABRE-RELAY.54 Now, a SABRE-hyperpolarised intermediary, such as ammonia, is able to readily transfer polarisation into agents such as isopropylamine, benzylamine and aniline via either direct proton exchange or mediated by residual water present in the sample. This approach expands the range of amines that can be hyperpolarised without changing their chemical identity through interactions with p-H2.
Given the increase in signal intensity that is observed for the amines in this study, we are now working towards their use as agents for mechanistic study58–64 in transfer hydrogenation,65,66 hydroamination,67,68 and vitally important N2 fixation reactions.69–71 Additionally, since phenylethylamine is a naturally occurring monoamine based alkaloid that acts as a promoter of catecholamine (dopamine and norepinephrine) release in plants and animals we expect these observations to be of wide interest.72,73 Furthermore, the SABRE-RELAY method54 has recently been shown to offer a route to hyperpolarise an even larger range of hydrogen transfer acceptors using OH functional groups. Optimisation of the intermediaries NH polarisation level reflects a key part to optimisation of this technique and hence these results will be of interest to any potential developer.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by The Wellcome Trust (Grants 092506 and 098335), the EPSRC (EP/R51181X/1) and the University of York.References
- S. B. Duckett and R. E. Mewis, Acc. Chem. Res., 2012, 45, 1247–1257 CrossRef CAS PubMed.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- J. Natterer and J. Bargon, Prog. Nucl. Magn. Reson. Spectrosc., 1997, 31, 293–315 CrossRef.
- R. A. Green, R. W. Adams, S. B. Duckett, R. E. Mewis, D. C. Williamson and G. G. R. Green, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 67, 1–48 CrossRef CAS PubMed.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645–2648 CrossRef CAS PubMed.
- J. Colell, P. Türschmann, S. Glöggler, P. Schleker, T. Theis, M. Ledbetter, D. Budker, A. Pines, B. Blümich and S. Appelt, Phys. Rev. Lett., 2013, 110, 137602 CrossRef PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. Lopez-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- P. J. Rayner and S. Duckett, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201710406.
- R. W. Adams, S. B. Duckett, R. A. Green, D. C. Williamson and G. G. R. Green, J. Chem. Phys., 2009, 131, 194505 CrossRef PubMed.
- A. N. Pravdivtsev, A. V. Yurkovskaya, H. M. Vieth, K. L. Ivanov and R. Kaptein, ChemPhysChem, 2013, 14, 3327–3331 CrossRef CAS PubMed.
- A. N. Pravdivtsev, K. L. Ivanov, A. V. Yurkovskaya, P. A. Petrov, H. H. Limbach, R. Kaptein and H. M. Vieth, J. Magn. Reson., 2015, 261, 73–82 CrossRef CAS PubMed.
- K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, J. Lopez-Serrano and A. C. Whitwood, J. Am. Chem. Soc., 2009, 131, 13362–13368 CrossRef CAS PubMed.
- N. Eshuis, R. Aspers, B. J. A. van Weerdenburg, M. C. Feiters, F. Rutjes, S. S. Wijmenga and M. Tessari, J. Magn. Reson., 2016, 265, 59–66 CrossRef CAS PubMed.
- M. L. Truong, T. Theis, A. M. Coffey, R. V. Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Phys. Chem. C, 2015, 119, 8786–8797 CAS.
- T. Theis, M. L. Truong, A. M. Coffey, R. V. Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Am. Chem. Soc., 2015, 137, 1404–1407 CrossRef CAS PubMed.
- R. V. Shchepin, L. Jaigirdar, T. Theis, W. S. Warren, B. M. Goodson and E. Y. Chekmenev, J. Phys. Chem. C, 2017, 121, 28425–28434 CAS.
- M. J. Cowley, R. W. Adams, K. D. Atkinson, M. C. R. Cockett, S. B. Duckett, G. G. R. Green, J. A. B. Lohman, R. Kerssebaum, D. Kilgour and R. E. Mewis, J. Am. Chem. Soc., 2011, 133, 6134–6137 CrossRef CAS PubMed.
- M. Fekete, O. Bayfield, S. B. Duckett, S. Hart, R. E. Mewis, N. Pridmore, P. J. Rayner and A. Whitwood, Inorg. Chem., 2013, 52, 13453–13461 CrossRef CAS PubMed.
- P. J. Rayner, M. J. Burns, A. M. Olaru, P. Norcott, M. Fekete, G. G. R. Green, L. A. R. Highton, R. E. Mewis and S. B. Duckett, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3188–E3194 CrossRef CAS PubMed.
- R. V. Shchepin, D. A. Barskiy, D. M. Mikhaylov and E. Y. Chekmenev, Bioconjugate Chem., 2016, 27, 878–882 CrossRef CAS PubMed.
- H. F. Zeng, J. D. Xu, J. Gillen, M. T. McMahon, D. Artemov, J. M. Tyburn, J. A. B. Lohman, R. E. Mewis, K. D. Atkinson, G. G. R. Green, S. B. Duckett and P. C. M. van Zijl, J. Magn. Reson., 2013, 237, 73–78 CrossRef CAS PubMed.
- P. Norcott, P. J. Rayner, G. G. R. Green and S. B. Duckett, Chem.–Eur. J., 2017, 23, 16990–16997 CrossRef CAS PubMed.
- D. A. Barskiy, R. V. Shchepin, A. M. Coffey, T. Theis, W. S. Warren, B. M. Goodson and E. Y. Chekmenev, J. Am. Chem. Soc., 2016, 138, 8080–8083 CrossRef CAS PubMed.
- E. B. Ducker, L. T. Kuhn, K. Munnemann and C. Griesinger, J. Magn. Reson., 2012, 214, 159–165 CrossRef PubMed.
- A. W. J. Logan, T. Theis, J. F. P. Colell, W. S. Warren and S. J. Malcolmson, Chem.–Eur. J., 2016, 22, 10777–10781 CrossRef CAS PubMed.
- T. Theis, G. X. Ortiz, A. W. J. Logan, K. E. Claytor, Y. Feng, W. P. Huhn, V. Blum, S. J. Malcolmson, E. Y. Chekmenev, Q. Wang and W. S. Warren, Sci. Adv., 2016, 2, e1501438 Search PubMed.
- R. E. Mewis, R. A. Green, M. C. R. Cockett, M. J. Cowley, S. B. Duckett, G. G. R. Green, R. O. John, P. J. Rayner and D. C. Williamson, J. Phys. Chem. B, 2015, 119, 1416–1424 CrossRef CAS PubMed.
- R. E. Mewis, K. D. Atkinson, M. J. Cowley, S. B. Duckett, G. G. R. Green, R. A. Green, L. A. R. Highton, D. Kilgour, L. S. Lloyd, J. A. B. Lohman and D. C. Williamson, Magn. Reson. Chem., 2014, 52, 358–369 CrossRef CAS PubMed.
- J. F. P. Colell, A. W. J. Logan, Z. J. Zhou, R. V. Shchepin, D. A. Barskiy, G. X. Ortiz, Q. Wang, S. J. Malcolmson, E. Y. Chekmenev, W. S. Warren and T. Theis, J. Phys. Chem. C, 2017, 121, 6626–6634 CAS.
- D. A. Barskiy, R. V. Shchepin, C. P. N. Tanner, J. F. P. Colell, B. M. Goodson, T. Theis, W. S. Warren and E. Y. Chekmenev, ChemPhysChem, 2017, 18, 1493–1498 CrossRef CAS PubMed.
- V. V. Zhivonitko, I. V. Skovpin and I. V. Koptyug, Chem. Commun., 2015, 51, 2506–2509 RSC.
- M. J. Burns, P. J. Rayner, G. G. R. Green, L. A. R. Highton, R. E. Mewis and S. B. Duckett, J. Phys. Chem. B, 2015, 119, 5020–5027 CrossRef CAS PubMed.
- A. M. Olaru, A. Burt, P. J. Rayner, S. J. Hart, A. C. Whitwood, G. G. R. Green and S. B. Duckett, Chem. Commun., 2016, 52, 14482–14485 RSC.
- K. D. Atkinson, M. J. Cowley, S. B. Duckett, P. I. P. Elliott, G. G. R. Green, J. Lopez-Serrano, I. G. Khazal and A. C. Whitwood, Inorg. Chem., 2009, 48, 663–670 CrossRef CAS PubMed.
- D. A. Barskiy, K. V. Kovtunov, I. V. Koptyug, P. He, K. A. Groome, Q. A. Best, F. Shi, B. M. Goodson, R. V. Shchepin, A. M. Coffey, K. W. Waddell and E. Y. Chekmenev, J. Am. Chem. Soc., 2014, 136, 3322–3325 CrossRef CAS PubMed.
- S. Knecht, A. S. Kiryutin, A. V. Yurkovskaya and K. L. Ivanov, J. Magn. Reson., 2018, 287, 74–81 CrossRef CAS PubMed.
- S. S. Roy, G. Stevanato, P. J. Rayner and S. B. Duckett, J. Magn. Reson., 2017, 285, 55–60 CrossRef CAS PubMed.
- T. Theis, M. Truong, A. M. Coffey, E. Y. Chekmenev and W. S. Warren, J. Magn. Reson., 2014, 248, 23–26 CrossRef CAS PubMed.
- A. N. Pravdivtsev, A. V. Yurkovskaya, H.-M. Vieth and K. L. Ivanov, J. Phys. Chem. B, 2015, 119, 13619–13629 CrossRef CAS PubMed.
- M. Carravetta, O. G. Johannessen and M. H. Levitt, Phys. Rev. Lett., 2004, 92, 1177–1182 CrossRef PubMed.
- T. Theis, G. X. Ortiz, A. W. J. Logan, K. E. Claytor, Y. Feng, W. P. Huhn, V. Blum, S. J. Malcolmson, E. Y. Chekmenev, Q. Wang and W. S. Warren, Sci. Adv., 2016, 2, e1501438 Search PubMed.
- A. M. Olaru, S. S. Roy, L. S. Lloyd, S. Coombes, G. G. R. Green and S. B. Duckett, Chem. Commun., 2016, 52, 7842–7845 RSC.
- S. S. Roy, P. Norcott, P. J. Rayner, G. G. R. Green and S. B. Duckett, Angew. Chem., Int. Ed., 2016, 55, 15642–15645 CrossRef CAS PubMed.
- S. S. Roy, P. J. Rayner, P. Norcott, G. G. R. Green and S. B. Duckett, Phys. Chem. Chem. Phys., 2016, 18, 24905–24911 RSC.
- S. S. Roy, P. Norcott, P. J. Rayner, G. G. R. Green and S. B. Duckett, Chem.–Eur. J., 2017, 23, 10496–10500 CrossRef CAS PubMed.
- Z. Zhou, J. Yu, J. F. P. Colell, R. Laasner, A. Logan, D. A. Barskiy, R. V. Shchepin, E. Y. Chekmenev, V. Blum, W. S. Warren and T. Theis, J. Phys. Chem. Lett., 2017, 8, 3008–3014 CrossRef CAS PubMed.
- V. Daniele, F.-X. Legrand, P. Berthault, J.-N. Dumez and G. Huber, ChemPhysChem, 2015, 16, 3413–3417 CrossRef CAS PubMed.
- N. Eshuis, B. J. A. van Weerdenburg, M. C. Feiters, F. P. J. T. Rutjes, S. S. Wijmenga and M. Tessari, Angew. Chem., Int. Ed., 2015, 54, 1481–1484 CrossRef CAS PubMed.
- L. S. Lloyd, R. W. Adams, M. Bernstein, S. Coombes, S. B. Duckett, G. G. R. Green, R. J. Lewis, R. E. Mewis and C. J. Sleigh, J. Am. Chem. Soc., 2012, 134, 12904–12907 CrossRef CAS PubMed.
- I. Reile, R. L. E. G. Aspers, J.-M. Tyburn, J. G. Kempf, M. C. Feiters, F. P. J. T. Rutjes and M. Tessari, Angew. Chem., Int. Ed., 2017, 56, 9174–9177 CrossRef CAS PubMed.
- W. N. O. Wylie, A. J. Lough and R. H. Morris, Organometallics, 2013, 32, 3808–3818 CrossRef.
- Y. Sato, Y. Kayaki and T. Ikariya, Organometallics, 2016, 35, 1257–1264 CrossRef CAS.
- M.-L. Yuan, J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, ACS Catal., 2016, 6, 3665–3669 CrossRef CAS.
- W. Iali, P. J. Rayner and S. B. Duckett, Sci. Adv., 2018, 4, eaa06250 Search PubMed.
- L. S. Lloyd, A. Asghar, M. J. Burns, A. Charlton, S. Coombes, M. J. Cowley, G. J. Dear, S. B. Duckett, G. R. Genov, G. G. R. Green, L. A. R. Highton, A. J. J. Hooper, M. Khan, I. G. Khazal, R. J. Lewis, R. E. Mewis, A. D. Roberts and A. J. Ruddlesden, Catal. Sci. Technol., 2014, 4, 3544–3554 CAS.
- R. V. Shchepin, M. L. Truong, T. Theis, A. M. Coffey, F. Shi, K. W. Waddell, W. S. Warren, B. M. Goodson and E. Y. Chekmenev, J. Phys. Chem. Lett., 2015, 6, 1961–1967 CrossRef CAS PubMed.
- M. Fekete, P. J. Rayner, G. G. R. Green and S. B. Duckett, Magn. Reson. Chem., 2017, 55, 944–957 CrossRef CAS PubMed.
- O. G. Salnikov, K. V. Kovtunov, D. A. Barskiy, A. K. Khudorozhkov, E. A. Inozemtseva, I. P. Prosvirin, V. I. Bukhtiyarov and I. V. Koptyug, ACS Catal., 2014, 4, 2022–2028 CrossRef CAS.
- M. Leutzsch, L. M. Wolf, P. Gupta, M. Fuchs, W. Thiel, C. Fares and A. Furstner, Angew. Chem., Int. Ed., 2015, 54, 12431–12436 CrossRef CAS PubMed.
- R. V. Shchepin, D. A. Barskiy, A. M. Coffey, B. M. Goodson and E. Y. Chekmenev, ChemistrySelect, 2016, 1, 2552–2555 CrossRef CAS PubMed.
- C. Godard, S. B. Duckett, S. Polas, R. Tooze and A. C. Whitwood, Dalton Trans., 2009, 2496–2509 RSC.
- D. J. Fox, S. B. Duckett, C. Flaschenriem, W. W. Brennessel, J. Schneider, A. Gunay and R. Eisenberg, Inorg. Chem., 2006, 45, 7197–7209 CrossRef CAS PubMed.
- D. Blazina, S. B. Duckett, P. J. Dyson and J. A. B. Lohman, Angew. Chem., Int. Ed., 2001, 40, 3874–3877 CrossRef CAS PubMed.
- S. A. Colebrooke, S. B. Duckett, J. A. B. Lohman and R. Eisenberg, Chem.–Eur. J., 2004, 10, 2459–2474 CrossRef CAS PubMed.
- J. S. M. Samec, J. E. Backvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC.
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS.
- M. Patel, R. K. Saunthwal and A. K. Verma, Acc. Chem. Res., 2017, 50, 240–254 CrossRef CAS PubMed.
- T. E. Muller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
- H. P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- S. Kuriyama, K. Arashiba, K. Nakajima, H. Tanaka, N. Kamaru, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2014, 136, 9719–9731 CrossRef CAS PubMed.
- R. T. Premont, R. R. Gainetdinov and M. G. Caron, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9474–9475 CrossRef CAS PubMed.
- I. A. Paterson, A. V. Juorio and A. A. Boulton, J. Neurochem., 1990, 55, 1827–1837 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, characterisation data and example spectra. See DOI: 10.1039/c8sc00526e |
| This journal is © The Royal Society of Chemistry 2018 |