
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free C–H alkylation of heteroarenes with alkyltrifluoroborates: a general protocol for 1°, 2° and 3° alkylation†
Jennifer K.
Matsui
 ,
David N.
Primer
and
Gary A.
Molander
*
,
David N.
Primer
and
Gary A.
Molander
*
Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 S. 34th Street, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: gmolandr@sas.upenn.edu
First published on 6th March 2017
Abstract
A photoredox-catalyzed C–H functionalization of heteroarenes using a variety of primary, secondary, and tertiary alkyltrifluoroborates is reported. Using Fukuzumi's organophotocatalyst and a mild oxidant, conditions amenable for functionalizing complex heteroaromatics are described, providing a valuable tool for late-stage derivatization. The reported method addresses the three major limitations of previously reported photoredox-mediated Minisci reactions: (1) use of superstoichiometric amounts of a radical precursor, (2) capricious regioselectivity, and (3) incorporation of expensive photocatalysts. Additionally, a number of unprecedented, complex alkyl radicals are used, thereby increasing the chemical space accessible to Minisci chemistry. To showcase the application in late-stage functionalization, quinine and camptothecin analogues were synthesized. Finally, NMR studies were conducted to provide a rationalization for the heteroaryl activation that permits the use of a single equivalent of radical precursor and also leads to enhanced regioselectivity. Thus, by 1H and 13C NMR a distinct heteroaryl species was observed in the presence of acid catalyst and BF3.
Introduction
Heteroaryl subunits are among the most commonly employed, privileged structures in medicinal chemistry.1 Unfortunately, the synthesis of many of these structures is often predicated on cyclization reactions2 from simple starting materials (e.g., aldehydes, hydrazines, alkynes, etc.). Although these methods often prove advantageous when the precursors needed for a particular molecular target are inexpensive and commercially available, these sequences are untenable when attempting to access a diverse library of structurally rich heteroarenes with a common core (Fig. 1). | ||
| Fig. 1 Complex heteroarenes poised for diversification. | ||
To overcome this challenge, two predominant anionic strategies have emerged for diversifying common heteroaryl motifs (Scheme 1): fluorination to enable facile SNAr-based chemistry3 and halogenation (bromination,4 chlorination5) to provide a site for cross-coupling.6 These methods are highly effective for many systems, but in numerous cases installation of the halide at the desired site is unselective or impossible, requiring re-evaluation of the synthetic strategy. Furthermore, when successful incorporation of halides has occurred, heteroarenes are often obstinate partners in cross-coupling chemistry,7 requiring careful screening to achieve fruitful union. Most importantly, from an efficiency standpoint, these strategies are sub-optimal as they first require a functional handle to be installed, only to be immediately replaced in a subsequent displacement reaction.
 | ||
| Scheme 1 Approaches to heteroarene functionalization. | ||
Radical alkylations of heteroarenes (Minisci reactions)8 represent a more direct functionalization of specific C–H bonds. For this reason, radical alkylation strategies have recently risen to prominence for the late-stage functionalization of heteroaryl systems. Although Minisci and coworkers initially used silver oxidants at elevated temperatures to generate alkyl radicals from the corresponding carboxylic acids,9 advances in alkyl radical generation have enabled much milder methods to be developed (Scheme 1). These have included methods using boronic acids,10 peresters,11 and other precursors12 under much less forcing conditions. Most notably, the Baran laboratory has pioneered the use of sulfinate salts13 that have enabled the extremely facile introduction of functionalized alkyl radicals into an impressive array of heteroarenes. These methods allow the formal C–H alkylation of heterocycles in a mild, metal-free manner – drawing rapid adoption by medicinal chemists for the late stage diversification of pharmaceutically relevant compounds.14
Unfortunately, even with these advances, current methods are not without their limitations, particularly with regard to the continued requirement for high loadings of both the radical partner and oxidant. Even the milder methods employing sulfinate salts often require a significant excess of an expensive and synthetically demanding precursor (3–6 equiv.) and oxidant (5–10 equiv.) to achieve good yields.13 To date, methods grounded on stoichiometric radical generation have been continually limited by parasitic reactivity (e.g., homocoupling, H-atom abstraction, and chain processes) that appear to outcompete the desired Minisci alkylation. As such, often the only solution has been to push reactions to completion with increased reagent loadings or successive dosing (Scheme 1A).
To combat the disproportionate stoichiometry and typically harsh conditions, other groups have turned to photoredox catalysis for the efficient generation of radical.15 Contributions by the MacMillan,16 DiRocco,17 Chen,18 and Barriault19 groups have all highlighted the advantages afforded under this paradigm. Although a variety of alkyl radicals precursors were presented in these photoredox contributions, each method exhibits some inherent limitations. For example, the MacMillan group targets weak C–H bonds, limited to α-heteroatom bonds. Barriault and coworkers demonstrated that alkyl bromides could be reduced to generate the corresponding alkyl radicals, but 3 equivalents of these precursors and a UVA LED light source were required. Additionally, MacMillan and Barriault used expensive iridium or gold photocatalysts, respectively. The recent report by Chen addressed many of these issues with the use of sustainable boronic acid partners and inexpensive Ru(bpy)3, but their method employs the use of a stoichiometric iodine oxidant that seems uniquely required for activation.
In light of these advances, we recognized the potential to marry photoredox Minisci chemistry with our laboratory's interest in alkyltrifluoroborate reagents, especially given reports by our laboratory and others20 on the favorable single electron oxidation potentials of these salts to form alkyl radicals (Ered = +1.10 V vs. SCE for 1° benzylic20a and +1.50 V vs. SCE for 2° alkyltrifluoroborates20b). Here, with appropriate organic photocatalyst selection (for 1: *Ered = +2.06 V vs. SCE21), all representative radical classes could be activated under identical reaction conditions (Scheme 1B). In this scenario, one can envision photocatalyst excitation serving as a chaperone for steady, synchronized, catalytic radical generation. Such a protocol ensures that an excess of radicals are not generated, minimizing at least some of the byproducts formed through stoichiometric processes. The photoredox catalyst-coordinated generation of radicals in this manner leads to improvement in reagent and oxidant loading, thus enhancing the efficiency and sustainability of these methods. Serendipitously, these aforementioned concepts, in conjunction with the generation of Lewis acidic BF3 byproduct, also led to enhanced reactivity and selectivity in these transformations.
Results and discussion
Mechanistically, we anticipated that visible light irradiation of organic photocatalyst 1 would generate the excited complex 1*, which is highly oxidizing and capable of oxidizing an alkyltrifluoroborate to release the desired alkyl radical 3 and BF3 (Scheme 2). The generated radical 3 can then intercept the protonated heteroarene to form the radical cation 5. Reduction of persulfate (Ered = >+0.35 V vs. SCE22) by the reduced form of the photocatalyst 4 (Ered = +0.49 V vs. SCE21) generates the sulfate dianion and sulfate radical anion, regenerating photocatalyst 1.21 Finally, H-atom abstraction of 5 by the sulfate radical anion leads to rearomatization, affording the desired alkylated heteroarene 6.16 Quantum yield studies indicate this is not a radical chain process as evidenced by a ϕ of 0.31 (see ESI†). Furthermore, addition of excess allyl acetate (a known sulfate radical anion trap23) did not interfere with reaction efficiency or conversion. | ||
| Scheme 2 Proposed mechanism. | ||
To begin this optimization, we keyed in on tertiary radical partners for our initial studies. In particular, tert-butyltrifluoroborate as the appropriate radical precursor and isoquinoline carboxylate as the heteroaryl partner. Before screening various photocatalysts, controls probing the need for oxidant (Table 1, entry 2), light (entry 3), and photocatalyst (entry 4) were completed. Additionally, the absence of acid (entry 5) afforded 30% conversion to product, although as alkyl radical 3 is generated, BF3 (an electron-deficient Lewis acid) is concomitantly being formed. Performing the reaction in air provided a comparable yield (entry 6) to a reaction carried out under inert conditions (entry 1).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | % yieldb |
a Optimization reactions were run with heteroarene (1.0 equiv.), tert-butyltrifluoroborate (1.0 equiv.), photocatalyst (5 mol%), K2S2O8 (2.0 equiv.), trifluoroacetic acid (1.0 equiv.) in MeCN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) on 0.1 mmol scale.
b Yields were obtained by HPLC using a calibration curve. :1) on 0.1 mmol scale.
b Yields were obtained by HPLC using a calibration curve.
|
||
| 1 | None | 57 |
| 2 | No persulfate | <5 |
| 3 | No light | <5 |
| 4 | No photocatalyst | 0 |
| 5 | No acid | 30 |
| 6 | Open to air (needle puncture) | 53 |
| 7 | Ru(bpy)3Cl2 (photocatalyst) | 8 |
| 8 | Ru(bpz)3Cl2 (photocatalyst) | 16 |
| 9 | lr[dFCF3ppy]2(bpy)PF6 (photocatalyst) | 86 |
| 10 | 4CzIPN (photocatalyst) | 96 |
| 11 | 2.0 equiv. trifluoroborate | 45 |
| 12 | 4.0 equiv. persulfate | 84 |
|
||
| Photocatalysts | ||
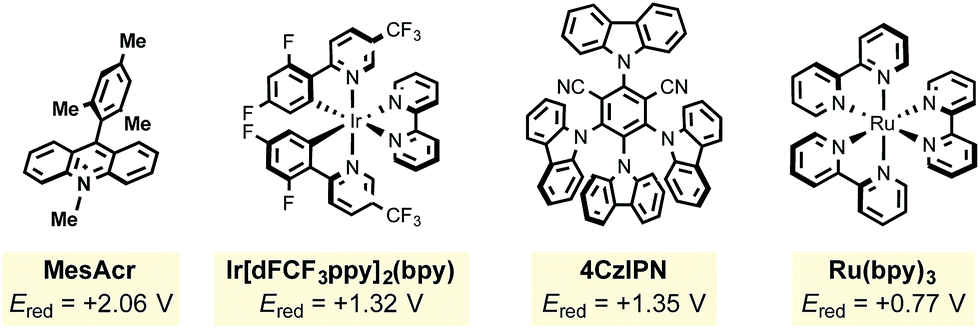
|
||

The mesityl acridinium photocatalyst was then compared to other photocatalysts (entries 7–10). Noting the need for a relatively high oxidizing potential of the photocatalyst, it was expected that ruthenium photocatalysts (with significantly lower oxidation potentials than the acridinium dyes) would experience diminished yields, which proved to be the case (entries 7 and 8). During the period that photoredox catalysis has gained traction in the organic chemistry community, one major criticism has been the high cost of the key metal-based photocatalysts (i.e., those based on iridium and ruthenium). Nicewicz24 and Zhang25 have explored alternative organophotocatalysts that have been shown to exhibit similar reactivity to their metal-based counterparts. Surprisingly, 4CzIPN (entry 10) significantly outperformed MesAcr over a 12 h period. Neither excess alkyltrifluoroborate (entry 11) nor excess oxidant (entry 12) provided any perceived advantages.
In view of their low cost and ease of access, going forward we focused on the two organophotocatalysts, MesAcr and 4CzIPN. To determine the most suitable organic photocatalyst for our purposes, we scaled up the reactions described in entries 1 and 10, and discovered that running the reaction for 16 h afforded similar yields, consistent with the small scale reactions. It was important for this protocol to be general for primary, secondary, and tertiary alkyltrifluoroborates. Because primary alkyltrifluoroborates have a markedly higher oxidation potential (Ered = +1.90 V vs. SCE20k) than secondary or tertiary alkyltrifluoroborates, MesAcr and 4CzIPN were compared with a primary alkyltrifluoroborate (Scheme 3). In this assay, MesAcr outperformed CzIPN with 76% versus <10% isolated yields, respectively.
 | ||
| Scheme 3 Comparing organophotocatalysts. | ||
Tricoordinate organoboron compounds are isoelectronic with carbocations, and thus would not be expected to be easily oxidized by single electron transfer (SET) processes. To confirm that this chemistry was unique to alkyltrifluoroborates, a variety of organoboron reagents were synthesized and analyzed by cyclic voltammetry to determine their relative oxidation potentials (Table 2). As expected, the trivalent organoboron variants exhibit very high oxidation potentials as compared to their tetravalent, “ate” complex analogues. Consequently, under the reaction conditions, no radical formation from the boronic acids and/or esters was observed. For the tetravalent species, triolborates exhibit comparatively low reduction potentials. Unfortunately, the swift hydrolysis of triolborates under the acidic, aqueous reaction conditions forms the redox inactive boronic acid.26 By contrast, alkyltrifluoroborates are both stable to the reaction conditions and redox amenable, resulting in an excellent 72% yield with methyl isoquinoline carboxylate after 16 h.
|
|
|||
|---|---|---|---|
| Alkylboron | Reduction potential | Result | Drawbacks |
|
a Reactions were run with heteroarene (1.0 equiv.), alkylboron reagent (1.0 equiv.), MesAcr (5 mol%), K2S2O8 (2.0 equiv.), trifluoroacetic acid (1.0 equiv.) in MeCN/H2O (1:1) on 0.2 mmol scale.
|
|||
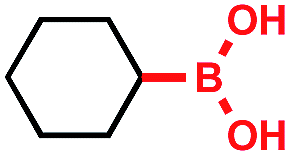
|
>2.5 V vs. SCE | No conversion | • Air sensitive |
| • Poor redox properties | |||

|
>2.5 V vs. SCE | No conversion | • Limited commercial availability |
| • Poor atom economy | |||
| • Poor redox properties | |||

|
>2.5 V vs. SCE | <5% conversion | • Air sensitive |
| • Poor atom economy | |||
| • Poor redox properties | |||

|
+1.1 V vs. SCE | No conversion | • Limited commercial availability |
| • Hydrolytically unstable | |||
| • Poor atom economy | |||

|
|||

tert-Butyltrifluoroborate was selected to examine the amenability of various heteroarene partners (Table 3). Regioselectivity was probed using quinoline itself, leaving both the C2 and C4 positions available for alkylation. Currently there are two methods for installing tertiary radicals regioselectively into the quinoline core structure. Namgoong and coworkers reported the use of excess TMP-zincate to deprotonate selectively at the C2 position, followed by incorporation of the tert-butyl substituent.27 This was a singular transformation, with no demonstrated scope. Additionally, Minisci and coworkers have reported selective C2 C–H substitution on quinolines using a 3-fold excess of alkyl iodide radical precursors.8b Although efficient, drawbacks of this approach included using peroxide as the oxidant, thereby limiting functional group tolerance, and the limited number of commercially available, complex alkyl iodides. Therefore, we were pleased to observe regioselective addition to the C2 position of the heteroarene in 72% yield (1a). With substitution at the C4 position of quinoline, the reaction reached full conversion to product within 16 h as determined by GC-MS analysis (1b). When isolated, a 95% yield of the desired product was achieved. Beyond alkyl substituents, halides and trifluoromethyl groups incorporated within the heteroaromatics allowed excellent conversion to product (1c, 1d).

In addition to quinoline cores, indazole (1e), isoquinoline (1g), and quinoxaline (1h, 1i) moieties were successful partners under standard conditions. Indazole 1e is particularly notable because of the prevalence in medicinal chemistry1 and, to the best of our knowledge, the absence of Minisci examples in the literature. As expected, the use of the benzimidazole core resulted in no conversion (1f) because of the electron-rich nature of the C2 site. Additionally, N-methyl benzimidazole was similarly unreactive. Pyridines also exhibited selective mono-addition (1j–1l, 1n). There has appeared only one report of nicotinamide alkylation via Minisci chemistry, wherein three equiv. of the corresponding carboxylic acid were necessary under silver catalyzed conditions.28 A medicinally relevant core, quinazolinone 1p, afforded product in 90% yield. Purine 1q contains two sites of potential radical addition, but substitution was observed only on the pyrimidine subunit. Overall, this method is broadly tolerant of a wide range of functional groups as demonstrated with quinine 1r (possessing alkene, alcohol, tertiary amine groups). Notably, the tertiary amine embedded within quinine did not interfere with the photocatalytic cycle despite its propensity for competitive oxidation.
Currently, there are a limited number of tertiary alkylboranes commercially available and no alkyltrifluoroborates. Presumably, this is a result of narrow synthetic utility compared to their primary and secondary counterparts. Although we demonstrated that tert-butyltrifluoroborate was viable, more highly elaborated systems could also be incorporated. Cook and coworkers recently reported a manganese-catalyzed borylation of alkyl bromides. Using Cook's procedure,29 >1 g of pinacolborane intermediate 1t was synthesized from the corresponding alkyl bromide 1s. This borylation was followed by treatment with saturated KHF2 to afford the corresponding alkyltrifluoroborate 1u (Scheme 4).
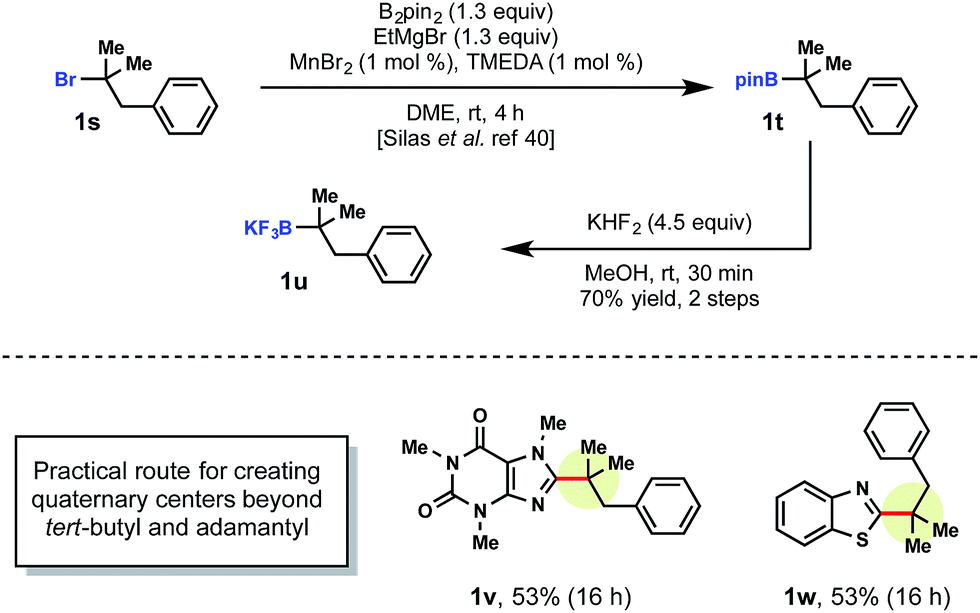 | ||
| Scheme 4 Synthesis of tertiary alkyltrifluoroboratea. aReactions were run with heteroarene (1.0 equiv.), tertiary alkyltrifluoroborate (1.0 equiv.), MesAcr (5 mol%), K2S2O8 (2.0 equiv.), trifluoroacetic acid (1.0 equiv.) in MeCN/H2O (1:1) on 0.3 mmol scale. | ||
With tertiary alkyltrifluoroborate 1u in hand, radical addition to caffeine 1v and benzothiophene 1w was carried out. The development of the Cook approach to 3° alkyltrifluoroborates thus allows access to an even greater array of coupling partners.
In further studies, a wide range of alkyltrifluoroborates were found to be suitable partners in the developed photoredox Minisci conditions (Table 4). First, a benzyl protected α-alkoxyalkyltrifluoroborate afforded 2a in 58% yield. Additional examples of unactivated, secondary alkyl radical precursors could be appended as in 2b–2j. For 2b, the successful addition of fluorinated isosteres is encouraging, given the well-documented propensity these subunits have for modulating solubility and binding affinity in medicinal chemistry.30 For 2c–2e, the tetrahydropyranyl and piperidinyl moieties are commonly introduced ring structures that serve as useful probes for H-bond donors/acceptors in SAR efforts.31

To the best of our knowledge, primary alcohols appended to the radical coupling partner have not been reported in Minisci-type reactions, perhaps owing to their propensity to undergo H-atom abstraction alpha to the hydroxyl group. Therefore, we were pleased to obtain 2f, albeit in low yield. Of note, this selectivity is complementary to the MacMillan group's work on alcohols as radical precursors in photoredox-Minisci couplings.16a A further exploration of 5- and 4-membered heterocycles (2g–2i) was conducted with promising results, where tetrahydrofuran 2g was afforded in 34% yield, but access to pyrrolidine 2h was not successful. For azetidine 2i, standard solvent conditions provided only trace amounts of product, but switching to DCE/H2O (1:1) proved to be beneficial. Cyclopropyltrifluoroborate, although unproductive in previously reported photoredox/Ni dual cross-coupling methods,20b afforded 2j, albeit in 20% yield. 1-Adamantyltrifluoroborate has never been used in Minisci-type alkylations. Using the standard conditions, 2k was isolated in 45% yield. Lastly, chemoselective radical addition to the isoquinoline was observed for 2l despite containing an alternate site for alkylation within the alkyltrifluoroborate partner.
Moving forward, this Minisci process was anticipated to be broadly applicable even to primary alkyltrifluoroborates, which possess relatively high oxidation potentials (Table 5). As an initial foray, we first investigated whether stabilized primary α-alkoxymethyltrifluoroborates could be competent partners in this process (3a–3d). Non-stabilized alkyl radicals were also demonstrated in examples 3e–3l. Notably, 3j contains an ortho bromide functional handle for further elaboration. Unfortunately, electron deficient 3,3,3-trifluoropropyltrifluoroborate afforded 3l in diminished yield, likely because of its high oxidation potential and resultant destabilization of the generated radical. These trends track with other recent reports specifically targeting more electrophilic radicals.13e,16b
|
a Reactions were run with 1-methyl isoquinolinecarboxylate (1.0 equiv.), primary alkyltrifluoroborate (1.0 equiv.), MesAcr (5 mol%), K2S2O8 (2.0 equiv.), trifluoroacetic acid (1.0 equiv.) in MeCN/H2O (1:1) on 0.3 mmol scale.
|
|---|
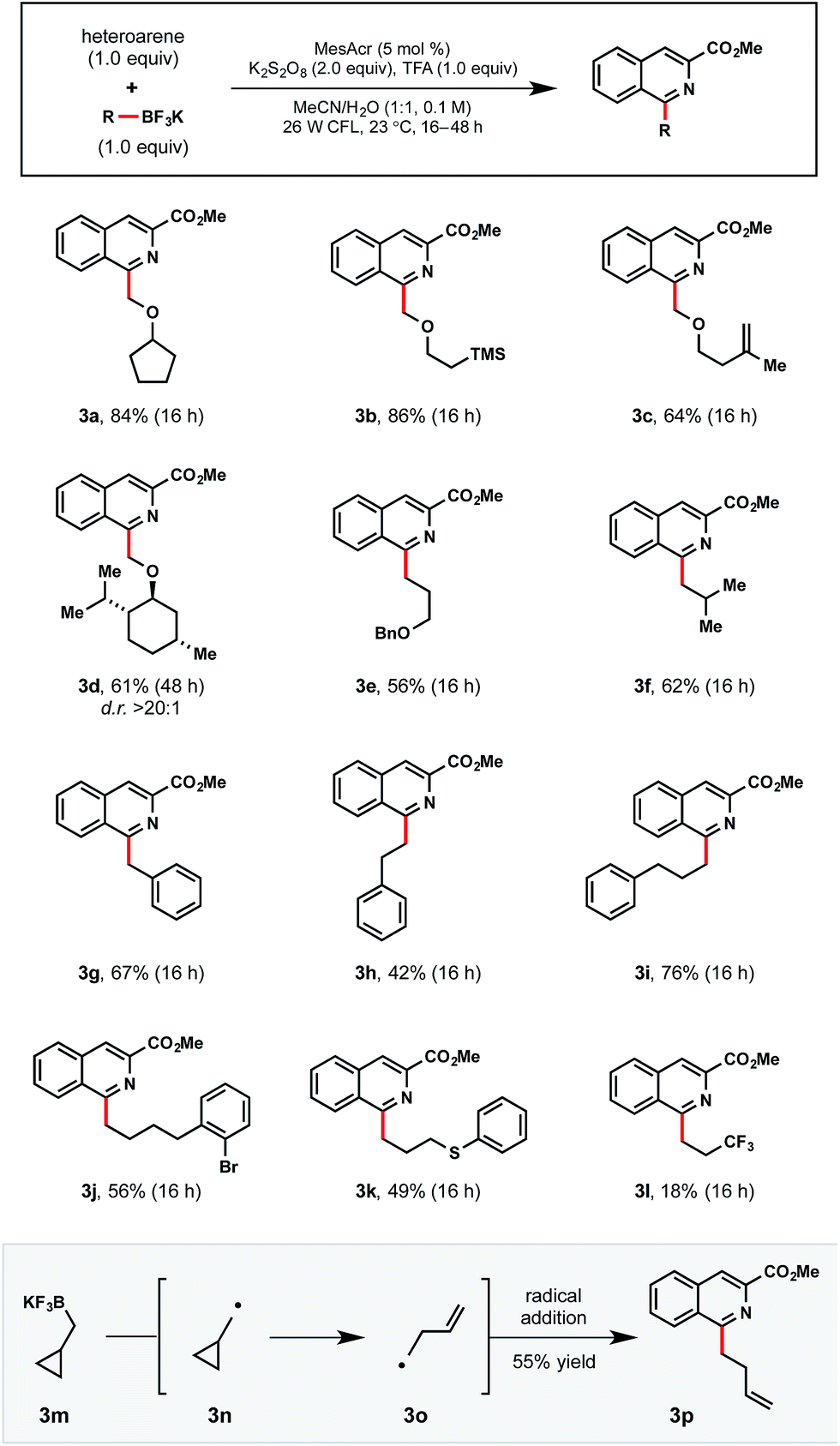
|
As would be anticipated for the radical-based mechanism of the transformation, cyclopropylcarbinyltrifluoroborate 3m formed the corresponding radical 3o after SET oxidation and subsequent rearrangement. Following radical addition to the activated heteroaromatic, 3p was isolated in 55% yield.
The diversity of the process was expanded to underline the potential for widespread utility (Table 6). Lepidine was coupled with a variety of stabilized α-alkoxy (4a–4c) and unstabilized (4d–4f) radicals. With quinoline, C2 regiospecificity was again observed (4d, 4e), albeit in diminished yields. This is particularly noteworthy given the challenges associated with avoiding complex mixtures of isomers in prior Minisci-type reaction reports.32 With heteroarene cores such as pyridine (4f), benzothiazole (4g), indazole (4h), and quinazolinone (4i, 4j), exceptional yields were observed. When exploring reactivity with caffeine, methylcyclopentyltrifluoroborate afforded 4k in 37% yield. Additionally, a secondary alkyl radical was successfully appended to functionally rich quinine as demonstrated in 4l.
|
a Reactions were run with heteroarene (1.0 equiv.), alkyltrifluoroborate (1.0 equiv.), MesAcr (5 mol%), K2S2O8 (2.0 equiv.), trifluoroacetic acid (1.0 equiv.) in MeCN/H2O (1:1) on 0.3 mmol scale.
|
|---|

|
Practical applications
Given the success of this method, the use of this technology for selective ligand modifications was envisioned (Scheme 5). As a case study, difunctionalization of a bipyridine-based ligand was selected for further investigation. Commonly these ligands are synthesized through the SNAr reaction of strong nucleophiles (e.g., tert-butyllithium) to a fluorinated bipyridine precursor, which is unfortunate given the popularity of these ligands in Cu and Ni catalysis.33 In contrast to the use of such pyrophoric alkylating agents, bench-stable alkyltrifluoroborates represent an attractive alternative. Gratifyingly, unfunctionalized substructures 5a and 5c were converted to the desired products in 84% and 68% yields, respectively. Pyridine ligands are also valuable for the synthesis of iridium photocatalysts. Under the same reaction conditions, heteroarene 5e afforded mono-alkylated product 5f in 82% yield. When tridentate ligand terpyridine 5g was subjected to the standard conditions with two equivalents of tert-butyltrifluoroborate, mono-alkylated 5h was formed exclusively.34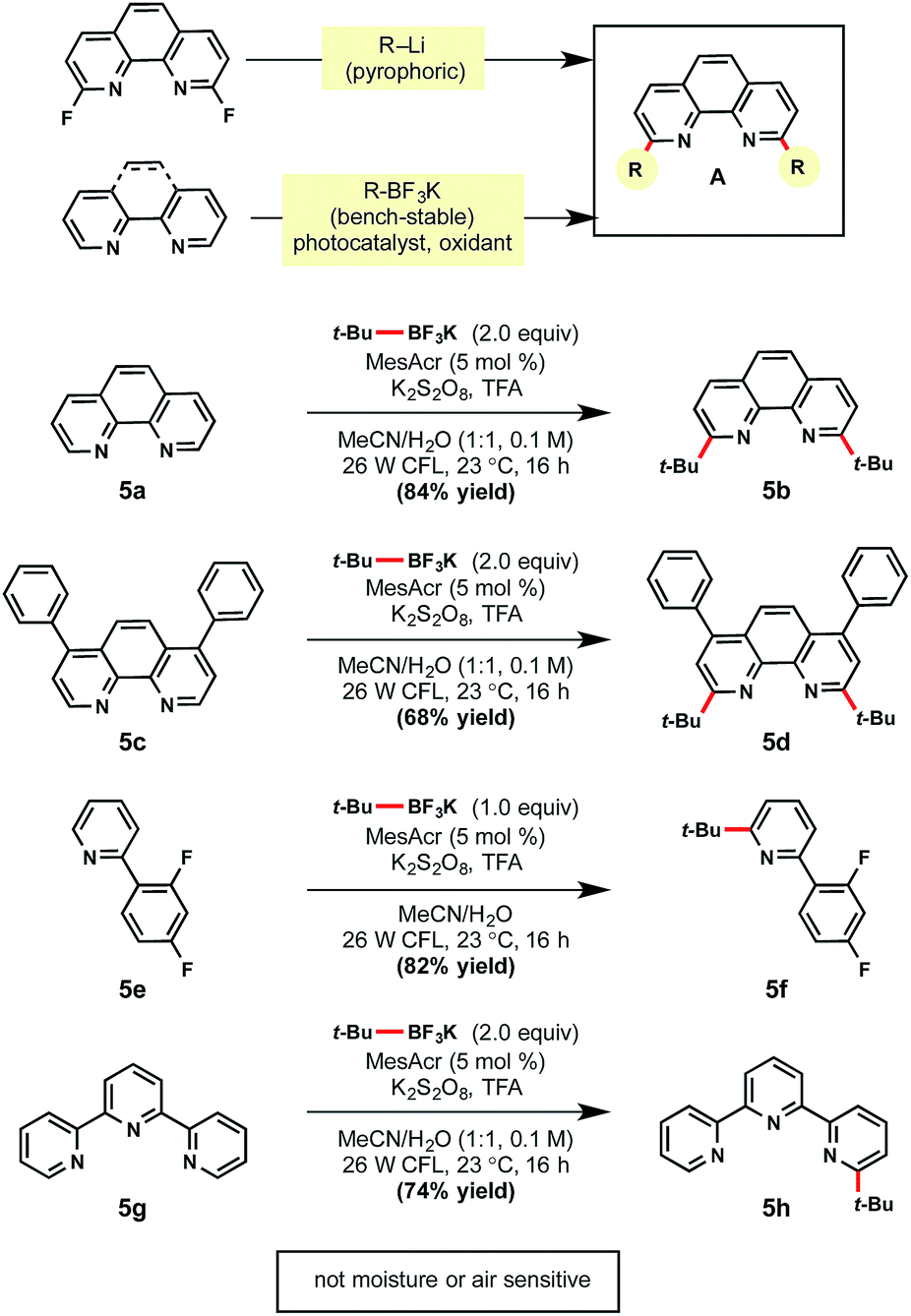 | ||
| Scheme 5 Mild alternative for ligand synthesis. aReactions were run with heteroarene (1.0 equiv.), alkyltrifluoroborate (1.0 or 2.0 equiv.), MesAcr (5 mol%), K2S2O8 (2.0 equiv.), trifluoroacetic acid (1.0 equiv.) in MeCN/H2O (1:1) on 0.3 mmol scale. | ||
Although medicinal chemists have principally capitalized on late-stage Minisci heteroarene alkylation,9 it can be advantageous to introduce alkyl substitution earlier in a synthetic sequence.35 Using the present protocol, the installation of alkyl subunits on brominated heteroarenes provides an excellent opportunity to highlight divergency in molecular synthesis. Furthermore, the halides installed within a variety of heteroarenes can serve a dual purpose: they may serve as an electronic bias for regioselective alkylations ortho to the halide,13d and they provide a functional handle for cross-coupling reactions, etc.6 To initiate studies to explore these concepts, a number of brominated heteroarenes were selected (Table 7). First, pyrimidine 6a was paired with an α-alkoxyalkyltrifluoroborate to yield exclusively the C4-substituted product. When moving to pyridines, a mixture of C2 and C6 alkylation occurred with tosyl-protected piperidinyltrifluoroborate (6b). Conversely, with primary alkyltrifluoroborates, only the C2-substituted products were observed in examples 6c and 6d. Chloride substitution was also well tolerated as demonstrated in 6d. When 4-bromoisoquinoline was utilized as a substrate, a mixture of regioisomers 6e and 6f was generated. Based on these studies, 6a and 6c contrast favorably with previous findings in the Baran laboratory wherein regiomeric isomers were observed for heteroaryl bromides and chlorides.13d

|
As a final highlight of the potential for late-stage functionalization, camptothecin (CPT) was selected as an appropriate, complex heteroarene. In medicinal chemistry, CPT was identified as an anti-cancer drug candidate because of its cytotoxic activity in a number of cancer cell lines. Unfortunately, poor solubility was a systemic issue and prevented moving it forward as a drug candidate.36 To tackle this challenge, various alkyl substitutions on the C7 position of the natural alkaloid were introduced, resulting in a more highly-active clinical candidate.37 Although SAR studies were conducted to confirm the promising properties of alkyl decoration, the alkyl substituents were primarily limited to simple alkyl groups (e.g., methyl, ethyl) using the corresponding alkyl bromides, peroxide, and acid under thermal conditions.38 To demonstrate the advantages of using the currently developed, mild photoredox conditions, secondary and primary alkyltrifluoroborates were selected as reagents for generation of analogues. As displayed in Scheme 6, primary α-alkoxyalkyl subunits could be incorporated, along with secondary alkyl moieties (7a, 7b).
 | ||
| Scheme 6 Camptothecin analogues: examples of late-stage functionalization. | ||
Mechanistic investigations
From our previous reports on Minisci chemistry12 and the results provided herein, it became clear that the use of alkyltrifluoroborates provided clear advantages in terms of both reactivity (permitting a single equivalent of radical precursor to be employed) and regioselectivity in heteroarene alkylation. Given these observations, it seemed that there might be some inherent advantage to the generation of BF3 byproduct during the course of the reaction. Two distinct possibilities were considered: (1) the BF3 produced was intimately involved with the generated radical, providing a shepherding effect in delivering the radical with enhanced selectivity, or (2) the BF3 was coordinating to the heteroarene prior to the radical addition, activating the heteroaryl substrate and enhancing preference for one site beyond the activation provided by the added protic acid.To assess the first hypothesis, DFT calculations were performed to determine the effective “bond order” for radical association with BF3 in solution. This potential interaction was of particular interest as this escorting effect has little precedent in the chemical literature; currently, there are only examples of nitrogen- and oxygen-centered radicals interacting with BF3 in solution.39 Indeed, preliminary calculations suggest that a strong preference exists for the free radical over a BF3-bound complex in solution (see ESI†). Noting this lack of association, we were quick to investigate an alternative hypothesis.
To probe each of the possible heteroarene complexes, we prepared stoichiometric solutions of one of our successful, selective substrates, quinoline, with no additive, trifluoroacetic acid (TFA) alone, BF3 alone, and finally TFA with added BF3, hoping to probe the effect of coordination on the proton and carbon shifts of heteroarene and provide some rationale for the differences between our chemistry and previously established protocols.
For trifluoroacetic acid, a clear downfield shift was observed in the C2 and C4 protons of quinoline (Fig. 2). Likewise, for the addition of BF3, a slightly less pronounced, but clear, downfield shift was also observed for the same protons. However, the combination of both additives together produced a wholly different shift in resonances to a unique heteroarene species.
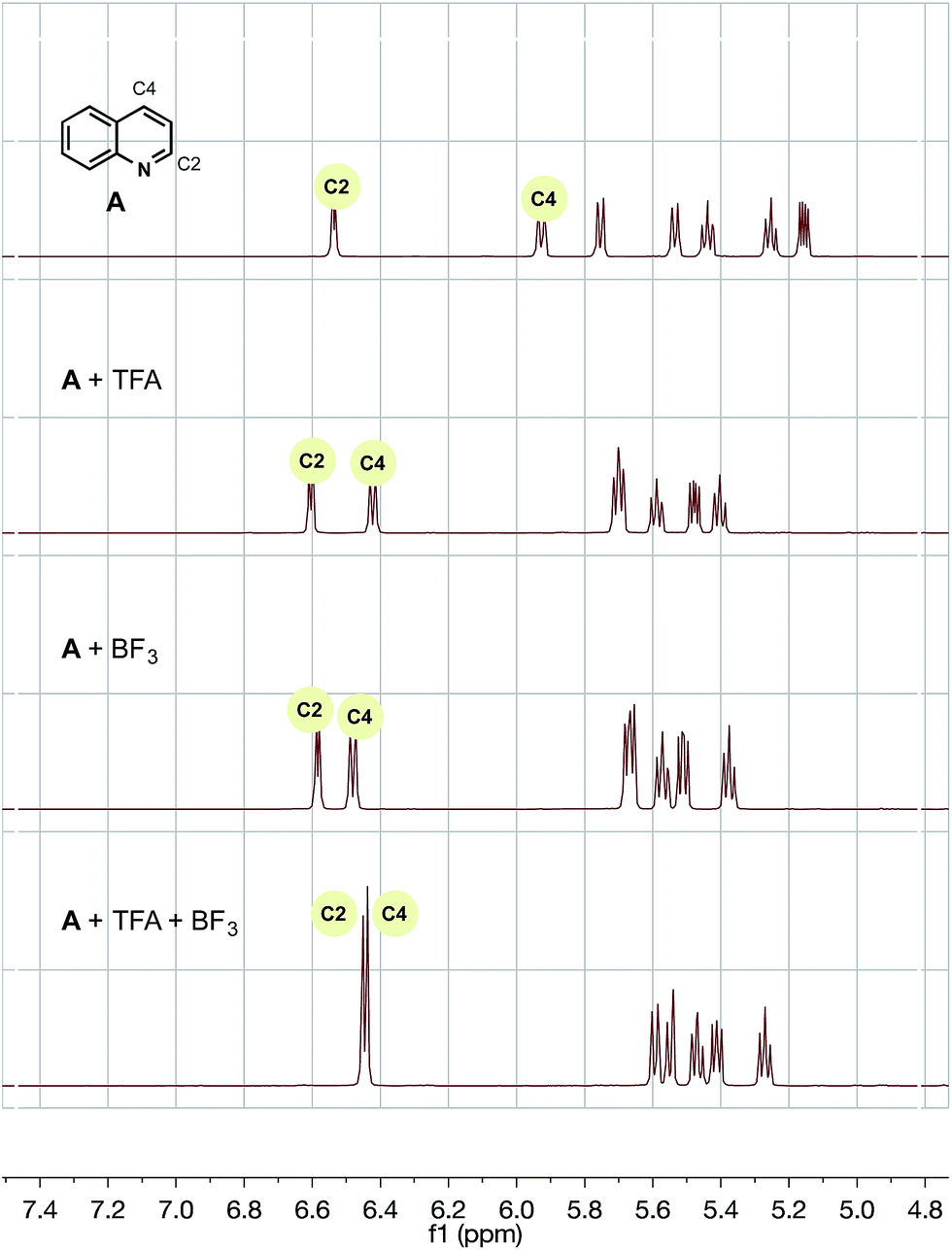 | ||
| Fig. 2
1H NMR of A with trifluoroacetic acid and/or BF3 additive in CD3CN/H2O (1:1). TMS was used as a reference. | ||
Most importantly, the carbon shifts of C2 relative to C4 (the best indicator of relative radicophilicity) possess enhanced differentiation in relative downfield shift (∼3 ppm) in comparison to the BF3 and trifluoroacetic acid complexes alone (∼1 ppm) (Fig. 3). Based on these chemical shifts, it is reasonable to suggest the C2 position is preferentially activated over the C4 position40 under the reaction conditions, resulting in the enhanced regioselective mono-alkylation observed for the quinoline system and, by analogy, many of the other systems in which alkyltrifluoroborates appear uniquely reactive and selective (vida infra).
 | ||
| Fig. 3
13C NMR of A with trifluoroacetic acid and/or BF3 additive in CD3CN/H2O (1:1). CD3CN solvent peak was used as a reference. | ||
To probe these findings further, we hoped that the use of alternative radical precursors (e.g., sulfinate salts or carboxylic acids) could be used in the presence and/or absence of BF3 to achieve the same desired regioselectivity. Unfortunately, under our standard conditions, sulfinate salts were universally ineffective, and carboxylic acids gave the same regiomixture, even with added BF3. Given this current ambiguity, this enhanced reactivity and regioselectivity appears to remain unique to the trifluoroborate precursors (see ESI†).
At this time, although it is impossible to propose a structure for this distinct, activated species, we speculate that BF3 activation of either the heteroarene π-cloud41 or polarization of the trifluoroacetic acid42 to increase its effective pKa may be responsible. Intriguingly, this type of dual activation could perhaps be more broadly operative in the selective alkylation of other heteroaryl cores.
Conclusions
In summary, a mild, room temperature method for the introduction of a diverse palette of alkyl groups has been developed. Under a set of unified reaction conditions, primary, secondary, and tertiary alkyltrifluoroborates can be employed in photoredox Minisci chemistry for the first time. This chemistry makes use of an inexpensive, mild oxidant, organic photocatalyst, and requires only one equivalent of alkyl radical partner, providing one of the most cost efficient and sustainable approaches to date. Additionally, enhanced regioselectivity was observed compared to current “state of the art” Minisci-type alkylations. Both of these phenomena may be related to the synchronized, catalytic generation of radicals in conjunction with the in situ generation of BF3 during the course of the reaction. The resultant method has been demonstrated broadly with respect to the heteroarene and alkyltrifluoroborate partners. Furthermore, access to bipyridine-type ligands as well as the late stage diversification of medicinally relevant substructures has been showcased. Finally, NMR studies provided support for a unique, highly activated species caused by the formation of BF3 upon the single-electron oxidation of alkyltrifluoroborates. Currently, further mechanistic studies are underway to elucidate the nature of this complex.Given the wide array of commercially available alkyltrifluoroborates, in addition to those synthesized by our laboratory and emerging from other groups, this approach provides a valuable contribution to the Minisci-type alkylation literature that is both highly competitive with, and complementary to, existing protocols. As alkyltrifluoroborates are well behaved, bench stable salts, these reagents will continue to serve as useful “radicals in a bottle” for synthetic chemists.
Acknowledgements
We thank the National Institutes of Health (NIGMS 1 R01 GM113878) for support. Dr Mirna El Khatib and Professor Sergei Vinogradov (University of Pennsylvania) are thanked for assisting in the quantum yield studies and providing the necessary equipment. Dr Niki Patel (University of Pennsylvania) is recognized for suggesting the use of allyl acetate as a sulfate radical anion trap. Ms Shorouk Badir (University of Pennsylvania) is acknowledged for the preparation of tertiary alkyltrifluoroborate 1u. Frontier Scientific is thanked for the generous donation of alkyltrifluoroborates. Pfizer graciously donated organophotocatalyst MesAcr. Lastly, Mr Sergei Tcyrulnikov (University of Pennsylvania) is thanked for performing DFT calculations.Notes and references
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS PubMed.
- (a) D. Yang, S. Burugupalli, D. Daniel and Y. Chen, J. Org. Chem., 2012, 77, 4466 CrossRef CAS PubMed; (b) L. Zheng, J. Ju, Y. Bin and R. Hua, J. Org. Chem., 2012, 77, 5794 CrossRef CAS PubMed; (c) G. Zhang, H. Ni, W. Chen, J. Shao, H. Liu, B. Chen and Y. Yu, Org. Lett., 2013, 15, 5967 CrossRef CAS PubMed; (d) J.-A. Jiang, W.-B. Huang, J.-J. Zhai, H.-W. Liu, Q. Cai, L.-X. Xu, W. Wang and Y.-F. Ji, Tetrahedron, 2013, 69, 627 CrossRef CAS; (e) M. R. Kumar, A. Park, N. Park and S. Lee, Org. Lett., 2011, 13, 3542 CrossRef CAS PubMed.
- (a) J. F. Bunnett and R. E. Zahler, Chem. Rev., 1951, 49, 273 CrossRef CAS; (b) O. F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley-VCH, Weinheim, Germany, 2013 Search PubMed; (c) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 10139 CrossRef CAS PubMed.
- G. Joshi and S. Adimurthy, Ind. Eng. Chem. Res., 2011, 50, 12271 CrossRef CAS.
- R. A. Rodriguez, C.-M. Pan, Y. Yabe, Y. Kawamata, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 6908 CrossRef CAS PubMed.
- (a) K. L. Billingsley, K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 3484 CrossRef CAS PubMed; (b) K. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3358 CrossRef CAS PubMed.
- S. Ge and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 12837 CrossRef CAS PubMed.
- (a) F. Minisci, Acc. Chem. Res., 1975, 8, 165 CrossRef CAS; (b) F. Minisci, E. Vismara and F. Fontana, J. Org. Chem., 1989, 54, 5224 CrossRef CAS; (c) W.-M. Zhao, X.-L. Chen, J.-W. Yuan, L.-B. Qu, L.-K. Duan and Y.-F. Zhao, Chem. Commun., 2014, 50, 2018 RSC; (d) R. Xia, M.-S. Xie, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Org. Lett., 2014, 16, 444 CrossRef CAS PubMed.
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575 CrossRef CAS.
- I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194 CrossRef CAS PubMed.
- K. Foo, E. Sella, I. Thome, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5279 CrossRef CAS PubMed.
- (a) G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852 CrossRef CAS PubMed; (b) M. Presset, N. Fleury-Brégeot, D. Oehlrich, F. Rombouts and G. A. Molander, J. Org. Chem., 2013, 78, 4615 CrossRef CAS PubMed.
- (a) R. Gianatassio, S. Kawamura, C. L. Eprile, K. Foo, J. Ge, A. C. Burns, M. R. Collins and P. S. Baran, Angew. Chem., Int. Ed., 2014, 53, 9851 CrossRef CAS PubMed; (b) Q. Zhou, A. Ruffoni, R. Gianatassio, Y. Fujiwara, E. Sella, D. Shabat and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 3949 CrossRef CAS PubMed; (c) Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411 CrossRef CAS PubMed; (d) F. O'Hara, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2013, 135, 12122 CrossRef PubMed; (e) Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herle, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95 CrossRef CAS PubMed.
- M. A. Duncton, MedChemComm, 2011, 2, 1135 RSC.
- J.-P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924 CrossRef CAS PubMed.
- (a) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS PubMed; (b) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224 CrossRef CAS PubMed; (c) J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565 CrossRef CAS PubMed.
- D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802 CrossRef CAS PubMed.
- G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407 RSC.
- T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4754 RSC.
- (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (b) D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195 CrossRef CAS PubMed; (c) I. Karakaya, D. N. Primer and G. A. Molander, Org. Lett., 2015, 17, 3294 CrossRef CAS PubMed; (d) Y. Yamashita, J. C. Tellis and G. A. Molander, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12026 CrossRef CAS PubMed; (e) M. El Khatib, R. A. Serafim and G. A. Molander, Angew. Chem., Int. Ed., 2016, 55, 254 CrossRef CAS PubMed; (f) D. Ryu, D. N. Primer, J. C. Tellis and G. A. Molander, Chem.–Eur. J., 2016, 22, 120 CrossRef CAS PubMed; (g) J. Amani, E. Sodagar and G. A. Molander, Org. Lett., 2016, 18, 732 CrossRef CAS PubMed; (h) R. Karimi-Nami, J. C. Tellis and G. A. Molander, Org. Lett., 2016, 18, 2572 CrossRef CAS PubMed; (i) J. C. Tellis, J. Amani and G. A. Molander, Org. Lett., 2016, 18, 2572 CrossRef PubMed; (j) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414 CrossRef CAS; (k) T. Chinzei, K. Miyazawa, Y. Yasu, T. Koike and M. Akita, RSC Adv., 2015, 5, 21297 RSC; (l) G. Sorin, R. M. Mallorquin, Y. Contie, A. Baralle, M. Malacria, J. P. Goddard and L. Fensterbank, Angew. Chem., Int. Ed., 2010, 49, 8721 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS PubMed.
- R. J. Memming, J. Electrochem. Soc., 1969, 116, 785 CrossRef CAS.
- (a) A. J. Kalb and T. L. Allen, J. Am. Chem. Soc., 1964, 86, 5107 CrossRef CAS; (b) N. R. Patel and R. A. Flowers II, J. Am. Chem. Soc., 2013, 135, 4672 CrossRef CAS PubMed.
- (a) D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577 CrossRef CAS PubMed; (b) P. D. Morse and D. A. Nicewicz, Chem. Sci., 2015, 6, 270 RSC; (c) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef CAS; (d) J. Grandjean and D. A. Nicewicz, Angew. Chem., Int. Ed., 2013, 52, 3967 CrossRef CAS PubMed; (e) M. Riener and D. A. Nicewicz, Chem. Sci., 2013, 4, 2625 RSC; (f) D. J. Wilger, N. J. Gesmundo and D. A. Nicewicz, Chem. Sci., 2013, 4, 3160 RSC; (g) T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588 CrossRef CAS PubMed; (h) A. J. Perkowski and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 10334 CrossRef CAS PubMed; (i) D. A. Nicewicz and D. S. Hamilton, Synthesis, 2014, 25, 1191 Search PubMed; (j) T. M. Nguyen, N. Manohar and D. A. Nicewicz, Angew. Chem., Int. Ed., 2014, 53, 6198 CrossRef CAS PubMed; (k) N. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024 CrossRef CAS PubMed; (l) M. A. Zeller, M. Riener and D. A. Nicewicz, Org. Lett., 2014, 16, 4810 CrossRef CAS PubMed; (m) N. J. Gesmundo, J.-M. M. Grandjean and D. A. Nicewicz, Org. Lett., 2015, 17, 1316 CrossRef CAS PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 CrossRef CAS.
- M. Butters, J. N. Harvey, J. Jover, A. J. J. Lennox, G. C. Lloyd-Jones and P. M. Murray, Angew. Chem., Int. Ed., 2010, 49, 5156 CrossRef CAS PubMed.
- H. J. Seo, S. J. Yoon, S. H. Jang and S. K. Namgoong, Tetrahedron Lett., 2011, 52, 3747 CrossRef CAS.
- M. Tada and Y. Yokoi, J. Heterocycl. Chem., 1989, 26, 45 CrossRef CAS.
- T. C. Atack and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 6139 CrossRef CAS PubMed.
- (a) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- K. E. Murphy-Benenato, N. Olivier, A. Choy, P. L. Ross, M. D. Miller, J. Thresher, N. Gao and M. R. Hale, ACS Med. Chem. Lett., 2014, 5, 1213 CrossRef CAS PubMed.
- (a) G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407 RSC; (b) Y. Siddaraju, M. Lamani and K. R. Prabhu, J. Org. Chem., 2014, 79, 3856 CrossRef CAS PubMed.
- P. G. Sammes and G. Yahioglu, Chem. Soc. Rev., 1994, 23, 327 RSC.
- We suspect that for the bipyridine system, initial alkylation enhances the rate of alkylation to the adjacent pyridine. For the larger, more diffuse terpyridine ligand, this rate acceleration is not seen due to both stereo- and electronic effects.
- R. Gianatassio, S. Kawamura, C. L. Eprile, K. Foo, J. Ge, A. C. Burns, M. R. Collins and P. S. Baran, Angew. Chem., Int. Ed., 2014, 53, 9851 CrossRef CAS PubMed.
- V. J. Venditto and E. E. Simanek, Mol. Pharm., 2010, 7, 307 CrossRef CAS PubMed.
- C. D. Britten, S. G. Hilsenbeck, S. G. Eckhardt, J. Marty, G. Mangold, J. R. MacDonald, E. K. Rowinsky, D. D. Von Hoff and S. Weitman, Cancer Res., 1999, 59, 1049 CAS.
- M. Li, W. Jin, C. Jiang, C. Zheng, W. Tang, T. You and L. Lou, Bioorg. Med. Chem. Lett., 2009, 19, 4107 CrossRef CAS PubMed.
- (a) S. Sekiguchi, K. Akiyama and S. Tero-Kubota, J. Chem. Soc., Perkin Trans. 2, 1997, 1619 RSC; (b) Y. Y. Lim and R. S. Drago, Inorg. Chem., 1972, 11, 1334 CrossRef CAS.
- Note that allthough enhanced C2/C4 differentiation is also observed for the heteroaryl in the absence of Bronsted/Lewis acid, the radical addition and/or oxidation steps are substantially slowed by their absence.
- (a) H. B. Mansaray, A. D. L. Rowe, N. Phillips, J. Niemeyer, M. Kelly, D. A. Addy, J. I. Bates and S. Aldridge, Chem. Commun., 2011, 47, 12295 RSC; (b) P. Tarakeshwar, S. J. Lee, J. Y. Lee and K. S. Kim, J. Phys. Chem. B, 1999, 103, 184 CrossRef CAS.
- (a) J. Ren, C. J. Cramer and R. R. Squires, J. Am. Chem. Soc., 1999, 121, 2633 CrossRef CAS; (b) H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. See DOI: 10.1039/c7sc00283a |
| This journal is © The Royal Society of Chemistry 2017 |