
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMixed valency in ligand-bridged diruthenium frameworks: divergences and perspectives
Arijit Singha Hazari
a,
Arindam Indra
*b and
Goutam Kumar Lahiri
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai-400076, India. E-mail: lahiri@chem.iitb.ac.in
bDepartment of Chemistry, Indian Institute of Technology (Banaras Hindu University), Varanasi, Uttar Pradesh-221005, India. E-mail: arindam.chy@iitbhu.ac.in
First published on 14th August 2018
Abstract
The present review article illustrates the mixed valence aspects of ligand-bridged symmetric and unsymmetric diruthenium complexes beyond the textbook example of the Creutz–Taube ion as well as the Robin and Day classification by citing representative examples based on our recent observations. The consideration of varied coordination situations involving bridging and ancillary ligands of diverse electronic and steric demands extended important fundamental events including (i) the influence of ancillary ligands besides the bridge in the intermetallic coupling process, (ii) varying profile of the intervalence charge transfer (IVCT) transition in RuIIIRuII (d5d6) and RuIIIRuIV (d5d4) mixed valence set up, (iii) divergence between the electrochemical (Kc = comproportionation constant) and electronic (IVCT) coupling and (iv) occurrence of the hybrid class II-class III situation. Furthermore, additional challenges due to the introduction of redox non-innocent ligands in assigning valence and spin distributions at the metal–ligand interface as well as in differentiating the emerging alternatives of the radical-derived state and the mixed valence situation along the redox chain have been addressed.
 Arijit Singha Hazari | Arijit Singha Hazari received his B.Sc. degree from University of Calcutta and M.Sc. degree from Birla institute of technology and Science. He then joined IIT Bombay in 2013 to pursue his Ph.D. degree under the supervision of Prof. G. K. Lahiri. His research work primarily focusses on the electronic structural aspects of transition metal complexes involving redox-active ligands and their applications in homogeneous catalysis. |
 Arindam Indra | Dr. Arindam Indra received his Ph.D. from IIT Bombay (India) in solid supported catalysis. After completing his postdoctoral research from Technische Universitat Berlin (Germany) and Hanyang University (Korea), he joined IIT (BHU), Varanasi (India) as an assistant professor. His group designs metal organic framework (MOF)-derived functional materials for energy conversion and storage. |
 Goutam Kumar Lahiri | Prof. Goutam Kumar Lahiri received his Ph.D degree from Jadavpur University (IACS) in 1990 under the supervision of Prof. Animesh Chakravorty. After completing his postdoctoral studies at West Virginia University, USA, he joined the department of chemistry at IIT Bombay in 1993, where he is currently an Institute Chair Professor. The main theme of his research is centered around the electron transfer aspects and catalytic features of transition metal complexes. |
1. Introduction
1.1. Mixed valence
Mixed-valence compounds are those that feature an element in more than one formal oxidation state. A sub-set of mixed valence compounds includes those in which two (or more) metal centers are linked through a bridging ligand. In particular cases, the bridge can mediate intermetallic electron transfer from the reduced metal ion to the oxidized one.1 The electron transfer at the mixed valence state is guided by various intriguing factors such as molecular conformation, separation of the redox centers, nature of the spacer and their redox potentials.2In biological systems, the occurrence of polynuclear structures with metal ions in different oxidation states can be found in the oxygen evolving center (OEC) of photosystem II (PSII),3 cytochrome-c-oxidase,4 ferredoxins,5 etc. In PS II, four electrons and four protons are transferred through multistep processes involving high-energy intermediates. The proton-coupled electron transfer to produce molecular oxygen from water is eased by the mixed valent MnIII–MnIV cluster in OEC (Fig. 1).6 Similarly, dioxygen activation is attained by mixed valent CuI/CuII centers in cytochrome-c-oxidase.7
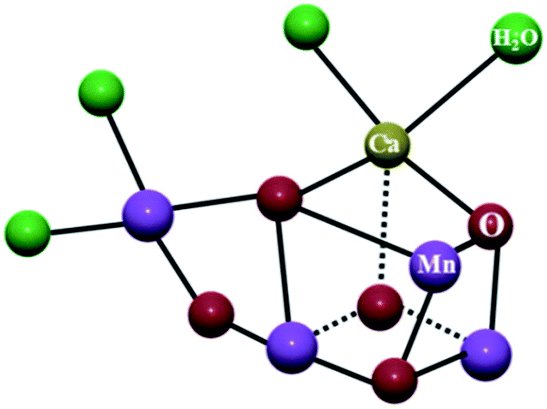 | ||
| Fig. 1 Oxygen evolving center (OEC) in photosystem II represents oxide-bridged mixed valent MnIII and MnIV ions. Reprinted from ref. 6 with the permission of Wiley VCH. | ||
Naturally occurring metal oxides (Spinels (AIIBIII2O4)) such as Fe3O4 (magnetite),8 Co3O4, Mn3O4 (ref. 9) and cyanide-bridged Prussian blue (FeIII4[FeII(CN)6]3·14H2O, coordination polymer) or its analogues (PBA) also contain metal ions in different oxidation states.10
The mixed valency in the designed molecular framework was first described by Creutz and Taube11 in the pyrazine-bridged diruthenium complex [(NH3)5Ru(μ-pyrazine)Ru(NH3)5]5+ (Fig. 2).
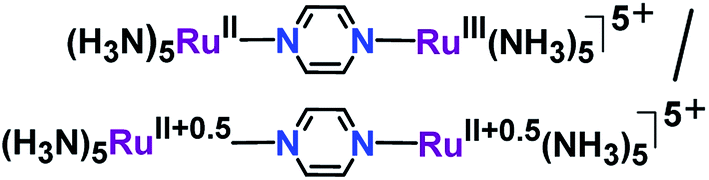 | ||
| Fig. 2 Alternate electronic forms of the Creutz–Taube ion. | ||
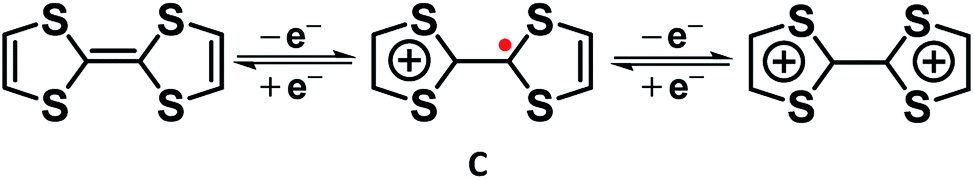 | ||
| Fig. 3 Electron transfer series of tetrathiafulvalene. | ||
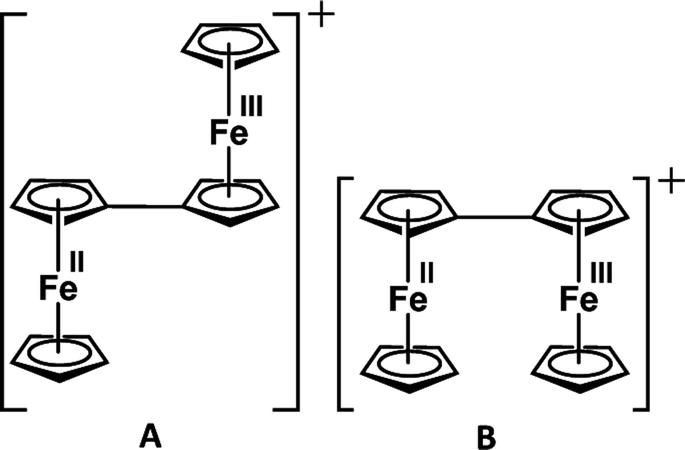
Similarly, Cowan's group12 described the mixed valency in biferrocene (A) and biferrocenylene (B). Later, they introduced the tetrathiafulvalene radical cation (C in Fig. 3) as an odd-electron organic compound to show the mixed valency in an organic set up.13
Under the purview of the broader context of mixed valency, diruthenium complexes have been extensively investigated to understand the bridge-mediated electron transfer in mixed valent RuII(d6)RuIII(d5) systems primarily due to better kinetic stability of varying (+2, +3 or even +4) oxidation states of the ruthenium ion.14
Mixed valence complexes were initially synthesized to understand the fundamental aspects of intermetallic electronic coupling processes.15 Later, the focus was also directed towards exploring potential applications of the mixed valence compounds in molecular electronics, molecular wires and quantum automata.16 In nanochemistry, the intramolecular electron transfer can be employed to demonstrate the nanojunction device of bulk metal-single molecule-bulk metal.17 A mixed valence compound that derived molecular switch for long-range electron transfer (15–20 Å) was also investigated.18
1.2. Classification of mixed valence complexes
In a ligand-bridged diruthenium(II/III) complex [(AL)RuII(μ-BL) RuIII(AL)] (BL = Bridging ligand and AL = ancillary or peripheral ligand), the electron transfer is initiated from the starting metal center to the arrival site through a bridging ligand (BL). Thus, the bridging ligand plays the role of a mediator to exchange the odd electron between the metal centers at the termini. Hence, the electronic nature and the composition of the bridge play important roles in governing the rate of electron transfer. This provides useful information regarding the separation of the redox potential between the metal centers under chemically identical environments (symmetric set up). In this context, superexchange mechanism provides a better perception about the bridge-mediated intermetallic interaction, which involves efficient overlap of the metal d-orbitals and the π-orbitals of the spacer (through bond interaction). It can be conceived as a sequential electron transfer between the molecular orbitals of the ligand (π type) and the metal (dπ type), resulting in delocalization of the electron density across the metal–bridge–metal domain to some extent, and it occurs via either of the following two pathways (Fig. 4):19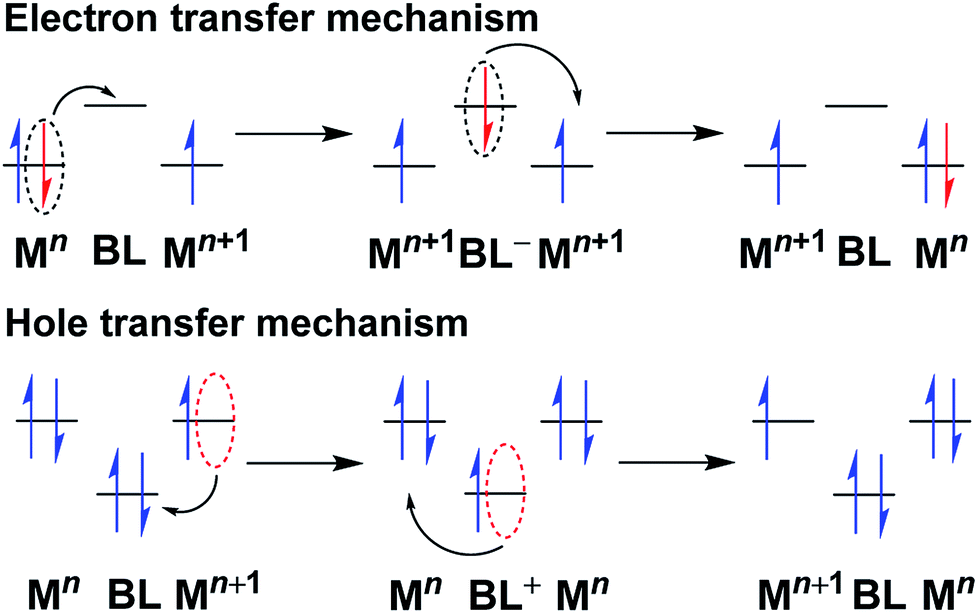 | ||
| Fig. 4 Bridging ligand-mediated electron transfer and hole transfer pathways in mixed valence complexes.19c,d | ||
Robin and Day classified20 the mixed valence complexes based on the extent of intermetallic interaction. A very weak to negligible interaction between the metal centers is defined as a class I system, whereas a moderate coupling results in a localized valence-trapped class II ([(AL)RuII(μ-BL)RuIII(AL)]) system. On the other hand, strong intermetallic coupling leads to a valence delocalized [(AL)Ru2.5(μ-BL) Ru2.5(AL)] class III set up. Therefore, detailed studies with varied molecular frameworks have been conducted over the past decades to understand the role of metal ions and ligands (ancillary and bridging) in modulating the intermetallic electronic coupling at the mixed valence state.21
1.3. Comproportionation constant
Comproportionation constant, Kc, implies the thermodynamic stability of the intermediate redox state. For a two-step redox reaction (eqn (1)), the comproportionation constant (Kc) can be calculated from the potential difference (ΔE) of the successive redox couples.22
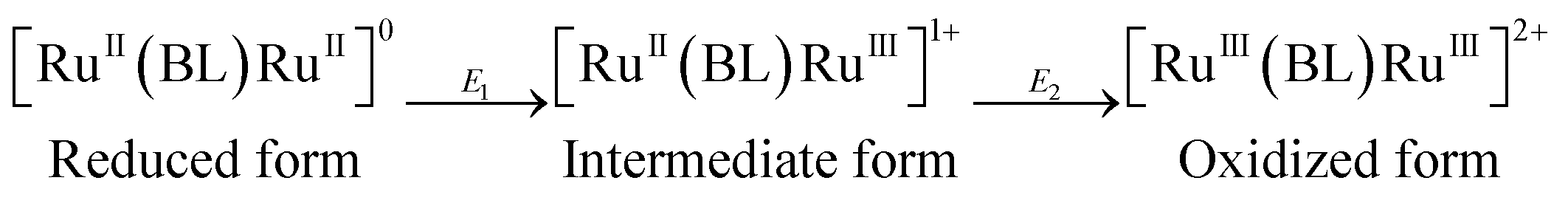 | (1) |
| Kc = [intermediate form]2/{[reduced form][oxidized form]} |
ΔGc = −RT(ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kc) = −nF(ΔE0); (ΔE = E2 − E1) Kc) = −nF(ΔE0); (ΔE = E2 − E1) |
For a well-defined metal-based redox process, Kc signifies the thermodynamic stability of the mixed valence state. For example, successive RuII/RuIII redox couples in the Creutz–Taube ion are well separated by 0.39 V (Kc = 106.6), leading to a stable mixed valence state with respect to the disproportionation reaction that produces isovalent RuII/RuII and RuIII/RuIII.11a,23
According to the Robin and Day classification, a genuine non-interacting class I system is defined by a statistical Kc value of 4, whereas Kc values close to 105 and greater than 106 correspond to class II and class III systems, respectively.23 In a qualitative sense, Kc values ranging in between the valence localized class II and the valence delocalized class III may represent a borderline class II/III hybrid situation. Despite the Robin and Day classification of the intermetallic interaction based on the value of Kc, the equilibrium constant alone cannot provide an evaluation of the metal–metal electronic coupling since the free energy of the comproportionation reaction (ΔGc) consists of several interplaying factors (eqn (2)).24
| ΔGc = ΔGs + ΔGe + ΔGi + ΔGr + ΔGaf + ΔGip | (2) |
In eqn (2), ΔGs corresponds to the statistical distribution, ΔGe reflects the electrostatic repulsion between the positively charged metal ions, ΔGi accounts for the inductive factor related to the competitive coordination of the bridging ligand by the metal ions, ΔGr is the free energy of the resonance exchange, ΔGaf relates to the antiferromagnetic exchange stabilization of the isovalent higher analogue state Mn+1Mn+1 over that of its mixed valent form MnMn+1 and ΔGip represents the ion-pairing effect, which changes its magnitude depending on the charge of the complex moiety. However, in the case of hole transfer mechanism, the value of ΔGaf becomes almost insignificant. Although Kc extends the prima facie information regarding the extent of the intermetallic interaction in a three-state arrangement, precautionary measures need to be taken into account. This is primarily due to the dependence of redox potentials on the nature of the solvent (dielectric constant or nucleophilicity) as well as counter ions (ion-pairing ability), which can affect the value of ΔGc appreciably.24b Thus, to extract the extent of electronic coupling leading to valence localization or delocalization at the mixed valence state, the collective consideration of Kc and the intervalence charge transfer (IVCT) feature are rather necessary.
1.4. Intervalence charge transfer (IVCT) transition
In the case of a weakly coupled valence localized system, the charge transfer occurs from the metal in a lower oxidation state (electron rich) to the metal in a higher oxidation state (electron deficient), resulting in bridge-mediated uni-directional intervalence charge transfer transition (IVCT band) at the low-energy NIR region of electron absorption spectroscopy.25 The correlation of IVCT band with electronic coupling between the metal centers at the mixed valence state follows the Hush formula.26 For class II type complexes, the bandwidth at half-height (Δν1/2) of IVCT is related to the energy maximum (eqn (3)):| Δν1/2 = [2.31 × 103 (Emax)]1/2 | (3) |
In the case of a symmetrical band, IVCT can be correlated with the electronic coupling constant (Vab) by eqn (4),
Vab = {[2.05 × 10−2(εmax![[small nu, Greek, macron]](https://www.rsc.org/images/entities/char_e0ce.gif) max1/2)1/2]/R} max1/2)1/2]/R}
| (4) |
max is the absorption maximum in wavenumbers, 1/2 is the bandwidth at half-height in wavenumbers and R is the metal–metal distance in Å. However, the effective metal–metal distance can also vary significantly depending on the extent of the mixing of orbitals of the bridge and the metal ions at the termini.27
For a fully delocalized class III system, the calculated Δν1/2 value of the IVCT band using the Hush equation is always higher than the experimentally obtained value, and Vab amounts to max/2.
Please note that the valence localized class II system exhibits a double well potential. On the contrary, the extensive charge delocalization between the donor and acceptor sites in the class III system results in the disappearance of separate minima, exhibiting single minimum in the energy surface instead.
The larger bandwidth at half-height (Δν1/2) of IVCT for the weakly coupled (class II) system than that for the strongly coupled (class III) system can be due to the transition from the flat potential energy (PE) surface to a steep one in the former and the transition from the flat PE surface to another flat PE surface in the latter.15b
In fact, the correlation between the IVCT band and the intermetallic coupling is not so straightforward as the band shape, bandwidth and intensity vary appreciably depending on the solvent polarity and electronic and vibrational motions.28,29 In the case of class III-type mixed valency, the motions from the solvent and vibrations are averaged to delocalize the electron density. This results in narrow solvent-independent IVCT bands. In class II-type, the IVCT band is broad and depends on the dielectric constant of the solvent as the motions from the exchange of electrons and the solvent are localized. On the contrary, in the borderline (hybrid) class II–III system, the solvent dynamic parameters play influential roles in controlling the rate of the intermolecular electron transfer by minimizing the impact of the solvent reorganization energy or polarity.23b Additionally, the parameter “Γ” (Γ = 1 − (Δν1/2)/(16ln(2)RT)1/2) introduced by Sutin and co-workers has been widely acknowledged in identifying the intriguing situation of the class II–III hybrid system.15b The room-temperature limiting value of Γ can distinguish between the weakly coupled class I (0 < Γ < 0.1), moderately coupled class II (0.1 < Γ < 0.5), class II–III hybrid (Γ ≈ 0.5) and class III (Γ > 0.5) systems.
1.5. Vibrational and electron spin resonance spectroscopy
In this context, vibrational (IR and Raman) spectroscopic techniques have been considered to be very effective for the selectively designed molecular frameworks containing suitable functional groups such as CO, NO, and CN to extract more precise information relating to the valence localization or delocalization process at the mixed valence state with a faster picosecond to femtosecond time scale.30The correlation of EPR g parameters (g-anisotropy, Δg = g1 − g3 and 〈g〉={1/3(g12 + g22 + g32)}1/2,31 hyperfine splitting) with the DFT calculated spin density distribution at the metal–bridge–metal domain of the paramagnetic mixed valence state provides additional justification in favor of the valence localization or delocalization process.32 However, a mixed valence state without any EPR response is not so uncommon, particularly due to the rapid relaxation process. This situation may improve upon moving to the liquid helium temperature (4 K).33
It should be noted that the time scale of a particular spectroscopic technique also plays a critical role in establishing the mixed valence feature. It may give rise to contradictory observations based on the rate of the electron transfer, solvent reorganization process and solvent dynamics. The slower time scale of the EPR spectroscopic technique (10−5 to 10−9 s) often fails to distinguish the valence localized and delocalized situations, whereas a faster vibrational spectroscopy technique (∼10−12 s) is found to be more precise in this regard.34
1.6. Diruthenium complexes involving a redox active bridge
In a specialized situation relating to the selective participation of bridge-based Frontier orbitals in the redox processes of the complex moieties, an additional complexity arises due to the probable alternate formation of a radical bridged isovalent state ([(AL)RuII(μ-BL)RuII(AL)] − e− → [(AL)RuII(μ-BL˙)RuII(AL)]+) upon electron transfer instead of the otherwise expected mixed valence state ([(AL)RuII(μ-BL)RuIII(AL)]).35 Therefore, appropriate care needs to be taken in formulating the electronic structure of the redox states as well. In this context, EPR g-anisotropy in conjunction with DFT-calculated spin distribution at the paramagnetic intermediate state can facilitate to differentiate the alternative mixed valence state versus the radical-bridged isovalent state.The present review article highlights our recent observations concerning the divergent issues in addressing mixed valency in ligand-bridged diruthenium systems by citing the following representative cases.
2. Metal-centered mixed valency
2.1. Role of the ancillary ligand
Pyrazine-mediated facile intramolecular electron transfer in the mixed valent Creutz–Taube ion essentially leads to the development of analogous bis-tridentate non-planar 2,3,5,6-tetrakis(2-pyridyl) pyrazine (tppz)-bridged isovalent diruthenium(II) complexes ([(AL)(Cl)RuII(μ-tppz)RuII(Cl)(AL)]n, 1n).36 The impact of a set of ancillary ligands (AL) with varying electronic demands in the mixed valent RuIIRuIII state (1n) was explored (Table 1) via an experimental investigation in conjunction with DFT calculations. Successive two-step reversible electrochemical oxidations of isovalent RuIIRuII (1n) complexes resulted in a mixed valent {RuII(μ-tppz)RuIII} intermediate (1(n+1)) followed by the isovalent higher {RuIII(μ-tppz)RuIII}I congener (1(n+2)). The detailed structural, electrochemical, spectroscopic and theoretical investigations revealed that in addition to the bridge, the electronic nature (σ-donating to π-acidic to strongly π-acidic) of the ancillary ligand (AL) also played an important role in tuning the mixed valence feature (Kc as well as IVCT, Table 1). The enhanced electron density on the metal ions via the insertion of an electron-rich ancillary ligand facilitated the intramolecular electron transfer process with larger Kc and intense IVCT (borderline class II–III to class III), whereas a reverse pattern (smaller Kc and weak IVCT) was apparent for the acceptor-type ancillary ligand. Please note that to the best of our knowledge, a well-defined valence-localized (weakly coupled) class II mixed valence system involving a tppz-bridged diruthenium system is not reported so far.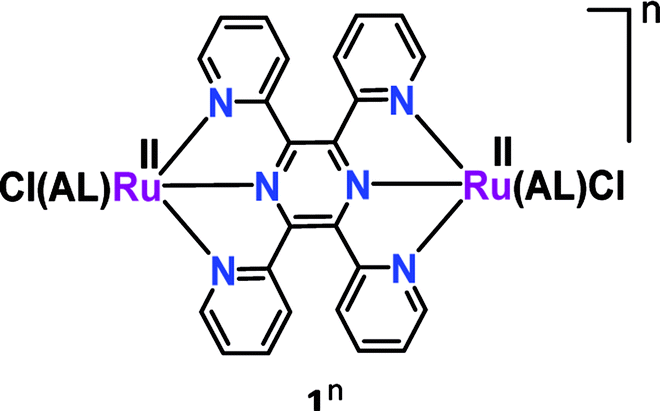
| [Cl(AL)RuII(μ-tppz)RuIII(AL)Cl]n (1n) | |||
|---|---|---|---|
| AL | Kc | νIVCT/nm (ε/M−1 cm−1) | Classa |
| a The classification (class III or class II–III) was defined primarily based on the Kc and IVCT band features by following the Hush formula as stated in Sections 1.3 and 1.4. | |||
| 2-Phenylazopyridine | 4.0 × 103 | 1890 (3800) | II–III |
| 2-[(4-Methylphenyl)azo]-pyridine | 2.1 × 104 | 1890 (4600) | II–III |
| 2-[(4-Chlorophenyl)azo]-pyridine | 2.2 × 103 | 1890 (3000) | II–III |
| 2,2/-Dipyridylamine | 2.7 × 106 | 1700 (2250) | III |
| 2-(2-Pyridyl)benzoxazole | 3.8 × 104 | 1635 (1400) | II–III |
| 2-(2-Pyridyl)benzthiazole | 5.6 × 104 | 1465 (440) | II–III |
| 1-Methyl-2-(2-pyridyl)-1H-benzimidazole | 2.6 × 104 | 1555 (423) | II–III |
| 2,2/-Dipyridylketone | 2.5 × 104 | 1800 (1500) | II–III |
| 2,2/-bis(1-methylimidazolyl) ketone | 1.2 × 105 | 1637 (1200) | II–III |
| 2-Picolinate | 4.0 × 106 | 1670 (2600) | III |
| 2-Quinolinecarboxylate | 8.9 × 106 | 1575 (1200) | III |
| 8-Quinolinecarboxylate | 1.9 × 107 | 1875 (2600) | III |
| Acetylacetonate | 1010 | 1680 (2900) | III |
| 3,5-Heptanedionate | 1.5 × 1010 | 1688 (3300) | III |
| 2,2,6,6-Tetramethyl-3,5-heptanedionate | 1.1 × 1012 | 1750 (3000) | III |
| 3-Methyl-2,4-pentanedionate | 1010 | 1660 (2300) | III |
| 3-Ethyl-2,4-pentanedionate | 1.5 × 1010 | 1674 (2239) | III |
2.2. RuIIRuIII (d6d5) versus RuIIIRuIV (d5d4) mixed valence systems
Though numerous examples of ligand-bridged RuIIRuIII mixed valence systems are available in the literature,37 only a few well-defined RuIIIRuIV mixed valence systems (D and E) are reported so far.38b,c
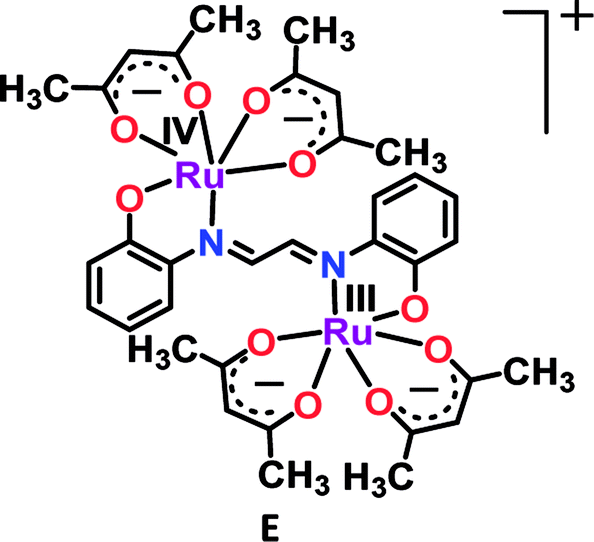
In this context, bis-chelating dianionic oxamidato-bridged diruthenium(III) complex 2n (n = 0) in S = 1 ground state (J = −40 cm−1) exhibited two successive oxidative (RuIIIRuIII (2) → RuIIIRuIV (2+) → RuIVRuIV (22+), O1/O2) and reductive (RuIIIRuIII (2) → RuIIIRuII (2−)→RuIIRuII (22−), R1/R2) couples with moderate Kc values of 105.8 and 105.9 for mixed valent RuIIIRuIV and RuIIIRuII, respectively (Fig. 5).
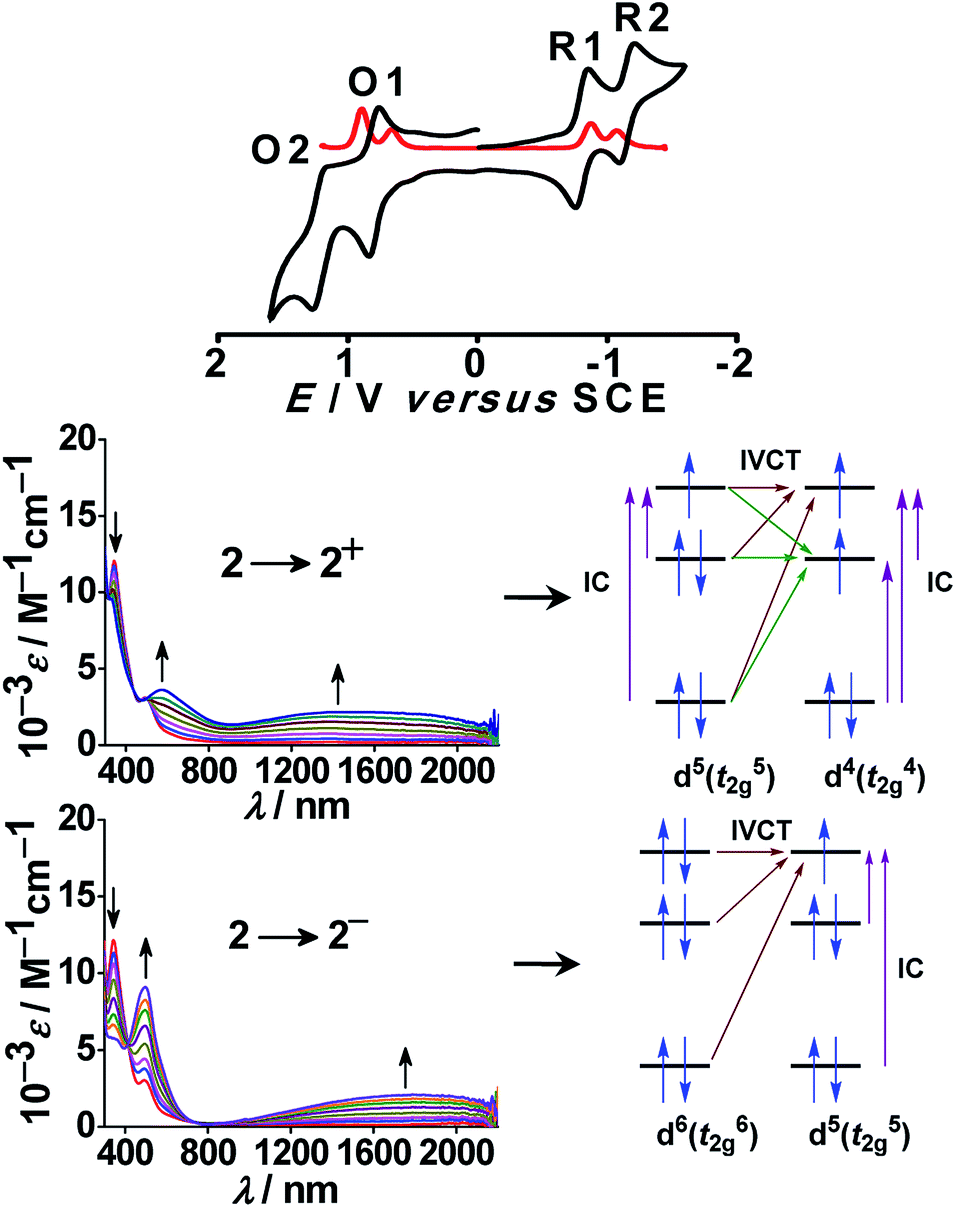 | ||
| Fig. 5 Cyclic (black) and differential pulse (red) voltammograms of 2 in CH3CN (top). UV-vis-NIR spectroelectrochemistry in CH3CN and the sketch for IVCTT and IC transitions (bottom). | ||
Both the electrochemically generated mixed valence states RuIIIRuII (2−) and RuIIIRuIV (2+) displayed moderately intense broad IVCT bands in the NIR region at 1800 nm (ε/M−1 cm−1: 2100) and 1500 nm (ε/M−1 cm−1: 2200), respectively, corresponding to the class II system. The appearance of a relatively broader IVCT band for the RuIIIRuIV (d5d4) mixed valence state compared to that for the RuIIIRuII (d5d6) mixed valence state could possibly be due to more number of intervalence (IVCT) and d–d (IC) transitions in the former with respect to that in the latter because of the presence of three and one holes at metal t2g levels, respectively (Fig. 5). Thus, 2n represents a novel example demonstrating two mixed valence intermediates RuIIIRuII (via reductive pathway) and less common RuIIIRuIV (via oxidative pathway).38
2.3. Paradox between Kc and IVCT
According to the Robin and Day classification and Hush formalism, a symmetric class III molecular set is expected to display large Kc and an intense IVCT band in the NIR region.26,39 However, the mixed valent (RuIIRuIII) complex 3+ failed to display the expected low-energy IVCT band up to 2500 nm despite the very large Kc value of 1013.9.40 On the other hand, the previously reported analogous complex 4 exhibited a solvent–independent and very weak IVCT band (ε/M−1 cm−1: 20) with a similar Kc value of 1013.6.41 The absence of the IVCT band despite the very large Kc value in 3+ appeared to be rather puzzling. Though we were unable to offer any simple explanation, very weak to undetectable IVCT bands were also observed earlier for mixed valent ruthenium complexes with appreciably large Kc values.42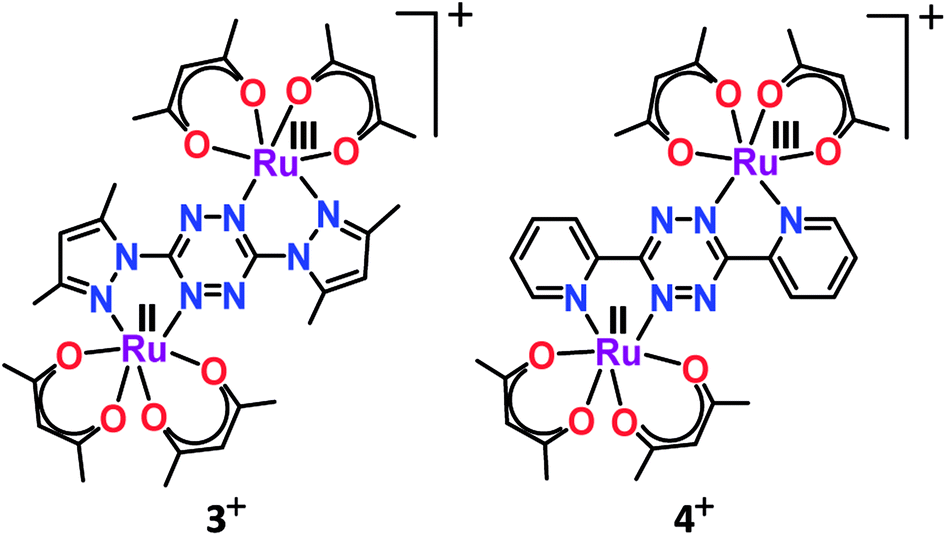
On the other hand, 2,5-pyrazine-dicarboxylate (BL)-bridged isovalent diruthenium(III) complex (5n (n = 0)) (acac = actylacetonate) in S = 1 ground state (J = −1.78 cm−1) displayed successive Ru(III/II) redox couples with Kc of 107.43 Stepwise one-electron reductions facilitated the generation of a mixed-valent intermediate [(acac)2RuIII(μ-BL)RuII(acac)2]− (5−) followed by isovalent [(acac)2RuII(μ-BL)RuII(acac)2]2− (52−) in lower oxidation states. The mixed valent 5− exhibited an RuIII-based anisotropic EPR spectrum (Δg = 0.32). However, the very weak IVCT band of the intermediate 5− at 1040 nm (ε = 380 M−1 cm−1) implied limited electronic communication between the two metal centers ({RuIII(μ-BL)RuII}, class II) despite the appreciably high Kc value of 107. The diverse inference regarding the mixed valence feature of 5− based on Kc (class III) and IVCT (class II) was however addressed via IR spectroelectrochemistry (Fig. 6). The unsplit ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bands of the carboxylate groups of BL for mixed valent 5− as those for isovalent 5 or 52− indeed suggested a delocalized (Ru2.5Ru2.5, class III to borderline class II–III) situation even at the vibrational time scale.
O) bands of the carboxylate groups of BL for mixed valent 5− as those for isovalent 5 or 52− indeed suggested a delocalized (Ru2.5Ru2.5, class III to borderline class II–III) situation even at the vibrational time scale.
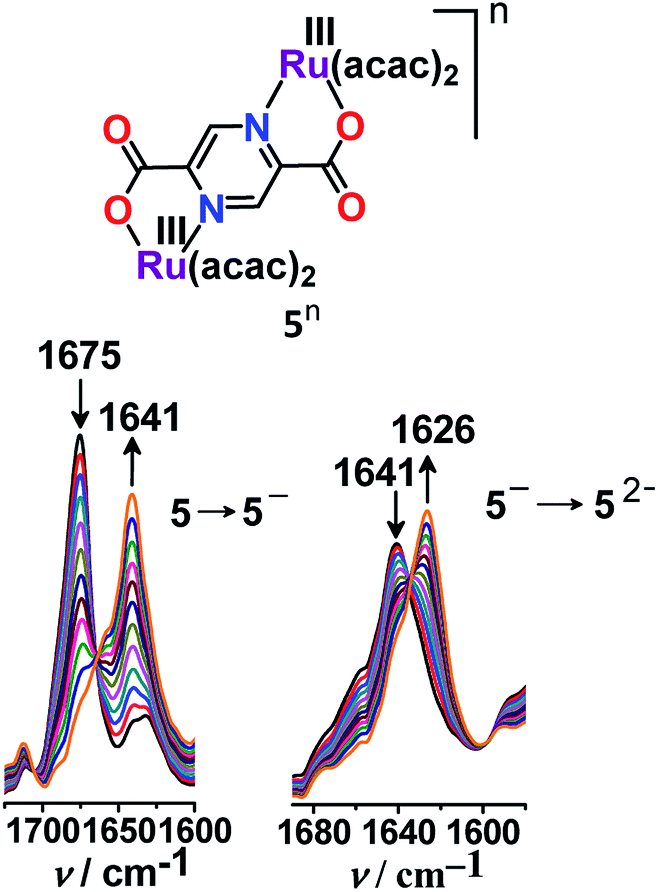 | ||
| Fig. 6 Shift in the ν(CO) frequency on moving from 5 → 5− and 5− → 52− in CH3CN. | ||
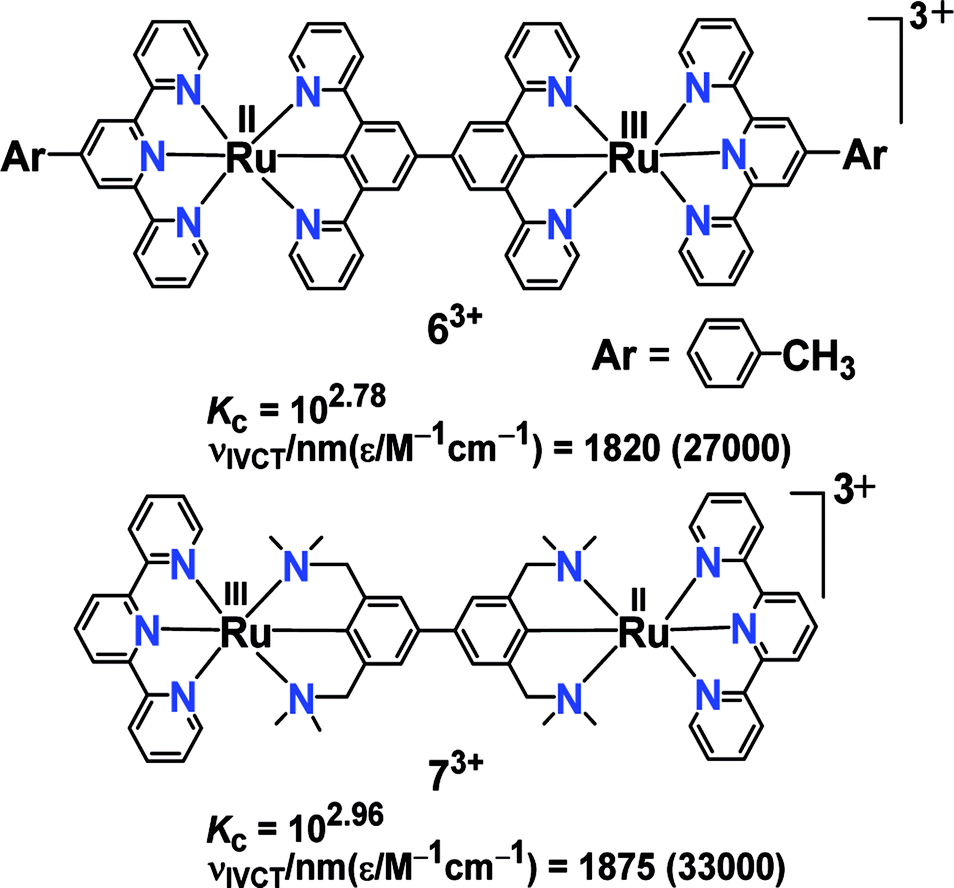
On the contrary, 63+ and 73+ exhibited intense IVCT bands despite small Kc values.44
2.4. Unsymmetrical diruthenium complexes
Asymmetry in dinuclear frameworks can be introduced by using different ancillary ligands at the metal sites or by using an unsymmetrical bridge or even by coordination to two different metal ions.45 Unlike that in a symmetrical system, the donor center asymmetry (ancillary or bridging ligand) around the metal ions in an unsymmetrical system makes it rather challenging to quantify the contribution of intermetallic electrochemical coupling (Kc) out of the potential separation of the successive redox processes.In this context, unsymmetrical diruthenium(II) complexes 8a2+, 8b2+ and 8c2+ exhibited two successive oxidation processes (RuIIRuII/RuIIRuIII and RuIIRuIII/RuIIIRuIII) via the intermediate mixed valent RuIIRuIII state (Fig. 7).46 The separation in potential between the stepwise redox processes led to the Kc values of 104, 102 and 1014 for the intermediates 8a3+, 8b3+ and 8c3+, respectively. The 100-fold larger Kc value of 8a3+ with respect to that of 8b3+ was due to moderate and strong π-acceptor features of bpy and pap, respectively. On the other hand, the very large Kc value of 1014 for 8c3+, which comprised metal fragments with two distinctly different acceptor ligands bpy (moderate π acceptor) and pap (strong π acceptor) could be the reflection of three simultaneously operating effects: (i) effect of unsymmetrical ancillary ligand, (ii) effect of unsymmetrical bridging ligand and (iii) bridging ligand-mediated electrochemical coupling between the reduced and oxidized metal ions. The very weak IVCT band at the near IR region corresponding to 8a3+ and 8b3+ in conjunction with low Kc values of 104 and 102, respectively, suggested the class II mixed valence state. However, the unstable nature of the electrochemically generated 6c3+ restricted us from assigning its mixed valence feature.
 | ||
| Fig. 7 Cyclic voltammograms of 82+ in CH3CN. | ||
Similarly, a cyano-bridged unsymmetric mixed valence45g dinuclear complex [(NH3)5–Ru-(μ-CN)Fe(CN)5]− involving different metal ions and ancillary ligands was assigned to be a valence-localized class-II system (eqn (5)) primarily based on its optical (IVCT) features and electrochemical aspects as a function of the solvent polarity and the charges of cations and anions of the electrolytes.45f
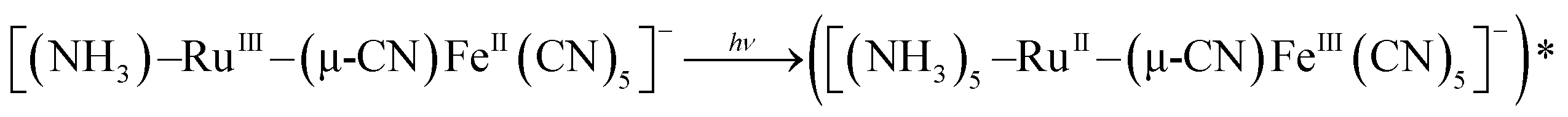 | (5) |
3. Involvement of redox non-innocent ligands
The closeness of the Frontier orbitals of ruthenium and redox active (non-innocent) ligands leads to the feasibility of alternate electronic structural forms including resonating configuration47 instead of any precise valence description along the redox chain.48 In certain instances, the redox-induced electron-transfer process (RIET) is also considered to be operational.49 Appropriate care is therefore needed in assigning the valence distribution process at the metal–ligand interface of such molecular forms.A few representative cases involving multi-redox sites (metal, bridge or ancillary ligands) within the discrete molecular set up are therefore highlighted below.
3.1. Active bridge and ancillary ligand
Experimental and theoretical investigations of two successive reversible oxidation processes (O1/O2) of the deprotonated ellagate-bridged diruthenium(II) complex 9n (n = 0) (Fig. 8) revealed selective participation of bridge-based orbitals in the redox processes (9 → 9+ (O1) and 9+ → 92+ (O2) in Fig. 8), i.e., a catecholate/semiquinone-based redox system instead of otherwise expected metal-based (RuII/RuIII) oxidations.50 The intermediate radical-bridged isovalent metal (RuII) centers in 9+ exhibited free radical EPR (g1 = 2.011, g2 = 2.002, g3 = 1.987; Δg = 0.024, 〈g〉 = 2.000), which was corroborated by the calculated bridge-dominated spin (96%) and the intense LLCT (donor radical bridge to π-accepting pap) transitions in the NIR region around 2000 nm (ε/M−1 cm−1: 1843) (Fig. 8). Successive reductions (R1 (92−), R2 (94−), two-electron each) of the fully reduced bridge-based 9 were however associated with the azo (–NN–, pap0/pap˙−/pap2−, as shown above) function of pap (Fig. 9).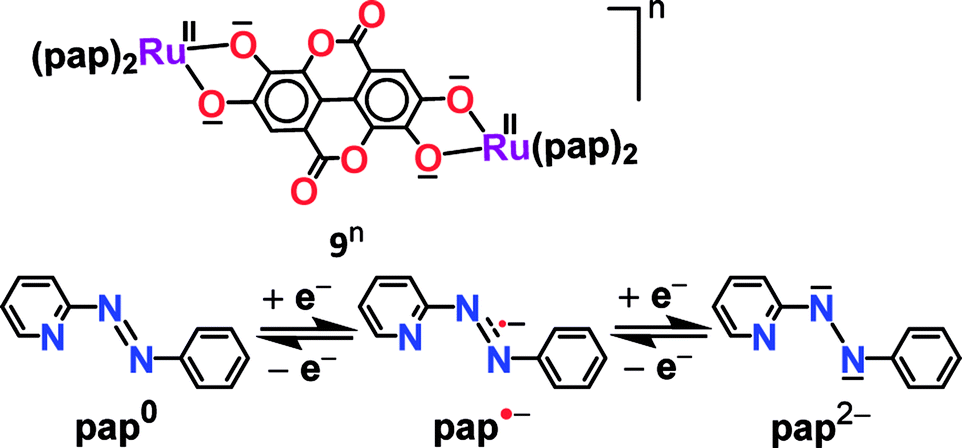
 | ||
| Fig. 8 Cyclic voltammograms of 9 in CH3CN. | ||
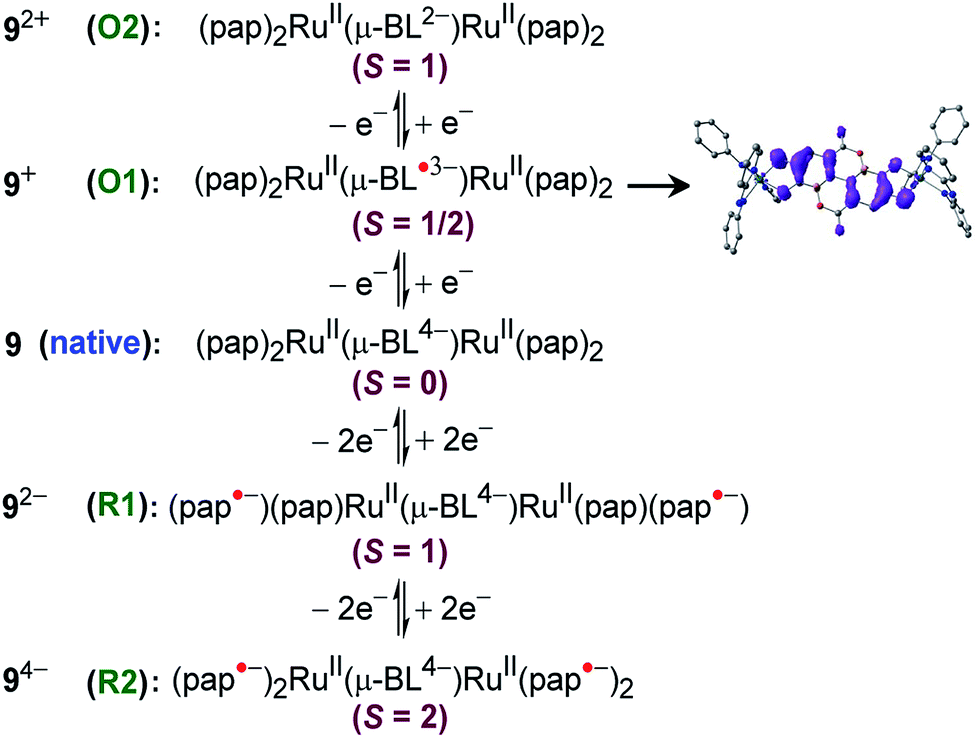 | ||
| Fig. 9 Electronic structural forms along the redox chain including the Mulliken spin density plot for 9+. | ||
3.2. Active ancillary ligand
Structural, spectroscopic and magnetic studies showed that the uncoupled semiquinone ancillary ligands stabilized the tppz-bridged diruthenium(II) complex 10n (n = 0) in the triplet (S = 1, J = −3.07 cm−1) ground state.51 Stepwise oxidations (O1, O2) and reductions (R1, R2) of complex 10 yielded the calculated Kc values of 102 and 104, respectively, for the intermediate states (Fig. 10). The pertinent question regarding the involvement of ancillary ligand (quinone (Q), semiquinone (Q˙−) and catecholate (Q2−)), bridge (tppzo, tppz˙−, tppz2−) and metal (RuII, RuIII, RuIV) in accessible redox processes of 10n was ascertained via the collective consideration of experimental and theoretical investigations. It revealed the preferential participation of ancillary ligands in both oxidation (Q˙− to Q0) and reduction ((Q˙− to Q2−)) processes, leaving the metal ion as the silent spectator (Fig. 11).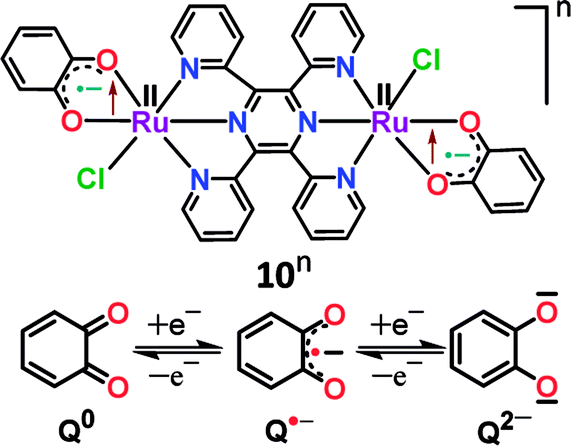
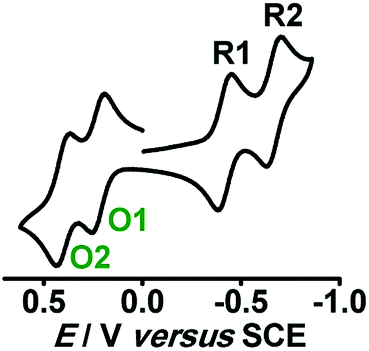 | ||
| Fig. 10 Cyclic voltammogram of 10 in CH3CN. | ||
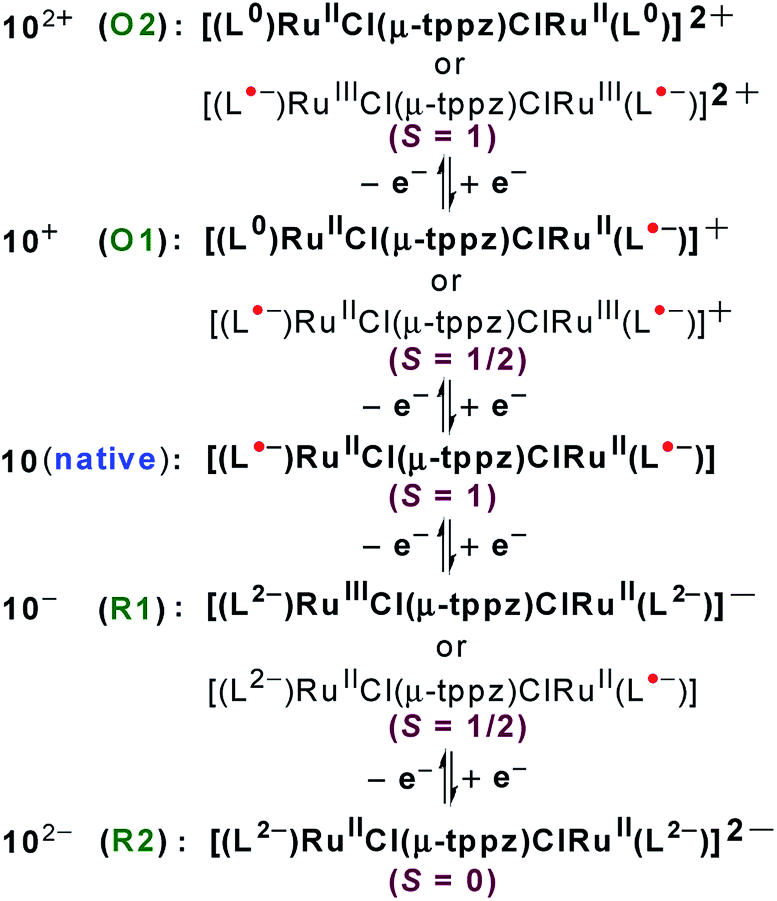 | ||
| Fig. 11 Electronic structural forms of 10n in accessible redox states. Boldface refers to the dominating forms. | ||
3.3. Active bridge, ancillary ligand and metal
Dianionic DPPP2− (H2DPPP = 2,5-dihydro-3,6-di-2-pyridylpyrrolo[3,4-c]pyrrole-1,4-dione)-bridged diruthenium(II) complexes 112+ and 122+ displayed two successive oxidations and multiple reductions within the potential window of ±2.0 V versus SCE (Fig. 12).52 DFT and TD-DFT-supported experimental studies established bridge-based first oxidation (DPPP2− → DPPP˙− (O1, Fig. 12)), followed by metal oxidation (O2, Fig. 12), whereas the first two reversible reductions were associated with DPPP2− (DPPP2− → DPPP˙3−) or the ancillary ligand based on the relative π-accepting strength of the latter (bpy versus pap) (Fig. 13).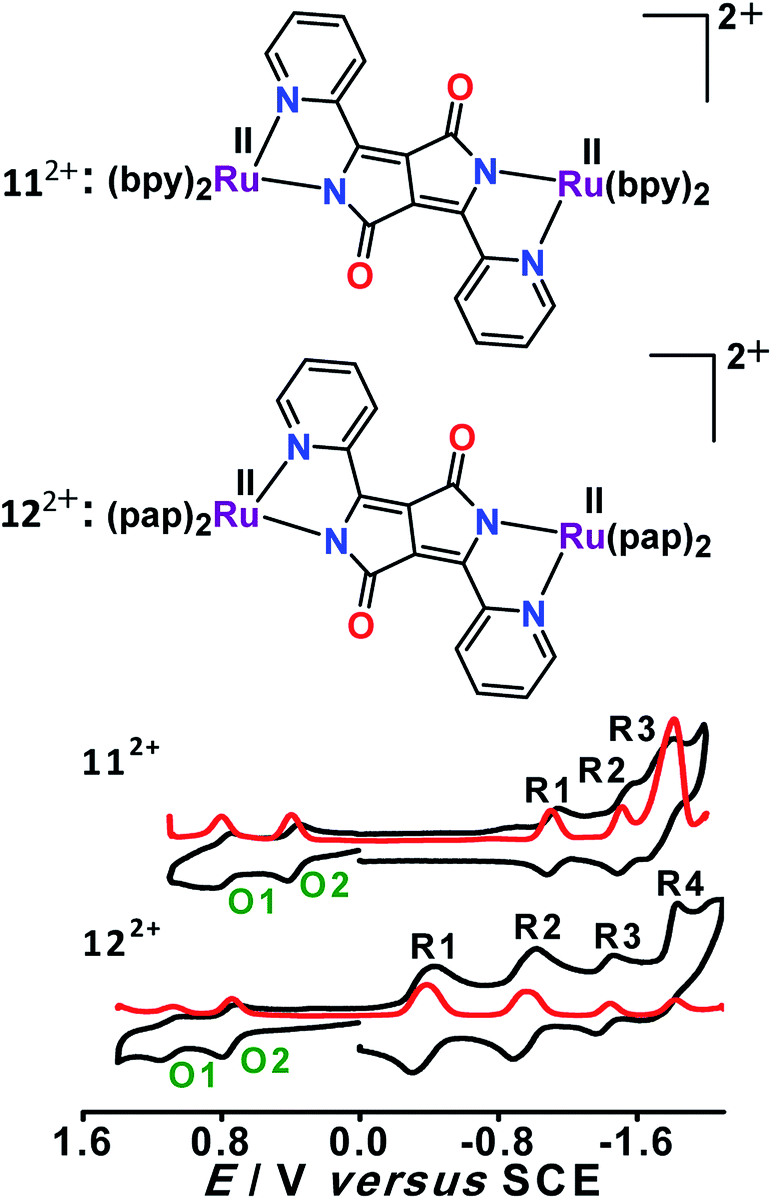 | ||
| Fig. 12 Cyclic (black) and differential pulse (red) voltammograms in CH3CN. | ||
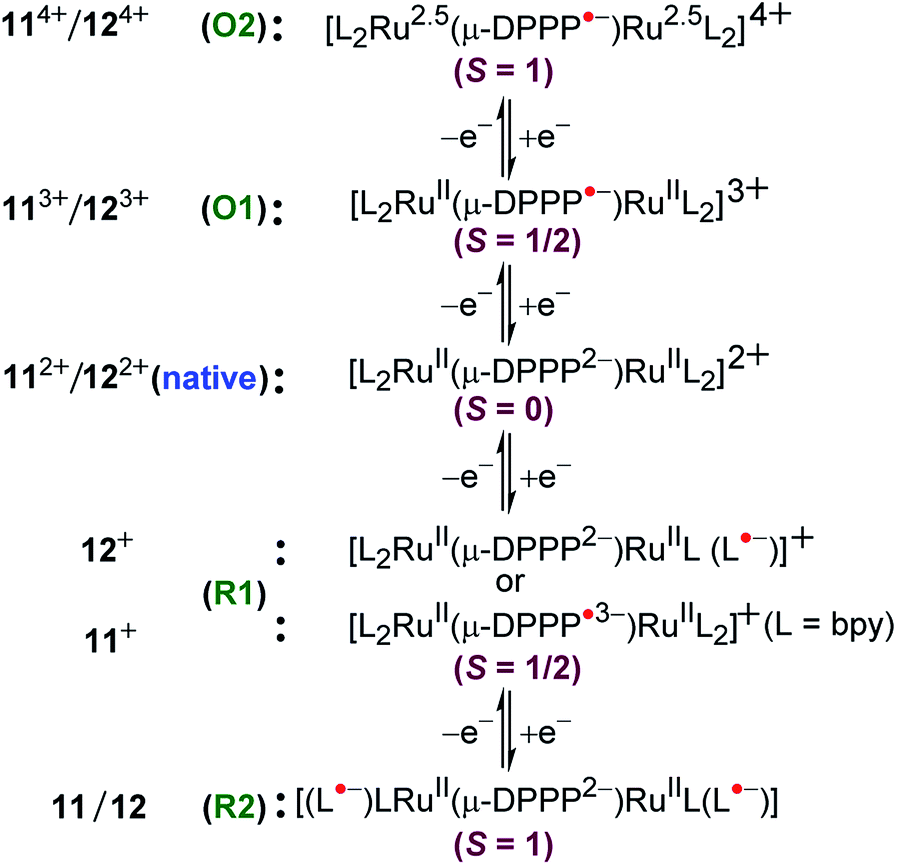 | ||
| Fig. 13 Electronic structural forms of 11n and 12n. | ||
The occurrence of the radical-bridged isovalent situation along the redox chain was also evident by free radical EPR (giso = ∼2.005) and the moderately intense inter-ligand (LLCT) low-energy band in the NIR region (2036 nm, (ε/M−1 cm−1: 2410).
3.4. Unusual spin interaction
Twisted doubly reduced BL2− (BL2− = 1,4-dioxido-2,3-bis(3,5-dimethylpyrazol-1′-yl)benzene)-bridged [(acac)2RuIII(μ-BL)RuIII (acac)2]n (13n (n = 0)) (acac = acetylacetonate) in S = 1 state (J = −37 cm−1) exhibited two successive oxidations (O1, O2) and reductions (R1, R2) (Fig. 14).53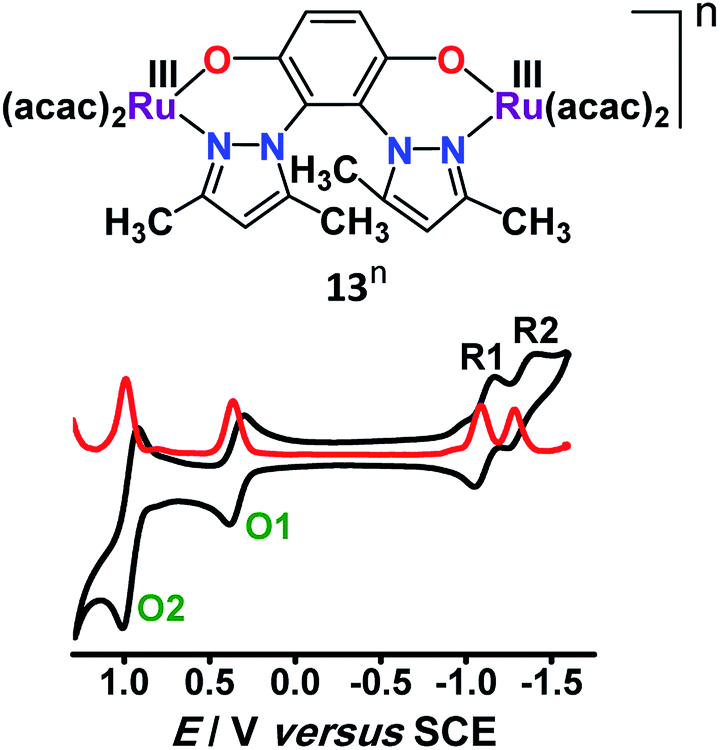 | ||
| Fig. 14 Cyclic (black) and differential pulse (red) voltammograms of 13 in CH3CN. | ||
UV-vis-NIR spectroelectrochemistry (Fig. 15) and EPR (Fig. 16) collectively corroborated metal-based reductions, RuIIIRuIII (13) → RuIIIRuII (13−, R1) → RuIIRuII (132−, R2). The mixed valent 13− displayed an anisotropic RuIII-based EPR spectrum (g1 = 2.40, g2 = 2.19, g3 = 1.79; Δg = 0.061, 〈g〉 = 2.14) and a broad IVCT band at 1520 nm (ε/M−1 cm−1: 1000; Δν1/2 (calculated/experimental): 3898/5000 cm−1), corresponding to the valence localized class II system, as also suggested by Kc of 103.7. However, the IVCT band of 13− disappeared on further reduction to the isovalent RuIIRuII state in 132−. A reversible first oxidation (O1, 13 → 13+) resulted in an intense NIR band at 892 nm (ε/M−1 cm−1: 11600) and a free radical EPR at g = 2.005. The observed free radical EPR of 13+ could be rationalized by considering the electronic form of [(acac)2RuIII(μ-BL˙−)RuIII(acac)2]+ with an up-up-down (↑↑↓) spin configuration, where the antiferromagnetic coupling between the remote metal spins left the unpaired spin on the bridge. The alternate three-spin arrangement with the up-down-up (↑↓↑) spin configuration in the ground state would favor metal-centered spin through antiferromagnetic coupling between the spins associated with one of the ruthenium ions and the radical bridge.
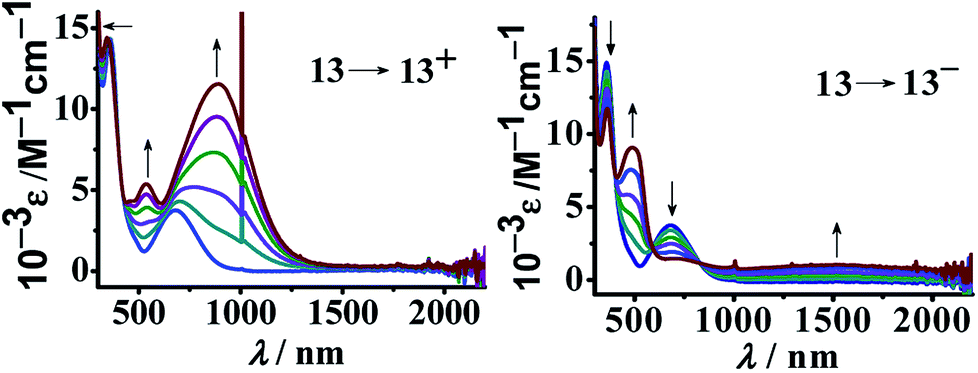 | ||
| Fig. 15 UV-vis-NIR spectroelectrochemistry of 13n in CH3CN. | ||
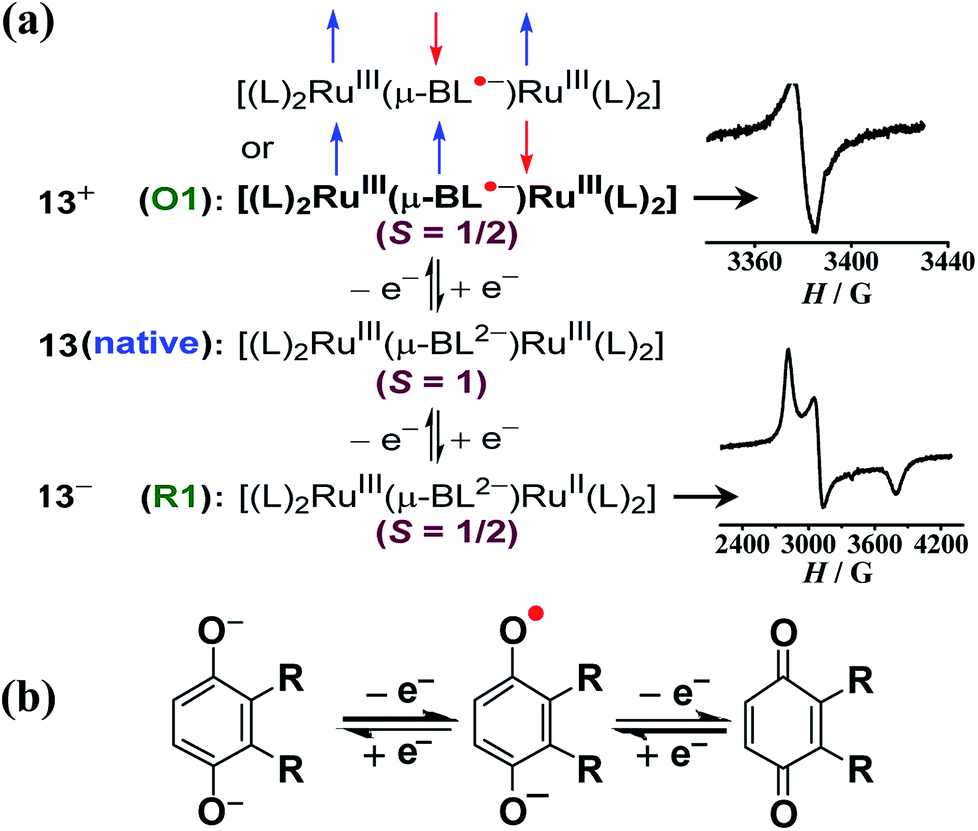 | ||
| Fig. 16 (a) Electronic structural forms of 13n with EPR for the S = 1/2 state in CH3CN. Boldface refers to the correct form. (b) Redox states of the para-quinone unit. | ||
4. Conclusion and perspectives
Although mixed valence aspects of transition metal complexes have been covered quite extensively in existing literature, the present deliberation indicates the further scope of expansion depending on the conspicuous nature of the bridge in a three-state mixed valence system.The present review article essentially highlighted the emerging diverse fundamental aspects relating to the mixed valency in ligand-bridged symmetric and unsymmetric diruthenium complexes by citing selective recent cases. It demonstrated the impact of the electronic nature of both ancillary and bridging ligands in tuning the intramolecular electron transfer process at the mixed valence state (RuIIRuIII or RuIIIRuIV). It also addressed the pertinent additional issues regarding valence and spin situations due to the introduction of redox active ligands in complex frameworks. Moreover, the necessary cautions with special references to the comproportionation constant (Kc) and the possible low-energy (NIR) ligand-to-ligand charge transfer (LLCT) transition instead of the usual metal-based IVCT transition for redox-active ligand-derived systems were noted.
Besides the well explored application potential of mixed valence complexes for molecular wires,54 molecular switches55 or telecommunication devices,56 the recent revelations of superior conductivity of transition metal-derived metal–organic frameworks (beyond the classical discrete metal complexes) due to better orbital overlapping at the mixed valent state57 as well as involvement of mixed valent state in the water oxidation process58 would provide additional impetus in developing more fundamentally challenging or attractive mixed valent systems.
Thus, further research activities with fundamentally rich and diversified mixed valence compounds would likely expand the scope of designing new generation molecular electronic devices and provide a better understanding of the electron transfer phenomenon in a complex molecular set up.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support received from Science and Engineering Research Board (SERB, Department of Science and Technology) and University Grant Commission (Fellowship to A. S. H), New Delhi, India, is gratefully acknowledged. Contributions of all the co-authors are also gratefully acknowledged.Notes and references
- (a) D. B. Brown, Mixed Valency Systems—Applications in Chemistry, Physics and Biology, ed. K. Prassides, Kluwer Academic Publishers, Dordrecht, 1991 Search PubMed; (b) S. F. Nelsen, Chem.–Eur. J., 2000, 6, 581 CrossRef PubMed; (c) P. Aguirre-Etcheverry and D. O'Hare, Chem. Rev., 2010, 110, 4839 CrossRef PubMed; (d) P. J. Low and N. J. Brown, J. Cluster Sci., 2010, 21, 235 CrossRef; (e) M. H. Chisholm and B. J. Lear, Chem. Soc. Rev., 2011, 40, 5254 RSC; (f) A. Hildebrandt and H. Lang, Organometallics, 2013, 32, 5640 CrossRef; (g) W. Kaim, A. Klein and M. Glöckle, Acc. Chem. Res., 2000, 33, 755 CrossRef PubMed.
- (a) L. L. Miller and K. R. Mann, Acc. Chem. Res., 1996, 29, 417 CrossRef; (b) D.-L. Sun, S. V. Rosokha, S. V. Lindeman and J. K. Kochi, J. Am. Chem. Soc., 2003, 125, 15950 CrossRef PubMed; (c) J. Casado, K. Takimiya, T. Otsubo, F. J. Ramirez, J. J. Quirante, R. P. Ortiz, S. R. Gonzalez, M. M. Oliva and J. T. L. Navarrete, J. Am. Chem. Soc., 2008, 130, 14028 CrossRef PubMed; (d) D. Cornelis, E. Franz, I. Asselberghs, K. Clays, T. Verbiest and G. Koeckelberghs, J. Am. Chem. Soc., 2011, 133, 1317 CrossRef PubMed; (e) S. P. Jagtap, S. Mukhopadhyay, V. Coropceanum, G. L. Brizius, J.-L. Bredas and D. M. Collard, J. Am. Chem. Soc., 2012, 134, 7176 CrossRef PubMed; (f) S. T. Schneebeli, M. Frasconi, Z. Liu, Y. Wu, D. M. Gardner, N. L. Strutt, C. Cheng, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 13100 CrossRef PubMed.
- (a) V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676 RSC; (b) A. Klauss, M. Haumann and H. Dau, Proc. Natl. Acad. Sci., 2012, 109, 16035 CrossRef PubMed; (c) T. A. Roelofs, W. Liang, M. J. Latimer, R. M. Cinco, A. Rompel, J. C. Andrews, K. Sauer, V. K. Yachandra and M. P. Klein, Proc. Natl. Acad. Sci., 1996, 93, 3335–3340 CrossRef PubMed.
- (a) R. Boelens and R. Wever, FEBS Lett., 1980, 116, 223 CrossRef PubMed; (b) D. A. Proshlyakov, M. A. Pressler and G. T. Babcock, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8020 CrossRef PubMed.
- (a) H. Beinert, R. H. Holm and E. Münck, Science, 1997, 277, 653 CrossRef PubMed; (b) S. Subramanian, E. C. Duin, S. E. J. Fawcett, F. A. Armstrong, J. Meyer and M. K. Johnson, J. Am. Chem. Soc., 2015, 137, 4567 CrossRef PubMed; (c) S. Rumpel, J. F. Siebel, M. Diallo, C. FarÀs, E. J. Reijerse and W. Lubitz, ChemBioChem, 2015, 16, 1663–1669 CrossRef PubMed; (d) M.-E. Pandelia, D. Bykov, R. Izsak, P. Infossi, M.-T. G. Orticoni, E. Bill, F. Neese and W. Lubitz, Proc. Natl. Acad. Sci., 2013, 110, 483 CrossRef PubMed.
- (a) H. Dau, A. Grundmeier, P. Loja and M. Haumann, Philos. Trans. R. Soc., B, 2008, 363, 1237–1244 CrossRef PubMed; (b) A. Indra, P. W. Menezes and M. Driess, ChemSusChem, 2015, 8, 776 CrossRef PubMed.
- (a) J. M. D. Coey and M. Viret, Adv. Phys, 1999, 48, 167 CrossRef; (b) G. Subías, J. García, J. Blasco, M. G. Proietti, H. Renevier and M. C. Sánchez, Phys. Rev. Lett., 2004, 93, 156408 CrossRef PubMed; (c) T. Mochida, E. Nagabuchi, M. Takahashi and H. Mori, Chem. Commun., 2014, 50, 2481 RSC; (d) A. Dey, X. Xie and E. I. Solomon, Chem. Soc. Rev., 2008, 37, 623 RSC.
- H. He, Y. Zhong, X. Liang, W. Tan, J. Zhu and C. Y. Wang, Sci. Rep., 2015, 5, 10139 CrossRef PubMed.
- (a) J. M. Herrera, A. Bachschmidt, F. Villain, A. Bleuzen, V. Marvaud, W. Wernsdorfer and M. Verdaguer, Philos. Trans. R. Soc., A, 2008, 366, 127 CrossRef PubMed; (b) D. M. Rogers and J. O. Johansson, Mater. Sci. Eng. B, 2018, 227, 28 CrossRef; (c) P. W. Menezes, A. Indra, D. González-Flores, N. R. Sahraie, I. Zaharieva, M. Schwarze, P. Strasser, H. Dau and M. Driess, ACS Catal., 2015, 5, 2017 CrossRef; (d) A. Indra, P. W. Menezes, C. Das, D. Schmeißer and M. Driess, Chem. Commun., 2017, 53, 8641 RSC; (e) P. W. Menezes, A. Indra, V. Gutkin and M. Driess, Chem. Commun., 2017, 53, 8018 RSC; (f) A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeißer, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530 CrossRef PubMed; (g) A. Indra, P. W. Menezes, C. Das, C. Göbel, M. Tallarida, D. Schmeißer and M. Driess, J. Mater. Chem. A, 2017, 5, 5171 RSC.
- (a) M. B. Solomon, T. L. Church and D. M. D'Alessandro, CrystEngComm, 2017, 19, 4049 RSC; (b) A. Indra, U. Paik and T. Song, Angew. Chem., Int. Ed., 2018, 57, 1241 CrossRef PubMed.
- (a) C. Creutz and H. Taube, J. Am. Chem. Soc., 1969, 91, 3988 CrossRef; (b) C. Creutz and H. Taube, J. Am. Chem. Soc., 1973, 95, 1086 CrossRef; (c) U. Fuerholz, H. B. Buergi, F. E. Wagner, A. Stebler, J. H. Ammeter, E. Krausz, R. J. H. Clark, M. J. Stead and A. Ludi, J. Am. Chem. Soc., 1984, 106, 121 CrossRef.
- (a) C. Levanda, K. Bechgaard and D. Cowan, J. Org. Chem., 1976, 41, 2700 CrossRef; (b) D. O. Cowan and F. Kaufman, J. Am. Chem. Soc., 1970, 92, 219–220 CrossRef; (c) T. M. Westerhoff, Angew. Chem., Int. Ed., 1986, 25, 702–717 CrossRef.
- R. D. McCullough, G. B. Kok, K. A. Lerstrup and D. O. Cowan, J. Am. Chem. Soc., 1987, 109, 4115 CrossRef.
- W. Kaim and G. K. Lahiri, Angew. Chem., Int. Ed., 2007, 46, 1778 CrossRef PubMed and references therein.
- (a) H. Taube, Angew. Chem., Int. Ed., 1984, 23, 329 CrossRef; (b) B. S. Brunschwig, C. Creutz and N. Sutin, Chem. Soc. Rev., 2002, 31, 168 RSC; (c) B. S. Brunschwig and N. Sutin, Coord. Chem. Rev., 1999, 187, 233 CrossRef; (d) D. E. Richardson and H. Taube, Coord. Chem. Rev., 1984, 60, 107 CrossRef; (e) D. O. Cowan, C. LeVanda, J. Park and F. Kaufman, Acc. Chem. Res., 1973, 6, 1 CrossRef.
- (a) S. B. Braun-Sand and O. Wiest, J. Phys. Chem. A, 2003, 107, 285 CrossRef; (b) C. S. Lent, B. Isaksen and M. Lieberman, J. Am. Chem. Soc., 2003, 125, 1056 CrossRef PubMed; (c) A. Burgun, B. G. Ellis, T. Roisnel, B. W. Skelton, M. I. Bruce and C. Lapinte, Organometallics, 2014, 33, 4209 CrossRef.
- (a) M. Sturza, C. D. Malliakas, D. E. Bugaris, F. Han, D. Y. Chung and M. G. Kanatzidis, Inorg. Chem., 2014, 53, 12191 CrossRef PubMed; (b) K. Himoto, S. Suzuki, T. Okubo, M. Maekawa and T. Kuroda-Sowa, New J. Chem., 2018, 42, 3995 RSC.
- (a) J. P. Launay, Chem. Soc. Rev., 2001, 30, 386 RSC; (b) S. Bock, O. A. Al-Owaedi, S. G. Eaves, D. C. Milan, M. Lemmer, B. W. Skelton, H. M. Osorio, R. J. Nichols, S. J. Higgins, P. Cea, N. J. Long, T. Albrecht, S. Martín, C. J. Lambert and P. J. Low, Chem.–Eur. J., 2017, 23, 2133 CrossRef PubMed; (c) B. S. Kim, J. M. Beebe, C. Olivier, S. Rigaut, D. Touchard, J. G. Kushmerick, X.-Y. Zhu and C. D. Frisbie, J. Phys. Chem. C, 2007, 111, 7521 CrossRef; (d) L. Luo, A. Benameur, P. Brignou, S. H. Choi, S. Rigaut and C. D. Frisbie, J. Phys. Chem. C, 2011, 115, 19955 CrossRef; (e) N. A. Wasio, R. C. Quardokus, R. P. Forrest, S. A. Corcelli, Y. Lu, C. S. Lent, F. Justaud, C. Lapinte and S. A. Kandel, J. Phys. Chem. C, 2012, 116, 25486 CrossRef; (f) E. Wuttke, F. Pevny, Y.-M. Hervault, L. Norel, M. Drescher, R. F. Winter and S. Rigaut, Inorg. Chem., 2012, 51, 1902 CrossRef PubMed; (g) F. Lissel, T. Fox, O. Blacque, W. Polit, R. F. Winter, K. Venkatesan and H. Berke, J. Am. Chem. Soc., 2013, 135, 3826 CrossRef PubMed.
- (a) A. B. Ricks, G. C. Solomon, M. T. Colvin, A. M. Scott, K. Chen, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2010, 132, 15427 CrossRef PubMed; (b) P. J. Mosher, G. P. A. Yap and R. J. Crutchley, Inorg. Chem., 2001, 40, 1189 CrossRef PubMed; (c) R. H. Laye, S. M. Couchman and M. D. Ward, Inorg. Chem., 2001, 40, 4089 CrossRef PubMed; (d) M. D. Todd, A. Nitzan and M. A. Ratner, J. Phys. Chem., 1993, 97, 29 CrossRef.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247 CrossRef.
- (a) S. Dammers, T. P. Zimmermann, S. Walleck, A. Stammler, H. Bögge, E. Bill and T. Glaser, Inorg. Chem., 2017, 56, 1779 CrossRef PubMed; (b) S. Ghumaan and G. K. Lahiri, J. Chem. Sci., 2006, 118, 537 CrossRef; (c) Y.-W. Zhong, Z.-L. Gong, J.-Y. Shao and J. Yao, Coord. Chem. Rev., 2016, 312, 22 CrossRef; (d) H.-N. Nie, J.-Y. Shao, C.-J. Yao and Y.-W. Zhong, Chem. Commun., 2014, 50, 10082 RSC.
- (a) M. D. Ward, Chem. Soc. Rev., 1995, 24, 121 RSC; (b) J. A. McCleverty and M. D. Ward, Acc. Chem. Res., 1998, 31, 842 CrossRef; (c) D. Astruc, Acc. Chem. Res., 1997, 30, 383 CrossRef.
- (a) D. E. Richardson and H. Taube, J. Am. Chem. Soc., 1983, 105, 40 CrossRef; (b) L. T. Zhang, J. Ko and M. J. Ondrechen, J. Am. Chem. Soc., 1987, 109, 1666 CrossRef; (c) S. D. Glover, J. C. Goeltz, B. J. Lear and C. P. Kubiak, Coord. Chem. Rev., 2010, 254, 331 CrossRef; (d) S. D. Glover, J. C. Goeltz, B. J. Lear and C. P. Kubiak, Eur. J. Inorg. Chem., 2009, 585 CrossRef; (e) C. P. Kubiak, Inorg. Chem., 2013, 52, 5663 CrossRef PubMed; (f) B. J. Lear, S. D. Glover, J. C. Salsman, C. H. Londergan and C. P. Kubiak, J. Am. Chem. Soc., 2007, 129, 12772 CrossRef PubMed.
- (a) R. J. Crutchley, Adv. Inorg. Chem., 1994, 41, 273 CrossRef; (b) C. E. B. Evans, M. L. Naklicki, A. R. Rezvani, C. A. White, V. V. Kondratiev and R. J. Crutchley, J. Am. Chem. Soc., 1998, 120, 13096 CrossRef; (c) M. Al-Noaimi, G. P. A. Yap and R. J. Crutchley, Inorg. Chem., 2004, 43, 1770 CrossRef PubMed; (d) R. J. Crutchley, Angew. Chem., Int. Ed., 2005, 44, 6452 CrossRef PubMed; (e) D. M. D'Alessandro and F. R. Keene, Dalton Trans., 2004, 3950 RSC; (f) R. F. Winter, Organometallics, 2014, 33, 4517 CrossRef; (g) S. Santi, A. Bisello, R. Cardena and A. Donoli, Dalton Trans., 2015, 44, 5234 RSC.
- (a) K. D. Demadis, D. C. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655 CrossRef PubMed; (b) M. A. Fox, B. L. Guennic, R. L. Roberts, D. A. Brue, D. S. Yufit, J. A. K. Howard, G. Manca, J. Halet, F. Hartl and P. J. Low, J. Am. Chem. Soc., 2011, 133, 18433 CrossRef PubMed; (c) F. Ding, H. Wang, Q. Wu, T. V. Voorhis, S. Chen and J. P. Konopelski, J. Phys. Chem. A, 2010, 114, 6039 CrossRef PubMed; (d) E. Göransson, R. Emanuelsson, K. Jorner, T. F. Markle, L. Hammarström and H. Ottosson, Chem. Sci., 2013, 4, 3522 RSC; (e) D. M. D'Alessandro and F. R. Keene, Chem. Soc. Rev., 2006, 35, 424 Search PubMed.
- (a) N. S. Hush, Prog. Inorg. Chem., 1967, 8, 391 Search PubMed; (b) C. Creutz, M. D. Newton and N. Sutin, J. Photochem. Photobiol., A, 1994, 82, 47 CrossRef.
- D. H. Oh, M. Sano and S. G. Boxer, J. Am. Chem. Soc., 1991, 113, 6880 CrossRef.
- (a) N. S. Hush, Coord. Chem. Rev., 1985, 64, 135 CrossRef; (b) N. A. Lewis, R. R. McNeer and D. V. Taveras, Inorg. Chim. Acta, 1994, 225, 89 CrossRef.
- (a) J. T. Hupp, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier Science, Amsterdam, 2003, p. 709 Search PubMed; (b) P. Chen and T. J. Meyer, Chem. Rev., 1998, 98, 1439 CrossRef PubMed.
- (a) J. C. Salsman and C. P. Kubiak, J. Am. Chem. Soc., 2005, 127, 2382 CrossRef PubMed; (b) C. H. Londergan and C. P. Kubiak, Chem.–Eur. J., 2003, 9, 5962 CrossRef PubMed; (c) C. H. Londergan and C. P. Kubiak, J. Phys. Chem. A, 2003, 107, 9301 CrossRef; (d) C. H. Londergan, J. C. Salsman, S. Ronco, L. M. Dolkas and C. P. Kubiak, J. Am. Chem. Soc., 2002, 124, 6236 CrossRef PubMed; (e) T. Ito, T. Hamaguchi, H. Nagino, T. Yamaguchi, J. Washington and C. P. Kubiak, Science, 1997, 277, 660 CrossRef; (f) T. Ito, T. Hamaguchi, H. Nagino, T. Yamaguchi, H. Kido, I. S. Zaverine, T. Richmond, J. Washington and C. P. Kubiak, J. Am. Chem. Soc., 1999, 121, 4625 CrossRef.
- (a) W. Kaim and B. Sarkar, Coord. Chem. Rev., 2013, 257, 1650 CrossRef; (b) S. Patra, B. Sarkar, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2003, 42, 6469 CrossRef PubMed.
- P. Mondal, F. Ehret, M. Bubrin, A. Das, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2013, 52, 8467 CrossRef PubMed.
- (a) W. Kaim and V. Kasack, Inorg. Chem., 1990, 29, 4696 CrossRef; (b) T. Scheiring, W. Kaim, J. A. Olabe, A. R. Parise and J. Fiedler, Inorg. Chim. Acta, 2000, 125, 300 Search PubMed.
- C. G. Atwood and W. E. Geiger, J. Am. Chem. Soc., 2000, 122, 5477 CrossRef.
- (a) M. Chatterjee, P. Mondal, K. Beyer, A. Paretzki, W. Kaim and G. K. Lahiri, Dalton Trans., 2017, 46, 5091 RSC; (b) M. A. Ansari, A. Mandal, K. Beyer, A. Paretzki, B. Schwederski, W. Kaim and G. K. Lahiri, Dalton Trans., 2017, 46, 15589 RSC; (c) P. Mondal, M. Chatterjee, A. Paretzki, K. Beyer, W. Kaim and G. K. Lahiri, Inorg. Chem., 2016, 55, 3105 CrossRef PubMed; (d) M. Moscherosch, E. WaldhIr, H. Binder, W. Kaim and J. Fiedler, Inorg. Chem., 1995, 34, 4326 CrossRef; (e) S. Ghumaan, S. Mukherjee, S. Kar, D. Roy, S. M. Mobin, R. B. Sunoj and G. K. Lahiri, Eur. J. Inorg. Chem., 2006, 4426 CrossRef.
- (a) T. Kundu, D. Schweinfurth, B. Sarkar, T. K. Mondal, J. Fiedler, S. M. Mobin, V. G. Puranik, W. Kaim and G. K. Lahiri, Dalton Trans., 2012, 41, 13429 RSC; (b) T. Kundu, B. Sarkar, T. K. Mondal, J. Fiedler, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2010, 49, 6565 CrossRef PubMed; (c) M. Koley, B. Sarkar, S. Ghumaan, E. Bulak, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2007, 46, 3736 CrossRef PubMed; (d) S. Ghumaan, B. Sarkar, N. Chanda, M. Sieger, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2006, 45, 7955 CrossRef PubMed; (e) N. Chanda, B. Sarkar, S. Kar, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2004, 43, 5128 CrossRef PubMed; (f) N. Chanda, B. Sarkar, J. Fiedler, W. Kaim and G. K. Lahiri, Dalton Trans., 2003, 3550 RSC; (g) N. Chanda, R. H. Laye, S. Chakraborty, R. L. Paul, J. C. Jeffery, M. D. Ward and G. K. Lahiri, J. Chem. Soc., Dalton Trans., 2002, 3496 RSC.
- (a) R. Hage, J. G. Haasnoot, H. A. Nieuwenhuis, J. Reedijk, D. J. A. D. Ridder and J. G. Vos, J. Am. Chem. Soc., 1990, 112, 9245 CrossRef; (b) T. Ito, N. Imai, T. Yamaguchi, T. Hamaguchi, C. H. Londergan and C. P. Kubiak, Angew. Chem., Int. Ed., 2004, 43, 1376 CrossRef PubMed; (c) J. C. Salsman, S. Ronco, C. H. Londergan and C. P. Kubiak, Inorg. Chem., 2006, 45, 547 CrossRef PubMed; (d) C. H. Londergan, J. C. Salsman, B. J. Lear and C. P. Kubiak, Chem. Phys., 2006, 324, 57 CrossRef; (e) P. V. Bernhardt, F. Bozoglián, M. Font-Bardía, M. Martínez, A. P. Meacham, B. Sienra and X. Solans, Eur. J. Inorg. Chem., 2007, 5270 CrossRef; (f) S. D. J. McKinnon, B. O. Patrick, A. B. P. Lever and R. G. Hicks, Inorg. Chem., 2013, 52, 8053 CrossRef PubMed; (g) D. M. Dattelbaum, C. M. Hartshorn and T. J. Meyer, J. Am. Chem. Soc., 2002, 124, 4938 CrossRef PubMed; (h) M. Dürr, J. Klein, A. Kahnt, S. Becker, R. Puchta, B. Sarkar and I. Ivanović-Burmazović, Inorg. Chem., 2017, 56, 14912 CrossRef PubMed; (i) A. Das, T. M. Scherer, A. D. Chowdhury, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2012, 51, 1675 CrossRef PubMed.
- (a) H. Agarwala, T. Scherer, S. Maji, T. K. Mondal, S. M. Mobin, J. Fiedler, F. A. Urbanos, R. Jiménez-Aparicio, W. Kaim and G. K. Lahiri, Chem.–Eur. J., 2012, 18, 5667 CrossRef PubMed; (b) S. Kar, B. Sarkar, S. Ghumaan, D. Roy, F. A. Urbanos, J. Fiedler, R. B. Sunoj, R. Jiménez-Aparicio, W. Kaim and G. K. Lahiri, Inorg. Chem., 2005, 44, 8715 CrossRef PubMed; (c) Y. Hoshino, S. Higuchi, J. Fiedlar, C. Y. Su, A. Knödler, B. Schwederski, B. Sarkar, H. Hartmann and W. Kaim, Angew. Chem., Int. Ed., 2003, 42, 674 CrossRef PubMed.
- T. D. Glover, B. J. Lear, J. C. Salsman, C. H. Londergan and C. P. Kubiak, Philos. Trans. R. Soc., A, 2008, 366, 177 CrossRef PubMed.
- S. Patra, B. Sarkar, S. Ghumaan, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2004, 43, 6108 CrossRef PubMed.
- S. Chellamma and M. Lieberman, Inorg. Chem., 2001, 40, 3177 CrossRef PubMed.
- (a) M. D. Ward, Inorg. Chem., 1996, 35, 1712 CrossRef PubMed; (b) B. D. Yeomans, L. S. Kelso, P. A. Tregloan and F. R. Keene, Eur. J. Inorg. Chem., 2009, 585 Search PubMed; (c) M. D. Ward and J. A. McCleverty, J. Chem. Soc., Dalton Trans., 2002, 275 RSC; (d) S. Patra, B. Sarkar, S. Ghumaan, J. Fiedler, W. Kaim and G. K. Lahiri, Dalton Trans., 2004, 754 RSC.
- A. Das, T. Scherer, S. Maji, T. K. Mondal, S. M. Mobin, F. A. Urbanos, R. Jiménez-Aparicio, W. Kaim and G. K. Lahiri, Inorg. Chem., 2011, 50, 7040 CrossRef PubMed.
- (a) J.-P. Sutter, D. M. Grove, M. Beley, J.-P. Collin, N. Veldman, A. L. Spek, J.-P. Sauvage and G. van Koten, Angew. Chem., Int. Ed., 1994, 33, 1282 CrossRef; (b) J. P. Collin, P. Laine, J. P. Launay, J. P. Sauvage and A. Sour, J. Chem. Soc., Chem. Commun., 1993, 434 RSC.
- (a) S. Chakraborty, R. H. Laye, P. Munshi, R. L. Paul, M. D. Ward and G. K. Lahiri, J. Chem. Soc., Dalton Trans., 2002, 2348 RSC; (b) Y. Halpin, D. Dini, H. M. Y. Ahmed, L. Cassidy, W. R. Browne and J. G. Vos, Inorg. Chem., 2010, 49, 2799 CrossRef PubMed; (c) H. M. Y. Ahmed, N. Coburn, D. Dini, J. J. D. Jong, C. de Villani, W. R. Browne and J. G. Vos, Inorg. Chem., 2011, 50, 5861 CrossRef PubMed; (d) M. Heilmann, S. Frantz, W. Kaim, J. Fiedler and C. Duboc, Inorg. Chim. Acta, 2006, 359, 821–829 CrossRef; (e) A. das, T. M. Scherer, S. M. Mobin, W. Kaim and G. K. Lahiri, Chem.–Eur. J., 2012, 18, 11007 CrossRef PubMed; (f) F. Scandola, R. Argazzi, C. A. Bignozzi, C. Chiorboli, M. T. Indelli and M. A. Rampi, Coord. Chem. Rev., 1993, 125, 283 CrossRef; (g) H. Vahrenkamp, A. Geiss and G. N. Richardson, J. Chem. Soc., Dalton Trans., 1997, 3643 RSC; (h) N. Zhu and H. Vahrenkamp, Chem. Ber., 1997, 130, 1241 CrossRef.
- A. Das, T. Kundu, S. M. Mobin, J. L. Priego, R. Jiménez-Aparicio and G. K. Lahiri, Dalton Trans., 2013, 42, 13733 RSC.
- (a) C. Remenyi and M. Kaupp, J. Am. Chem. Soc., 2005, 127, 11399 CrossRef PubMed; (b) S. Roy, B. Sarkar, D. Bubrin, M. Niemeyer, S. Záliš, G. K. Lahiri and W. Kaim, J. Am. Chem. Soc., 2008, 130, 15230 CrossRef PubMed; (c) A. Das, P. Ghosh, S. Plebst, B. Schwederski, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 3376 CrossRef PubMed.
- (a) P. Ghosh, S. Panda, S. Banerjee and G. K. Lahiri, Inorg. Chem., 2017, 56, 10735 CrossRef PubMed; (b) S. Panda, A. Mandal, P. Ghosh and G. K. Lahiri, Inorg. Chem., 2017, 56, 14900 CrossRef PubMed; (c) P. Ghosh, S. Banerjee and G. K. Lahiri, Inorg. Chem., 2017, 55, 12832 CrossRef PubMed; (d) A. S. Hazari, A. Paretzki, J. Fiedler, S. Zalis, W. Kaim and G. K. Lahiri, Dalton Trans., 2016, 45, 18241 RSC; (e) M. A. Ansari, A. Mandal, A. Paretzki, K. Beyer, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2016, 55, 5655 CrossRef PubMed; (f) P. Mondal, A. Das and G. K. Lahiri, Inorg. Chem., 2016, 55, 1208 CrossRef PubMed; (g) P. Ghosh and G. K. Lahiri, Dalton Trans., 2016, 45, 5240 RSC; (h) A. Mandal, B. Schwederski, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 8126 CrossRef PubMed; (i) P. Mondal, R. Ray, A. Das and G. K. Lahiri, Inorg. Chem., 2015, 54, 3012 CrossRef PubMed; (j) P. Ghosh, R. Ray, A. Das and G. K. Lahiri, Inorg. Chem., 2014, 53, 10695 CrossRef PubMed; (k) P. Mondal, S. Plebst, R. Ray, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2014, 53, 9348 CrossRef PubMed; (l) H. Masui, A. L. Freda, M. C. Zerner and A. B. P. Lever, Inorg. Chem., 2000, 39, 141 CrossRef PubMed; (m) N. Roy, S. Sproules, T. Weyhermueller and K. Wieghardt, Inorg. Chem., 2009, 39, 141 Search PubMed; (n) B. Sarkar, S. Patra, J. Fiedler, R. B. Sunoj, D. Janardanan, S. M. Mobin, M. Niemeyer, G. K. Lahiri and W. Kaim, Angew. Chem., Int. Ed., 2005, 44, 5655 CrossRef PubMed; (o) B. Sarkar, S. Patra, J. Fiedler, R. B. Sunoj, D. Janardanan, G. K. Lahiri and W. Kaim, J. Am. Chem. Soc., 2008, 130, 3532 CrossRef PubMed.
- (a) J. S. Miller and K. S. Min, Angew. Chem., Int. Ed., 2009, 48, 262 CrossRef PubMed; (b) S. Ghumaan, B. Sarkar, S. Patra, J. V. Slageren, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2005, 44, 3210 CrossRef PubMed.
- A. Mandal, A. Grupp, B. Schwederski, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 10049 CrossRef PubMed.
- (a) T. Kundu, B. Sarkar, T. K. Mondal, S. M. Mobin, F. A. Urbanos, J. Fiedler, R. Jiménez-Aparicio, W. Kaim and G. K. Lahiri, Inorg. Chem., 2011, 50, 4753 CrossRef PubMed; (b) A. K. Das, B. Sarkar, J. Fiedler, S. Zális, I. Hartenbach, S. Strobel, G. K. Lahiri and W. Kaim, J. Am. Chem. Soc., 2009, 131, 8895 CrossRef PubMed.
- A. S. Hazari, A. Mandal, K. Beyer, A. Paretzki, W. Kaim and G. K. Lahiri, Inorg. Chem., 2017, 56, 2992 CrossRef PubMed.
- D. Kumbhakar, B. Sarkar, S. Maji, S. M. Mobin, J. Fiedler, F. A. Urbanos, R. Jiménez-Aparicio, W. kaim and G. K. Lahiri, J. Am. Chem. Soc., 2008, 130, 17575 CrossRef PubMed.
- (a) A. Klein, O. Lavastre and J. Fiedler, Organometallics, 2006, 25, 635 CrossRef; (b) G.-L. Xu, G. Zou, Y.-H. Ni, M. C. DeRosa, R. J. Crutchley and T. Ren, J. Am. Chem. Soc., 2003, 125, 10057 CrossRef PubMed; (c) J.-H. Tang, J.-H. Shao, Y.-Q. He, Si.-H. Wu, J. Yao and Y.-W. Zhong, Chem.–Eur. J., 2016, 22, 10341 CrossRef PubMed; (d) W. P. Forrest, Z. Cao, K. M. Hassell, B. M. Prentice, P. E. Fanwick and T. Ren, Inorg. Chem., 2012, 51, 3261 CrossRef PubMed.
- (a) D. Chong, X. Wan and J. Zhang, J. Mater. Chem. C, 2017, 5, 6442 RSC; (b) J. A. Christie, R. P. Forrest, S. A. Corcelli, N. A. Wasio, R. C. Quardok, R. Brown, S. A. Kandel, Y. Lu, C. S. Lent and K. W. Henderson, Angew. Chem., Int. Ed., 2015, 54, 15448 CrossRef PubMed; (c) T. Matsumoto, G. N. Newton, T. Shiga, S. Hayami, Y. Matsui, H. Okamoto, R. Kumai, Y. Murakami and H. Oshio, Nat. Commun., 2014, 5, 3865 CrossRef PubMed.
- G. LeClair and Z. Y. Wand, J. Solid State Electrochem., 2009, 13, 365 CrossRef.
- (a) R. Murase, C. F. Long and D. M. D'Alessandro, Inorg. Chem., 2017, 56, 14373 CrossRef PubMed; (b) L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566 CrossRef PubMed.
- (a) P. Zhang, X. Hou, L. Liu, J. Mi and M. Dong, J. Phys. Chem. C, 2015, 119, 28028 CrossRef; (b) F. Bozoglian, S. Romain, M. Z. Ertem, T. K. Todorova, C. Sens, J. Mola, M. Rodriguez, I. Romero, J. B.-. Buchholz, X. Fontrodona, C. J. Cramer, L. Gagliardi and A. Llobet, J. Am. Chem. Soc., 2009, 131, 15176 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |