
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkali metal complexes based on bisheterocyclomethanide ligands†‡
Johannes
Kretsch
,
Anne
Kreyenschmidt
,
Regine
Herbst-Irmer
 and
Dietmar
Stalke
*
and
Dietmar
Stalke
*
Institut für Anorganische Chemie, Universität Göttingen, Tammannstraße 4, 37077 Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de
First published on 12th June 2018
Abstract
A series of new alkali metal complexes containing deprotonated bisheterocyclomethanide were synthesised and structurally as well as spectroscopically characterised. Starting from the neutral bis-(benzoxazol-2-yl)methane ({NCOC6H4}2CH2), the complexes were obtained by facile deprotonation of the methylene bridge with nbutyllithium (nBuLi) or potassium hydride. Each of the resulting complexes [({NCOC6H4}2CH)Li(OEt2)2] (1), [({NCOC6H4}2CH)K(thf)]n (2) and [({NCOC6H4}2CH)K(18-crown-6)]n (3) (n → ∞) was analysed in the solid state by single crystal X-ray diffraction and in solution by various NMR techniques. All the compounds show a nearly planar ligand with the alkali metal ion chelated by the two ring nitrogen atoms of the conjugated C3N2 moiety. Furthermore, water NMR titration experiments revealed that both compounds 2 and 3 are examples of water-stable reactive organo potassium compounds. Due to their hydrolysis stability, these complexes are under further investigations in deep eutectic solvents.
Introduction
At the end of the 19th century and the early 20th century, Grignard reagents and organolithiums were developed.1 Today metal organics of the s-block have become the most commonly used compounds in the syntheses of organic non-natural and natural products.2 However, because of their highly polarized M–C, or in the case of metal hydride M–H bonds, these compounds are prone to hydrolysis and need to be handled under strictly anhydrous conditions. Air- and water-free reaction media are required. Stoichiometric or catalytic amounts of water are long known to be effective in increasing the reaction rate, in favouring a lithium/halogen exchange reaction or in affecting the stereochemistry of certain synthetic routes. On the other hand, the use of bulk water and other unconventional reaction media (e.g., deep eutectic solvents, glycerol) in the chemistry of Grignard and organolithium reagents is a recent scientific breakthrough that is heavily investigated.3In 1975, one of the first examples with an unexpected influence of water on the outcome of an alkali organometallic reaction was published by Taylor. It was observed that the most convenient method for the synthesis of tritiated arenes was the addition of nBuLi to a bromoarene in T2O wetted sodium-dried Et2O. The formation of labelled arenes was explained by a lithium/bromide exchange reaction that has to be at least as fast as, and most probably even faster than, the expected reaction between nBuLi and T2O.4
Another landmark was the lithiation of 2-mercaptobenzoxazole in the presence of N,N,N′,N′-tetramethylethylenediamine (TMEDA) and H2O and was reported by Wright and co-workers. The obtained complex [(C6H4OCSN)Li(TMEDA)(H2O)] shows a strong hydrogen bonding between one proton of a coordinated H2O molecule and the polarised sulfur atom of the anion rather than a protonated Li–OH⋯SH species.5 In 1992, Schleyer et al. presented the crystal structure of a lithium H2O-containing complex of an organic molecule bearing an acidic C–H unit. The addition of TMEDA and H2O to lithiated malonodinitrile in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 molar ratio led to the formation of [LiCH(CN)2(H2O)(TMEDA)]∞. This polymeric structure includes some interesting features e.g. the lack of TMEDA–Li interactions which is unprecedented for a bidentate ligand as well as the ability of each H2O molecule to simultaneously mould hydrogen bonding to TMEDA (donor function) and lithium ions (acceptor function).6 Recently we found [(18-crown-6)K{(4,6-tBu-OCNC6H2)2CH}·H2O] as a water-containing organopotassium complex based on the bulky deprotonated bis(4,6-tBu-benzoxazol-2-yl)methane. Its crystal structure revealed a rare example of a bis(benzoxazole-2-yl)methanide featuring solely O-coordination to a potassium ion. Furthermore, its unparalleled water-stability was investigated by NMR titration and DOSY spectroscopy. The complex shows full protonation only after seven days and 114 equivalents of water and might turn out to be a sustainable synthetic synthon in deep eutectic solvents.7,8
:1:1 molar ratio led to the formation of [LiCH(CN)2(H2O)(TMEDA)]∞. This polymeric structure includes some interesting features e.g. the lack of TMEDA–Li interactions which is unprecedented for a bidentate ligand as well as the ability of each H2O molecule to simultaneously mould hydrogen bonding to TMEDA (donor function) and lithium ions (acceptor function).6 Recently we found [(18-crown-6)K{(4,6-tBu-OCNC6H2)2CH}·H2O] as a water-containing organopotassium complex based on the bulky deprotonated bis(4,6-tBu-benzoxazol-2-yl)methane. Its crystal structure revealed a rare example of a bis(benzoxazole-2-yl)methanide featuring solely O-coordination to a potassium ion. Furthermore, its unparalleled water-stability was investigated by NMR titration and DOSY spectroscopy. The complex shows full protonation only after seven days and 114 equivalents of water and might turn out to be a sustainable synthetic synthon in deep eutectic solvents.7,8
In this paper, the preparation of one organolithium and two organopotassium compounds is presented. All complexes reported herein were characterised by single crystal X-ray diffraction. Additionally, the potassium complexes were tested for hydrolysis stability and monitored by NMR experiments.
Results and discussion
Synthesis and characterisation of [({NCOC6H4}2CH)Li(OEt2)2] (1)
Following similar protocols to those of Stalke9 and Roesky10 [({NCOC6H4}2CH)Li(OEt2)2] (1) was prepared by facile deprotonation of bis-(benzoxazol-2-yl)methane with nBuLi in diethyl ether (Et2O) at 0 °C (Scheme 1). Once the addition of nBuLi solution was complete the reaction mixture was stirred for 2 h at ambient temperature, volatiles were removed under reduced pressure and the lithiated species 1 was isolated in high yields (90%).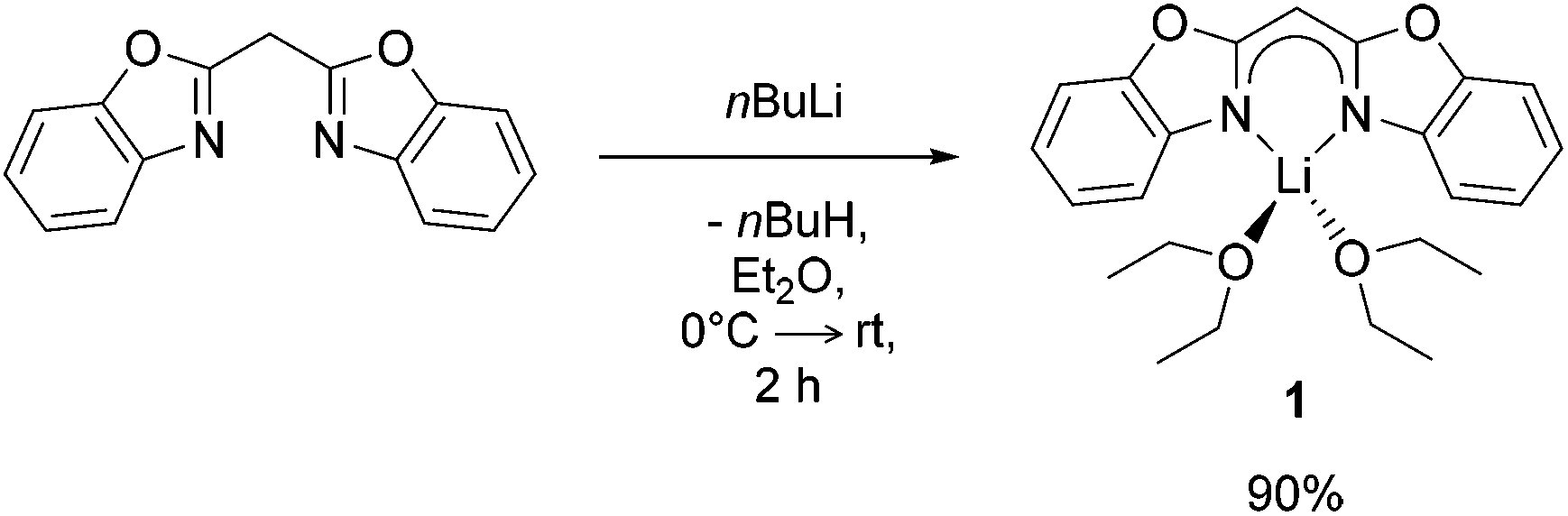 | ||
| Scheme 1 Synthesis of [({NCOC6H4}2CH)Li(OEt2)2] (1) by deprotonation with nBuLi solution in Et2O. | ||
Crystals of 1 suitable for X-ray diffraction analysis were grown from a saturated Et2O solution at 2 °C. It crystallises in the monoclinic space group P21/c with one molecule in the asymmetric unit. Mononuclear [({NCOC6H4}2CH)Li(OEt2)2] (1) (Fig. 1) consists of a lithium ion which is N,N-chelated by the negatively charged ligand and two diethyl ether molecules in a distorted tetrahedral fashion. The two imine nitrogen atoms of the bis-(benzoxazol-2-yl)methanide coordinate the metal ion equidistantly (1.995(2) Å for Li(1)–N(1) and 2.004(2) Å for Li(1)–N(2)). The bite angle N(1)–Li(1)–N(2) of 94.1(1)° is much more acute than the ideal tetrahedral angle. The oxygen atoms of the diethyl ether molecules coordinate at Li(1) in distances of 1.980(2) Å (Li(1)–O(3)) and 1.978(2) Å (Li(1)–O(4)), respectively. The included O(3)–Li(1)–O(4) angle of 103.0(1)° is much wider than the aforementioned angles. The nacnac-like planar structural C3N2 unit in [({NCOC6H4}2CH)Li(OEt2)2] (1) shows bond lengths between 1.54 Å (C(sp3)–C(sp3)) and 1.34 Å (C(sp2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(sp2)) as well as between 1.47 Å (C(sp3)–N(sp3)) and 1.29 Å (C(sp2)–N(sp2)), indicating a fully conjugated system extended throughout the virtually planar ligand.11
C(sp2)) as well as between 1.47 Å (C(sp3)–N(sp3)) and 1.29 Å (C(sp2)–N(sp2)), indicating a fully conjugated system extended throughout the virtually planar ligand.11
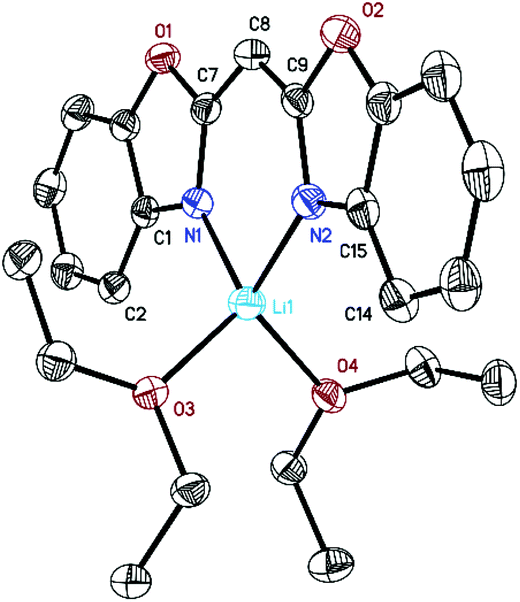 | ||
| Fig. 1 Molecular structure of [({NCOC6H4}2CH)Li(OEt2)2] (1). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Table 1 and the Experimental. | ||
| 1 | 2 | 3 | |
|---|---|---|---|
| N–M | 1.995(2), 2.004(2) | 2.762(2), 2.754(2) | 3.37(9) (sof 0.39(3)), 2.87(2) (sof 0.82(3)) |
| O–M | — | — | 3.42(5) (sof 0.61(3)), 3.0(1) (sof 0.18(3)) |
| C(7)–C(8) | 1.394(2) | 1.403(3) | 1.384(4) |
| C(8)–C(9) | 1.390(2) | 1.403(3) | 1.398(3) |
| C(7)–N(1) | 1.330(2) | 1.318(2) | 1.34(2) (sof 0.61(3)), 1.33(2) (sof 0.39(3)) |
| C(9)–N(2) | 1.333(2) | 1.324(2) | 1.320(9) (sof 0.82(3)), 1.35(3) (sof 0.18(3)) |
| N–M–N angle | 94.1(1) | 68.35(5) | 56(1) |
| M⋯C3N2 plane | 0.050(3) | 0.784(3) | — |
| Folding angle | 8.04(5) | 7.25(7) | 18.0(8) |
The lithium ion is only 0.050(3) Å dislocated from the central chelating C3N2 plane. This almost ideal in-plane arrangement is also observed in [({4,6-iPr-NCOC6H2}2CH)Li(thf)2]12 (0.00(3) to 0.07(5) Å). Furthermore, a butterfly folding angle of 8.04(5)° between both benzoxazole moieties indicates only a slightly bent ligand system. In addition, complex 1 was analysed by NMR spectroscopy. The 7Li NMR spectrum shows a signal at 2.19 ppm (thf-d8) which suggests the formation of a single lithium complex in solution. The 1H NMR spectrum of 1 reveals that in contrast to the uncharged ligand a de-shielding and simultaneous downfield shift from the methane to the methanide bridge from δ(–H2C–) = 4.70 ppm to δ(–HC(8)–) = 4.80 ppm occurs. The aromatic protons experience an up-field shift. This correlates with an increased electron density at these atoms due to charge delocalization from the C3N2 bridge towards the aryl systems in the ligand periphery and indicates mainly σ-bonding of the lithium cation to the electron-rich imine atoms.13
Synthesis and characterisation of potassium complexes 2 and 3
The syntheses of the closely related organopotassium compounds [({NCOC6H4}2CH)K(thf)]n (2) and [({NCOC6H4}2CH)K (18-crown-6)]n (3) (Scheme 2) can be achieved in a clean reaction of bis-(benzoxazol-2-yl)methane with potassium hydride. The deprotonation at the acidic methylene bridge indicated by the release of hydrogen and coordination of a potassium ion starts already at −30 °C. In the case of complex 2 thf was used as solvent, while the synthesis of 3 was conducted in toluene. Both reaction mixtures were allowed to warm to ambient temperature and stirred for about one day. After filtration and evaporation of all volatiles, both complexes were obtained as powders in good yields.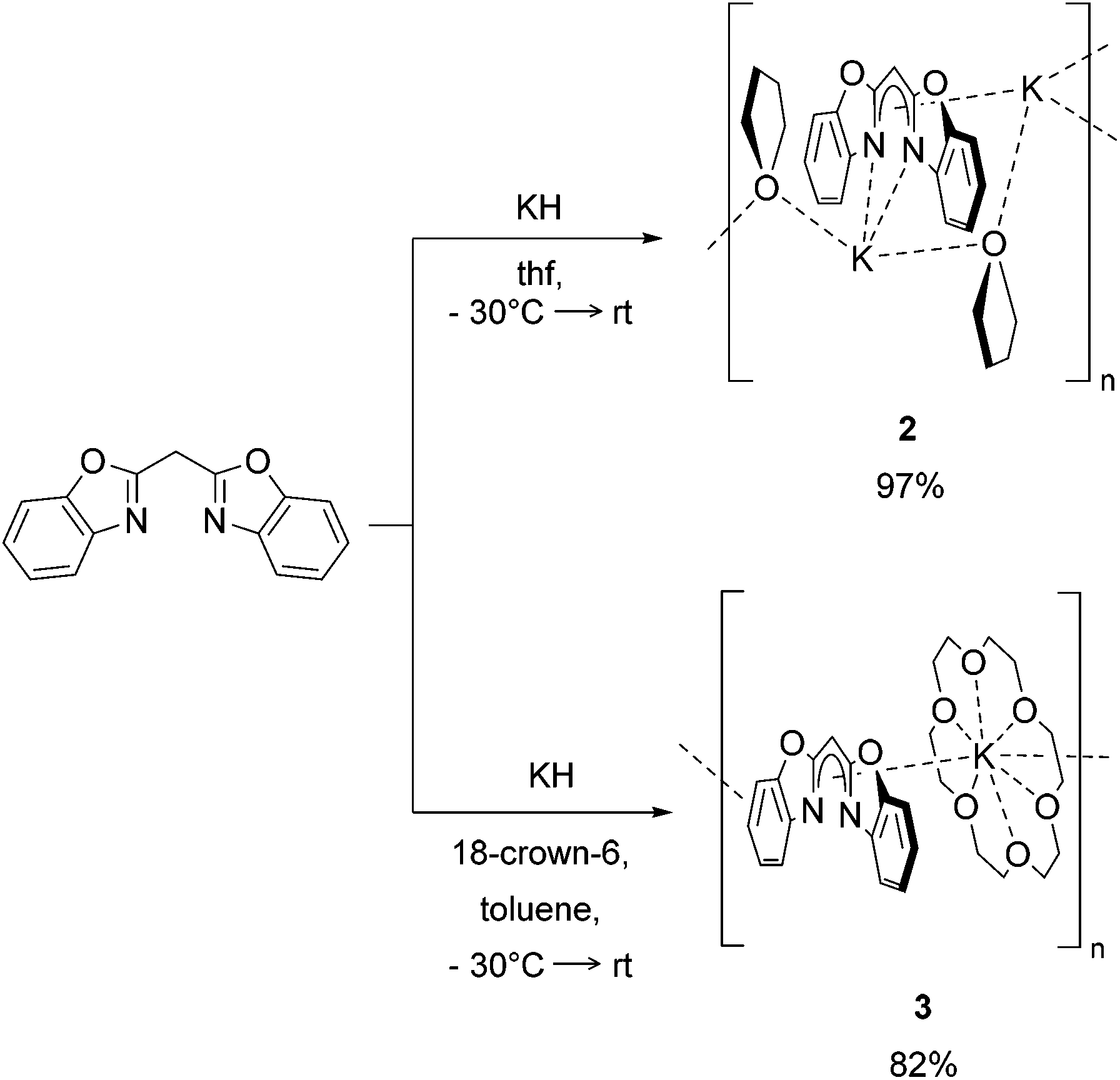 | ||
| Scheme 2 Syntheses of [({NCOC6H4}2CH)K(thf)] (2) and [({NCOC6H4}2CH)-K(18-crown-6)] (3) by deprotonation with KH. | ||
Solid-state structures of potassium complexes 2 and 3
Crystals suitable for single crystal X-ray diffraction were grown from a concentrated solution of the related solvents. [({NCOC6H4}2CH)K(thf)]n (2) formed yellowish crystalline needles in the orthorhombic space group Pna21 with one molecule in the asymmetric unit. It forms polymeric zig-zag strands with potassium ions N,N-σ-coordinated in the plane of one ligand and η5-C3N2 π-coordinated to a second one (Fig. 2). In addition, each potassium cation is coordinated to a μ-bridging thf molecule.14 Due to the formation of a conjugated π-system and the re-hybridisation of the carbon atoms at the methylene bridge from sp3 to sp2 upon deprotonation, a decrease in C–C bond lengths from 1.489(2) Å in the uncharged ligand ({NCOC6H4}2CH2)15 to 1.403(3) Å in C(8)–C(9) and C(7)–C(8) is observed. The opposite is true for the distances between N(1)–C(7) (1.318(2) Å) and N(2)–C(9) (1.324(2) Å), which elongate in comparison with the bis-(benzoxazol-2-yl)methane (1.286(2) Å).15 To facilitate the η5-coordination to a second ligand, the metal ion experiences a strong deviation of 0.784(3) Å from the coordinating C3N2 plane that was also shown for the corresponding potassium in the complex [({4-MeNCOC6H4}2CH)K(thf)] (1.33 Å).16 Structural features like the K–N distances (2.754(2), 2.762(2) Å) and the N–K–N bite angle (68.35(5)°) as well as the butterfly folding angle (7.25(7)°) between both benzoxazole moieties and best O(1)–N(1)–C(1–7) planes in regard to O(2)–N(2)–C(9–15) are also almost identical to those of [({4-MeNCOC6H4}2CH)K(thf)].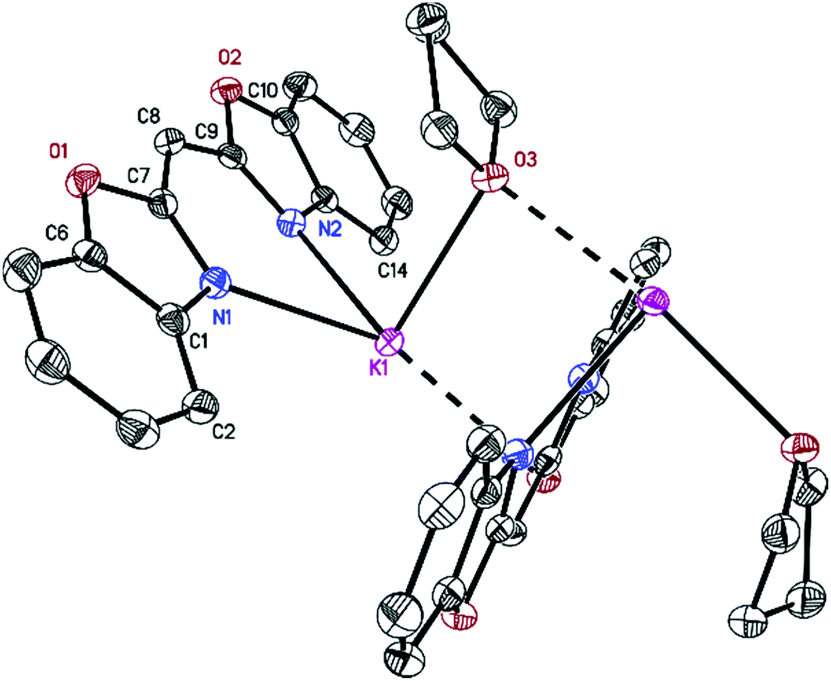 | ||
| Fig. 2 Polymeric structure of [({NCOC6H4}2CH)K(thf)]n (2). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms and the positional disorder are omitted for clarity. Structural data are given in Table 1 and the Experimental section. | ||
[({NCOC6H4}2CH)K(18-crown-6)]n (3) forms pale yellow crystalline needles in the orthorhombic space group Pca21 with one molecule in the asymmetric unit. Its solid-state structure shows the formation of a molecular contact ion pair (Fig. 3). Due to the high affinity of potassium to 18-crown-6 all six oxygen atoms of the crown coordinate to the cation. Additionally, a slipped η5-C3N2 π-coordination of the anionic ligand is formed to one side of the embraced metal, while the other is η2-coordinated to a benzyl moiety of an adjacent ligand, on this part already η5-C3N2 π-coordinated to another carbanion. In the π-coordination, the N(2)–C(9) bond is closer to the metal than the opposite N(1)–C(7) bond. Additionally, both positions are N/O disordered. In the closer N(2)/O(2)–C(9) residue (K(1)–N(2) 2.87(2), K(1)–O(2) 3.0(1) Å) the nitrogen has a much higher occupancy of 0.82(3) relative to only 0.18(3) oxygen, while in the far N(1)/O(1)–C(7) moiety (K(1)–N(1) 3.37(9), K(1)–O(1) 3.42(5) Å) the oxygen occupies 61(3)% of the site. In [({NCOC6H4}2CH)K(18-crown-6)]n (3) the potassium metal is in demand of the much better donor nitrogen17 different to the similar complex [({4,6-tBu-NCOC6H4}2CH)K(18-crown-6)·H2O]8 in which the metal is exclusively oxygen coordinated. In the latter, an additional lattice thf molecule provides density to the metal which is much more beneficial than the η2-coordination to the adjacent benzyl moiety in 3. Additionally, the rotation about the C–C bond in 3 is much facilitated because the benzoxazolyl group is not further substituted. The K(1)–O(18-crown-6) distance in the range of 2.751(2) to 2.995(2) Å is in good agreement with the average value of related compounds.18,19
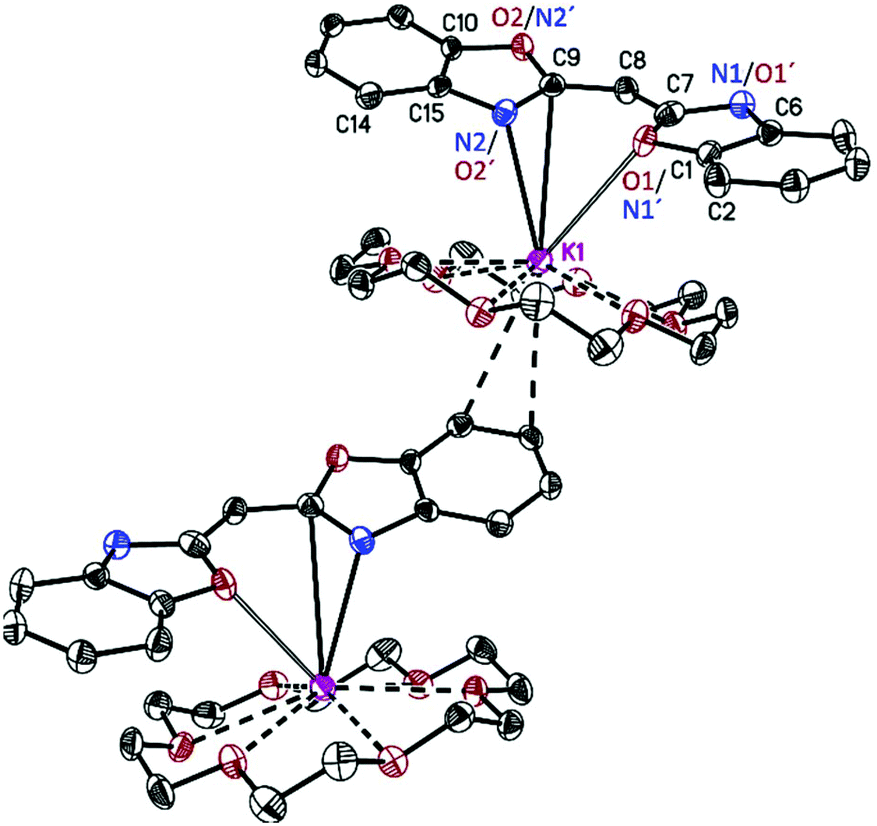 | ||
| Fig. 3 Polymeric structure of [({NCOC6H4}2CH)K(18-crown-6)]n (3). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Table 1 and the Experimental. | ||
NMR spectroscopic and hydrolysis studies of 2–3
The 1H NMR spectra (thf-d8) of complexes 2 and 3 (ESI‡) show a set of signals in the aromatic region from 7.06 to 6.57 ppm which can be assigned to 2-H to 5-H as well as to 11-H and 14-H of the ({NCOC6H4}2CH) ligand. These signals are shifted up-field in comparison with 7.69–7.27 ppm of the aromatic protons of the free uncharged ({NCOC6H4}2CH2) ligand. In particular, 2-H and 14-H experienced a significant deshielding that leads to a down-field shift of 7.03–7.01 ppm (2) and 7.06–7.03 ppm (3), respectively. In addition, the 1H NMR spectra of compounds 2 and 3 reveal that in relation to the protonated ({NCOC6H4}2CH2) a slight shielding and up-field shift of the methanide bridge Δδ = 0.60 ppm (δ(–HC(8)–) = 4.64 ppm, δ(–H2C–) = 4.70 ppm) is observed. Furthermore, multiplets which can be assigned to the thf molecules of [({NCOC6H4}2CH)K(thf)]n (2), occur at 4.64 ppm (O–CH2) and 1.77 ppm (–CH2). In contrast, the methylene functions of 18-crown-6 in 3 are observed at 3.46 ppm, which fits the chemical shift of related compounds.19 Fuelled by the unexpected stability of [({4,6-tBu-NCOC6H4}2CH)-K(18-crown-6)·H2O] towards hydrolysis we embarked on 1H NMR water titration experiments for compounds 2 and 3. Well-defined amounts of water (0.2 to 0.8 μL; 0.18 to 7.21 eq.) were added to a NMR sample and the effect of the successively increasing water content was monitored by 1H NMR spectroscopy. Both complexes [({NCOC6H4}2CH)K(18-crown-6)] (3) (ESI‡) and particularly surprisingly [({NCOC6H4}2CH)K(thf)] (2) are stable towards hydrolysis. Here the umbrella effect against hydrolysis recently observed by Gimbert et al. by complexing the lithium amide of 3-aminopyrrolidine with lithium halides or MeLi cannot be operational.20 It is shown in Fig. 4 that the addition of a vast excess of water has virtually no effect neither on the chemical shifts nor the signal line shape of complex 2. The water signal is first observed at 2.99 ppm (0.2 μL, 0.18 eq.) while shifting to 2.86 ppm (2.0–2.4 μL, 1.80–2.16 eq.). Even after adding 7.21 equivalents of water and waiting for one week still no protonation of 2 and formation of bis-(benzoxazol-2-yl)methane were detected. A slight deshielding and shift of 2-H and 14-H towards the down-field of Δδ = 0.06 ppm can be observed. Due to 1H NMR water titration experiments of 2 and 3, these compounds seem to be interesting examples of water-stable organopotassium compounds. Apparently the pKa of the ligand is on par with that of water. This is why the pKa values in this ligand family are of urgent interest and will be determined soon. Further experiments to exploit the reaction behaviour of alkali metal bis-(benzoxazol-2-yl)methanide complexes are on the way.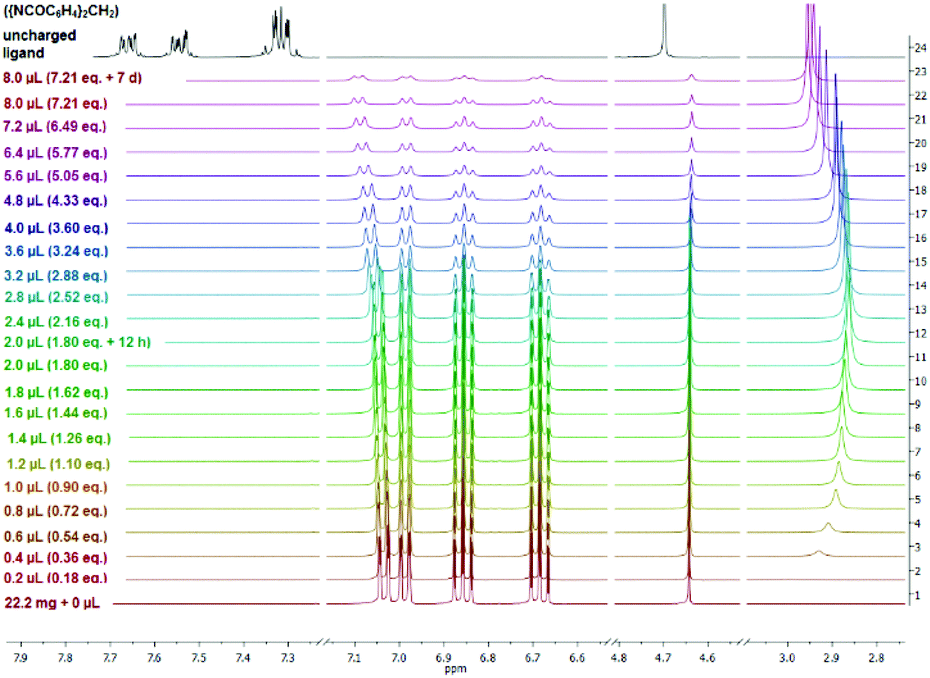 | ||
| Fig. 4 Stacked 1H NMR spectra (thf-d8) of a titration experiment of [({NCOC6H4}2CH)K(thf)]n (2) with water and a free uncharged ({NCOC6H4}2CH2) ligand (black). | ||
Conclusions
In this work the successful syntheses of three alkali metal bis-(benzoxazol-2-yl)-methanides 1–3 are presented. These complexes were obtained by the concerted deprotonation-metalation reaction of bis-(benzoxazol-2-yl)methane with typical alkali–metal reagents such as nBuLi or KH. Each of the resulting complexes [({NCOC6H4}2CH)Li(OEt2)2] (1), [({NCOC6H4}2CH)K(thf)]n (2) and [({NCOC6H4}2CH)K(18-crown-6)]n (3) (n → ∞) was characterised in the solid state by X-ray single diffraction and in solution by different NMR techniques. It was shown by 1H NMR water titration experiments that both organopotassium complexes 2 and 3 show surprising stability against hydrolysis. All complexes can be used as precursors for salt elimination and transmetalation reactions. Due to their hydrolysis stability, these complexes are under further investigations in deep eutectic solvents.Experimental
General procedures
All reactions were carried out under an atmosphere of N2 and Ar by Schlenk techniques.21 All solvents were distilled from Na or K before using for synthesis. Starting materials were purchased commercially and used without further purification. 1H, 13C NMR spectroscopic data were recorded on a Bruker Avance 401 MHz and a Bruker Avance 300 MHz spectrometer and referenced to the deuterated solvent (thf-d8). Deuterated solvents were dried over activated molecular sieves (3 Å) and stored in an argon dry box. Elemental analyses (C, H, N) were carried out on a Vario EL3 at the Mikroanalytisches Labor, Institut für Anorganische Chemie, University of Göttingen. LIFDI-MS spectra were measured on a Jeol AccuTOF spectrometer.Crystal data for 1 at 100(2) K: C23H29LiN2O4, Mr = 404.42 g mol−1, 0.467 × 0.352 × 0.265 mm, monoclinic, P21/c, a = 17.284(3) Å, b = 9.272(2) Å, c = 13.883(2) Å, β = 97.03(2)°, V = 2208.1(8) Å3, Z = 4, μ(Mo Kα) = 0.082 mm−1, Θmax = 25.547°, 24547 reflections measured, 4129 independent (Rint = 0.0271), R1 = 0.0334 [I > 2σ(I)], wR2 = 0.0816 (all data), res. density peaks: 0.210 to −0.155 e Å−3.
Crystal data for 2 at 100(2) K: C19H17KN2O3, Mr = 360.44 g mol−1, 0.364 × 0.177 × 0.126 mm, orthorhombic, Pna21, a = 12.200(2) Å, b = 19.897(3) Å, c = 7.745(2) Å, V = 1725.9(6) Å3, Z = 4, μ(Mo Kα) = 0.328 mm−1, Θmax = 26.111°, 26470 reflections measured, 3432 independent (Rint = 0.0238), R1 = 0.0210 [I > 2σ(I)], wR2 = 0.0566 (all data), absolute structure parameter 0.04(1),22 res. density peaks: 0.184 to −0.250 e Å−3.
Crystal data for 3 at 100(2) K: C27H33KN2O8, Mr = 552.65 g mol−1, 0.226 × 0.172 × 0.098 mm, orthorhombic, Pca21, a = 15.913(3) Å, b = 10.981(2) Å, c = 15.309(3) Å, V = 2675.1(9) Å3, Z = 4, μ(Mo Kα) = 0.251 mm−1, Θmax = 26.427°, 33321 reflections measured, 5489 independent (Rint = 0.0478), R1 = 0.0343 [I > 2σ(I)], wR2 = 0.0727 (all data), absolute structure parameter 0.01(2),22 res. density peaks: 0.288 to −0.268 e Å−3.
Shock cooled crystals were selected from a Schlenk flask under an argon atmosphere using the X-TEMP2 device.23 The data were collected with an Mo-IμS microfocus source.24 All data were integrated with SAINT25 and a multiscan absorption correction (SADABS)26 was applied. For structure 2, a 3λ correction was applied.27 The structures were solved by direct methods (SHELXT)28 and refined on F2 using the full-matrix least-squares methods of SHELXL29 within the SHELXLE GUI.30 The Crystallographic Information Files (CIF) using the reference numbers 1839909, 1839910 and 1839911.
[({NCOC6H4}2CH)Li(OEt2)2] (1)
Bis-(benzoxazol-2-yl)methane (530 mg, 2.12 mmol, 1.00 eq.) was dissolved in Et2O (8 mL) and cooled to 0 °C. At this temperature, a solution of nBuLi (921 μL, 136 mg, 2.12 mmol, 1.00 eq.) in hexane was slowly added. After the reaction mixture was stirred for 2 h at room temperature, the solvent was removed under reduced pressure and a yellowish-white powder (771 mg, 1.91 mmol, 90%) was obtained. Crystals suitable for single crystal X-ray diffraction were grown from a saturated Et2O solution at 2 °C.1H NMR (300 MHz, thf-d8, ppm): 7.13–7.10 (m, 2 H, 2-H, 14-H), 7.09–7.06 (m, 2 H, 5-H, 11-H), 7.01–6.95 (m, 2 H, 3-H, 13-H), 6.84–6.79 (m, 2 H, 4-H, 14-H), 4.80 (s, 1 H, 8-H), 3.39 (q, 3JHH = 7.0 Hz, 8 H, CH2 Et2O), 1.12 (t, 3JHH = 7.0 Hz, 12 H, CH3 Et2O); 13C NMR (75 MHz, thf-d8, ppm): 171.14 (C-7, C-9), 150.33 (C-6, C-10), 145.46 (C-1, C-15), 123.52 (C-3, C-13), 119.67 (C-4, C-12), 113.36 (C-5, C-11), 108.54 (C-2, C-14), 66.38 (CH2 Et2O), 57.38 (C-8), 15.74 (CH3 Et2O); 7Li NMR (116 MHz, toluene-d8, ppm): 2.19; MS (LIFDI[+], toluene): m/z = 256.08 (100) [M − 2Et2O]+, 506.1 (4) [M + ({NCOC6H4}2CH2)]+; C15H9AlI2N2O2: C 68.31; H 7.23; N 6.93%; found C 68.20; H 6.68; N 7.58%.
[({NCOC6H4}2CH)K(thf)] (2)
Bis-(benzoxazol-2-yl)methane (49.3 mg, 197 μmol, 1.00 eq.) was dissolved in thf (4 mL) and cooled to −30 °C. At this temperature potassium hydride (15 mg, 374 μmol, 1.90 eq.) was added. While stirring, the reaction mixture was warmed to room temperature. After 18 h, the solution was filtered, and solvents were removed under reduced pressure. Crystals from the yellowish solid (55 mg, 191 μmol, 97%) were grown from a concentrated solution of 2 in thf. 1H NMR (300 MHz, thf-d8, ppm): 7.03–7.01 (m, 2 H, 2-H, 14-H), 7.00–6.97 (m, 2 H, 5-H, 11-H), 6.86–6.83 (m, 2 H, 3-H, 13-H), 6.71–6.65 (m, 2 H, 4-H, 12-H), 4.64 (s, 1 H, 8-H), 3.62 (m, 4 H, O–CH2 thf), 1.77 (m, 4 H, CH2 thf); 13C NMR (75 MHz, thf-d8, ppm): 170.94 (C-7, C-9), 150.53 (C-6, C-10), 147.86 (C-1, C-15), 122.69 (C-3, C-13), 118.61 (C-4, C-12), 113.29 (C-2, C-14), 107.76 (C-5, C-11), 68.40 (O–CH2 thf), 56.98 (C-8), 26.55 (CH2 thf); MS (LIFDI[+], toluene): m/z = 249.0 (11) [M − K − thf]+, 288.0 (100) [M − thf]+, 327.0 (28) [M + K − thf]+; a satisfactory CHN analysis could not be obtained because the coordinating thf leaves the lattice and gave varying not reproducible results.[({NCOC6H4}2CH)K(18-crown-6)] (3)
Bis-(benzoxazol-2-yl)methane (100 mg, 400 μmol, 1.00 eq.) was dissolved in toluene (5 mL) and cooled to −30 °C. At this temperature potassium hydride (24 mg, 600 μmol, 1.50 eq.) was added. While stirring, the reaction mixture was warmed to room temperature. After 24 h, the solution was filtered, and solvents were removed under reduced pressure. Crystals suitable for X-ray diffraction were grown from a concentrated solution of 3 (181 mg, 328 μmol, 82%) in toluene. 1H NMR (300 MHz, thf-d8, ppm): 7.06–7.03 (m, 2 H, 2-H, 14-H), 6.98–6.96 (m, 2 H, 5-H, 11-H), 6.80–6.75 (m, 2 H, 5-H, 11-H), 6.62–6.57 (m, 2 H, 4-H, 12-H), 4.64 (s, 1 H, 8-H), 3.46 (m, 24 H, 18-crown-6); 13C NMR (75 MHz, thf-d8, ppm): 170.74 (C-7, C-9), 151.06 (C-6, C-10), 148.35 (C-1, C-15), 122.41 (C-3, C-13), 117.41 (C-4, C-12), 114.06 (C-2, C-14), 107.12 (C-5, C-11), 70.91 (18-crown-6), 57.34 (C-8); MS (LIFDI[+], toluene): m/z = 303.1 (100) [M − ({NCOC6H4}2CH)]+; MS (ESI[+], thf): m/z = 303.1 (100) [M − ({NCOC6H4}2CH)]+; MS (ESI[−], thf): m/z = 249.1 (100) [M − K@18-crown-6]−, 513.2 (12) [M − K]−, 521.2 (21) [M]−; C27H33KN2O2: C 56.86; H 6.02; N 5.07%; found C 56.96; H 5.65; N 5.70%.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Danish National Research Foundation (DNRF93) funded Center for Materials Crystallography (CMC) for partial support and the Federal State of Lower Saxony, Germany, for providing fellowships in the GAUSS PhD programmes.Notes and references
- (a) RMgX: V. Grignard, C. R. Hebd. Seances Acad. Sci., 1900, 130, 1322–1324 Search PubMed; (b) RLi: W. Schlenk and J. Holtz, Ber. Dtsch. Chem. Ges., 1917, 50, 262–274 CrossRef; (c) J. J. Eisch, Organometallics, 2002, 21, 5439–5463 CrossRef; (d) D. Seyferth, Organometallics, 2009, 28, 1598–1605 CrossRef; (e) Lithium compounds in organic synthesis. From fundamentals to applications, ed. R. Luisi and V. Capriati, Wiley, Weinheim, 2014 Search PubMed.
- (a) R. E. Mulvey and S. D. Robertson, Angew. Chem., 2013, 125, 11682–11700 CrossRef; (b) R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2013, 52, 11470–11487 CrossRef PubMed; (c) T. L. Rathman and J. A. Schwindeman, Org. Process Res. Dev., 2014, 18, 1192–1210 CrossRef; (d) V. Capriati, F. M. Perna and A. Salomone, Dalton Trans., 2014, 43, 14204–14210 RSC; (e) T. P. Hanusa, The Lightest Metals. Science and Technology from Lithium to Calcium, Wiley, 1st edn, 2015 Search PubMed; (f) E. Carl and D. Stalke, Structure-Reactivity Relationship in Organolithium Compounds in Lithium Compounds in Organic Synthesis – From Fundamentals to Applications, Wiley-VCH, Weinheim, 1st edn, 2014 Search PubMed.
- (a) V. Mallardo, R. Rizzi, F. C. Sassone, R. Mansueto, F. M. Perna, A. Salomone and V. Capriati, Chem. Commun., 2014, 50, 8655–8658 RSC; (b) C. Vidal, J. Garcia-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., 2014, 53, 5969–5973 CrossRef PubMed; (c) J. García-Álvarez, E. Hevia and V. Capriati, Eur. J. Org. Chem., 2015, 6779–6799 CrossRef; (d) F. C. Sassone, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Commun., 2015, 51, 9459–9462 RSC; (e) L. Cicco, S. Sblendorio, R. Mansueto, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Sci., 2016, 7, 1192–1199 RSC; (f) C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., 2016, 55, 16145–16148 CrossRef PubMed; (g) L. Cicco, M. J. Rodríguez-Álvarez, F. M. Perna, J. García-Álvarez and V. Capriati, Green Chem., 2017, 19, 3069–3077 RSC; (h) G. Dilauro, M. Dell'Aera, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2017, 56, 10200–10203 CrossRef PubMed; (i) G. Dilauro, M. Dell'Aera, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., 2017, 129, 10334–10337 CrossRef; (j) M. J. Rodríguez-Álvarez, J. García-Álvarez, M. Uzelac, M. Fairley, C. T. O'Hara and E. Hevia, Chem. – Eur. J., 2018, 24, 1720–1725 CrossRef PubMed.
- R. Taylor, Tetrahedron Lett., 1975, 16, 435–436 CrossRef.
- D. Barr, P. R. Raithby, P. v. R. Schleyer, R. Snaith and D. S. Wright, J. Chem. Soc., Chem. Commun., 1990, 22, 643–645 RSC.
- (a) C. Lambert, P. v. R. Schleyer, U. Pieper and D. Stalke, Angew. Chem., Int. Ed. Engl., 1992, 31, 77–79 CrossRef; (b) C. Lambert, P. v. R. Schleyer, U. Pieper and D. Stalke, Angew. Chem., 1992, 104, 78–79 CrossRef.
- I. Koehne, S. Bachmann, T. Niklas, R. Herbst-Irmer and D. Stalke, Chem. – Eur. J., 2017, 23, 13141–13149 CrossRef PubMed.
- (a) I. Koehne, S. Bachmann, R. Herbst-Irmer and D. Stalke, Angew. Chem., Int. Ed., 2017, 56, 15141–15145 CrossRef PubMed; (b) I. Koehne, S. Bachmann, R. Herbst-Irmer and D. Stalke, Angew. Chem., 2017, 129, 15337–15342 CrossRef.
- T. Kottke and D. Stalke, Chem. Ber./Recl., 1997, 130, 1365–1374 CrossRef.
- J. Prust, K. Most, I. Müller, A. Stasch, H. W. Roesky and I. Usón, Eur. J. Inorg. Chem., 2001, 2001, 1613–1616 CrossRef.
- P. Rademacher, Strukturen organischer Moleküle, VCH, Weinheim, 2005.
- I. Koehne, N. Graw, T. Teuteberg, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2017, 56, 14968–14978 CrossRef PubMed.
- F. Engelhardt, C. Maaß, D. M. Andrada, R. Herbst-Irmer and D. Stalke, Chem. Sci., 2018, 9, 3111–3121 RSC.
- S. Brooker, F. T. Edelmann, T. Kottke, H. W. Roesky, G. M. Sheldrick, D. Stalke and K. H. Whitmire, J. Chem. Soc., Chem. Commun., 1991, 325, 144–146 RSC.
- D.-R. Dauer and D. Stalke, Dalton Trans., 2014, 43, 14432–14439 RSC.
- I. Koehne, R. Herbst-Irmer and D. Stalke, Eur. J. Inorg. Chem., 2017, 2017, 3322–3326 CrossRef.
- (a) F. Ragaini, M. Pizzotti, S. Cenini, A. Abbotto, G. A. Pagani and F. Demartin, J. Organomet. Chem., 1995, 489, 107–112 CrossRef; (b) A. Abbotto, S. Bruni, F. Cariati and G. A. Pagani, Spectrochim. Acta, Part A, 2000, 56, 1543–1552 CrossRef; (c) H. Arii, M. Matsuo, F. Nakadate, K. Mochida and T. Kawashima, Dalton Trans., 2012, 41, 11195–11200 RSC; (d) B. D. Ward and L. H. Gade, Chem. Commun., 2012, 48, 10587–10599 RSC.
- (a) C. Schade and P. v. R. Schleyer, Adv. Organomet. Chem., 1987, 27, 169–278 CrossRef; (b) J. D. Smith, Adv. Organomet. Chem., 1999, 43, 267–348 CrossRef.
- (a) R. W. Saalfrank, N. Löw, S. Kareth, V. Seitz, F. Hampel, D. Stalke and M. Teichert, Angew. Chem., Int. Ed., 1998, 37, 172–175 CrossRef; (b) R. W. Saalfrank, N. Löw, S. Kareth, V. Seitz, F. Hampel, D. Stalke and M. Teichert, Angew. Chem., 1998, 110, 182–184 CrossRef.
- Y. Gimbert, D. Lesage, C. Fressigné and J. Maddaluno, J. Org. Chem., 2017, 82, 8141–8147 CrossRef PubMed.
- Virtuelles Labor I, http://www.stalke.chemie.uni-goettingen.de/virtuelles_labor/advanced/13 de.html.
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 249–259 CrossRef PubMed.
- (a) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef; (b) D. Stalke, Chem. Soc. Rev., 1998, 27, 171 RSC; (c) Virtuelles Labor III, http://www.stalke.chemie.uni-goettingen.de/virtuelles_labor/special/22_de.html.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef.
- SAINT v8.30C. Bruker Apex CCD, Bruker AXS Inc., Madison, WI, USA, 2013 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef PubMed.
- L. Krause, R. Herbst-Irmer and D. Stalke, J. Appl. Crystallogr., 2015, 48, 1907–1913 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 238–240 CrossRef PubMed.
Footnotes |
| † Dedicated to Prof. Bernt Krebs on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Representation of 1H, 7Li, 13C NMR spectra of 1, 1H, 13C NMR spectra of 2 and 3, stacked 1H NMR spectra of a titration experiment of 2 and 3. LIFDI spectrometry data of compounds 1–3. Crystallographic data of compounds 1–3. CCDC 1839909–1839911. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt01678j |
| This journal is © The Royal Society of Chemistry 2018 |