
Isotachophoresis applied to biomolecular reactions
C.
Eid†
 * and
J. G.
Santiago
*
* and
J. G.
Santiago
*
Department of Mechanical Engineering, Stanford University, USA. E-mail: juan.santiago@stanford.edu
First published on 11th October 2017
Abstract
This review discusses research developments and applications of isotachophoresis (ITP) to the initiation, control, and acceleration of chemical reactions, emphasizing reactions involving biomolecular reactants such as nucleic acids, proteins, and live cells. ITP is a versatile technique which requires no specific geometric design or material, and is compatible with a wide range of microfluidic and automated platforms. Though ITP has traditionally been used as a purification and separation technique, recent years have seen its emergence as a method to automate and speed up chemical reactions. ITP has been used to demonstrate up to 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold acceleration of nucleic acid assays, and has been used to enhance lateral flow and other immunoassays, and even whole bacterial cell detection assays. We here classify these studies into two categories: homogeneous (all reactants in solution) and heterogeneous (at least one reactant immobilized on a solid surface) assay configurations. For each category, we review and describe physical modeling and scaling of ITP-aided reaction assays, and elucidate key principles in ITP assay design. We summarize experimental advances, and identify common threads and approaches which researchers have used to optimize assay performance. Lastly, we propose unaddressed challenges and opportunities that could further improve these applications of ITP.
000-fold acceleration of nucleic acid assays, and has been used to enhance lateral flow and other immunoassays, and even whole bacterial cell detection assays. We here classify these studies into two categories: homogeneous (all reactants in solution) and heterogeneous (at least one reactant immobilized on a solid surface) assay configurations. For each category, we review and describe physical modeling and scaling of ITP-aided reaction assays, and elucidate key principles in ITP assay design. We summarize experimental advances, and identify common threads and approaches which researchers have used to optimize assay performance. Lastly, we propose unaddressed challenges and opportunities that could further improve these applications of ITP.
I. Introduction and background
Although the term “isotachophoresis” was coined only 47 years ago, similar techniques have existed for nearly a century. In 1923, Kendall and Crittenden1 described a technique to separate acids and metals, which they called the “ion migration method”. After that, several studies in the 1930s and later described “moving boundary electrophoresis”2 and “displacement electrophoresis”,3 processes nearly identical to isotachophoresis. It wasn't until 1970 that Haglund4 introduced the term “isotachophoresis” (ITP), based on the fact that all ITP zones migrate at the same velocity at steady state (“isos” meaning equal and “takhos” meaning velocity in Greek). In the following years, ITP enjoyed a period of significant popularity, owing in part to the fact that it could be performed in capillaries larger than those used for capillary electrophoresis (CE).5 The 1980s saw a decrease in ITP's popularity, but also the rise of a new area of application of ITP. Up to this point, ITP had mostly been limited to a preconcentration and separation technique. Then, in a number of publications led by researchers like Bocek6,7 and Furst,8,9 ITP applications were expanded to include the initiation and control of chemical reactions, particularly those involving enzymes and peptides. In those assays, ITP's high-resolving capability would be used to detect concentration changes in the enzyme-catalyzed mixture of reactants and products at several time points of the reaction.ITP re-emerged in the spotlight in the 1990s through its coupling to CE. Researchers designed assays which incorporated ITP focusing followed by a disruption of ITP and initiation of CE separation. This combination provides dramatic increase in sensitivity while preserving the excellent separation efficiency of CE.10–12 In recent years, ITP has found increased adoption in microfluidic formats, which leverage its various advantages (including self-sharpening zones, insensitivity to errors in injection or disturbances, and purification capabilities) in a wide array of applications. For readers interested in learning more about the history and development of ITP, we refer to several excellent electrophoresis and isotachophoresis reviews.12–17 Indeed, today there are typically 40–50 papers published per year using ITP, the vast majority (>90%) of which being in microfluidic formats. These publications cover a wide range of applications, such as preconcentration of analytes prior to CE separation,12,18,19 purification from complex samples,20–25 analytical and computational modeling,26–28 and fractionation of biological and chemical species.29–31
Unlike the majority of electrophoretic methods, ITP uses a two-buffer system consisting of a high-mobility leading electrolyte (LE) buffer and a low-mobility trailing electrolyte (TE) buffer. Sample ions with effective mobility magnitudes greater than the TE (in the TE zone) and less than the LE (in the LE zone) focus at an interface between these two co-ions.32,33 Preconcentration factors of up to one-million have been achieved,34 although 1000 to 15000-fold preconcentration is typically observed with more complex biological samples such as nucleic acids in blood.22 The LE and TE zones have respectively high and low conductivity, and so a relatively high electric field is established in the TE zone and a low field in the LE zone. In accordance with the conservations of species and current, the TE and LE travel at the same rate. The strong electric field gradient establishes a self-sharpening and translating TE-to-LE interface which makes ITP robust to disturbances like pressure-driven flow, rough channel surfaces, and changes in channel geometry.
In Fig. 1, we demonstrate qualitatively the self-sharpening feature of ITP. TE ions which diffuse into the lower electric field LE zone are overtaken by neighboring higher-mobility LE ions and thus fall back into their original TE zone. Conversely, higher mobility LE ions diffusing into the higher electric field TE zone are restored since they migrate faster than the TE. Importantly, TE and LE mobilities are chosen such that sample ions in the TE (LE) migrate faster (slower) than neighboring TE (LE) ions and are driven toward the TE-to-LE interface. See for example Khurana et al.35 and Garcia-Schwarz et al.36 for more detailed and quantitative descriptions (including models and experimental studies) of the diffusion- and dispersion-limited focusing dynamics of ITP sample ions.
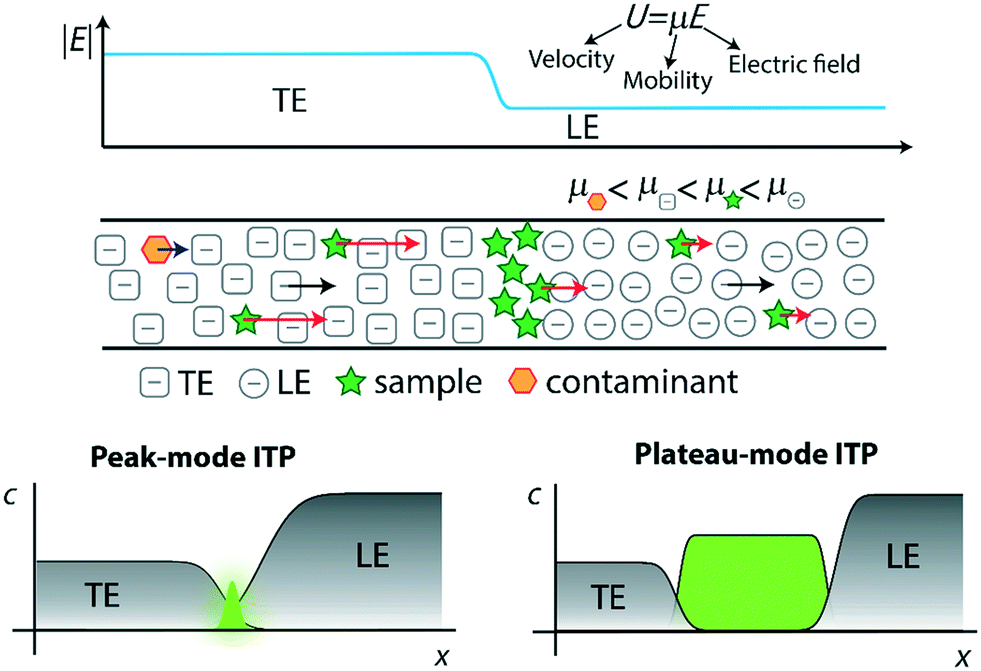 | ||
| Fig. 1 Top: Schematic representation of selective focusing in ITP. Sample species with intermediate mobilities migrating in the TE zone overspeed neighboring TE ions, while those migrating in the LE zone are overtaken by neighboring LE ions. Sample species therefore focus at the TE–LE interface, where their velocity will match that of the LE and TE zones (the so-called ITP velocity). Species that have a mobility lower than that of the TE electromigrate but fall behind the ITP interface, whereas species with higher mobility than the LE overtake the LE. Bottom: Schematic of peak and plateau ITP modes. In the former, dilute sample ions focus in a Gaussian-like peak. Multiple sample ions can co-focus in partially or entirely overlapping peaks, depending on their relative mobilities. The typical ITP peak magnitude is exaggerated considerably for visualization purposes. In plateau mode, sample ions at sufficiently high concentration to form plateaus of constant (in time) and locally uniform concentration, and contribute significantly to local conductivity. | ||
ITP processes can conveniently be categorized as either peak-mode or plateau-mode. Peak-mode ITP is associated with sample ions present in trace concentrations. Such samples focus into the TE-to-LE interface region but there is insufficient time (and equivalently distance along the channel) for the sample ions to appreciably influence local ionic conductivity in the channel.37 In peak-mode ITP, the sample species respond solely to the electric field established by the dynamics of the TE and LE. Importantly, multiple sample ions can co-focus within and significantly overlap within the same sharp ITP interface. In an approximate sense, well-focused sample ions accumulate into Gaussian peaks with continuously increasing area and significant spatial overlap. The focusing and relative positions of these peaks is determined solely by the electric field established by the TE and LE and the relative mobilities of the TE, the LE, and each sample species.
The second useful category for ITP is plateau-mode ITP. Above a certain threshold concentration (and duration of the ITP process), sample ions accumulate and reach a local maximum concentration. Here, multiple sample ions will reach respective maximum concentration and segregate into respective, multiple plateau-like zones of locally uniform (and constant) concentration. If there is a continuous influx of sample ions (e.g., from a reservoir), these plateaus increase in length in proportion to the amount of electrical charge run through the system.38 For the rare case of ITP of fully-ionized species, this threshold is determined by the Kohlrausch regulating function (KRF).39 For the common case of weak electrolytes (e.g., LE and TE solutions which are pH buffers), the threshold is governed by the Alberty40 and Jovin41 functions instead. Plateau-mode ITP has been leveraged for many applications, including separation and indirect detection of toxins, amino acids, and others.42–44 Briefly, peak-mode ITP is well-suited for mixing and driving reaction kinetics due to co-focusing of trace sample ions into high-concentration, overlapping peaks; while plateau-mode ITP is better-suited for separation of species into distinct zones for the purpose of purification or identification.
In this review, we outline and discuss an emerging use and field of application of ITP: the initiation (via mixing), control, and acceleration of chemical reactions involving at least one ionic species. Accordingly, we will specifically consider applications of ITP wherein at least one reagent in a chemical reaction is focused at an ITP interface, and this focusing is used to control a chemical reaction involving that reagent and at least one other reagent. We first briefly summarize simple concepts of second-order reactions. We then review a series of papers in which ITP was used to preconcentrate and mix reagents, and describe mixing time scales for two adjoining zones. We then review papers using ITP to accelerate biomolecular reactions, and separately discuss homogeneous and heterogeneous assay configurations. For each class of application, we review and describe physical modeling and scaling of ITP-aided reaction assays and summarize relevant literature. In Table 1, we summarize the studies discussed in this review, classify these in a manner consistent with our discussions, and briefly mention their major contributions. In Table 2, we characterize the reactant species, kinetics, and performance of the reactions described in these studies. Lastly, we discuss unaddressed challenges and make recommendations for promising areas for future work.
| Authors | Year | Primary contribution |
|---|---|---|
| ITP to mix and control chemical reactions | ||
| Kawabata et al.46 | 2008 | First work to use ITP to speed-up chemical reactions |
| Park et al.47 | 2008 | |
| First commercial system to use ITP | ||
| Kagebayashi et al.48 | 2009 | |
| ITP to preconcentrate, mix, and accelerate homogeneous reactions | ||
| Persat and Santiago61 | 2011 | First experimental demonstration of ITP-aided enhancement of nucleic acid hybridization |
| Bercovici et al.63 | 2011 | First quantitative demonstration of reaction acceleration using ITP |
| Bercovici et al.52 | 2012 | First analytical model of ITP reaction assays, framework for several future models |
| Bahga et al.64 | 2013 | Coupling ITP-based reaction with capillary electrophoresis for clean-up of excess reactants |
| Eid et al.65 | 2013 | Inline reaction-separation assay using ionic spacer with sub-picomolar detection limit |
| Rubin et al.58 | 2014 | Analytical model focusing on sample distribution within ITP zones, and its effect on reaction rates |
| Shintaku et al.59 | 2014 | First demonstration of ITP hybridization in bead-based assays |
| Schwartz and Bercovici70 | 2014 | First work to use ITP to speed up reactions with whole cell reactants |
| Eid et al.57 | 2015 | Recruiting non-focusing protein reactant into ITP using modified aptamer probes |
| Eid et al.53 | 2016 | Analytical modeling of influx and reaction rates in ITP as a function of initial sample placement |
| Phung et al.73 | 2017 | Coupling ITP with FISH for detection of whole bacterial cells |
| Van Kooten et al.66 | 2017 | Focusing samples from 50 μL initial volume down to 500 pL |
| ITP to preconcentrate, mix, and accelerate heterogeneous reactions | ||
| Garcia-Schwarz and Santiago79,80 | 2012 | ITP reaction coupled with gel-based excess reactant removal |
| 2013 | ||
| Karsenty et al.75 | 2014 | First experimentally-validated model for ITP-aided surface hybridization |
| Han et al.76 | 2014 | Demonstration of ITP-aided DNA microarray hybridization with sub-picomolar limit of detection |
| First demonstration of truly multiplexed ITP detection | ||
| Shkolnikov and Santiago78,81 | 2014 | Coupling ITP preconcentration to affinity chromatography purification |
| Comprehensive and highly predictive analytical modeling of ITP-aided capture kinetics | ||
| Khnouf et al.85 | 2014 | Demonstration of ITP enhancement for surface-based immunoassays |
| Moghadam et al.87 | 2015 | First demonstration of ITP accelerating lateral flow assays |
| Paratore et al.77 | 2017 | Lowest limit of detection achieved by an ITP immunoassay to date |
| Authors | Species | Reaction type (kon, koff, KD if known) | Species controlling completion time | Reaction time | Sensitivity |
|---|---|---|---|---|---|
| Kawabata et al.46 | Probe 1: DNA anti-AFP WA1 antibody | Homogeneous | Probes 1 and 2 | 2 min | 1 pM |
| Park et al.47 | Probe 2: anti-AFP WA2 antibody (L) | ||||
| Kagebayashi et al.48 | |||||
| Target: alpha feto protein | |||||
| Persat and Santiago61 | Probe: molecular beacon (L) | Homogeneous | Probe | 2 min | 10 pM |
| Target: miRNA | |||||
| Bercovici et al.63 | Probe: molecular beacon (L) | Homogeneous | Probe | 30 pM | |
| Target: 16S rRNA | (kon = 4.75 × 103 M−1 s−1) | ||||
| Bahga et al.64 | Probe: molecular beacon (L) | Homogeneous | Probe | 3 min | 3 pM |
| Target: ssDNA oligo | |||||
| Eid et al.65 | Probe: ssDNA oligo (L) | Homogeneous | Probe | 5 min | 230 fM |
| Target: ssDNA oligo | |||||
| Shintaku et al.59 | Probe: beads conjugated with ssDNA oligo | Homogeneous | Probe | 20 min | 100 fM |
| Target: ssDNA oligo | (kon = 4.4 × 10−6 M−1 s−1KD = 7.3 × 10−12 M) | ||||
| Schwartz and Bercovici70 | Probe: antimicrobial peptides (L) | Homogeneous | Probe | Up to 1 h | 2 × 104 cfu mL−1 |
| Target: E. coli cells | |||||
| Eid et al.57 | Probe: SOMAmer (L) | Homogeneous | Probe | 5 min | 2 nM |
| Target: C-reactive protein | (KD = 10−9 M) | ||||
| Van Kooten et al.66 | Probe: molecular beacon (L) | Homogeneous | Probe | 7.5 min | 1 pM |
| Target: ssDNA oligo | |||||
| Phung et al. | Probe: ssDNA (L) | Homogeneous | Probe | 30 min | 6 × 104 cfu mL−1 |
| Target: E. coli and P. aeruginosa cells | |||||
| Garcia-Schwarz and Santiago79,80 | Reporter: hairpin oligo | Gel | Reporter and probe | 15 min | 2 pM |
| 10 pM | |||||
| Probe: ssDNA oligo (i) | |||||
| Target: let-7a miRNA | |||||
| Karsenty et al.75 | Probe: molecular beacon (i) | Surface | Target | 3 min | 1 nM |
| Target: ssDNA oligo | |||||
| Han et al.76 | Probe: ssDNA oligo (i) | Microarray (surface) | Target | 30 min | 100 fM |
| (kon = 7.6 × 104 mol−1 s−1koff = 4.4 × 10−5 s−1KD = 5.7 × 10−10 M) | |||||
| Target: ssDNA oligo | |||||
| Shkolnikov and Santiago81 | Probe: ssDNA oligo (i) | Affinity column | Probe | 10 min | — |
| Target: ssDNA oligo | (kon = 1.5 × 103 M−1 s−1KD = 10−11 M) | ||||
| Khnouf et al.85 | Probe: antibody-coated magnetic beads (i) or antibodies (i) | Surface | Probe | 2 min | 18 pM |
| Target: bovine serum albumin | |||||
| Moghadam et al.87 | Probe: rabbit anti goat IgG | Surface | Probe | 7 min | 0.7 nM |
| Target: goat anti-rabbit IgG (L) | (koff = 1.75 × 10−3 s−1KD = 1.42 × 10−4 M) | ||||
| Paratore et al.77 | Probe: anti-GFP antibody | Surface | Target | 6 min | 220 fM |
| Target: EGFP antigen | (kon = 2.1 × 105 M−1 s−1) |
II. Earliest work involving ITP to control chemical reactions
A common theme in the first set of papers on ITP-aided reaction acceleration is the use of ITP to mix and control reactants in an ITP zone. We first provide a brief and simple scaling analysis to provide some physical context for this process. Unlike other types of microfluidic mixing using stirring or chaotic flows, mixing in ITP is typically accomplished via a deterministic electrophoretic process wherein one species is electromigrated into a region occupied by a second species. As mentioned above, migration velocity is the product of the electrophoretic mobility and the local electric field,| Ui = μiE | (1) |
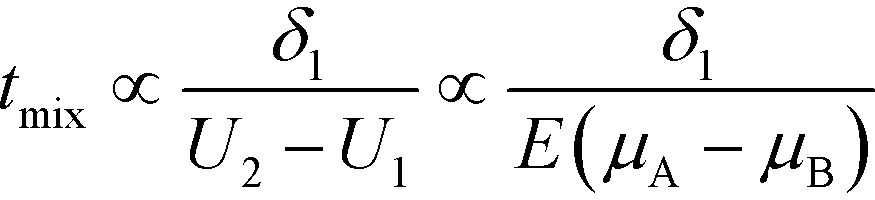 | (2) |
To our knowledge, the first demonstrated use of ITP to mix and initiate chemical reactions came in 2008 from scientists working at Wako Pure Chemical Industries, in a series of papers describing the development of the assay concept, its optimization, and its development into a commercial instrument. The result of this work, the μTASWako i30,45 was the first commercially-available instrument that uses ITP.
In their first paper, Kawabata et al.46 described an assay that they called the Electrokinetic Analyte Transport Assay. The assay leveraged ITP to focus a DNA-coupled antibody and increase its concentration while reacting with a target protein, α-fetoprotein (AFP). Conjugating the antibody with a DNA molecule increased its electrophoretic mobility and enabled the DNA-antibody to focus in ITP. The differential velocity between the ITP-focused DNA-antibody complex and the AFP (which was not focused in ITP) was used to initiate the reaction. The DNA-antibody complex reacted with AFP and Kawabata hypothesized that the ITP preconcentration of the DNA-antibody complex accelerated these reaction kinetics (neither quantitative data nor analysis supporting accelerated kinetics was provided). Further, the reaction resulted in recruitment of unfocused AFP into ITP mode, increasing product concentration by up to 140-fold. Applied voltages where then reconfigured on their chip to initiate CE and separate the immune complex of interest from background fluorescent signal, as shown in Fig. 2. The plastic microfluidic chip was designed as a straight channel with several branches to allow the introduction of the various buffers and reagents. Their LE and TE buffers contained Tris-HCl and Tris-HEPES, respectively, and additional components like polymers, albumin, salts, and surfactants to improve assay performance. They achieved a limit of detection of 5 pM with this assay, impressively nearly 2 orders of magnitude below clinically-relevant limits.
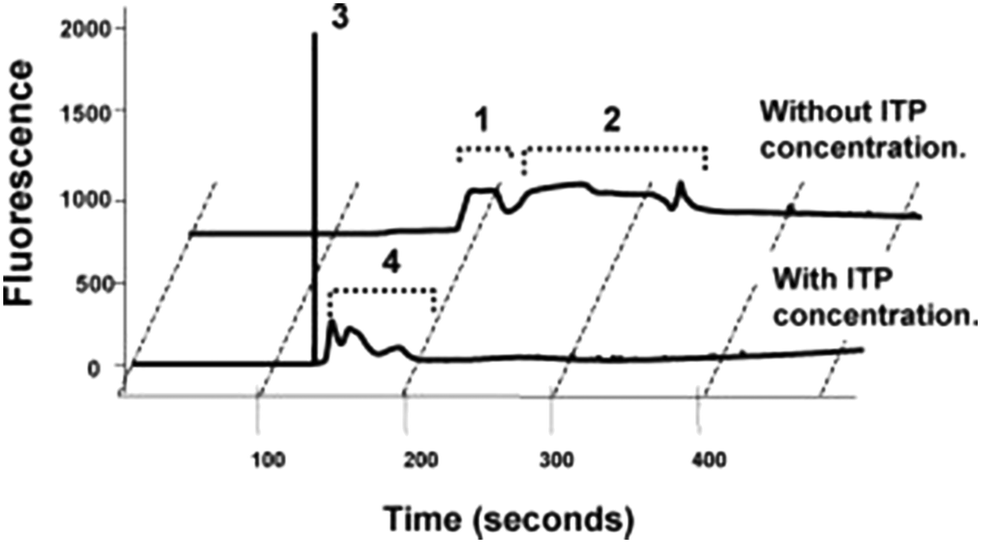 | ||
| Fig. 2 Electropherogram from Kawabata et al.46 using of ITP preconcentration and DNA-labeled antibodies to control an ITP assay for AFP, a protein target. Peaks 1 and 3 correspond to the DNA-antibody–AFP complex, whereas peaks 2 and 4 correspond to unreacted antibodies, trailing the immune complex. ITP resulted in a 140-fold increase in fluorescent signal, enabling lower detection limits. | ||
Simultaneously (the papers were published within the same week), Park et al.47 published a paper on improving the reproducibility of the assay. They studied peak intensity and separation, and their dependence on “handoff time”, the moment at which voltage switching causes the assay to transition from ITP stacking to CE separation. Interestingly, they found that changes in buffer concentration or small manufacturing defects in the devices caused noticeable variation in arrival times, which in turn affected handoff and adversely affected data quality. To combat this, Park introduced automated handoff and timing mechanisms which relied on computer monitoring of voltage, in order to adjust for external factors, and to achieve highly precise control of signal intensity and peak separation.
The final paper in this series was published in 2009, by Kagebayashi et al.48 In it, they described the automated AFP-L3 assay, and the μTASWako i30 immunoanalyzer which evolved from the previous two papers. Kagebayashi et al. described its mechanism and characterized its performance. In addition to quantifying total AFP levels, they also incorporated an affinity-based separation step to simultaneously quantify the L3 isoform of AFP, AFP-L3. AFP-L3% is a clinically-relevant biomarker that is specific to hepatocellular carcinoma and other pathologies.49,50 By specifically interacting with lectin in the CE separation channel, the DNA-AFP-L3 immunocomplex is separated from AFP-L1 isoform, allowing the quantitation of both isoforms and their ratio using laser-induced fluorescence. They validated their assay in spiked serum samples, and achieved a limit of detection of 1 pM, with 2% coefficient of variation. Their test demonstrated good correlation with a commercially-available reference assay. The μTASWako i30 immunoanalyzer received FDA 510(k) clearance in 2011,51 and remains commercially available as of this publication.
III. ITP to preconcentrate, mix, and accelerate homogeneous reactions
III.a. Homogeneous reactions: theory and models
In this section, we will briefly review analytical modeling and scaling of homogeneous chemical reactions (i.e. all reactants suspended in solution) using ITP. Standard second-order chemical reactions can be expressed as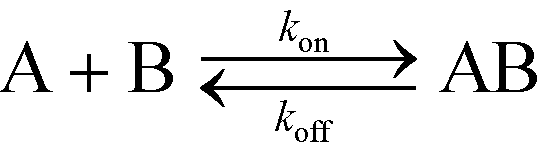 | (3) |
 | (4) |
Bercovici et al.52 developed the first analytical model examining ITP-aided chemical reactions wherein both reacting species are focused in peak-mode ITP. This initial model assumed a perfectly overlapped Gaussian reactant peaks, and used volume-averaged concentrations, given by
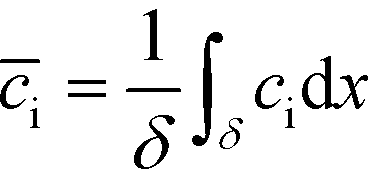 | (5) |
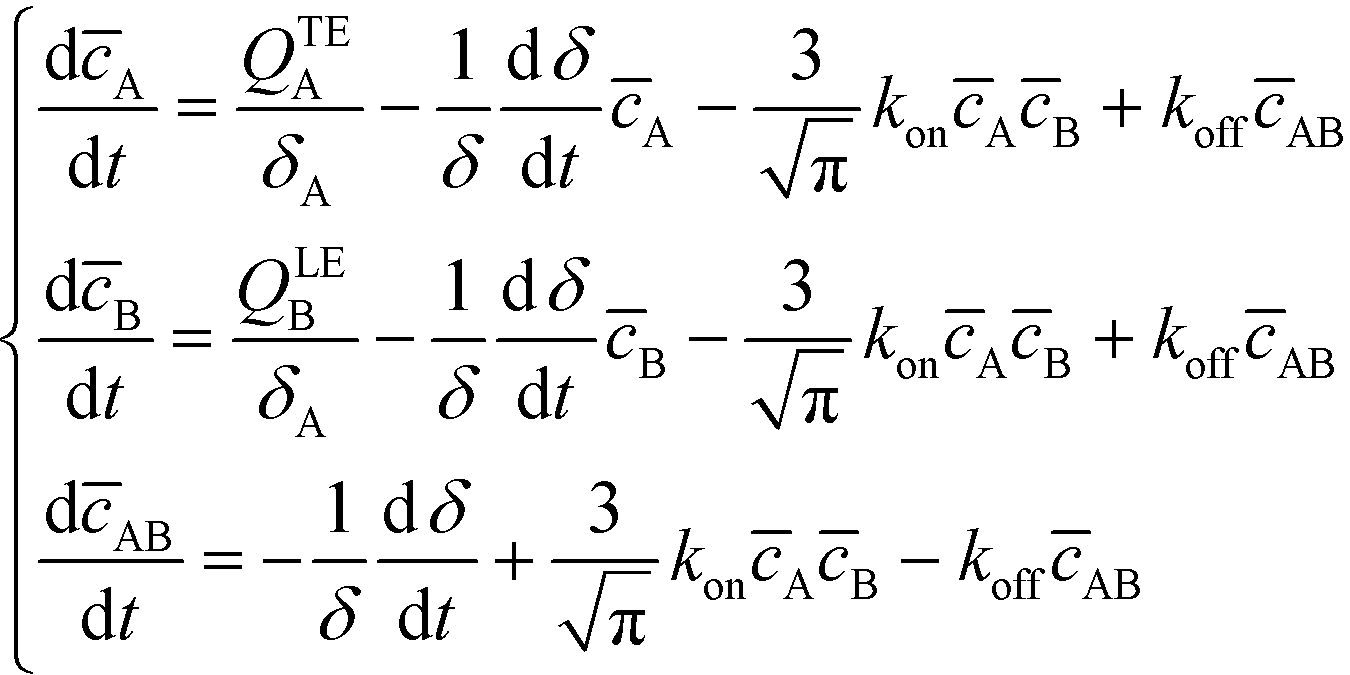 | (6) |
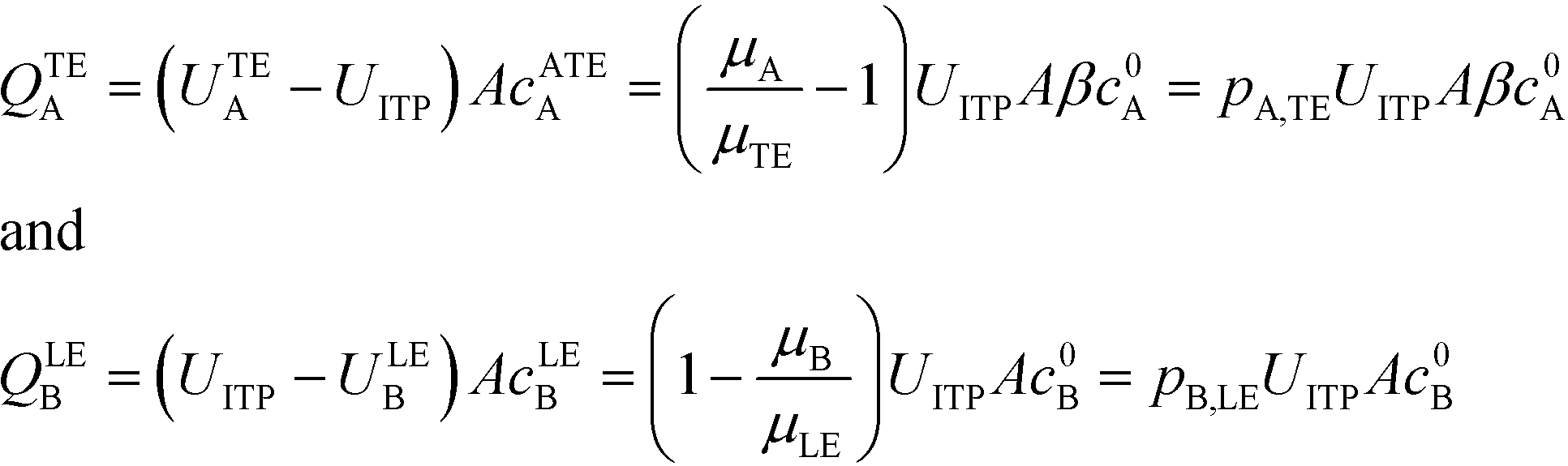 | (7) |
U ITP is the velocity of the ITP zone, A the channel area, c0A and c0B the respective reservoir or initial concentrations of species A and B, and β is the ratio of TE ion concentrations in the adjusted TE and TE zones. For more on adjusted TE zones, we refer interested readers to Khurana et al.35 and to Eid and Santiago.53δ is determined by the balance of dispersion effects (e.g., diffusion which acts to mix species and broaden the peak) and electromigration (which acts to sharpen the interface). In ideally, diffusion-limited conditions this width can be estimated as54
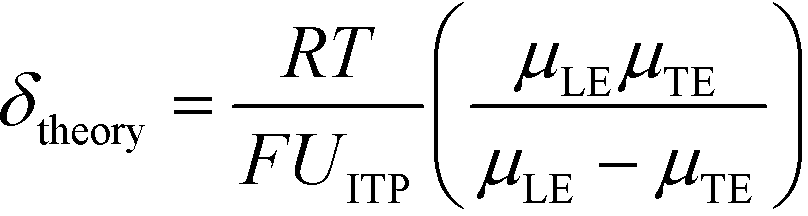 | (8) |
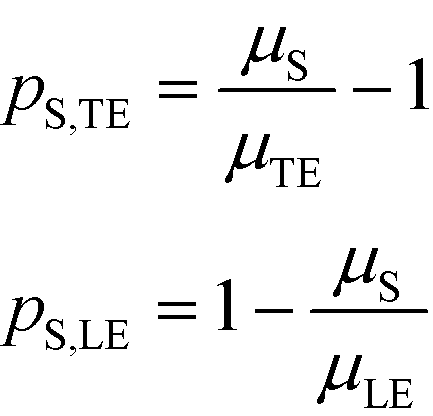 | (9) |
Separabilities quantify the relative mobilities of a species and the buffer it is in, and are useful in estimating the focusing rate of species.53
By assuming that one of the two reactants is in relative excess at the ITP interface, as well as an equilibrium constant which is low compared to the local concentration of the species in excess, an analytical solution to the system in eqn (6) can be obtained. Under those circumstances, the above system of equations simplifies to a single ordinary differential equation, with the following exact and approximate solutions:
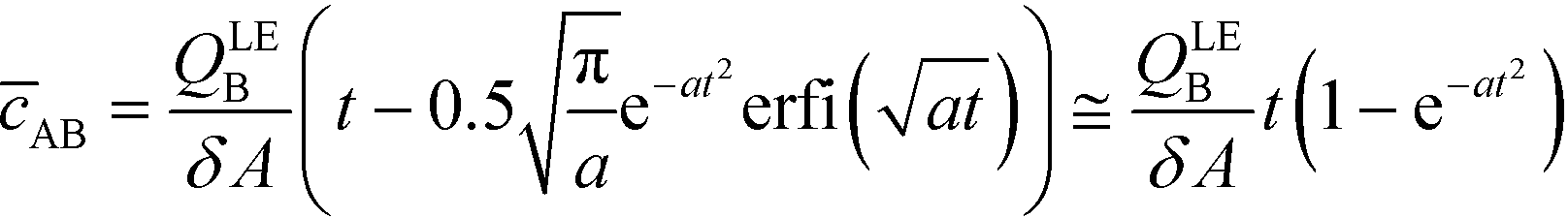 | (10) |
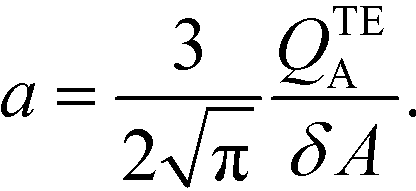
The simplest version of this volume-averaged unsteady product concentration can be compared to the value for a well-stirred reaction under similar assumptions, cAB ≅ cBo(1 − e−cAokont). We see that the effect of ITP can be interpreted as a pseudo-first order reaction wherein the initial value of the excess reactant c0A and the low-abundance concentration c0B (which normally limits the maximum level of cAB) are now functions directly proportional to time. Hence, this work revealed a new characteristic timescale for ITP-aided reaction kinetics, inversely proportional to square-root of initial concentration (compared to standard incubation, where the timescale is inversely proportional to initial concentration), as well as preconcentration due to ITP, and is given by
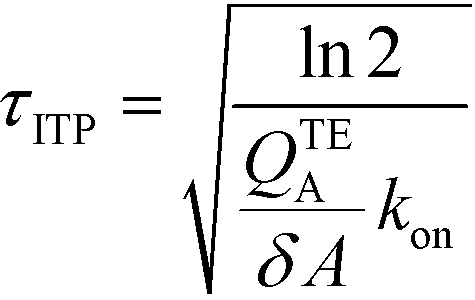 | (11) |
Putting the two reaction timescales from eqn (4) and (11) together elucidates the effect of ITP on acceleration of reaction kinetics:
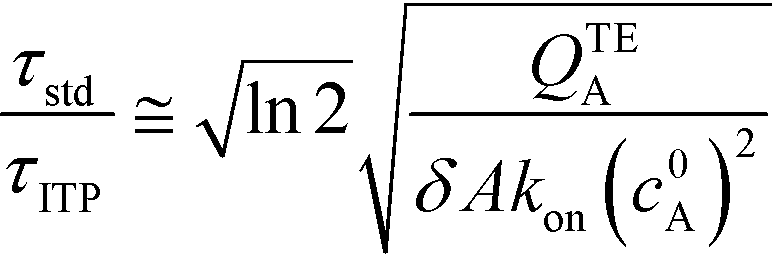 | (12) |
They supported their model with experimental validation using a molecular beacon probe and oligo target. We summarize some of these results in Fig. 3. ITP enhancement was more pronounced at lower reactant concentration (14000-fold reduced reaction time at 500 pM target concentration). Though their work nominally focused on DNA hybridization, it is theoretically applicable to any ITP-aided reaction assay in which both reactants are preconcentrated in ITP.
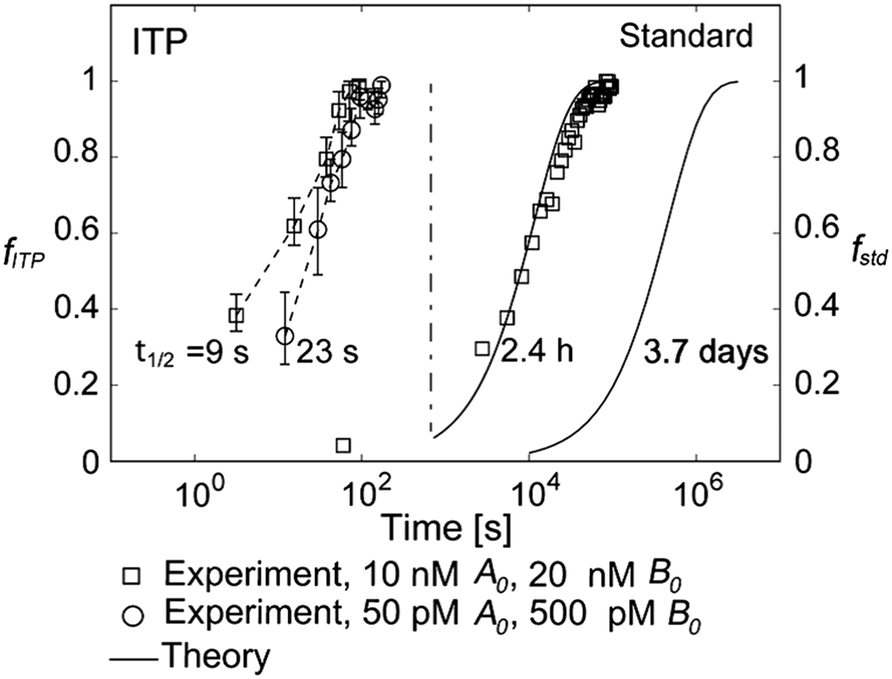 | ||
| Fig. 3 Experimental demonstration of ITP-aided hybridization acceleration from Bercovici et al.52 Fraction of reactants hybridized is shown for both standard and ITP-aided hybridizations at two different reactant concentrations. 960- and 14000-fold hybridization acceleration is demonstrated for limiting species concentrations of 10 nM and 100 pM, respectively. The theoretical model (discussed in section III.a) agreed with experimental data, and captured the significant trends resulting from ITP-aided hybridization. This analytical model of ITP was the first of its kind and served as the foundation for several subsequent analytical studies. | ||
Eid et al.57 presented a modified version of Bercovici's model for cases in which only one species is focused in ITP. Namely, Eid considered loading of two reactants into the LE buffer, but where only one of them focused in ITP. This resulted in a reduction of ITP-enhanced reaction rate. Under these conditions, the net reaction rate of the not-yet-focused species within the LE zone may be comparable (on a moles per second basis) to that in the ITP zone, due to the significantly larger volume of the LE zone. They modeled the latter effect as a shrinking reactor, and introduced a dimensionless parameter λ which incorporates several of the key variables which influence product formation
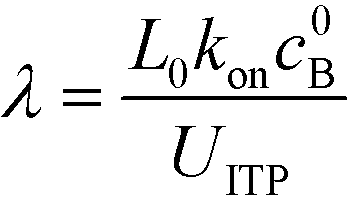 | (13) |
All the models discussed above make the simplifying assumption that ITP zones have a Gaussian profile and were perfectly overlapped, which allows the use of volume-averaged concentrations. Garcia-Schwarz et al.36 first showed that samples focused in ITP peak mode may exhibit species-specific and non-Gaussian/asymmetric profiles. In their work, they found that sample ion properties contributed greatly to dispersion and ITP peak shapes. In particular, ITP peaks wherein sample ions had mobilities near those of the TE or LE exhibited significant tailing into these respective zones and an associated asymmetry. Rubin et al.58 presented a study focused on sample distribution within ITP zones, and the effect of these species specific distributions on reaction rates. They presented closed-form solutions for peak shapes and production rates for the case of ITP dynamics dominated by pure diffusion (e.g., no advective dispersion) and electromigration. To account for sample zone shape asymmetry, they defined an effective association rate constant of the form
| keffon = konkform | (14) |
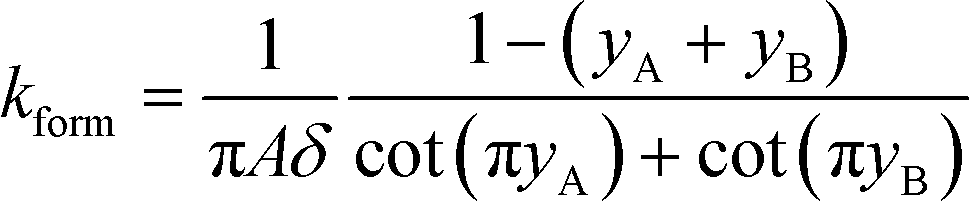 | (15) |
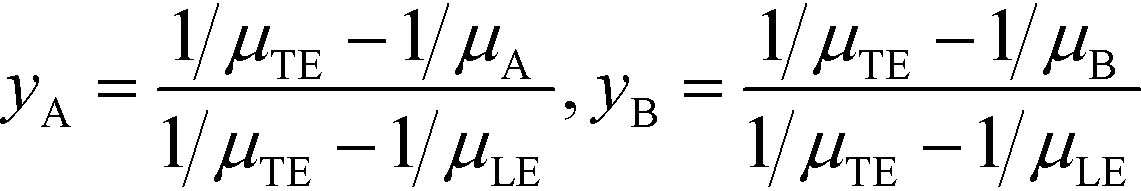 | (16) |
Interestingly, they found that reaction rate is not necessarily maximized when concentration profiles of two reacting species perfectly overlap, and instead varies depending on the relative mobilities of the species. As a result, production rates calculated while accounting for sample distribution are typically lower than those computed with volume-averaged concentration models.
Recently, Eid and Santiago53 considered the design parameters that govern the performance of peak-mode ITP assays. They incorporated the results of Rubin's more comprehensive species overlap model into their analysis. This analysis showed that for reaction times longer than the characteristic ITP reaction time scale τ, the number of molecules of product AB formed depends solely on the relative influx rates of reactants A and B, and defined a dimensionless production rate such that
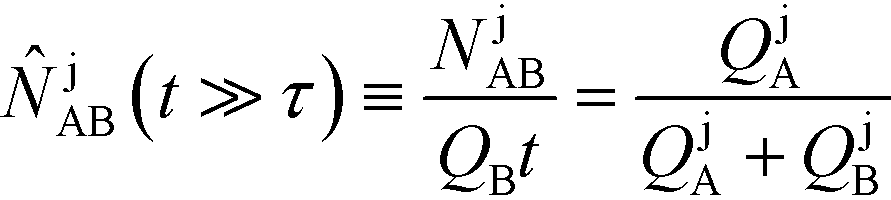 | (17) |
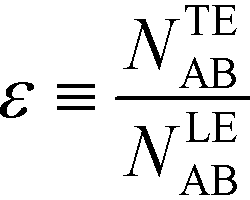 | (18) |
For long reaction times, they found that ε depends solely on ϕA, the influx ratio of the limiting species,
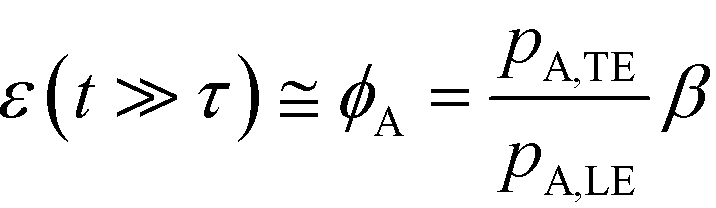 | (19) |
Eid and Santiago concluded that for sufficient reaction times, the optimal production rate of species AB in an ITP-aided reaction assay is obtained by simply maximizing the influx rate of the rate-limiting species.
III.b. Homogeneous reactions: experimental studies and assays
To our knowledge, Persat and Santiago61 were the first to experimentally demonstrate ITP-aided hybridization of nucleic acids. They used ITP and molecular beacons to selectively profile among seven microRNA (miRNA) species in total RNA samples from human liver and kidney. This is also the first published quantitative demonstration of high specificity reaction using ITP; and signal selectivity to target miR-26 (22 nt), compared with its relative, miR-126 (22 nt), and its precursor, pre-mir-26a (77 nt). Molecular beacons are single-stranded DNA (ssDNA) probes with a unique hairpin structure that places a fluorophore and quencher in close proximity.62 Upon binding to a specific target, the structure of the probe is disrupted, separating the fluorophore from its quencher, and resulting in increased fluorescent signal. Persat and Santiago developed a multistage assay wherein the channel contained three discrete regions with varying amounts of sieving matrix (polyvinylpyrrolidone, PVP), magnesium chloride, and denaturant. In the first region, all RNA molecules in the total RNA sample were preconcentrated in ITP. The second region contained a high concentration of sieving matrix in order to defocus large RNA and selectively retain miRNA. The third region applied stringent conditions to promote specific hybridization between the molecular beacons and miRNA target. Fig. 4 shows a schematic of their multi-stage assay. Persat verified both sequence- and size-selectivity of this assay; the former by titrating with mismatched miRNA, and the latter by titrating with larger precursor miRNA. They demonstrated initial biological relevance of this technique by achieving a 10 pM limit of detection of miR-122 in kidney and liver total RNA samples.
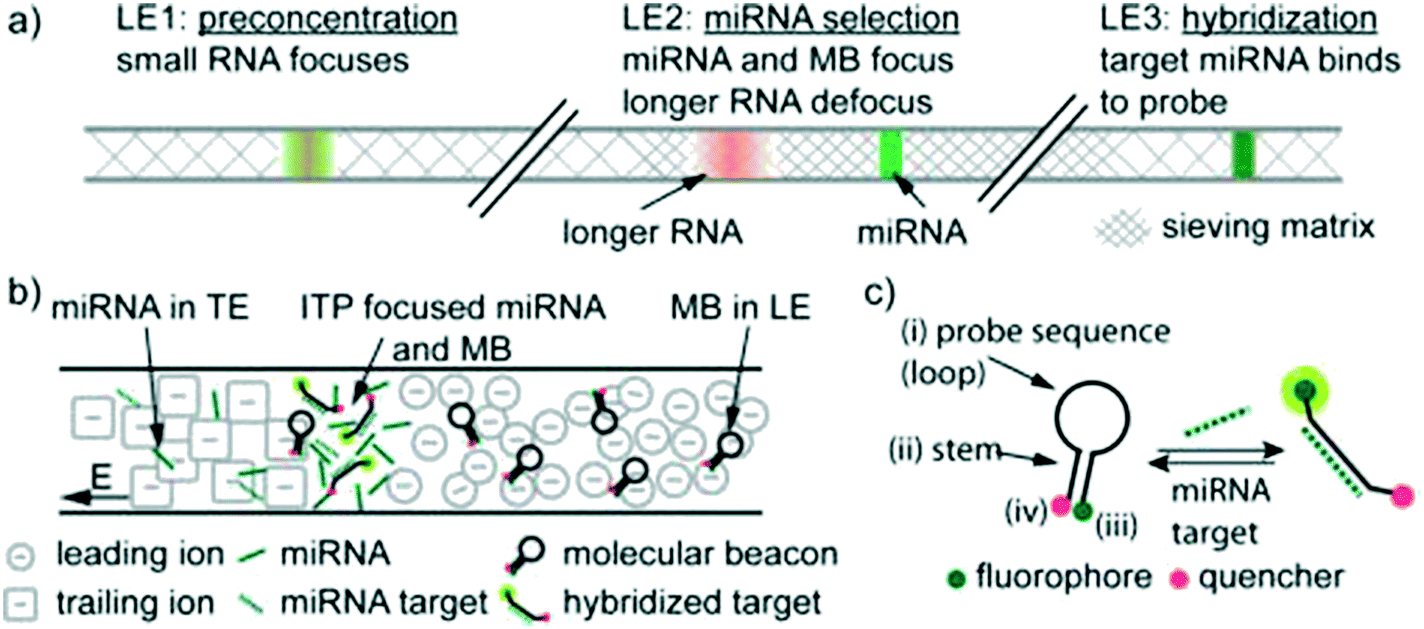 | ||
| Fig. 4 Schematic representation of the first experimental ITP hybridization assay from Persat and Santiago.61 a) Sieving matrix was used to divide the channel into three regions to respectively promote ITP preconcentration, size selectivity, and highly stringent hybridization. As shown in b) and c) the assay used molecular beacons, which are fluorescently (imperfectly) quenched in their native state but unravel and emit higher fluorescence upon binding to their target. This assay was able to selectively detect mature miRNA target in tissue total RNA samples, and is the first of many ITP hybridization studies to use molecular beacons as probes. | ||
Bercovici et al.63 combined ITP-aided reactions with molecular beacons, but used it to detect a significantly larger target, 16S rRNA from bacteria in cultures and patient urine samples. To our knowledge, this is the first quantitative demonstration of reaction acceleration using ITP in addition to the first quantitative theory and validation thereof. They successfully demonstrated the applicability of this approach in real patient samples at clinically-relevant levels. Another significant contribution of the latter work is the design of a photomultiplier tube (PMT) system for high sensitivity fluorescent signal quantification. The assay achieved a limit of detection of 106 cfu mL−1, or 30 pM, of E. coli in human urine samples. This work and that of Persat and Santiago61 demonstrated that molecular beacons can be used for selective assays; but also that ITP assays with molecular beacon based detection sacrifice sensitivity and dynamic range (due to large background signal of unreacted MBs). These limitations highlighted the need for removal (e.g., physical separation) of background signal following hybridization, and subsequent work in the area has devised and demonstrated various solutions to this issue.
Bahga et al.64 introduced a homogeneous ITP reaction assay for removing excess background signal of unreacted molecular beacons. They designed an assay in which ITP-aided DNA hybridization was coupled to the high-resolving power of CE using bidirectional ITP (see review by Bahga and Santiago12). Following ITP-aided hybridization, CE was triggered, and unbound molecular beacons separated from the larger (lower mobility) beacon–target complex. They successfully demonstrated sequence-specific detection of a 39 nt ssDNA target, with a 3 pM limit of detection. Though Bahga et al. improved sensitivity of ITP and molecular beacon assays with this approach, the use of CE resulted in more dispersed signal peaks, limiting sensitivity and downstream analysis of reaction products. Another drawback of this approach is the complexity of assay chemistry needed for effective bidirectional ITP.
A few months later, Eid et al.65 introduced a new method for homogeneous post-hybridization clean-up. Their multistage approach used ITP to accelerate hybridization between a 26 nt linear ssDNA probe and 149 nt ssDNA target, and an ionic spacer to subsequently separate reaction products. In a similar vein to Persat et al.,61 the first stage focused all probe and target, promoting rapid mixing and hybridization. The second stage contained high-concentration sieving matrix, which allowed the ionic spacer to overtake and separate the unbound probes from the slower probe–target complex. Fig. 5 shows the different stages of the reaction-separation assay. Eid et al.65 used 20 mM HEPES as TE, 1 mM MOPS as spacer, 1.8% hydroxyethyl cellulose (HEC) as sieving matrix. This resulted in two focused ITP peaks, one at the LE–spacer interface, and one at the spacer–TE interface. They demonstrated the advantage of this approach by achieving a 220 fM limit of detection in 10 min, with a 3.5 decade dynamic range. This technique is fairly simple and flexible, and produces two ITP-focused peaks; this improves signal and is compatible with downstream manipulation. However, the method lacks high selectivity, and requires sieving matrix and spacer optimization for targets of different sizes.
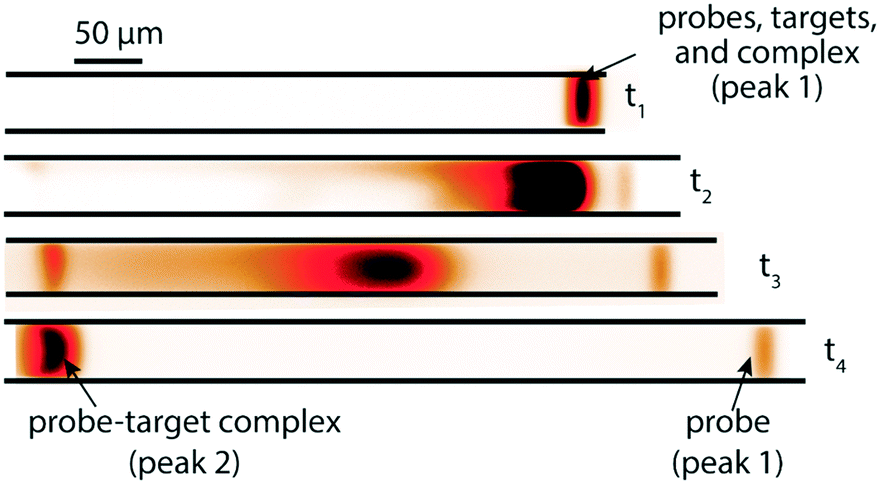 | ||
| Fig. 5 Experimental visualization from DNA reaction and separation assay using ITP and ionic spacers.65 The probe is a 26 nt fluorescently-labeled ssDNA oligo and target a 149 nt ssDNA oligo. In the first stage (t1), all reactants and products were co-focused in an ITP zone, which promoted rapid hybridization. In the second stage (t2 and t3), the ITP zone entered a region with sieving matrix, causing the ionic spacer to overspeed the larger probe–target complex but not the unreacted probe molecules. By t4, all species had reached equilibrium, and the two ITP peaks migrated in parallel. | ||
Most recently, van Kooten et al. presented a new microfluidic chip design that enables ITP focusing from initial sample volumes of 50 μL down to 500 pL using a gradual reduction of channel width.66 They estimated a 300000-fold increase in peak concentration in the narrow region (width of 100 μM) as compared with the wide region (with of 8 mm). They briefly demonstrated the application of this device to homogeneous hybridization assays using molecular beacons and ssDNA oligo as target, and achieved a 100-fold improvement in limit of detection as compared with standard microfluidic chip (the often-used NS-95 chip from Perkin Elmer, MA). Interestingly, they observed that even a few seconds of hybridization in the narrow channel region corresponded to significant acceleration of reaction kinetics.
Eid et al.57 devised a workaround, using high-mobility modified aptamers to bind to and recruit typically non-focusing C-reactive protein (CRP) into ITP. The modified aptamers are called SOMAmers (slow off-rate modified aptamer), and unlike traditional aptamers that are DNA or RNA oligonucleotides, SOMAmers have modified bases to increase their hydrophobicity.68,69 Eid here loaded both the protein and fluorescently-labeled SOMAmers into LE buffer. The ITP chemistry was chosen so as to preconcentrate the relatively high mobility SOMAmers and the SOMAmer/protein complex but not the unbound protein target. Hence ITP preconcentrated SOMAmers (increasing reaction rate) and simultaneously recruited proteins into the ITP zone upon binding. They extended the approach introduced by Eid and colleagues65 combining ITP and ionic spacers to separate unbound SOMAmers from SOMAmer–protein complex to remove excess background signal. In clean buffer, this technique achieved a limit of detection of 2 nM, well within the clinically-relevant range of CRP. However, when extended to 20-fold diluted serum, assay sensitivity suffered, decreasing by an order of magnitude. Eid hypothesized this was due to the complexity of serum, due to its high protein content and associated nonspecific binding. The presence of many ionic species which act as native spacer molecules may have also contributed.
 | ||
| Fig. 6 A) Schematic and B) experimental visualization of the first assay to use ITP to accelerate reactions with whole dispersed cells. Positively-charged antimicrobial peptides are focused in ITP then held stationary using counterflow, while whole bacterial cells migrate through the channel by pressure-driven flow. Peptides label the flowing cells, which are then detected downstream using a fluorescent detector. The experimental set-up was stable over the 1 h detection window. | ||
Phung et al.73 leveraged the use of counterflow pressure as well as an ionic spacer and sieving matrix to perform an ITP-aided fluorescence in situ hybridization (FISH) assay on bacterial cells. In the first step of their assay, bacterial cells and fluorescently labeled oligonucleotide probes are co-focused in peak-mode ITP. The nature of their synthetic cDNA hybridization probes makes this, to our knowledge, the first example of a high specificity reaction between cells and a reactant. Phung used counter flow to immobilize the ITP zone and prolong the sequence-specific hybridization process. In a second stage of the assay, a sieving matrix was used to separate stained cells from excess probe. Probes specific to target species E. coli and P. aeruginosa were respectively labeled with FAM (excitation wavelength of 488 nm) and Cy5 (excitation wavelength of 635 nm). Similar to Eid et al.,65 Phung used 1.8% HEC as sieving matrix, but used MES as an ionic spacer to separate stained cells from excess probe. They tested the selectivity of their technique by testing probes designed for different bacterial species, and found that selectivity heavily depends on probe design and the number of matched nucleotides with the bacterial target. The in-line ITP–FISH assay required 30 min to complete, a significant improvement over the 2.5 h typical of conventional FISH assays. However, the performance suffered due to reduced staining efficiency (approximately 50% of the 2.5 h conventional FISH offline hybridization reaction), and the assay achieved a limit of detection of 6 × 104 cells per mL. While this concentration range excludes several interesting applications, ITP–FISH is a faster, more convenient, and automation-friendly alternative to conventional FISH assays.
IV. ITP to preconcentrate, mix, and accelerate heterogeneous reactions
IV.a. Heterogenous reactions: theory and models
We here refer to heterogeneous assays as assays where one or more of the reactants is immobilized on a solid (stationary) surface, such as a functionalized gel or antibody-coated channel wall heterogeneous assays offer high multiplexing capability, with the possibility of detecting hundreds of targets simultaneously. A second important feature is the easy integration of post-hybridization washes and buffer exchanges. On the other hand, heterogeneous assays typically require more sophisticated fabrication, assembly, and implementation.Karsenty et al.75 developed the first analytical model for surface-based hybridization assays using ITP. They considered the case of a single target species migrating over a finite segment of functionalized magnetic beads immobilized using an external magnet. Through scaling analysis, they showed that the problem can be assumed to be one-dimensional, meaning that the analyte concentration at each point x along the channel can be effectively represented by its area-averaged concentration. Karsenty also showed that the spatial distribution of target ions remains unchanged as they react with the immobilized probes. They developed the following one-dimensional ordinary differential equation to describe the concentration of surface probes bound by target molecules
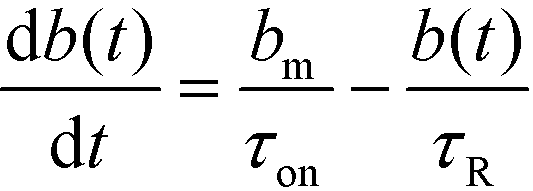 | (20) |
| τ−1R = τ−1on + τ−1off; τ−1on = αc0kon; τ−1off = koff | (21) |
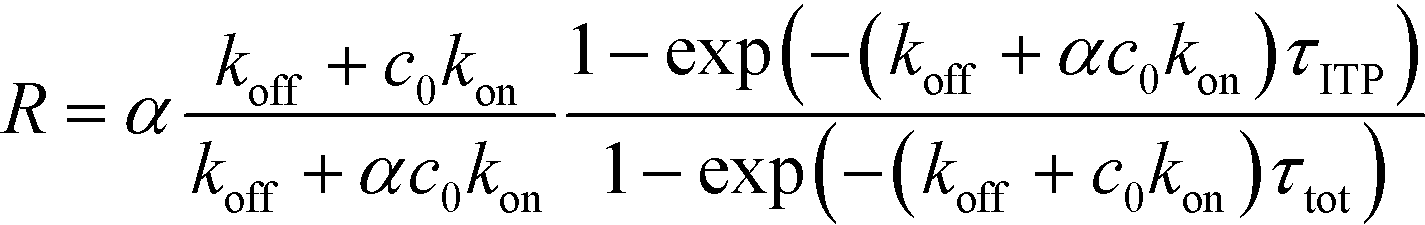 | (22) |
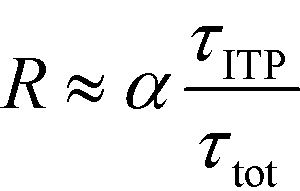 | (23) |
The above results indicate that at low concentrations, ITP-based enhancement depends on the preconcentration factor as well as the fraction of the total assay time in which the two reacting species overlap through ITP. At higher concentrations, the reaction rate, even without ITP, is sufficiently high to saturate the probes, and the enhancement ratio decays until it reaches unity.
The above model has been adapted by several subsequent works since the original publication. Han et al.,76 in their work on ITP-aided acceleration of microarray assays, provided a similar analysis of ITP-aided surface hybridization. They define the fraction of surface probes hybridized following ITP reaction by
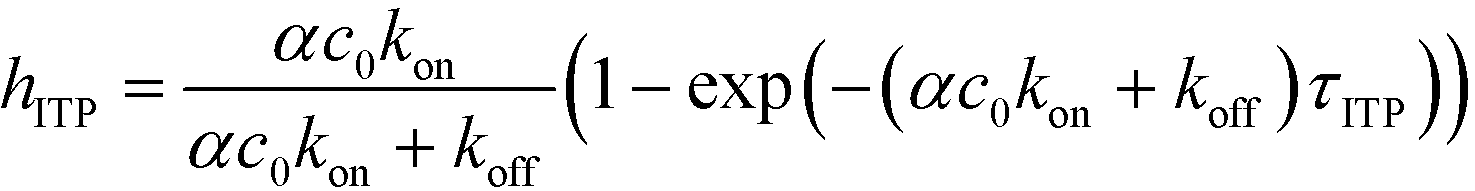 | (24) |
Han et al. also identified the Damkohler number as an important dimensionless parameter governing these reactions. Damkohler number, Da, is the ratio of reaction rate and diffusion rate, and can be used to determine whether a system is reaction- or diffusion-limited. Typical ITP assays have Da ≪ 1, indicating that reaction rates are much lower than diffusion, and thus these assays are reaction-limited. Moghadam et al.87 also used the analysis of Karsenty75 in their work on ITP-aided lateral flow assays. They too found that their assay was reaction-limited, and their limit of detection was governed by the preconcentration and off-rate constant, similarly to Bercovici et al.52
Recently, Paratore et al.77 expanded on the works of Karsenty and Han, and further investigated surface-based ITP reaction kinetics. They defined three operation modes for ITP: pass-over ITP (PO-ITP), stop and diffuse ITP (SD-ITP), and counterflow ITP (CF-ITP). The first of the three is the mode used in Karsenty's and Han's previous works, and consists of focused target passing over a reaction site containing immobilized probes, and reacting during a brief period of spatial overlap. In SD-ITP, once the focused target reaches the reaction site, the electric field was turned off, and the target diffused while reacting over the reaction site. To characterize reaction kinetics in this mode, the authors defined two characteristic time scales for depletion and axial diffusion, respectively:
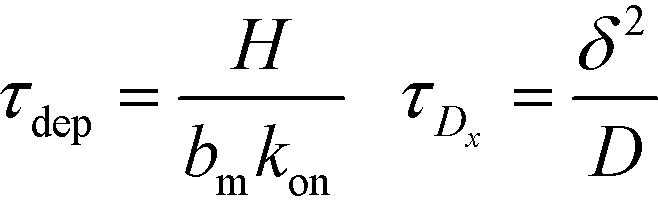 | (25) |
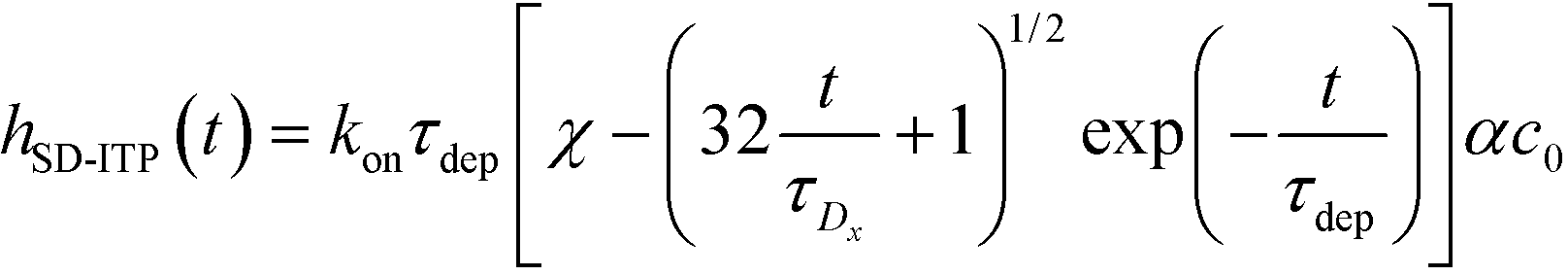 | (26) |
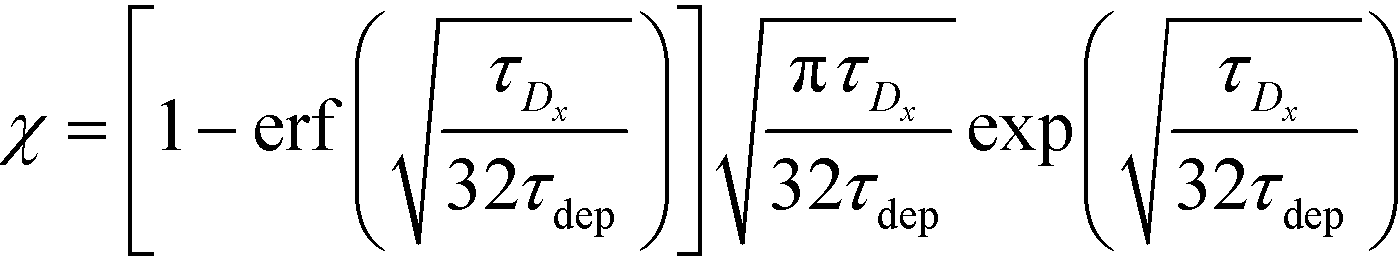 | (27) |
For sufficiently long times, longer than either characteristic time scales, the solution to eqn (26) approaches a steady state of χαc0. The third and final mode of ITP they discussed is counterflow ITP (CF-ITP), in which the ITP peak was held steady over the reaction site by applying counterflow pressure. There, the fraction of surface molecules bound is given by
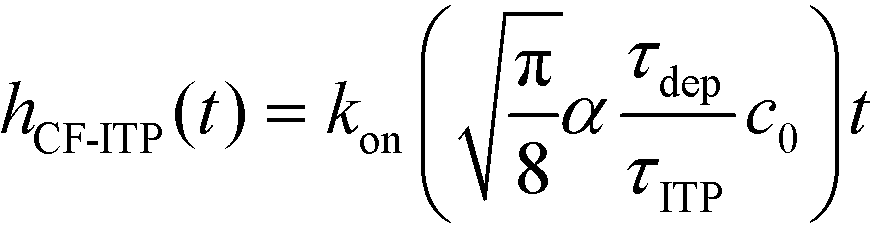 | (28) |
Paratore's model revealed that SD-ITP and CF-ITP result in significantly more reaction acceleration compared with PO-ITP. SD-ITP is highly dependent on the diffusion behavior of the focused species, which can be improved by increasing viscosity. On the other hand, CF-ITP is dependent on the ratio of ITP accumulation and species depletion time scales, which can be controlled by optimizing ITP focusing conditions.
Shkolnikov and Santiago78 developed a model for ITP coupled with affinity chromatography (AC), wherein target molecules are focused in ITP and probes are immobilized in an affinity capture region. Their analytical model they developed was broad and comprehensive. It contained fully-reversible second-order reaction kinetics, allowed for spatially variable bound substrate distribution, and accounted for zone dynamics of ITP-focused analytes. They found that key parameters like probe and target concentrations, forward and reverse reaction constants, and others collapsed into three dimensionless parameters that governed the different limiting regimes in the ITP-AC problem: the Damkohler number (Da), the ratio of peak target concentration scaled by the initial probe concentration (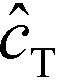 ), and a dimensionless equilibrium constant (
), and a dimensionless equilibrium constant (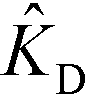 ),
),
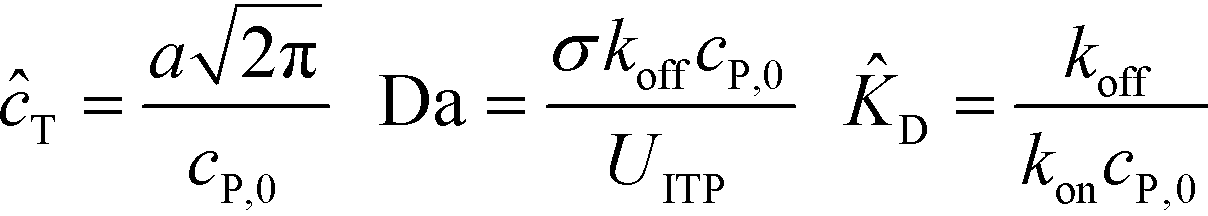 | (29) |
IV.b. Heterogeneous reactions: experimental studies and assays
In the second of their two-part work, Shkolnikov and Santiago78,81 presented a technique that coupled ITP preconcentration with affinity chromatography for sequence-specific capture and purification of target DNA molecules. The found that the three key parameters which they identified in their first paper (Da, 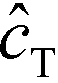 , and
, and 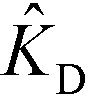 ) (and section IV.a above) showed remarkable consistency when compared with experimental results, accurately predicting the spatiotemporal behavior of the DNA. In addition to their analytical contributions (described in the previous section), they synthesized and functionalized a custom porous polymer monolith (PPM), made of poly(glycidyl methacrylate-co-ethylene dimetharcylate). They then functionalized this monolith with probe oligos, and characterized its performance. The PPM had little non-specific binding, was nonsieving, and compatible with ITP. Schkolnikov demonstrated the capability of this approach, purifying 25 nt Cy5-labeled target DNA from 10000-fold more abundant background DNA in less than a minute, and their technique had a 4 order of magnitude dynamic range.
) (and section IV.a above) showed remarkable consistency when compared with experimental results, accurately predicting the spatiotemporal behavior of the DNA. In addition to their analytical contributions (described in the previous section), they synthesized and functionalized a custom porous polymer monolith (PPM), made of poly(glycidyl methacrylate-co-ethylene dimetharcylate). They then functionalized this monolith with probe oligos, and characterized its performance. The PPM had little non-specific binding, was nonsieving, and compatible with ITP. Schkolnikov demonstrated the capability of this approach, purifying 25 nt Cy5-labeled target DNA from 10000-fold more abundant background DNA in less than a minute, and their technique had a 4 order of magnitude dynamic range.
Han et al.76 were the first to demonstrate multiplexed ITP-accelerated heterogenous reactions. They applied ITP to focus nucleic acids and accelerate a DNA hybridization microarray assay. A key feature of DNA array technologies is their massive multiplexing capacity, capable of detecting thousands of targets simultaneously.82,83 Up until the work of Han, ITP studies were limited to the detection of one or two targets. Han detected 20 target sequences using 60 microarray spots. To achieve this, Han et al. designed a PDMS microfluidic superstructure containing a single channel that they bonded to a commercially available microarray on a glass substrate, exposing 60 spots to the path of the ITP zone. The resulting device was 40 μm deep, 8 cm long, and 500 μm wide, except for a short constriction that had width of 200 μm. The electric field was varied to optimize both ITP preconcentration and DNA hybridization (Fig. 7). In the first part of the assay, they applied high electric field to accumulate all the ssDNA targets. Once the ITP zone reached the constriction, the electric field was turned off to allow target to redistribute through diffusion and minimize Joule heating effects. Finally, in the hybridization step, they applied low electric field values to avoid electrokinetic instabilities37,84 as the sample migrated over the immobilized array spots. The 30 min assay achieved a 100 fM limit of detection with a dynamic range of 4 orders of magnitude. Compared with a conventional 15 h microarray hybridization assay, the 30 min ITP assay also demonstrated 8-fold increase in sensitivity. Furthermore, this improvement in sensitivity was achieved without any loss in specificity, as measured using the specificity index introduced earlier by Shintaku et al.59 This work demonstrated the potential for ITP to integrate into DNA microarrays and other multiplexed surface reactions.
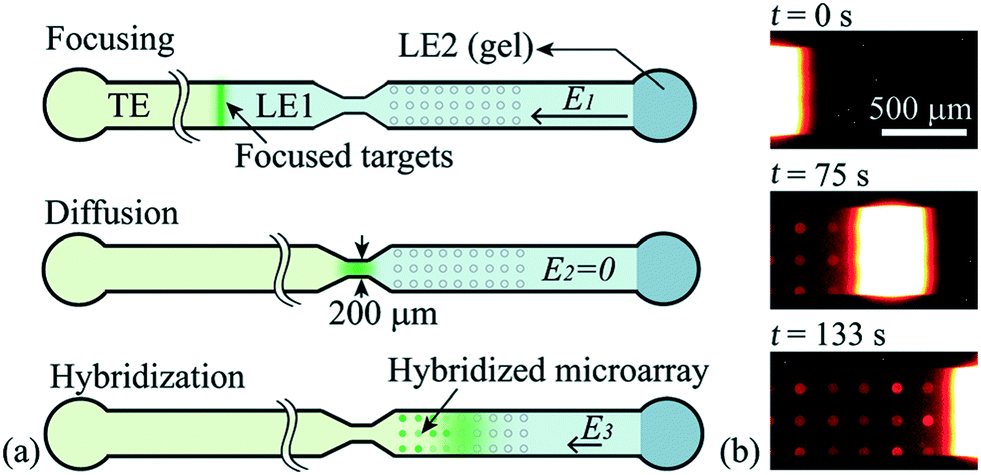 | ||
| Fig. 7 Schematic of the ITP-based multiplexed microarray assay designed by Han et al.76 First, labeled ssDNA targets were focused in ITP. Upon reaching the constriction, the electric field was turned off to minimize Joule heating and allow redistribution of target DNA through diffusion. The ssDNA targets then migrated over 60 immobilized probe sites representing 20 unique sequences, and fluorescence was measured after the ITP zone had swept by. The 30 min assay achieved a 100 fM limit of detection, the lowest for any heterogeneous ITP reaction assay. The array signal was quantified using a standard DNA array scanner. | ||
Moghadam et al.87 further demonstrated the applicability of ITP to one of the most widely-used immunoassay formats, lateral flow assays (LFA). Despite their convenience and popularity for qualitative applications, LFAs are often constrained by poor detection limits. Using ITP preconcentration, Moghadam demonstrated 2 orders of magnitude improvement in sensitivity in detecting IgG from a clean buffer. In conventional LFAs, samples migrate solely under capillary action, whereas they migrate electrophoretically in ITP-LFA (Fig. 8). In addition to reaction enhancement achieved by ITP, Moghadam estimated an extraction efficiency of nearly 30%, more than an order of magnitude improvement over conventional LFA. Additionally, Moghadam offered significant insight into the general design of ITP-LFA. Capture efficiency in conventional LFA depends on the normalized initial target concentration. In ITP-LFA, however, capture efficiency also depends on preconcentration factor, time, and dissociation rate constant. As a result, optimal assay performance can be achieved by carefully designing buffer chemistry and device geometry.
 | ||
| Fig. 8 Illustration of the various stages of the ITP-LFA designed by Moghadam et al.87 by using ITP to preconcentrate target molecules, ITP-LFA addresses the poor detection limits typical of conventional LFA. The assay demonstrated a two order of magnitude improvement in detecting IgG, and maintained the short assay times (5 min) typical of LFA. | ||
Most recently, Paratore et al.77 developed an assay in which they used different modes of ITP to accelerate surface-based immunoassays. They guided paramagnetic beads coated with anti-GFP antibodies to a desired location in the microfluidic channel, similar to Karsenty et al. They used enhanced GFP (EGFP) as a model protein, and demonstrated three separate operation modes of ITP: pass-over ITP (PO-ITP), stop and diffuse ITP (SD-ITP), and counterflow ITP (CF-ITP). For all assays, the total assay time was nearly 30 min, only 6 of which were for antibody–antigen binding. They found that SD-ITP, in which the electric field was turned off when the focused protein reached the reaction site, and CF-ITP, in which counterflow was applied to keep the focused proteins over the reaction site, resulted in significant decrease in LoD as compared with standard mode SD-ITP. Paratore reduced the LoD from 300 pM (for a standard flow immunoassay) to 220 fM using CF-ITP. They noted the influence of pH and salt concentration on the performance of the ITP immunoassay, and highlighted the heterogeneity of target proteins, which in turn requires careful optimization for each target. Despite the typical issues associated with ITP immunoassays (e.g. impact of extreme pH values and salt concentrations on focusing dynamics and reaction kinetics), Paratore's results are promising for the integration of ITP with protein detection assays.
V. Conclusions and recommendations
We reviewed a number of theoretical and experimental studies wherein ITP focusing and mixing is used to initiate and accelerate chemical reactions. ITP preconcentration has been shown to increase reaction rate by up to 14000-fold. By selectively focusing species within a designed electrophoretic mobility range, ITP can be used to remove contaminants and interfering species prior to a reaction, increase concentration during the reaction, and remove background signal or excess species following a reaction. Assays using ITP have demonstrated the ability to seamlessly integrate with various detection methods and other downstream assay steps. These assays leverage a variety of analytical chemistry tools including microfabrication, electric field control, a variety of cDNA-type probes, aptamer-type probes, antibodies, sieving matrices, spacer ions, denaturing conditions, microarrays and associated scanners, enzymatic reactions, and multispectral emissions and color detection. Downstream assays shown to be compatible with ITP include PCR, Luminex, and Bioanalyzer quantitation. Together, these studies demonstrate ITP's versatility and potential to control, enhance and accelerate chemical reactions. Furthermore, the continued near-decade existence of a commercial ITP-based immunoanalyzer suggests ITP's potential beyond the academic realm.
There remain several areas of unaddressed challenges and opportunities. There have been only a handful of immunoassays using ITP, and those have been limited to a few, particularly well-characterized proteins. Though the heterogeneity of protein mobilities, solubilities, and sensitivity to pH and ionic strength presents a significant challenge, the opportunities to incorporate ITP into proteomic analysis are significant and worth exploring. Integrating ITP with enzymatic processes, particularly amplification techniques like PCR and RPA, is also a significant opportunity for future ITP assays. Another area of opportunity is increasing the level of multiplexing in ITP-aided reactions from 20 species to thousands, and increasing throughput by analyzing and controlling samples in parallel.
The studies up to this point have been fairly limited in the input amount of sample that is processed. Increasing channel dimensions would potentially increase sensitivity and allow the processing and detection of rare species like viral DNA and circulating tumor cells. It may also encourage more widespread adoption of the technique. In particular, further incorporating ITP reaction process with complex sample extraction and purification (e.g. from blood, urine, tumor tissue, etc.) and with next-generation sequencing library preparation and target enrichment protocols would address key needs of those techniques (long incubation times, need for several washes). Such integration has immense potential.
Conflicts of interest
There are no conflicts to declare.References
- J. Kendall and E. D. Crittenden, Proc. Natl. Acad. Sci. U. S. A., 1923, 9, 75–78 CrossRef CAS.
- L. G. Longsworth, Ann. N. Y. Acad. Sci., 1939, 39, 187–202 CrossRef.
- A. J. P. Martin, Unpublished results, 1942 Search PubMed.
- H. Haglund, Sci. Tools, 1970, 17, 2–13 CAS.
- P. Smejkal, D. Bottenus, M. C. Breadmore, R. M. Guijt, C. F. Ivory, F. Foret and M. Macka, Electrophoresis, 2013, 34, 1493–1509 CrossRef CAS PubMed.
- V. Dolnik and P. Bocek, J. Chromatogr. A, 1982, 246, 343–345 CrossRef CAS.
- V. Dolnik and P. Bocek, J. Chromatogr. A, 1982, 246, 340–342 CrossRef CAS.
- P. Stehle and P. Furst, J. Chromatogr. A, 1985, 346, 271–279 CrossRef CAS.
- P. Stehle, H. S. Bahsitta and P. Furst, J. Chromatogr. A, 1986, 370, 131–138 CrossRef CAS.
- D. Kaniansky and J. Marak, J. Chromatogr. A, 1990, 498, 191–204 CrossRef CAS.
- F. Foret and E. Szoko, J. Chromatogr. A, 1992, 608, 3–12 CrossRef CAS.
- S. S. Bahga and J. G. Santiago, Analyst, 2013, 138, 735–754 RSC.
- P. Gebauer and P. Bocek, Electrophoresis, 2002, 23, 3858–3864 CrossRef CAS PubMed.
- P. Gebauer, Z. Mala and P. Bocek, Electrophoresis, 2009, 30, 29–35 CrossRef CAS PubMed.
- Z. Mala, P. Gebauer and P. Bocek, Electrophoresis, 2013, 34, 19–28 CrossRef CAS PubMed.
- P. G. Righetti, J. Chromatogr. A, 2005, 1079, 24–40 CrossRef CAS PubMed.
- L. Krivankova, P. Gebauer and P. Bocek, Methods Enzymol., 1996, 270, 375–401 CAS.
- P. Mikus, K. Marakova, L. Veizerova, J. Piestansky, J. Galba and E. Havranek, J. Chromatogr. Sci., 2012, 50, 849–854 CAS.
- D. W. Wegman, F. Ghasemi, A. S. Stasheuski, A. Khorshidi, B. B. Yang, S. K. Liu, G. M. Yousef and S. N. Krylov, Anal. Chem., 2016, 88, 2472–2477 CrossRef CAS PubMed.
- M. D. Borysiak, K. W. Kimura and J. D. Posner, Lab Chip, 2015, 15, 1697–1707 RSC.
- A. Persat, L. A. Marshall and J. G. Santiago, Anal. Chem., 2009, 81, 9507–9511 CrossRef CAS PubMed.
- A. Rogacs, L. A. Marshall and J. G. Santiago, J. Chromatogr. A, 2014, 1335, 105–120 CrossRef CAS PubMed.
- C. Eid and J. G. Santiago, Analyst, 2016, 142, 48–54 RSC.
- J. Sadecka and J. Polonsky, J. Chromatogr. A, 1996, 735, 403–408 CrossRef CAS PubMed.
- R. B. Schoch, M. Ronaghi and J. G. Santiago, Lab Chip, 2009, 9, 2145–2152 RSC.
- M. Bercovici, S. K. Lele and J. G. Santiago, J. Chromatogr. A, 2009, 1216, 1008–1018 CrossRef CAS PubMed.
- O. Dagan and M. Bercovici, Anal. Chem., 2014, 86, 7835–7842 CrossRef CAS PubMed.
- D. Bottenus, T. Z. Jubery, P. Dutta and C. F. Ivory, Electrophoresis, 2011, 32, 550–562 CrossRef CAS PubMed.
- T. Jacroux, D. Bottenus, B. Rieck, C. F. Ivory and W. J. Dong, Electrophoresis, 2014, 35, 2029–2038 CrossRef CAS PubMed.
- K. Kuriyama, H. Shintaku and J. G. Santiago, Electrophoresis, 2015, 36, 1658–1662 CrossRef CAS PubMed.
- C. Eid, S. S. Branda and R. J. Meagher, Analyst, 2017, 142, 2094–2099 RSC.
- F. M. Everaerts, J. L. Beckers and P. E. M. Verheggen, Isotachophoresis: Theory, instrumentation, and applications, 1976 Search PubMed.
- G. Garcia-Schwarz, A. Rogacs, S. S. Bahga and J. G. Santiago, J. Visualized Exp., 2012, e3890, DOI:10.3791/3890.
- B. Jung, R. Bharadwaj and J. G. Santiago, Anal. Chem., 2006, 78, 2319–2327 CrossRef CAS PubMed.
- T. K. Khurana and J. G. Santiago, Anal. Chem., 2008, 80, 6300–6307 CrossRef CAS PubMed.
- G. Garcia-Schwarz, M. Bercovici, L. A. Marshall and J. G. Santiago, J. Fluid Mech., 2011, 679, 455–475 CrossRef.
- C. H. Chen, H. Lin, S. K. Lele and J. G. Santiago, J. Fluid Mech., 2005, 524, 263–303 CrossRef.
- A. J. P. Martin and F. m. Everaert, Proc. R. Soc. London, Ser. A, 1970, 316, 493–514 CrossRef CAS.
- W. Kohlrausch, Verh. Dtsch. Ges. Kreislaufforsch., 1951, 17, 303–305 Search PubMed.
- J. Alberty, Klin. Wochenschr., 1950, 28, 786–787 CrossRef CAS PubMed.
- T. M. Jovin, Biochemistry, 1973, 12, 871–879 CrossRef CAS PubMed.
- M. Bercovici, G. V. Kaigala and J. G. Santiago, Anal. Chem., 2010, 82, 2134–2138 CrossRef CAS PubMed.
- M. Bercovici, G. V. Kaigala, C. J. Backhouse and J. G. Santiago, Anal. Chem., 2010, 82, 1858–1866 CrossRef CAS PubMed.
- R. D. Chambers and J. G. Santiago, Anal. Chem., 2009, 81, 3022–3028 CrossRef CAS PubMed.
- Wako, μTASWako® i30 Automated Platform microfluidic-based clinical immunoanalyzer, http://www.wakodiagnostics.com/utaswako_i30.html.
- T. Kawabata, H. G. Wada, M. Watanabe and S. Satomura, Electrophoresis, 2008, 29, 1399–1406 CrossRef CAS PubMed.
- C. C. Park, I. Kazakova, T. Kawabata, M. Spaid, R. L. Chien, H. G. Wada and S. Satomura, Anal. Chem., 2008, 80, 808–814 CrossRef CAS PubMed.
- C. Kagebayashi, I. Yamaguchi, A. Akinaga, H. Kitano, K. Yokoyama, M. Satomura, T. Kurosawa, M. Watanabe, T. Kawabata, W. Chang, C. Li, L. Bousse, H. G. Wada and S. Satomura, Anal. Biochem., 2009, 388, 306–311 CrossRef CAS PubMed.
- D. Li, T. Mallory and S. Satomura, Clin. Chim. Acta, 2001, 313, 15–19 CrossRef CAS.
- Y. J. Zhao, Q. Ju and G. C. Li, Mol. Clin. Oncol., 2013, 1, 593–598 CrossRef CAS PubMed.
- FDA, uTASWako i30 Analyzer and Test Systems 510(k) Summary, 2011 Search PubMed.
- M. Bercovici, C. M. Han, J. C. Liao and J. G. Santiago, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11127–11132 CrossRef CAS PubMed.
- C. Eid and J. G. Santiago, Anal. Chem., 2016, 88, 11352–11357 CrossRef CAS PubMed.
- D. A. MacInnes and L. G. Longsworth, Chem. Rev., 1932, 11, 171–230 CrossRef CAS.
- P. Bocek, P. Gebauer, V. Dolnik and F. Foret, J. Chromatogr., 1985, 334, 157–195 CrossRef CAS PubMed.
- L. Marshall, PhD, Stanford University, 2013 Search PubMed.
- C. Eid, J. W. Palko, E. Katilius and J. G. Santiago, Anal. Chem., 2015, 87, 6736–6743 CrossRef CAS PubMed.
- S. Rubin, O. Schwartz and M. Bercovici, Phys. Fluids, 2014, 26 DOI:10.1063/1.4861399.
- H. Shintaku, J. W. Palko, G. M. Sanders and J. G. Santiago, Angew. Chem., Int. Ed., 2014, 53, 13813–13816 CrossRef CAS PubMed.
- G. Goet, T. Baier and S. Hardt, Lab Chip, 2009, 9, 3586–3593 RSC.
- A. Persat and J. G. Santiago, Anal. Chem., 2011, 83, 2310–2316 CrossRef CAS PubMed.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS PubMed.
- M. Bercovici, G. V. Kaigala, K. E. Mach, C. M. Han, J. C. Liao and J. G. Santiago, Anal. Chem., 2011, 83, 4110–4117 CrossRef CAS PubMed.
- S. S. Bahga, C. M. Han and J. G. Santiago, Analyst, 2013, 138, 87–90 RSC.
- C. Eid, G. Garcia-Schwarz and J. G. Santiago, Analyst, 2013, 138, 3117–3120 RSC.
- X. F. van Kooten, M. Truman-Rosentsvit, G. V. Kaigala and M. Bercovici, Sci. Rep., 2017, 7, 10467 CrossRef PubMed.
- N. C. Stellwagen, C. Gelfi and P. G. Righetti, Biopolymers, 1997, 42, 687–703 CrossRef CAS PubMed.
- L. Gold, D. Ayers, J. Bertino, C. Bock, A. Bock, E. N. Brody, J. Carter, A. B. Dalby, B. E. Eaton, T. Fitzwater, D. Flather, A. Forbes, T. Foreman, C. Fowler, B. Gawande, M. Goss, M. Gunn, S. Gupta, D. Halladay, J. Heil, J. Heilig, B. Hicke, G. Husar, N. Janjic, T. Jarvis, S. Jennings, E. Katilius, T. R. Keeney, N. Kim, T. H. Koch, S. Kraemer, L. Kroiss, N. Le, D. Levine, W. Lindsey, B. Lollo, W. Mayfield, M. Mehan, R. Mehler, S. K. Nelson, M. Nelson, D. Nieuwlandt, M. Nikrad, U. Ochsner, R. M. Ostroff, M. Otis, T. Parker, S. Pietrasiewicz, D. I. Resnicow, J. Rohloff, G. Sanders, S. Sattin, D. Schneider, B. Singer, M. Stanton, A. Sterkel, A. Stewart, S. Stratford, J. D. Vaught, M. Vrkljan, J. J. Walker, M. Watrobka, S. Waugh, A. Weiss, S. K. Wilcox, A. Wolfson, S. K. Wolk, C. Zhang and D. Zichi, PLoS One, 2010, 5, e15004 CAS.
- J. C. Rohloff, A. D. Gelinas, T. C. Jarvis, U. A. Ochsner, D. J. Schneider, L. Gold and N. Janjic, Mol. Ther.--Nucleic Acids, 2014, 3, e201 CrossRef CAS PubMed.
- O. Schwartz and M. Bercovici, Anal. Chem., 2014, 86, 10106–10113 CrossRef CAS PubMed.
- F. Oukacine, L. Garrelly, B. Romestand, D. M. Goodall, T. Zou and H. Cottet, Anal. Chem., 2011, 83, 1571–1578 CrossRef CAS PubMed.
- S. C. Phung, Y. H. Nai, S. M. Powell, M. Macka and M. C. Breadmore, Electrophoresis, 2013, 34, 1657–1662 CrossRef CAS PubMed.
- S. C. Phung, J. M. Cabot, M. Macka, S. M. Powell, R. M. Guijt and M. Breadmore, Anal. Chem., 2017, 89, 6513–6520 CrossRef CAS PubMed.
- M. D. Borysiak, A. T. Bender, D. S. Boyle and J. D. Posner, presented in part at the 2016 IEEE Healthcare Innovation Point-Of-Care Technologies Conference, Cancun, Mexico, 2016 Search PubMed.
- M. Karsenty, S. Rubin and M. Bercovici, Anal. Chem., 2014, 86, 3028–3036 CrossRef CAS PubMed.
- C. M. Han, E. Katilius and J. G. Santiago, Lab Chip, 2014, 14, 2958–2967 RSC.
- F. Paratore, T. Zeidman Kalman, T. Rosenfeld, G. V. Kaigala and M. Bercovici, Anal. Chem., 2017, 89(14), 7373–7381 CrossRef CAS PubMed.
- V. Shkolnikov and J. G. Santiago, Anal. Chem., 2014, 86, 6220–6228 CrossRef CAS PubMed.
- G. Garcia-Schwarz and J. G. Santiago, Anal. Chem., 2012, 84, 6366–6369 CrossRef CAS PubMed.
- G. Garcia-Schwarz and J. G. Santiago, Angew. Chem., Int. Ed., 2013, 52, 11534–11537 CrossRef CAS PubMed.
- V. Shkolnikov and J. G. Santiago, Anal. Chem., 2014, 86, 6229–6236 CrossRef CAS PubMed.
- D. T. Okou, K. M. Steinberg, C. Middle, D. J. Cutler, T. J. Albert and M. E. Zwick, Nat. Methods, 2007, 4, 907–909 CrossRef CAS PubMed.
- D. T. Ross, U. Scherf, M. B. Eisen, C. M. Perou, C. Rees, P. Spellman, V. Iyer, S. S. Jeffrey, M. Van de Rijn, M. Waltham, A. Pergamenschikov, J. C. Lee, D. Lashkari, D. Shalon, T. G. Myers, J. N. Weinstein, D. Botstein and P. O. Brown, Nat. Genet., 2000, 24, 227–235 CrossRef CAS PubMed.
- J. D. Posner, C. L. Perez and J. G. Santiago, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14353–14356 CrossRef CAS PubMed.
- R. Khnouf, G. Goet, T. Baier and S. Hardt, Analyst, 2014, 139, 4564–4571 RSC.
- D. Milanova, R. D. Chambers, S. S. Bahga and J. G. Santiago, Electrophoresis, 2011, 32, 3286–3294 CrossRef CAS PubMed.
- B. Y. Moghadam, K. T. Connelly and J. D. Posner, Anal. Chem., 2015, 87, 1009–1017 CrossRef CAS PubMed.
Footnote |
| † Currently at Driver, Inc, San Francisco, CA, USA. |
| This journal is © The Royal Society of Chemistry 2018 |