
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of ortho- and polyphosphates on lead speciation in drinking water
Benjamin F.
Trueman
 ,
Wendy H.
Krkošek
and
Graham A.
Gagnon
*
,
Wendy H.
Krkošek
and
Graham A.
Gagnon
*
Department of Civil & Resource Engineering, Dalhousie University, 1360 Barrington St, Halifax, NS, B3H 4R2 Canada. E-mail: graham.gagnon@dal.ca; Fax: +902 494 3108; Tel: +902 494 3268
First published on 16th February 2018
Abstract
Lead is a potent neurotoxin and drinking water represents an important route of exposure, especially where legacy lead pipe is widespread. Polyphosphates are often added to drinking water to sequester iron and calcium, but they may form coordination complexes with lead, increasing its solubility. This risk is not well characterized in practice: the prevalence of lead-polyphosphate complexes in drinking water systems is not known. We used size-exclusion chromatography (SEC) with multi-element (ICP-MS) detection to compare the speciation of lead below 0.45 μm in two low-alkalinity water systems dosing phosphate-based corrosion inhibitors at different ortho![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :poly ratios and doses. In one system dosing polyphosphate at 0.05 mg P L−1 (3:1 ratio), it reverted almost completely to orthophosphate during distribution. In another system dosing polyphosphate at 0.20 mg P L−1 (1:1 ratio), it was detectable at points of use. Under the influence of polyphosphate, lead and iron were present as dissolved species strongly associated with phosphorus. Moreover, experiments with sodium hexametaphosphate confirmed our ability to detect lead as a coordination complex using SEC. In the absence of polyphosphate, lead and iron were present as colloidal particles. Orthophosphate on its own also appeared to reduce lead solubility, as estimated using paired sample profiles collected at seventeen single-unit residences. Increasing orthophosphate from 0.5 to 1.0 mg PO4 L−1 accompanied decreases in total lead concentrations of 38% (95% CI: 15–51%) within eight months. Analysis of recovered lead pipe corrosion scale was consistent with the presence of an insoluble lead phosphate compound: hydroxypyromorphite (Pb5(PO4)3OH).
:poly ratios and doses. In one system dosing polyphosphate at 0.05 mg P L−1 (3:1 ratio), it reverted almost completely to orthophosphate during distribution. In another system dosing polyphosphate at 0.20 mg P L−1 (1:1 ratio), it was detectable at points of use. Under the influence of polyphosphate, lead and iron were present as dissolved species strongly associated with phosphorus. Moreover, experiments with sodium hexametaphosphate confirmed our ability to detect lead as a coordination complex using SEC. In the absence of polyphosphate, lead and iron were present as colloidal particles. Orthophosphate on its own also appeared to reduce lead solubility, as estimated using paired sample profiles collected at seventeen single-unit residences. Increasing orthophosphate from 0.5 to 1.0 mg PO4 L−1 accompanied decreases in total lead concentrations of 38% (95% CI: 15–51%) within eight months. Analysis of recovered lead pipe corrosion scale was consistent with the presence of an insoluble lead phosphate compound: hydroxypyromorphite (Pb5(PO4)3OH).
Water impactPolyphosphates are widely used in drinking water systems for iron and calcium sequestration, but they may increase lead solubility via complexation. In practice, this potential health risk is not well understood. Separation of environmental samples via size-exclusion chromatography with multi-element (ICP-MS) detection represents a promising approach to understanding the prevalence of lead complexation by polyphosphates. |
1 Introduction
Lead service lines (LSLs) were installed widely throughout the first half of the twentieth century; in the United States, millions remain in place.1 Legacy lead pipe is a significant public health concern because lead in drinking water is a risk factor for elevated lead in blood.2–4 High blood lead levels in childhood are strongly associated with lasting deficits in cognitive and academic skills.5–8 The effect of childhood lead exposure is non-linear: at lower levels, increases in blood lead accompany steeper declines in cognitive performance.5,6,8Orthophosphate is added to drinking water to limit corrosion and release of lead from distribution networks.9 Orthophosphate inhibits lead release by forming insoluble lead phosphate compounds: possibilities include hydroxypyromorphite (Pb5(PO4)3OH), chloropyromorphite (Pb5(PO4)3Cl), and tertiary lead phosphate (Pb3(PO4)2).10,11 Orthophosphate may also inhibit dissolution of lead carbonates or other lead compounds by adsorbing to and passivating mineral surfaces,12 as has been suggested in the case of calcite.13 However, certain chemical species can interfere with these mechanisms. Successful formation of a phosphate-rich corrosion scale requires lead-phosphate supersaturation: species that form soluble complexes with lead—including carbonate and natural organic matter (NOM)14,15—can increase the equilibrium solubility of lead-phosphates (e.g., hydroxypyromorphite solubility increases with increasing dissolved inorganic carbon).12 Furthermore, precipitation of other metal-phosphates (e.g., hydroxyapatite, Ca5(PO4)3OH, in hard waters) may interfere with lead solubility control.10
Orthophosphate has been shown to inhibit lead release to drinking water in both laboratory and pilot studies.16,17 Field data have provided additional confirmation—using random daytime,18–20 30 minute stagnant,21 or 6 hour stagnant sampling22,23—that orthophosphate is effective in limiting lead release under at least some conditions (e.g., alkalinity less than 30 mg L−1 as CaCO3). In other circumstances (e.g., at higher alkalinity), orthophosphate-based inhibitors have yielded ambiguous, insignificant, or adverse results with respect to lead corrosion control.22,24 Nevertheless, orthophosphate treatment has been an important component of the response to severe instances of drinking water lead contamination, including those that occurred in Washington, DC (2001–2004)3 and Flint, MI (2014–2016).25 After orthophosphate dosing had been initiated, lead pipe corrosion scale recovered from the Washington, DC distribution system was dominated by hydroxypyromorphite.26
Orthophosphate is frequently dosed with polyphosphate as a blended formulation.9 Polyphosphates form stable complexes with many metals and are useful as sequestering agents for the prevention of coloured water and scaling due to iron oxidation and calcium carbonate precipitation, respectively.9,27,28 Polyphosphates may form soluble complexes with lead, increasing solubility over a wide alkalinity range by maintaining low activity of the metal cation and undersaturation of the solubility-controlling phase.27 Polyphosphates have been shown to enhance metal release from lead pipes in laboratory studies.16,29 These have been corroborated by observations of elevated lead in systems dosing polyphosphates.22,30 Nevertheless, polyphosphates revert to orthophosphate over time, and blends could be effective in hard waters—where reversion is predicted to be relatively fast28—or when metals other than lead are preferentially complexed.29 Inhibition of copper release by polyphosphates has been attributed in previous work to orthophosphate reversion.31
We evaluated the effect of ortho- and polyphosphates on lead speciation and the effect of orthophosphate on lead release/solubility. Data were collected in three separate water distribution systems via point-of-use sampling. These systems were characterized by low alkalinity along with several risk factors for elevated lead release: (1) distributed water pH in the circumneutral range (pH 7.2–7.3), (2) distribution by unlined cast iron mains,32,33 and (3) in two systems, a chloride-to-sulfate mass ratio above the critical threshold of 0.5–0.77 identified in previous work as a driver of galvanic corrosion.34,35
2 Materials and methods
2.1 Study area
The study area comprised both single-unit residences and non-residential buildings that were known or suspected to be at risk for high lead levels in drinking water due to building age and the presence of lead service lines. Residential study sites were supplied by one of two water systems, denoted A and B. Non-residential sites were supplied by system C. Participating residences within system A had full (12 sites) or partial LSLs (6 sites, including one site with a public LSL and a private line of unknown composition). Among the sites with partial LSLs, none had recently undergone replacement. Within system B, service line configurations at participating residences were not known. However, based on a threshold of 1 μg Pb L−1 in flushed samples collected at near-peak water temperatures,36 six sites were suspected to have service lines composed at least partially of lead. Non-residential outlets in system C included fountains and faucets but none of the service line configurations were known.Sample sites in system A were supplied by cast iron distribution mains. In system B, two sites were supplied by cast iron, and the rest were supplied by either cement-lined mains or those of unknown composition. While unlined cast iron mains were common in system C, the composition of distribution mains supplying study sites was not known. Sample sites in systems A and B received distributed water from facilities described elsewhere.37 These facilities employ enhanced coagulation by alum and free chlorine disinfection. System C was supplied by a facility employing enhanced coagulation with poly-aluminum chloride, membrane microfiltration, and free chlorine disinfection. Treated water quality is summarized in Table 1 for systems A–C.38 Initially, all three systems applied blended ortho/polyphosphate corrosion inhibitors (systems A and B dosed a 3:1 and system C a 1:1 ortho:poly formulation). In September 2015 A and B switched to formulations that did not include polyphosphate, and in April 2016, A and B increased the orthophosphate dose from 0.5 to 1.0 mg PO4 L−1. Comparisons of lead release at 0.5 and 1.0 mg PO4 L−1 reflect the influence of orthophosphate without polyphosphate. SEC separations, on the other hand, compare two polyphosphate doses: 0.05 (system A) and 0.20 mg P L−1 (system C).
| Parameter | System A | System B | System Ca |
|---|---|---|---|
| a Values represent flushed samples collected at a central location within the distribution system. b Concentrations are discussed within the text. | |||
| Alkalinity (as CaCO3) | 22.5 mg L−1 | 19.0 mg L−1 | 13 mg L−1 |
| Free chlorine | 1.2 mg L−1 | 1.3 mg L−1 | <0.5 mg L−1 |
| Hardness (as CaCO3) | 10.1 mg L−1 | 32.0 mg L−1 | 5.0 mg L−1 |
| Total organic carbon | 1.6 mg L−1 | 1.8 mg L−1 | 1.9 mg L−1 |
| pH | 7.3 | 7.2 | 7.4 |
| Chloride | 8.9 mg L−1 | 7.5 mg L−1 | 7 mg L−1 |
| Sulfate | 7.9 mg L−1 | 25.0 mg L−1 | 3 mg L−1 |
| Turbidity | <0.10 NTU | <0.04 NTU | 0.5 NTU |
| Iron | <0.05 mg L−1 | <0.05 mg L−1 | <0.05 mg L−1 |
| Lead | <0.5 μg L−1 | <0.5 μg L−1 | <0.5 μg L−1 |
| Total phosphorus | —b | —b | 0.34 mg L−1 |
| Orthophosphate (as P) | —b | —b | 0.19 mg L−1 |
2.2 Sample collection
Following a random daytime protocol,18 we collected 250 mL samples at non-residential buildings in system C. In order to compare orthophosphate doses, residents supplied by systems A and B collected profiles of six sequential 1 L samples (L1–L6) from kitchen cold-water taps without preflushing. These began with the first draw following a minimum 6 hour standing period and were followed by a 10 minute flush of each outlet. Immediately after flushing, a seventh 1 L sample (L7) was collected. Profiles were collected in April 2016 at an orthophosphate dose of 0.5 mg PO4 L−1 (no polyphosphate), in June 2016 at an orthophosphate dose of 1.0 mg PO4 L−1, and at approximate two-month intervals through April 2017. Residents were instructed to record exact stagnation times, to sample at maximum flow, and not to remove faucet aerators. Data were excluded from analysis when these instructions were not followed; sample sizes differ by round for this reason and also due to incomplete resident participation. Polyethylene (HDPE) bottles and caps were immersed in 2 M reagent-grade HNO3 for a minimum of 24 h and rinsed thoroughly with ultrapure water prior to use. As described elsewhere,39 system A samples for SEC analysis were collected by residents as 4 × 1 L profiles.2.3 Size-exclusion chromatography with UV and ICP-MS detection
The size-exclusion chromatography ICP-MS (SEC-ICP-MS) and SEC-UV methods applied for the separation of field and experimental samples have been detailed in previous work.39 All paired SEC-UV and SEC-ICP-MS separations were performed on an agarose-dextran stationary phase (Superdex 200, 10 × 300 mm, 13 μm nominal bead size, GE Healthcare) with a 50 mM Tris-HCl mobile phase (pH 7.3) at a flow rate of 0.5 mL min−1. Injection volumes for SEC-UV and SEC-ICP-MS were 180 and 212 μL, respectively. Thyroglobulin (monitored as 127I) and ovalbumin (31P) were purchased from GE Healthcare and the humic isolate (monitored as 208Pb) from Alfa Aesar. Molecular weight determination for the humic isolate is described elsewhere.32 All samples were filtered at 0.45 μm prior to size-separation; recovery of lead by cellulose nitrate membrane filters was 78.9%.39 SEC analysis was performed within 36 h of sample collection.2.4 Scale analysis
Crystalline phases in lead pipe scale recovered from system A were identified using an X-ray diffractometer (Siemens D500) equipped with CuKα radiation (λ = 1.54 Å) at 35 kV and 30 mA. Scans were performed with a step size of 0.05° (2θ) and a count time of 10.0 s per step. Scale was collected as a single layer from the interior of a recovered LSL section. Samples were held in a desiccator for 24 h and then finely ground with a mortar and pestle prior to X-ray diffraction (XRD). To determine elemental composition, scale samples were digested according to EPA method 3050B,40 with modifications to accommodate the available sample mass and equipment. Digested samples were centrifuged at 3000 rpm for 10 m prior to dilution and analysis.2.5 Other analytical methods
Total lead, iron, copper, aluminum, and phosphorus were measured by ICP-MS (Thermofisher X Series II, Standard Methods 3125 and 3030).41 Reporting limits were 0.4, 6.0, 0.7, 4.0, and 4.9 μg L−1, respectively. Orthophosphate was measured in a subset of samples, over the first two sampling rounds only, using a colorimeter (Standard Method 4500-P E).41 Distributed water temperature data were obtained from the utility monitoring stations nearest the study sites.2.6 Data analysis
System A and B profiles collected before and after the increase in orthophosphate were paired by civic address. Metals concentrations in each of the seven litres were summed, and total masses over each profile were compared using two-tailed signed rank tests by follow-up round.42 Natural log transformations were applied to the paired data to promote symmetry in the distribution of differences. Multiplicative differences in lead levels were quantified using a Hodges–Lehmann estimator, c, where c = median(Aij) and Aij = (Di + Dj)/2 for all i = 1, …, n and j ≥ i. The variable D = ln(xi) − ln(yi) denotes the set of pairwise (log-transformed) differences, where x and y represent lead levels at n sites before and after the increase in orthophosphate to 1.0 mg PO4 L−1. The quantity c estimates the ratio of lead levels, where y = exp(c) x.433 Results and discussion
3.1 Lead sequestration by polyphosphate
The apparent size distributions of lead and iron were strongly influenced by the presence or absence of polyphosphate. This is illustrated in Fig. 1, via size-exclusion chromatograms representing typical separations of point-of-use samples collected in two water systems (A and C). These data provide little evidence of metal complexation within system A (Fig. 1a). Lead in particular was never observed at low apparent molecular weight—indicating complexation—in anything more than trace quantities. This is probably because polyphosphate in system A (dosed at 0.05 mg P L−1) reverted to orthophosphate during distribution; phosphorus in system A samples eluted in the same retention volume as the orthophosphate anion. Data from a pilot-scale model of system A46 were also consistent with the apparent reversion of polyphosphate in field samples: as a linear predictor, phosphorus in LSL effluent explained 99% of the variation in orthophosphate (β1 = 2.8 ± 0.3, 95% CI). Under the assumption of complete reversion, the expected slope (β1 = 3.1) was within the confidence bound of the linear model. Furthermore, SEC data provided assurance that polyphosphate complexation was not significant within the model system: lead eluted in an entirely separate fraction from phosphorus in typical separations.46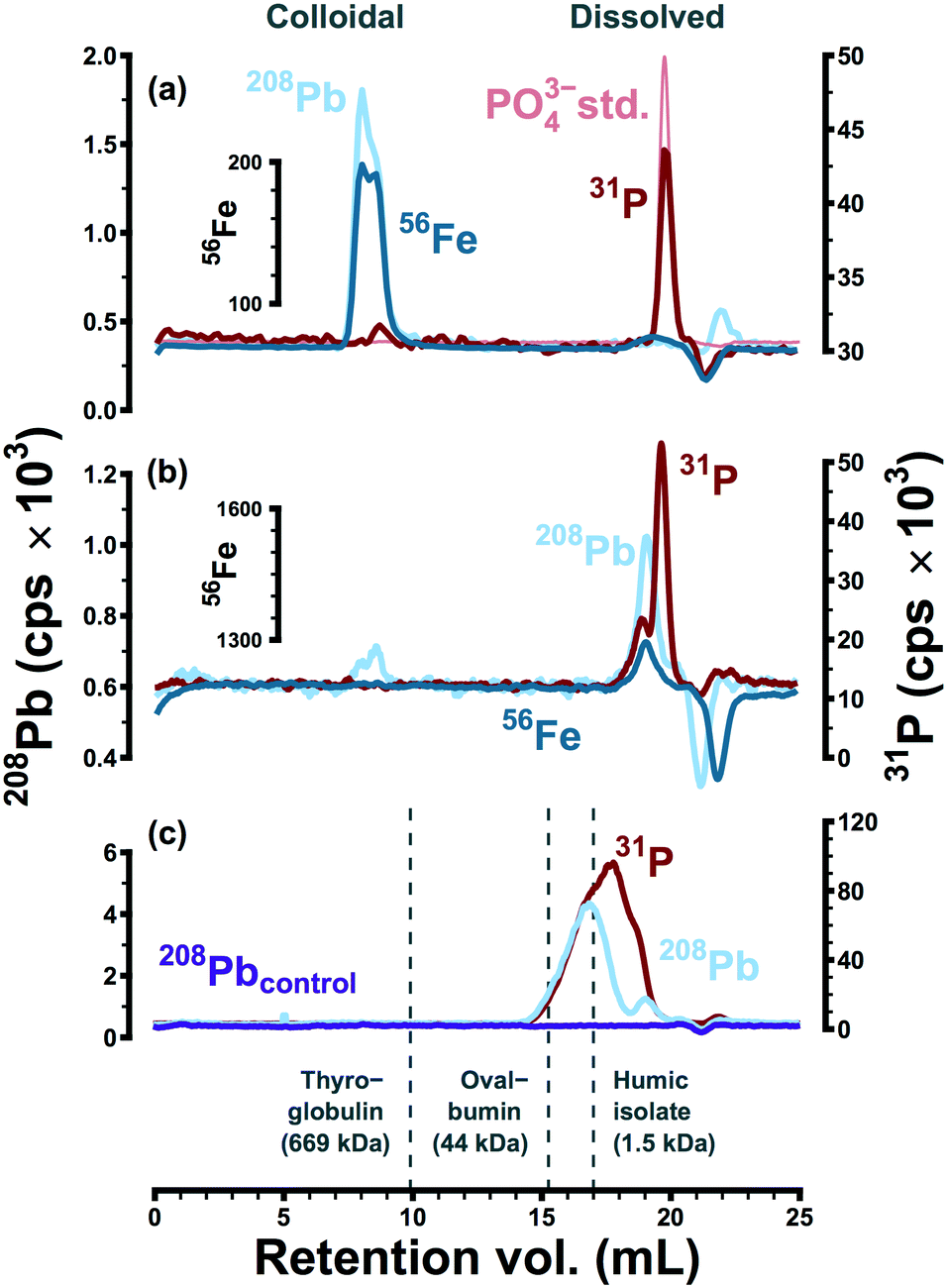 | ||
| Fig. 1 (a) In system A size-separations (<0.05 mg P L−1 polyphosphate), 208Pb eluted in a high molecular weight colloidal fraction—along with 56Fe—while 31P eluted in the same retention volume as the orthophosphate anion. (b) In system C size-separations (0.20 mg P L−1 polyphosphate), 208Pb and 56Fe eluted in a low molecular weight (dissolved) fraction along with 31P and consistent with sequestration by polyphosphate. (c) 208Pb and 31P coeluted at low apparent molecular weight in deionized water with sodium hexametaphosphate, a strong complexing agent for lead. Peak retention volumes of thyroglobulin (hydrodynamic diameter: 17 nm), ovalbumin (diameter: 6 nm), and a Pb-tagged humic isolate are provided as qualitative points of reference. | ||
In the absence of detectable polyphosphate (system A), lead and iron consistently coeluted as colloidal particles at high apparent molecular weight (Fig. 1a). These particles may have originated from an iron-rich lead corrosion scale or they may represent adsorption of lead to suspended colloidal iron (oxyhydr)oxides.39,44–46 Consistent with the occurrence of colloidal particles rich in these elements, filtration of system A field samples at 0.1 or 0.05 μm altered high molecular weight lead and iron peaks significantly and in near-identical fashion.39 Furthermore, high molecular weight lead and iron eluted consistently on stationary phases with opposite surface charges, while dissolved metal standards were retained completely at environmentally relevant concentrations.39 In previous work, lead and iron elution profiles and peak areas were highly correlated (Raverage2 = 0.96 and R2 = 0.82, respectively).39
Polyphosphate was persistent within system C: at points of use, only 55% of total phosphorus was present as orthophosphate (Table 1). Under the influence of polyphosphate (dosed at 0.20 mg P L−1), lead and iron were consistently observed at low molecular weight, strongly associated with the phosphorus signal (Fig. 1b). The apparent size distribution of phosphorus in system C was bimodal, and lead coeluted with the higher apparent molecular weight peak. This was interpreted as a consequence of sequestration/complexation: polyphosphates form stable coordination complexes with many metals, including lead,27 and polyphosphates can increase lead solubility due to complexation.16,27 Random daytime samples collected within system C had a 90th percentile total lead concentration of 33 μg L−1 (n = 14), and lead and iron concentrations in 0.45 μm filtrate ranged from 0.9–17.6 and >6.0 to 211.3 μg L−1, respectively.
The ability of SEC to detect lead-polyphosphate complexes was confirmed by separating deionized water with sodium bicarbonate (60 mg L−1), lead nitrate (65 μg Pb L−1), and sodium hexametaphosphate (2 mg P L−1), a strong complexing agent for lead. After 16 hours at pH 7.9, lead and phosphorus coeluted at low apparent molecular weight (Fig. 1c). Without sodium hexametaphosphate, lead was retained on the stationary phase as expected. While lead did elute in the presence of polyphosphate, lead and phosphorus peaks did not correspond entirely. This may have been due to the complex speciation of sodium hexametaphosphate.28 That is, the 31P signal may represent multiple poorly resolved peaks, only one of which was associated with 208Pb. Secondary interactions (e.g., charge exclusion) may also have been an important factor in observed molecular weight distributions. While high apparent molecular weight peaks representing colloidal metals were relatively consistent on stationary phases with opposite surface charges,39 soluble species may be more susceptible to electrostatic interactions with the stationary phase, as reported elsewhere in the case of iodide,47 vanadate (H2VO4−),48 and for a range of low molecular weight organic compounds.49
Given that increased lead solubility due to polyphosphate complexation is predicted over a wide alkalinity range,27 our speciation approach would be expected to apply to high alkalinity distributed waters. However, field data reported here represent low alkalinity conditions exclusively and further investigation would be required to validate the approach for high alkalinity water systems.
3.2 Complexation of lead by natural organic matter
Complexation of lead by humic or fulvic acids can also increase lead solubility by maintaining undersaturation of the solubility-controlling phase (e.g., hydrocerussite or hyroxypyromorphite).14 SEC with UV and multi-element detection has been used previously to characterize complexation of lead with NOM, and a strong correspondence between lead and UV absorption is typical in the presence of lead–NOM complexes.32,50,51 Previous size-separation of a humic isolate that contacted metallic lead over 48 h was consistent with formation of an organic lead complex.32 System A field data, in contrast, suggest that the strongest complexing fractions of NOM were removed during coagulation and filtration.52 In separations of system A samples, lead never eluted in the same fraction as the principal UV254 signal at appreciable signal-to-noise ratios.39 Complexation of lead by NOM within system C was considered unlikely as well: as in system A, removal of complexing NOM fractions via enhanced coagulation/filtration was expected. NOM may, however, have influenced lead mobility by promoting colloidal dispersion of lead-rich particles.53 This possibility is consistent with system A size-separations: colloidal lead and NOM (as UV254) consistently eluted together at high apparent molecular weight.393.3 Controlling lead solubility with orthophosphate
While polyphosphate in system C appeared to sequester/complex lead in solution, orthophosphate may have had the opposite effect in systems A and B by reducing lead solubility. Seven months after switching to an inhibitor formulation that did not include polyphosphate, the orthophosphate dose in these water systems was increased from 0.5 to 1.0 mg PO4 L−1. This change accompanied significant decreases in lead release (Fig. 2). | ||
| Fig. 2 System A and B lead concentrations at 1.0 mg PO4 L−1 were an estimated 67% of concentrations at 0.5 mg PO4 L−1 (paired measurements at 17 residences comparing Apr. 2016 with an average of Dec., Mar., and Apr. 2017). The gray shaded region represents the 95% CI around the group difference estimate of 0.67. | ||
Available data suggest that observed lead levels were a strong linear function of temperature within system A at 0.5 mg PO4 L−1 (raverage = 0.88).54 Group comparisons were structured to account for this source of variation: a significant reduction in lead was observed over the shortest interval for which temperature was approximately constant. December 2016 lead levels, at 1.0 mg PO4 L−1, were 62% of paired observations at 0.5 mg PO4 L−1 the previous April (Table 2, 95% CI: 49–85%). Lead levels in March and April 2017 were lower than paired observations in April 2016 by similar magnitudes: 68 and 71%, respectively. Averaging by site over the last three sampling rounds, lead levels at 1.0 mg PO4 L−1 were 67% of those observed at 0.5 mg L−1 (Fig. 2 and Table 2, final row, 95% CI: 54–82%). Despite the probable influence of seasonally varying water quality, variation in lead release over the 6 sampling rounds at 1.0 mg PO4 L−1 was generally low: standard deviations by site ranged from median 0.5 (L7) to 3.3 μg L−1 (L3) (systems A and B).
| Date (yyyy-mm) | % of initial Pb remaining | Distributed water temp. (°C) | % of initial Cu remaining | Paired sample size | ||
|---|---|---|---|---|---|---|
| Estimatea | 95% CI | Estimate | 95% CI | |||
| a Statistical significance: y: p < 0.01, z: p < 0.001. b Average of last three sampling rounds (if a study site was only sampled once over this period, this profile replaced the average in the calculation). | ||||||
| 2016-04 | — | — | 9.6 | — | — | — |
| 2016-06 | 91 | 67–120 | 15.1 | 79 y | 71–87 | 19 |
| 2016-08 | 89 | 64–130 | 21.8 | 73 z | 58–85 | 19 |
| 2016-10 | 79 | 43–110 | 16.7 | 73 z | 62–83 | 14 |
| 2016-12 | 62 y | 49–85 | 9.4 | 68 z | 57–80 | 16 |
| 2017-03 | 68 y | 55–87 | 6.4 | 67 y | 55–88 | 14 |
| 2017-04 | 71 y | 52–84 | 8.1 | 79 y | 65–93 | 15 |
| Averageb | 67 z | 54–82 | 8.0 | 71 z | 62–84 | 17 |
In agreement with the expected co-precipitation of lead and phosphate or the adsorption of phosphate to corrosion scale, phosphorus declined during stagnation. Precipitation of phosphate with copper is also possible: compared with the 0.5 mg PO4 L−1 dose, lower copper levels were observed at 1.0 mg PO4 L−1 (Table 2). Orthophosphate reduces copper release by promoting formation of relatively insoluble copper–phosphates, such as Cu3(PO4)2, over the more soluble cupric hydroxide (Cu(OH)2).55–57 Iron levels did not exhibit a negative trend (α = 0.05): orthophosphate has reduced iron release from corroded iron pipes,58,59 but in other cases it has increased or had little effect on iron levels in water.60 Orthophosphate demand at each study site was estimated as the largest difference between phosphorus in the flushed sample (L7) and each of the standing samples (L1–L6). By this estimate, median phosphorus demand at 0.5 mg PO4 L−1 was 13.3 μg L−1. Increasing orthophosphate accompanied higher phosphorus demand, from 59 μg L−1 in June to 35 μg L−1 the following April.
While increasing orthophosphate to 1.0 mg PO4 L−1 accompanied a decrease in lead release over the study sites as a group, peak lead levels remained high. The highest concentration at 0.5 mg PO4 L−1 was 58.8 μg L−1 and the highest at 1.0 mg PO4 L−1 was 60.3 μg L−1. At comparable pH and alkalinity levels to those characterizing systems A and B, equilibrium lead solubility has been predicted at approximately 30 and 20 μg L−1 at 0.5 and 1.0 mg PO4 L−1, respectively.10 Excess lead may be attributed to the presence of particles: particulate and colloidal lead in system A were predominant.39,54 Both equilibrium solubility predictions and observed lead levels suggest that orthophosphate might offer additional benefits at doses higher than 1.0 mg PO4 L−1. Due to the positive effect of carbonate complexation on lead solubility, systems distributing high alkalinity water are predicted to require higher orthophosphate doses than those reported here to achieve comparable effects.10
3.4 Identification of elements and crystalline phases in lead corrosion scale
Lead pipe corrosion scale collected from system A was characterized by a primary and a secondary crystalline phase: hydrocerussite and hydroxypyromorphite, respectively (Pb3(CO3)2(OH)2 and Pb5(PO4)3OH) (Fig. 3). The former phase is thermodynamically favoured under the expected pH and alkalinity conditions and the latter is expected under orthophosphate control of lead solubility. Elemental analysis was consistent with the presence of hydroxypyromorphite as a secondary phase: phosphorus represented 2.6% of scale mass while the theoretical phosphorus content of hydroxypyromorphite is 6.9%. Despite the presence of hydroxypyromorphite in scale, other phases may have also played a role in controlling lead solubility.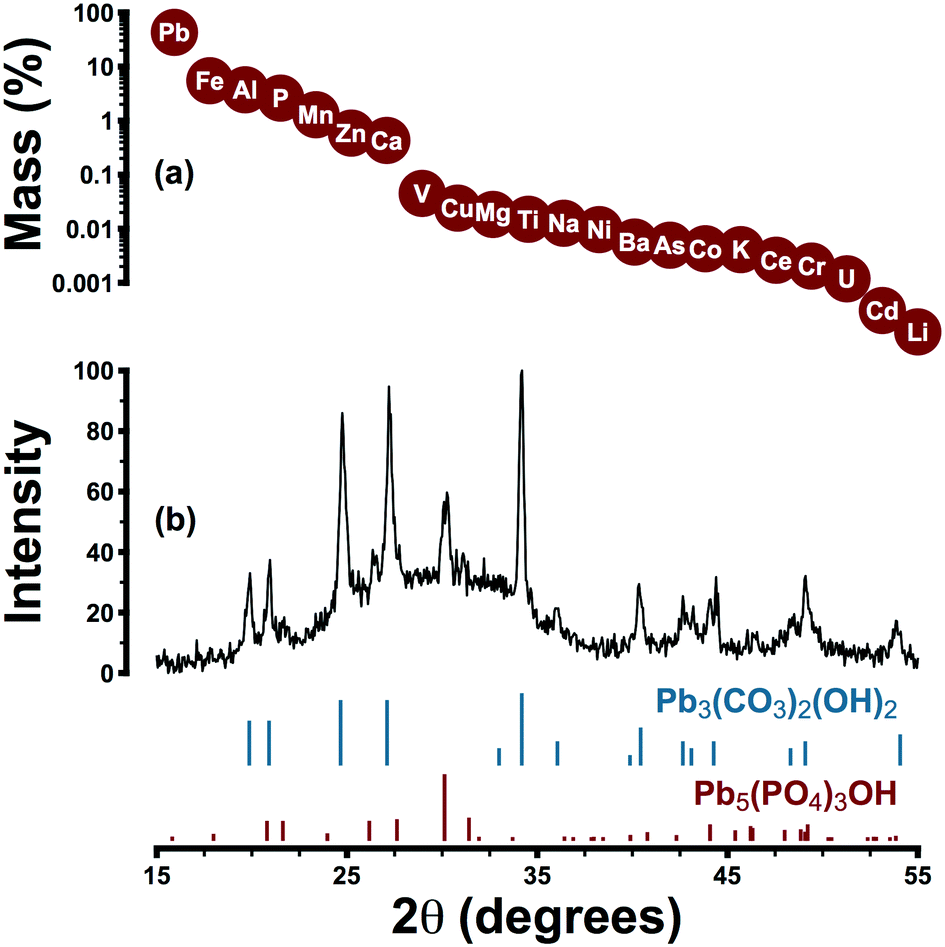 | ||
| Fig. 3 (a) Median mass fraction by element for triplicate scale samples representing an LSL recovered from system A. (b) XRD analysis was consistent with the presence of hydrocerussite, Pb3(CO3)2(OH)2, and hydroxypyromorphite, Pb5(PO4)3OH. | ||
The irregular baseline of the XRD pattern may be attributed to the amorphous content of the sample: iron, aluminum, manganese, zinc, and calcium were detected in amounts ranging from 0.4–5.5% (by mass) but were not present as distinct crystalline phases. Rather, these elements—especially iron and manganese—probably occurred as poorly crystalline (oxyhydr)oxides.61 While scale layers were aggregated for analysis, in place they were visually consistent with descriptions offered elsewhere: a layer composed predominantly of hydrocerussite beneath a surface layer of amorphous iron or manganese (oxyhydr)oxides.61 This stratification may explain the near-universal occurrence of colloidal particles rich in lead and iron in system A samples. Traces of vanadium, copper, nickel, barium, chromium, arsenic, and cadmium in scale samples are consistent with previous reports,61 and the zinc content (0.6%) was attributable to the phosphate formulation (zinc was present in distributed water at a median concentration of 65 μg L−1).
3.5 Potential influence of aluminum on lead release
Aluminum is a potential determinant of lead levels62,63 and may represent a source of seasonal variation. While median aluminum in system B was low and relatively constant at 8.3 μg L−1 with an interquartile range (IQR, lower to upper quartile) of 7.1–9.3 μg L−1, aluminum in system A varied seasonally, with higher levels occurring in winter (IQR: 37.8–125.3 μg L−1). At 0.5 mg PO4 L−1, median lead in system A first-draw samples was greater by 1.3 μg L−1 at the higher winter aluminum levels (signed rank test, paired data, n = 32 sites, 95% CI: 0.6–2.7 μg L−1).64 At these sites, first-draw sample temperatures were lower in winter by 0.7–4.3 °C (IQR).While the effect of aluminum on lead release is not well characterized, colloidal aluminum and lead in system A field samples—as determined by SEC-ICP-MS—were often present in a common size fraction.39 This may be explained by adsorption of lead to suspended colloidal iron and aluminum or to simultaneous release of these metals from corrosion scale. Lead adsorption to suspended particles could promote lead release by reducing the activity of lead in solution, prolonging undersaturation of the dissolving phase. Alternatively, aluminosilicate particles at pipe surfaces may buffer against increases in pH that would otherwise reduce lead solubility.63 In previous work,65 manganese appeared to inhibit formation of protective lead pipe scale (PbO2) in favour of an amorphous layer, and aluminum could also function in a similar manner. Conversely, some evidence suggests that aluminum deposits may act as a diffusion barrier to lead release.66
Despite the apparent effect of aluminum on lead release from premises plumbing, aluminum did not exert a primary influence on lead release over the full sample profile in system A. After controlling for changes in phosphorus, temperature, and iron via linear regression, aluminum was only weakly correlated with lead (r = 0.15). Nonetheless, relevant comparisons of lead release over time in system A were structured to minimize aluminum variation: the mass of aluminum over system A profiles in April 2017 (1.0 mg PO4 L−1) was 87% of the same mass measured in April 2016 (0.5 mg PO4 L−1, 95% CI: 78–96%).
4 Conclusion
This study demonstrated the use of size-exclusion chromatography with multi-element detection to better comprehend factors influencing lead mobility in drinking water systems. Orthophosphate was associated with colloidal particulate lead and iron in point-of-use samples and an overall reduction in lead release, while polyphosphate appeared to sequester lead as an aqueous complex. Previous studies have highlighted the risks of polyphosphate in systems with legacy lead plumbing, but these have largely relied on lab-scale experimental work or solubility modelling. Elemental speciation of environmental samples by SEC-UV-ICP-MS may allow for a more direct understanding of the potential for lead sequestration—by polyphosphate or NOM—in drinking water distribution systems.Conflicts of interest
The author's have no conflicts to declare.Acknowledgements
This work was funded by the Natural Sciences and Engineering Research Council (NSERC) Industrial Research Chair in Water Quality and Treatment (Grant No. IRCPJ 349838-11). We also acknowledge the technical support of Nicole Allward, Heather Daurie, Dr. Yuri Park, Michael Brophy, and water utility staff.References
- D. A. Cornwell, R. A. Brown and S. H. Via, J. - Am. Water Works Assoc., 2016, 108(4), E182–E191 CrossRef.
- G. C. Watt, A. Britton, W. H. Gilmour, M. R. Moore, G. D. Murray, S. J. Robertson and J. Womersley, Br. Med. J., 1996, 313(7063), 979–981 CrossRef CAS PubMed.
- M. Edwards, S. Triantafyllidou and D. Best, Environ. Sci. Technol., 2009, 43(5), 1618–1623 CrossRef CAS PubMed.
- M. Hanna-Attisha, J. LaChance, R. C. Sadler and A. Champney Schnepp, Am. J. Public Health, 2016, 106(2), 283–290 CrossRef PubMed.
- R. L. Canfield, C. R. Henderson Jr., D. A. Cory-Slechta, C. Cox, T. A. Jusko and B. P. Lanphear, N. Engl. J. Med., 2003, 348(16), 1517–1526 CrossRef CAS PubMed.
- B. P. Lanphear, K. Dietrich, P. Auinger and C. Cox, Public Health Rep., 2000, 115(6), 521–529 CrossRef CAS PubMed.
- M. L. Miranda, D. Kim, M. A. O. Galeano, C. J. Paul, A. P. Hull and S. P. Morgan, Environ. Health Perspect., 2007, 115(8), 1242–1247 CrossRef CAS PubMed.
- A. Evens, D. Hryhorczuk, B. P. Lanphear, K. M. Rankin, D. A. Lewis, L. Forst and D. Rosenberg, Environ. Health, 2015, 14(21), 1–9 CAS.
- L. S. McNeill and M. Edwards, J. - Am. Water Works Assoc., 2002, 94(7), 57–63 CrossRef CAS.
- M. R. Schock, J. - Am. Water Works Assoc., 1989, 81(7), 88–100 CrossRef CAS.
- J. D. Hopwood, R. J. Davey, M. O. Jones, R. G. Pritchard, P. T. Cardew and A. Booth, J. Mater. Chem., 2002, 12(6), 1717–1723 RSC.
- J. D. Noel, Y. Wang and D. E. Giammar, Water Res., 2014, 54, 237–246 CrossRef CAS PubMed.
- U. Svensson and W. Dreybrodt, Chem. Geol., 1992, 100(1–2), 129–145 CrossRef CAS.
- F. Debela, J. M. Arocena, R. W. Thring and T. Whitcombe, J. Environ. Manage., 2013, 116, 156–162 CrossRef CAS PubMed.
- C. E. Martinez, A. R. Jacobson and M. B. McBride, Environ. Sci. Technol., 2004, 38, 5584–5590 CrossRef CAS PubMed.
- M. Edwards and L. S. McNeill, J. - Am. Water Works Assoc., 2002, 94(1), 79–90 CrossRef CAS.
- C. Cartier, E. Doré, L. Laroche, S. Nour, M. Edwards and M. Prévost, Water Res., 2013, 47(2), 661–671 CrossRef CAS PubMed.
- C. R. Hayes, S. Incledion and M. Balch, J. Water Health, 2008, 6(2), 177–185 CrossRef CAS PubMed.
- P. T. Cardew, Water Res., 2003, 37(12), 2821–2832 CrossRef CAS PubMed.
- W. N. Richards and M. R. Moore, J. - Am. Water Works Assoc., 1984, 76(8), 60–67 CrossRef CAS.
- J. H. Colling, B. T. Croll, P. A. E. Whincup and C. Harward, Water Environ. J., 1992, 6(4), 259–268 CrossRef CAS.
- D. M. Dodrill and M. Edwards, J. - Am. Water Works Assoc., 1995, 87(7), 74–85 CrossRef CAS.
- R. G. Lee, W. C. Becker and D. W. Collins, J. - Am. Water Works Assoc., 1989, 81(7), 52–62 CrossRef CAS.
- P. C. Karalekas Jr., C. R. Ryan and F. B. Taylor, J. - Am. Water Works Assoc., 1983, 75(2), 92–95 CrossRef.
- K. J. Pieper, M. Tang and M. A. Edwards, Environ. Sci. Technol., 2017, 51(4), 2007–2014 CrossRef CAS PubMed.
- Y. Wang, V. Mehta, G. J. Welter and D. E. Giammar, J. - Am. Water Works Assoc., 2013, 105(7), E337–E351 CrossRef.
- T. R. Holm and M. R. Schock, J. - Am. Water Works Assoc., 1991, 83(7), 76–82 CrossRef CAS.
- T. R. Holm and M. Edwards, J. - Am. Water Works Assoc., 2003, 95(4), 172–178 CrossRef CAS.
- A. F. Cantor, D. Denig-Chakroff, R. R. Vela, M. G. Oleinik and D. L. Lynch, J. - Am. Water Works Assoc., 2000, 92(2), 95–102 CrossRef CAS.
- B. L. Ramaley, J. - Am. Water Works Assoc., 1993, 85(2), 64–67 CrossRef CAS.
- M. Edwards, L. Hidmi and D. Gladwell, Corros. Sci., 2002, 44(5), 1057–1071 CrossRef CAS.
- B. F. Trueman, G. A. Sweet, M. D. Harding, H. Estabrook, D. P. Bishop and G. A. Gagnon, Environ. Sci. Technol., 2017, 51(12), 6812–6820 CrossRef CAS PubMed.
- E. Camara, K. R. Montreuil, A. K. Knowles and G. A. Gagnon, J. - Am. Water Works Assoc., 2013, 105(8), E423–E431 CrossRef.
- M. Edwards and S. Triantafyllidou, J. - Am. Water Works Assoc., 2007, 99(7), 96–109 CrossRef CAS.
- C. K. Nguyen, B. N. Clark, K. R. Stone and M. A. Edwards, Corrosion, 2011, 67(6), 1–9 CrossRef.
- E. Deshommes, A. Bannier, L. Laroche, S. Nour and M. Prevost, J. - Am. Water Works Assoc., 2016, 108(11), E555–E570 CrossRef.
- L. E. Anderson, W. H. Krkosek, A. K. Stoddart, B. F. Trueman and G. A. Gagnon, Environ. Sci. Technol., 2017, 51(3), 1414–1422 CrossRef CAS PubMed.
- Halifax Regional Water Comission, 2015/2016 Annual Report, https://www.halifax.ca/sites/default/files/doc`uments/home-property/water/HalifaxWaterAnnualReport2016.pdf Search PubMed.
- B. F. Trueman and G. A. Gagnon, J. Hazard. Mater., 2016, 311, 151–157 CrossRef CAS PubMed.
- U.S. EPA, SW-846 Test Method 3050B: Acid Digestion of Sediments, Sludges, and Soils, https://www.epa.gov/sites/production/files/2015-12/documents/3050b.pdf Search PubMed.
- American Public Health Association, American Waterworks Association and Water Pollution Control Federation, Standard Methods For the Examination of Water and Wastewater, American Public Heath Association, Washington, DC, 22nd edn, 2012 Search PubMed.
- R Core Team, R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2017https://www.R-project.org/ Search PubMed.
- D. R. Helsel and R. M. Hirsch, Statistical Methods in Water Resources, Elsevier, Amsterdam, 1992, ch. 6, pp. 137–156 Search PubMed.
- S. Masters and M. Edwards, Environ. Eng. Sci., 2015, 32(5), 361–369 CrossRef CAS.
- M. McFadden, R. Giani, P. Kwan and S. H. Reiber, J. - Am. Water Works Assoc., 2011, 103(4), 76–89 CrossRef CAS.
- B. F. Trueman and G. A. Gagnon, Environ. Sci. Technol., 2016, 50(17), 9053–9060 CrossRef CAS PubMed.
- N. Kozai, T. Ohnuki and T. Iwatsuki, Water Res., 2013, 47(4), 1570–1584 CrossRef CAS PubMed.
- K. De Cremer, R. Cornelis, K. Strijckmans, R. Dams, N. Lameire and R. Vanholder, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 757(1), 21–29 CrossRef CAS.
- M. Edwards and M. M. Benjamin, J. - Am. Water Works Assoc., 1992, 84(6), 56–66 CrossRef CAS.
- L. Rottmann and K. G. Heumann, Anal. Chem., 1994, 66(21), 3709–3715 CrossRef CAS.
- F. Laborda, E. Bolea, M. P. Górriz, M. P. Martín-Ruiz, S. Ruiz-Beguería and J. R. Castillo, Anal. Chim. Acta, 2008, 606(1), 1–8 CrossRef CAS PubMed.
- G. V. Korshin, J. F. Ferguson, A. N. Lancaster and H. Wu, Corrosion and metal release for lead-containing materials: influence of NOM, AWWA Research Foundation, USA, 1999 Search PubMed.
- G. V. Korshin, J. F. Ferguson and A. N. Lancaster, Water Res., 2005, 39, 811–818 CrossRef CAS PubMed.
- B. F. Trueman, E. Camara and G. A. Gagnon, Environ. Sci. Technol., 2016, 50(14), 7389–7396 CrossRef CAS PubMed.
- S. H. Reiber, J. - Am. Water Works Assoc., 1989, 81(7), 114–122 CrossRef CAS.
- M. Edwards, L. Hidmi and D. Gladwell, Corros. Sci., 2002, 44(5), 1057–1071 CrossRef CAS.
- M. R. Schock and A. M. Sandvig, J. - Am. Water Works Assoc., 2009, 101(7), 71–82 CrossRef CAS.
- P. Sarin, J. A. Clement, V. L. Snoeyink and W. M. Kriven, J. - Am. Water Works Assoc., 2003, 95(11), 85–96 CrossRef CAS.
- D. A. Lytle, P. Sarin and V. L. Snoeyink, J. Water Supply: Res. Technol.--AQUA, 2005, 54(5), 267–281 CAS.
- L. S. McNeill and M. Edwards, J. Environ. Eng., 2000, 126(12), 1096–1102 CrossRef CAS.
- M. R. Schock, R. N. Hyland and M. M. Welch, Environ. Sci. Technol., 2008, 42(12), 4285–4291 CrossRef CAS PubMed.
- A. D. Knowles, C. K. Nguyen, M. A. Edwards, A. Stoddart, B. McIlwain and G. A. Gagnon, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2015, 50(4), 414–423 CrossRef CAS PubMed.
- S. Kvech and M. Edwards, J. - Am. Water Works Assoc., 2001, 93(11), 104–112 CrossRef.
- B. McIlwain, M.A.Sc. Thesis, Dalhousie University, 2013 Search PubMed.
- M. R. Schock, A. F. Cantor, S. Triantafyllidou, M. K. DeSantis and K. G. Scheckel, J. - Am. Water Works Assoc., 2014, 106(7), E336–E349 CrossRef.
- V. L. Snoeyink, M. R. Schock, P. Sarin, L. Wang, A. S. C. Chen and S. M. Harmon, J. Water Supply: Res. Technol.--AQUA, 2003, 52(7), 455–474 CAS.
| This journal is © The Royal Society of Chemistry 2018 |