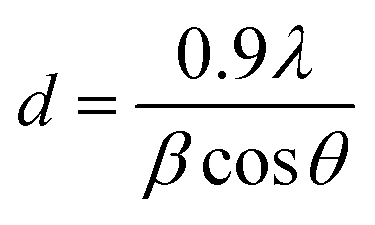DOI:
10.1039/C7RA06012B
(Paper)
RSC Adv., 2017,
7, 36594-36604
The effect of reduced graphene oxide on the catalytic activity of Cu–Cr–O–TiO2 to enhance the thermal decomposition rate of ammonium perchlorate: an efficient fuel oxidizer for solid rocket motors and missiles
Received
29th May 2017
, Accepted 6th July 2017
First published on 24th July 2017
Abstract
Reduced graphene oxide (rGO) modified transition metal oxide based composites were successfully synthesized via a sol–gel assisted Hummers' method. The present study includes the synthesis of CuCr2O4·0.7TiO2, the synthesis of rGO and the synthesis of rGO modified CuCr2O4·0.7TiO2. In order to synthesize the desired catalyst, rGO and Cu–Cr–O–0.7TiO2 were synthesized individually. The CuCr2O4·0.7TiO2 composite was synthesized via a sol–gel method. Reduced graphene oxide (rGO) used as a modifier in the catalyst, was also synthesized in the laboratory and was calcined at high temperature (1050 °C) to improve its activity. Finally, Cu–Cr–O–0.7TiO2 was modified with 10 wt% rGO. The post synthesis characterizations were performed using various instrumental techniques including X-ray diffraction (XRD) for phase analysis, Fourier transform infrared (FTIR) and Raman spectroscopy for molecular interactions, scanning electron microscopy (SEM) for surface morphology, energy dispersive X-ray analysis (EDX), elemental analysis and X-ray photoelectron spectroscopy (XPS) for binding energy. The catalytic efficiency of the synthesized composite catalyst samples based thermal decomposition of the host material (i.e. AP) was determined by differential thermal analysis (DTA) and thermogravimetric analysis (TGA). The rGO modification into the Cu–Cr–O–0.7TiO2 tri-metallic composition made it the most promising catalyst for the thermal decomposition of AP, due to the tremendously high electrical and thermal conductivity of rGO. Different amounts (2.5, 5.0, 7.5 and 10 wt%) of Cu–Cr–O–0.7TiO2–rGO were added to ammonium perchlorate (AP) to investigate its effect on the thermal decomposition of AP, which is a well known oxidizer used worldwide in the solid composite propellant (SCP) in modern rocketry. The 5 wt% of catalyst (Cu–Cr–O–0.7TiO2–rGO) addition into AP exhibited the remarkably enhanced thermal decomposition of AP. Finally, the burn rate of SCP was examined with 5 wt% catalyst modified AP. The 5 wt% of catalyst modified AP exhibited 175.31% higher burn rate of SCP, compared to the burn rate of pure AP added SCP. Furthermore, when it was compared with an industrial catalyst, i.e. activated copper chromite (ACR), it showed 133.61% higher burn rate of SCP. The SCP exhibited excellent ballistic performance with 0.6% of catalyst in AP, which enhanced the burn rate from 4.866 mm s−1 (for SCP having pure AP) to 8.531 mm s−1 (for SCP having catalyst added AP) and 6.385 mm s−1 (for SCP having industrial catalyst added AP) at 33 bar.
1. Introduction
Solid composite propellant (SCP) is the most reliable energy source in the era of solid rocket motors. SCP is composed of metallic fuel–aluminum powder (ALP), inorganic oxidizer–ammonium perchlorate (AP): NH4ClO4, and other additives as plasticizers and burn rate modifiers, etc. ALP and AP are embedded into the polymer matrix of hydroxyl terminated poly-butadiene (HTPB) and toluene di-isocyanate (TDI).1–7 The combustion of solid composite propellant releases chemical energy, which is converted into kinetic energy by means of the nozzle, and in turn it provides thrust to the solid rocket motor of a space vehicle or missile. The combustion efficiency of the SCP is a function of the stoichiometry of the propellant ingredients and strongly depends on the thermal decomposition of AP, as it is the major ingredient of SCP. The thermal decomposition of AP is greatly dependent on the composition (presence of any catalyst), particle size and shape of AP, which ultimately determine the burn rate of SCP. A higher burn rate with a lower pressure exponent is the desirable requirement; thus, thermal decomposition of AP is the key deciding factor to obtain the desired burn rate of SCP. The thermal decomposition of AP is greatly affected by the additives of transition metals, transition metal oxides and their mixtures. The decomposition mechanism has been understood by the concept of electron and proton exchange at elevated temperatures, as reported by Boldyrev et al. (2006).8 They studied the governing mechanism of the thermal decomposition of AP and proposed electron and proton transfer processes. AP is decomposed in two stages: low temperature decomposition, governed by the electron transfer process, and high temperature decomposition, initiated by the proton transfer process.8 The transition metals, transition metal oxides, and composites of ternary materials such as MnO2, CuO, NiO, CoFe2O4, Fe2O4, CuCr2O4, etc. are common catalysts that are used to enhance the thermal decomposition of AP.9–20
Recent developments in the area of carbon based materials such as carbon nanotubes (CNT), graphene oxide (GO) and reduced graphene oxide (rGO) have promised better catalysts and catalyst support due to their high specific surface area, high thermal and electrical conductivity. The dispersion difficulty of CNT leads to its restricted applications, whereas the hydrophilic nature of GO/rGO and the ease of synthesis provide the potential precursor material for the synthesis of graphene, which is a two dimensional sheet of carbon with the honeycomb structure that provides high specific surface area and superior electrical and thermal conductivity.21–23 The rGO has tremendous electron transfer properties. These properties of graphene have created remarkable scope for its potential applications in catalysis, particularly in the field of thermal decomposition of AP, promoted by the effect of the electron and proton transfer processes.
It is quite interesting to exploit both the catalytic properties of transition metal oxide based materials and graphene based materials in a single system. Recently, graphene based transition metal oxide composites (Fe2O3, TiO2, Co3O4 and MnO2 etc.) have been prepared and their effects on the thermal decomposition of AP have been studied.24–29 Li et al. (2012) synthesized the Ni/graphene based nanocomposites using the simple one-pot method and observed the single step decomposition of AP.7 In 2013, the same group observed the negligible effect of pure graphene on the modification of the thermal decomposition of AP, whereas nanoparticles of Mn3O4 dispersed on GO showed tremendous effects.25 Yuan et al. (2014) studied the thermal decomposition of AP in the presence of Fe2O3/GO and the single step decomposition of AP at much lower temperatures, with high energy release against the individual effects of Fe2O3 and GO.24
According to the literature, copper chromite (CuO·CuCr2O4)18–20 and titanium oxide (TiO2)21–23 are the most efficient metal oxide catalysts that considerably decrease the onset temperature of AP decomposition when they are used individually as additives in the AP. Despite the extensive and individual utilization of copper chromite and titanium oxide to manipulate the burn rate of AP, the physics and chemistry of their catalytic roles are not fully known. There is almost no literature on the combined effect of copper chromite and titanium oxide on the thermal decomposition of AP. Furthermore, the effect of rGO on the previously optimized Cu–Cr–O–TiO2 system to enhance the burn rate of AP is a very interesting idea. We have recently reported, for the first time, the optimization of the concentration of TiO2 in CuO·CuCr2O4 to get the best composite (Cu–Cr–O–0.7TiO2) for the improved thermal decomposition of AP.31 Herein, we investigate the effect of rGO in the Cu–Cr–O–0.7TiO2 system to enhance the thermal decomposition of AP as well as the burn rate of SCP. The aim of the present investigation is to achieve the highest burn rate of fuel at the minimum possible temperature. Efforts have been made to synthesize rGO and conduct rGO modification on ultrafine particles of CuCr2O4·0.7TiO2 as synthesized by the sol–gel method. Several methods such as the hydrothermal method, ceramic method, co-precipitation, solid state, solution combustion method and sol–gel method have been reported to prepare the catalysts. However, due to having better control on the homogeneity, and the ease of preparation, the sol–gel method was employed.
2. Experimental
2.1 Sample preparation
2.1.1 Materials. The chemical reagents for the synthesis of graphene oxide and those used in the present study were of analytical grade and high (>99.9%) purity. The graphite rod was purchased from Graphite India Ltd., whereas, the other ingredients, viz. sulfuric acid (H2SO4), ortho-phosphoric acid (H3PO4), potassium permanganate (KMnO4), hydrogen peroxide (H2O2), hydrochloric acid (HCl) and ethyl alcohol (C2H5OH) were purchased from Merck, Germany. Ammonium perchlorate (AP: NH4ClO4), with average particle size of 300 μm, was purchased from Pandian Chemicals Ltd., Cuddalore. Copper nitrate trihydrate (Cu (NO3)·3H2O) of extra pure grade was purchased from Titan Biotech Ltd., India, whereas chromium nitrate nonahydrate (Cr(NO3)3·9H2O) was purchased from Central Drug House (P) Ltd., India. Anhydrous citric acid (C6H8O7) was purchased from Fisher Scientific, India. TiO2 (P25) of nanometric size was purchased from Evonik Industries, Germany. All other reagents, except graphite powder, were used without further purification. The graphite in powder form was obtained by machining and grinding the as purchased graphite rod. The powder thus obtained was sieved and brought to the average particle size of 40 μm. The average particle size of ammonium perchlorate was reduced to 45 μm using an air classifying mill.
2.1.2 Methods.
(a) Synthesis of reduced graphene oxide (rGO). In order to get 1 g of rGO product, 120 ml of H2SO4 and 20 ml of H3PO4 were added to a beaker, with continuous stirring using magnetic stirrer. After 10 minutes of stirring, 1 g of graphite powder was dispersed into the beaker having a mixture of two different acids. The dispersion of the added graphite powder of average particle size of 40 μm was allowed a further 30 minutes of continuous stirring. The color of the mixture in the beaker was black due to graphite. After the homogeneous mixing of graphite particles, 3 g of KMnO4 was added slowly to the beaker, which led to the color change of graphite added acids from black to greenish blue. In due time, the color change from greenish blue to pink was observed. Vigorous stirring of the obtained solution was allowed for a further 36 h at room temperature. The color of the solution became muddy yellow. Distilled water (100 ml) was then added to the obtained yellowish solution, followed by the addition of 30 ml of 30% v/v H2O2. An exothermic reaction was observed and finally, golden yellow particles were seen in the solution. The impurities were removed from the solution by washing with water, followed by 30% v/v of HCl and C2H5OH. The solid particles were removed from the solution by centrifuging it at 5000 rpm. The obtained solid particles were dried in the oven at 60 °C for 6 h to get GO, which was transformed into rGO by a thermal treatment method as reported in literature.30 Finally, rGO was calcined at 1050 °C to improve its activity by reducing the attached functional groups. The rGO thus obtained was ready for further use.
(b) Synthesis of the Cu–Cr–O–0.7TiO2 composite. Tri-metallic oxide (Cu–Cr–O–0.7TiO2) was prepared using the sol–gel method. The molar ratio of Cu/Cr was kept at 0.7 and very interestingly, the molar concentration of TiO2, also 0.7 mol%, was found to be best among the three concentrations (0.5, 0.7 and 0.9 mol%) of TiO2 according to our previous study.31 The synthesis procedure for Cu–Cr–O–0.7TiO2 followed in the present investigation is exactly the same as in our previous report.31 Among the six compositions of tri-metallic oxide with three varying concentrations (0.5, 0.7 and 0.9 mol%) of TiO2 calcined at two different temperatures, 300 and 1050 °C, the Cu–Cr–O–0.7TiO2 composite calcined at 300 °C was found to be the best catalyst, which remarkably enhanced the burn rate of AP. The AP with particle size of 45 μm (AP45 μm) exhibited better thermal decomposition behavior than the AP having particle size of 300 μm (AP300 μm) as reported in literature (revealed in our previous work);31 therefore, we used AP45 μm in our present study and it is represented as AP. When 0.7 mol% of TiO2 added Cu–Cr–O composite, calcined at 300 °C, was used as the catalyst in AP, a very sharp single step decomposition of AP45 μ at 306 °C was obtained, which was 79 °C lower than that of pure AP. Since the catalyst sample Cu–Cr–O.0.7TiO2, calcined at 300 °C, had achieved remarkable enhancement in the thermal decomposition of AP when used as modifier in pure AP, the further modification by rGO in this composition may offer more significant results.
(c) Synthesis of Cu–Cr–O–0.7TiO2–rGO. In order to synthesize the rGO incorporated Cu–Cr–O–0.7TiO2 composite, 10 wt% of rGO was added to 90 wt% of Cu–Cr–O–0.7TiO2. Already synthesized rGO (10 g) was poured into 400 ml ethanol in a beaker. The rGO suspension was subjected to a magnetic stirrer for continuous stirring, and the Cu–Cr–O·0.7TiO2 powder was then added slowly to this suspension. The rGO modified Cu–Cr–O–0.7TiO2 mixture was continuously stirred at 80 °C until a paste-like material was obtained. The paste was dried in an oven at 130 °C for 3 h. The material thus obtained was then crushed using a mortar and pestle to get a homogeneous fine powder of Cu–Cr–O–0.7TiO2–rGO. This powder was further calcined at 300 °C for 4 h in the N2 atmosphere to form the desired Cu–Cr–O–0.7TiO2 composite materials having excellent catalytic properties on thermal decomposition of AP.
2.2 Sample characterization
The powder X-ray diffraction method was employed to record the diffraction pattern of the synthesized catalyst using an advanced X-ray diffractometer (Rigaku Miniflex II, Desktop X-ray D) with CuKα radiation (λ = 1.5418 Å) with Ni filter at a scanning speed of 3° min−1 and scanning range of 20–80°, operated at 30 kV and 15 mA. The surface morphology and particle size analyses were carried out using scanning electron microscopy (SEM) operated at 10 kV (ZEISS EVO 18). Elemental analysis of synthesized powder was carried out via the energy dispersive X-ray analysis method, using the INCA 250 EDS instrument with X-max of 20 mm detector, coupled with SEM (ZEISS EVO 18). Fourier Transform Infrared (FTIR) spectroscopy analysis was performed at room temperature using an FTIR spectrometer (Perkin Elmer with model no. 577 FT-IR). The FT-IR spectrum was recorded in the wavenumber range 4000–400 cm−1 using the KBr pellet method. The Raman spectrum of the GO sample was recorded using a Raman spectrometer (Renishaw, UK) with an excitation source of 514.5 nm wavelength. To confirm the formation of GO, the presence of D and G bands is mandatory in the Raman spectrum. The oxidation state and elemental analysis of the samples were determined using X-ray photon spectroscopy (XPS-model PHI5000, Versa Probe II, physical Electronics) with AlKα radiation for monochromatic X-ray of energy E = 1486.6 eV, Ar ion gun sputtering, beam diameter of 10 μm, 6–25 W and 15 kV, surface clean sputtering 2 kV, 2 mm × 2 mm for 1 min. The XPS patterns of various compositions were taken at 187.5 eV pass energy for the detection of all possible elements with atomic concentration limit above 0.5%. To confirm the oxidation states of constituent elements, the core level analysis was done at 11.75 eV pass energy at a step size of 0.1 eV with X-ray rating of 100 micron in diameter, 25 W and 15 kV. The electron neutralizer and Ar ion gun were used during the experiment for charge loss compensation, which occurred during the XPS process. The core level spectra were recorded under ultra-high vacuum (UHV) conditions of about 10−10 Torr. The adventitious carbon was recorded for reference from the surface and then sputtered by Ar ion gun at a rating of 2 kV, 2 × 2 at a sputter rate of 10 nm min−1 of SiO2 standard. Thermogravimetric and differential thermogravimetric analysis (TG-DTA) curves were obtained with LABSYSTM Setaram Instrumentation (Serial no. 1-3347-1 DTA/TGA/DSC) in nitrogen atmosphere at a heating rate of 10 °C min−1. Thermal decomposition of AP and modified AP was determined using DTA curves.
2.3 Catalytic activity measurements
The catalytic activity of the as synthesized composite materials was determined over the thermal decomposition of AP. In order to analyze the catalytic efficiency of the materials, the measured parameters were the burn rate of the solid composite propellant, the temperature of crystallographic change, low temperature decomposition and high temperature decomposition. A known quantity of AP (AP45 μm) powder was mixed with varying amounts (2.5, 5.0, 7.5 and 10 wt%) of synthesized catalyst (Cu–Cr–Ti–O–rGO). These samples were prepared homogenously (by the ball mill method) to see the effect of the synthesized catalyst on the burn rate of AP. The resultant compositions were analyzed by thermogravimetric and differential thermal (TG-DTA) characterizations at the heating rate of 10 °C min−1 in a N2 environment. Nichrome wire was used to supply the 24 volt of power supply, and the produced noise signal was captured through the acoustic measurement sensor. The burn rate of the SCP was achieved using the strand burner, and testing was conducted at 33 bar in water.
3. Results and discussion
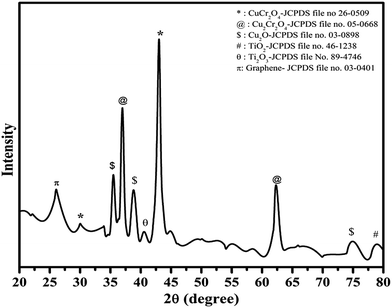
3.1 X-ray diffraction (XRD) pattern of synthesized catalyst
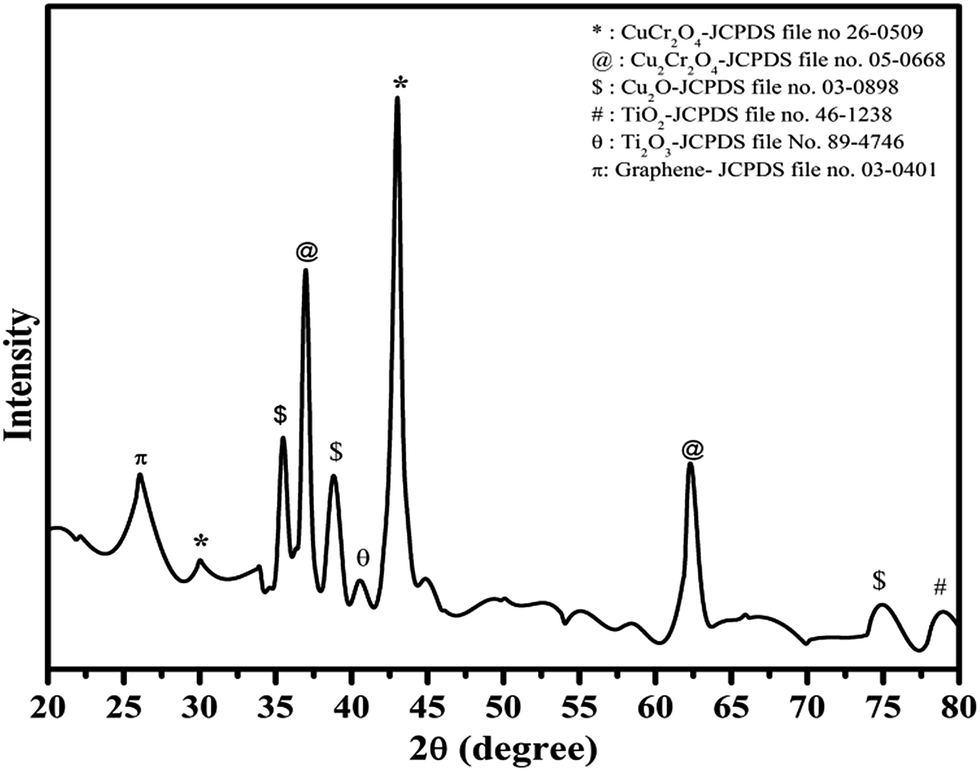
The crystalline phase and phase identification were carried out using the XRD pattern of the Cu–Cr–O–0.7TiO2–rGO composition as shown in Fig. 1. The characteristic diffraction peak for rGO with (002) as (hkl) values appears at the diffraction angle (2θ) of 26.1°, matching the JCPDS File no. 03-0401.32,33 The interlayer distance (d-spacing) between two layers is about 0.33 nm. The strongest diffraction peak at 43.02° is due to copper chromite CuCr2O4, corresponding to the (400) plane (see the JCPDS File no. 26-0509). However, a weak peak at 30.02° in the pattern is due to the (220) plane of the CuCr2O4 copper chromite.
 |
| | Fig. 1 XRD pattern of the synthesized catalyst. | |
Secondary phase of copper chromite, i.e. Cu2Cr2O4, was also identified by the presence of the diffraction peaks at 36.98° and 62.30°, corresponding to planes (100) and (hkl) respectively, matching the JCPDS File no. 05-0668.
Diffraction peaks due to Cu2O appeared at 35.48°, 38.82° and 75.23°, corresponding to (111), (110) and (220), respectively, as (hkl) values indexed to JCPDS File no. 80–4746. Titanium oxide was observed in two phases, i.e. Ti2O3 (JCPDS File no. 89-4746) and TiO2 (JCPDS File no. 89-4203) at the diffraction angles of 40.54° and 78.94°, with corresponding planes (113) and (117), respectively. The crystallite size of the synthesized catalyst was calculated by full width at half maximum (FWHM), using the Scherrer formula as given below in eqn (i).
| |
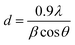 | (i) |
d = crystallite size in nm,
λ = wavelength in nm,
β = full width at half maximum and
θ = half of Bragg's angle. The crystallite size of the sample was observed to be 32 nm.
3.2 Infrared spectroscopy of synthesized catalyst
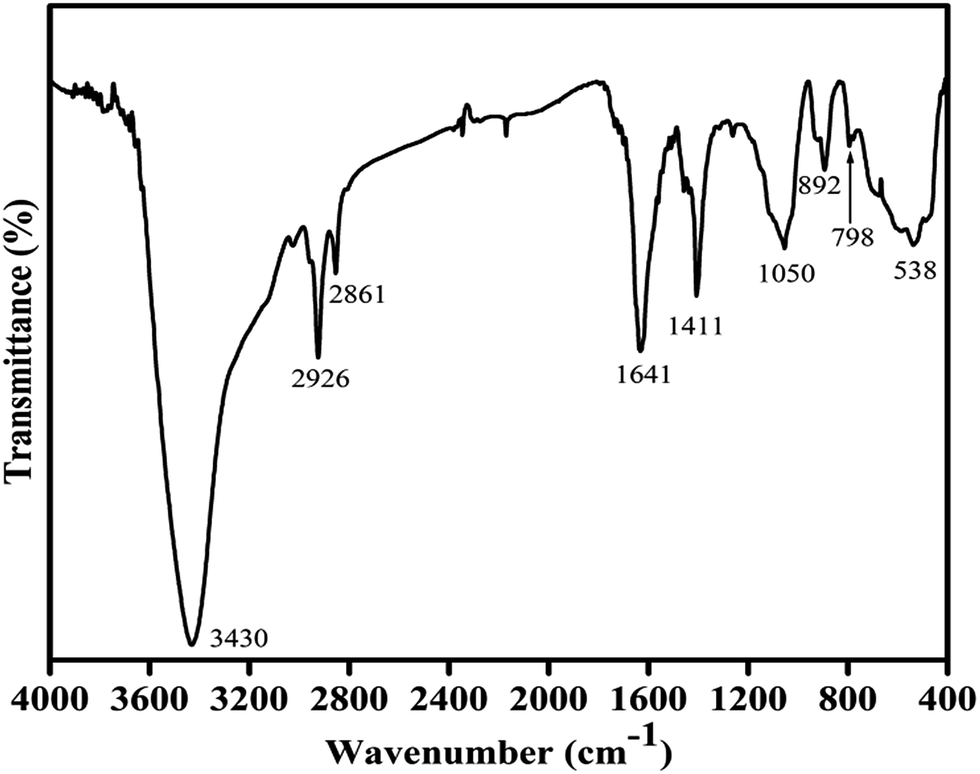
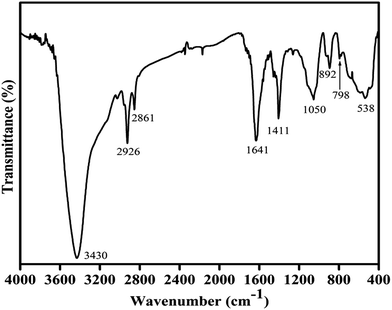
The IR spectrum of the Cu–Cr–O–0.7TiO2–rGO catalyst sample is shown in Fig. 2. The strongest absorption envelope at 3430 cm−1 is assigned to the symmetric stretching mode of OH−.34,35 Another strong shoulder observed at wavenumber 1641 cm−1 is due the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, which is the characteristic shoulder of rGO. The position of this band may vary depending on the host molecule to which H2O is attached. The medium intensity bands recorded at 2926 and 2861 cm−1 are attributed to the stretching vibrations of ν(C–H) of citric acid. A medium and sharp shoulder appearing at 1411 cm−1 may be assigned to the symmetric vibrations of surface hydroxyl groups. However, this band may also be attributed to ν3 carbonate ions (CO32–). An un-indexed weak shoulder appearing at 1278 cm−1 is attributed to the coupled bending and stretching vibrations of COO− of citric acid. Notably, citric acid has its melting point at 310 °C, which is just above 300 °C; this is the reason why citric acid is not destroyed at 300 °C.31 The shoulder at 1050 cm−1 can be assigned to the bending vibrations of C–H of citric acid. The weak absorption bands observed at 892, 798 and 697 cm−1 are attributed to the different vibrational modes of TiO2,31 which exhibit strong absorption in the range 600–890 cm−1 when treated with some other metal ions, as in the present case. A broad absorption band centered at 538 cm−1 is assigned to the characteristic vibrational band (asymmetric) of Cu–O in CuO; it also contains stretching bands of Cu–O–Cr.31
C, which is the characteristic shoulder of rGO. The position of this band may vary depending on the host molecule to which H2O is attached. The medium intensity bands recorded at 2926 and 2861 cm−1 are attributed to the stretching vibrations of ν(C–H) of citric acid. A medium and sharp shoulder appearing at 1411 cm−1 may be assigned to the symmetric vibrations of surface hydroxyl groups. However, this band may also be attributed to ν3 carbonate ions (CO32–). An un-indexed weak shoulder appearing at 1278 cm−1 is attributed to the coupled bending and stretching vibrations of COO− of citric acid. Notably, citric acid has its melting point at 310 °C, which is just above 300 °C; this is the reason why citric acid is not destroyed at 300 °C.31 The shoulder at 1050 cm−1 can be assigned to the bending vibrations of C–H of citric acid. The weak absorption bands observed at 892, 798 and 697 cm−1 are attributed to the different vibrational modes of TiO2,31 which exhibit strong absorption in the range 600–890 cm−1 when treated with some other metal ions, as in the present case. A broad absorption band centered at 538 cm−1 is assigned to the characteristic vibrational band (asymmetric) of Cu–O in CuO; it also contains stretching bands of Cu–O–Cr.31
 |
| | Fig. 2 FT-IR pattern of the synthesized catalyst. | |
3.3 Raman spectroscopy of the synthesized catalyst
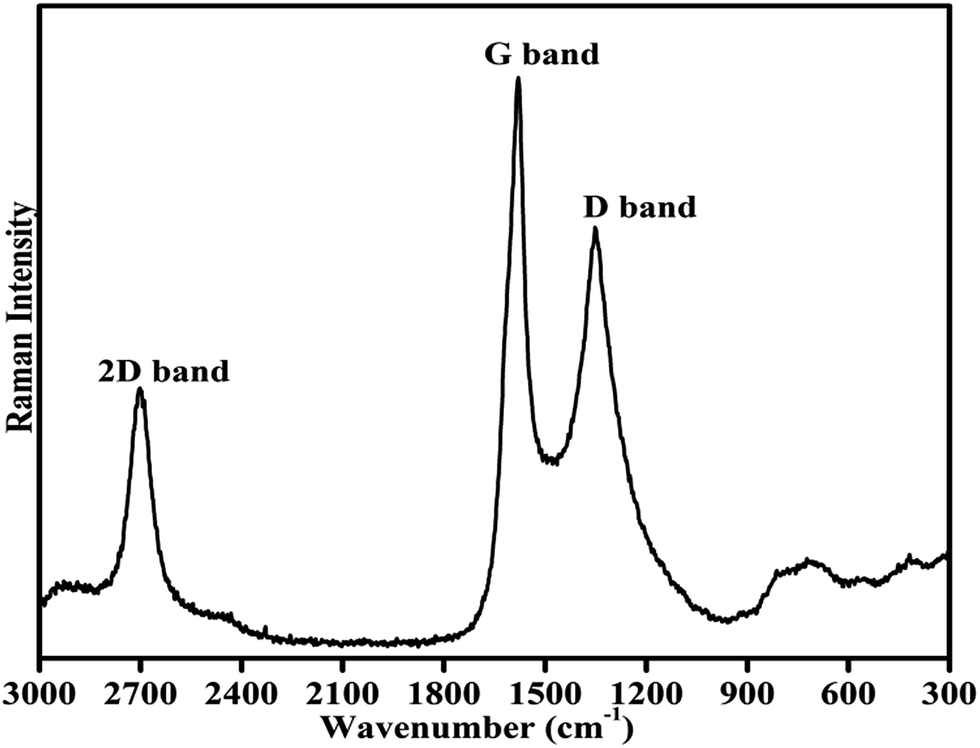
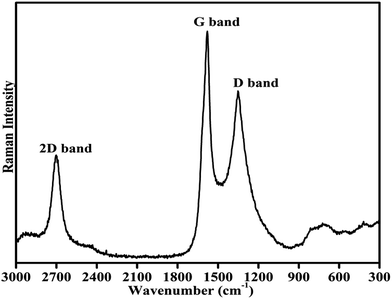
Fig. 3 shows the room temperature Raman spectrum of the synthesized composites of Cu–Cr–Ti–O/rGO. To avoid the decomposition of the sample, a laser of low power (5 mW) was employed for recording the Raman spectrum. The spectrum was recorded in the wavenumber range of 3000 to 300 cm−1. The two most intense features of the Raman spectrum of rGO are the presence of a G band and D bands. The G band is observed at ∼1580 cm−1, and this band always appears as the strongest band in the Raman spectrum, as is clearly shown in Fig. 4.36,37 Another significant mark of the Raman profile of rGO is the 2D band appearing at ∼2700 cm−1, as usually observed in graphitic structures. The zone-boundary phonons violate the Raman fundamental selection rule and consequently give rise to a peak at ∼1352 cm−1 in defective graphite, called the D-band, rather than the appearance of a Raman peak in the first order Raman spectrum of defect-free graphite.36,38,39 The Raman spectrum of the graphene has a single sharp 2D peak; the number of layers in graphene is estimated based on the intensity of the 2D peak. Literature shows that as the number of layers of graphene sheets increases, the intensity of the G band increases significantly, whereas the intensity of the 2D band decreases.36,39–41
 |
| | Fig. 3 Raman spectrum of the synthesized catalyst. | |
 |
| | Fig. 4 SEM images of the synthesized catalyst at (a) 1 μm and (b) 2 μm magnifications. | |
The position of the D band in the present spectrum is exactly at 1352 cm−1. The G and D bands appear due to the doubly degenerate zone centre E2g mode at ∼1580 cm−1 and the breathing modes of six atom rings at 1352 cm−1 because of the defects in graphite. The recorded Raman profile containing the conventional and characteristic G, D and 2D bands at their defined positions with defined intensity confirm the presence of graphene oxide in the prepared catalyst composite.
3.4 Scanning electron microscopy (SEM) and associated energy dispersive X-ray (EDX) analysis of the synthesized catalyst
The surface morphology of the synthesized catalyst sample is shown in Fig. 4(a and b). The microstructures of the sample at different magnifications (a) 2 μm and (b) 1 μm were determined using scanning electron microscopy, operated at 10 kV. The micrographs show that the particles are spherical in shape, and the particle size is in the range of 200–300 nm. Elemental compositions of the rGO added Cu–Cr–O–TiO2 catalyst sample were determined using EDX, and the observed atomic wt% of the constituent elements is shown in Table 1. This analysis confirmed the presence of carbon, copper, chromium, titanium and oxygen. The increasing weight% of oxygen with an increase in the temperature may occur due to the transformation of the intermediate compounds of Cu–Cr–O-citric acid into metal oxides. Interestingly, CuO already showed its presence in XRD patterns of the samples calcined at 1050 °C, but not in the samples calcined at 300 °C.
Table 1 Elemental composition of the synthesized catalyst
| Element |
Weight% |
| C |
38.33 |
| O |
13.21 |
| Ti |
26.13 |
| Cr |
13.89 |
| Cu |
8.44 |
3.5 Transmission electron microscopy (TEM) of the synthesized catalyst
Fig. 5 shows the transmission electron microscopic images of the Cu–Cr–O·0.7TiO2–rGO composite sample calcined at 300 °C. Two images of the composite at two different magnifications (a) 200 nm and (b) 20 nm are shown; however, the inset image (c) represents the selected area diffraction pattern (SAED) pattern. At first glance, in image (a), the Cu–Cr–O–0.7TiO2 particles of size around 100 nm can be seen easily in yellow dotted yellow rings as shown by yellow arrows and rest is rGO as shown by bright brown arrows. More clear view of rGO sheets is captured at higher magnification as shown in image (b). The Cu–Cr–O–0.7TiO2 particles are scattered on the rGO sheets to form an organic–inorganic blend.
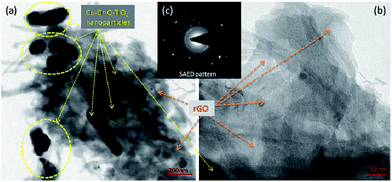 |
| | Fig. 5 TEM images of synthesized catalyst at two different magnifications (a) 200 nm and (b) 20 nm; (c) SAED pattern as inset image. | |
3.6 X-ray photoelectron spectroscopy (XPS)
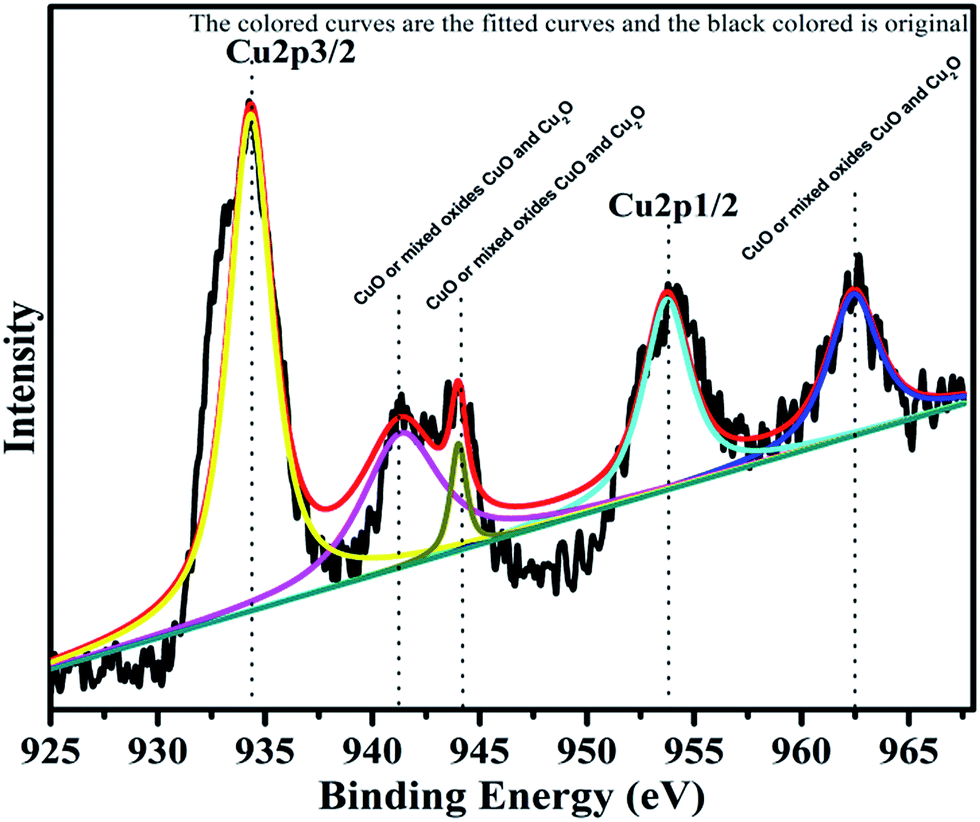
The surface composition and oxidation states of the constituent elements of synthesized catalyst were represented by the characteristic core level spectra of Cu, Cr, Ti, O and C. The XPS pattern of Cu is shown below in Fig. 6.
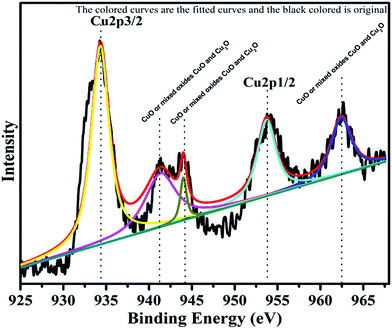 |
| | Fig. 6 XPS Pattern of copper with fitted curves. | |
Copper is observed in two oxidation states namely Cu+ and Cu2+ in 72.24% and 27.76%, respectively. The XPS spectrum of Cu2+ is characterized by two spin orbit doublets with strong satellite peaks. The binding energies for Cu 2p3/2 and Cu 2p1/2 are 933.5 eV and 953.5 eV, respectively. Three satellite peaks with binding energies, 941.5 eV, 944.03 eV and 962 eV support the presence of CuO, or mixed oxides CuO and Cu2O. The low energy component with Cu 2p3/2 at 933.8 eV is associated with Cu2+ in octahedron sites, whereas the high energy component at 935.3 eV is assigned to Cu2+ in tetrahedral sites.
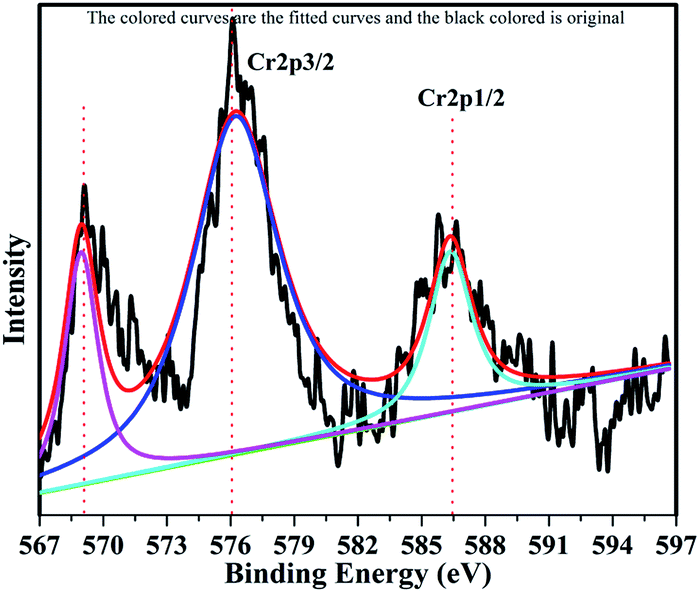
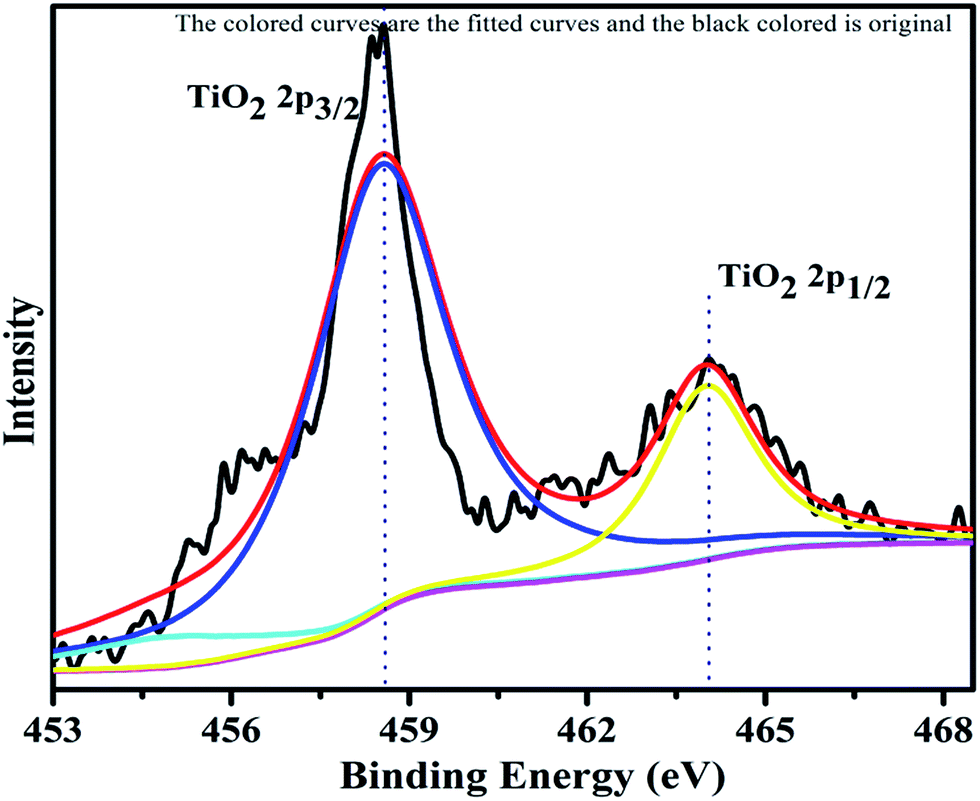
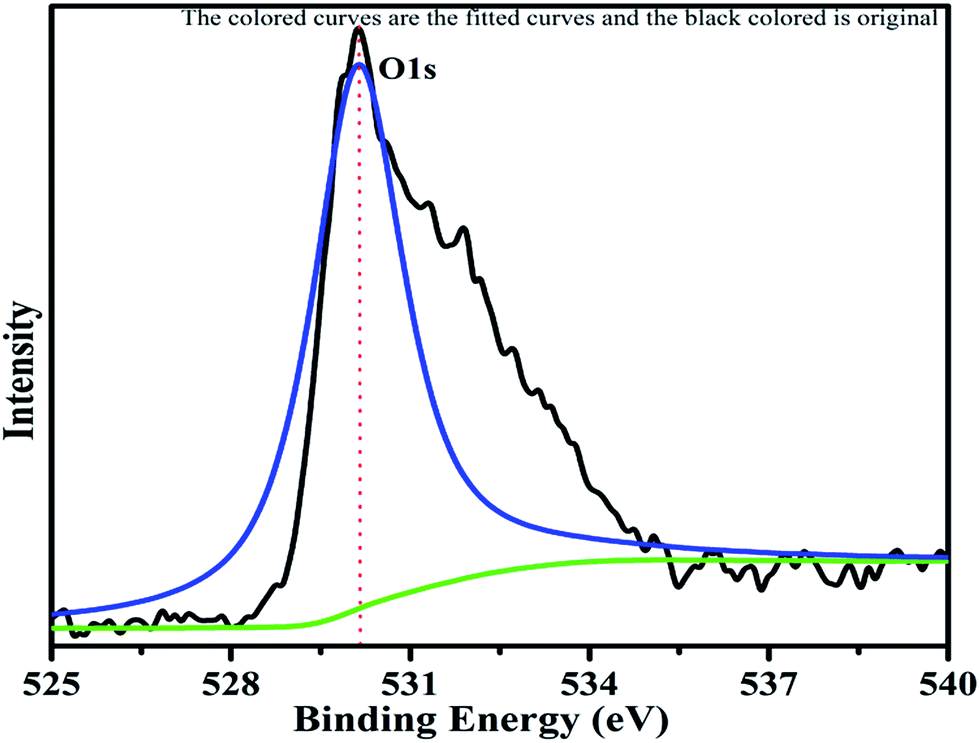
The XPS profile of Cr is shown in Fig. 7. The XPS profile shows three peaks, two of which appear with binding energies of 586.4 eV and 576.07 eV, corresponding to Cr 2p1/2 and Cr 2p3/2.42 The third peak with binding energy of 568.98 eV could not be assigned. The XPS profile of TiO2 is represented in Fig. 8. Titanium is observed in two different oxidation states, i.e., Ti2+ and Ti4+ in 1.68% and 77.7%, respectively. TiO2 exhibits two peaks having binding energies of 458.5 eV and 464.05 eV, attributed to TiO2 2p3/2 and TiO2 2p1/2. Fig. 9 exhibits the XPS pattern of oxygen (O). Oxygen shows a peak with binding energy 530.16 eV, which is assigned to O 1s. This peak is the main oxygen peak, ascribed to the lattice oxygen in TiO2.
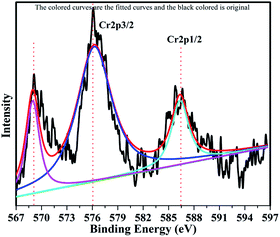 |
| | Fig. 7 XPS Pattern of chromium with fitted curves. | |
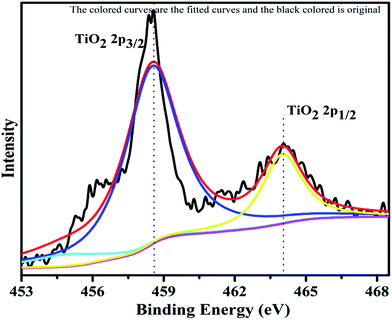 |
| | Fig. 8 XPS Pattern of titanium dioxide with fitted curves. | |
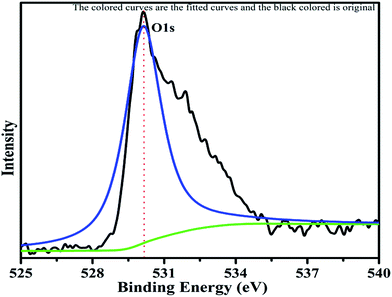 |
| | Fig. 9 XPS pattern of oxygen with fitted curves. | |
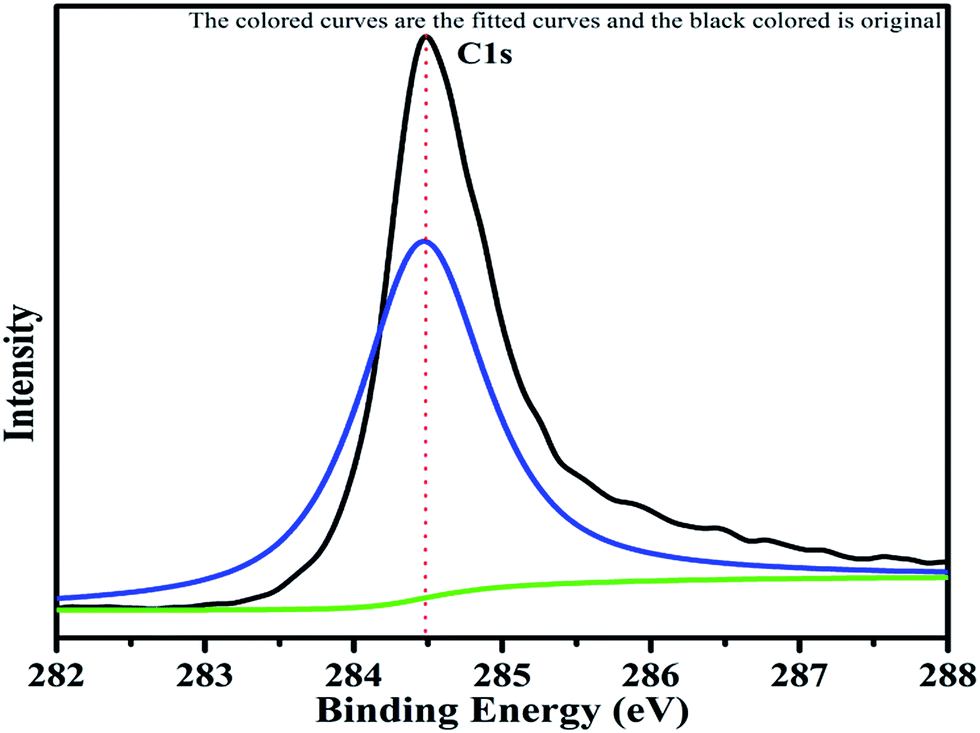
The XPS spectrum of carbon is shown in Fig. 10. The C 1s peak with binding energy of 284.4 eV can be clearly seen in the pattern. This is the carbon of rGO used as additive in the catalyst. This C 1s spectrum is taken as the reference standard for the peak shift correction of other elements because of the peak at 284.4 eV serves as the reference for the C–C bond. The carbon was observed as an asymmetric single bonded carbon (C–C) with sp2 hybridization, the characteristic state of graphene. The XPS study also confirmed the presence of rGO, due to the maximum intensity peak at 284.4 eV (see Fig. 10), which is associated with the asymmetric single bond of sp2 hybridization of carbon.43,44
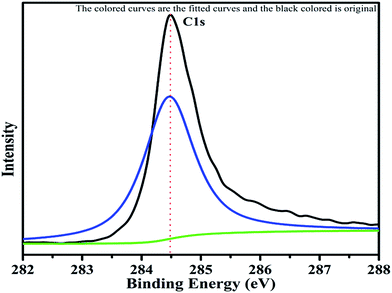 |
| | Fig. 10 XPS pattern of carbon with fitted curves. | |
3.7 Catalytic efficiency
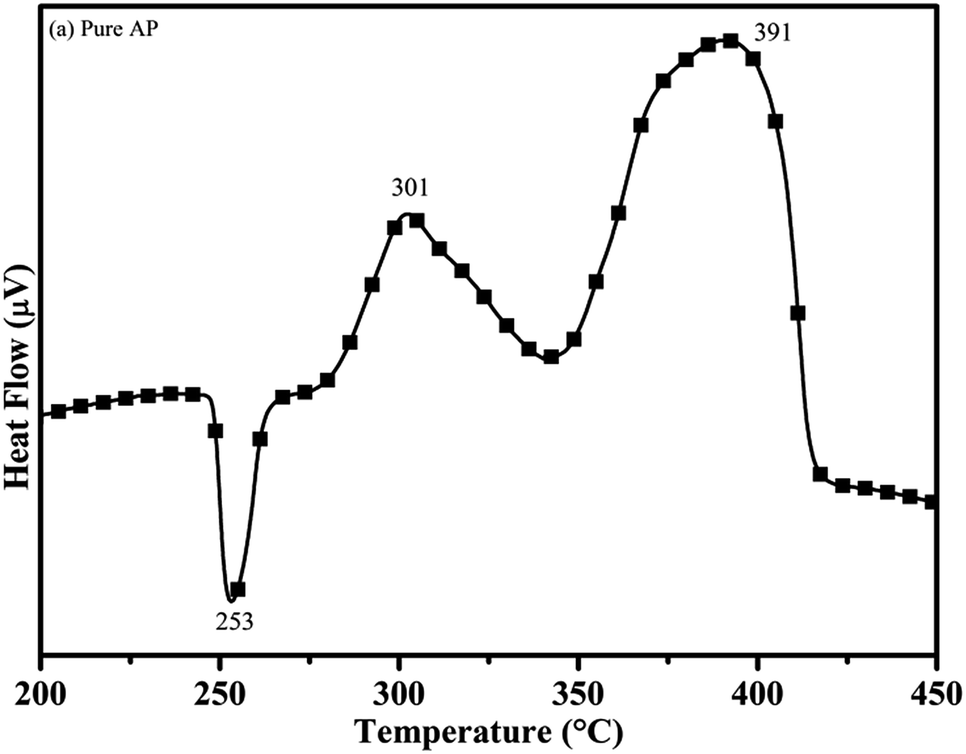
3.7.1 The effect of the synthesized catalyst on the thermal decomposition of ammonium perchlorate (AP). The catalytic activity of the as-synthesized material was carried out by the thermogravimetric and differential thermal analysis (TG-DTA) technique. The DTA curve of pure AP is shown in Fig. 11.30 According to the curve, pure AP exhibits the first endothermic peak at the temperature of 253 °C, associated with the crystallographic change of AP from orthorhombic to cubic. This is the characteristic crystallographic conversion of the AP30, which exists in the orthorhombic structure up to 230 °C, beyond which it gets transformed into the cubic structure. The cubic structure of AP provides a comparatively dense packing and crystal defects resulting in the faster rate of thermal decomposition and in turn, releases high energy during the decomposition process.45–49 The first exothermic peak or low temperature decomposition (LTD) peak is observed at 301 °C. LTD is assigned to the formation of intermediates, i.e., the decomposition of AP into NH4+ and ClO4−, which consequently form ammonia (NH3) and perchloric acid (HClO4), respectively. The low temperature decomposition is governed by the electron transfer process. This decomposition is due to dissociation and sublimation and may be more magnified in the reaction eqn (ii):| | |
NH4ClO4 → NH4(s)+ + ClO4(s)− ↔ NH3(g) + HClO4(g)
| (ii) |
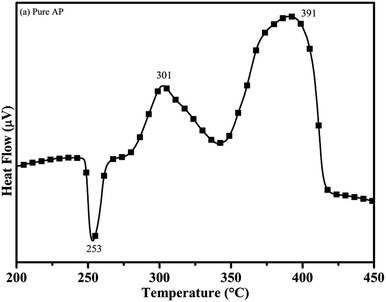 |
| | Fig. 11 DTA curve of pure ammonium perchlorate (AP). | |
The critical and most important factor governing the decomposition of (NH4ClO4) is the above-mentioned electron transfer process. The systematic scheme is given as follows:
| NH4+ + e− → NH3 + H (atomic hydrogen) |
ClO3 acts as an electron scavenger and is converted into ClO3−.
The tri-metal composite of Cu–Cr–Ti–O (i.e. Cu–Cr–O–0.7TiO2) exhibits excellent properties, due the following proposed mechanism. Cu–Cr–O heterogeneously couples with TiO2, due to the suitable energy band gap. Cu–Cr–O has a 3d orbital structure and can simultaneously participate in the process of electron transfer. Cu–Cr–O releases the electron under stimulated conditions and it induces the conduction band of TiO2 resulting in the active participation of both Cu–Cr–O and TiO2. The mechanism of electron release is as follows:
| Cr3+ + ClO4− → Cr2+ + ClO4 |
| Cr3+ + Cu2+ (3d9) → Cr3+ + Cu+ (3d10) |
| Cr4+ + ClO4− → Cr3+ + ClO4 |
However, in the case of the Cu–Cr–O–TiO2–rGO composite, rGO exhibits excellent electrical and optoelectrical properties with n-type semiconducting nature,50 which allows it to accept the electrons released from ClO4−, and intermediates are formed during the decomposition of ammonium perchlorate (NH4ClO4). A continuous electron transfer process from metal oxides and ClO4− takes place. In the Cu–Cr–O–TiO2–rGO system, the Cu–Cr–O–TiO2 being the p-type semiconductor releases the electron and rGO being the n-type semiconducting material accepts and facilitates the motion of those electrons released from Cu–Cr–O–TiO2 more smoothly. Consequently, the electron transfer process was tremendously enhanced, due to having high electron mobility, compared to the Cu–Cr–O–TiO2 system.
The second exothermic peak (or third i.e. final decomposition) of AP occurred at the comparatively higher temperature of 391 °C. This is known as the high temperature decomposition (HTD) peak. HTD behavior is also exothermic in nature like LTD. HTD peak at 391 °C is associated with the further decomposition of H2O, HCl, N2, H2 and O.51 At high temperature decomposition, HClO4 gets converted into small molecules, and the reaction is governed by photon transfer process. The probable reactions are shown below in eqn (iii) and (iv).
| | |
NH4ClO4 → H2O + NOx + Cl2+ N2 +HCl
| (iii) |
| | |
HClO4 → H2 + Cl2 + O2 +H2O
| (iv) |
The particles of CuCr2O4·0.7TiO2 reduced the peak temperature of high decomposition with the merging of the low temperature decomposition peak at a faster rate. CuCr2O4·0.7TiO2 initiated the decomposition process by the electron transfer process, followed by the deprotonation of HClO4 on a solid surface in the gaseous phase. During the decomposition, rGO plays a vital role in accelerating the electron transfer process.
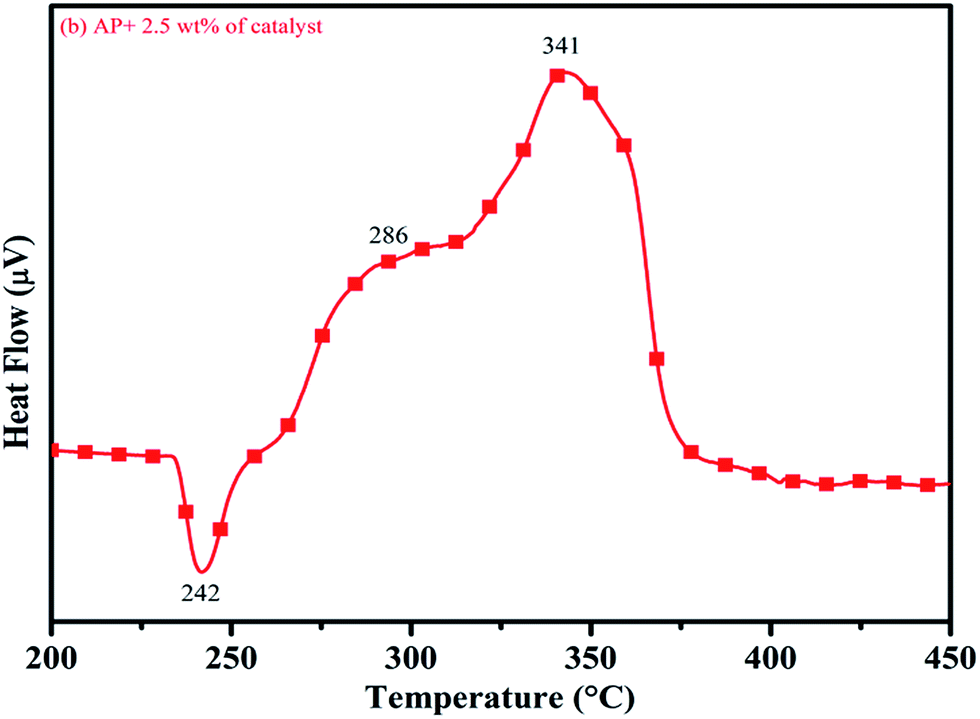
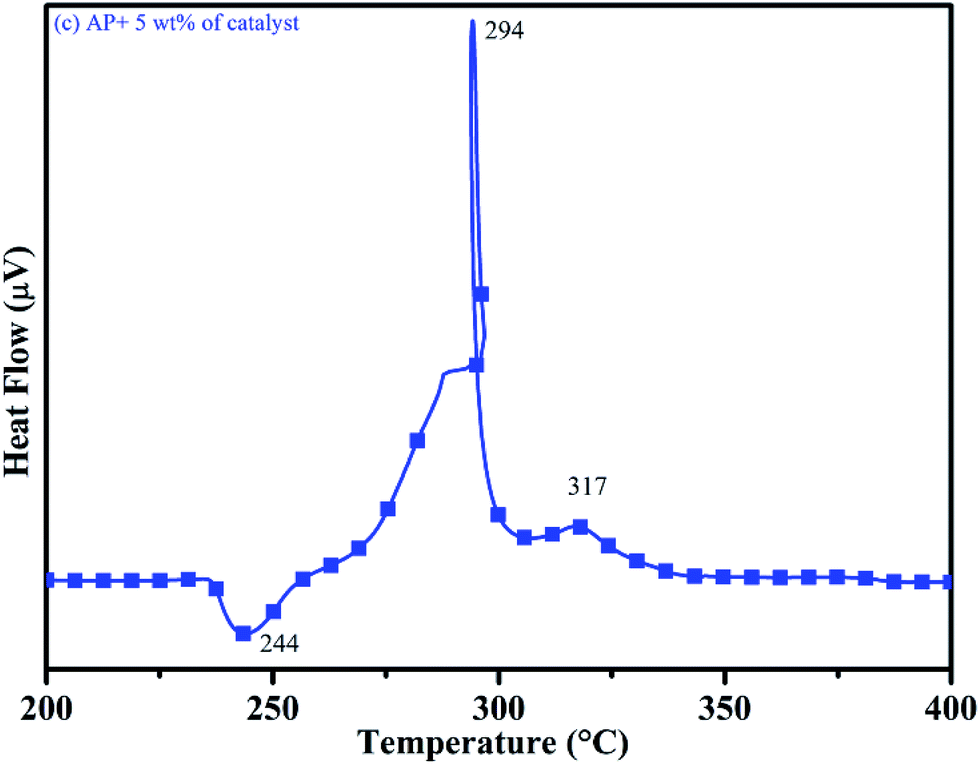
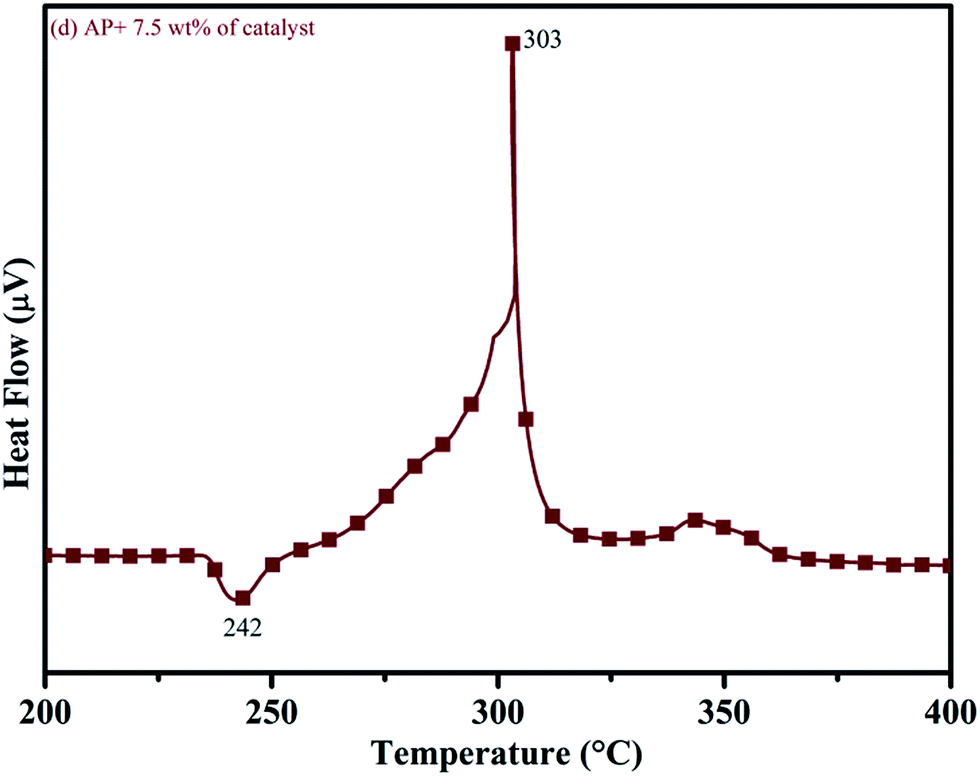
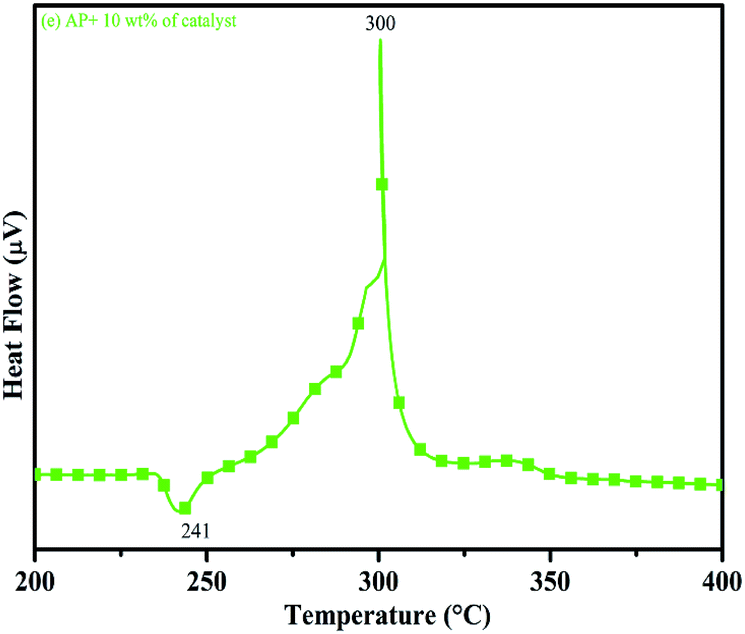
DTA curves of AP modified with different amounts (2.5, 5.0, 7.5 and 10 wt%) of synthesized catalyst (Cu–Cr–O–TiO2–GO) are shown in Fig. 12, 13, 14 and 15, respectively. A careful examination of the first endothermic peaks in the DTA curves of all the samples, i.e. pure as well as modified AP, concluded that the incorporation of catalyst additive into pure AP significantly reduced the crystallographic transition temperature from 253 °C to 242 °C. However, the study of this peak does not fall within the scope of the present study, yet the results are discussed briefly; however, another two exothermic peaks, LTD and HTD, are of interest in the present study. A significant effect of different wt% of synthesized catalyst in pure AP has been observed on the low as well as high temperature decomposition peak positions of AP. In the AP sample modified with the least amount (2.5 wt%) of catalyst (see Fig. 12), the partial decomposition occurred at 286 °C rather than 301 °C, and complete decomposition took place at 341 °C rather than 391 °C as for pure AP. Peak positions of LTD and HTD were reduced significantly by 15 °C and 50 °C, respectively, compared to the those of pure AP. On the other hand, the separation between LTD and HTD was also reduced. More interesting results were observed when the amount of catalyst was increased. As the 5 wt% of the synthesized catalyst was added into pure AP, interestingly, both the LTD and HTD merged, followed by single peak decomposition at 294 °C as shown in Fig. 13. A tremendous lower shift of 97 °C of HTD and 7 °C lower shift of LTD were noticed. The single stage decomposition of AP assisted by the catalyst shows the spontaneous process of decomposition occurring within an exceptionally lower time interval. This graph contains a very small hump at 317 °C, which may have appeared due to an infinitesimally small amount of NH4ClO4 or HClO4 left in the sample. When the amount of catalyst was further increased to 7.5 wt% and 10 wt% in AP, the recorded thermal decomposition profiles of AP showed single decomposition peaks at 303 °C (see Fig. 14) and 300 °C (see Fig. 15), respectively. Single stage peak decomposition positions for higher concentrations of catalyst are shifted towards higher comparatively temperatures, which is against one of our requirements, since our objective is to reduce the thermal decomposition of AP with single stage decomposition. The addition of larger amounts (7.5 wt% and 10 wt%) of catalyst does not have a significant impact on the thermal decomposition of AP, compared to the addition of 5 wt% of catalyst into AP. However, these samples (7.5 and 10 wt% of catalyst into AP) have much better performance than the industrial catalyst (ACR) as well as the previously reported catalysts.31
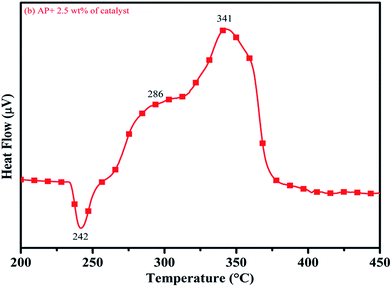 |
| | Fig. 12 DTA curve of AP modified with 2.5 wt% of catalyst. | |
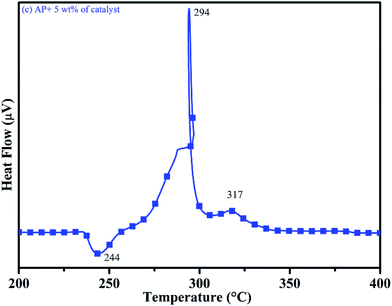 |
| | Fig. 13 DTA curve of AP modified with 5.0 wt% of catalyst. | |
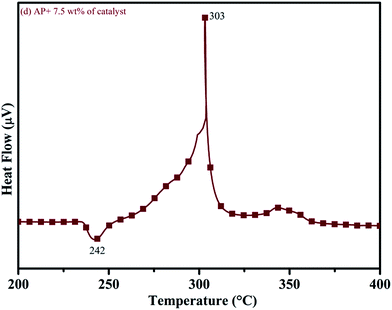 |
| | Fig. 14 DTA curve of AP modified with 7.5 wt% of catalyst. | |
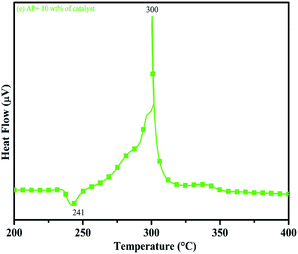 |
| | Fig. 15 DTA curve of AP modified with 10 w% catalyst. | |
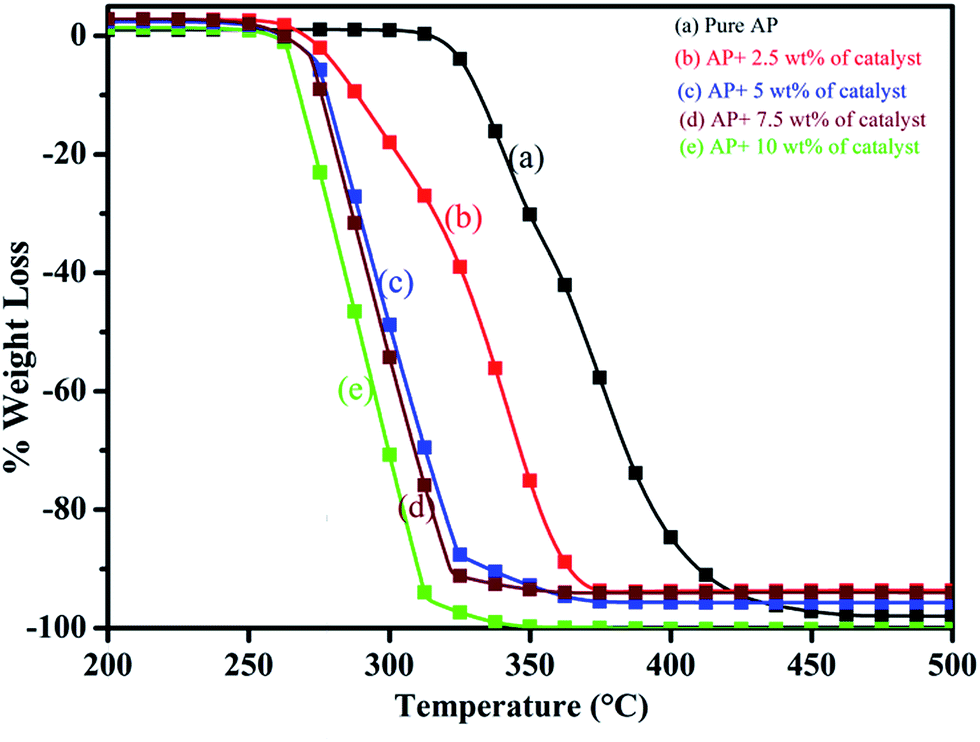
Thus, excellent single stage decomposition of AP was achieved at minimum temperature (294 °C), with maximum decomposition rate (burn rate) in the presence of 5.0 wt% of synthesized catalyst material in AP. Fig. 16(a–e) represents the TGA curves of (a) pure AP and AP with (b) 2.5, (c) 5, (d) 7 and (e) 10 wt% of synthesized catalyst (Cu–Cr–O–TiO2–GO) recorded in the temperature range of 200–500 °C. TGA curves of all the samples are in line with the corresponding DTA curve. TGA curves show the change in % weight loss of the sample in the temperature range of 260 to 380 °C. Beyond 380 °C, the TGA curve becomes straight, which clearly explains the completion of the thermal decomposition of ammonium perchlorate.
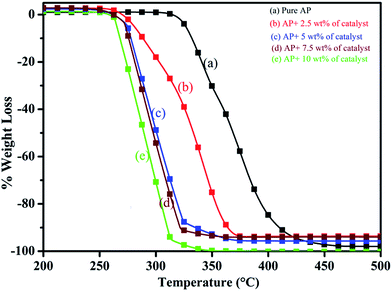 |
| | Fig. 16 (a–e): TGA curves of (a) pure AP, and AP with (b) 2.5, (c) 5, (d) 7 and (e) 10 wt% of the catalyst. | |
3.8 Burn rate of solid composite propellant
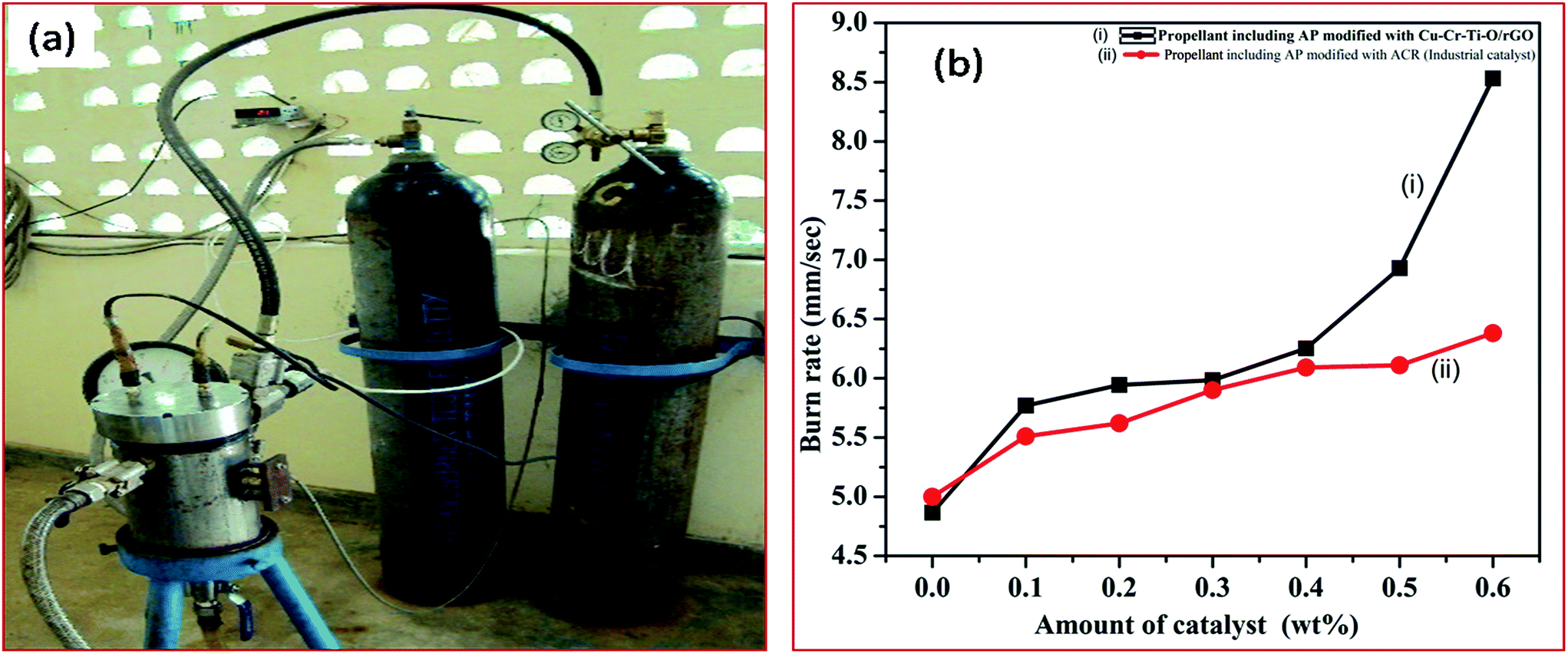
3.8.1 Effect of the synthesized catalyst (modified AP) on the burn rate of solid composite propellant (SCP). The combustion efficiency of the SCP is the function of the stoichiometry of the propellant ingredients. Since the major ingredient (68 wt%) of SCP is AP, the thermal decomposition of AP is the major deciding factor for the combustion efficiency of SCP. Furthermore, the thermal decomposition of AP is strongly dependent on its composition, particularly on the presence of catalyst, as well as on particle size and the shape of AP. A change in the thermal decomposition of AP will ultimately affect the burn rate of SCP. The better the thermal decomposition of AP, the higher the burn rate of SCP. In this study, the thermal decomposition of AP was remarkably enhanced using different concentrations of synthesized catalyst in AP. The AP modified by the synthesized catalyst is used in SCP to enhance the burn rate of SCP. The efficiency of the synthesized catalyst in terms of the burn rate of SCP is comparable to that of the industrial catalyst (ACR). The higher burn rate at low temperature with the lower pressure exponent is our requirement. The burn rate of the SCP is evaluated at combustion pressure of 33 bar using the acoustic measurement method through the strand burner, as shown below in Fig. 17(a).
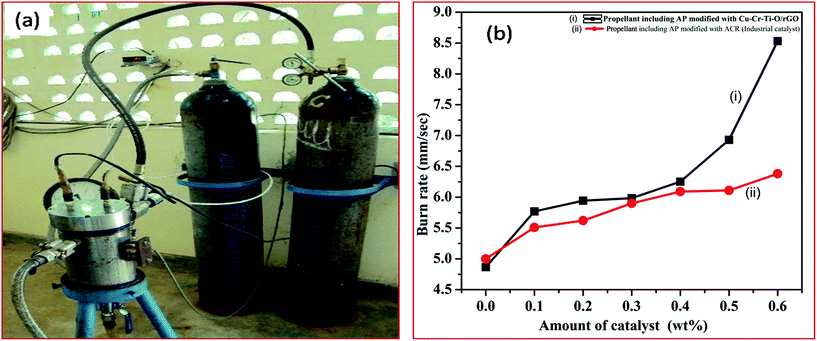 |
| | Fig. 17 (a) Images of the strand burner. (b) Burn rate of (i) SCP having AP modified with synthesized catalyst and (ii) SCP having AP modified with industrial catalyst. | |
The governing burn rate equation for the strand burner is shown below.
where
r is the burn rate in mm s
−1, ‘
a’ is the function of the initial temperature,
p is the combustion pressure in bar and
n represents the combustion index.
The burn rate curves of SCP having AP modified with synthesized catalyst and AP modified with industrial catalyst are shown in Fig. 17(b). The burn rate was achieved using the strand burner. The excellent catalytic effect is observed in presence of 5 wt% of CuCr2O4·0.7TiO2–rGO catalyst in AP. A tremendous increase (175.31%) in the burn rate of SCP is observed at 0.6 wt% of synthesized catalyst (8.531 mm s−1) in AP compared to pure AP (4.866 mm s−1). The burn rate of SCP is noticed to be 133.61% for AP modified with industrial catalyst ACR (6.385 mm s−1). The obtained faster rate decomposition of AP in the single stage can be considered as the explosive reaction, which is supported by the auto-ignition process. The synthesized rGO added Cu–Cr–O–0.7TiO2 catalyst has potential for use as a burn rate modifier in SCP for space vehicles, ballistic and tactical missiles.
4. Conclusions
A reduced graphene oxide (rGO) modified transition metal oxide based catalyst (Cu–Cr–O–7TiO2–rGO) was synthesized successfully via the sol–gel assisted Hummers' method. The rGO and Cu–Cr–O–7TiO2 were synthesized individually to form the desired composition of catalyst. Different concentrations (2.5, 5.0, 7.5 and 10 wt%) of Cu–Cr–O–0.7TiO2–rGO catalyst were added to AP, and their individual catalytic behavior in the thermal decomposition of AP was investigated extensively.
The 5.0 wt% of Cu–Cr–O–0.7TiO2–rGO catalyst added to AP exhibited excellent catalytic activity, accelerating the decomposition behavior of AP. This novel composition of catalyst added in AP was further examined on the burn rate of SCP. The burn rate of SCP having AP modified with the synthesized catalyst was found to be tremendously enhanced as compared to the SCP having AP modified with industrial catalyst (ACR) as well as pure AP. The burn rate of the synthesized catalyst assisted SCP was observed to be 133% and 175%, as compared to the industrial catalyst and pure AP assisted SCP. Further application of novel catalyst synthesized in the present study will led to the revolution in the field of missiles technology for next generation.
Conflict of interest
Authors declare no conflict of interest.
Acknowledgements
H. Kumar is thankful to J. Ram Mohan, General Manager SF Complex, DRDO, for his techno-commercial support.
Notes and references
- C. Huang, Q. Liu, W. Fan and X. Xiu, Sci. Res., 2015, 5, 16736 CAS.
- R. A. Chandru, S. Patra, C. Oommen, N. Munichandraiah and B. N. Rgahunandan, J. Mater. Chem., 2012, 22, 6536–6538 RSC.
- D. L. Reid, R. Draper, D. Richardson, A. Demko, T. Allen, E. L. Petersen and S. Seal, J. Mater. Chem. A, 2014, 2, 2313–2322 CAS.
- K. D. Grossman, T. S. Sakthivel, C. Dillier, E. L. Petersen and S. Seal, RSC Adv., 2016, 6, 89635–89641 RSC.
- D. L. Reid, A. E. Russo, R. V. Caro, M. A. Stephens, A. R. LePage, T. C. Spalding, E. L. Petersen and S. Seal, Nano Lett., 2007, 7, 2157–2161 CrossRef CAS.
- M. A. Stephens, E. L. Petersen, R. Carro, D. L. Reid and S. Seal, Propellants, Explos., Pyrotech., 2010, 35, 143–152 CrossRef CAS.
- N. Li, M. Cao, Q. Wu and C. Hu, CrystEngComm, 2012, 14, 428–434 RSC.
- V. V. Boldyrev, Thermochim. Acta, 2006, 443, 1–36 CrossRef CAS.
- P. R. Patil, V. N. Krishnamurthy and S. S. Joshi, Propellants, Explos., Pyrotech., 2008, 33, 266–270 CrossRef CAS.
- V. G. Dedgaonkar and D. B. Sarwade, J. Therm. Anal., 1990, 36, 223–229 CrossRef CAS.
- K. Kishore and M. R. Sunitha, AIAA Pap., 1979, 17, 1118–1125 CrossRef CAS.
- L. Liu, F. Li, L. Tan, L. Ming and Y. Yi, Propellants, Explos., Pyrotech., 2004, 29(1), 34–38 CrossRef CAS.
- L. Song, S. Zhang, B. Chen, J. Ge and X. Jia, Colloids Surf., A, 2010, 360, 1–5 CrossRef CAS.
- E. A. Gheshlaghi, B. Shaabani, A. Khodayari, Y. A. Kalandaraghand and R. Rahimi, Powder Technol., 2012, 217, 330–339 CrossRef.
- Y. Zhnag, X. Liu, J. Nie, L. Yu, Y. Zhong and C. Huang, J. Solid State Chem., 2011, 184, 387–390 CrossRef.
- H. Xu, X. Wang and L. Zhang, Powder Technol., 2008, 185, 176–180 CrossRef CAS.
- R. A. Chandru, S. Patra, C. Oommen, N. Munichandraiah and B. N. Raghunandan, J. Mater. Chem., 2012, 22(14), 6536–6538 RSC.
- T. Liu, L. Wang and B. Hu, Mater. Lett., 2008, 62, 4056–4058 CrossRef CAS.
- S. Zhao and D. Ma, J. Nanomater., 2010, 2010, 1–5 Search PubMed.
- L. Chen, P. Li and G. Li, J. Alloys Compd., 2008, 464, 532–536 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80(6), 1339 CrossRef CAS.
- S. Pei and H. M. Cheng, Carbon, 2012, 50(9), 3210–3228 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4(4), 217–224 CrossRef CAS PubMed.
- Y. Yuan, W. Jiang, Y. Wang, P. Shen, F. Li, P. Li, F. Zhaoand and H. Gao, Appl. Surf. Sci., 2014, 303, 354–359 CrossRef CAS.
- N. Li, Z. Geng, M. Cao, L. Ren, X. Zhao, B. Liu, Y. Tian and C. Hu, Carbon, 2013, 54, 124–132 CrossRef CAS.
- Z. B. Lei, F. H. Shi and L. Lu, ACS Appl. Mater. Interfaces, 2012, 4(2), 1058–1064 CAS.
- P. Wang, Y. M. Zhai, D. J. Wang and S. J. Dong, Nanoscale, 2011, 3(4), 1640 RSC.
- W. Zhang, Q. Luo, X. Duan, Y. Zhou and C. Pei, Mater. Res. Bull., 2014, 50, 73–78 CrossRef CAS.
- X. Guan, L. Li, J. Zheng and G. Li, RSC Adv., 2011, 1, 1808–1814 RSC.
- D. Zhan, Z. Ni, W. Chen, L. Sun, Z. Luo, L. Lai, T. Yu, A. Thye, S. Wee and Z. Shen, Carbon, 2011, 49, 1362–1366 CrossRef CAS.
- H. Kumar, P. N. Tengli, V. K. Mishra, P. Tripathi, D. B. Pal and P. K. Mishra, RSC Adv., 2017, 7, 12486–12495 RSC.
- H. K. Jeong, Y. P. Lee, M. H. Park, K. H. An and I. J. Kim, et al., J. Am. Chem. Soc., 2008, 130, 1362 CrossRef CAS PubMed.
- F. Yang, M. Zhao, B. Zheng, D. Xiao, L. Wu and Y. Guo, J. Mater. Chem., 2012, 22, 25471–25479 RSC.
- V. K. Mishra, B. N. Bhattacharjee, D. Kumar, S. B. Rai and O. Parkash, New J. Chem., 2016, 40, 5432–5441 RSC.
- V. K. Mishra, S. B. Rai, B. P. Asthana, O. Parkash and D. Kumar, Ceram. Int., 2014, 40, 11319–11328 CrossRef CAS.
- A. Kaniyoor and S. Ramaprabhu, AIP Adv., 2012, 22, 032183 CrossRef.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8(1), 36–41 CrossRef CAS PubMed.
- R. J. Nemanich and S. A. Solin, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 392 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- A. Das, B. Chakraborty and A. K. Sood, Bull. Mater. Sci., 2008, 31, 579 CrossRef CAS.
- L. G. Cancado, K. Takai, T. Enoki, M. Endo, Y. A. Kim, H. Mizusaki, N. L. Speziali, A. Jorio and M. A. Pimenta, Carbon, 2008, 46, 272 CrossRef CAS.
- M. Aronniemi, J. Sainio and J. Lahtinen, Surf. Sci., 2005, 578, 108–123 CrossRef CAS.
- A. H. C. Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–163 Search PubMed.
- D. S. Sutar, G. Singh and V. Divakar Botcha, Appl. Phys. Lett., 2012, 101, 103103–103107 Search PubMed.
- G. P. Sutton and O. Biblarge, Rocket Propulsion Elements, A Wiley Inter-Science Publication, New York, 7th edn, 2000 Search PubMed.
- P. W. M. Jacob and H. M. Whitehead, Chem. Rev., 1969, 69, 551–590 CrossRef.
- P. Wang, Y. M. Zhai, D. J. Wang and S. J. Dong, Nanoscale, 2011, 3(4), 164011 Search PubMed.
- C. Ping, F. Li, Z. Jian and J. Wei, Propellants, Explos., Pyrotech., 2006, 31, 452–455 CrossRef CAS.
- W. Zhang, P. Li, H. Xu, R. Sun, P. Qing and Y. Zhang, J. Hazard. Mater., 2014, 268, 273–280 CrossRef CAS PubMed.
- G. Giovannetti, P. A. Khomyakov, G. Brocks, V. M. Karpan, J. van den Brink and P. J. Kelly, Phys. Rev. Lett., 2008, 101, 026803 CrossRef CAS PubMed.
- A. G. Keenan and R. F. Q. Siegmund, Chem. Soc. Rev., 1969, 23, 430–452 RSC.
Footnotes |
| † Present Address: Defence Materials and Stores Research and Development Establishment (DMSRDE), Defence Research and Development Organization, (DRDO) G. T. Road Kanpur-208013, India. |
| ‡ Present Address: Endocrinology Division, CSIR-Central Drug Research Institute, Lucknow-226031, India. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Prahalad N. Tenglic,
Vijay Kumar Mishra‡
ab,
Prahalad N. Tenglic,
Vijay Kumar Mishra‡