Control of catalytic nanoparticle synthesis: general discussion
Aldiar
Adishev
,
Rosa
Arrigo
,
Francesca
Baletto
,
Alexis
Bordet
,
Valerii
Bukhtiyarov
,
Michele
Carosso
,
Richard
Catlow
,
Matthew
Conway
,
Josh
Davies
,
Philip
Davies
 ,
Déborah
De Masi
,
Cansunur
Demirci
,
Jennifer K.
Edwards
,
Cynthia
Friend
,
Simone
Gallarati
,
Justin
Hargreaves
,
Haoliang
Huang
,
Graham
Hutchings
,
Stanley
Lai
,
Carlo
Lamberti
,
Margherita
Macino
,
David
Marchant
,
Toru
Murayama
,
Yaroslav
Odarchenko
,
Jennifer
Peron
,
Laura
Prati
,
Jonathan
Quinson
,
Nia
Richards
,
Scott
Rogers
,
Andrea
Russell
,
Parasuraman
Selvam
,
Parag
Shah
,
Mzamo
Shozi
,
Chris-Kriton
Skylaris
,
Katerina
Soulantica
,
Federico
Spolaore
,
Bob
Tooze
,
Laura
Torrente-Murciano
,
Annette
Trunschke
,
Baldassarre
Venezia
,
James
Walker
and
Keith
Whiston
,
Déborah
De Masi
,
Cansunur
Demirci
,
Jennifer K.
Edwards
,
Cynthia
Friend
,
Simone
Gallarati
,
Justin
Hargreaves
,
Haoliang
Huang
,
Graham
Hutchings
,
Stanley
Lai
,
Carlo
Lamberti
,
Margherita
Macino
,
David
Marchant
,
Toru
Murayama
,
Yaroslav
Odarchenko
,
Jennifer
Peron
,
Laura
Prati
,
Jonathan
Quinson
,
Nia
Richards
,
Scott
Rogers
,
Andrea
Russell
,
Parasuraman
Selvam
,
Parag
Shah
,
Mzamo
Shozi
,
Chris-Kriton
Skylaris
,
Katerina
Soulantica
,
Federico
Spolaore
,
Bob
Tooze
,
Laura
Torrente-Murciano
,
Annette
Trunschke
,
Baldassarre
Venezia
,
James
Walker
and
Keith
Whiston
First published on 9th August 2018
Déborah De Masi opened the discussion of the paper by Laura Prati: Can you explain why you have better results with ruthenium nanoparticles than with bimetallic RuAu NPs? Is it due to a change of the crystallinity of Ru when it is on the surface of Au?Laura Prati replied: There is not a direct explanation but we experienced that the synergistic effect is not always positive. It depends on the structure, on the electronic distribution, on the matching between reactant, etc.
Aldiar Adishev asked: Can you explain how the introduction of gold affects reactions tested with bimetallic systems? Or do you have future plans to further study the effect of gold?
Laura Prati answered: We have seen that Au can have an effect on both electronic or geometric effects. However the balance between these two factors depends on the structures of the bimetallic particles you are synthesizing.
In the case of Pd, for example, we have shown that the effect of gold mainly affects the O2 interaction with the catalyst thus in turn reducing the occurrence of Pd–O species.
Baldassarre Venezia remarked: In this work it has also been emphasized how the Pd/AC and AuPd/AC catalyst were selective in the oxidation of benzyl alcohol to benzaldehyde. However, might this result be due to the low concentration of benzyl alcohol (0.3 M in cyclohexane) employed in the oxidation reaction? The low amount of benzyl alcohol in the autoclave might have hindered the disproportionation reaction of benzyl alcohol into toluene and benzaldehyde, whose kinetics is generally proportional to benzyl alcohol concentration and reaction temperature.
Laura Prati responded: Actually we used quite dilute solution (0.3 M). Of course this could have an impact on the selectivity of the reaction.
However we established that in the case of AuPd compared to pure Pd there is a decreased adsorption of oxygen on the catalyst surface which can explain a different selectivity. Please refer to previous work in ref. 1 and 2.
1 A. Savara, C. E. Chan-Thaw, I. Rossetti, A. Villa and L. Prati, ChemCatChem, 2014, 6, 3464.
2 A. Savara, C. E. Chan-Thaw, J. E. Sutton, D. Wang, L. Prati and A. Villa, ChemCatChem, 2017, 9, 253.
Parag Shah asked: Have you done any post-reaction analysis of the bimetallic nanoparticles? Is there any change in the particle size/composition?
Laura Prati replied: We have performed (not in this case) some characterisation of used catalysts. Only when you are using a basic environment can you observe some migration of Pd thus changing the initial composition.
Valerii Bukhtiyarov said: I have two questions. What are the main products of benzaldehyde oxidation? In the paper, I can find the data for benzaldehyde hydrogenation only. My other question concerns the much higher activity of palladium compared with monometallic Pt and Ru. Do you have any idea what the reasons for this behavior are?
Laura Prati answered: Benzaldehyde can be normally transformed into benzoic acid under oxidative conditions. However starting from benzyl alcohol without any addition of a base, we selectively produce benzaldehyde. For more detail on the mechanism you can see ref. 50 and 51 in the paper.
Nia Richards commented: You have stated that there is segregation in the copper gold catalyst. However, the catalytic activity of the catalyst (Fig. 11 in the paper) is still increasing when the reaction was stopped. Do you think that if you were to continue the reaction the activity would stabilise or deactivate as further segregation takes place?
Laura Prati replied: We stopped the reaction around full conversion (Fig. 11 in the paper) in the case of the AuCu catalyst.
Michele Carosso commented: The catalytic performance of alloyed bimetallic nanoparticles depends on a wide range of parameters among which are the chosen metals, the composition (atomic ratio between the two metals), the particles’ size and shape, the sample homogeneity, the employed support, the operating conditions and also the reaction chosen to test the catalytic performance. Do you expect a similar behaviour also for other selective oxidation reactions?
Laura Prati answered: As you rightly stated there are a lot of parameters playing a role in determining the catalytic activity. From my experience, what I can say is that the nature of metals is very important to determine a positive or negative synergistic effect, especially in relation to the substrate. In other words, I only expected similar behaviour for strictly correlated reactions.
Jonathan Quinson remarked: The experimental section states the use of CuCl3. Later on it is Cu(NO3)2 that is used. Which one is the right one to use?
Laura Prati replied: It should be Cu(II) nitrate in the paper. It was a mistake.
Nia Richards opened the discussion of the paper by Jennifer Edwards: Have you performed post analysis characterisation of your catalysts as they show stability based on the reaction data?
Jennifer Edwards responded: XPS analysis of the used material indicated no change. Had the materials shown deactivation a detailed STEM study may have been conducted, but with limited access to the microscope no further analysis was done on the reproducible, stable materials.
Annette Trunschke remarked: In your synthesis, the mixture of your support with aqueous solutions of your metal salt precursors was treated at 150 °C for 15 minutes in a closed vessel in a microwave system. Under these conditions an autogenous pressure is certainly developed. You compared your microwave synthesis with a synthesis at the same temperature (150 °C, normal heating) in an open system (ambient pressure). What is the effect of pressure without application of energy by microwaves? Did you compare your microwave synthesis with a synthesis under “normal” hydrothermal conditions at the same temperature and pressure?
Jennifer Edwards answered: The microwave system we use to make the catalysts also monitors the pressure, and we see no significant pressure rise during the preparation (perhaps 1 bar) hence we felt that the “blank” comparison catalyst prepared in a stirred, open system was OK. We can, and will, investigate the catalyst preparation via hydrothermal synthesis to be sure.
Jennifer Peron asked: Do you attribute the enhanced catalyst reusability/stability to its strong interaction/good dispersion with/onto the support or to its core–shell structure with the Pd shell preventing core-metals dissolution?
Jennifer Edwards replied: Typically, with the catalysts we prepare by impregnation a high calcination temperature (400 °C) is required to provide a stable catalyst that does not leach under reaction conditions. It is possible that the localised temperature at the nanoparticle in the microwave is much higher than the pre-set temperature, affording a more stable structure/strong interaction with the support. We have made similar core–shell structures before that are unstable, so I don’t think the Pd shell is causing the stabilisation in this case.
Margherita Macino asked: Regarding the particles’ size, do you think it is possible to control their size and distribution by varying the microwave preparation method?
Jennifer Edwards responded: Absolutely, and this is currently something we are investigating.
Matthew Conway asked: In your paper you suggest that hydrogenation occurs at a higher rate on smaller nanoparticles, but how do the rates of hydrogenation compare if you normalise for metal surface area? You also observe an extremely high hydrogenation activity; have you investigated if you are working in a mass transfer limited regime? Could it be that the hydrogen peroxide formed has retarded diffusion resulting in its hydrogenation?
Jennifer Edwards answered: We have thus far been unable to quantify the surface area of the AuPd surface using standard techniques, so normalising degradation rates to the metal surface has not been possible. In terms of mass transport, the hydrogen peroxide synthesis reaction using the experimental details outlined in the paper operates outside any mass limiting conditions. There are computational models (cited in the paper) that indicate a pure Pd surface is more adept at retaining H2O2 than a Pd surface interrupted with an Au atom; it is possible that the configuration of the surface plays a role in the increased hydrogenation of hydrogen peroxide over these catalysts.
Alexis Bordet said: As outlined during your talk and in the corresponding paper, the syntheses of nanoparticles under microwave irradiation have the potential to be very quick and reproducible. Could you please comment on the ease of scale up of reactions involving microwave irradiation?
On which maximum scale were you able to synthesize the AuPd@Pd nanoparticles?
Do you see any technical limitation to the microwave synthesis of nanoparticles on a large-scale (> kg)?
Jennifer Edwards responded: We have prepared the catalyst on a 1 g scale so far. Larger autoclave systems do exist, as do multiport reactors where 10 or more catalysts can be prepared in tandem. At the present time we are focusing on preparing the most active catalyst in the small-scale reactor. Once we have this right we will start investigating the possibility and practicalities of scaling up.
Graham Hutchings addressed Jennifer Edwards and Laura Torrente-Murciano: Can you combine the microwave synthesis method with a continuous flow reactor? That could provide an additional way to control the particle size.
Laura Torrente-Murciano responded: This is a good idea with high potential. Indeed, there are few reported studies for the continuous synthesis of nanoparticles using microwaves. I would highlight a recent paper by Prof Graviilidis.1 Further control could be achieved by combining this method with the fluid dynamic control within the reactor as mentioned in our paper presented in this symposium.
1 M. K. Bayazit, J. Yue, E. Cao, A. Gavriilidis and Junwang Tang, ACS Sustainable Chem. Eng., 2016, 4, 6435–6442.
Jennifer Edwards responded: Indeed, once we feel we have fully optimised the small-scale system (in terms of catalyst activity, stability and selectivity) we will focus our efforts on further developing the methodology into a flow reactor system.
James Walker continued the discussion of the paper by Jennifer Edwards: Your method affords good control over particle size and morphology. Your 1 wt% metal loading is lower than might be required for other catalytic systems though. In terms of scalability, can you please comment on how this method might translate to systems with higher metal loadings, other bimetallics or other catalyst supports?
Jennifer Edwards replied: We have scaled up the synthesis to 1.5 wt% without a loss in the linear TOF as presented in the paper, however at higher loadings the activity starts to drop. The surface area of TiO2 is 50–60 m2, and the literature states that the microwave methodology is good for preparing Pt/C fuel cell catalysts over 20 wt%. I expect with higher surface area supports the linear increase in TOF may be better. We have data for a number of activated carbon support materials, and we are preparing a number of follow up publications.
David Marchant asked: You claim that this preparation method should be scalable. What is the largest batch of catalyst that you have prepared using this microwave synthesis technique in the laboratory?
Jennifer Edwards responded: We have prepared 1g of catalyst. There are dozens of examples in the literature where organic reactions have been scaled up from bench to 1 L scale, and as such larger reactors are available to buy (or build). Once we have the “right” catalyst in the small-scale, we will shift focus on how to scale it up either in a larger reactor or in a flow system.
Bob Tooze said: In your verbal summary of your paper you included some indication that you have recently produced catalysts with improved performance; can you comment on how this was achieved and was it by a planned enhancement of the catalyst synthesis based on previous results?
Jennifer Edwards replied: Given the high throughput nature of the catalyst synthesis procedure, we have been able to optimise catalyst performance for the direct synthesis (DS) reaction using the microwave technique in a much faster timeframe (5–6 months) than it’s taken with catalysts prepared by sol-immobilisation and impregnation. We are lucky to have access on site to high resolution SEM, so we can get accurate particle sizes and compositions quickly, which then helps us assign attributes to certain solvents (glycerol, ethylene glycol) and the resulting nanoparticle composition. This has allowed a more iterative, results-led methodology for the catalyst preparation, though unfortunately we are still some way from a complete molecular understanding of the system (which is something we are working on!).
Mzamo Shozi said: I have seen a lot of literature where mostly TiO2 and carbon are used for sol-immobilization synthesis. Are these the ideal supports for this type of synthesis or could the traditional SiO2 and Al2O3 supports also be used?
Jennifer Edwards answered: As I understand, Au based catalysts on SiO2 are very unstable, with the nanoparticles prone to washing off the catalyst during catalytic testing. Indeed Al2O3 may be used as a support; for H2O2 synthesis when we prepare catalysts by impregnation on alumina we get very low activity given the high decomposition activity over the support.
Simone Gallarati opened the discussion of the paper by Laura Torrente-Murciano: One of the limitations of the coiled flow inverter reactors used to synthesise the silver–palladium nanoparticles seems to be that only relatively small amounts of reagents (mM solutions) can be used, thus limiting the amount of NPs that can be produced via this method. Many catalytic tests/experimental conditions and setups require that larger amounts (grams rather than milligrams) of catalysts need to be available for testing. How could this synthetic method be scaled up and what would the challenges then be?
Laura Torrente-Murciano answered: It is right to say that small amounts of reagents are used to avoid the agglomeration of the particles during the synthesis due to the absence of capping ligands as we have previously reported,1 however, the limits of this approach in terms of concentrations has not been fully explored.
Having said this, the low inlet concentration of reagents is not a limitation to producing large quantities of catalysts in continuous reactors (contrary to the case of batch systems) as one would only need to collect samples for the required amount of time to match their requirements. In addition, the scale up of these manufacturing technologies is relatively easy, not by increasing the size of the reactors but instead by increasing the number of reactors working in parallel to ensure that full control of the fluid dynamics is maintained.
1 K.-J. Wu, G. M. De Varine Bohan and L. Torrente-Murciano, React. Chem. Eng., 2017, 2, 116–128.
Cansunur Demirci asked: Is the ratio of Pd/Ag fixed or can you change it? Can you control the thickness of the hollow particles, i.e. thinner shells? Are you able to obtain porous structures?
Laura Torrente-Murciano responded: The Pd/Ag ratio of the hollow particles can be easily varied. In this particular case, a Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag ratio of 4:1 was used to enhance the kinetics of the galvanic displacement reaction by increasing the concentration of Pd precursor in the final reactor. Higher Pd:Ag ratios would further enhance the rate of this stage while lower Pd:Ag ratios would require longer reactors to provide residence time.
:Ag ratio of 4:1 was used to enhance the kinetics of the galvanic displacement reaction by increasing the concentration of Pd precursor in the final reactor. Higher Pd:Ag ratios would further enhance the rate of this stage while lower Pd:Ag ratios would require longer reactors to provide residence time.
The thickness of the hollow particles is the result of two parameters, firstly the size of the initial core and secondly, the concentration of the second metal. A decrease of both parameters would lead to thinner shells while the opposite is true for an increase of the core size or the concentration of the second metal.
Following the discussion during this session, it is difficult to evaluate the nature of the shell structure and whether porous shells are formed or not. As it can be seen in Fig. 6 (DOI: 10.1039/c8fd00001h), the shell of the hollow particles is not smooth, suggesting the presence of multi-crystals and thus, the potential presence of porosity, however, further characterisation would be required to confirm it.
Graham Hutchings asked: How short can the reactors be? This will be important with respect to scale out to make large quantities of the supported nanoparticles. Also do you have any problems with the nanoparticles coating the tubing?
Laura Torrente-Murciano replied: The reactors should be long enough to ensure the residence time needed for the full conversion of the metal precursors to avoid continuation of the reactions downstream which would lead to the loss of size control. Having said this, our reported reactors are relatively small (from a few centimetres to ∼1 m).We are quite conservative in this regard for the reasons mentioned above and further optimisation could be done if the system is scaled up for manufacturing purposes. In any case, it is important to mention that the scale up of this technology should be carried out by adding reactors in parallel rather than by conventional increment of the reactor dimensions, which would lose the control in the fluid dynamics achieved by the coiled inverted reactors. Although this approach would require a considerable capital cost in conventional industrial systems (using stainless steel), the low cost of these micro-tubular reactors would make the technology feasible.
Finally, regarding the fouling of the tube, appropriate material selection and changes on the wettability of the walls of the reactor can successfully avoid this problem. In our case, we could only observe a small fouling on the entrance of the reactor, where localised turbulence takes place even after hours of continuous operation. In any case, it did not affect the stability and reproducibility of the process.
Laura Prati asked: During the preparation of NPs (at least in the first step) you declared you didn’t use any capping agent. I was wondering, what is the stability of the NP suspension you obtained? Indeed also the catalytic test you ran indicates a very quick reaction.
Laura Torrente-Murciano answered: That is right, the production of nanoparticles in continuous coiled flow reactors does not need the presence of organic capping ligands to avoid their agglomeration by exploitation of the opportunities offered by the laminar flow regime in addition to the formation of Dean vortexes to lead to narrow size distributions. Further information can be found in ref. 1.
This means that the particles are only stabilised electronically by BH4− and citrate in the different stages of the presented process and thus, prone to agglomeration once they leave the reactor. In this particular case, the hollow bimetallic particles were stable, judging by UV-vis spectroscopy, for over 2 h after reaction, after that, agglomeration was observed. This timescale was sufficient to demonstrate the enhanced catalytic activity achieved by hollow particles respect to solid or monometallic ones.2
It is true that higher stability would be required for other catalytic applications, especially if this technology is to be implemented as a new manufacturing route for catalysts. In this regard, the weak electronic stabilisation of the particles and the absence of organic ligands on their surface would facilitate their deposition on a range of supports.3–5
1 K.-J. Wu and L. Torrente-Murciano, React. Chem. Eng., 2018, 3, 267–276.
2 L. Torrente-Murciano, T. Villager and D. Chadwick, ChemCatChem, 2015, 7 925–927.
3 R. E. Owen, P. Plucinski, D. Mattia. L. Torrente-Murciano, V. P. Ting and M. D. Jones, J. CO2 Util., 2016, 16, 97–103.
4 L. Torrente-Murciano, R. S. L. Chapman, A. Narvaez-Dinamarca, D. Mattia and M. D. Jones, Phys. Chem. Chem. Phys., 2016, 18, 15496–15500.
5 F. C. Walsh, D. V. Bavykin, L. Torrente-Murciano, A. A. Lapkin and B. A. Cressey, Trans. Inst. Met. Finish., 2006, 84, 293–299.
Katerina Soulantica asked: Concerning the hollow nanoparticles, could the internal (concave) and external (convex) surfaces exhibit different catalytic properties?
Laura Torrente-Murciano answered: This is a very interesting point and I believe that the internal and external surfaces of the hollow bimetallic particles would have different reactivities based on the configuration of the atoms in both surfaces. Although I do not have any experimental evidence in the case of the hollow particles (and I don’t even know whether the reactants would have access to the interior of the particles), it is true that we have reported similar effects of the configuration of the surface atoms for Ru1 and Co particles2 on their final activity.
1 A. K. Hill and L. Torrente-Murciano, Int. J. Hydrogen Energy, 2014, 39, 7646–7654.
2 L. Torrente-Murciano, A. K. Hill and Tamsin E. Bell, Catal. Today, 2017, 286, 131–140.
Cansunur Demirci asked: Is this technique expandable to other metals, i.e. Au/Ag? If yes, which precursor would you use for gold? A common precursor is chloroauric acid (for gold) but using this would lead to AgCl. So which precursor would you use?
Laura Torrente-Murciano replied: This technique will be applicable to any combination of metals where the reduction potentials are appropriate to enable the galvanic displacement reaction, leading to hollow particles. In other words, the standard electrode potential of the second metal needs to be higher than that of the initial core metal to allow the displacement reaction to take place.
As shown in the paper (DOI: 10.1039/c8fd00001h), the choice of metal precursors would be critical to determine this. For example, while PdCl42+ has a lower reduction potential than Ag+/Ag, the use of Pd2+ is feasible for this particular system.
Regarding a Au/Ag system, the reduction potential of both metals indicate the feasibility of the synthesis of hollow Au/Ag when using a Ag core, followed by displacement reduction by a gold precursor. Au+, Au3+, AuOH2+, AuBr2+ or Au(OH)3 could be alternative options to chloroauric acid to avoid the use of Cl− and the consequent formation of AgCl. The solubility of different Au precursors should be evaluated to identify the best gold precursor for the system.
Chris-Kriton Skylaris remarked: In simulations we have done on Pt nanoparticles when we raise the temperature, we observe that the surface starts melting while the core reamins solid. Have you examined at high temperatures how your hollow nanoparticles behave and how stable they are?
Laura Torrente-Murciano answered: We haven’t examined the effect of temperature on the stability of the hollow bimetallic nanoparticles but, as mentioned, it is expected that the surface will present liquid-like properties, especially at high temperatures. The surface melting of the nanoparticles has a detrimental effect on their stability due to their high mobility when supported, as we have demonstrated for Au–Pd bimetallic nanoparticles1 and thus, it should be further explored following this comment.
1 T. García, S. Agouram, A. Dejoz, J. F. Sánchez-Royo, L. Torrente-Murciano and B. Solsona, Catal. Today, 2015, 248, 48–57.
Margherita Macino commented: You mentioned you could support the hollow particles on a support. Could you characterise them? Are the hollow structures maintained?
Laura Torrente-Murciano replied: That’s right, the absence of organic capping ligands on the surface of the nanoparticles facilitates their deposition on supports downstream in the reactor. Although we have probed this approach using monometallic solid particles, we haven’t done it for the hollow bimetallic particles reported here, however, we would expect a very similar outcome as both present similar properties and chemical environments.
Once supported, further characterisation of the particles should be done to test whether the hollow structure is maintained, but again, we cannot see why it shouldn’t.
Francesca Baletto asked: Could you provide more information, including if possible any details at the atomistic level, on the two-step processes for the synthesis of Ag-based NPs? Could you detect what is the minimum size at which you can stabilise the hollow structure? What is the role of temperature? Could high T hinder the stabilisation of a hollow structure? What is the size of the ‘hole’ in relation to the diameter of the whole nanoparticle?
Laura Torrente-Murciano responded: The formation of hollow particles by galvanic displacement has been reported before. In this work, the formation of silver seeds and their growth take place in the first two reactors, respectively. In the third reactor, and triggered by the addition of Pd(NO3)2, galvanic displacement takes place where Pd is reduced on the surface of the nanoparticles by re-dissolving Ag. The reactions taking place are presented in the paper (reactions (6–9)). The presence of a mild reducing agent such as hydroquinone, promotes the kinetics of this process by continually reducing Ag on the surface of the newly formed hollow particles. The motion of the metals within the shell through the Kirkendall effect is responsible of the formation of the hollow structure.
The size of the hollow structure will be determined by the size of the silver particle. Although we haven’t determined the minimum feasible size, the bigger the starting particles, the higher the surface area, promoting the galvanic reactions mentioned above.
Temperature has a role in the kinetics of the process but also in the capability of the nucleation to form monometallic particles. At mild temperatures, as reported here, reduction of silver can only take place on the surface of the already formed particles, while an increase in the temperature would lead to its nucleation and thus, the simultaneous formation of solid monometallic and hollow bimetallic particles.
Keith Whiston said: What is contained within the hollow particles that you presented? Is it simply water or might other salts be present? Is it possible to determine this?
Laura Torrente-Murciano replied: This is an interesting question that we did not consider during this work, which was focused on the continuous synthesis of hollow bimetallic particles in a single system rather than the multi-step batch protocols presented in the literature for such complex particles.
In our opinion, if the galvanic displacement of the silver core by palladium is complete, the particles are completely hollow and one would expect that the solvent (in this case water) and any salts would be inside, however, further advanced characterisation would be necessary to confirm this.
Rosa Arrigo commented: The synthesis of hollow NPs presented in this paper is very interesting on many levels. I am intrigued by how the alloying process occurs and how the inner void is formed. From the transmission electron micrographs presented in this contribution, it appears in some cases that the metal shell has a different thickness (different contrast), thus, one could imagine that the shell is porous and/or presents open channels connecting the inner hollow core to the external surface. This is an effective way of enhancing the surface area of the metal nanoparticles provided that they are stable under reaction conditions. What evidence do you have that the shell is indeed closed?
Laura Torrente-Murciano replied: This comment/question is related to the previous question by Francesca Baletto.
The synthesis of the hollow bimetallic nanoparticles consists of different steps carried out in the microreactors connected in series. Initially, silver seeds with sizes ∼5 nm and narrow size distribution are formed using a strong reducing agent. In the second reactor, the seeds are grown to ∼9 nm following a seed-mediated mechanism. These solid silver particles are then introduced into the final reactor where, in the presence of Pd(NO3)2, the galvanic reaction (eqn (5) in the paper) takes place where Pd is reduced by re-dissolving Ag into the solution. In addition, due to the presence of a mild reducing agent such as hydroquinone, a number of alloying and dealloying reactions (described in reactions (6–8) also take place. The formation of the hollow structure is facilitated by the Kirkendall effect related to the motion of the two metals within the shell. This mechanism of formation of hollow particles has been reported before, however, the novelty of this work is related to the continuous synthesis rather than the complex multi-step protocols previously reported.
It is right to say that the TEM pictures of the hollow particles (Fig. 6 in the paper) suggest a rough surface, probably formed by multi-crystals, however, further characterisation would be needed to determine its porosity.
Federico Spolaore said: Dear Dr Laura Torrente-Murciano, thanks for your presentation. During the discussion you mentioned that you didn’t use any capping agent or polymer to stabilise the nanoparticles. Have you tried without the sodium citrate? Is the reported ratio of Ag/citrate the minimum amount to have stable nanoparticles in suspension?
Also, the second step of your process is a seeded growth that required the same ratio of Ag/citrate. Again, is it possible to obtain stable nanoparticles without making use of citrate but maybe another reducing agent that doesn’t act as a stabilising agent?
My last question is on the overall process itself. Do you see any possibility to grow the silver nanoparticles from very beginning, without the second step of the seeded growth?
Laura Torrente-Murciano answered: Dear Federico, thanks for your comment.
You are right to say that during our continuous synthesis of nanoparticles in microreactors, we do not use any organic compounds or polymers to stabilise the particles, but instead we use a weak electronic interaction by BH4− and citrate which allows us to achieve control over size.
In the presented work, we have used a sodium citrate/silver molar ratio of 7:1, however, we have not done any optimisation to explore whether the ratio could be further decreased. It is important to highlight that sodium citrate does not only act as a stabiliser but also as a reducing agent in the second reactor where the silver seeds synthesised in the first reactor are grown to sizes of 9.4 nm, which are then used as core particles for the subsequent galvanic reaction in the final reactor in series. Further information about this can be found in a recent publication.1
Although we haven’t tried to replace the sodium citrate by another reducing agent that does not act as a stabilising agent, I believe that doing so would lead to agglomeration of particles due to the Brownian motion of the nanoparticles in suspension.
Finally, we haven’t been able to grow silver nanoparticles with sizes above ∼5–6 nm in a single stage while maintaining a narrow size distribution as it won’t be possible to separate the nucleation and growth of the particles, leading to a wide range size distribution. Instead, microreactors enable the development of single systems where reactors are connected in series to achieve the above-mentioned separation of nucleation and growth stages as we have recently reported by quick changes in the reaction conditions (e.g. temperature) and chemical environments (e.g. reducing agents and concentrations).1
1 K.-J. Wu and L. Torrente-Murciano, React. Chem. Eng., 2018, 3, 267.
Andrea Russell asked: To follow on from the earlier question about what is inside the hollow particles, I wonder if they are actually hollow or if the stoichiometry of the galvanic displacement reaction produces a lower density silver core to the particles, akin to a sponge. A way of exploring this would first be to determine the Ag:Pd stoichiometry of the product by an alternative analytical technique such as ICP-OES. Does the stoichiometry really match that of the hollow core or is there more Ag present that you’d predict. Your data in Fig. 7 of the paper do show Ag throughout the particle, just less of it than the Pd. A second way of exploring this would be to use a different element as the core, preferably one with a similar charge to the Pd ions so that the stoichiometry of the galvanic displacement is more 1:1, such as a Cu core.
Laura Torrente-Murciano replied: The stoichiometry of the galvanic displacement reaction between Ag and Pd2+, in addition to the excess of Pd in this particular system (Pd/Ag molar ratio 4:1) could in principle lead to the full re-dissolution of the initial silver particles. In addition, the presence of hydroquinone has the capability to further reduce palladium and silver precursors on the surface of the particles (note that nucleation and the formation of solid particles is not possible under the conditions used herein). Having said this, it is possible that the core of the particle is a silver sponge-like type, although unlikely considering the Kirkendall effect.
I agree that further analysis by ICP-OES would be useful to find out the actual Pd:Ag ratio but not useful to probe the formation of hollow versus sponge-type cores. Regarding Fig. 7 in the paper, it is important to note that the lower concentration of Ag related to Pd is likely to be related to the Pd:Ag ratio mentioned above.
The idea of exploring this system by using different core metals is definitely worth pursuing.
Graham Hutchings remarked: Following on concerning the discussion of how to make a porous hollow spherical nanoparticle; if you made a bimetallic nanoparticle you might be able to leach out the second metal and create a porous sponge-like material. That could be a very interesting catalyst.
Laura Torrente-Murciano answered: Very interesting idea, especially if the size of the porous sponge particle is small (<5 nm) although the lack of crystallinity at these sizes might render them as unstable. Even at larger sizes, this would be a potential way of not only increasing the surface area of metals but also promote the formation of defects with high catalytic activity.
Carlo Lamberti commented: A possible way to solve the problem of what is inside the hollow nanoparticles (NPs) is to perform SAXS experiments, that are sensitive to the electron density of the internal and core parts of the NPs. By measuring the sample as such, under dynamic vacuum and in the presence of an equilibrium pressure of a high-Z gas (e.g. I2) one should understand whether the internal void of the hollow NPs is accessible or not: if the internal electron density changes in the three experiments then it is, if the internal electron density is constant then it is not.
Laura Torrente-Murciano responded: This is a good suggestion which would enable the determination of the content of the hollow interior only if the bimetallic shell is porous to allow the entrance of the high-Z gas during the analysis. It is not clear if this technique would provide definite proof if the core is partially hollow (e.g. sponge-like structure).
Baldassarre Venezia commented: I would like to follow up on Prof. Hutchings’ question on the reactor engineering issues. How could the fouling problem be tackled on a larger scale where the reactors are numbered up? Moreover, how would the progressive nanoparticle coverage of the reactor wall affect the overall process yield?
Laura Torrente-Murciano answered: While fouling of the wall of the microreactors can be a problem to ensuring constant and reproducible production of metal nanoparticles, thereby decreasing the yield at the same time, this can be avoided by: (i) appropriate selection of the reactor material, and (ii) modification of the wettability of the walls to avoid the heterogeneous nucleation of metals on the walls.
Both of these approaches are followed in this work and thus, fouling of the reactor was not observed, except in the inlet area due to localised turbulence, even after operation for a number of hours in continuous mode. These effective techniques can be directly applied independently of the number of reactors working in parallel.
Katerina Soulantica asked: Is there any evidence that it is only the external surface of the hollow nanoparticles that is accessible to the reactants during catalysis?
Laura Torrente-Murciano answered: Following the answer to another question, this work did not characterise the nature of the bimetallic shell and whether it is solid or porous. Judging from the TEM pictures shown in Fig. 6 (DOI: 10.1039/c8fd00001h), it is suggested that the shell is formed by polycrystals, however, there is not certainty whether the reactants have access to their interior.
Nia Richards asked: Have you considered the Kirkendall effect on your hollow nanoparticles?
Laura Torrente-Murciano responded: Yes, indeed, the motion of the metals within the particle (Kirkendall effect) is responsible for the formation of the hollow particles, otherwise the process would stop once a shell of Pd is formed by the galvanic reaction.
Andrea Russell opened a general discussion of the papers by Laura Prati, Jennifer Edwards and Laura Torrente-Murciano: It was really nice to have these three papers presented together, as you’ve all used very different reducing agents, ranging in scale from a harsh reagent, NaBH4, used by Laura Prati, to a less commonly employed reagent, DMF, used by Jennifer Edwards, to selecting different reagents for each step of the process as was done by Laura Torrente-Murciano. Would you like to discuss the reasons behind your choices and how these influence the particle size distributions or other attributes of the nanoparticles produced?
Laura Torrente-Murciano replied: The choice of reducing agent is of key importance in determining not only the resulting particle size but also the distribution. Indeed, in our work (DOI: 10.1039/c8fd00001h) we used different reducing agents depending on the aim of each stage. Initially, silver seeds are formed with a narrow size distribution using a strong reducing agent such as sodium borohydrate. However, a milder reducing agent such as sodium citrate as well as a different reaction temperature are used in the second reactor in series to ensure the growth of the seeds while avoiding the secondary nucleation that a strong reducing agent would trigger. Further details about this approach can be found in our recently published work.1 Finally, hydroquinone is used for the final reduction of palladium during the galvanic displacement stage to avoid the formation of Pd-only nanoparticles.
We would like to highlight that these rapid changes in reaction conditions are only possible due to the use of microreactors characterised by high mass and heat transfer rates.
1 K.-J. Wu and L. Torrente-Murciano, React. Chem. Eng., 2018, 3, 267.
Laura Prati replied: The choice of the reducing agent is very important and should be managed carefully in the synthesis of nanoparticles. The choice is of course directed by the E0 of the metal but there are also some experimental details that can play a crucial role. One of these is the rate of addition of the reducing agent and the temperature. We have always to keep in mind that the size/shape of our particles is determined by the relative kinetics of two elementary steps: nucleation and growth. Increasing nucleation (fast reduction) we decrease the particle size, while increasing nucleation (slow reduction) we increase the particle size. Obviously there are a lot of other parameters that have to be controlled.
Jennifer Edwards replied: For the microwave preparation, DMF is ideal given its permanent dipole (meaning heat is transferred extremely effectively as the microwave passes through the sample) whilst at the same time acting as a reducing agent. Other alcohols can be used, and give us much different activities in the catalysis (ethylene glycol produces a homogeneous alloy, for instance) and we are currently working on trying to explain these subtle differences.
Carlo Lamberti commented: Some 20 years ago, a few scientists (among them: M. A. Bañares, A. Brückner, M. Daturi, F. Thibault-Starzyk, I. E. Wachs and B. M. Weckhuysen) gave birth to the concept of “operando spectroscopy”, immediately extended also to scattering techniques. Since then, a strong community has grown around this concept and they were able to have more than a hundred delegates at the 6 successive operando spectroscopy conferences held so far: 2003 Lunteren (NL), 2006 Toledo (S); 2009 Rostock (D); 2012 Brookhaven National Laboratory (BNL, Upton, NY, USA); 2015 Deauville (F), 2018 Estepona (S). In these last two decades, important improvements have been made in the understanding of the structural, electronic and vibrational properties of the active sites of catalysts under reaction conditions. I wonder if it is not the right time to push our scientific community in the direction of “construerendo spectroscopy”, (or “aedificando spectroscopy”) i.e. in the development of ad hoc cells able to follow the catalyst preparation along all its steps and to shed light, at the molecular level, to the synthesis process from the precursors to the final catalyst. Indeed, very few attempts are present in the literature along this line,1–4 and a much larger effort is needed in my opinion.
1 F. Rey, G. Sankar, J. M. Thomas, P. A. Barrett, D. W. Lewis, C. R. A. Catlow, S. M. Clark and G. N. Greaves, Chem. Mater., 1995, 7, 1435–1436.
2 G. Sankar, J. M. Thomas, F. Rey and G. N. Greaves, J. Chem. Soc., Chem. Commun., 1995, 0, 2549–2550.
3 D. Grandjean, A. M. Beale, A. V. Petukhov and B. M. Weckhuysen, J. Am. Chem. Soc., 2005, 127, 14454–14465.
4 G. Agostini, E. Groppo, A. Piovano, R. Pellegrini, G. Leofanti and C. Lamberti, Langmuir, 2010, 26, 11204–11211.
Laura Prati replied: Your observation is absolutely right. To be honest we had some attempts to “look” at the metallic sol during its formation but unfortunately up to now we failed. Reasons can be found in experimental difficulties, such as bubble formation or the detrimental effect of the beam high energy on the sol formation.
This topic requires much more effort to solve practical problems but I’m confident that in the future we’ll be able to give an answer.
Jennifer Edwards replied: I absolutely agree, there are very few, if any, reports in the literature that focus on determining the stepwise mechanism of nanoparticle synthesis. This is an area we are currently active in.
Stanley Lai asked: Expanding on the previous question by Prof. Andrea Russell, how significant would you consider the impact of nanoparticles to be depending on the synthetic method? For example, in the paper presented by Prof. Prati, only AuCu shows a tremendous synergistic effect, whereas the effect of addition of Au to Pd, Ru and Pt is modest. However, AuCu was prepared in a different manner than the other bimetallics. In this case, could it be that the difference in synthesis method plays a significant role?
More philosophically, would this imply that a fair comparison between nanoparticles can only be made if they are prepared under the ‘exact’ same condition?
Laura Torrente-Murciano responded: This is a very interesting point that applies to most of the work done in catalysis and emphasises the need for standardisation methods for the understanding of the different effects affecting the resulting catalytic activity, beyond the chemical composition (e.g. bimetallic systems) and particle size. In this context, I believe that the method of preparation of supported nanoparticles is critical to determine the interaction between the metal nanoparticle and the support, which in turn would affect their reactivity. As an example, we have reported that a high AuPd metal–support interaction leads not only to enhanced reactivity, but also stability under reaction conditions.1
In this context, the continuous synthesis of nanoparticles in flow reactors presented in our work, followed by their deposition on supports, would enable the fundamental understanding of these parameters as the support can be functionalised to vary its interaction with the nanoparticles independently of the nanoparticle synthesis.
1 L. Torrente-Murciano, Q. He, G. J. Hutchings, C. J. Kiely and D. Chadwick, ChemCatChem, 2014, 6, 2531–2534.
Laura Prati replied: The characterisation of bimetallic particles is still a challenge in the sense that sometimes conclusions are made on the basis of few data. It could happen that we do have not a representative picture of our catalyst. Segregation, different ratio of metals, and different structures can occur.
So, I’m convinced that two different preparation methods can produce two different catalysts, but if different preparation methods produce the real same catalyst, in this case we should observe the same catalytic behaviour. In the case of AuCu, we are producing a catalyst that shows some inhomogeneity and no definite conclusion can be made.
Jennifer Edwards replied: I think Professor Prati may be better able to answer the questions specific to her paper, but I’m happy to add my views.
It is clear that the preparation method of the nanoparticle plays a huge role in determining the activity of the resulting catalyst. However, from my experience in sol immobilisation preparation, the reaction conditions vary depending on the type of alloy you want to make so, for instance, the reaction conditions to prepare a NiPd alloy (type of reductant, temperature) are very different to the ones required for AuPd. The role of potential impurities, nature of the alloy (core–shell vs homogeneous) and particle size should be kept in mind when making comparisons between different catalysts.
Bob Tooze returned to the discussion of the paper by Jennifer Edwards: A question was asked about the scale-up of microwave synthesis. I commented I was aware of a Company C-Tech Innovations in Cheshire that were involved in continuous microwave reactors.
Jennifer Edwards responded: We have worked with C-Tech previously on a large greywater project, thank you for this information!
Justin Hargreaves asked: In those cases where borohydride is applied as a reductant, please may I ask the extent to which it is possible to discount the possibility of boron species being associated with the resultant nanoparticles?
Jennifer Edwards replied: I don’t think it wise to discount the incorporation of boron into the catalysts prepared using NaBH4 as the reductant. In our studies we know that the boric acid produced following the reduction plays a key role in determining the structure we see (paper in preparation) but we have not detected boron impurities in the nanoparticles to date.
Katerina Soulantica opened a general discussion of the paper by Scott Rogers: The use of PVA during nanoparticle synthesis may impact the catalytic properties. Have you considered/measured the amount of organic capping agent present on the surface of the catalysts?
Scott Rogers replied: Indeed, the presence of PVA on the nanoparticle surface can limit the catalytic performance for some liquid phase reactions. In previous work, heat treatments and extensive washing processes have been used in order to remove the PVA,1,2 however it is almost inevitable that an increase in particle size is observed. In our study we used the as-synthesised catalysts washed only at room temperature to minimise particle growth so that a systematic particle size dependent study could be performed, and to maximise the aggregation of small Pd clusters. Quantifying the amount of PVA on the surface is a challenging process and is still under investigation.
1 J. Pritchard, M. Piccinini, R. Tiruvalam, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, D. J. Morgan, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 308–317.
2 J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556.
Carlo Lamberti asked: What is the CO surface coverage of the IR spectra shown in Fig. 3 of your paper (DOI: 10.1039/c7fd00216e)? Did you perform a coverage dependent IR study? Indeed, the evolution of the C–O stretching frequencies upon increasing the coverage can be very informative on the nature of the Pd nanoparticles, in terms of exposed faces, presence of defects, etc.1–4
1 C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001.
2 E. Groppo, S. Bertarione, F. Rotunno, G. Agostini, D. Scarano, R. Pellegrini, G. Leofanti, A. Zecchina and C. Lamberti, J. Phys. Chem. C, 2007, 111, 7021–7028.
3 G. Agostini, R. Pellegrini, G. Leofanti, L. Bertinetti, S. Bertarione, E. Groppo, A. Zecchina and C. Lamberti, J. Phys. Chem. C, 2009, 113, 10485–10492.
4 E. Groppo, G. Agostini, A. Piovano, N. B. Muddada, G. Leofanti, R. Pellegrini, G. Portale, A. Longo and C. Lamberti, J. Catal., 2012, 287, 44–54.
Scott Rogers responded: We did not perform any experiments in order to quantify the CO surface coverage as we did not have the facility to administer a specific quantity of CO to the surface – this is an interesting experiment that could be performed in future work.
Laura Prati commented: The role of capping agents during the catalysis should always be kept in mind. In fact, the capping agent is adsorbed on the surface through its active sites. This necessarily decreases the catalytic activity. If we want to use unsupported materials we need the presence of molecules able to stabilise, and sometimes compensate for, the interaction between particles. The use of a support can compensate for the attraction among particles and allows the removal of the capping agent, thus retrieving the active sites. It should be noted that a capping agent sometimes plays a useful role in driving selectivity.
Laura Torrente-Murciano replied: I would like to add that our contribution (DOI: 10.1039/c8fd00001h) demonstrates that the addition of capping agents adsorbed on the surface of the particles can be completely avoided by using continuous reactors under laminar flow. In this case, simple weak electronic stabilisation is enough to produce particles with narrow size distributions and also tuneable sizes. In these cases, the colloidal suspension can also be easily supported on different materials using the same approach (e.g. considering the zeta potential of the support).
Margherita Macino remarked: When changing the Pd loading of the catalysts during preparation did you keep the initial Pd concentration constant while changing the amount of support added, therefore avoiding possible formation of larger sol particles?
Scott Rogers responded: Yes, when preparing Pd catalysts of different metal loading the concentration of the sol remains constant at 1.25 mM [Pd], and the amount of support material is varied. The catalytic properties are ‘preformed’ in the sol, and so a higher concentration promotes nanoparticle growth.
Yaroslav Odarchenko said: Taking into account that the session is dedicated to the control of nanoparticle synthesis, how would you compare the scalability of the sol-immobilisation approach used in your paper with other preparation methods?
Scott Rogers replied: The current metal concentration used in the sol-immobilisation method is ∼0.1 mM, which is impractical when scaling up the synthesis (∼1 gram of catalyst requires ∼800 mL of solvent for Pd). However, we have recently investigated the influence of metal concentration on the particle properties for colloidal Au, to which these preliminary studies concluded that there was little effect on the properties when prepared between the concentrations of 0.1 to 1 mM [Au]. The results indicate that it could indeed be practical to prepare catalysts using the sol-immobilisation method on a large scale.
We are also developing the continuous flow preparation of metal nanoparticles prepared using the same reagents (PVA and NaBH4), which is a growing research area to achieve larger scale processing.
Jonathan Quinson remarked: Among the experimental parameters you studied here and in previous reports (temperature, solvent, etc.) I was wondering if you studied the influence of the nature of the precursor.
You also said that you varied the concentration of Pd precursor without seeing much effect on the size then obtained. I was wondering which range of concentration was investigated?
Scott Rogers answered: The palladium precursor used in this study is K2PdCl4 but we have not studied the influence of the nature of the precursor, although it is a future study worth considering. The nature of the Pd precursor affects solubility, the reduction rate and also the existence of species such as Na, Cl and K etc.
To clarify, we have recently investigated the influence of colloidal metal concentration on the particle size for Au, which concluded that, between the range of 0.1 to 1 mM [Au], the effect on the particle size within the colloidal Au was insignificant. The Pd concentration effect experiment has not been performed.
Matthew Conway remarked: Following up on your response to an earlier question: you have modified many variables in the self-immobilisation preparation procedure, but have you ever used a different hydride source?
Scott Rogers responded: Sodium borohydride is commonly used as the hydride source due to its strong reducing power making it possible to generate small nanoparticles. LiAlH4 is another strong reducing agent, however the high toxicity makes it impractical. We have not studied the effect of using other milder hydride sources, although we are considering the use of hydrazine, THPC/NaOH or citrate to study steric versus electrostatic stabilisation.
Valerii Bukhtiyarov asked: Did you study the influence of metal particle size on the nitrobenzene hydrogenation?
Scott Rogers replied: During our study, we can correlate the metal particle size with the catalytic activity in the hydrogenation of nitrophenol reaction.
Toru Murayama remarked: Fig. 4C in the paper showed that only 0.2Pd1C (same as 0.2PdA1?) had good reusability. The authors concluded that one of the reasons for the high catalytic activity and good reusability of 0.2PdA1 was due to the size of the Pd clusters. However, the average size of the Pd particle was not so different and they have the similar distribution in Table 2 in the paper. It is difficult to explain this phenomena due to the size of the particles. If the authors show the histogram of the size distribution of particles, the author could predict the ultra-small Pd clusters. Did the authors check the particle size after the reaction? That characterization is needed to discuss the results of the reusability test.
Scott Rogers answered: I agree that the average Pd particle sizes obtained by TEM are not so dissimilar, and this is a result of the low loading of the Pd (0.2 wt.%), and the limited resolution of the TEM microscope. The presence of ultra-small Pd clusters (<20 atoms) is not evidenced in this study and is only inferred from a previous study whereby only low temperature preparation generated ultra-small clusters. Observing any growth in particle size using TEM will only be possible if there has been significant growth, although it will not determine the cluster populations. To better understand the particle growth, XAFS would be a more suitable technique, however it was not possible for it to be performed.
Josh Davies remarked: With your catalysts prepared at 30, 50 and 75 °C there is very little difference between them. Is it simply possible that, as you have not tested for metal loading and are working off theoretical values, the variations seen between these samples are purely due to metal loading or slight differences in their preparation rather than temperature? Also, have you thought about looking in more detail between the 1 and 30 °C samples to attempt to understand in more detail what is causing the change? So for example preparing catalysts at 5 degrees?
Scott Rogers answered: We have confirmed the Pd metal loadings with microwave plasma-atomic emission spectroscopy (MP-AES), with the results shown in the supplementary information of the paper (Table S1). The catalytic activities were calculated using these values. Indeed, low temperature preparation yields the most exciting nanoparticles with regards to the stabilisation of small metal clusters, and thus investigating these temperatures would be interesting. However, obtaining particle size information using advanced characterisation techniques (STEM HAADF and XAS) is difficult due to the accessibility and cost of the facilities. The study also demonstrates a practical synthesis method to generate a range of particle sizes.
Richard Catlow commented: The approach and the results presented in your paper nicely illustrate the point made earlier that microscopy and X-ray spectroscopy provide complementary information.
Scott Rogers answered: I agree with the comment and anticipate there being more forthcoming research in this area.
Federico Spolaore commented: Since the need of analysing the nanoparticle size has been, up to now, addressed only considering TEM/HRTEM and XAS techniques, I would like to add a few methods from my own knowledge for analysing the particle size:
(1) XRD, for supported nanoparticles and when particles are sufficiently big (dozens of nm) with the Scherrer equation, to obtain an average particle size from the width of the peak.
(2) DISC-Centrifuges, when the particles are in a sol, to have both the average size and size distribution with a decent response close to reality (sonification could be used before to destroy possible aggregates).
(3) DLS, also in the case of colloidal solutions, is one of the most used techniques. It is not as precise at low sizes.
Scott Rogers responded: HRTEM and XAS were a focal point of the discussion in this paper as a result of the very small particle sizes (∼2 nm), which can’t be resolved using XRD and DLS. I do not have experience of DISC-Centrifuges, and thus it is a good consideration to perform the measurement if particles of all sizes are probed.
Bob Tooze asked: Does the molecular weight of the stabiliser (PVA) have an impact on the nanoparticles formed?
Scott Rogers replied: Polyvinyl alcohol (PVA) stabilises metal nanoparticles through steric effects, and thus it is highly probable that a change in the molecular weight of the stabiliser will affect the metal nanoparticle size as well as the covering of active sites. This is not something we can confirm, and it is an area we are keen to probe in future studies.
Jonathan Quinson remarked: In Table 1 of the paper, one of the experimental parameters is the nature of the solvent. This parameter was actually not changed in this specific study. Therefore I believe it should be removed from the description of Table 1 for clarity within the frame of this specific paper.
Scott Rogers replied: I agree with the comment.
Haoliang Huang opened a general discussion of the paper by Philip Davies: I have two questions about the treatment of HOPG: (1) Do the results from C(1s) XPS show the same oxygenated functionalities, especially in the sample treated by high concentration acid? (2) If HOPG is treated by alkaline solution, will it produce similar results?
Philip Davies responded: Using very strong acids causes too much damage to the graphite for us to study the surface with AFM. The extent of functionality we create with the mild acids we use (0.1–5 M) is not great enough to significantly alter the substrate C(1s) peak in the XPS. We have studied graphite surfaces treated with both sodium and ammonium hydroxide and in both cases see the formation of surface hydroxyls identical to those seen for the acids. This work will be published separately.
Carlo Lamberti remarked: Prof. Davies, in your presentation you mentioned the presence of C–OH and C=O groups at the surface of highly ordered pyrolytic graphite. What is the experimental evidence that allowed you to detect such groups?
Philip Davies responded: We used a selective derivitization approach with trifluoroacetic anhydride, trifluoroethyl hydrazine, triflouroethanol and N-N′-di-tert-butylcarbodiimide to identify the functional groups at the surface. Details are given in our previous work which are ref. 8 and 9 of the present paper.
Parasuraman Selvam asked: What is the nature of the carbon, as shown by C(1s) spectra, upon the various acid treatments? It would have been better if you have given a representative C(1s) spectra that will help us to understand the metal–support interaction. What is your comment on this issue? Alternatively, is the data already available/published? If not, I would very much appreciate it if you could give at least one C(1s) spectra figure before and after treatment with acids.
Philip Davies replied: The extent of surface functionalisation we achieve with our treatment is generally less than 20% of the surface. As a result changes to the C(1s) spectrum are minimal and indeed undetectable with our equipment. Fig. 1 shows two examples of C(1s) spectra. (a) is recorded after exposure of the carbon surface to a gold containing solution, no adsorption of gold was observed. (b) shows the same sample after exposure to 5 M HCl followed by the gold solution. There is no discernible difference between the two spectra.
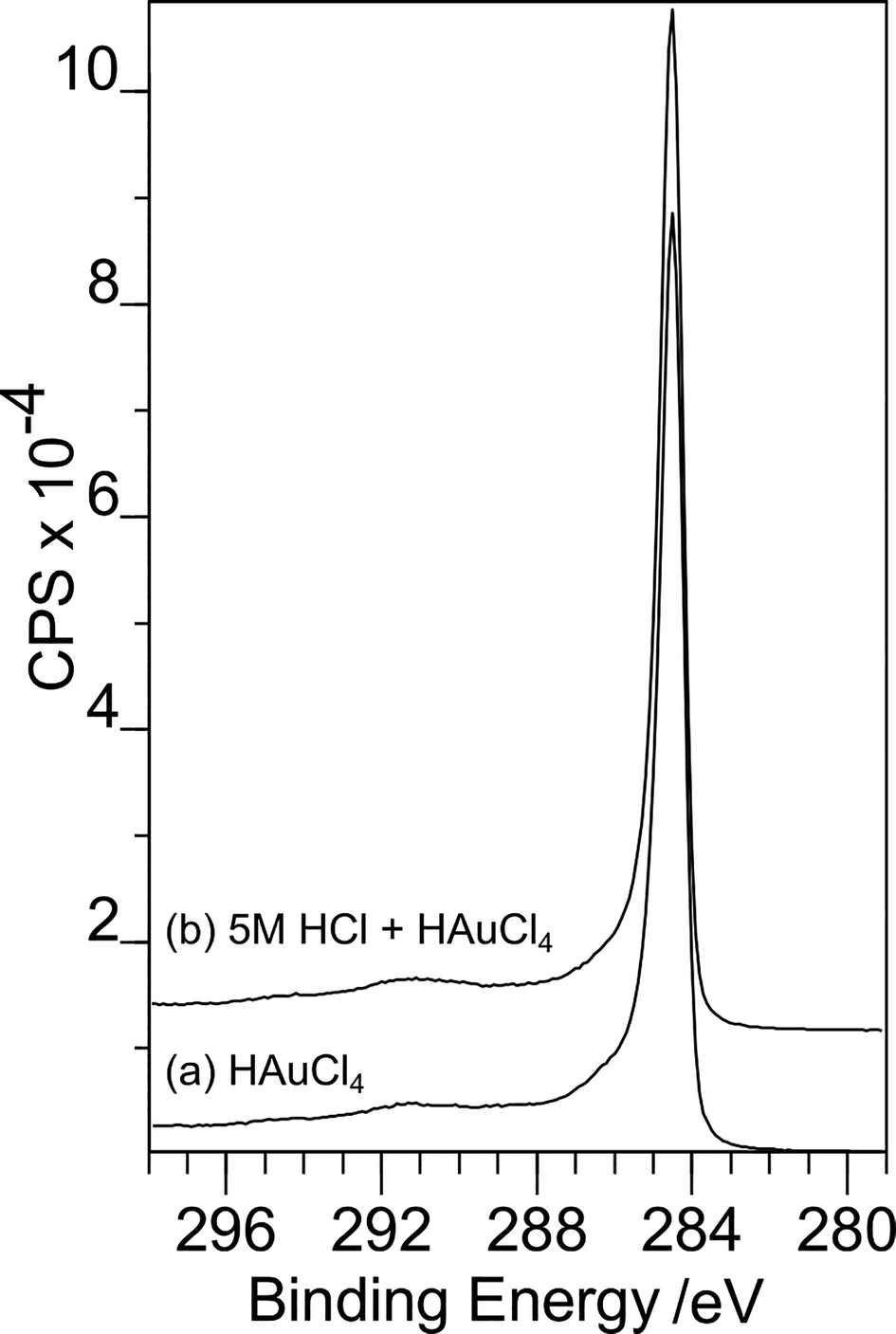 | ||
| Fig. 1 C(1s) spectra where (a) is recorded after exposure of the carbon surface to a gold containing solution, no adsorption of gold was observed, and (b) shows the same sample after exposure to 5 M HCl followed by the gold solution. | ||
Graham Hutchings remarked: You have investigated a number of acids. When using HCl/HNO3 one would presume that Cl− would be retained on the surface. Do you observe this and if not what is the reason for this? What about using less strong acids, e.g. acetic acid?
Philip Davies answered: At the relatively weak concentrations (0.1–5 M) we have used in our studies, neither HCl nor HNO3 generated any functional group at the graphite surface other than the hydroxyls reported in ref. 8 and 9 of the paper. We have not studied any weaker acids but we have examined the effect of bases such as sodium hydroxide and ammonium hydroxide, both of which generated similar hydroxyls to those seen with the acids, showing both the same XP binding energy of 532.7 eV and similar features in the AFM images.
Michele Carosso commented: I’m very interested in your paper contribution because in our research group we are also studying the surface chemistry of activated carbons employed as supports in industrially relevant catalysts.1–3 In particular, we are focusing our attention on the effect of the activation treatment (steam vs. H3PO4), and of the post-activation treatments in the presence of oxidizing agents, such as HNO3. In the latter case we found that carboxylic and carboxylate groups are mainly formed, the latter stabilized by the extended conjugation of the π electrons and being more abundant on small and irregular graphitic platelets. Carboxylates, in particular, have an influence on the metal dispersion and affect the catalytic properties of the final catalyst. Do you have some evidence for the existence of carboxylate groups also on your acid-treated HOPG material?
(1) A. Lazzarini, R. Pellegrini, A. Piovano, S. Rudić, C. Castan-Guerrero, P. Torelli, M. R. Chierotti, R. Gobetto, C. Lamberti and E. Groppo, Catal. Sci. Technol., 2017, 7, 4162–4172.
(2) A. Lazzarini, A. Piovano, R. Pellegrini, G. Agostini, S. Rudić, C. Lamberti and E. Groppo, Phys. Procedia, 2016, 85, 20–26.
(3) A. Lazzarini, A. Piovano, R. Pellegrini, G. Leofanti, G. Agostini, S. Rudić, M. R. Chierotti, R. Gobetto, A. Battiato, G. Spoto, A. Zecchina, C. Lamberti and E. Groppo, Catal. Sci. Technol., 2016, 6, 4910–4922.
Philip Davies responded: We have not managed to generate carboxylic acid groups on the HOPG surfaces using the relatively mild acid strengths we have investigated. ref. 8 and 9 in the paper give details of the selective derivatisation we used to establish this point. We plan to investigate the formation of such groups in more detail with alternative acid treatments such as formic acid.
Stanley Lai asked: In this paper you introduce chemical groups on the HOPG surface by acid treatments. Using a surface science approach, would you learn anything new by modifying the HOPG surface through the controlled introduction of a monolayer, for example through diazonium coupling. In this way, one could control the nature and density of chemical groups introduced.
Philip Davies responded: That is a very good suggestion which is very much in line with the approach we have taken; we think there are tremendous possibilities for this general approach to elucidate several aspects of catalysis.
Matthew Conway asked: Does the functional group affect gold dispersion?
Philip Davies answered: The dispersion of the Au3+ and Au0 states seem to be subtly different; the gold metal forms discrete, small islands illustrated nicely in Fig. 10(c) in the paper, whereas the oxidised gold state (which retains both hydroxy and chloride counter ions) forms what looks like networks across the surface best seen in Fig. 4(d) in the paper.
Bob Tooze commented: You report that treatment of HOPG with acids also leads to the formation of more macroscopic defects, which you ascribe to localised delamination. Are these significant in the deposition of metals and can you isolate the formation of these defects from the presence of specific functional groups such as OH or C=O?
Philip Davies answered: The number and size of features observed in the AFM correlates very well with the strength of the acid used to treat the surface (see ref. 8 and 9 in the paper for details) but, interestingly, changing the hydroxyls formed initially to carbonyls (either by heating or allowing the sample to stand in air for a few days) significantly reduces the number and size of features evident. We conclude that it is the hydroxyls that cause the features whereas the carbonyls do not. The presence of oxygen functionality on the graphite surface is a prerequisite for deposition of gold from solution in a reasonable time frame; our paper demonstrates that whereas the hydroxyls result in the deposition of reduced gold (at low concentrations), carbonyls give rise primarily to the deposition of Au3+ states.
Graham Hutchings remarked: In the discussion you mentioned that sometimes the gold was difficult to observe; is this because it is highly dispersed? If you add the gold together with the aqua regia is the dispersion improved?
Philip Davies answered: The features in the AFM images that we attribute to deposited gold metal or gold salts are much smaller (∼2–3 nm high) than the local delamination features that still remain. Imaging features at different scales can be tricky and there may also be issues with mobility of species under the AFM tip, which makes high resolution imaging difficult. Preliminary data in which the gold is deposited from an aqua regia solution suggest good dispersion but our data is not yet consistent enough to be published.
Valerii Bukhtiyarov commented: Acid treatment of HOPG used in your study can modify not only the surface of the pyrolitic graphite, but also the subsurface layers. What do you think about this possibility? I know that C(1s) spectra are quite insensitive for this. But as we showed earlier, carbon KVV Auger spectra can be used to study distortion of subsurface layers of HOPG. Indeed, we showed that Ar+ sputtering diminishes the intensity of the high energy component in the Auger spectrum. Since this line is an indicator of chemical interaction between two graphene layers of graphite, this result had lead us to conclusion about insertion of argon ions in subsurface HOPG layers and distortion of subsurface HOPG layers.
Philip Davies replied: That’s a very interesting point, we don’t have any evidence that enables us to comment on the extent of the effect of the acids. We have looked at the carbon Auger signal but did not see any change after acid treatment. This may be because of the relatively low concentration of acid used and the small extent of functionalisation that results.
Cynthia Friend opened a general discussion of the papers by Scott Rogers and Philip Davies: Many different methods for materials’ synthesis were described during the meeting. It would be very helpful to have more standardization in how the various materials are evaluated. We need methods to determine which perform best in terms of activity and selectivity. Currently, there is not standardization. The field would be advanced by having a standard catalyst tested under standard conditions to which new materials could be compared. This would need to be done for specific sets of reactions and may require a materials bank. The data obtained could be logged into an open access database for reference by the community as a whole.
Graham Hutchings said: Continuing the discussion on the use of standard catalysts and standard reaction conditions; in my group we have standard catalysts that new researchers prepare and test using standard procedures and we find this works really well. However, the suggestion reminds me of the situation in the 1980s with methane oxidative coupling, which was a field which rapidly expanded following the seminal paper by Lunsford.1 There were several hundreds of papers and many claimed to have the best paper but the reaction conditions were incredibly disparate as highlighted by Oyama.2 Although the community agreed that standard catalysts and conditions were needed it was not implemented as the different groups continued as before. I just raise this as an example of the difficulty facing this idea but we should not be deterred.
1 T. Ito and J. H. Lunsford, Nature, 1985, 314, 721–722.
2 J. S. Lee and S. T. Oyama, Catal. Rev.: Sci. Eng., 1988, 30, 249–280.
Carlo Lamberti remarked: I believe that the suggestion of Prof. Friend is an excellent idea. We should involve some industrial partners that are willing to prepare a huge amount of standardized catalysts for the different reactions that could be used in the different laboratories for a direct comparison with their own catalysts. We should try to have a standard catalyst for the most relevant catalytic reactions, as Degussa P25 has become the standard material in photocatalysis.
Laura Prati commented: The use of benchmark catalysts is an excellent idea for having reliable comparison of activity/selectivity. This is what happened in the past with gold-based catalysts when the World Gold Council provided samples of gold catalyst to everyone as a benchmark. Obviously this could be helpful for any single reaction.
Yaroslav Odarchenko said: I have commented on the discussion opened by Prof. Cynthia Friend who has talked about standardization and creation of protocols for catalyst synthesis and testing as one of the important challenges for the heterogeneous catalysis community in the future. Prof. Andrea Russell has suggested to create a sort of database with the benchmark catalysts for the common catalytic processes. I completely agree with this idea and would like to share my experience from Li-ion batteries where, for example, any potential material for the anode has to be tested against the benchmark graphite anode (that is readily available on the market) using a defined industrial battery cycling test. Regarding the catalyst’s synthesis, in my opinion there are not many suppliers of metal nanoparticles with the controlled size and particle size distribution. Therefore it would be very helpful to have a possibility to test any new catalysts (or improved synthesis procedure) against the existing (previously reported) well-defined system by following the established protocol (pressure, temperature, reactor setup, etc.).
Conflicts of interest
There are no conflicts to declare.| This journal is © The Royal Society of Chemistry 2018 |