
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homologation chemistry with nucleophilic α-substituted organometallic reagents: chemocontrol, new concepts and (solved) challenges
Laura
Castoldi
,
Serena
Monticelli
,
Raffaele
Senatore
,
Laura
Ielo
and
Vittorio
Pace
 *
*
University of Vienna, Department of Pharmaceutical Chemistry, Althanstrasse, 14, A-1090, Vienna, Austria. E-mail: vittorio.pace@univie.ac.at
First published on 17th May 2018
Abstract
The transfer of a reactive nucleophilic CH2X unit into a preformed bond enables the introduction of a fragment featuring the exact and desired degree of functionalization through a single synthetic operation. The instability of metallated α-organometallic species often poses serious questions regarding the practicability of using this conceptually intuitive and simple approach for forming C–C or C–heteroatom bonds. A deep understanding of processes regulating the formation of these nucleophiles is a precious source of inspiration not only for successfully applying theoretically feasible transformations (i.e. determining how to employ a given reagent), but also for designing new reactions which ultimately lead to the introduction of molecular complexity via short experimental sequences.
 Laura Castoldi | Laura Castoldi (Pavesako) obtained her master's degree in Pharmaceutical Chemistry and Technology in 2011 from the University of Pavia, followed by a postgraduate master's in Drug Design and Development. She received a doctoral degree in 2018 at the University of Vienna (Austria) under the guidance of Priv. Doz. Dr V. Pace and Prof. W. Holzer. She was a visiting PhD student at the Complutense University of Madrid (Spain, Prof. A. R. Alcántara). Her main research activities focused on the development of new synthetic strategies in homologation reactions with lithium carbenoids. |
 Serena Monticelli | Serena Monticelli received a degree cum laude in Pharmacy in 2013 from the University of Bari. In 2015, she obtained a postgraduate master's in Drug Design and Development from the University of Pavia. Later on, she started doctoral studies at the University of Vienna, Austria under the supervision of V. Pace and T. Langer working on organometallic chemistry. Her research interests include the use of organolithium and organomagnesium methods with vistas to applications in synthetic medicinal chemistry. |
 Raffaele Senatore | Raffaele Senatore received a master's degree in Pharmacy in 2015 from the University of Perugia. In 2016 he was awarded with the OeAD Ernst Mach postgraduate fellowship to join Pace's group at the University of Vienna. In 2017, he started his PhD with Dr Vittorio Pace and Prof. E. Urban, being currently a University Assistant at the Department of Pharmaceutical Chemistry. His principal scientific interests include organometallic chemistry and NMR elucidation studies. |
 Laura Ielo | Laura Ielo graduated cum laude in Pharmaceutical Chemistry and Technology in 2015 from the University of Messina where she started doctoral studies with Prof. De Luca afterwards. In 2017, she spent a PhD placement in Pace's group at Vienna developing novel homologation methodologies based on the use of halomethyllithium reagents. Besides this interest in organometallic chemistry, her research encompasses the design and the synthesis of tyrosinase inhibitors. |
 Vittorio Pace | Vittorio Pace received a PhD in Chemical Sciences cum laude from the Complutense University of Madrid in 2010 working with Profs Alcántara and Sinisterra. After postdoctoral training at Vienna (Prof. Holzer), Manchester (Prof. Procter) and Stockholm (Prof. Olofsson), he obtained a group leader position at the University of Vienna and a tenure track in 2018. In 2016, he received the Habilitation in Pharmaceutical Chemistry from the University of Vienna and, in 2017, the Habilitation for Full Professor of Organic Chemistry. He has received several awards including the Ciamician Medal of the Italian Chemical Society, the Caglioti Prize of the Accademia Nazionale dei Lincei, and the Young Investigator Award of the Faculty of Life Sciences at Vienna. His research core is represented by the design and development of new transformations with functionalized organolithiums. |
Introduction
The introduction of C1 units into organic frameworks – usually referred to as homologation – represents a versatile tool with widespread applications in organic synthesis.1 Historically, the prototypical example of homologation transformations is represented by the diazomethane-mediated Arndt–Eistert reaction2 which opened the field for introducing analogous agents displaying a similar nucleophilic reactivity such as Corey–Chaykovsky sulfur ylides3 and metal carbenoids.4 The particular properties featured by the latter class of reagents make them unique entities in the synthetic panorama, in primis for their constitutive ambiphilicity enabling them to manifest a nucleophilic or electrophilic behaviour, depending on the reaction conditions.4,5 Two main parameters are considered the domini governing such reactivity-switching equilibrium: (1) the nature of the metal and, (2) the temperature. Nowadays, it is well established that carbenoids of truly electron-positive metals (e.g. Li) usually behave as nucleophiles,5a while less-positive ones (Zn) show a predominant electrophilic reactivity.6 This is evidently a property arising from the fact that the carbon atom of a carbenoid is concomitantly linked to the metal and to the (usually) electron-negative group, e.g. halogen. In fact, the Achilles heel of carbenoids is the limited stability, the result of α-elimination triggered by the internal coordination of the metal with the halogen, finally leading to a free carbene and a metal halide.4,7 As a result, the chemistry of carbenoids is somewhat conditioned by this intrinsic limitation and, in this context, the proper management of carbenoid preparation and stabilization becomes crucial for their synthetic exploitation.8 Studies conducted by pioneers of the field (Villieiras and Barluenga)9 revealed the beneficial activity of lithium halides and ethereal solvents in disrupting the internal coordination responsible for the undesired α-elimination (Scheme 1). The chemical modification of the halocarbenoid structure through the introduction of a stabilizing silicon-containing group is an equally effective solution as reported by Magnus, though an additional silicon-removal step should be considered.10 | ||
| Scheme 1 General features of carbenoids. | ||
Thus, it becomes evident that the preparation of these unstable species requires carefully controlled techniques and, with the judicious – but simple – selection of the methodologies, chemocontrolled strategies can be designed.
In this feature article, we will critically analyze selected examples from our own and others’ research dealing with the modulation of carbenoidic reactivity by tuning mainly the reactive species formation event (i.e. how the preparation of the reagent influences the overall reactivity). In agreement with classical methods for preparing organometallic species, the following strategies are usually employed: (a) metal-halide exchange, (b) metal-proton exchange, (c) metal-sulfinyl exchange, and (d) tin-metal exchange.11 Considering that lithium and magnesium halocarbenoids (although at a lesser extent) present nucleophilic attitude, our attention will be limited to these species.
Vistas and potential in homologation chemistry with nucleophilic carbenoids
The versatility of lithium carbenoids as nucleophilic species can be advantageously employed for introducing a CH2 group as a nucleophile (i.e. LiCH2X) and, then, it reacts as an electrophile at a later stage of the synthetic sequence. Classical examples of this synthetic philosophy are the epoxidation9c,12 or aziridination13 of carbonyls and imines or the Matteson homologation of boronic esters14 with (eventually) chiral carbenoids.15 In the former case, the intermediate alkoxide (or amide) attacks the electrophilic halomethyl group, thus forming a three-membered cycle. On the other hand, the ate complex rearrangement causes the expulsion of the halide furnishing the homologated structure.14a Ultimately, there is a possibility to open the door for further chemistry on the now electrophilic CH2X group. Accordingly, the design of new processes relies on: (1) generation of the nucleophilic reagent; (2) a deep understanding of mechanistic aspects which could play a determinant role (Scheme 2).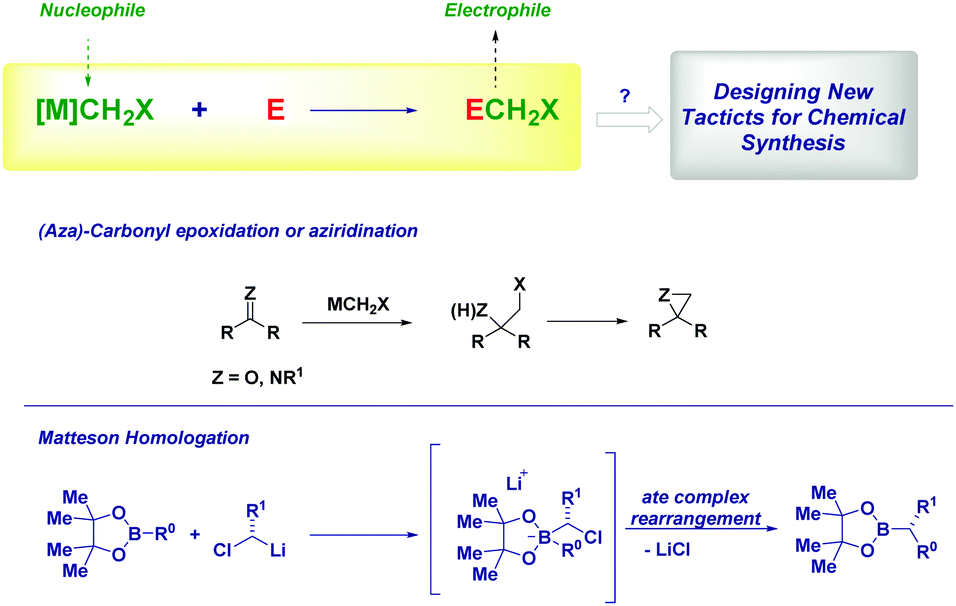 | ||
| Scheme 2 Exploiting the introduction of a CH2X fragment: general considerations. | ||
Lithiation–borylation has emerged as a powerful tool to control the stereochemistry along a carbon chain and to build up multiple stereogenic centers with high stereocontrol,16 by using – inter alia – lithium carbenoids, lithium carbamates and benzoates.17 Aggarwal and coworkers carried out a short synthesis of (+)-kalkitoxin and (+)-hydroxyphthioceranic acid by the iterative homologation of boronic esters, alternating chiral lithiated benzoate esters and chloromethyllithium, to insert the required methylene units (Scheme 3).18
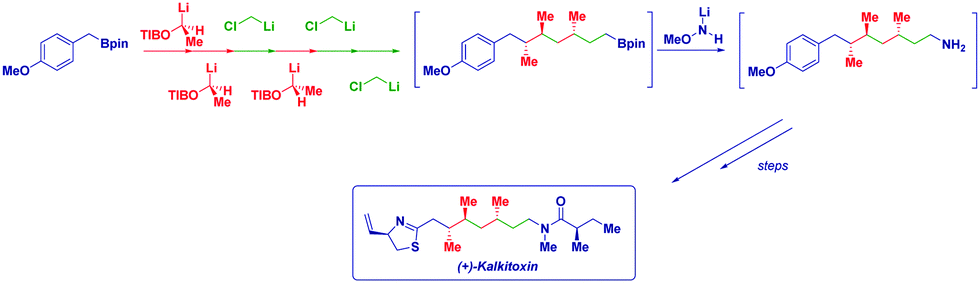 | ||
| Scheme 3 Aggarwal's assembly-line synthesis of (+)-kalkitoxin. | ||
Notably, purification between several homologations could be avoided, thus considerably improving the overall sequence. In the case of (+)-kalkitoxin, six iterative homologations were performed on the commercially available boronic ester to build up the carbon array before the C–B linkage was transformed into the required C–N bond without purification of the intermediates.
Thus, the assembly line strategy enabled the chain extension with full stereocontrol and, the simple increase of the temperature was found to be sufficient for decomposing the carbenoid in excess and, as a consequence, allowing the next homologation step to be performed directly.19
Reagents of LiCH2X and LiCHX2 type: preparation
The metal halogen exchange – formally an oxidative addition – run on a given dihalomethane precursor is undoubtedly the most common method for preparing halocarbenoids for synthetic purposes.8a In line with the general theory that heavier halogens are exchanged first, iodo-containing derivatives represent excellent manifolds for generating carbenoids. Accordingly, an alkyl or aryl-lithium reagent promotes the exchange on XCH2I(Br) furnishing the corresponding Li carbenoid. Because of the acidity of dihalomethanes conferred by the electron-withdrawing halogens, the proton abstraction is easily achieved with a lithium amide base such as LDA and LTMP (Scheme 4).20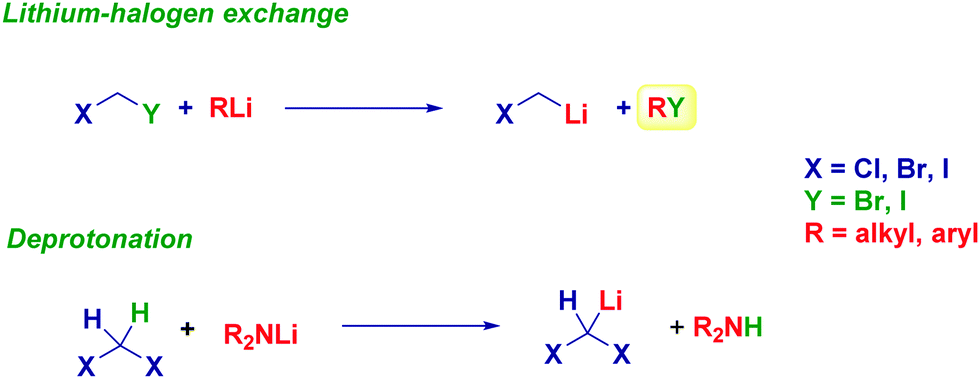 | ||
| Scheme 4 Preparation of monohalolithium and dihalolithium carbenoids via halogen-Li and proton-Li exchange. | ||
Lithium halocarbenoids are regarded as extremely labile entities because of the aforementioned α-eliminationthey suffer: however, operating in batch mode by adopting Barbier-type conditions makes it possible to efficiently form these reagents and, therefore to react them with the appropriate electrophile.8 It should be observed that the extremely rapid halogen-lithium exchange is so fast that no concomitant attack of the nucleophilic organolithium reagent to the electrophile already present in the reaction medium takes place. Indeed, as suggested by Matteson, the generation of a carbenoid in the presence of LiBr further decreases the occurrence of such undesired events.21
Carbon-electrophilic partners for LiCH2X reagents
The homologation of Weinreb amides with carbenoids emerged as a versatile tool for preparing different α-haloketones (α-amino and α,β-unsaturated) under controlled conditions.23 In particular, no undesired phenomena such as the overaddition of the organometallic reagent (observed with esters), Michael-type processes or, the reaction with sensitive functionalities decorating the structure could be noticed. We employed this technique for preparing an important scaffold for the synthesis of the HIV inhibitor Nelfinavir (Scheme 5).24
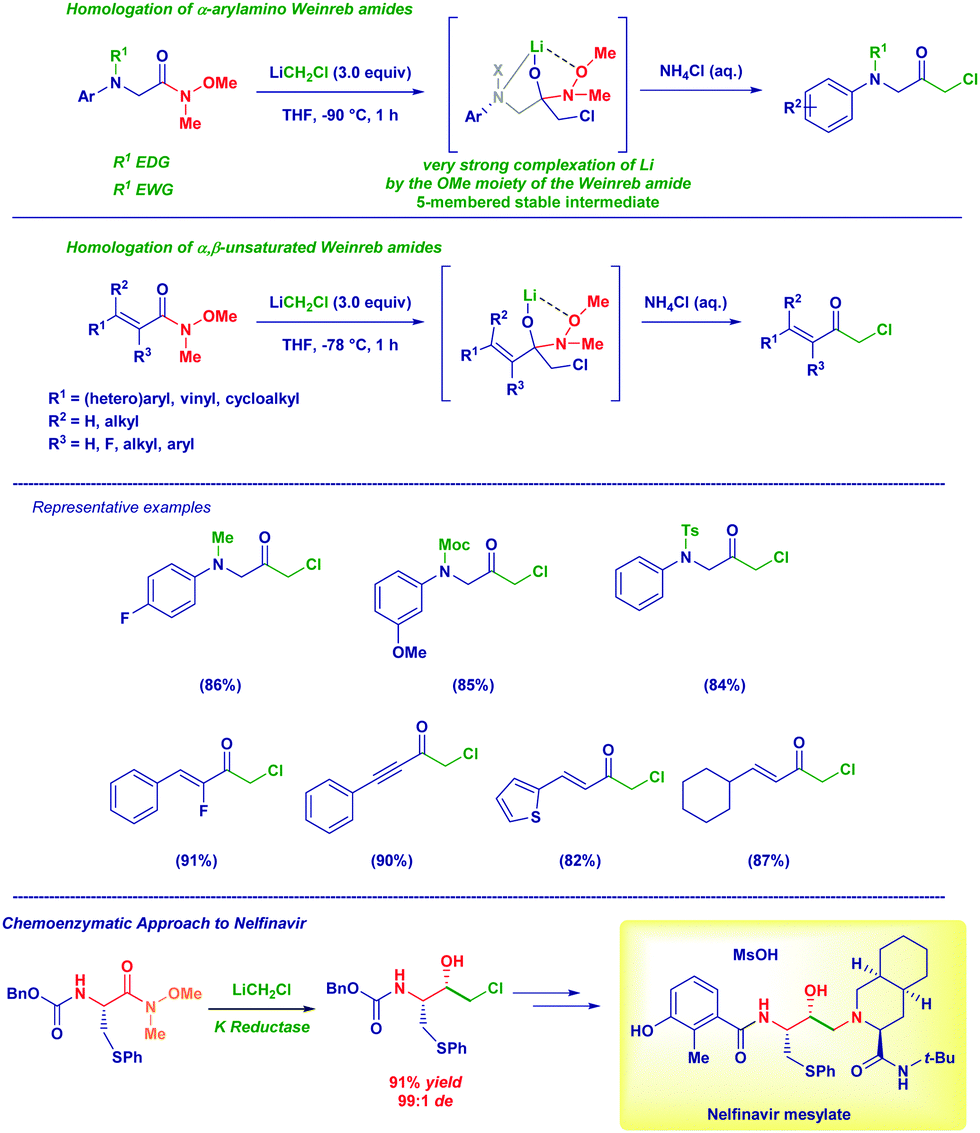 | ||
| Scheme 5 Versatility of Weinreb amides for preparing α-haloketones. | ||
The excellent stability of tetrahedral intermediates formed upon the addition of lithium (di)halocarbenoids to Weinreb amides is showcased by their isolation as O-TMS protected hemiaminals (Scheme 6).25 As documented by our group in 2017, such labile species constitute the first example of isolated and fully characterized intermediates from Weinreb amides and organolithiums known since their introduction in 1981.22a Again, the process preserves the chemoselectivity in the presence of different other reactive functionalities, including a potentially exchangeable aromatic iodide. Analogous intermediates formed from N-acylpyrroles26 could be isolated under identical conditions, even in the presence of sterically demanding moieties.25 Crucial factors enabling the isolation of these intermediates where the use of TMS-imidazole (Im-TMS) as the silylating agent and deactivated neutral alumina (Brockmann grade 3) for chromatography.
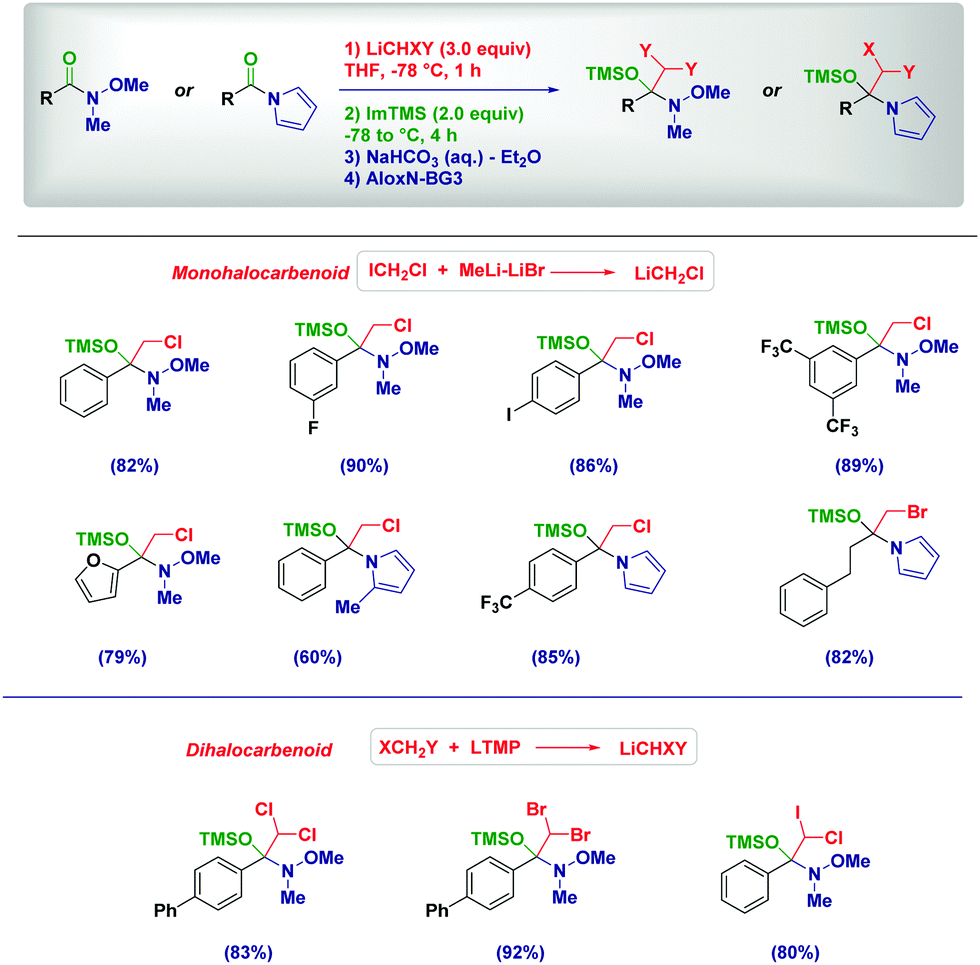 | ||
| Scheme 6 Trapping of tetrahedral intermediates formed from Weinreb amides and (di)halocarbenoids. | ||
Given the nucleophilicity of nitrile-type carbanions27 and the unique acylating properties of Weinreb amides, we have developed a simple, efficient, protocol for the synthesis of variously functionalized α-cyanoketones (Scheme 7).28 Key features of the method are: (a) uniformly high yields, employing both different substituted acetonitriles and/or Weinreb amides; (b) easy work-up procedure when reactions are carried out in the presence of an excess of LiCH2CN; (c) possibility to access multi-substituted cyanomethylketones by simply selecting the proper R1R2CLiCN carbanion; and (d) excellent chemoselectivity observed in particular systems such as α,β-unsaturated Weinreb amides.
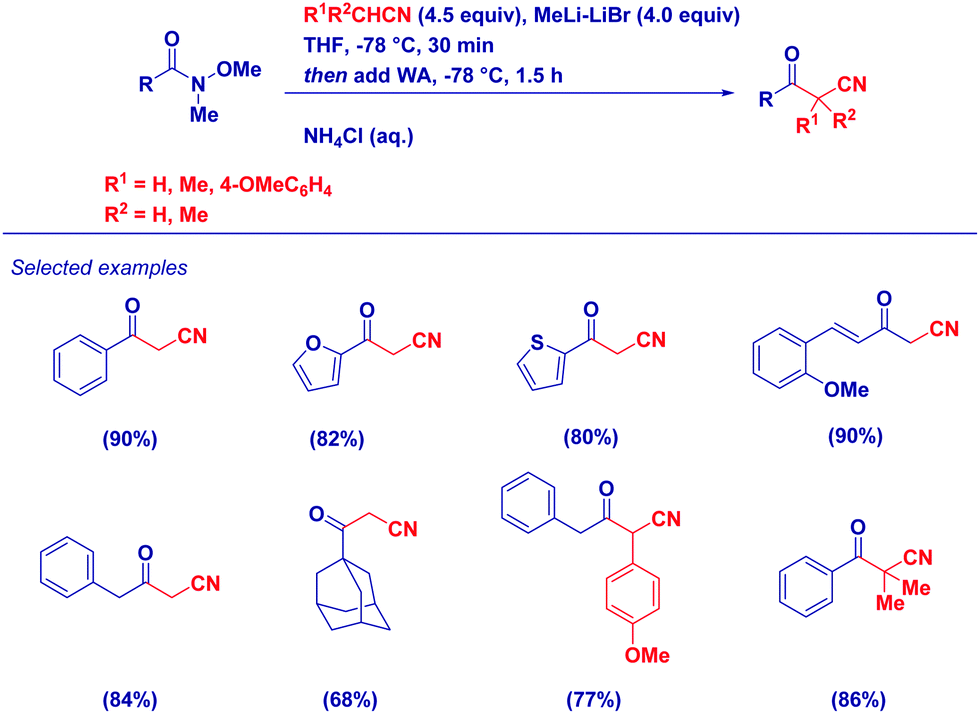 | ||
| Scheme 7 Synthesis of α-cyanoketones from Weinreb amides and lithiated acetonitriles. | ||
The Weinreb amides-based approach is also amenable for the preparation of α-oxyketones, through the generation of nucleophiles of type LiCH2OR29via Yus’ arene catalyzed reductive lithiation of chloromethyl ethers.30 Broad scope in terms of acylating agents and carbanions used was observed without any detrimental effect displayed by electronic and steric factors. Despite the basic conditions, the stereochemical information could be fully transferred from the Weinreb amide or from a chiral α-alkoxyorganolithium (Scheme 8). Similarly, the proper generation of α-thiomethyllithium reagents via reductive lithiation or deprotonation conditions (for systems of LiCHRAr type) followed by the reaction with variously functionalized Weinreb amides represents an excellent method to access β-oxo thioethers.31 The procedure, adaptable to the case of both alkyl- or aryl-thiomethyllithiums, combines the conceptual simplicity and versatility of the addition of organometallics to Weinreb amides with the possibility to fully preserve the optical information contained in the starting materials.
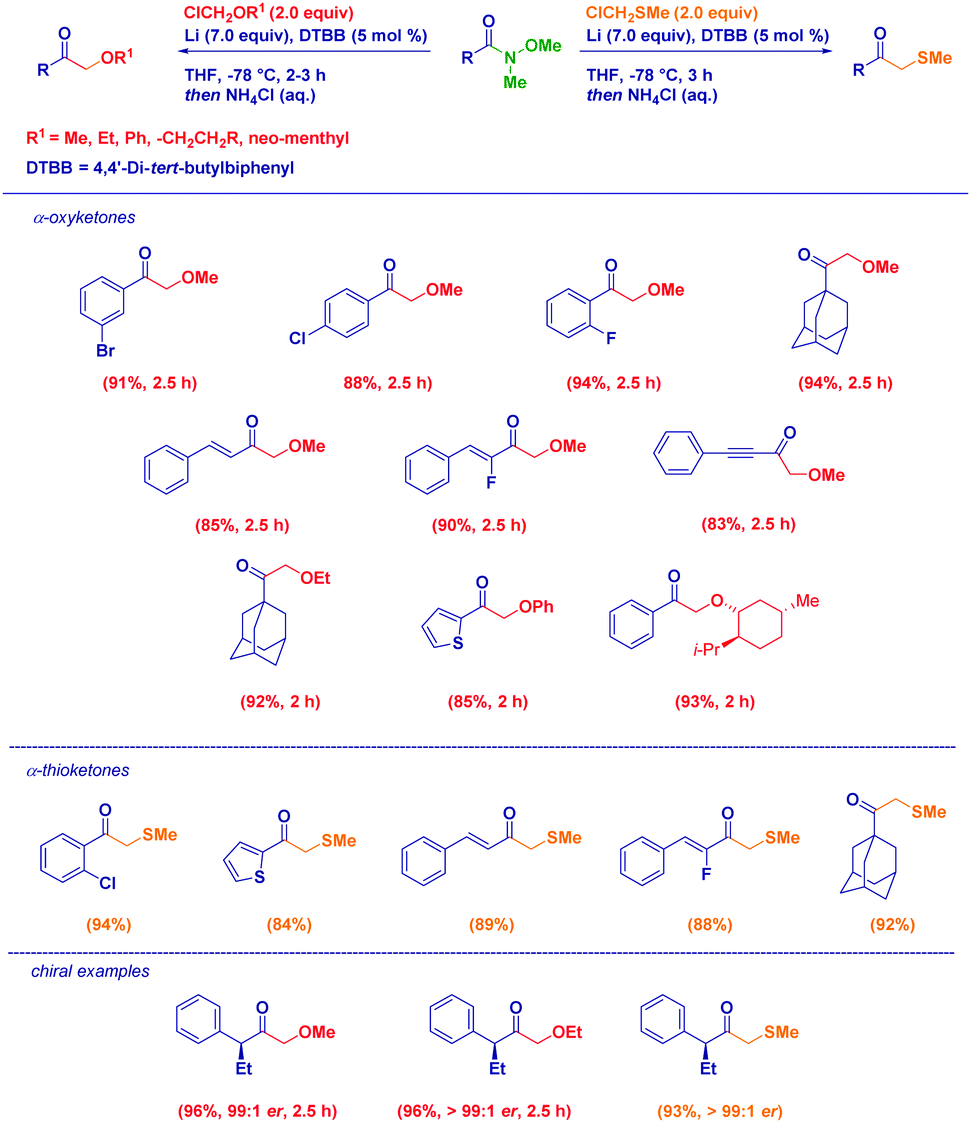 | ||
| Scheme 8 Synthesis of α-oxo- and α-thio methylketones from Weinreb amides and organolithiums generated via reductive lithiation. | ||
These tactics represent a practical and valuable solution for access to α-substituted ketones which can be problematic when direct nucleophilic substitutions on α-haloketones32 or enolate trapping methods with the corresponding electrophiles are attempted.33
Very recently, our group demonstrated the feasibility of such an approach for preparing challenging α-aryl- and α-alkyl-selenomethyl ketones under full chemocontrol from Weinreb amides and diselenoacetals (Scheme 9).34 The straightforward lithium/selenium exchange these compounds undergo represents an excellent entry to selenomethyllithium reagents (LiCH2SeR), which then are trapped with several Weinreb amides to give – upon hydrolysis – the desired compounds. Remarkably, previous work by Back in the 1980s on the Cu-catalyzed diazomethane homologation of selenoesters35 (to α-selenoketones) pointed out that diselenoacetals were by-products of the reaction. In our tactic – just cognizant of the easiness of the Li/Se exchange36 – these materials were identified as valuable sources for the corresponding carbanions.
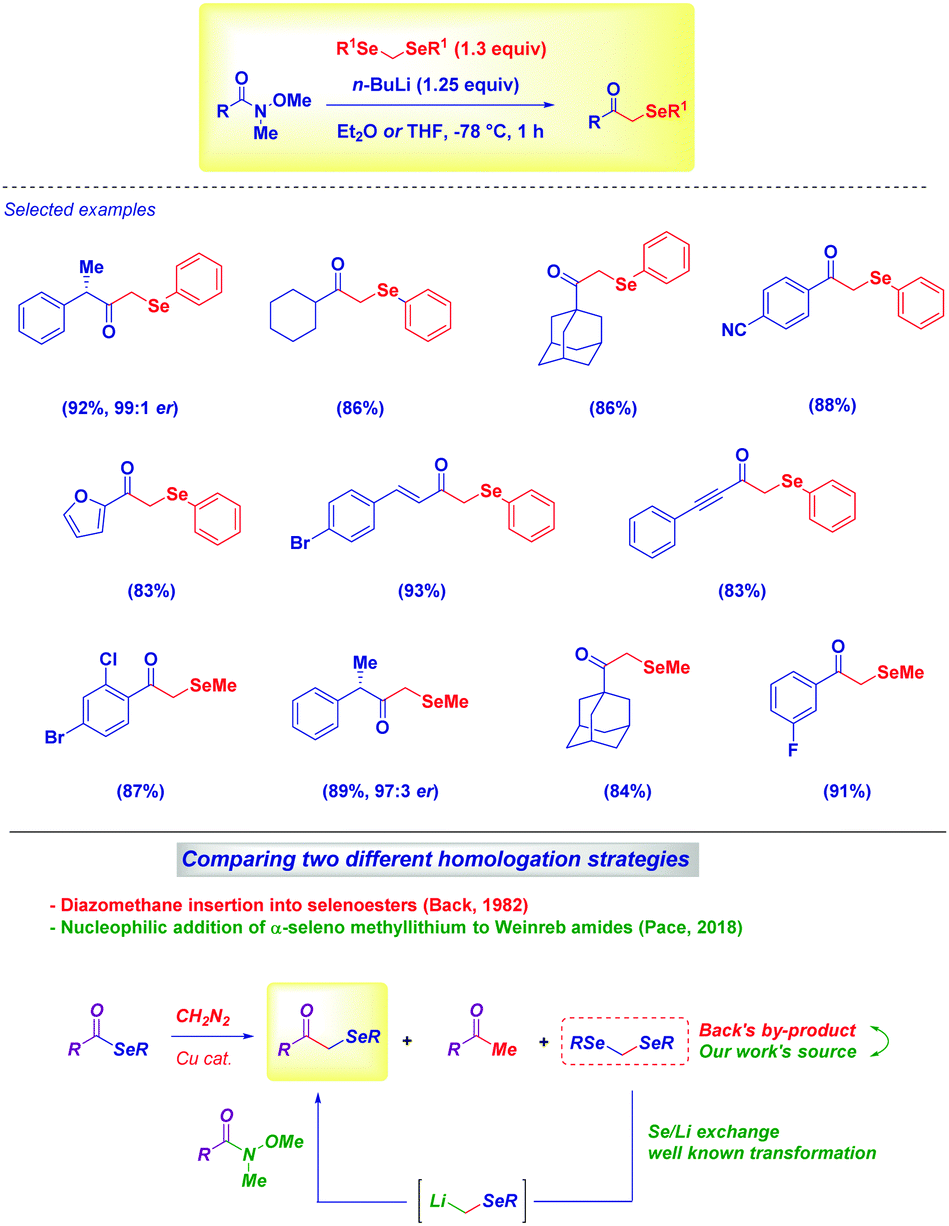 | ||
| Scheme 9 Synthesis of α-seleno methylketones from Weinreb amides using diselenoacetals as precursors. | ||
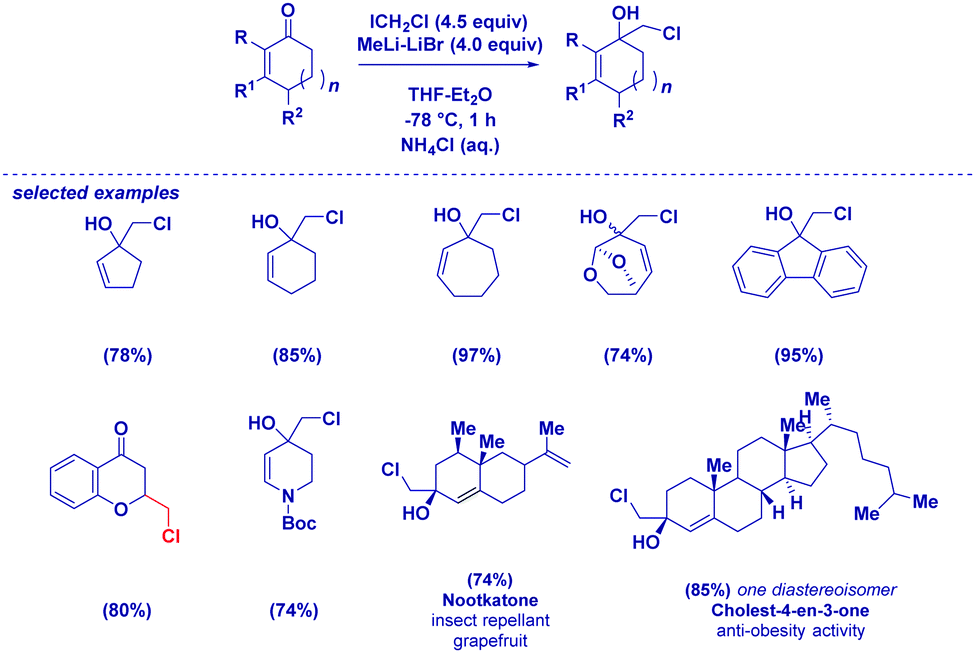 | ||
| Scheme 10 Chemoselective addition of lithium carbenoids to α,β-unsaturated cyclic ketones. | ||
Isatins are particularly reactive unsaturated carbonyl compounds, featuring an extremely high electrophilic C3 carbon due to the constitutive presence of the electron-withdrawing lactam.41 Halocarbenoids can be added to C3 with precise chemoselectivity, even in the presence of functionalities which are known to be placeholders for halomethylating reagents (Scheme 11).42 Notoriously, organometallic sensitive groups such as Weinreb amides, esters, nitriles, alkenes and alkynes do not alter the chemoselectivity of the process. Pleasingly, also the incorporation of bromine on aromatic or olefinic residues is tolerated, as well as an acidic N–H lactam proton. This is particularly relevant and indicative of the excellent electrophilicity of the C3 of isatins compared to other electrophiles. In fact, Kunisuke,43 Hilpert44 and our own group23a evidenced the difficulties of homologating esters or Weinreb amides in systems presenting free (i.e. unprotected) amidic or aminic NH groups. The carbenoid addition, ensued by the rapid base-triggered ring-closure of the so-formed halohydrin, enables valuable spiro-epoxyoxindoles to finally be produced through an extremely simple and reliable procedure. It should be observed that the key halohydrin-alkoxide required for cyclization was previously prepared by Zhu via a more complex intramolecular Friedel–Crafts strategy.45 However, considering also the effectiveness of Corey–Chaykovsky strategies recently presented by Nair46 and Hajra,47 it appears preferable to prepare unsubstituted spiro-epoxyoxindoles through a homologation tactic.
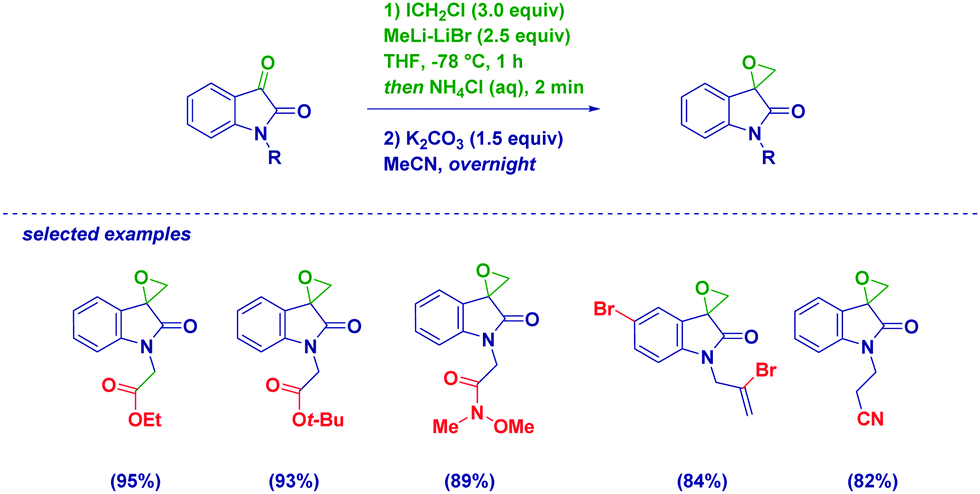 | ||
| Scheme 11 Preparation of spiro-epoxyoxindoles from isatins and LiCH2X reagents. | ||
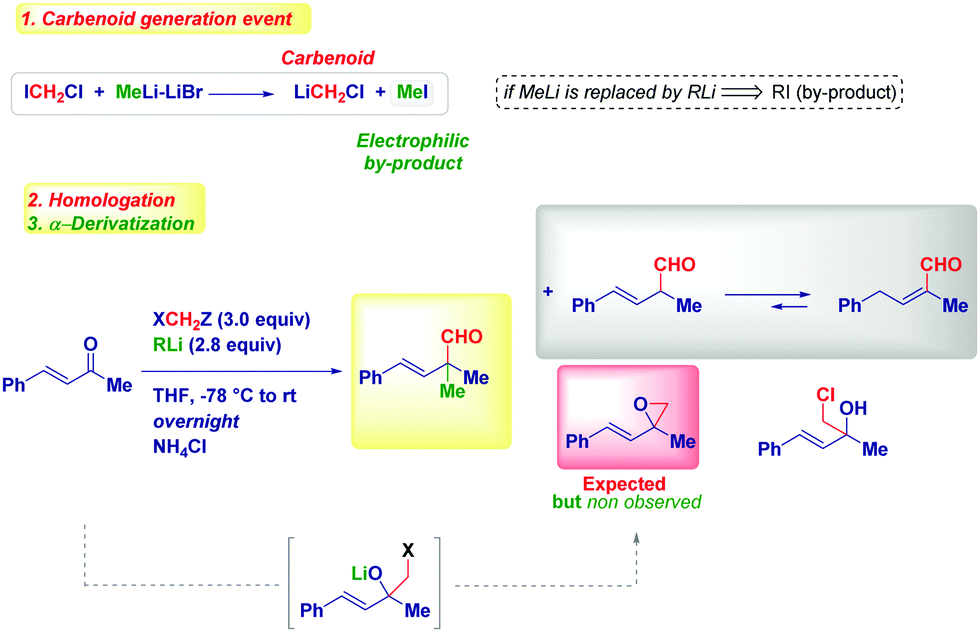 | ||
| Scheme 12 Surprising discovery of the formation of full-quaternary α-aldehydes instead of vinyl epoxides. | ||
This unexpected outcome spurred us to critically analyse the transformation. The presence of an additional carbon (i.e. the aldehydic one) was diagnostic of the homologation process, while the additional α-methyl group could not be rationalized at first sight. The carbenoid formation event has a deep influence on the reaction mechanism, as evidenced by properly selecting the reaction conditions. In fact, by considering the carbenoid generation equation via lithium/halogen exchange run with MeLi–LiBr, the formation of an electrophilic by-product (MeI) seems plausible and thus, it can indicate the origin of the α-methyl group in the final product. Simply by replacing MeLi–LiBr with n-BuLi, PhLi or TMSCH2Li, we observed the formation of a homologated α,β-unsaturated aldehyde showing no quaternarization at the α-position. Should the hypothesized carbenoid formation event be the key to control the process, the less electrophilic by-products (compared to MeI formed in the presence of MeLi–LiBr) obtained with these organolithiums – n-BuI, PhI or TMSCH2I – would not be able to promote the α-functionalization, in agreement with the experimental result. A similar reactivity was deduced by generating LiCH2Cl via Sn/Li exchange. Moreover, two additional parameters were considered fundamental for the transformation: (a) the presence of LiBr as a mild Lewis acid and, (b) the increase of temperature from −78 °C to room temperature. Taken together these three aspects (organolithium employed, the presence of LiBr, temperature increasing), it is conceivable assuming a mechanism constituted by three-different separation processes: (1) LiCH2X-mediated homologation followed by ring-closure to an epoxide; (2) LiBr-mediated Meinwald-type rearrangement of the epoxide into an aldehyde50 and, (3) electrophilic functionalization of a putative enolate. Notably, the formation of this enolate via 1,2-hydride shift51 on the allylic carbocation D, is in agreement with the impossibility to observe such transformation in saturated ketones, thus identifying α,β-alkenyl type carbonyls as privileged substrates. As a consequence, the enolate-intermediate F is amenable for further treatment with electrophilic species, finally leading to α-quaternary aldehydes or homologated α,β-unsaturated aldehydes (via tautomerization) by simply selecting the quenching reagent (alkyl halide or acid) (Scheme 13).
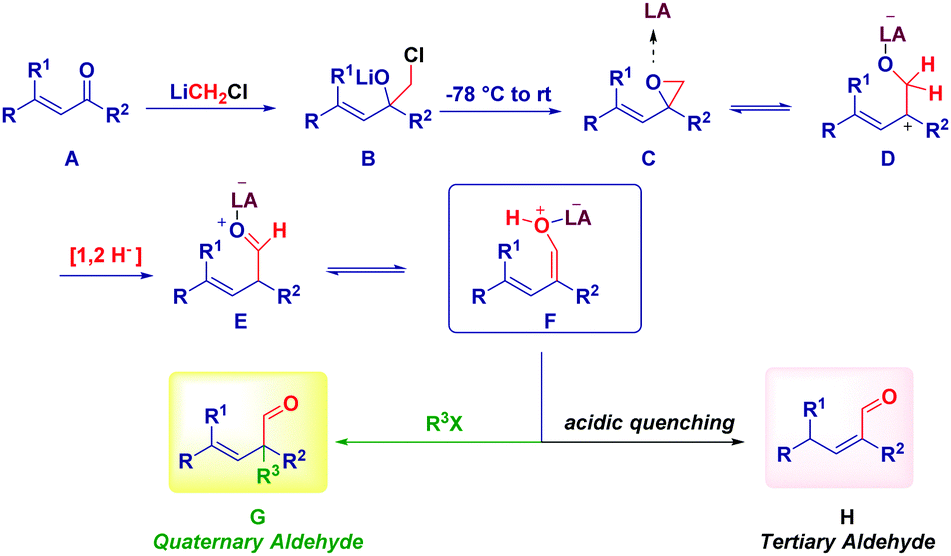 | ||
| Scheme 13 Plausible rational for the transformation. | ||
The use of labelled carbenoids and/or labelled methyl iodide provided convincing evidence for the hypothesized enolate-type mechanism: (i) the use of deuterated carbenoid LiCD2Br generated from CD2Br2 and MeLi–LiBr afforded the aldehyde presenting a α-CH3 group and a deuterated carbonyl. (ii) The employment of CD3Li·LiI and CH2Br2 – which evidently forms LiCH2Br – gave the H-aldehyde featuring the CD3 group at the α-position. (iii) Homologation with LiCD2Br – formed from CD2Br2 and CD3Li·LiI – yielded the aldehyde showing deuteration at both sites: the aldehyde and the α-position. (iv) Homologating with LiCD2Br generated with TMSCH2Li and quenching with an acid, delivered the deuterated α,β-unsaturated aldehyde (Scheme 14). Thus, it appeared demonstrated that the whole reactivity is determined by the general equation for forming the nucleophilic carbenoid.
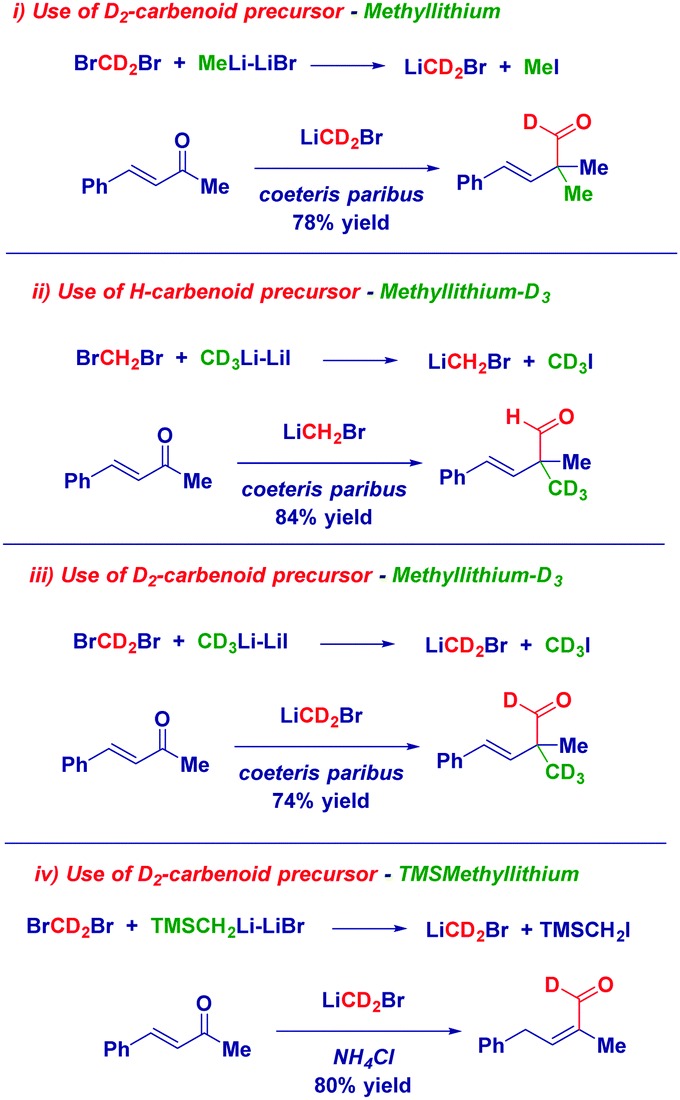 | ||
| Scheme 14 Labelled experiments supporting the mechanism. | ||
Based on these mechanistic indications, we designed a direct and finely tuned transformation by simply selecting the carbenoid precursor, the organolithium and the electrophilic trapping agent. For example, generating LiCH2Cl from ICH2Cl and MeLi–LiBr allowed the preparation of a series of cyclic and acyclic α-methyl aldehydes, including derivatives of natural products. On the other hand, TMSCH2Li–LiBr is the reagent of choice for introducing at the α-position a residue from a different electrophile subsequently added to the reaction mixture (Scheme 15). Reactive alkyl-type halides (i.e. benzyl, allyl, crotyl and propargyl) are excellent partners for the transformation and, the concomitant presence of sensitive groups such as exchangeable halides or esters does not affect the chemoselectivity. Although a heteroatom electrophile such as a disulfide can be used for the trapping of the enolate, unfortunately no reactivity was observed with acyl-type partners (carbonyls, amides, isocyanates and carboxylic derivatives). In the absence of an external electrophile, the simple acidic quenching of the enolate results in the formation of α,β-unsaturated aldehydes through the spontaneous tautomerization of the corresponding (less stable) β,γ-unsaturated analogues. Thus, our method introducing the concept of synthetic equivalence between a vinyl oxirane and a β,γ-unsaturated aldehyde, complements previously available tactics for accessing homoallylic-type aldehydes such as the α-allylation of aldehydes via transition metal-catalyzed chemistry52 or via an aldehyde enolate approach.53 It should be mentioned that these strategies, based on the formal allylation of a pre-existing aldehyde motif, could display full stereocontrol under careful selection of the conditions. In this context, a nice and elegant selective fragmentation of diastereomerically pure and enantioenriched cyclopropanols was introduced by Marek for accessing n-butenals with all-carbon quaternary centers.54
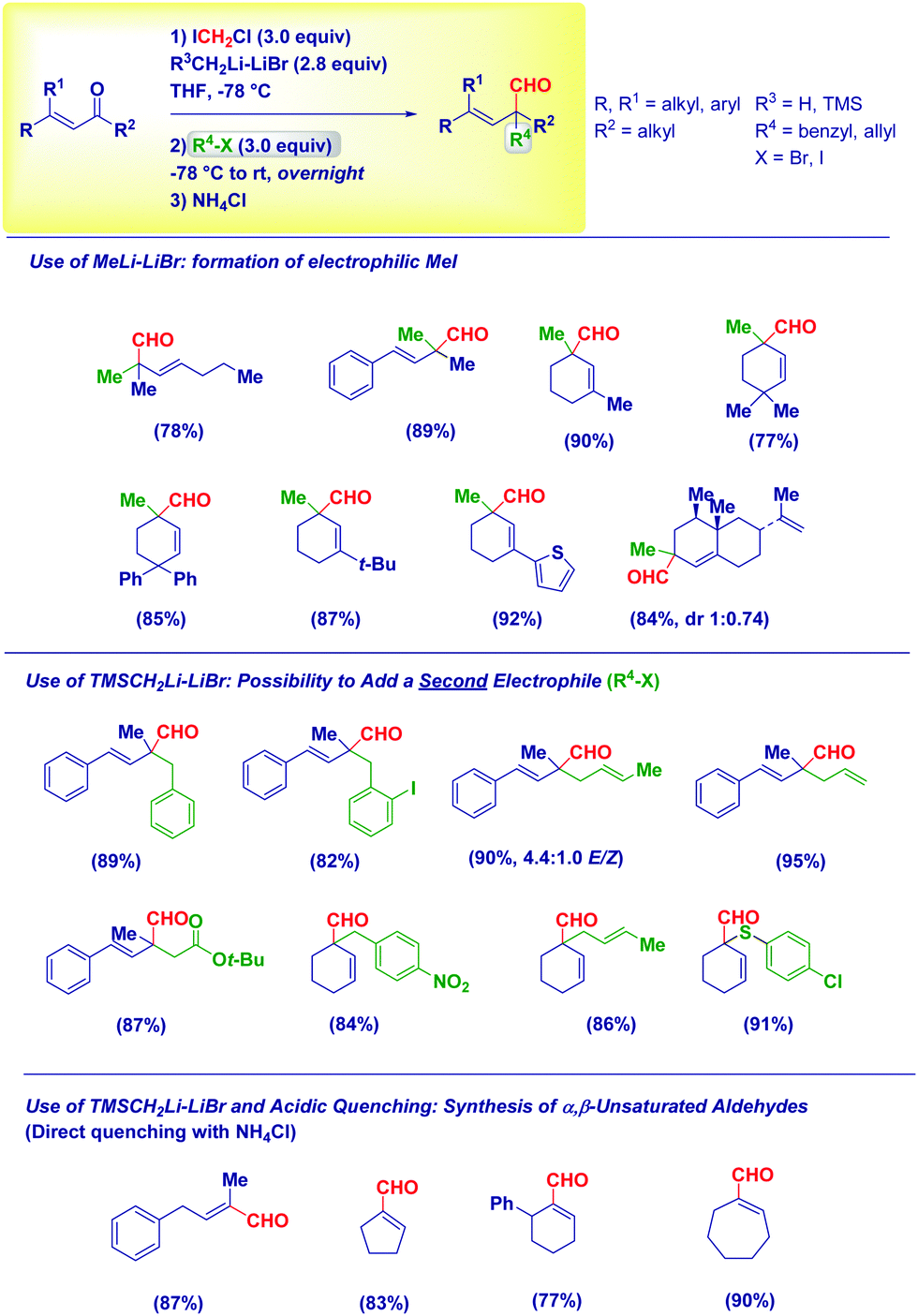 | ||
| Scheme 15 Scope of the all-carbon quaternary and tertiary α-functionalized homoallyl-type aldehydes. | ||
Our group exploited this tactic for the chemoselective preparation of α-haloacetamides through the addition of halocarbenoids to isocyanates (Scheme 16).61 The protocol features a robust scope, as observed in the case of both aromatic and aliphatic isocyanates. As underlined above, common problematic elements limiting the synthesis of sterically hindered amides via classical strategies – e.g. the condensation of an amine and a carboxylic derivative – are practically suppressed by using such an approach. Some additional points merit mention: (1) chemoselectivity is still preserved in the case of bromine-containing aryl nuclei by just reducing temperature; (2) enantiopure isocyanates undergo the transformation under full retentive conditions; (3) no concomitant Simmons–Smith type cyclopropanations could be observed in the presence of an olefinic functionality; (4) uniformly high yields demonstrate the robustness of the methodology. The methodology could also be extended to the synthesis of α,α-dihaloamides, which is a demanding task because of the chemoselective difficulties associated with the use of halogenation procedures. The generation of dihalolithium carbenoids (LiCHXY) through the deprotonation of a dihalomethane with a lithium amide base under Barbier conditions in the presence of isocyanate gives the expected compounds in high yields and chiral integrity.20 We found LTMP as the optimal base for selectively generating dihalocarbenoids.
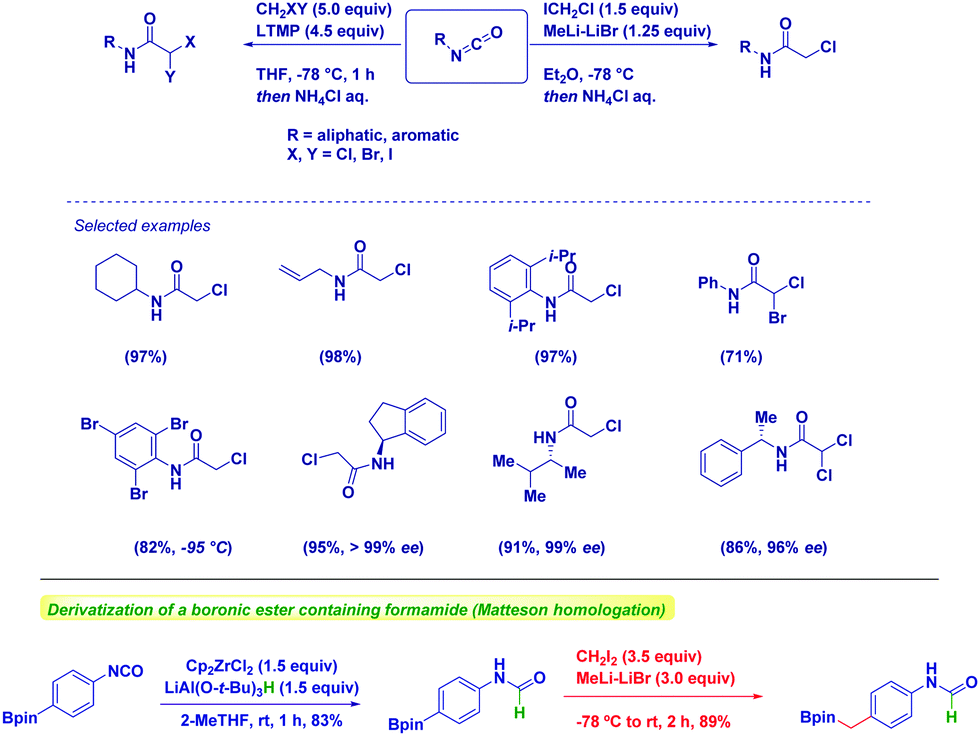 | ||
| Scheme 16 Synthesis of α-haloamides from isocyanates and carbenoids. | ||
Moreover, we employed the hydride-transfer via nucleophilic addition to isocyanates en route to formamides.62 The Schwartz reagent proved to be the optimal H− source in terms of chemoselectivity. In fact, this long-waiting transformation was previously limited by the full reduction of isocyanates to N-methyl amines.63 Moreover, full chemocontrol emerged in the Matteson homologation of a boronic ester featuring a formamide unit, thus indicating how selecting the order of the transformation is critical for the target-oriented synthesis.
Intriguingly, attempts to halomethylate the corresponding isothiocyanates for preparing α-halothioamides were unsuccessful presumably because of the instability of these species, as indicated by the few reports available on this class.64
However, it spurred us to deeply investigate the reactivity of isothiocyanates, bearing in mind that accessing thioamides through such an approach would have allowed the severe limitations related to the synthesis of these building blocks (e.g. harsh conditions, long reaction times and the use of non-pleasant thionating agents) to be overcome.65 The procedure is applicable for a set of different combinations of isothiocyanates and α-substituted organolithiums giving thioamides in excellent yields (Scheme 17). We were delighted in applying the protocol to the straightforward synthesis of complex thioamides including highly sterically demanding structures. It is important to note that the transmetallation of a lithium carbanion to a Gilman reagent (R2CuLi)66 enables the chemoselective addition exclusively at the isothiocyanate moiety, thus leaving the ester group untouched.
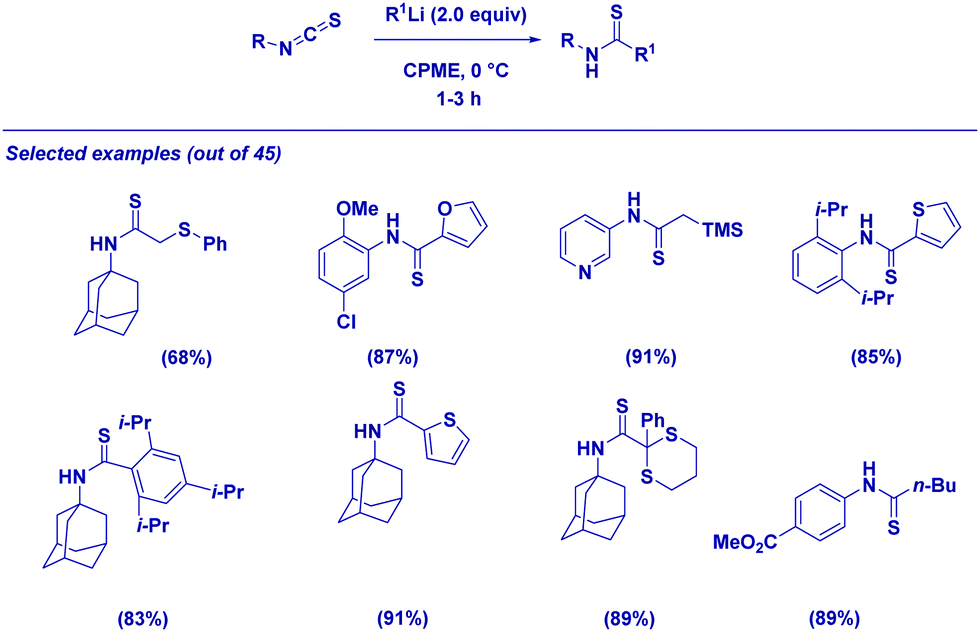 | ||
| Scheme 17 Synthesis of thioamides from isothiocyanates and organolithiums. | ||
No racemization takes place upon the addition of various organolithium reagents to optically active isothiocyanates. The formation of enantiopure N-Boc-pyrrolidine or Hoppe's carbamate,11c,67 through deprotonation in the presence of chiral ligands (−)- or (+)-sparteine68 affords the corresponding thioamides with full retention of stereochemistry and in excellent diastereomeric ratio (Scheme 18). As expected, by switching from one enantiomer of sparteine to the other, it is possible to get both asymmetric forms of a given thioamide with comparable optical integrity. The simplicity of the reaction conditions developed, and the results in terms of enantiopurity allow the conclusion to be drawn that the present method shows better performances compared with sulfurating protocols in which ee's erosion can be problematic.69
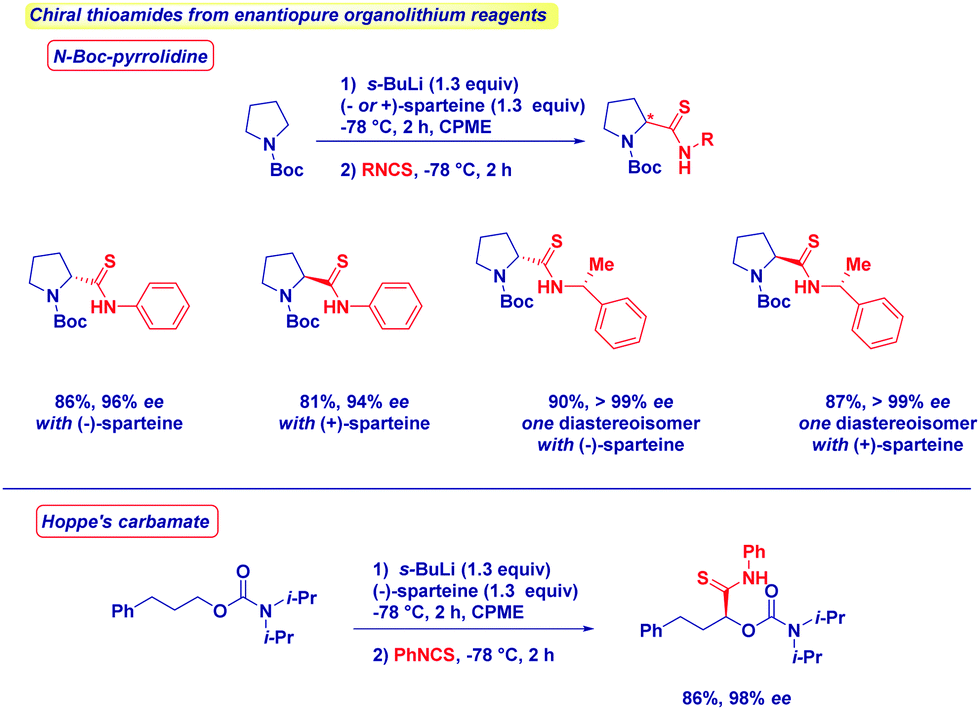 | ||
| Scheme 18 Preparation of optically active thioamides from isothiocyanates. | ||
An indicative example of the good nucleophilicity of lithium carbenoids – towards heteroatom electrophiles – is the homologation of disulfides to the corresponding dithioacetals (Scheme 19).72 Inspired by the known synthesis of thioethers from a disulfide and an organolithium reagent,73 we were pleased to observe its adaptability to the more challenging homologation. In fact, such a transformation, previously realized with different nucleophilic homologating reagents (diazomethane74 and sulfur ylides75) or with electrophilic zinc carbenoids,76 proved to be in both instances highly difficult and, in general, low yielding. Our idea of employing the highly nucleophilic Li carbenoid consisted of forming an (isolable) α-halomethyl thiosulfide intermediate, whose halide was then displaced by the mercapto anion (RS−) released during the first homologation step. We noticed a beneficial activity of TMSCl in promoting such a second nucleophilic displacement, presumably via a coordination effect. This strategy illustrates the true concept of homologation with CH2X reagents: the introduction of this reactive unit is followed by the subsequent expulsion of the halide, thus finally leading to the formal insertion of the methylene moiety. In analogy, such an overall process recalls the Matteson reaction of boronic esters or the epoxidation of carbonyls, in which the halogen of the carbenoid is only present at an intermediate level, but not in the final compound. The protocol shows an excellent scope, and the chemoselectivity is fully maintained in the presence of additional sensitive functionalities such as aryl halides, heterocycles, esters and a secondary amide (i.e., again the NH group does not affect homologations!). Importantly, the procedure could be employed for homologating an asymmetric disulfide and aryl or alkyl diselenides.72
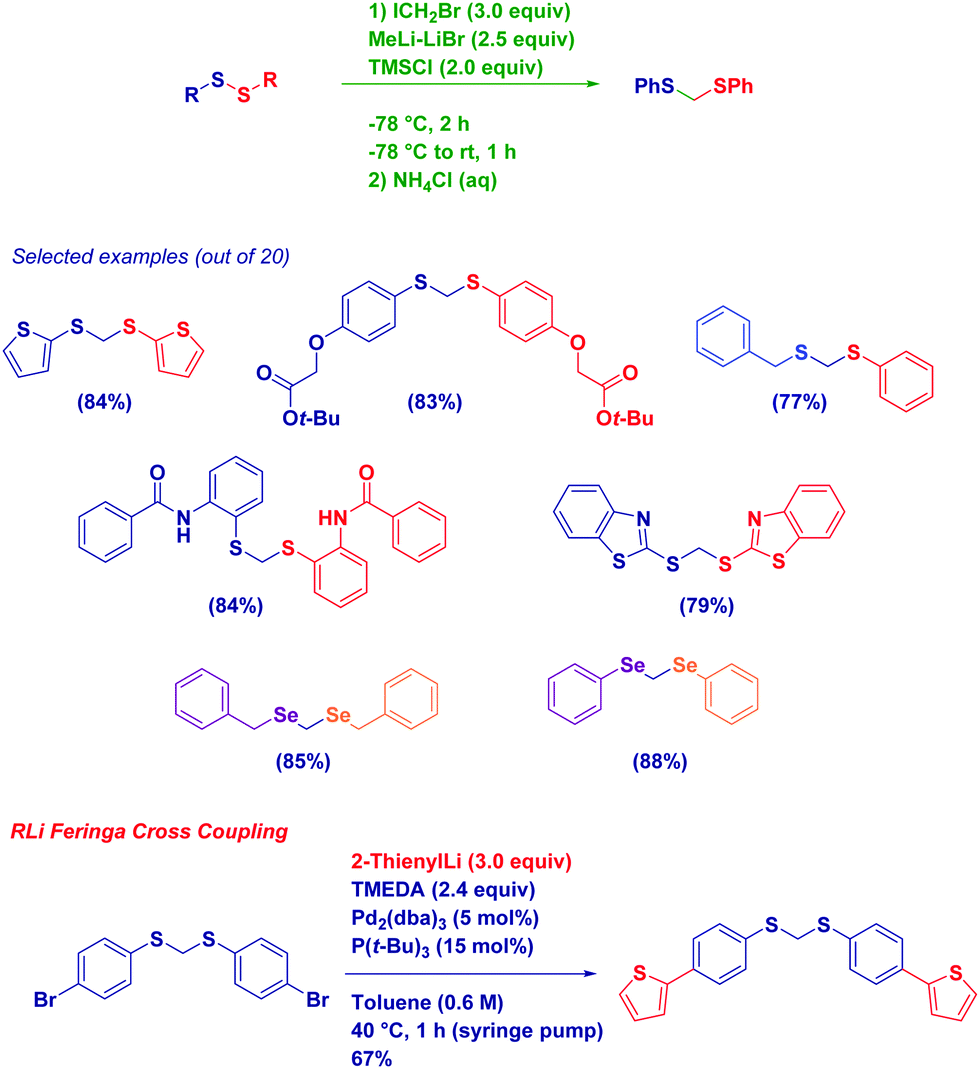 | ||
| Scheme 19 Homologation of disulfides and diselenides. | ||
The presence of a reactive aromatic bromide opened up the field to evaluate the Pd-catalyzed coupling of an aryl bromide-containing substrate with an organolithium reagent reported by Feringa as an effective method for generating C–C bonds with lithium carbanions.77
Nucleophilic magnesium carbenoids
Considering that taming the nucleophilicity can be a fine expedient for improving chemoselectivity – as, for example, seen above in the case of a Gilman reagent – magnesium carbenoids are regarded as more stable, but less reactive nucleophiles than the corresponding lithium analogues.78 That is, the use of non-Barbier conditions is compatible with these carbenoids and, from a chemoselective perspective, it is worth mentioning the excellent discriminating capability between two distinct reactive carbonyls. Clososky reported a highly chemocontrolled synthesis of chlorohydrins – in the presence of ketones – using ClCH2MgC·LiCl prepared by the metallation of ICH2Cl with the Turbo Grignard reagent (Scheme 20).79 Notably, the chemoselectivity of the protocol was further demonstrated in the cases of other sensitive functionalities, such as nitrile, ester, and alcohol.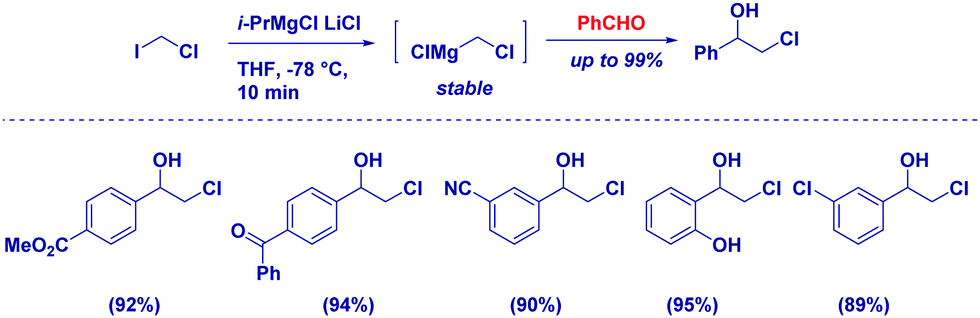 | ||
| Scheme 20 Chemoselective addition of magnesium carbenoids to aldehydes. | ||
Knochel and co-workers prepared diastereopure multi-substituted cyclopropane carboxylates through the selective metalation of halogen cis to the ester (Scheme 21). The stereochemical integrity is maintained after trapping with different electophiles.80 Similarly, starting from gem-dibromoalkene – possessing an alkoxycarbonyl group – they were able to prepare β-bromobutenolide.81 Mechanistically, the magnesium carbenoid generated through bromo/magnesium exchange reacted with a carbonyl compound, giving an alkoxide intermediate that attacks the ester furnishing the cyclization product.
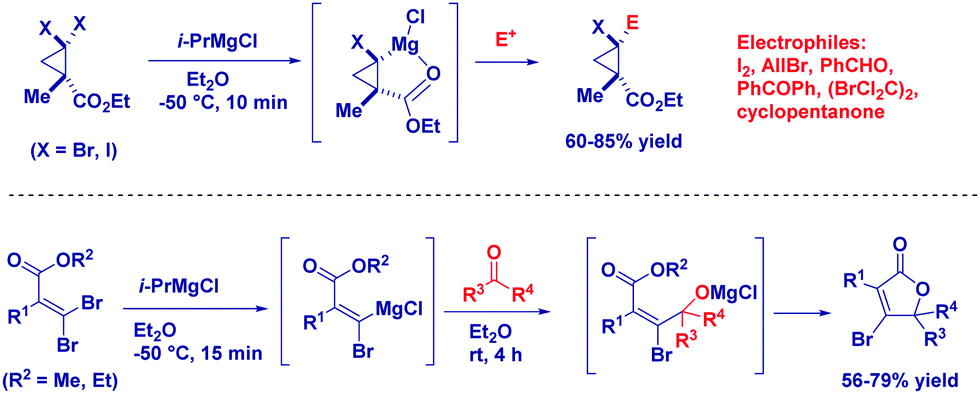 | ||
| Scheme 21 Chemoselective elaboration of complex architectures with magnesium carbenoids. | ||
An excellent alternative for the preparation of magnesium carbenoids is based on the metal exchange on a α-halosulfoxide moiety.82 It should be observed that magnesium carbenoids manifest an excellent electrophilic behaviour, as recently reviewed by Satoh78a and Kimura.78c Bach employed Mg-sulfoxide exchange for preparing secondary magnesium carbenoids which upon reaction with aldehydes, followed by Dess–Martin oxidation, produced a crucial α-chloroketone for the synthesis of geldamycin analogues.83 Hoffmann and co-workers focused their attention on the generation of chiral magnesium carbenoids from optically active 1-chloroalkyl aryl sulfoxides. The treatment of pure 1-chloroalkyl aryl sulfoxides with EtMgBr at −78 °C affords configurationally stable magnesium carbenoids that promptly react with benzaldehyde. The stereochemical information contained in the reagent is fully transferred to the product, allowing the synthesis of an enantioenriched epoxide via a homologation/ring closure sequence (Scheme 22).82,84
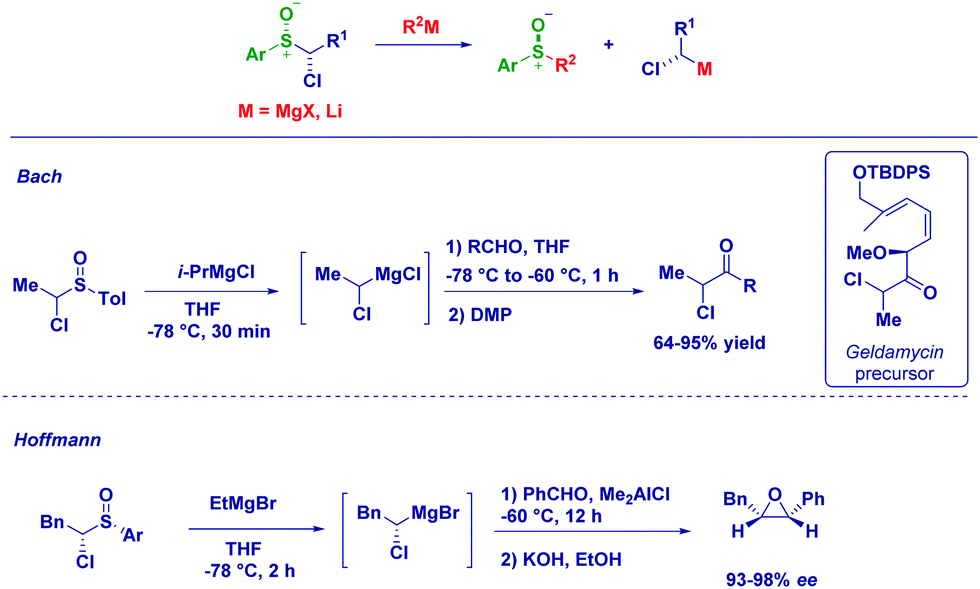 | ||
| Scheme 22 Use of the sulfoxide-metal exchange for preparing carbenoids. | ||
New directions in halocarbenoids
Only very recently, in the course of a collaboration with R. Luisi, we addressed this long standing challenge on the use of LiCH2F at preparative scale.91
The use of a sulfoxide as the precursor of the lithium carbenoid provides the model fluorohydrin in 23% yields, together with side reaction products. On the other hand, the corresponding magnesium fluorocarbenoids generated via sulfoxide-magnesium exchange suffered from halogen-scrambling similarly to zinc fluorocarbenoids noticed by Charette.92 However, these results further confirm the existence of LiCH2F, though it appears evident that neither tin- nor sulfoxide-fluoromethylstannanes are the ideal carbenoid precursors (Scheme 23).
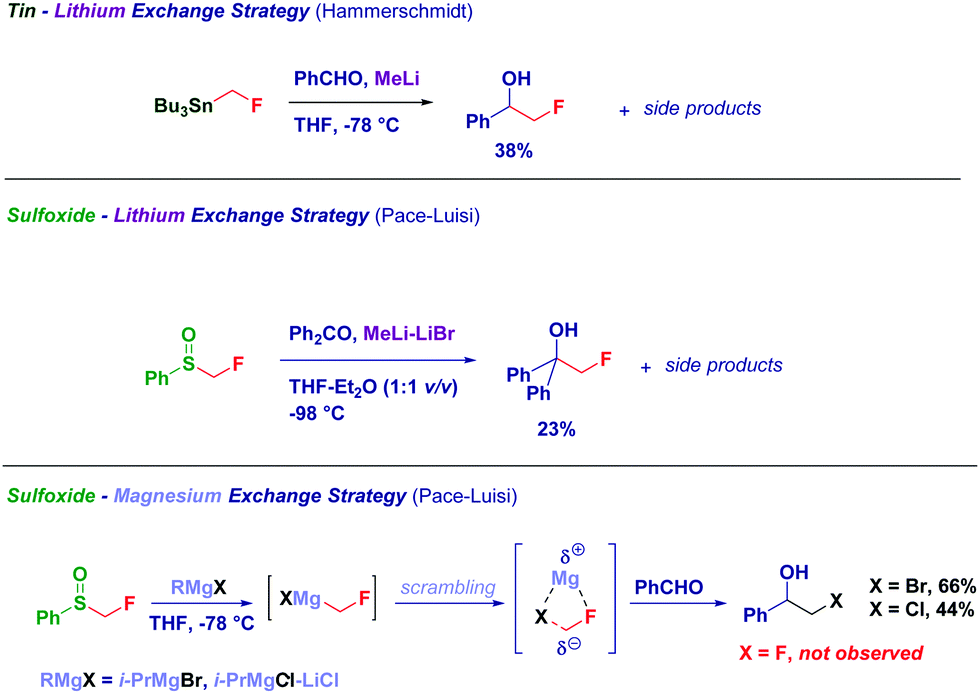 | ||
| Scheme 23 Formation of α-fluoromethyl metal carbanions from tin and sulfoxide precursors. | ||
An effective solution was finally found by adopting a highly effective iodine–lithium exchange on the currently commercially available and easy to manipulate (liquid, bp 53 °C) fluoroiodomethane (Scheme 24).93 The fine tuning of the reaction conditions allows the model compounds to be obtained in an excellent 88% isolated yield, thus making it suitable for preparative processes.91 In depth optimization studies defined the crucial factors accounting for the successful preparation of LiCH2F: (1) MeLi–LiBr as the lithiating agent; (2) generation of the carbenoid under Barbier conditions; (3) stoichiometry (electrophile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeLi–LiBr:ICH2F = 1:1.5:2); and (4) solvent mixture THF:Et2O 1:1 v/v.
:MeLi–LiBr:ICH2F = 1:1.5:2); and (4) solvent mixture THF:Et2O 1:1 v/v.
 | ||
| Scheme 24 Fluoromethyllithium: generation and use in synthesis. | ||
Our fluoromethylation strategy presents an excellent scope as indicated by the variety of functionalized electrophiles used (e.g. carbonyls, Weinreb amides and imines). Ketones and aldehydes were converted into α-fluoro alcohols in very high yields and remarkable chemocontrol, even in the presence of a double bond or potentially exchangeable halogens. Biologically relevant and complex scaffolds such as 3-O-methylestrone and 4-colesten-3-one efficiently undergo the transformation furnishing the corresponding fluorohydrins as single stereoisomers. The reactivity of LiCH2F was also evaluated towards α-, β- or γ-halo ketones giving mixed fluoro-halohydrins which upon base-mediated cyclization resulted in practically quantitative yields of interesting fluoromethyl-oxygenated 3, 4 or 5 membered cycles.
Luisi and coworkers elegantly demonstrated how microfluidic systems are effective in the generation of the thermolabile LiCH2Cl, followed by trapping with electrophiles at high temperatures (up to −20 °C).94 This methodology – constituting a significant advancement in the field – works quite well with different electrophiles from carbonyl compounds, imines, Weinreb amides and isocyanates, thus paralleling the substrate scope observed under classical Barbier-type conditions (Scheme 25).
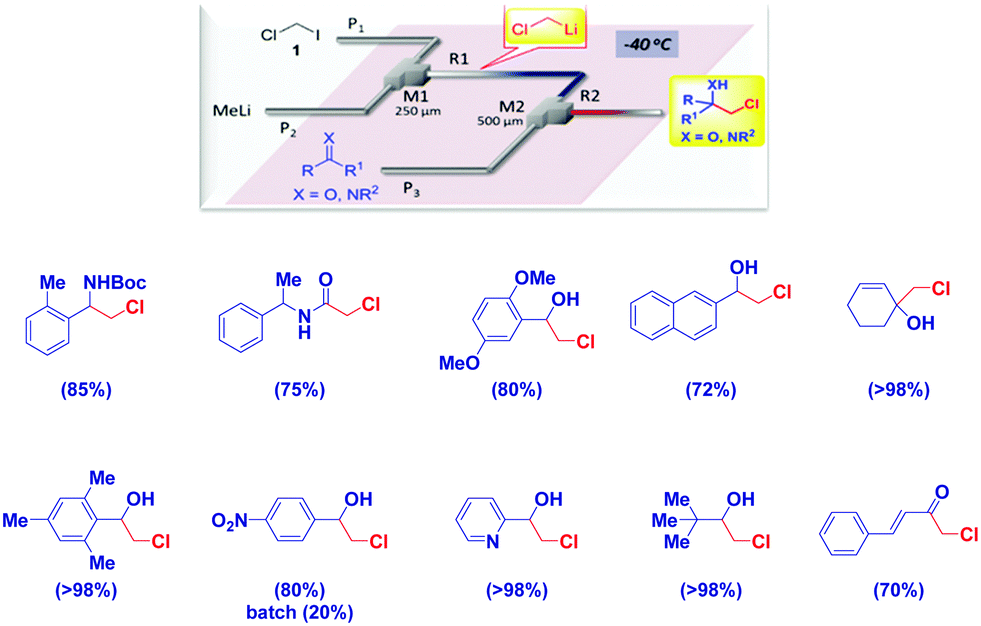 | ||
| Scheme 25 In flow generation of chloromethyllithium and trapping with electrophiles under non Barbier conditions. | ||
Sedelmeier's group employed a similar technique to generate dichloromethyllithium, by treating DCM with n-butyllitium at −30 °C, followed by the addition of aldehydes to form dichlorocarbinols or boronic esters to access the corresponding α-chloro analogues.95 The flow procedure is also amenable for preparing α,α-dibromoketones from esters and LiCHBr2 – which were practically unreactive under classical batch conditions – as shown by Ley.96
Conclusions
The addition of a nucleophilic CHRX fragment to an electrophile can not only be conveniently used for obtaining the result of the (expected) transfer, but also can be envisaged as the first step of a sequence leading to more complex molecular architectures. The instability of α-organometallic substituted species has represented in the past a severe limitation for the design of novel strategies. Fortunately, nowadays the understanding of the mechanisms regulating the formation and the decomposition of these species, contributes to expanding the synthetic versatility of these tactics. The demonstration that analogous reagents are configurationally stable clearly defines one of the next huge challenges in the field: the use at the preparative level of such chiral carbenoids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Vienna for financial support and for a Unidocs fellowship to S. M., and ABCR Germany for the generous supply of fluoroiodomethane (ref. AB291947). L. C. acknowledges the Austrian Academy of Sciences for a postdoctoral L’OREAL grant. We deeply appreciate the collaboration of Prof. Dr Renzo Luisi (University of Bari) for breakthrough studies we conducted on fluorinated organometallics.Notes and references
- J. J. Li, Name Reactions for Homologation, Wiley, Hoboken, 2009 Search PubMed.
- (a) F. Arndt, B. Eistert and W. Ender, Chem. Ber., 1929, 62, 44 CrossRef; (b) N. R. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937 CrossRef PubMed; (c) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef PubMed; (d) V. Pace, G. Verniest, J. V. Sinisterra, A. R. Alcántara and N. De Kimpe, J. Org. Chem., 2010, 75, 5760 CrossRef PubMed; (e) G. Maas, Angew. Chem., Int. Ed., 2009, 48, 8186 CrossRef PubMed; (f) L. Castoldi, L. Ielo, W. Holzer, G. Giester, A. Roller and V. Pace, J. Org. Chem., 2018, 83, 4336 CrossRef PubMed.
- (a) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1962, 84, 867 CrossRef; (b) V. K. Aggarwal and C. L. Winn, Acc. Chem. Res., 2004, 37, 611 CrossRef PubMed.
- G. Boche and J. C. W. Lohrenz, Chem. Rev., 2001, 101, 697 CrossRef PubMed.
- (a) M. Braun, in The Chemistry of Organolithium Compounds, ed. Z. Rappoport and I. Marek, John Wiley and Sons, Chichester, 2004, vol. 1, p. 829 Search PubMed; (b) V. Capriati and S. Florio, Chem. – Eur. J., 2010, 16, 4152 CrossRef PubMed; (c) S. Molitor and V. H. Gessner, Chem. – Eur. J., 2013, 19, 11858 CrossRef PubMed; (d) S. Molitor and V. H. Gessner, Synlett, 2015, 861 Search PubMed; (e) S. Molitor and V. H. Gessner, Angew. Chem., Int. Ed., 2016, 55, 7712 CrossRef PubMed.
- M. Pasco, N. Gilboa, T. Mejuch and I. Marek, Organometallics, 2013, 32, 942 CrossRef.
- (a) W. Kirmse, Angew. Chem., Int. Ed. Engl., 1965, 4, 1 CrossRef; (b) G. Köbrich, Angew. Chem., Int. Ed. Engl., 1972, 11, 473 CrossRef.
- (a) V. Pace, W. Holzer and N. De Kimpe, Chem. Rec., 2016, 16, 2061 CrossRef PubMed; (b) V. Pace, Aust. J. Chem., 2014, 67, 311 Search PubMed.
- (a) R. Tarhouni, B. Kirschleger, M. Rambaud and J. Villieras, Tetrahedron Lett., 1984, 25, 835 CrossRef; (b) J. Barluenga, B. Baragaña, A. Alonso and J. M. Concellón, J. Chem. Soc., Chem. Commun., 1994, 969 RSC; (c) J. Barluenga, B. Baragaña and J. M. Concellón, J. Org. Chem., 1995, 60, 6696 CrossRef; (d) J. Barluenga, B. Baragaña, J. M. Concellón, A. Piñera-Nicolás, M. R. Díaz and S. García-Granda, J. Org. Chem., 1999, 64, 5048 CrossRef.
- C. Burford, F. Cooke, E. Ehlinger and P. Magnus, J. Am. Chem. Soc., 1977, 99, 4536 CrossRef.
- (a) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302 CrossRef PubMed; (b) J. Clayden, Organolithiums: Selectivity for Synthesis, Pergamon, Oxford, 2002 Search PubMed; (c) Lithium Compounds in Organic Synthesis: From Fundamentals to Applications, ed. R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 2014 Search PubMed.
- (a) J. M. Concellón, H. Cuervo and R. Fernández-Fano, Tetrahedron, 2001, 57, 8983 CrossRef; (b) T. J. Michnick and D. S. Matteson, Synlett, 1991, 631 CrossRef; (c) A. K. Yudin, Aziridines and Epoxides in Organic Synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed.
- (a) J. M. Concellón, H. Rodríguez-Solla and C. Simal, Org. Lett., 2008, 10, 4457 CrossRef PubMed; (b) J. M. Concellón, H. Rodríguez-Solla, P. L. Bernad and C. Simal, J. Org. Chem., 2009, 74, 2452 CrossRef PubMed; (c) D. Savoia, G. Alvaro, R. Di Fabio, A. Gualandi and C. Fiorelli, J. Org. Chem., 2006, 71, 9373 CrossRef PubMed; (d) J. A. Bull, T. Boultwood and T. A. Taylor, Chem. Commun., 2012, 48, 12246 RSC.
- (a) D. S. Matteson, J. Org. Chem., 2013, 78, 10009 CrossRef PubMed; (b) D. S. Matteson and D. Majumdar, J. Am. Chem. Soc., 1980, 102, 7588 CrossRef.
- (a) P. R. Blakemore, S. P. Marsden and H. D. Vater, Org. Lett., 2006, 8, 773 CrossRef PubMed; (b) P. R. Blakemore and M. S. Burge, J. Am. Chem. Soc., 2007, 129, 3068 CrossRef PubMed; (c) A. L. Barsamian and P. R. Blakemore, Organometallics, 2011, 31, 19 CrossRef; (d) C. R. Emerson, L. N. Zakharov and P. R. Blakemore, Chem. – Eur. J., 2013, 19, 16342 CrossRef PubMed; (e) Z. Wu, X. Sun, K. Potter, Y. Cao, L. N. Zakharov and P. R. Blakemore, Angew. Chem., Int. Ed., 2016, 55, 12285 CrossRef PubMed; (f) P. R. Blakemore and R. W. Hoffmann, Angew. Chem., Int. Ed., 2018, 57, 390 CrossRef PubMed.
- D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174 CrossRef PubMed.
- (a) J. L. Stymiest, G. Dutheuil, A. Mahmood and V. K. Aggarwal, Angew. Chem., Int. Ed., 2007, 46, 7491 CrossRef PubMed; (b) G. Dutheuil, M. P. Webster, P. A. Worthington and V. K. Aggarwal, Angew. Chem., Int. Ed., 2009, 48, 6317 CrossRef PubMed.
- S. Balieu, G. E. Hallett, M. Burns, T. Bootwicha, J. Studley and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 4398 CrossRef PubMed.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183 CrossRef PubMed.
- V. Pace, L. Castoldi, A. D. Mamuye and W. Holzer, Synthesis, 2014, 2897 CrossRef.
- K. M. Sadhu and D. S. Matteson, Tetrahedron Lett., 1986, 27, 795 CrossRef.
- (a) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815 CrossRef; (b) S. Balasubramaniam and I. S. Aidhen, Synthesis, 2008, 3707 Search PubMed; (c) V. Pace, W. Holzer and B. Olofsson, Adv. Synth. Catal., 2014, 356, 3697 CrossRef.
- (a) V. Pace, W. Holzer, G. Verniest, A. R. Alcántara and N. De Kimpe, Adv. Synth. Catal., 2013, 355, 919 CrossRef; (b) V. Pace, L. Castoldi and W. Holzer, J. Org. Chem., 2013, 78, 7764 CrossRef PubMed.
- L. Castoldi, L. Ielo, P. Hoyos, M. J. Hernáiz, L. De Luca, A. R. Alcántara, W. Holzer and V. Pace, Tetrahedron, 2018, 74, 2211 CrossRef.
- L. Castoldi, W. Holzer, T. Langer and V. Pace, Chem. Commun., 2017, 53, 9498 RSC.
- (a) D. A. Evans, G. Borg and K. A. Scheidt, Angew. Chem., Int. Ed., 2002, 41, 3188 CrossRef; (b) A. M. Goldys and C. S. P. McErlean, Eur. J. Org. Chem., 2012, 1877 CrossRef.
- (a) F. F. Fleming, Z. Zhang and P. Knochel, Org. Lett., 2004, 6, 501 CrossRef PubMed; (b) F. F. Fleming, Z. Zhang, G. Wei and O. W. Steward, Org. Lett., 2005, 7, 447 CrossRef PubMed; (c) D. Nath and F. F. Fleming, Chem. – Eur. J., 2013, 19, 2023 CrossRef PubMed.
- A. D. Mamuye, L. Castoldi, U. Azzena, W. Holzer and V. Pace, Org. Biomol. Chem., 2015, 13, 1969 Search PubMed.
- V. Pace, I. Murgia, S. Westermayer, T. Langer and W. Holzer, Chem. Commun., 2016, 52, 7584 RSC.
- (a) A. Guijarro and M. Yus, Tetrahedron Lett., 1993, 34, 3487 CrossRef; (b) A. Guijarro, B. Mancheño, J. Ortiz and M. Yus, Tetrahedron, 1996, 52, 1643 CrossRef.
- R. Senatore, L. Ielo, E. Urban, W. Holzer and V. Pace, Eur. J. Org. Chem., 2018 DOI:10.1002/ejoc.201800095.
- (a) N. De Kimpe and R. Verhé, in The Chemistry of α-Haloketones, α-Haloaldehydes and α-Haloimines, ed. S. Patai, Wiley, Chichester, 1988, p. 1 Search PubMed; (b) F. Colpaert, S. Mangelinckx, M. T. Rocchetti and N. De Kimpe, Org. Biomol. Chem., 2011, 9, 549 RSC; (c) N. de Kimpe, W. de Cock and C. Stevens, Tetrahedron, 1992, 48, 2739 CrossRef.
- P. Mizar and T. Wirth, Angew. Chem., Int. Ed., 2014, 53, 5993 CrossRef PubMed.
- R. Senatore, L. Castoldi, L. Ielo, W. Holzer and V. Pace, Org. Lett., 2018, 20, 2685 CrossRef PubMed.
- (a) T. G. Back and R. G. Kerr, Tetrahedron Lett., 1982, 23, 3241 CrossRef; (b) T. G. Back and R. G. Kerr, Tetrahedron, 1985, 41, 4759 CrossRef.
- (a) H. J. Reich, S. K. Shah and F. Chow, J. Am. Chem. Soc., 1979, 101, 6648 CrossRef; (b) T. Wirth, Organoselenium Chemistry: Synthesis and Reactions, Wiley-VCH, Weinheim, 2011 Search PubMed.
- V. Pace, L. Castoldi and W. Holzer, Adv. Synth. Catal., 2014, 356, 1761 CrossRef.
- Z. Ke, Y. Zhou, H. Gao, C. Zhao and D. L. Phillips, Chem. – Eur. J., 2007, 13, 6724 CrossRef PubMed.
- M. J. Duran-Pena, M. E. Flores-Giubi, J. M. Botubol-Ares, L. M. Harwood, I. G. Collado, A. J. Macias-Sanchez and R. Hernandez-Galan, Org. Biomol. Chem., 2016, 14, 2731 Search PubMed.
- V. Pace, L. Castoldi, P. Hoyos, J. V. Sinisterra, M. Pregnolato and J. M. Sánchez-Montero, Tetrahedron, 2011, 67, 2670 CrossRef.
- (a) A. D. Mamuye, S. Monticelli, L. Castoldi, W. Holzer and V. Pace, Green Chem., 2015, 17, 4194 RSC; (b) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef PubMed.
- V. Pace, L. Castoldi, A. D. Mamuye, T. Langer and W. Holzer, Adv. Synth. Catal., 2016, 358, 172 CrossRef.
- (a) T. Onishi, N. Hirose, T. Nakano, M. Nakazawa and K. Izawa, Tetrahedron Lett., 2001, 42, 5883 CrossRef; (b) T. Onishi, T. Nakano, N. Hirose, M. Nakazawa and K. Izawa, Tetrahedron Lett., 2001, 42, 5887 CrossRef.
- W. Gohring, S. Gokhale, H. Hilpert, F. Roessler, M. Schlageter and P. Vogt, Chimia, 1996, 50, 532 Search PubMed.
- I. Gorokhovik, L. Neuville and J. Zhu, Org. Lett., 2011, 13, 5536 CrossRef PubMed.
- M. Chouhan, K. R. Senwar, R. Sharma, V. Grover and V. A. Nair, Green Chem., 2011, 13, 2553 RSC.
- (a) S. Hajra, S. Maity and R. Maity, Org. Lett., 2015, 17, 3430 CrossRef PubMed; (b) S. Hajra, S. Maity and S. Roy, Adv. Synth. Catal., 2016, 358, 2300 CrossRef; (c) S. Hajra, S. Roy and S. Maity, Org. Lett., 2017, 19, 1998 CrossRef PubMed.
- J. He, J. Ling and P. Chiu, Chem. Rev., 2014, 114, 8037 CrossRef PubMed.
- V. Pace, L. Castoldi, E. Mazzeo, M. Rui, T. Langer and W. Holzer, Angew. Chem., Int. Ed., 2017, 56, 12677 CrossRef PubMed.
- (a) J. Meinwald, S. S. Labana and M. S. Chadha, J. Am. Chem. Soc., 1963, 85, 582 CrossRef; (b) B. Rickborn and R. M. Gerkin, J. Am. Chem. Soc., 1971, 93, 1693 CrossRef; (c) M. E. Jung and D. C. D'Amico, J. Am. Chem. Soc., 1995, 117, 7379 CrossRef.
- M. Lautens, S. G. Ouellet and S. Raeppel, Angew. Chem., Int. Ed., 2000, 39, 4079 CrossRef PubMed.
- (a) M. Kimura, Y. Horino, R. Mukai, S. Tanaka and Y. Tamaru, J. Am. Chem. Soc., 2001, 123, 10401 CrossRef PubMed; (b) A. R. Brown, W.-H. Kuo and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 9286 CrossRef PubMed; (c) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef PubMed; (d) B. Yann, T. Brodie, F. Vincent and S. Mathieu, Angew. Chem., Int. Ed., 2017, 56, 7460 CrossRef PubMed.
- T. B. Wright and P. A. Evans, J. Am. Chem. Soc., 2016, 138, 15303 CrossRef PubMed.
- (a) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682 CrossRef PubMed; (b) M. Simaan, P.-O. Delaye, M. Shi and I. Marek, Angew. Chem., Int. Ed., 2015, 54, 12345 CrossRef PubMed; (c) D. S. Muller and I. Marek, Chem. Soc. Rev., 2016, 45, 4552 RSC.
- (a) The Amide Linkage. Selected Structural Aspects in Chemistry, Biochemistry and Materials Science, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, John Wiley & Sons Inc., New York, 2000 Search PubMed; (b) R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029 CrossRef PubMed; (c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- H. Gilman and C. R. Kinney, J. Am. Chem. Soc., 1924, 46, 493 CrossRef.
- (a) I. Stefanuti, S. A. Smith and R. J. K. Taylor, Tetrahedron Lett., 2000, 41, 3735 CrossRef; (b) M. I. Antczak and J. M. Ready, Chem. Sci., 2012, 3, 1450 RSC.
- (a) G. Schäfer, C. Matthey and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9173 CrossRef PubMed; (b) G. Schäfer and J. W. Bode, Chimia, 2014, 68, 252 CrossRef PubMed.
- (a) X. Wang, M. Nakajima, E. Serrano and R. Martin, J. Am. Chem. Soc., 2016, 138, 15531 CrossRef PubMed; (b) A. Correa and R. Martin, J. Am. Chem. Soc., 2014, 136, 7253 CrossRef PubMed.
- V. Pace, S. Monticelli, K. de la Vega-Hernandez and L. Castoldi, Org. Biomol. Chem., 2016, 14, 7848 Search PubMed.
- V. Pace, L. Castoldi and W. Holzer, Chem. Commun., 2013, 49, 8383 RSC.
- V. Pace, K. de la Vega-Hernández, E. Urban and T. Langer, Org. Lett., 2016, 18, 2750 CrossRef PubMed.
- M. M. Więcław and S. Stecko, Eur. J. Org. Chem., 2018 DOI:10.1002/ejoc.201701537.
- B. Yde, N. M. Yousif, U. Pedersen, I. Thomsen and S. O. Lawesson, Tetrahedron, 1984, 40, 2047 CrossRef.
- V. Pace, L. Castoldi, S. Monticelli, S. Safranek, A. Roller, T. Langer and W. Holzer, Chem. – Eur. J., 2015, 21, 18966 CrossRef PubMed.
- P. Knochel, X. Yang and N. Gommermann, in Handbook of Functionalized Organometallics, ed. P. Knochel, Wiley-VCH, Weinheim, 2005, vol. 2, p. 379 Search PubMed.
- D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 36, 2282 CrossRef.
- S. T. Kerrick and P. Beak, J. Am. Chem. Soc., 1991, 113, 9708 CrossRef.
- T. S. Jagodziński, Chem. Rev., 2003, 103, 197 CrossRef PubMed.
- (a) S. Dixon, S. M. Fillery, A. Kasatkin, D. Norton, E. Thomas and R. J. Whitby, Tetrahedron, 2004, 60, 1401 CrossRef; (b) E. Negishi, K. Akiyoshi, B. O'Connor, K. Takagi and G. Wu, J. Am. Chem. Soc., 1989, 111, 3089 CrossRef; (c) A. Kasatkin and R. J. Whitby, Tetrahedron Lett., 1997, 38, 4857 CrossRef; (d) A. N. Kasatkin and R. J. Whitby, Tetrahedron Lett., 2000, 41, 6201 CrossRef.
- T. Boddaert, C. François, L. Mistico, O. Querolle, L. Meerpoel, P. Angibaud, M. Durandetti and J. Maddaluno, Chem. – Eur. J., 2014, 20, 10131 CrossRef PubMed.
- V. Pace, A. Pelosi, D. Antermite, O. Rosati, M. Curini and W. Holzer, Chem. Commun., 2016, 52, 2639 RSC.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley-VCH, Weinheim, 5th edn, 2001 Search PubMed.
- N. Petragnani and G. Schill, Chem. Ber., 1970, 103, 2271 CrossRef.
- L. Field and H.-K. Chu, J. Org. Chem., 1977, 42, 1768 CrossRef.
- L. Field and C. H. Banks, J. Org. Chem., 1975, 40, 2774 CrossRef.
- (a) M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667 CrossRef PubMed; (b) V. Pace and R. Luisi, ChemCatChem, 2014, 6, 1516 CrossRef.
- (a) T. Satoh, Chem. Soc. Rev., 2007, 36, 1561 RSC; (b) T. Satoh, Heterocycles, 2012, 85, 1 CrossRef; (c) T. Kimura, Synthesis, 2017, 5105 CrossRef.
- (a) R. H. V. Nishimura, V. E. Murie, R. A. Soldi, J. L. C. Lopes and G. C. Clososki, J. Braz. Chem. Soc., 2015, 26, 2175 Search PubMed; (b) R. H. V. Nishimura, V. E. Murie, R. A. Soldi and G. C. Clososki, Synthesis, 2015, 1455 Search PubMed; (c) R. H. V. Nishimura, F. T. Toledo, J. L. C. Lopes and G. C. Clososki, Tetrahedron Lett., 2013, 54, 287 CrossRef.
- V. A. Vu, I. Marek, K. Polborn and P. Knochel, Angew. Chem., Int. Ed., 2002, 41, 351 CrossRef PubMed.
- V. A. Vu, I. Marek and P. Knochel, Synthesis, 2003, 1797 Search PubMed.
- R. W. Hoffmann and P. G. Nell, Angew. Chem., Int. Ed., 1999, 38, 338 CrossRef PubMed.
- T. Hampel, T. Neubauer, T. van Leeuwen and T. Bach, Chem. – Eur. J., 2012, 18, 10382 CrossRef PubMed.
- (a) R. W. Hoffmann, P. G. Nell, R. Leo and K. Harms, Chem. – Eur. J., 2000, 6, 3359 CrossRef PubMed; (b) R. W. Hoffmann, Chem. Soc. Rev., 2003, 32, 225 RSC.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (b) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef PubMed; (c) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef PubMed.
- C. Ni and J. Hu, Synlett, 2011, 770 Search PubMed.
- (a) V. H. Gessner, Chem. Commun., 2016, 52, 12011 RSC; (b) S. Molitor, K.-S. Feichtner and V. H. Gessner, Chem. – Eur. J., 2017, 23, 2527 CrossRef PubMed.
- W. Zhang, C. Ni and J. Hu, Top. Curr. Chem., 2011, 308, 25 CrossRef.
- X. Shen, M. Zhou, C. Ni, W. Zhang and J. Hu, Chem. Sci., 2014, 5, 117 RSC.
- (a) D. C. Kail, P. Malova Krizkova, A. Wieczorek and F. Hammerschmidt, Chem. – Eur. J., 2014, 20, 4086 CrossRef PubMed; (b) P. Malova Krizkova and F. Hammerschmidt, Eur. J. Org. Chem., 2013, 5143 CrossRef PubMed; (c) D. C. Kapeller and F. Hammerschmidt, J. Am. Chem. Soc., 2008, 130, 2329 CrossRef PubMed.
- G. Parisi, M. Colella, S. Monticelli, G. Romanazzi, W. Holzer, T. Langer, L. Degennaro, V. Pace and R. Luisi, J. Am. Chem. Soc., 2017, 139, 13648 CrossRef PubMed.
- L.-P. B. Beaulieu, J. F. Schneider and A. B. Charette, J. Am. Chem. Soc., 2013, 135, 7819 CrossRef PubMed.
- (a) S. Monticelli and V. Pace, Aust. J. Chem., 2018 DOI:10.1071/CH18045 ; For the preparation of ICH2F, see: ; (b) J. Hu, B. Gao, L. Li, C. Ni and J. Hu, Org. Lett., 2015, 17, 3086 CrossRef PubMed.
- L. Degennaro, F. Fanelli, A. Giovine and R. Luisi, Adv. Synth. Catal., 2015, 357, 21 CrossRef.
- A. Hafner, V. Mancino, M. Meisenbach, B. Schenkel and J. Sedelmeier, Org. Lett., 2017, 19, 786 CrossRef PubMed.
- J. Hartwig, J. B. Metternich, N. Nikbin, A. Kirschning and S. V. Ley, Org. Biomol. Chem., 2014, 12, 3611 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |