DOI:
10.1039/C7NH00167C
(Communication)
Nanoscale Horiz., 2018,
3, 305-311
A programmable lipid-polymer hybrid nanoparticle system for localized, sustained antibiotic delivery to Gram-positive and Gram-negative bacterial biofilms
Received
17th October 2017
, Accepted 29th January 2018
First published on 8th February 2018
Abstract
Bacteria enmeshed in an extracellular matrix, biofilms, exhibit enhanced antibiotic tolerance. Coupled with the rapid emergence of multidrug-resistant strains, the current cohorts of antibiotics are becoming ineffective. Alternative antimicrobial approaches are therefore urgently needed to overcome recalcitrant biofilm infections. Here, we propose the use of a non-toxic lipid-polymer hybrid nanoparticle (LPN) system composed of a solid polymer core (i.e. PLGA; poly lactic-co-glycolic acid) and a cationic lipid shell (i.e. DOTAP) for localized, sustained release of antimicrobial agents to bacterial biofilms. LPNs were synthesized through a simple, robust self-assembly approach. LPNs of uniform particle size (i.e. 100–130 nm), efficiently encapsulated (up to 95%) bioimaging molecules or antibiotics and provided controlled release of the latter. The cationic lipid coating enabled the LPN to anchor onto surfaces of a diverse range of Gram-positive and Gram-negative bacterial pathogens, either in the planktonic or biofilm form. Consistently, the LPN formulations reduced more than 95% of biofilm activity at concentrations that were 8 to 32-fold lower than free antibiotics. These data clearly indicate that these novel formulations could be a useful strategy to enhance the efficacy of antimicrobials against planktonic cells and biofilms of diverse species.
Conceptual insights
Nanoparticle-based drug delivery has generated profound impact on the medicinal and healthcare systems. Liposomal and polymeric nanoparticles, in particular, have been explored intensively for antimicrobial applications, and many are under various stages of pre-clinical and clinical trials. Despite some promising outcomes, these individual formulations suffer from various degrees of limitations such as low drug loading efficiency, stability, costly production and scalability. Herein, we report for the first time, the formulation of a robust nanoscale carrier that integrates the liposomal and the polymeric systems for the delivery of different classes of antibiotics. This hybrid nanoparticle system not only retains the features of liposomes, such as superior biofilm binding affinity and penetrating ability, but also provides controlled and sustained release of drugs, at enhanced encapsulation efficiencies. More importantly, this hybrid formulation can effectively inactivate biofilms of diverse species at concentrations dramatically lower than the free-drug counterparts. Given the threat of the global rise in multidrug-resistance, a versatile, economical, scalable and efficient delivery system, such as these lipid-polymer hybrid nanoparticles as demonstrated here, is thus critically and timely fitting to be the next-generation, universal antimicrobial carrier to treat various forms of biofilm infections while mitigating the development of resistance.
|
Introduction
Microbial infection is emerging as one of the most deadly infectious diseases due to the rapid evolution of antimicrobial tolerant and resistant strains.1 The National Institute of Health (NIH) estimated that 65–80% of the microbial infections occurring in the human body are biofilm-mediated. Biofilms are represented as structured groups of bacterial cells housed within self-secreted extracellular matrix.2 This biopolymeric matrix provides numerous fitness benefits to bacteria, including protection from environmental stresses, enhanced nutrient availability and increased resilience via phenotypic diversification.3,4 Thus, the biofilm mode of growth is generally recognized as an adaptive survival mechanism that allows bacteria to thrive in many environments.
Several mechanisms have been identified for biofilm-mediated antimicrobial resistance and tolerance.5,6 Clinical studies suggest that the biofilm matrix is the key defending barrier responsible not only for shielding bacteria from the immune phagocytic cells, but also for protecting them from antibiotics by reducing the diffusivity of antimicrobial agents into the biofilms.6 Antimicrobial agents can also be inactivated via binding to biofilm matrix components or by extracellular enzymatic degradation. Bacteria growing within a biofilm matrix can become physiologically stratified, where some cells have lower metabolic activities, rendering bacterial cells less susceptible to certain antibiotics and hence leading to recalcitrant infections.7–9 The high density of bacteria within biofilms, on the other hand, provides an ideal niche for horizontal gene transfer, allowing the transmission of resistance genes across different species and thus contributing to the development of multidrug-resistant strains.10 Although the conventional use of antibiotics has been highly successful in combating bacterial infections in the last century, innovative strategies are now urgently needed to treat biofilm-mediated infections.
Nanoparticle-based antimicrobial delivery is one possible approach that has been widely explored to manage biofilm infections.11–16 Advantages of nanoparticle delivery systems include enhanced drug solubility, tuneable drug release, multiple drug co-delivery, reduced systemic toxicity and targeted drug delivery.11 It is hypothesized that an efficient antimicrobial delivery system will not only enhance the therapeutic potency of antimicrobials but will also be a practical and sustainable solution to mitigate the emergence and development of drug resistance.11 Liposomal and polymeric nanoparticles are among the most commonly used carriers for antimicrobial delivery owing to their biocompatibility, versatility for surface modification, and capability to encapsulate diverse range of small hydrophobic and hydrophilic molecules.11,15–22 Liposomal formulations are particularly primed for their ability to fuse with bacterial outer membrane to deliver antibiotics directly to the interior of bacteria (e.g. Fluidosomes™), while polymeric encapsulation strategies allow for sustained and controlled drug release.17 Despite the attractive features for antimicrobial treatment, the development of liposomal formulations is constrained by its packaging stability, low drug loading efficiency, scalability and costly production.15 In comparison, polymeric formulations have limited affinity for bacterial cells/biofilms, low biofilm penetration and are ineffective in the encapsulation of hydrophilic molecules.17 The concept of hybridizing liposomal and polymeric systems, which would synergize the biofilm binding affinity of liposomes and the structural integrity afforded by the core polymer, is thus appealing as a promising, next-generation anti-biofilm drug carrier.
In this study, we have developed a highly programmable, reproducible and scalable lipid-polymer hybrid nanoparticle (LPN) system via a simple and economical emulsion approach. The LPNs (i.e. PLGA/DOTAP) were able to bind to a diverse range of bacterial species, including both Gram-negative and Gram-positive pathogens in either planktonic or biofilm mode with superior affinity compared to the PLGA nanoparticles. These LPNs were highly uniform in size with an ideal biofilm penetrating dimension of 100–130 nm, capable of entrapping different antibiotics at high drug loading efficiencies, and with sustained and controlled releasing features. Compared to the free antibiotics, the lipid-polymer formulations achieved a remarkable decrease in MICs as well as a significant increase in biofilm inhibition regardless of the bacterial species. This highlights the potential of LPN system for antimicrobials delivery against recalcitrant bacterial biofilms.
Experimental section
Materials
PLGA (lactide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolide molar ratio 50:50, intrinsic viscosity: 1.18) was purchased from Purac. DOTAP and DSPE-PEG were purchased from Avanti Polar Lipids (Alabaster, AL, USA). All antibiotics and lecithin were purchased from Sigma-Aldrich. Additional chemicals were obtained commercially at analytical grade.
:glycolide molar ratio 50:50, intrinsic viscosity: 1.18) was purchased from Purac. DOTAP and DSPE-PEG were purchased from Avanti Polar Lipids (Alabaster, AL, USA). All antibiotics and lecithin were purchased from Sigma-Aldrich. Additional chemicals were obtained commercially at analytical grade.
Bacterial strains
Streptococcus thermophilus LMD-9, Enterococcus faecalis OG1RF, Staphylococcus aureus ATCC25923, Staphylococcus aureus USA300, Escherichia coli BL21, Escherichia coli UTI89, Pantoea stewartii R067d and Pseudomonas aeruginosa PAO1 (Table 1) were routinely maintained in tryptic soy broth (TSB) or agar (TSA; 1.5% w/v) (Oxoid, UK). P. stewartii R067d was cultivated at 30 °C for 24 h while other bacteria were grown at 37 °C for 24 h.
Table 1 Characterization of bacterial strains
| Species |
Strain |
Gram |
Characteristic |
Reference or source |
|
American type culture collection (atcc) number.
|
|
S. thermophilus
|
LMD-9 |
Positive |
Probiotics |
ATCC BAA-491a |
|
E. faecalis
|
OG1RF |
Positive |
Clinical isolate |
Dunny et al.23 |
|
S. aureus
|
Seattle 1945 |
Positive |
Clinical isolate |
ATCC 25923a |
|
S. aureus
|
USA300 |
Positive |
Clinical isolate |
Diep et al.24 |
|
E. coli
|
BL21 |
Negative |
Laboratory strain |
Studier and Moffatt25 |
|
E. coli
|
UTI89 |
Negative |
Clinical isolate |
Chen et al.26 |
|
P. stewartii
|
R067d |
Negative |
Environmental isolate |
Tan et al.27 |
|
P. aeruginosa
|
PAO1 |
Negative |
Environmental isolate |
ATCC BAA-47a |
Fabrication of lipid-polymer hybrid nanoparticles
Encapsulation of free antibiotics in LPN was performed using a self-assembly method. In brief, 30 mg PLGA and a smaller quantity (1.0–3.3 mg) of cationic lipid were dissolved in 1 mL acetone solvent. A total of 100 μL antibiotics (20 mg mL−1) solution was mixed with the acetone solution. Next, the polymer solution was added dropwise into a 20 mL aqueous solution containing lipid-PEG (e.g. DSPE-PEG5K) and lecithin under ultra-sonication for 10 s. The nanoparticles formed instantly upon mixing. Residual acetone in the suspension was evaporated by a continuous stirring of the suspension at room temperature for 4 h. Nanoparticles were washed three times in distilled water in Amicon tubes (MWCO 100 kDa; Millipore) to remove any remaining organic solvent and free antibiotics before concentrating in phosphate buffered saline (PBS) solution.
Characterization of lipid-polymer hybrid nanoparticles
The field emission scanning electron microscopy (FESEM 6340F) was used to analyze the morphology of LPNs. Briefly, freeze-dried LPNs were uniformly mounted onto metal stubs and sputter coated with gold. Transmission electron microscopy (TEM, JEOL 2100F) was used to observe shell/core structure of PLGA/DOTAP nanoparticles. TEM samples were prepared by the addition of the nanoparticle solution onto a hydrophilic Formvar-coated copper grid for 3 min. Particle size and polydispersity indices were determined by dynamic light scattering (DLS) using HORIBA Nano Particle Analyzer SZ-100. Prior to DLS, the LPNs were suspended in dH2O and ultrasonicated for 10 s to disperse the LPNs uniformly.
Release profile of lipid-polymer hybrid nanoparticles
In vitro release studies of different formulations were evaluated using a dialysis bag (molecular weight cut-off of 7000 (Membra-Cel; Viskase, Inc., Chicago, IL, USA)) filled with LPNs (10 mg). The dialysis bag was then immersed in PBS (pH 7.4, 20 mL) or TSB (pH 7.2, 20 mL). The release medium (5 mL) was collected and replaced with the same volume of fresh medium at pre-determined time points. The amount of the released drug was measured by HPLC system.
Determination of binding affinity using fluorescent dye labeling
To determine the binding affinity of LPN, antibiotic was replaced with propidium iodide (PI) for encapsulation in LPNs. It is assumed here that the amount of PI-labelled nanoparticle attached to the bacteria is equivalent to the binding efficiency of the nanoparticles to the bacteria. For planktonic cells, 180 μL of an overnight culture (i.e. ∼1–5 × 109 cells mL−1) was co-incubated with 20 μL of LPN (272 μM PI in 100 mg mL−1 nanoparticles) for 4 h at 30 °C or 37 °C. All cultures were counterstained by Syto9. The labeling efficiency of bacteria was examined under a confocal laser scanning microscope (CLSM) (Zeiss LSM 780, Carl Zeiss Singapore) at excitation/emission wavelengths of 480/500 nm and 490/635 nm for Syto9 and PI, respectively. The binding affinity of nanoparticles to the bacteria was determined by normalizing the number of bacteria labeled with PI over the total number of bacteria labeled with Syto9. For biofilm study, a 24 h biofilm culture was pre-established on an 8-well glass chamber prior to exposure to the nanoparticles, and subsequently viewed under CLSM.
Viability assay for planktonic cultures
Free or nanoparticle-encapsulated antibiotics were prepared in serial concentrations using TSB. The initial concentration of antibiotics encapsulated in LPNs was determined by HPLC. An appropriate dilution of planktonic culture was added to the antibiotic suspensions to a final bacterial concentration of 8 × 106 cells mL−1. Bacteria were exposed to the antibiotics for 24 h and the bacterial density was determined at optical density 600 nm (OD600). The minimum inhibitory concentration (MIC) was defined as the antibiotic concentration where no visible bacterial growth was observed or the OD600 was <5% compared to the untreated control after 24 h of antibiotic exposure.
Viability assay for biofilm cultures
Actively growing biofilms for S. aureus USA300 and E. fecaelis OG1RF were pre-established in a 96-well plate by seeding 200 μL of bacterial culture at a final bacterial density of 8 × 106 cells mL−1 for 8 h. E. coli UTI89 biofilms were established on a PEG-lid for 48 h. The spent liquid medium was discarded and the biofilms was washed gently using TSB to remove the planktonic bacteria. Free or nanoparticle-encapsulated antibiotics prepared in serial concentrations in TSB was added to the biofilms and incubated for 16 h at 37 °C. The initial concentration of antibiotics encapsulated in LPNs was determined by HPLC. The viability of bacterial biofilms was assessed based on the ATP content of the biofilms using the BacTiter-Glo microbial cell viability assay (Promega, Singapore), according to the manufacturer's instructions. Briefly, the spent liquid medium was removed and 200 μL of diluted reagent was added immediately to each well. The plate was incubated with constant shaking (i.e. 200 rpm) for 10 min at room temperature. A total of 150 μL liquid suspension was transferred to a white 96-well plate for luminescence measurement. Minimum metabolic inhibitory concentration (MMIC) was defined as ≥95% reduction in biofilm ATP content compared to the untreated control after 16 h of antibiotic exposure.
Results and discussion
Fabrication and characterization of lipid-polymer hybrid nanoparticles
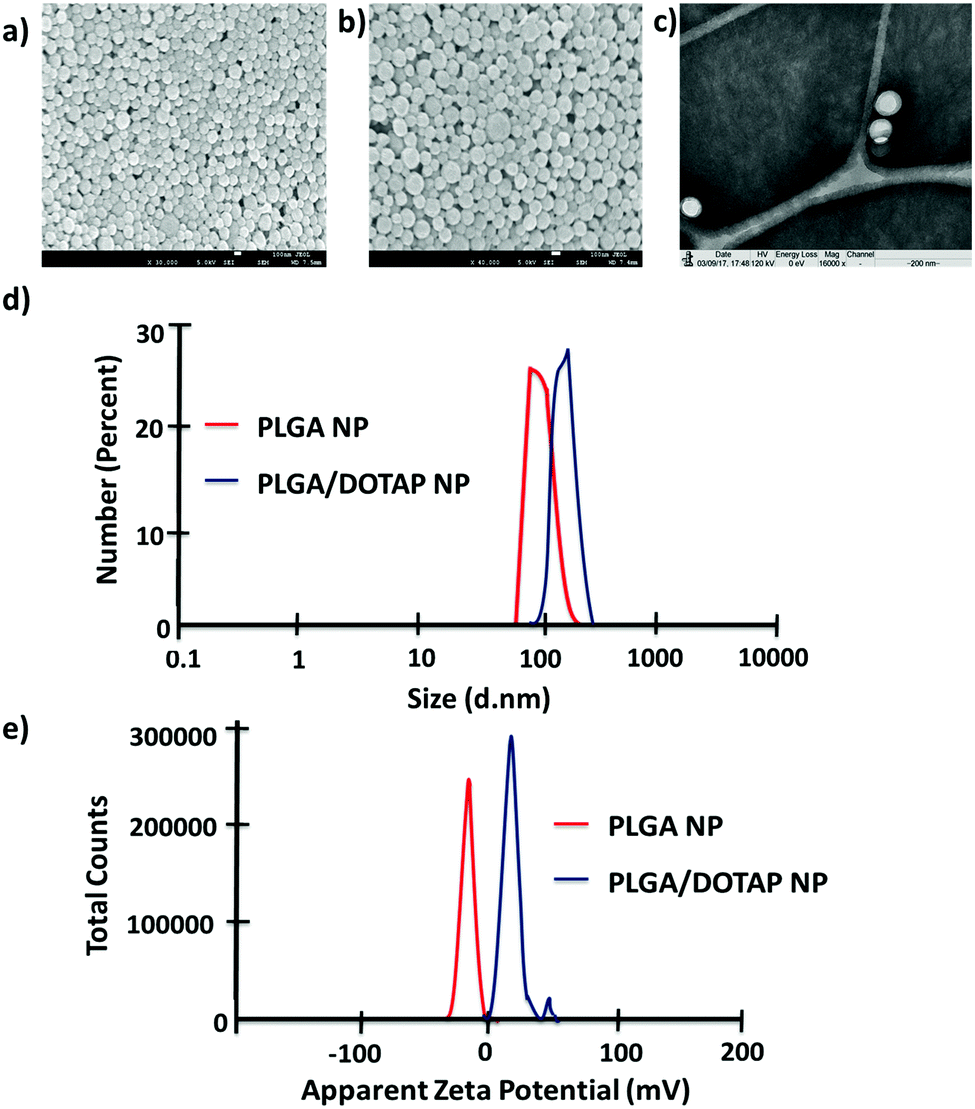
In this study, LPN was prepared through a simple, economical, single step technique. LPN can be consisted of polymer and lipid, forming a core/shell structure while the polymer/lipid ratio can be modified to achieve different particle size and release profile for different purposes (data not shown). Here, we have extended this approach to load the DOTAP-modified PLGA nanoparticles with different antibiotics. The organic acetone solution consisting of cationic lipid (i.e. DOTAP) and PLGA polymer was mixed with an aqueous solution containing lecithin and lipid-PEG. Upon mixing, the PLGA polymer was co-precipitated to form a solid nanoparticle core that is enveloped by a lecithin/lipid-PEG shell. PLGA nanoparticles were ∼90 nm in size (Fig. 1), while PLGA/DOTAP nanoparticles showed a slight increase in particle size (i.e. ∼110 nm), due to the lipid coating (Fig. 1c). Both PLGA and PLGA/DOTAP nanoparticles were spherical in shape. With DOTAP as the surface cationic lipid coating, PLGA/DOTAP nanoparticles exhibited a positively charged zeta potential (i.e. 15.2 ± 3.6 mV), while a negative zeta potential (i.e. −19.2 ± 3.6 mV) was measured for naked PLGA nanoparticles. This suggests that the cationic DOTAP lipid molecules were coated onto the surface of the PLGA nanoparticles.
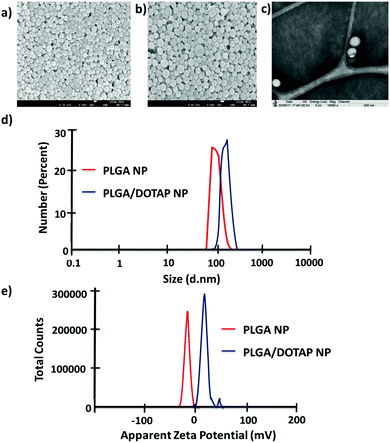 |
| | Fig. 1 Physicochemical properties of PLGA nanoparticles and PLGA/DOTAP nanoparticles. SEM images of (a) PLGA nanoparticles and (b) PLGA/DOTAP nanoparticles. (c) TEM image of PLGA/DOTAP nanoparticles. (d) Dynamic laser scattering of PLGA nanoparticles and PLGA/DOTAP nanoparticles. (e) Zeta potential of PLGA nanoparticles and PLGA/DOTAP nanoparticles. | |
Bacteria tagging by lipid-polymer hybrid nanoparticles
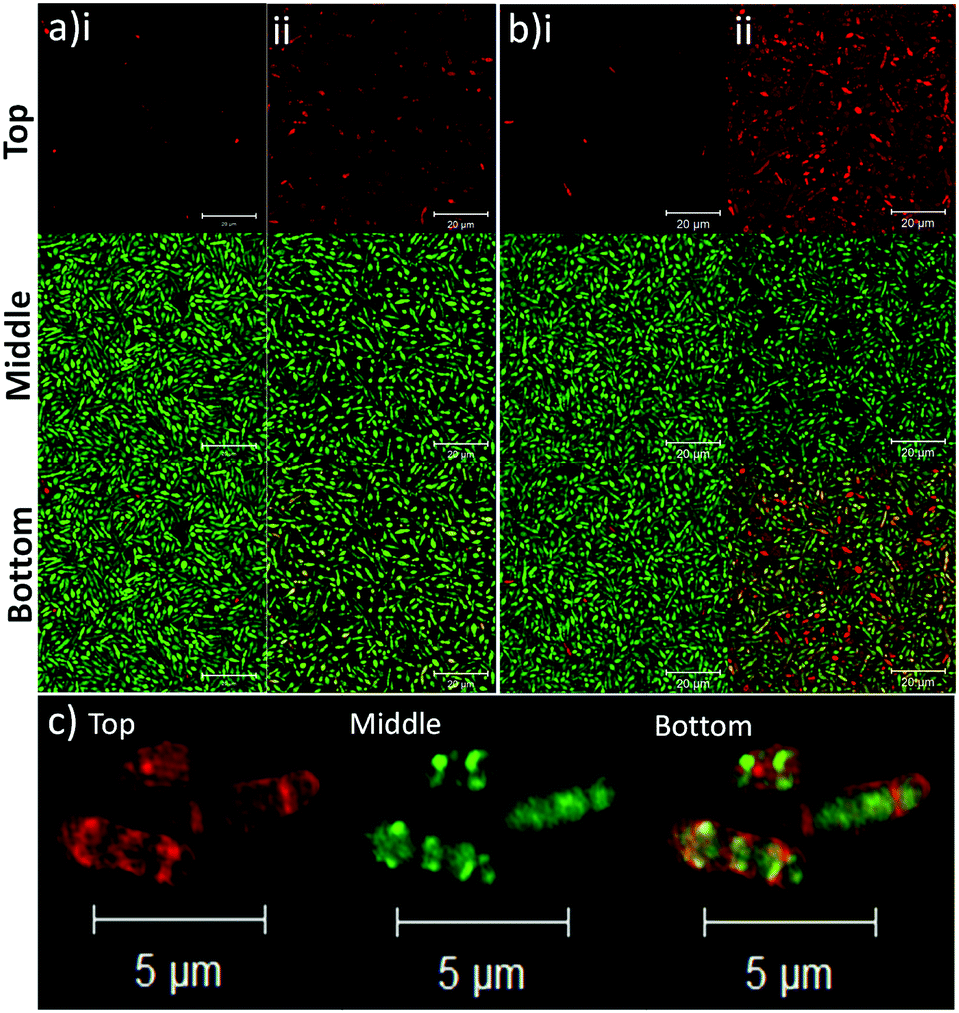
To determine the binding affinity of PLGA and PLGA/DOTAP nanoparticles to bacteria, propidium iodide (PI) encapsulated nanoparticles were prepared, and introduced to planktonic cells of S. thermophilus LMD-9 (Fig. 2). PI is generally recognized as an indicator of cell viability, by staining bacteria whose membranes are compromised.28 Consistent with this, free PI was not able to stain any viable bacteria even in the presence of the blank PLGA or PLGA/DOTAP nanoparticles (Fig. 2ai and bi; as negative controls). This suggests that neither the PLGA nor the DOTAP disrupt membrane integrity. In contrast, PI encased in either PLGA or PLGA/DOTAP nanoparticles could stain the live bacteria uniformly, with the latter resulting in considerably higher PI intensity of the bacterial cells (Fig. 2aii and bii). The increased PI intensity was likely attributed to the aggregation of the PLGA/DOTAP nanoparticles around the bacteria (Fig. 2c), due to charge–charge interaction between the positive DOTAP and the negative polyteichoic acid component of the Gram-positive bacterial cell wall.17,29 It is also possible that some bacterial cells may uptake the nanoparticles or the nanoparticles may penetrate the bacterial cell envelope to certain extent, resulting in the increased PI intensity within the cells. However, this remains to be further confirmed experimentally.
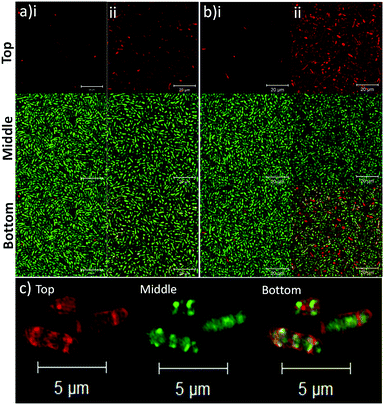 |
| | Fig. 2 Lipid coating (i.e. DOTAP) enhanced the binding affinity of PLGA nanoparticles to planktonic bacteria. The binding affinity of PLGA (a) and PLGA/DOTAP (b) nanoparticles to S. thermophilus LMD-9, was determined. The bacterial culture was incubated with propidium iodide (PI) in the presence of blank nanoparticles (i) or with PI encapsulated nanoparticles (ii), together with Syto9, for 4 h at 37 °C prior to imaging by confocal laser scanning microscope (CLSM). Close-up views of PI encapsulated PLGA/DOTAP and Syto9 co-staining for S. thermophilus LMD-9 (c). The top, middle and bottom panels represent the images of PI, Syto9 and the PI + Syto9 overlay, respectively. Scale bar: 20 μm (a and b) and 5 μm (c). | |
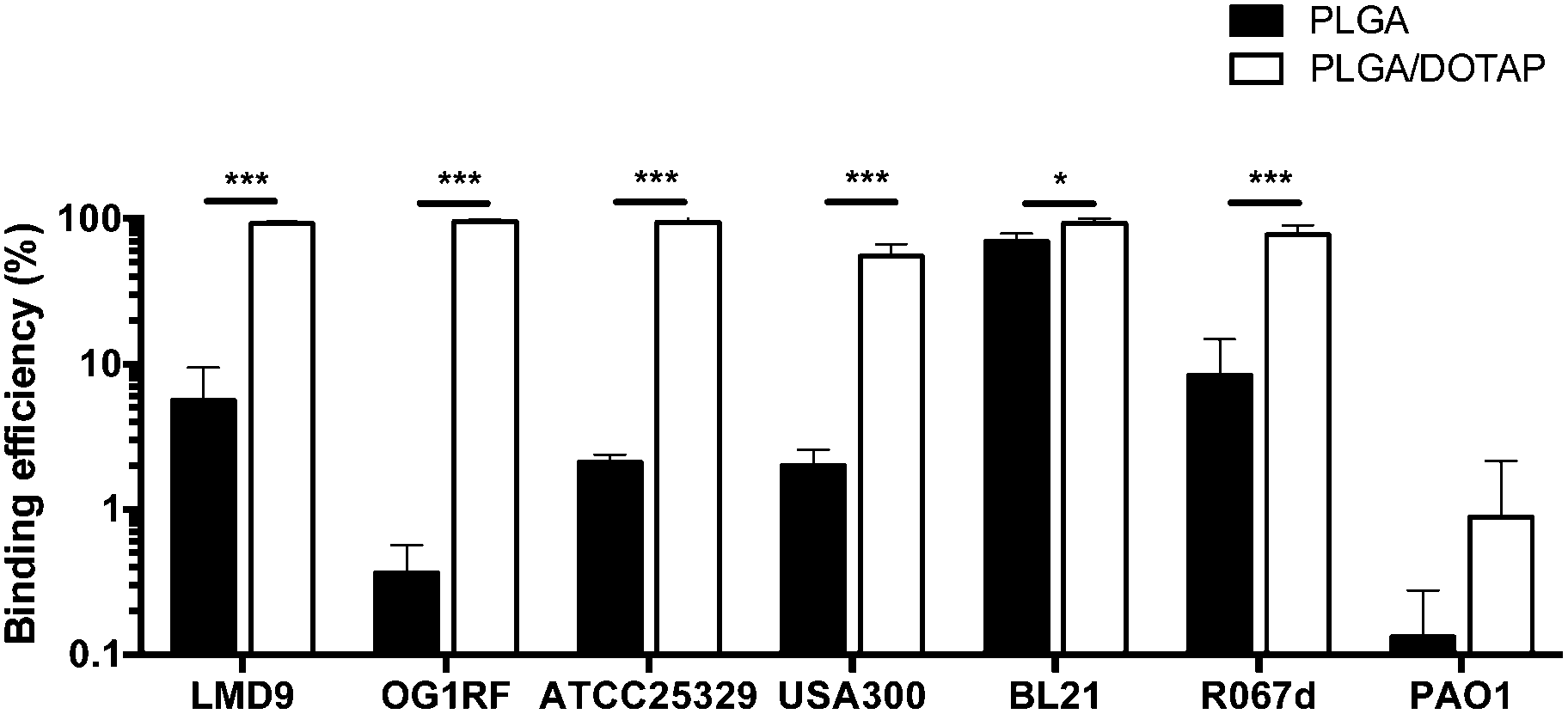
PLGA/DOTAP nanoparticles were also able to bind with high affinity, to a wide range of bacterial species, including E. faecalis OG1RF, S. aureus ATCC25923 and S. aureus USA300, as well as the Gram negative bacteria E. coli BL21 and P. stewartii R067d (Fig. 3). P. aeruginosa PAO1 was the only species examined that could not be efficiently labelled with the PLGA/DOTAP nanoparticles, for reasons yet to be determined. It was previously reported that different strains of P. aeruginosa might interact with DOTAP containing liposomes to different extent of affinity, depending on the outer membrane protein composition.29 While the staining efficiency of PLGA/DOTAP nanoparticles for P. aeruginosa PAO1 was low, it was higher than that of naked PLGA nanoparticles. Overall, it is evident that the DOTAP coating enhanced the binding affinity of PLGA nanoparticles onto bacterial cells and possibly, increased the penetrability of nanoparticles in a species independent fashion.
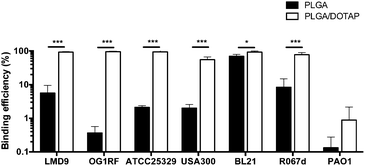 |
| | Fig. 3 PLGA/DOTAP nanoparticles can bind efficiently to both Gram-positive and Gram-negative bacteria. The binding efficiency of PI-encapsulated PLGA (solid bar) and PLGA/DOTAP (empty bar) to different bacterial species, including S. thermophilus LMD-9, E. faecalis OG1RF, S. aureus ATCC25923, S. aureus USA300, E. coli BL21, P. stewartii R067d and P. aeruginosa PAO1, was determined by normalizing the number of cells stained by PI against the total cell stain, Syto9 (n = 10, mean ± S.D). False discovery rate (FDR) corrections for multiple comparisons were performed and significant differences are indicated as follows: *P < 0.01 and ***P < 0.001. Scale bars: 20 μm. | |
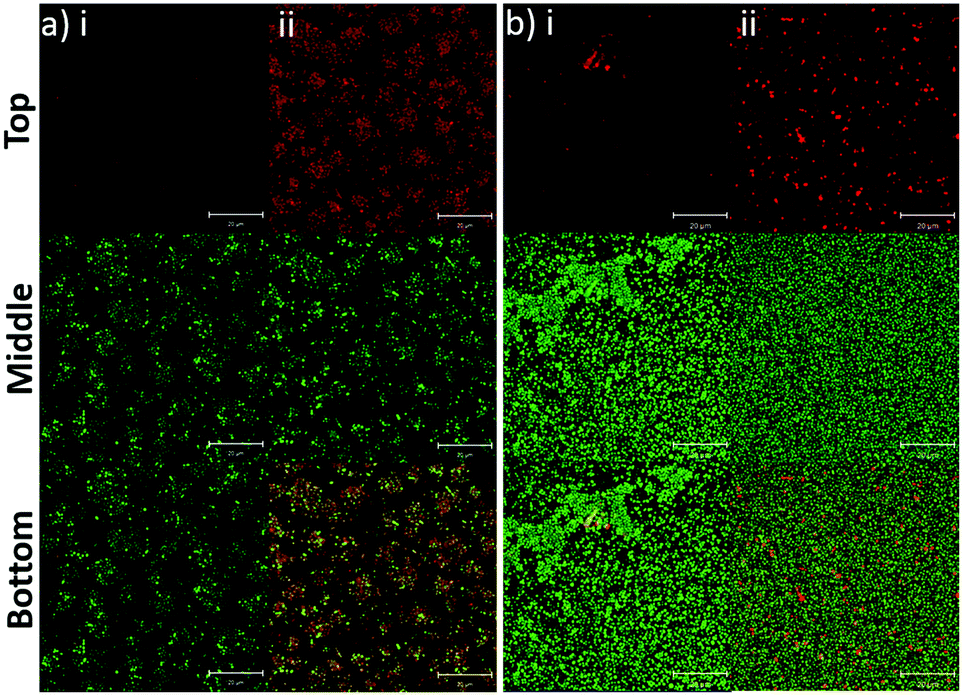
In addition to the planktonic bacteria, the binding capacity of nanoparticles to bacterial biofilms was also investigated (Fig. 4). Similar to the planktonic culture, PLGA/DOTAP nanoparticles appeared to bind to E. faecalis OG1RF and S. aureus USA300 biofilms with affinity higher than naked PLGA nanoparticles. The enhanced PI intensity appeared to be a consequence of accumulation of PLGA/DOTAP nanoparticles around the biofilms. However, it remains unclear if the increased binding efficiency was solely due to the attachment of nanoparticles to the biofilm matrix or bacterial cells, or both. Besides particle adsorption, the penetrability of the nanoparticles into biofilms is also crucial in determining the potency of a delivery system.30 Forier et al. reported that the optimal particle size for dense biofilm penetration was around 100–130 nm.31 Even though the interaction between DOTAP and the biofilm matrix or bacterial cell envelope may deter the diffusion of PLGA/DOTAP nanoparticles into biofilms, the narrow and uniform nano-size distribution of these particles (i.e. 110–130 nm) indicates a strong potential for deep penetration into biofilm clusters.32 However, it remains to be confirmed if these particles can indeed penetrate biofilms and if the penetration kinetics of these nanoparticles can be fine-tuned for subsequently drug delivery into biofilms. Similarly, it is possible that the nanoparticles maybe taken up by the biofilm cells, although further investigation is needed. Together, these data suggest that PLGA/DOTAP nanoparticles could be a suitable carrier to deliver drugs/substances in close proximity to the bacterial cells either in the planktonic or biofilm context to enhance treatment efficacy.
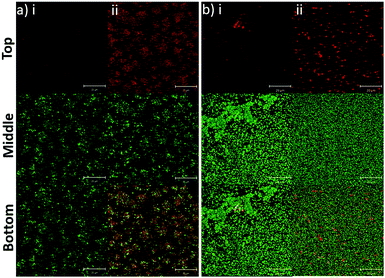 |
| | Fig. 4 PLGA/DOTAP nanoparticles can bind efficiently to bacterial biofilms. Confocal laser scanning microscopy (CLSM) images of E. faecalis OG1RF (a) and S. aureus USA300 (b) biofilms co-stained by Syto9 and propidium iodide (PI) encapsulated PLGA (i) or PLGA/DOTAP (ii). An 8 h biofilm culture was incubated with PI-encapsulated nanoparticles and Syto9 for 4 h at 37 °C prior to imaging by CLSM. The top, middle and bottom panels represent the images of the PI, Syto9 and the PI + Syto9 overlay, respectively. Scale bars: 20 μm. | |
Antibiotic release from lipid-polymer hybrid nanoparticles
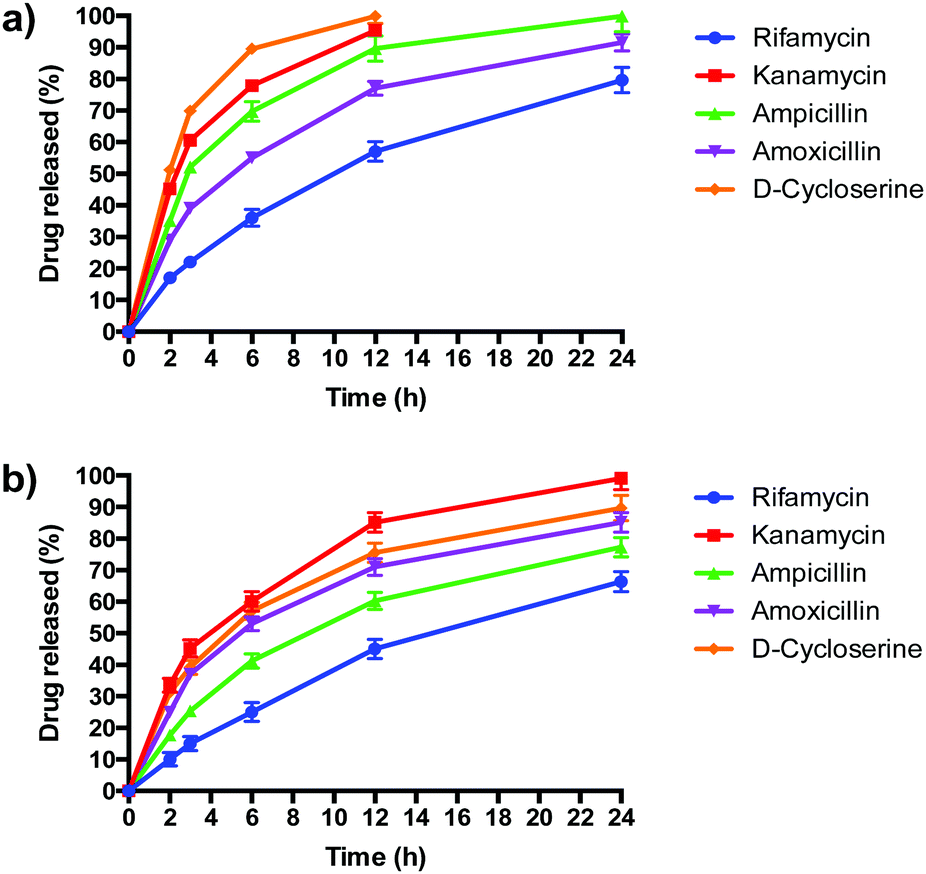
PLGA/DOTAP nanoparticles carrying antibiotics with different solubilities, ranging from highly hydrophobic to amphiphilic to highly hydrophilic, were fabricated (Table 2). The water solubility of the antibiotics influenced their encapsulation into (Table 2), and release profiles from (Fig. 5), PLGA and PLGA/DOTAP nanoparticles. PLGA and PLGA/DOTAP nanoparticles exhibited similar encapsulation efficiency of hydrophobic antibiotic (i.e. rifamycin). However, it is worth noting that the encapsulation efficiencies of amphiphilic (i.e. ampicillin) and hydrophilic drugs (i.e. ampicillin, kanamycin, D-cycloserine) of PLGA/DOTAP nanoparticles were significantly higher than that of PLGA nanoparticles. PLGA, a hydrophobic polymer, has been shown to have a high encapsulation efficiency and sustained release profile for hydrophobic drugs.33–35 While hydrophobic drugs interact well with PLGA, amphiphilic and hydrophilic drugs have weaker interactions. Hydrophilic drugs generally localize near the surface of PLGA nanoparticles, resulting in lower encapsulation efficiency and faster release rates.36 With a cationic lipid layer for PLGA/DOTAP nanoparticles, hydrophilic drugs can be condensed between the cationic lipid and PLGA matrix, thus increasing encapsulation efficiency. Subsequent in vitro release studies conducted in culture medium (i.e. TSB) showed similar release kinetics as for PBS (data not shown). While similar sustained release profiles were observed for rifamycin (hydrophobic) from both nanoparticles, PLGA/DOTAP nanoparticles exhibited a more sustained release profile for the amphiphilic and hydrophilic antibiotics as compared to PLGA nanoparticles. Thus, PLGA/DOTAP particles can be used for antibiotics irrespective of their water solubility and release the drugs over a longer time frame (Table 3).
Table 2 Physical properties of PLGA and PLGA/DOTAP nanoparticles
| Formulation |
Particle size (nm) |
Polydispersity index |
Zeta potential (mV) |
Encapsulation efficiency (%) |
| PLGA NPs |
| Rifamycin |
92.4 ± 5.1 |
0.120 ± 0.004 |
−21.3 ± 3.1 |
90.5 ± 5.2 |
| Kanamycin A |
102.4 ± 6.2 |
0.154 ± 0.010 |
−14.6 ± 6.1 |
39.6 ± 2.1 |
| Ampicillin |
105.4 ± 3.5 |
0.142 ± 0.014 |
−24.5 ± 5.5 |
33.5 ± 1.9 |
| Amoxicillin |
107.6 ± 3.8 |
0.168 ± 0.012 |
−29.6 ± 6.7 |
28.7 ± 1.3 |
|
D-Cycloserine |
103.7 ± 2.9 |
0.146 ± 0.016 |
−24.3 ± 4.7 |
31.2 ± 2.2 |
|
|
| PLGA/DOTAP NPs |
| Rifamycin |
111.3 ± 4.8 |
0.188 ± 0.002 |
15.2 ± 2.1 |
95.0 ± 6.1 |
| Kanamycin A |
119.6 ± 7.8 |
0.210 ± 0.012 |
16.6 ± 3.4 |
58.3 ± 3.8 |
| Ampicillin |
123.7 ± 6.9 |
0.234 ± 0.028 |
21.9 ± 5.3 |
48.3 ± 3.3 |
| Amoxicillin |
129.6 ± 7.1 |
0.216 ± 0.014 |
14.6 ± 4.7 |
51.6 ± 4.2 |
|
D-Cycloserine |
120.1 ± 9.3 |
0.192 ± 0.008 |
18.9 ± 6.6 |
53.3 ± 5.6 |
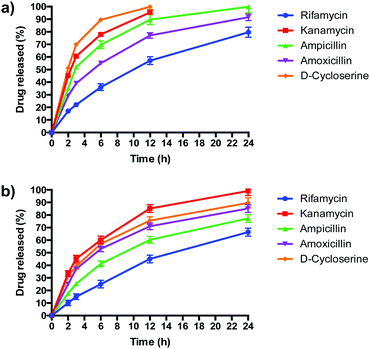 |
| | Fig. 5 The release profiles of antibiotics from PLGA nanoparticles and PLGA/DOTAP nanoparticles. In vitro release profiles of different antibiotics from (a) PLGA nanoparticles and (b) PLGA/DOTAP nanoparticles in PBS (pH 7.4) for 24 h at 37 °C (n = 5, mean ± S.D). | |
Table 3 Minimum inhibitory concentration (MIC) and minimum metabolic inhibitory concentration (MMIC) of free and PLGA/DOTAP-encapsulated antibiotics against different bacterial species
|
|
MIC (μg mL−1) |
MMIC (μg mL−1) |
| UTI89 |
USA300 |
OG1RF |
UTI89 |
USA300 |
OG1RF |
| UTI89: E. coli UTI89; USA300: S. aureus USA300; OG1RF: E. faecalis OG1RF. Free DOTAP was mixed with free ampicillin instead of encapsulating ampicillin within DOTAP. For carrier alone (i.e. blank nanoparticles), serial concentration of the respective carrier was prepared according to the amount of carrier used for ampicillin encapsulation in serial dilutions. In all cases, none of the carriers showed toxicity to all three species even at the highest carrier concentrations. |
| Carrier-antibiotics |
| Ampicillin (A) |
4 |
8 |
1 |
8 |
8 |
1 |
| DOTAP + ampicillin (DA)a |
4 |
8 |
1 |
8 |
8 |
1 |
| PLGA- − ampicillin (PA) |
2 |
4 |
0.5 |
4 |
1 |
0.25 |
| PLGA/DOTAP-ampicillin (LA) |
<0.125 |
0.25 |
<0.125 |
0.25 |
<0.125 |
<0.125 |
|
|
|
D-Cycloserine (C) |
>64 |
64 |
>64 |
16 |
32 |
>128 |
| PLGA/DOTAP-D-cycloserine (LC) |
16 |
8 |
64 |
2 |
4 |
16 |
|
|
| Carrier alone |
| DOTAP (D) |
>640 |
>640 |
>640 |
>640 |
>640 |
>640 |
| PLGA (P) |
>3686 |
>3686 |
>3686 |
>3686 |
>3686 |
>3686 |
| PLGA/DOTAP (L) |
>2560 |
>2560 |
>2560 |
>2560 |
>2560 |
>2560 |
Enhanced antimicrobial effects via lipid-polymer hybrid nanoparticles
Given the superior properties of PLGA/DOTAP over PLGA nanoparticles, including increased biofilm binding affinity (Fig. 4), enhanced encapsulation efficiency (Table 2) and extended drug release (Fig. 5), we hypothesized that the PLGA/DOTAP formulation could therefore be a better alternative to the classic free drug formulation for planktonic and biofilm treatments. Ampicillin and D-cycloserine were individually encapsulated into PLGA/DOTAP nanoparticles and their effect on planktonic and biofilm growth was compared with the free-form of the drug. Irrespective of the type of antibiotic used and the bacterial species, the MICs and MMICs of the PLGA/DOTAP formulations were at least 8 fold lower than for the non-encapsulated antibiotics. In some cases, as much as a 32 fold reduction could be achieved. In contrast, there was no significant reduction in MIC or MMIC when ampicillin was formulated into nanoparticles of either DOTAP or PLGA alone. Similarly, none of the nanoparticles were growth inhibitory to the bacteria in the absence of antibiotics. These results are comparable with liposomal formulations that have been shown to reduce MICs in 2–4 fold,19,37–40 depending on the physicochemical features of the liposomes (i.e. size, surface charges, encapsulation efficiency), characteristics of the antimicrobial agents and the bacterial species studied. Although complete biofilm eradication was not achieved even at the highest antibiotic concentrations examined (i.e. 64 and 256 μg mL−1; data not shown), the PLGA/DOTAP formulation was more effective than when the antibiotics were delivered in solution or in the PLGA nanoparticles. These results thus clearly suggest that the combination of PLGA and DOTAP, structurally as double-layered nanoparticles, is essential for antibiotic encapsulation and enhancement of antibiotic efficacy.
Conclusions
In this study, we have developed a simple, economical and scalable approach that hybridize the liposomal and polymeric systems to achieve a highly stable and programmable nano-drug carrier for localized and sustained release of antibiotics to both planktonic bacteria and biofilms. Our lipid-polymer hybrid nanoparticle system synergizes the desirable features of both liposomes and polymers in terms of drug encapsulation efficiency, stability, affinity to bacterial envelope, tunability of drug release profile and biofilm penetrability. It is evident that the lipid-polymer hybrid system is capable of enhancing the therapeutic efficacy of antibiotics, by protecting the drugs from inactivation by extracellular enzymes and prolonging their effects. Given the flexibility of modifying the individual component of the hybrid system, it is expected that the lipid-polymer nanoparticle system can be endorsed with more functions tailored for specific anti-biofilm applications in the near future.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
We would like to thank Associate Professor Kimberly Kline for providing bacterial strains, including E. coli UTI89, E. faecalis OG1RF and S. aureus USA300. This work was supported by the School of Materials Science and Engineering and the Singapore Centre for Environmental Life Sciences Engineering, whose research is supported by the National Research Foundation Singapore, Ministry of Education, Nanyang Technological University and National University of Singapore, under its Research Centre of Excellence Programme.
References
- C. L. Ventola, Pharm. Ther., 2015, 40, 277–283 Search PubMed.
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- C. H. Tan, K. W. K. Lee, M. Burmølle, S. Kjelleberg and S. A. Rice, Environ. Microbiol., 2017, 19, 42–53 CrossRef PubMed.
- H.-C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef CAS PubMed.
- A. Harms, E. Maisonneuve and K. Gerdes, Science, 2016, 334, aaf4268 CrossRef PubMed.
- H. Van Acker, P. Van Dijck and T. Coenye, Trends Microbiol., 2014, 22, 326–333 CrossRef CAS PubMed.
- K. Lewis, Annu. Rev. Microbiol., 2010, 64, 357–372 CrossRef CAS PubMed.
- T. Bjarnsholt, M. Alhede, M. Alhede, S. R. Eickhardt-Sørensen, C. Moser, M. Kühl, P. Ø. Jensen and N. Høiby, Trends Microbiol., 2013, 21, 466–474 CrossRef CAS PubMed.
- S. L. Chua, J. K. H. Yam, P. Hao, S. S. Adav, M. M. Salido, Y. Liu, M. Givskov, S. K. Sze, T. Tolker-Nielsen and L. Yang, Nat. Commun., 2016, 7, 10750 CrossRef CAS PubMed.
- J. S. Madsen, M. Burmølle, L. H. Hansen and S. J. Sørensen, FEMS Immunol. Med. Microbiol., 2012, 65, 183–195 CrossRef CAS PubMed.
- W. Gao, Y. Chen, Y. Zhang, Q. Zhang and L. Zhang, Adv. Drug Delivery Rev., 2017 DOI:10.1016/j.addr.2017.09.015.
- C. Y. Flores, A. G. Miñán, C. A. Grillo, R. C. Salvarezza, C. Vericat and P. L. Schilardi, ACS Appl. Mater. Interfaces, 2013, 5, 3149–3159 CAS.
- S. Goswami, D. Thiyagarajan, G. Das and A. Ramesh, ACS Appl. Mater. Interfaces, 2014, 6, 16384–16394 CAS.
- D. L. Slomberg, Y. Lu, A. D. Broadnax, R. A. Hunter, A. W. Carpenter and M. H. Schoenfisch, ACS Appl. Mater. Interfaces, 2013, 5, 9322–9329 CAS.
- Z. Drulis-Kawa and A. Dorotkiewicz-Jach, Int. J. Pharm., 2010, 387, 187–198 CrossRef CAS PubMed.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- K. Forier, K. Raemdonck, S. C. De Smedt, J. Demeester, T. Coenye and K. Braeckmans, J. Controlled Release, 2014, 190, 607–623 CrossRef CAS PubMed.
- A. F. Radovic-Moreno, T. K. Lu, V. A. Puscasu, C. J. Yoon, R. Langer and O. C. Farokhzad, ACS Nano, 2012, 6, 4279–4287 CrossRef CAS PubMed.
- L. Sande, M. Sanchez, J. Montes, A. J. Wolf, M. A. Morgan, A. Omri and G. Y. Liu, J. Antimicrob. Chemother., 2012, 67, 2191–2194 CrossRef CAS PubMed.
- U. S. Toti, B. R. Guru, M. Hali, C. M. McPharlin, S. M. Wykes, J. Panyam and J. A. Whittum-Hudson, Biomaterials, 2011, 32, 6606–6613 CrossRef CAS PubMed.
- P. Sabaeifard, A. Abdi-Ali, M. R. Soudi, C. Gamazo and J. M. Irache, Eur. J. Pharm. Sci., 2016, 93, 392–398 CrossRef CAS PubMed.
- F. Esmaeili, M. Hosseini-Nasr, M. Rad-Malekshahi, N. Samadi, F. Atyabi and R. Dinarvand, Nanomedicine, 2007, 3, 161–167 CrossRef CAS PubMed.
- G. M. Dunny, B. L. Brown and D. B. Clewell, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3479–3483 CrossRef CAS.
- B. A. Diep, S. R. Gill, R. F. Chang, T. H. Phan, J. H. Chen, M. G. Davidson, F. Lin, J. Lin, H. A. Carleton, E. F. Mongodin, G. F. Sensabaugh and F. Perdreau-Remington, J.-Lancet, 2006, 367, 731–739 CrossRef CAS.
- F. W. Studier and B. A. Moffatt, J. Mol. Biol., 1986, 189, 113–130 CrossRef CAS PubMed.
- S. L. Chen, C.-S. Hung, J. Xu, C. S. Reigstad, V. Magrini, A. Sabo, D. Blasiar, T. Bieri, R. R. Meyer, P. Ozersky, J. R. Armstrong, R. S. Fulton, J. P. Latreille, J. Spieth, T. M. Hooton, E. R. Mardis, S. J. Hultgren and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5977–5982 CrossRef CAS PubMed.
- C. H. Tan, K. S. Koh, C. Xie, J. Zhang, X. H. Tan, G. P. Lee, Y. Zhou, W. J. Ng, S. A. Rice and S. Kjelleberg, NPJ Biofilms and Microbiomes, 2015, 1, 15006 CrossRef PubMed.
- L. Shi, S. Günther, T. Hübschmann, L. Y. Wick, H. Harms and S. Müller, Cytometry, Part A, 2007, 71A, 592–598 CrossRef PubMed.
- Z. Drulis-Kawa, A. Dorotkiewicz-Jach, J. Gubernator, G. Gula, T. Bocer and W. Doroszkiewicz, Int. J. Pharm., 2009, 367, 211–219 CrossRef CAS PubMed.
- Z. Rukavina and Ž. Vanić, Pharmaceutics, 2016, 8, 18 CrossRef PubMed.
- K. Forier, A.-S. Messiaen, K. Raemdonck, H. Nelis, S. De Smedt, J. Demeester, T. Coenye and K. Braeckmans, J. Controlled Release, 2014, 195, 21–28 CrossRef CAS PubMed.
- D. Dong, N. Thomas, B. Thierry, S. Vreugde, C. A. Prestidge and P.-J. Wormald, PLoS One, 2015, 10, e0131806 Search PubMed.
- C.-G. Keum, Y.-W. Noh, J.-S. Baek, J.-H. Lim, C.-J. Hwang, Y.-G. Na, S.-C. Shin and C.-W. Cho, Int. J. Nanomed., 2011, 6, 2225–2234 CAS.
- H. Wang, W. Su, S. Wang, X. Wang, Z. Liao, C. Kang, L. Han, J. Chang, G. Wang and P. Pu, Nanoscale, 2012, 4, 6501–6508 RSC.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- T. S. J. Kashi, S. Eskandarion, M. Esfandyari-Manesh, S. M. A. Marashi, N. Samadi, S. M. Fatemi, F. Atyabi, S. Eshraghi and R. Dinarvand, Int. J. Nanomed., 2012, 7, 221–234 Search PubMed.
- C. Mugabe, M. Halwani, A. O. Azghani, R. M. Lafrenie and A. Omri, Antimicrob. Agents Chemother., 2006, 50, 2016–2022 CrossRef CAS PubMed.
- J. Gubernator, Z. Drulis-Kawa, A. Dorotkiewicz-Jach, W. Doroszkiewicz and A. Kozubek, Lett. Drug Des. Discovery, 2007, 4, 297–304 CrossRef CAS.
- Z. Drulis-Kawa, J. Gubernator, A. Dorotkiewicz-Jach, W. Doroszkiewicz and A. Kozubek, Int. J. Pharm., 2006, 315, 59–66 CrossRef CAS PubMed.
- M. Alhajlan, M. Alhariri and A. Omri, Antimicrob. Agents Chemother., 2013, 57, 2694–2704 CrossRef CAS PubMed.
Footnote |
| † Co-first authors. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Noele Kai Jing
Ng
b,
Yee Phan
Yeo
bc,
Scott A.
Rice
ab,
Noele Kai Jing
Ng
b,
Yee Phan
Yeo
bc,
Scott A.
Rice