
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pathway complexity in the self-assembly of a zinc chlorin model system of natural bacteriochlorophyll J-aggregates†
Soichiro
Ogi‡
ab,
Charlotte
Grzeszkiewicz
a and
Frank
Würthner
 *ab
*ab
aUniversität Würzburg, Institut für Organische Chemie, Am Hubland, 97074 Würzburg, Germany. E-mail: wuerthner@uni-wuerzburg.de
bUniversität Würzburg, Center for Nanosystems Chemistry (CNC), Bavarian Polymer Institute (BPI), Theodor-Boveri-Weg, 97074 Würzurg, Germany
First published on 14th February 2018
Abstract
Whilst bacteriochlorophyll c, d, and e dyes self-assemble into the most efficient light harvesting J-aggregate systems found in nature, their supramolecular packing arrangements are still a matter of debate and a significant number of models have been suggested for their local and long-range ordering. Here we reveal for a synthetic model system based on a zinc chlorin (ZnChl) dye an intriguing interplay of two competing aggregation pathways by kinetic and thermodynamic studies in MeOH/water solvent mixtures: the formation of kinetically controlled off-pathway nanoparticles consisting of excitonically coupled J-dimers versus the formation of thermodynamically more stable one-dimensional helical fibers consisting of J-coupled extended aggregates. The higher order of the latter is evidenced by atomic force microscopy and a more narrow absorption spectrum of the J-aggregates. Based on a recently developed thermodynamic model that combines the cooperative K2–K growth model with a competing dimerization model, an energy landscape could be derived that describes the pathway complexity of this biomimetic system. Our studies reveal that the kinetic stability of the off-pathway nanoparticles increases with increasing concentration of ZnChl or water content in a MeOH/water solvent mixture. For a water content >90% deeply trapped off-pathway nanoparticle products are formed that do not transform anymore to the more ordered thermodynamic product within reasonable time scales. Based on these observations, we hypothesize that out-of-equilibrium aggregate structures of natural BChl dyes may also exist in the natural chlorosomes of green bacteria.
Introduction
Despite the vast amount of research accomplished in the last few decades on self-assembly,1 there is still an apparent gap between the mechanistic understanding of self-assembly processes of synthetic systems so far achieved2 and those that occur in nature, e.g. the formation of tobacco mosaic virus,3 amyloid fibers4 or biological bilayer membranes.5 A prime example where chemistry and biology meet is that of chlorosomes.6 These are light-harvesting antenna systems of green bacteria composed of self-assembled metallochlorin dyes, in particular bacteriochlorophylls (BChls) c, d, and e that possess a chlorophyll scaffold bearing a hydroxyl group at the 31-position and a metal ion at the center (Fig. 1a).6c,7 The hydroxyl group is responsible for the slipped π-stacking arrangement into J-aggregates upon self-assembly of these natural BChl dyes driven by coordinative bonding to the metal ion of the neighbouring tetrapyrrole scaffold.8 Whilst this particular interaction is undisputed, the macroscopic morphology (sheets, tubes, and onions) and specific molecular arrangement of BChl dyes (parallel and antiparallel stacking arrangement, see Fig. 1b) in natural chlorosomal self-assemblies have been controversially discussed in the literature for decades.9 However, so far little attention has been paid to the self-assembly mechanism10 and the possibility of competing self-assembly pathways for natural BChl dyes7,8 or their semi-synthetic metallochlorin model compounds.11 The reason might be that it is very challenging to simulate the conditions that occur in natural environments and that many of the mathematical models for the characterization of self-assembly processes became available only in recent years.2 | ||
| Fig. 1 (a) Chemical structures of the semi-synthetic model compound ZnChl 1 and natural BChl c, d, and e molecules. (b) Schematic illustration of the antiparallel (left) and the parallel (right) one-dimensional stacking arrangement proposed for BChl c, d, and e aggregates. | ||
Motivated by significant recent progress in the mechanistic understanding of aggregation processes,2 we here elucidate the self-assembly mechanism of a semi-synthetic zinc chlorin model compound (ZnChl 1)11d (Fig. 1a) by kinetic and thermodynamic studies. This model compound is, like natural BChls, inherently chiral and contains a hydroxyl group at the 31-position and a central metal ion. We have previously reported that the hydroxyl group confines the self-assembly of ZnChl 1 into one-dimensional aggregates through the formation of coordinative bonds with the central zinc ions of neighbour molecules and directs the further growth of the one-dimensional stacks into tubes by hydrogen bonding to carbonyl groups of other stacks.8b,11d
We are aware that the self-assembly pathways given in natural chlorosomes are more complex than those of semi-synthetic model systems due to the presence of different BChl dyes, including variations in diastereomers,7b,11a,12,13 and additional possibilities of growth and the embedding of the natural dyes within a lipid monolayer membrane. However, due to the structural similarity of the model compound ZnChl 1 to natural BChls our present mechanistic exploration will also shed light on the natural counterparts, in particular with regard to the first level of hierarchical growth, i.e. the formation of slipped J-aggregate π-stacks. Here we report that ZnChl 1 self-assembles in methanol (MeOH)/water solvent mixtures into kinetically trapped nanoparticle aggregates that are structurally and spectroscopically distinct from the thermodynamically equilibrated J-aggregate helical fibers. At high concentrations of ZnChl 1 and with a high water content in the solvent mixtures the out-of-equilibrium nanoparticles prevail for a very long time as a deeply trapped state. Thermodynamic analysis performed for the higher methanol content, where equilibration is fast, suggests that the nanoparticle aggregates consisting of antiparallel ZnChl dimers are an off-pathway product and one-dimensional fibril aggregates with parallel stacks are a thermodynamic product.
Results and discussion
The self-assembly behaviour of ZnChl 1 was investigated by solvent-dependent UV/vis absorption and circular dichroism (CD) spectroscopy. For the sample preparation, ZnChl 1 was dissolved in methanol and then deionized water was added to the monomer solution in methanol until the water content reached 50–80 vol% with a total concentration (cT) of 1 × 10−5 M. The absorption spectrum of 1 in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 MeOH/water mixture showed a molecularly dissolved state (1mono) with the characteristic Soret band at 428 nm and the Qy band at 656 nm (Fig. 2a, orange line).11d Upon increasing the volume ratio of water in the MeOH/water mixture from 50:50 to 20:80, the monomer Qy band with a full width at half maximum (FWHM) of 450 cm−1 decreased and a new broad Qy band (FWHM = 850 cm−1) appeared at 738 nm (Fig. 2a, green line). The bathochromic shift of the Qy absorption maximum by 82 nm in the 20:80 MeOH/water mixture was accompanied by the appearance of bisignate CD signals with positive and negative maxima at around 725 and 749 nm, respectively (Fig. 2a, green line). These spectral features are indicative of the transition from 1mono to a J-aggregate state (denoted as 1J1) with chiral excitonic coupling14 of the Qy transition dipole moments.
:50 MeOH/water mixture showed a molecularly dissolved state (1mono) with the characteristic Soret band at 428 nm and the Qy band at 656 nm (Fig. 2a, orange line).11d Upon increasing the volume ratio of water in the MeOH/water mixture from 50:50 to 20:80, the monomer Qy band with a full width at half maximum (FWHM) of 450 cm−1 decreased and a new broad Qy band (FWHM = 850 cm−1) appeared at 738 nm (Fig. 2a, green line). The bathochromic shift of the Qy absorption maximum by 82 nm in the 20:80 MeOH/water mixture was accompanied by the appearance of bisignate CD signals with positive and negative maxima at around 725 and 749 nm, respectively (Fig. 2a, green line). These spectral features are indicative of the transition from 1mono to a J-aggregate state (denoted as 1J1) with chiral excitonic coupling14 of the Qy transition dipole moments.
 | ||
| Fig. 2 (a) Solvent-dependent CD and UV/vis absorption spectra of freshly prepared solutions of 1 in MeOH/water solvent mixtures with volume ratios of 50:50 (orange lines), 40:60 (blue lines), 30:70 (pink lines), and 20:80 (green lines) at a concentration of 1 × 10−5 M and a temperature of 293 K. The black lines indicate CD and UV/vis absorption spectra of ZnChl 1 in a 30:70 MeOH/water solvent mixture observed after 24 h at 293 K (cT = 1 × 10−5 M). (b and c) AFM height images of (b) 1J1 spin-coated onto a silicon wafer from a freshly prepared solution of ZnChl 1 in 30:70 MeOH/water and (c) 1J2 spin-coated onto a silicon wafer from this solution stored in the absence of light for a few weeks. The scale bar is 500 nm. The z scales are (b) 5 nm and (c) 4.5 nm. | ||
Interestingly, the absorption and CD spectra of 1J1 further changed over time with a higher water content of 30:70 in the MeOH/water mixture at a concentration of 1 × 10−5 M. Thus, the Qy absorption band of 1J1 at 738 nm was bathochromically shifted to 749 nm with a pronounced increase of intensity and a narrowing of the band (FWHM = 510 cm−1) after 24 h (Fig. 2a, black line). Additionally, a strongly intensified CD couplet was observed at 745 and 760 nm as positive and negative maxima, respectively. The time-dependent spectral changes clearly indicate that 1J1 is a kinetic self-assembly product, which is transformed into a thermodynamically equilibrated J-aggregate state (denoted as 1J2). This transformation from 1J1 to 1J2 is characterized by the anisotropy factor g (defined as Δε/ε) at 745 nm that increased from −2.7 × 10−3 to 9.4 × 10−3. The strong increase of the g value during the transformation from 11J to 1J2 suggests a different spatial arrangement of the dyes and a more extended and highly ordered aggregate structure in the thermodynamically equilibrated 1J2 state. Indeed, atomic force microscopy (AFM) images of a freshly prepared solution of 1J1 in the 30:70 MeOH/water mixture showed self-assembled nanoparticles (Fig. 2b), while fibrous nanostructures with a unimolecular width were observed for the 1J2 sample (Fig. 2c and S1 in the ESI†). Taking into account the structural proposals discussed in the literature for aggregates of similar metallochlorins based on UV/vis and CD spectroscopic studies,11 as well as scanning tunneling microscopy studies,15 it is reasonable to relate the 1J1 state to closely π-stacked antiparallel dimers (small CD effects are due to very small rotational displacements of the transition dipole moments) whose further aggregation into nanoparticles lacks high order. In contrast, the 1J2 state with its elongated helical fibrous structure, large CD amplitude and much sharper UV/vis absorption band is in accordance with the widely favoured parallel π-stack models (compare Fig. 1b) for natural BChl self-assemblies in the chlorosome,9 where the one-dimensional metallo-supramolecular chain is directed into a helical arrangement by the inherent chirality of the chlorin scaffold.
To gain insight into the transformation mechanism from 1J1 into 1J2, the aggregation kinetics at a temperature of 293 K was evaluated by monitoring the time-dependent changes of the UV/vis and CD spectra (ε and g values, respectively) at different total concentrations of ZnChl 1 in a 30:70 MeOH/water mixture (Fig. 3 and S2 in the ESI†). An important finding of this kinetic study is that the initial transformation rate decreases with an increased total concentration of 1 (Table S1 in the ESI†). This concentration dependence of the kinetics resembles those previously reported for porphyrin and perylene bisimide aggregate systems, in which the nanoparticles are off-pathway intermediates with regard to the thermodynamically stable supramolecular polymer,16 and is opposed to a system in which the nanoparticles are on-pathway aggregates that further grow into nanosheets.17 Thus, our concentration-dependent data support the conclusion that the kinetically formed 1J1 nanoparticle state is an off-pathway non-equilibrium aggregate,18 which is not used as a building block for the formation of thermodynamically stable fibrous J-aggregate 1J2.
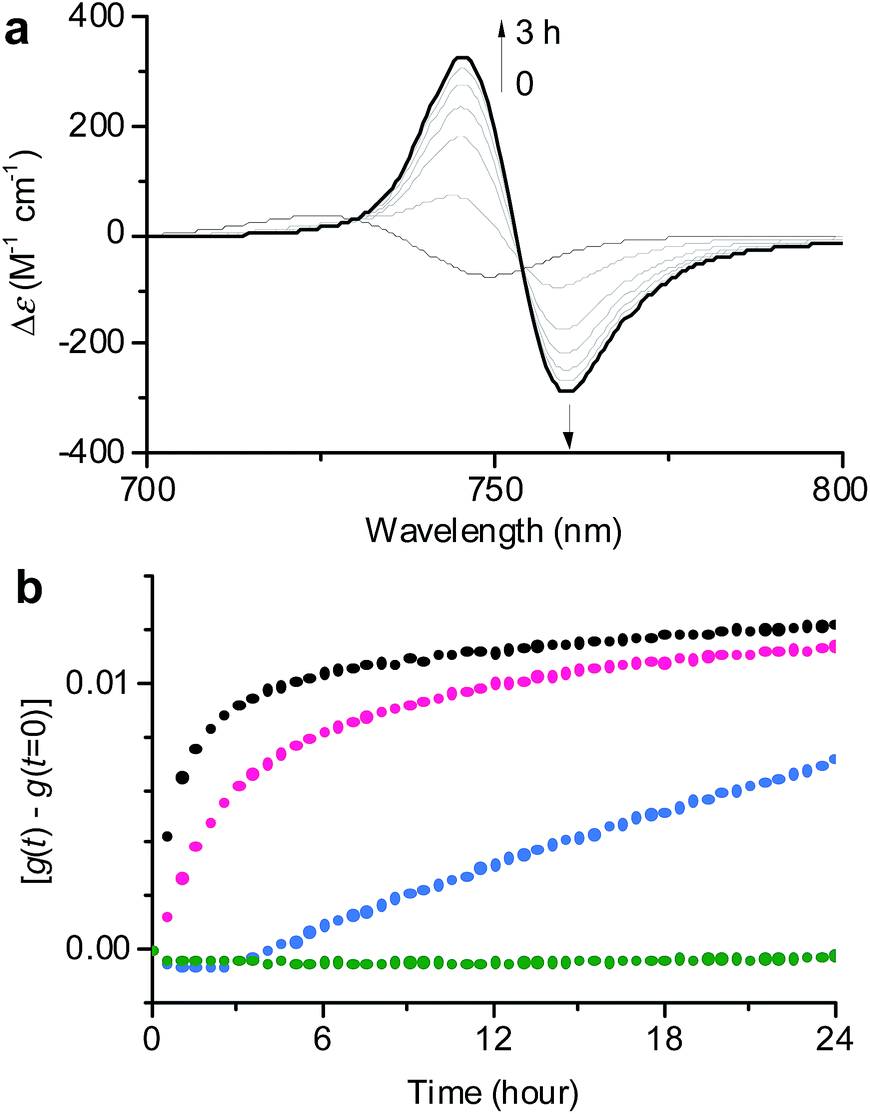 | ||
| Fig. 3 (a) Time-dependent CD spectral changes showing the transformation from 1J1 to 1J2 in a 30:70 MeOH/water solvent mixture at cT = 1 × 10−5 M and 293 K. The arrows indicate spectral changes with time increasing from 0 to 3 h. (b) Time course of the g value changes at 745 nm showing the transformation from 1J1 into 1J2 in various MeOH/water mixtures of 30:70 (black), 20:80 (pink), 10:90 (blue), and 1:99 (green) at a concentration of 1 × 10−5 M and a temperature of 293 K. The rates for the transformation from 1J1 to 1J2 are shown in Table S1 in the ESI.† | ||
In general, the thermodynamic stability of π-conjugated dye aggregates increases with a higher water content due to hydrophobic solvation,19 which in our case leads to a deeper kinetic trapping for the 1J1 species. This issue has been addressed by time-dependent CD spectroscopic measurements of 1 in various MeOH/water mixtures from 30:70 up to 1:99 at a concentration of 1 × 10−5 M and 293 K (Fig. 3). Indeed, at this concentration the kinetics for the transformation process (d(g)/dt) from 1J1 into 1J2 decreased from 8.6 × 10−3 h−1 to 2.5 × 10−3 h−1 with increasing water content from 70% to 80% (Fig. 3b, black and pink dots). A freshly prepared solution of 1 in the 10:90 MeOH/water mixture showed the absorption spectrum of 1J1 (Fig. S3a in the ESI†), which was kinetically trapped for 3 h and transformed into 1J2 with a rate (d(g)/dt) of only 0.4 × 10−3 h−1 (Fig. 3b, blue dots). For the 1:99 MeOH/water mixture no noticeable transformation was observed on the experimental timescale (Fig. 3b, green dots), clearly indicating that the off-pathway 1J1 has high kinetic stability in water (Fig. S3b in the ESI†).19 AFM images of a freshly prepared sample of 1 in the 1:99 MeOH/water solvent mixture showed the presence of nanoparticles, which remained unchanged for several days (Fig. S4 in the ESI†). Thus, the 1J1 aggregate represents a very stable dispersion of the metallochlorin that can be stored for a long period of time. This observation now explains why the preparation of the highly defined nanotubular aggregate of ZnChl 1 in water-rich solutions required a very long time of up to several months.8b
To verify the existence of two competing aggregation pathways, we have further investigated the concentration-dependent self-assembly behaviour of ZnChl 1 at 293 K based on thermodynamic analysis. In this study, a solvent mixture of 40:60 MeOH/water was chosen because in this solvent mixture 1 is fully dissolved monomerically at low concentrations and the equilibrium between 1mono, 1J1, and 1J2 is rapidly reached with a higher MeOH content even with higher concentrations. The concentration-dependent absorption spectral changes for fully equilibrated solutions confirmed the formation of 1J1 as characterized by the appearance of a weak Qy absorption band at 740 nm in the lower concentration (<7 × 10−6 M) range (Fig. 4a, black lines and black dots in the inset). In this concentration range the prevailing species are still monomers, which however start to self-assemble into dimeric species upon increasing the concentration. At concentrations above 1.2 × 10−5 M the Qy absorption band with its maximum now at 750 nm strongly intensified and the positive and negative CD signals at 745 and 761 nm intensified in the same manner (Fig. 4, red lines and red dots in the inset).
 | ||
| Fig. 4 (a) Concentration-dependent UV/vis absorption and (b) CD spectral changes showing the formation of 1J1 and 1J2 in a 40:60 MeOH/water solvent mixture at 293 K. The arrows indicate spectral changes with increasing concentration from 5 × 10−7 to 3 × 10−5 M−1. Insets: plots of ε values at 750 nm and Δε values at 746 nm as a function of concentration. | ||
The non-sigmoidal curves shown in the insets of Fig. 4 are indicative of a cooperative growth of monomeric species into 1J2. The biphasic behaviour of the competing aggregation processes could be fitted using the cooperative growth model combined with a recently introduced competing dimerization model,20 giving an off-pathway dimerization constant of KJ1 = 1.3 × 104 M−1, a very small nucleation binding constant of KN = 2.4 × 10−2 M−1, and an elongation binding constant of KJ2 = 1.0 × 105 M−1 (Fig. S5 in the ESI†). The Gibbs free energy ΔG° for each process was calculated by applying the values of KJ1, KN, and KJ2 to eqn (1), in which R is the ideal gas constant, T is the absolute temperature, and K is the equilibrium constant:
| ΔG° = −RTlnK | (1) |
By merging the thermodynamic data we can derive an energy landscape, in which the formation of nuclei is the most energetically unfavourable step for the formation of thermodynamically stable 1J2 nanofibers (Fig. 5). It is important to point out that the values shown in Fig. 5 are obtained for a high methanol content (40%), i.e. a situation where the hydrophobic effect is still small. With a higher water content the nucleation barrier increases significantly and therefore 1J1 constitutes a deeply trapped state as illustrated by our kinetic data for the 1:99 MeOH/water mixture (Fig. 3b, green dots). This raises the question: how is the self-assembly of BChl dyes in natural chlorosomes controlled? Because living systems are in general not in thermodynamic equilibrium, it is anticipated that within chlorosomes out-of-equilibrium aggregate structures may exist. Subtle changes in environmental conditions would, however, foster nucleation processes, as similarly demonstrated in seeded supramolecular polymerizations,16 and initiate a change in the packing arrangement. Such structural changes may indeed be nature’s way of adapting chlorosomal light harvesting systems to different light intensities and of modulating functional properties such as exciton transport.9h,21 Accordingly, different aggregate morphologies for wild type and mutant strains of green bacteria might have their origin in the selection of different aggregation pathways. However, in in vitro experiments they may also arise by unintentional nucleation processes during sample preparation.
 | ||
| Fig. 5 Schematic energy landscape of the interplay of two competing pathways for the formation of off-pathway nanoparticles composed of antiparallel dimers (1J1) and thermodynamically more stable helical nanofibers composed of parallel metallo-supramolecular π-stacks (1J2) of ZnChl 1 in a 40:60 MeOH/water mixture. | ||
Conclusions
In conclusion, we have investigated the self-assembly mechanism of a semi-synthetic model compound ZnChl dye 1, which possesses high structural similarity to the natural BChl c scaffold. Our kinetic studies on self-assembly processes of ZnChl 1 in various MeOH/water solvent mixtures at different concentrations revealed the formation of nanoparticles with short-range J-coupling as an off-pathway intermediate with regard to the formation of thermodynamically more stable one-dimensional helical J-aggregate nanofibers. Based on a thermodynamic analysis that combines the cooperative K2–K growth model with a competing dimerization model, an energy landscape could be derived that describes the pathway complexity property of this system. Importantly, since the kinetic stability of the off-pathway nanoparticles increases with increasing concentration of ZnChl 1 or the water content in the MeOH/water solvent mixture, the off-pathway nanoparticle product gets trapped to such an extent that no transformation to the more ordered thermodynamic product is observed anymore within reasonable time scales for a water content >90%. Therefore, we assume that out-of-equilibrium aggregate structures of natural BChl dyes may also be present in natural chlorosomes. Furthermore, our results should raise awareness regarding the preparation conditions for metallochlorin dye aggregates, independently of whether they are of synthetic or natural origin. In particular, in an aqueous environment π-scaffolds are likely to self-assemble into kinetically trapped aggregate structures whose functional properties may be significantly different from those of the thermodynamically most stable aggregate structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Vladimir Stepanenko for his help with the AFM measurements. S.O. thanks the JSPS Postdoctoral Fellowships for Research Abroad for their financial support. The funding from the State of Bavaria for the establishment of the KeyLab for Supramolecular Polymers of the Bavarian Polymer Institute (BPI) is gratefully acknowledged.Notes and references
- For recent reviews, see: (a) Self-Assembly and Supramolecular Devices, Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, Wiley, Chichester, 2012, vol. 5 Search PubMed; (b) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (c) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed; (d) J. Schill, A. P. H. J. Schenning and L. Brunsveld, Macromol. Rapid Commun., 2015, 36, 1306–1321 CrossRef CAS PubMed; (e) L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed; (f) S. Yagai, Bull. Chem. Soc. Jpn., 2015, 88, 28–58 CrossRef; (g) F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- For reviews, see: (a) T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed; (b) Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC; (c) P. A. Korevaar, T. F. A. de Greef and E. W. Meijer, Chem. Mater., 2014, 26, 576–586 CrossRef CAS; (d) M. R. Molla and S. Ghosh, Phys. Chem. Chem. Phys., 2014, 16, 26672–26683 RSC; (e) C. Rest, R. Kandanelli and G. Fernández, Chem. Soc. Rev., 2015, 44, 2543–2572 RSC; (f) E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed; (g) L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC.
- (a) S. J. Shire, J. J. Steckert and T. M. Schuster, J. Mol. Biol., 1979, 127, 487–506 CrossRef CAS PubMed; (b) S. J. Shire, J. J. Steckert, M. L. Adams and T. M. Schuster, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 2745–2749 CrossRef CAS PubMed; (c) A. Klug, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 531–535 CrossRef CAS PubMed.
- (a) P. Friedhoff, M. von Bergen, E.-M. Mandelkow, P. Davies and E. Mandelkow, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15712–15717 CrossRef CAS PubMed; (b) I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- (a) T. Kunitake, Angew. Chem., Int. Ed. Engl., 1992, 31, 709–726 CrossRef; (b) E. Reimhult, F. Höök and B. Kasemo, Langmuir, 2003, 19, 1681–1691 CrossRef CAS.
- (a) D. C. Brune, G. H. King, A. Infosino, T. Steiner, M. L. W. Thewalt and R. E. Blankenship, Biochemistry, 1987, 26, 8652–8658 CrossRef CAS PubMed; (b) J. T. Beatty, J. Overmann, M. T. Lince, A. K. Manske, A. S. Lang, R. E. Blankenship, C. L. Van Dover, T. A. Martinson and F. G. Plumley, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9306–9310 CrossRef CAS PubMed; (c) G. T. Oostergetel, H. van Amerongen and E. J. Boekema, Photosynth. Res., 2010, 104, 245–255 CrossRef CAS PubMed.
- (a) T. S. Balaban, H. Tamiaki and A. R. Holzwarth, Top. Curr. Chem., 2005, 258, 1–38 CrossRef CAS; (b) T. Miyatake and H. Tamiaki, J. Photochem. Photobiol., C, 2005, 6, 89–107 CrossRef CAS; (c) S. Sengupta and F. Würthner, Acc. Chem. Res., 2013, 46, 2498–2512 CrossRef CAS PubMed.
- (a) H. Tamiaki, M. Amakawa, Y. Shimono, R. Tanikaga, A. R. Holzwarth and K. Schaffner, Photochem. Photobiol., 1996, 63, 92–99 CrossRef CAS; (b) S. Sengupta, D. Ebeling, S. Patwardhan, X. Zhang, H. von Berlepsch, C. Böttcher, V. Stepanenko, S. Uemura, C. Hentschel, H. Fuchs, F. C. Grozema, L. D. A. Siebbeles, A. R. Holzwarth, L. Chi and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6378–6382 CrossRef CAS PubMed.
- (a) D. L. Worcester, T. J. Michalski and J. J. Katz, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 3791–3795 CrossRef CAS PubMed; (b) H. Tamiaki, A. R. Holzwarth and K. Schaffner, J. Photochem. Photobiol., B, 1992, 15, 355–360 CrossRef CAS; (c) A. R. Holzwarth and K. Schaffner, Photosynth. Res., 1994, 41, 225–233 CrossRef CAS PubMed; (d) T. Mizoguchi, K. Hara, H. Nagae and Y. Koyama, Photochem. Photobiol., 2000, 71, 596–609 CAS; (e) B.-J. van Rossum, D. B. Steensgaard, F. M. Mulder, G. J. Boender, K. Schaffner, A. R. Holzwarth and H. J. M. de Groot, Biochemistry, 2001, 40, 1587–1595 CrossRef CAS PubMed; (f) J. Pšenčík, T. P. Ikonen, P. Laurinmäki, M. C. Merckel, S. J. Butcher, R. E. Serimaa and R. Tuma, Biophys. J., 2004, 87, 1165–1172 CrossRef PubMed; (g) G. T. Oostergetel, M. Reus, A. G. M. Chew, D. A. Bryant, E. J. Boekema and A. R. Holzwarth, FEBS Lett., 2007, 581, 5435–5439 CrossRef CAS PubMed; (h) Y. Tian, R. Camacho, D. Thomsson, M. Reus, A. R. Holzwarth and I. G. Scheblykin, J. Am. Chem. Soc., 2011, 133, 17192–17199 CrossRef CAS PubMed; (i) L. M. Günther, M. Jendrny, E. A. Bloemsma, M. Tank, G. T. Oostergetel, D. A. Bryant, J. Knoester and J. Köhler, J. Phys. Chem. B, 2016, 120, 5367–5376 CrossRef PubMed.
- (a) T. S. Balaban, J. Leitich, A. R. Holzwarth and K. Schaffner, J. Phys. Chem. B, 2000, 104, 1362–1372 CrossRef CAS; (b) T. Miyatake, K. Shitasue, Y. Omori, K. Nakagawa, M. Fujiwara, T. Matsushita and H. Tamiaki, Photosynth. Res., 2005, 86, 131–136 CrossRef CAS PubMed.
- (a) K. M. Smith and L. A. Kehres, J. Am. Chem. Soc., 1983, 105, 1387–1389 CrossRef CAS; (b) J. Chiefari, K. Griebenow, N. Griebenow, T. S. Balaban, A. R. Holzwarth and K. Schaffner, J. Phys. Chem., 1995, 99, 1357–1365 CrossRef CAS; (c) V. Huber, M. Katterle, M. Lysetska and F. Würthner, Angew. Chem., Int. Ed., 2005, 44, 3147–3151 CrossRef CAS PubMed; (d) V. Huber, S. Sengupta and F. Würthner, Chem.–Eur. J., 2008, 14, 7791–7807 CrossRef CAS PubMed; (e) S. Shoji, T. Ogawa, T. Hashishin, S. Ogasawara, H. Watanabe, H. Usami and H. Tamiaki, Nano Lett., 2016, 16, 3650–3654 CrossRef CAS PubMed.
- K. M. Smith, C. W. Craig, L. A. Kehres and N. Pfennig, J. Chromatogr., 1983, 281, 209–223 CrossRef CAS.
- F. Fages, N. Griebenow, K. Griebenow, A. R. Holzwarth and K. Schaffner, J. Chem. Soc., Perkin Trans. 1, 1990, 10, 2791–2797 RSC.
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS PubMed.
- V. Huber, M. Lysetska and F. Würthner, Small, 2007, 3, 1007–1014 CrossRef CAS PubMed.
- (a) S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed; (b) S. Ogi, V. Stepanenko, J. Thein and F. Würthner, J. Am. Chem. Soc., 2016, 138, 670–678 CrossRef CAS PubMed; (c) W. Wagner, M. Wehner, V. Stepanenko, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 16008–16012 CrossRef CAS PubMed.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS PubMed.
- (a) P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed; (b) A. Das, B. Maity, D. Koley and S. Ghosh, Chem. Commun., 2013, 49, 5757–5759 RSC; (c) D. van der Zwaag, P. A. Pieters, P. A. Korevaar, A. J. Markvoort, A. J. H. Spiering, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 12677–12688 CrossRef CAS PubMed; (d) A. T. Haedler, S. C. J. Meskers, R. H. Zha, M. Kivala, H.-W. Schmidt and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 10539–10545 CrossRef CAS PubMed; (e) A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- (a) M. R. Molla and S. Ghosh, Chem.–Eur. J., 2012, 18, 9860–9869 CrossRef CAS PubMed; (b) Y. Tidhar, H. Weissman, D. Tworowski and B. Rybtchinski, Chem.–Eur. J., 2014, 20, 10332–10342 CrossRef CAS PubMed; (c) B. Narayan, S. P. Senanayak, A. Jain, K. S. Narayan and S. J. George, Adv. Funct. Mater., 2013, 23, 3053–3060 CrossRef CAS; (d) K. Jalani, S. Dhiman, A. Jain and S. J. George, Chem. Sci., 2017, 8, 6030–6036 RSC.
- F. Fennel, S. Wolter, Z. Xie, P.-A. Plötz, O. Kühn, F. Würthner and S. Lochbrunner, J. Am. Chem. Soc., 2013, 135, 18722–18725 CrossRef CAS PubMed.
- J. Dostál, J. Pšenčík and D. Zigmantas, Nat. Chem., 2016, 8, 705–710 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, supplementary figures and a table. See DOI: 10.1039/c7sc03725b |
| ‡ Current address: Department of Chemistry, Graduate School of Science, Nagoya University, Furo, Chikusa, Nagoya 464-8602, Japan. |
| This journal is © The Royal Society of Chemistry 2018 |