
Direct transformation of bijels into bicontinuous composite electrolytes using a pre-mix containing lithium salt†‡
Dongyu
Cai
 a,
Felix H.
Richter§
b,
Job H. J.
Thijssen
c,
Peter G.
Bruce
b and
Paul S.
Clegg
*c
a,
Felix H.
Richter§
b,
Job H. J.
Thijssen
c,
Peter G.
Bruce
b and
Paul S.
Clegg
*c
aKey Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University, 30 South PuZhu Road, Nanjing, Jiangsu 211816, China
bDepartment of Materials, University of Oxford, Parks Road, Oxford, OX1 3PH, UK
cSUPA School of Physics and Astronomy, University of Edinburgh, Peter Guthrie Tait Road, Edinburgh, EH9 3FD, UK. E-mail: paul.clegg@ed.ac.uk
First published on 20th February 2018
Abstract
We report a general strategy for making bicontinuous conducting composite materials in a controllable fashion. Our approach begins with a bicontinuous interfacially jammed emulsion gel (bijel) fabricated from a pre-mix containing a salt, here bis(trifluoromethane)sulfonimide lithium salt (LiTFSI). The resulting structure has interpenetrating ionic conducting and non-conducting domains composed of an ethylene carbonate (EC)-rich phase and a p-xylene (xylene)-rich phase of roughly equal volumes. This is the first time that bijel fabrication has been carried out using a pair of partially miscible liquids whose phase behaviour has been modified due to the addition of salt. Diffusing polystyrene (PS) into the xylene-rich phase enables the facile formation of a PS-filled bijel in place of a multi-step polymerization of added monomers. Drying the bijel results in the selective removal of xylene, reducing the total sample volume without compromising the morphology of the EC domain. Electrochemical impedance spectroscopy of the composite electrolytes confirms the existence of ion conducting pathways.
Conceptual insightsBattery electrolytes need to provide high ionic conductivity and at the same time have appropriate mechanical characteristics. Failure to meet this need results in dramatic safety problems. This challenge motivates our development of new composite electrolytes with the combination of the transport properties of a liquid and the mechanical stability of a solid. The underlying concept is the use of the bijel as a template, a novel gel with two tortuous interpenetrating fluid domains separated by a rigid interface. We successfully demonstrate a route for selectively incorporating ions and synthetic polymers into the two separated fluidic domains of the bijel template. Remarkably, the bijel structure is retained even when the liquid is removed from the polymer-filled fluidic domain. The final composite materials show a well-defined bicontinuous arrangement of liquid and solid phases which is tunable across a wide range. It is an ideal material design motif because two phases in this structure provide the conductivity and the rigidity characteristics as parallel but separate functionalities. This study points out a controllable strategy for designing such bicontinuous composite electrolytes. |
1. Introduction
Combining two components with contrasting or even contradictory properties into a composite is a major materials design challenge1,2 – for example, an improved composite containing a large volume of non-conductor but which continues to exhibit good conductivity, or a composite which combines the transport characteristics of a liquid with the mechanical properties of a solid. Ideally, the characteristics of both components would be captured in the final material with the composition and microstructure being tunable across a wide range. These requirements are unlikely to be realized if the components are fully miscible or one component is isolated as a dispersed phase in another. A bicontinuous arrangement of immiscible phases is an ideal material design motif because each phase in this structure is fully interconnected and able to function independently. Application areas include electrode materials, photovoltaic devices, membranes and cross-flow reaction media. However, making such an arrangement of domains permanent is challenging as the system tends to progress towards complete phase separation. For example, a recent study3 reports a curing-induced phase separation route for yielding bicontinuous ionic liquid–epoxy resin domains as structured electrolytes for supercapacitors. The rigidity of cross-linked epoxy resin maintains the stability of the composite. The success of this route is highly dependent on the choice of epoxy resins. Poor control of phase separation can result in the failure of domain connectivity across the cured sample. The challenge is that a novel process needs to be combined with a high level of control.Here, we report a route for preparing composite materials with interpenetrating conducting and structural domains in a well-controlled manner. The core concept is the use of a bicontinuous liquid–liquid gel (known as a bijel) as a scaffold to control the fabrication process. The bijel has two continuous interpenetrating fluid domains stabilized by a layer of colloidal particles jammed at the interface.4,5 The formation process of the bijel6 relies on the spinodal decomposition of a binary liquid mixture in the presence of colloidal particles with a three-phase contact angle close to 90°. These particles can become trapped on the percolating interface without imposing any preferred mean curvature.7,8 The coarsening of the spinodal structure is then arrested as a result of the particles jamming together as the area of the interface decreases. This novel soft material shows the rare combination of tunable domain sizes,9,10 contrasting phases within the domains11–13 and mechanical robustness of the complete composite.14,15 These characteristics make the bijel a desirable scaffold for creating bicontinuous composite materials for demanding applications.15–21
In this contribution, we present a novel route for incorporating a lithium salt and a synthetic high molecular weight polymer into the two liquid domains of a bijel scaffold. The bicontinuity of the bijel is successfully retained during processing. We describe a post-process step involving the selective removal of the host liquid phase containing the synthetic polymer, leaving behind a dense and robust polymer/particle scaffold. We show that the composite exhibits a good combination of tortuosity and ionic conductivity. Due to the wide choice of synthetic polymers, this physical approach offers the flexibility to tune the properties of the bicontinuous composite.
2. Results and discussion
2.1. Realization of bijel scaffolds with LiTFSI
Fig. 1 shows the schematic diagram of our material-fabrication route, which incorporates the salt and polymer into the two fluidic channels of a bijel scaffold. The salt, bis(trifluoromethane)sulfonimide lithium salt (LiTFSI), is incorporated into the bijel structure from the very beginning. Ethylene carbonate (EC) and p-xylene (xylene) are selected as a pair of partially miscible liquids for preparing the bijel because EC and xylene can be used to keep the salt and polymer apart. Our previous work22 has shown that EC/xylene bijels can be stabilized using fumed silica (Fig. S1, ESI‡) in the form of secondary particle clusters including the use of appropriate chemical treatment (silanization). The optimal conditions were pinpointed for creating neutrally wetting particles (see Methods), in which the key parameter is the ratio (by weight) of a silane-coupling agent, hexamethyldisilazane (HMDS), to fumed silica in a wet chemistry process. Fig. 2(a) shows a typical confocal image of an EC/xylene bijel without the salt. The xylene-rich phase is labelled with Nile red (light grey). When the HMDS/particle ratio is 0.4, particles with neutral wetting surfaces are achieved with good reproducibility. Developing a strategy for filling bijels with active/functional/high performance components is the key challenge for turning this novel soft matter into a useful material.18,20 Direct incorporation of the salt into the bijel is successfully realized in our case, where LiTFSI is pre-dissolved into EC to give a single fluid phase prior to adding xylene and silica particles; this step is then followed by thermally induced spinodal decomposition.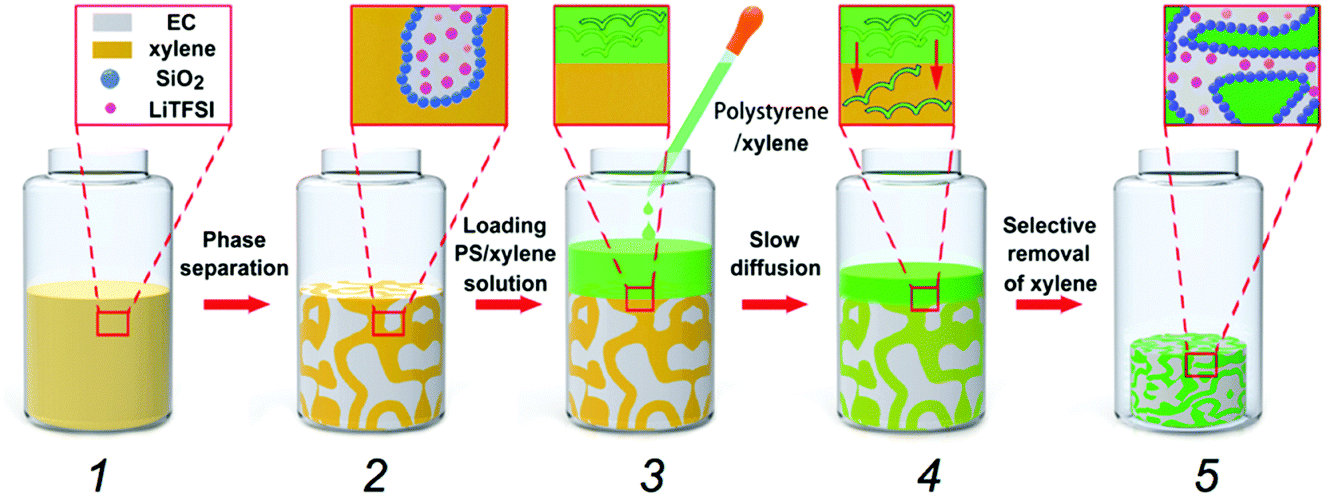 | ||
| Fig. 1 Schematic route for making a bicontinuous composite electrolyte: (1) formation of a single fluid phase of EC and xylene with LiTFSI and silica particles; (2) arrest of the spinodal decomposition of EC and xylene induced by thermal quenching; (3) loading of a PS/xylene solution (30 wt%) onto the bijel; (4) selective diffusion of polystyrene into the xylene-rich phase; and (5) selective removal of xylene to solidify the polystyrene phase. | ||
 | ||
| Fig. 2 Confocal images of arrested spinodal structures in glass cuvettes. (a)–(c) demonstrate the spinodal structures arrested by 2.6 vol% particles (HMDS/particle = 0.4); here samples (b) and (c) are filled with 0.1 M and 0.3 M LiTFSI, respectively; (d)–(f) show the confocal images of the bijels with 0.1 M lithium salt in EC stabilized by 0.6 vol%, 1.8 vol% and 3.6 vol% particles (HMDS/particle = 0.16), respectively. Note: The xylene-rich phase (light grey) is labelled with Nile red; the salt concentration is relative to the amount of pure EC that is added to form bijels. | ||
Fig. 2(a)–(c) shows the effect of LiTFSI concentration on bijel formation; the changes become more prominent with increasing salt concentration. Fig. 2(c) shows that although the spinodal structure is still arrested, a large number of EC-rich droplets (dark phase) have formed within the xylene-rich phase and the size of the two domains has grown considerably. This suggests that the addition of LiTFSI salts modifies the system such that the particles now slightly prefer the xylene-rich phase to the EC-rich phase. This is likely to have arisen from a change to the dielectric properties of the EC caused by the added salt.23 For this composite, the additional droplets are undesirable; hence, we produced a new batch of particles by reducing the HMDS/particle ratio from 0.4 to 0.16 via a process of very careful tuning that is not unusual in bijel fabrication.5 Reducing the amount of the silane coupling agent counter-balances the effect of the added salt, and Fig. 2(d)–(f) shows a series of “clean” bijels arrested by the new batch of particles.
Using these new particles, we can also tune the bijel domain size by changing the particle concentration, Fig. 2(d)–(f). This provides detailed control over the internal structure of the bicontinuous composite. It is notable that the bijel can be arrested at a very low particle concentration, here 0.6 vol%. Our observations suggest that the presence of the salt makes this possible. Since the salt is soluble in EC only, we assume that it remains in the EC-rich phase after phase separation. This is further verified by the impedance measurements below.
2.2. Selective partitioning of PS macromolecules into the bijel scaffold
Following the creation of a salt-filled bijel, the next step is the incorporation of polystyrene with an average molecular weight of ∼350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. Unlike small molecules, this highly entangled PS cannot diffuse into the xylene-rich phase directly. As shown in Fig. 1, we tackled this challenge by dispersing PS in xylene and loading the dispersion onto the bijel. PS macromolecules are insoluble in the EC-rich phase and they exclusively diffuse into the xylene-rich domain of the bijel due to the gradient in concentration. Anthracene-grafted PS is the fluorescent probe for tracking the movement of the macromolecules. The bijel used here is stabilized by 1.8 vol% particles. The inset in Fig. 3(a) shows the loading of a PS/xylene solution (clear liquid) onto the bijel (pink in colour), and no damage to the bijel on a macroscopic scale is observed. Fig. 3(a) and (b) shows the confocal images of the same region of the bijel after 24 h, in which the xylene-phase is labelled with Nile red (light grey) and anthracene-grafted PS (green), respectively. This confirms the selective entrance of fluorescent anthracene-grafted PS macromolecules (green) into the xylene-rich phase, and indicates that the microstructure of the bijel scarcely changes with the partitioning of the macromolecules. In this post-processing approach, the movement of macromolecules is dominated by the entanglements formed in the solution, which could be tuned either by the solution concentration or via the choice of macromolecular weight. In the following section, we will present a more detailed exploration of this aspect. Xylene plays no useful role in the final composite and hence its selective removal is the final step in our route. Removing this solvent leaves a PS phase in one channel, which improves the mechanical properties of the electrolyte. It is feasible to selectively remove the xylene because EC has a much lower vapor pressure than xylene.24,25Fig. 3(c) and (d) shows the confocal images collected from the composite templated using a bijel with 1.8 vol% particles. When xylene is removed, the PS (green) and EC (yellow) are labelled with anthracene-grafted PS and Nile red, respectively. This shows that the PS and EC phases are bicontinuously arranged, although the PS channel has been substantially compacted. The size of the PS phase is much smaller than the original xylene-rich phase due to the evaporation of xylene. We also observe that the volume reduction, relative to the bijel scaffold from which it was constructed, can be seen in the dried sample on a macroscopic scale (see the inset in Fig. 3(c)). Fig. 3(e) shows a bicontinuous PS–EC composite with much reduced domain size using a bijel scaffold stabilized with 3.6 vol% particles. This demonstrates that our approach of using bijels as scaffolds provides good control of the structure of the solid composites.
000 g mol−1. Unlike small molecules, this highly entangled PS cannot diffuse into the xylene-rich phase directly. As shown in Fig. 1, we tackled this challenge by dispersing PS in xylene and loading the dispersion onto the bijel. PS macromolecules are insoluble in the EC-rich phase and they exclusively diffuse into the xylene-rich domain of the bijel due to the gradient in concentration. Anthracene-grafted PS is the fluorescent probe for tracking the movement of the macromolecules. The bijel used here is stabilized by 1.8 vol% particles. The inset in Fig. 3(a) shows the loading of a PS/xylene solution (clear liquid) onto the bijel (pink in colour), and no damage to the bijel on a macroscopic scale is observed. Fig. 3(a) and (b) shows the confocal images of the same region of the bijel after 24 h, in which the xylene-phase is labelled with Nile red (light grey) and anthracene-grafted PS (green), respectively. This confirms the selective entrance of fluorescent anthracene-grafted PS macromolecules (green) into the xylene-rich phase, and indicates that the microstructure of the bijel scarcely changes with the partitioning of the macromolecules. In this post-processing approach, the movement of macromolecules is dominated by the entanglements formed in the solution, which could be tuned either by the solution concentration or via the choice of macromolecular weight. In the following section, we will present a more detailed exploration of this aspect. Xylene plays no useful role in the final composite and hence its selective removal is the final step in our route. Removing this solvent leaves a PS phase in one channel, which improves the mechanical properties of the electrolyte. It is feasible to selectively remove the xylene because EC has a much lower vapor pressure than xylene.24,25Fig. 3(c) and (d) shows the confocal images collected from the composite templated using a bijel with 1.8 vol% particles. When xylene is removed, the PS (green) and EC (yellow) are labelled with anthracene-grafted PS and Nile red, respectively. This shows that the PS and EC phases are bicontinuously arranged, although the PS channel has been substantially compacted. The size of the PS phase is much smaller than the original xylene-rich phase due to the evaporation of xylene. We also observe that the volume reduction, relative to the bijel scaffold from which it was constructed, can be seen in the dried sample on a macroscopic scale (see the inset in Fig. 3(c)). Fig. 3(e) shows a bicontinuous PS–EC composite with much reduced domain size using a bijel scaffold stabilized with 3.6 vol% particles. This demonstrates that our approach of using bijels as scaffolds provides good control of the structure of the solid composites.
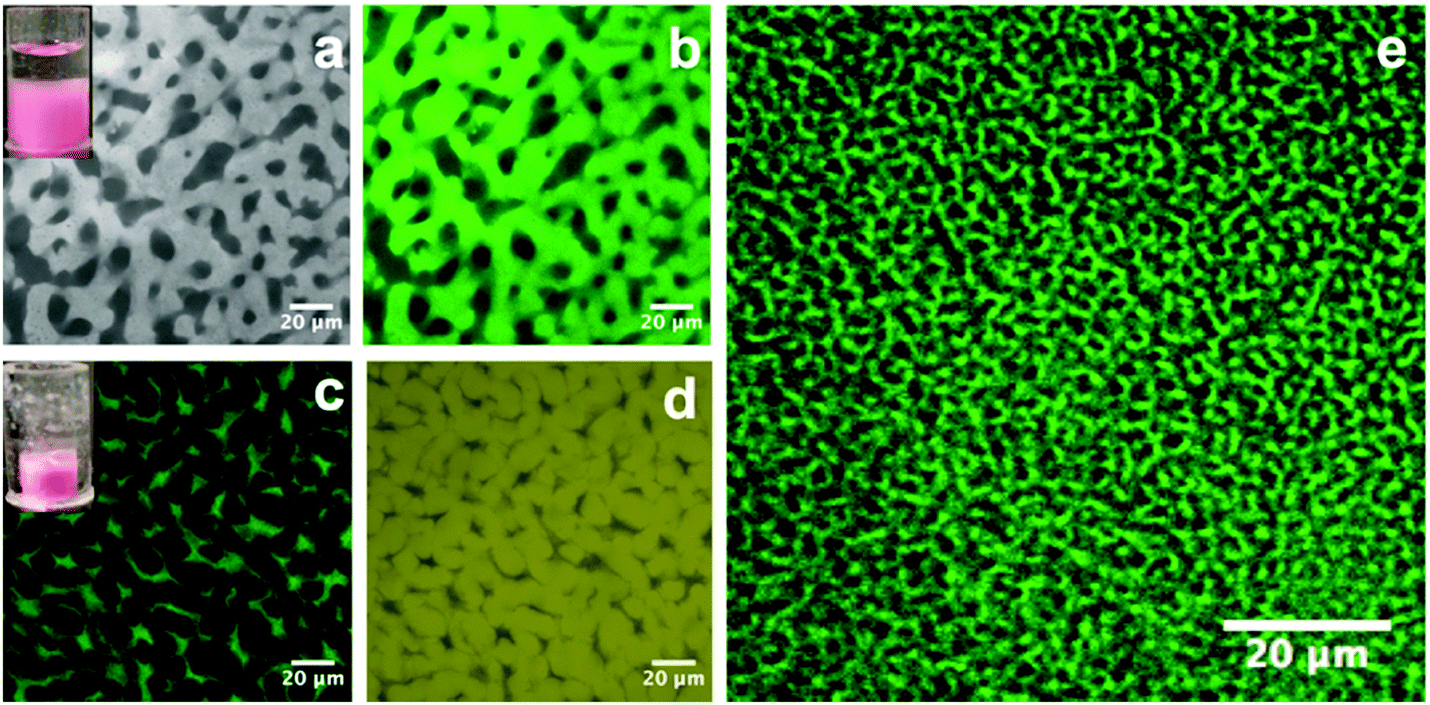 | ||
| Fig. 3 Evolution of the bijel microstructure during post-processing. (a) and (b) show the confocal images of the bijel with the partitioning of PS macromolecules, in which the xylene-rich phase (light grey) is labelled with Nile red and anthracene-grafted PS (green), respectively; (c) shows the confocal image of the solid PS–EC composite with xylene removed from the bijel scaffold, in which the PS phase (green) is labelled with anthracene-grafted PS; (d) shows the counterpart of (c) with the EC phase (yellow) labelled with Nile red. (e) shows the confocal image of the PS–EC composite prepared from a bijel with smaller liquid channels. The insets in (a) and (c) demonstrate the change to a bijel scaffold on a macroscopic scale during post-processing. | ||
Here we have demonstrated a very significant post-process procedure involving infiltrating PS and subsequently removing a large volume of xylene. The bicontinuous channel arrangement of the bijel scaffold has been robust in the face of this micro-engineering approach. It is known that the stability of the bijel structure results from the synergistic combination of interfacial tension and interparticle interactions, and these two factors are very sensitive to changes in the chemical composition.15 We observe slight shrinking of the bijel during the infiltration of PS (average molecular weight: ∼350000 g mol−1) into the xylene-rich domain (see Fig. S2, ESI‡). To better understand this, we selected two types of monodisperse PS with controlled molecular weight (PS1: 695000 g mol−1; PS2: 3116000 g mol−1; Mw/Mn ≈ 1). The PS concentration is fixed at 5 wt% in the xylene carrier solvent. Fig. 4 shows that PS2 causes a more significant decrease in the size of the xylene-rich domain, whereas the effect of PS1 is small. This suggests an explanation for the reduction in the volume of bijel. The release of xylene out of the bijel could, in part, be related to osmosis. Dispersing PS into xylene facilitates the mobility of PS macromolecules; however, entangled PS chains move very slowly in particular for PS2 with an extremely high molecular weight of about 3 million g mol−1. Our observation suggests that high molecular weight PS does not immediately diffuse into the xylene-rich phase. Thus, there is an outflow of xylene from the bijel driven by the osmotic pressure in the external xylene phase. The diffusion process is slow, but the big PS macromolecules still diffuse in eventually. Despite the shrinking, the bijel is sufficiently robust that it retains its initial bicontinuous structure. It has been demonstrated that interfacial particles remain trapped on bijel interfaces even when the overall structure is quite severely distorted.26 The stability of the bijel scaffold stems mostly from the jammed particle network at the interface, whereas the role of polystyrene in retaining the spinodal structure is more substantial in the composite electrolyte once the xylene has been removed.
 | ||
| Fig. 4 Effect of the molecular weight of PS on the size of the xylene-rich domain. (a) and (b) show the confocal images of the bijel before loading PS/xylene (5 wt%) solutions. Confocal images (c) and (d) show the bijel after loading the PS1 (695000 g mol−1) and PS2 (3116000 g mol−1) solution, respectively. The xylene-rich phase is dyed with Nile red (light grey). | ||
2.3. Characterization of the PS–EC composites
To demonstrate the stability of the bijel morphology and our control over it, we determined the interface separation for a range of bijels at different stages during sample preparation. Fig. 5(a) shows the channel width as a function of particle concentration with a post-processed sample included. This shows that we have good control over the channel width in these composite materials via our choice of the particle concentration. The channel width values were derived from the radial average of a fast Fourier transform of the confocal microscopy data (shown in the inset).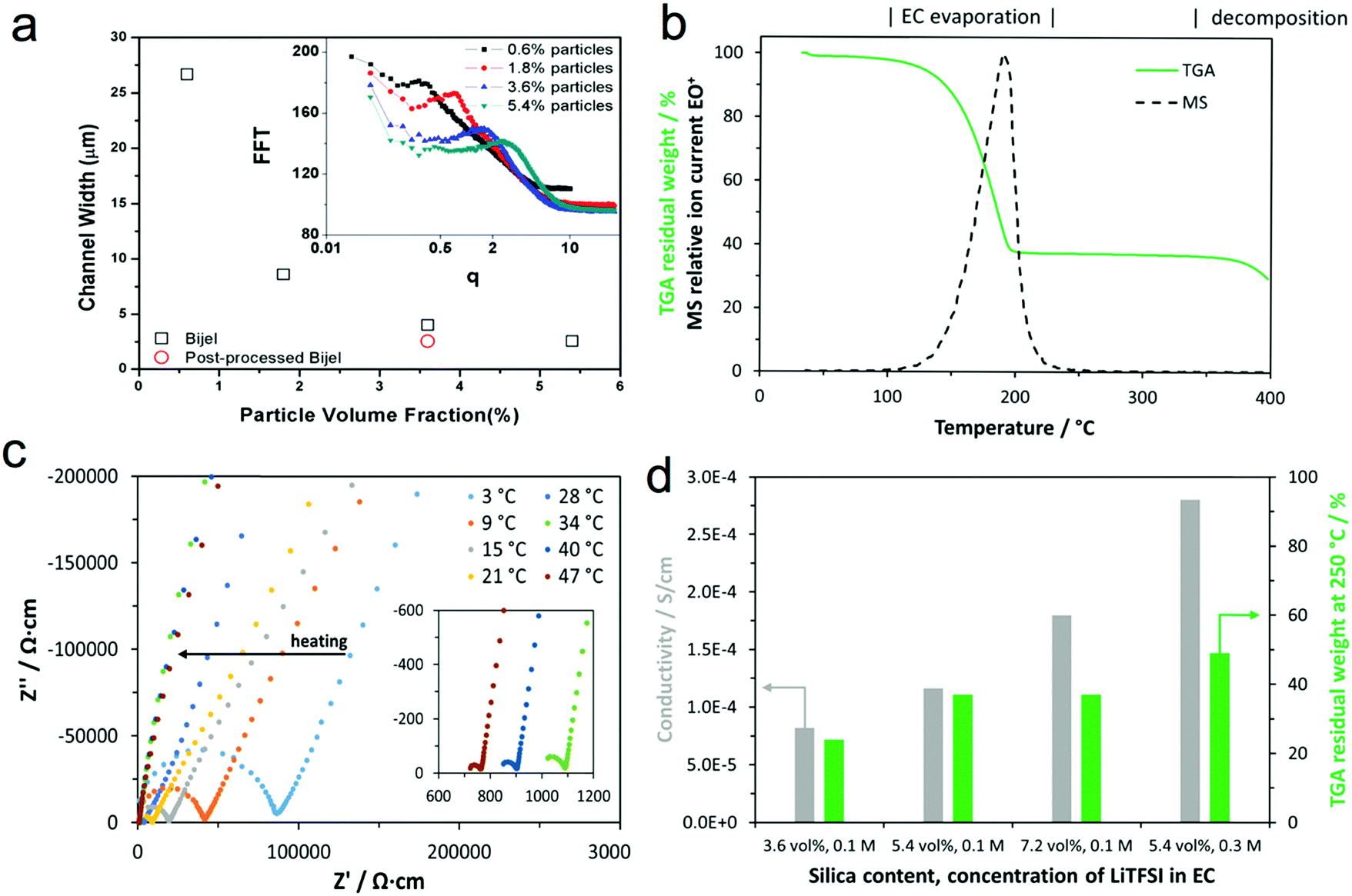 | ||
| Fig. 5 Characterization of the PS–EC composites. (a) The channel width of the bijel determined from confocal images for a range of sample compositions; the inset shows the FFT as a function of the spatial wavevector. (b) Thermogravimetric analysis exemplified by 5.4 vol% silica, 0.1 M EC(LiTFSI) coupled with mass spectrometry analysis of the exhaust gas at m/z = 88, demonstrating the evaporation of EC at a heating rate of 5 °C. (c) Impedance plots for increasing temperatures from 3 °C to 47 °C for the composite with 5.4 vol% silica and 0.3 M LiTFSI concentration; the inset shows a magnification of the low impedance region. (d) Ionic conductivities at ambient temperature resulting from fits of the impedance data and residual mass at 250 °C derived from thermogravimetric analysis for samples of the corresponding compositions. Note: The label ‘3.6 vol%, 0.1 M’ stands for the sample with 3.6 vol% silica particles and 0.1 M LiTFSI pre-mixed in EC; the other samples are described accordingly. | ||
We used thermogravimetric analysis coupled with mass spectrometry of the exhaust gas (TGA-MS) to determine the electrolyte content of the composite following post-processing (i.e. once the PS was infiltrated and the xylene was removed). Fig. 5(b) shows the thermogravimetric analysis (including mass spectrometry analysis of the exhaust gas at m/z = 88) of a composite prepared with 5.4 vol% silica and 0.1 M EC(LiTFSI) in the pre-mix. During the measurement in an argon atmosphere, weight-loss is dominated by the evaporation of EC below the temperature of 250 °C, which is indicated by the mass spectrometry signal in this temperature range at an m/z value of 88, which is attributed to EC+. As PS decomposes at temperatures above 350 °C, the residual weight at 250 °C is mostly made up of silica and PS since the LiTFSI content is small. No residual xylene is detected by MS, suggesting that xylene was effectively removed by the drying process. Thermogravimetric analysis indicates that the proportion of the EC phase decreases with increasing salt and silica content in the pre-mix (Fig. 5(d)), which is in line with the observations from confocal microscopy shown above (Fig. 2).
The impedance responses of the composites placed between stainless steel blocking electrodes are determined by alternating current electrochemical impedance spectroscopy and are shown in Fig. 5(c) in the form of impedance plots at different temperatures from 3 °C to 47 °C. We fitted the impedance plots using an equivalent circuit composed of a resistor in series with a parallel combination of a resistor and a constant phase element, and a constant phase element: R1–(R2‖CPE2)–CPE3. The first two elements model the response of the composite electrolyte, whereas the third element represents the capacitive response at the interfaces with the stainless steel blocking electrodes. From the total resistance (R1 + R2) and sample dimensions, we calculated the corresponding ionic conductivities at ambient temperature, which are summarized in Fig. 5(d). Thus, the total ionic conductivities of the composites at ambient temperature are in the range of 10−4 S cm−1, but this cannot solely be attributed to lithium ion conduction, as it is likely that the lithium transference number is smaller than one. Furthermore, the observed increase in conductivity with silica content points towards the silanol groups on the silica particles donating protons to the electrolyte,27 or may indicate residual moisture adsorbed on the silica particles causing the increase. The effect of the LiTFSI concentration is as expected: a higher salt content results in higher conductivity. As the thermogravimetric analysis shows that more silica particles and salt also yield higher residual mass, the conductivity is associated predominantly with the concentration and nature of ions and less with the mass fraction of the EC phase.
Fig. S3 (ESI‡) shows the temperature dependence of the conductivity moving from 20 °C, to 3 °C, to 47 °C, and back to 20 °C for sample composition 5.4 vol% silica and 0.3 M LiTFSI. The EC phase in the composite melts during heating above 30 °C, but hysteresis during cooling of the sample results from the supercooling of the EC phase. The ionic conductivity of the composite ranges between those of liquid and polymer electrolytes.28,29 Below the melting point of the EC phase, the lithium ion conductivity is likely to be too low for battery applications at ambient temperature. At temperatures above the melting point of the EC phase, the composite acts as an electrolyte and a separator because the polystyrene framework gives structural support and maintains separation of the electrodes.
3. Conclusion
In this work, we present the first example of a bijel formed from a pre-mix which contains a lithium salt. This, combined with the subsequent processing, demonstrates a direct and controllable route to preparing composite electrolytes made up of two bicontinuous domains, one providing a continuous ionic conduction pathway, and the other offering structural support. The bijel is prepared via arresting spinodal demixing of two partially miscible liquids: p-xylene and ethylene carbonate in the presence of a lithium salt (LiTFSI). The liquid–liquid interface is stabilized using silane-treated silica nanoparticles. Our results imply that the salt affects the three-phase contact angle of the nanoparticles at the interface; the size of the domains can be controlled by changing the content of silica particles. The bijel is directly transformed into a composite electrolyte by diffusion of polystyrene into the xylene phase and subsequent selective removal of xylene. The bijel structure is preserved during this multi-step processing. Our analysis by electrochemical impedance spectroscopy confirms the existence of ion conducting pathways in the composite electrolytes. In general, the ability to introduce additional components such as salt and synthetic polymers into bijels offers a facile route to turn bijels into functional multiphase materials.4. Experimental
Materials
Raw materials purchased from Sigma-Aldrich (UK) include ethylene carbonate (EC), p-xylene (xylene), hexamethyldisilazane (HMDS), ammonia solution (35%), ethanol, PS pellets (average Mw = ∼350000 g mol−1; average Mn = ∼170000 g mol−1) and bis(trifluoromethane)-sulfonamide lithium salt (LiTFSI). Hydrophobic fumed silica particles (HDK, H30), which have 50 wt% hydroxyl groups converted to methyl groups, were supplied by Wacker-Chemie. Monodisperse PS powders (PS1: Mw = 695000 g mol−1; PS2: Mw = 3116000 g mol−1; Mw/Mn ≈ 1) were provided by Polymer Laboratories. EC and p-xylene were dried over molecular sieves (4 Å) for three days before use and stored in a desiccator with silica gel.
Modification of H30 fumed silica
0.08 g of HMDS and 2 g of ammonia solution were dissolved in 15 g of ethanol and then 0.5 g of H3O particles was dispersed. The mixture was subjected to vortex mixing for 10 seconds, and then sonicated in an ultrasonic bath for half an hour. The particles were left immersed in the silanization solution for 20 hours at ambient temperature. The HMDS-modified particles were washed several times using ethanol, and the wet particles were dried at 50 °C for 24 hours. All the particles were further dried at 150 °C under vacuum for 24 hours to remove residual ethanol and moisture.Preparation of bicontinuous PS–EC composites
The preparation of bicontinuous PS–EC composites was carried out in a nitrogen-filled glove bag. All raw materials were transferred to the bag and purged with a flow of N2 for 30 min. In a typical experiment, LiTFSI (0.1 M) was dissolved in EC. Together, 0.1453 g of EC(LiTFSI), 0.1987 g of xylene and 0.024 g of treated particles were added to a glass vial. After heating up to 120 °C using a hot plate, the pre-mix, including EC and xylene in the single-fluid phase, was quickly sonicated using a tip ultrasonicator at a power of 5 watts for 10 seconds. Phase separation could be avoided due to the heat generated by sonication. Then the pre-mix was quickly transferred to a glass cell (see the inset in Fig. 3) preheated at 120 °C. The glass cell was constructed from a glass cylinder (ϕ = 6 mm) glued onto a glass cover slide. The bijel was formed when the pre-mix was subjected to a quench by contact with a cold metal plate. Afterwards, 0.15 g of a PS/xylene solution (30 wt%) was gently loaded on top of the bijel in the glass cell and sealed by gluing another glass slide on the top. After 24 hours, the cell was re-opened by peeling off the glass slide on the top; the composite was dried at 50 °C for 24 hours. The sample was carefully extracted from the glass cell and sealed in a larger glass vial before moving out of the glove bag.Composite characterization
Imaging was performed by confocal microscopy using a Zeiss Observer.Z1 inverted microscope in conjunction with a Zeiss LSM 700 scan head and a ×20, 0.4 NA air objective or a ×40, 1.3 NA oil immersion objective. The fluorescent dye, Nile red, was added to the xylene-rich phase and excited with a 488 nm laser. Though we observed partitioning of Nile red into both EC and xylene-rich phases, careful selection of emission filters allowed separate imaging of the xylene-rich phase (light grey). Alternatively, anthracene-labelled PS was used as a fluorescent dye to label the PS phase that was excited in the dried PS–EC composites using a 405 nm laser.Alternating Current Electrochemical Impedance Spectroscopy (AC EIS) measurements were carried out using a ModuLab XM instrument from Solartron Analytical. A specially designed cutting tool was used to prepare discs with parallel faces from the pristine samples using a razor blade. The sample discs were placed between two stainless steel electrodes. AC EIS measurements were carried out over the temperature range of 3 °C to 47 °C. The temperature was set using a Model F32-MA refrigerated/heating circulator obtained from Julabo GmbH. AC EIS measurements were recorded at an amplitude of 15 mV from a start to end frequency of 1 MHz to 0.1 Hz, respectively. Thermogravimetric analysis coupled to mass spectrometry was used to assess the compositions of the samples once fabrication was complete. The sample was heated from ambient temperature to 400 °C at a rate of 5 °C min−1 in an argon atmosphere; mass spectrometry was used to determine which component was leaving the sample.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the funding provided by the National Natural Science Foundation of China (21503110), EPSRC (EP/J007404/1, EP/L019469/1), and Deutsche Forschungsgemeinschaft (RI2614/1-1).Notes and references
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- N. Shirshova, A. Bismarck, S. Carreyette, Q. P. V. Fontana, E. S. Greenhalgh, P. Jacobsson, P. Johansson, M. J. Marczewski, G. Kalinka, A. R. J. Kucernak, J. Scheers, M. S. P. Shaffer, J. H. G. Steinke and M. Wienrich, J. Mater. Chem. A, 2013, 1, 15300–15309 CAS.
- M. E. Cates and P. S. Clegg, Soft Matter, 2008, 4, 2132–2138 RSC.
- J. Tavacoli, J. H. J. Thijssen and P. S. Clegg, in Particle-Stabilized Emulsions and Colloids: Formation and Applications, ed. T. Ngai and S. Bon, Royal Society of Chemistry, Cambridge, 2014, ch. 6, pp. 129–168 Search PubMed.
- E. M. Herzig, K. A. White, A. B. Schofield, W. C. K. Poon and P. S. Clegg, Nat. Mater., 2007, 6, 966–971 CrossRef CAS PubMed.
- M. Reeves, A. T. Brown, A. B. Schofield, M. E. Cates and J. H. J. Thijssen, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 92, 032308 CrossRef CAS PubMed.
- M. Reeves, K. Stratford and J. H. J. Thijssen, Soft Matter, 2016, 12, 4082–4092 RSC.
- J. W. Tavacoli, J. H. J. Thijssen, A. B. Schofield and P. S. Clegg, Adv. Funct. Mater., 2011, 21, 2020–2027 CrossRef CAS.
- J. A. Witt, D. R. Mumm and A. Mohraz, Soft Matter, 2013, 9, 6773–6780 RSC.
- H. Firoozmand, B. S. Murray and E. Dickinson, Langmuir, 2009, 25, 1300–1305 CrossRef CAS PubMed.
- M. M. Cui, T. Emrick and T. P. Russell, Science, 2013, 342, 460–463 CrossRef CAS PubMed.
- L. Bai, J. W. Fruehwirth, X. Cheng and C. W. Macosko, Soft Matter, 2015, 11, 5282–5293 RSC.
- E. Sanz, K. A. White, P. S. Clegg and M. E. Cates, Phys. Rev. Lett., 2009, 103, 255502 CrossRef PubMed.
- M. N. Lee, J. H. J. Thijssen, J. A. Witt, P. S. Clegg and A. Mohraz, Adv. Funct. Mater., 2013, 23, 417–423 CrossRef CAS.
- M. N. Lee and A. Mohraz, J. Am. Chem. Soc., 2011, 133, 6945–6947 CrossRef CAS PubMed.
- L. Imperiali, C. Clasen, J. Fransaer, C. W. Macosko and J. Vermant, Mater. Horiz., 2014, 1, 139–145 RSC.
- M. N. Lee, M. A. Santiago-Cordoba, C. E. Hamilton, N. K. Subbaiyan, J. G. Duque and K. A. D. Obrey, J. Phys. Chem. Lett., 2014, 5, 809–812 CrossRef CAS PubMed.
- M. F. Haase, K. J. Stebe and D. Lee, Adv. Mater., 2015, 27, 7065–7071 CrossRef CAS PubMed.
- J. A. Witt, D. R. Mumm and A. Mohraz, J. Mater. Chem. A, 2016, 4, 1000–1007 CAS.
- S. Zekoll, C. Marriner-Edwards, A. K. O. Hekselman, J. Kasemchainan, C. Kuss, D. E. J. Armstrong, D. Cai, R. J. Wallace, F. H. Richter, J. H. J. Thijssen and P. Bruce, Energy Environ. Sci., 2018, 11, 185–201 CAS.
- D. Y. Cai and P. S. Clegg, Chem. Commun., 2015, 51, 16984–16987 RSC.
- C. F. Holmes, J. Am. Chem. Soc., 1973, 95, 1014–1016 CrossRef CAS.
- L. S. Kassel, J. Am. Chem. Soc., 1936, 58, 670–671 CrossRef CAS.
- C. S. Hong, R. Wakslak, H. Finston and V. Fried, J. Chem. Eng. Data, 1982, 27, 146 CrossRef CAS.
- K. A. Rumble, J. H. J. Thijssen, A. B. Schofield and P. S. Clegg, Soft Matter, 2016, 12, 4375–4383 RSC.
- H. J. Walls, M. W. Riley, R. R. Singhal, R. J. Spontak, P. S. Fedkiw and S. A. Khan, Adv. Funct. Mater., 2003, 13, 710–717 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
Footnotes |
| † Data have been uploaded to Oxford Research Archive, DOI: 10.5287/bodleian:AmaNbjgdA |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7mh01038a |
| § Present address: Physikalisch-Chemisches Institut, Justus-Liebig-Universität Gießen, 35392 Gießen, Germany. |
| This journal is © The Royal Society of Chemistry 2018 |