
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of natural products containing benzofuran rings
Majid M. Heravi
 *,
Vahideh Zadsirjan
,
Hoda Hamidi
and
Parvin Hajiabbas Tabar Amiri
*,
Vahideh Zadsirjan
,
Hoda Hamidi
and
Parvin Hajiabbas Tabar Amiri
Department of Chemistry, School of Sciences, Alzahra University, Vanak, Tehran, Iran. E-mail: mmh1331@yahoo.com
First published on 5th May 2017
Abstract
Research on natural products containing benzofuran has remarkably increased during the past few decades. Newly isolated natural products with complex structures are being studied, characterized and screened for possible biological activities. Several of such compounds have exhibited various biological activities, thus their total syntheses have attracted much attention from synthetic organic chemists. In this review, we aim to highlight the origins, structures, biological potencies, and synthetic approaches of those natural products bearing at least one benzofuran in their complex structures. Furthermore, we especially focus on the step in which this key heterocycle is installed during the total synthesis of a natural product as the desired target.
 Majid M. Heravi | Majid M Heravi was born in 1952 in Mashhad, Iran. He received his B. Sc. degree from the National University of Iran in 1975 and his M. Sc. and Ph. D. degrees from Salford University, England in 1977 and 1980, respectively. He completed his doctoral thesis under the supervision of the late Jim Clarck in Salford University, England. He started his career as a research fellow in Daroupakhsh (a pharmaceutical company) in 1981 Tehran, Iran and joined as an assistant professor to Ferdowsi University of Mashhad, Iran in 1983, and was promoted to associate professor in 1993 and full professor in 1997 in the aforementioned university. In 1999 he moved to Alzahra University of Tehran, Iran as professor of chemistry where he still works. He has previously been a visiting professor at UC Riverside, California, USA and Hamburg University, Hamburg, Germany. His research interests focus on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. |
 Vahideh Zadsirjan | Vahideh Zadsirjan was born in Shiraz, Iran in 1979. She received her B. Sc. in Pure Chemistry from Kharazmi University in 2002, and her M. Sc by course in 2007 and Ph. D degrees in organic chemistry in 2016 under the supervision of Professor Majid M. Heravi from Alzahra University Tehran, Iran. His research is focused on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. |
 Hoda Hamidi | Hoda Hamidi was born in1984 in Ramsar, Mazandaran, Iran. She obtained her B.Sc. degree in applied chemistry from Mohaghdas Ardebili University, Ardebil, Iran, in 2006 and her M.Sc. degree in 2010 and Ph. D degree in 2015 in organic chemistry from Alzahra University, Tehran, Iran, in 2010 and 2015 under the supervision of Professor Hossein Abdi Oskooei and Professor Majid M. Heravi. Her research interests are in the area of the synthesis of organic compounds particularly heterocycles and synthetic methodology. |
 Parvin Hajiabbas Tabar Amiri | Parvin Hajiabbasi was born in1984 in Babol, Mazandaran, Iran. She received her B.Sc. degree in chemistry from Tehran University, Tehran, Iran in 2006, her M.Sc. degree in organic chemistry from Tehran University, Tehran, Iran, in 2010 and her Ph.D degree in organic chemistry at Alzahra University, Tehran, Iran under supervision of Professor Ghodsi Mohammadi Ziarani in 2015. She is presently enduring her researches in the synthesis of organic compounds, heterocycles, natural products and medicinal compounds. |
1 Introduction
Benzofuran and its derivatives are widely present as scaffolds in the complex molecules of natural products. These kinds of naturally occurring compounds have attracted much attention from synthetic organic chemists, due to their interesting biological and pharmacological activities.1–3 Several natural products bearing benzofuran and its derivatives as a moiety,4–6 exhibit diverse biological activities such as being potent antibacterial,7 antimicrobial,8 antitumor,9 anticonvulsant anti-inflammatory,10 antidiabetic11 and antineophobic agents.12 Furthermore, certain derivatives of benzofuran present in natural products show high cytotoxicity.13 The exceptional structural features of benzofuran and its wide assortment of biological as well as pharmacological activities make it a privileged structure in the field of drug discovery. Nowadays, several benzofurans are being prescribed for treatment of Alzheimer's disease.14 They have also been screened and found being acting as protein tyrosine phosphatase inhibitors (PTP-1B).15 Indeed benzofuran is a versatile scaffold for its synthetic pathways and functionalization; moreover, it exhibits a medicinal chemistry interest due to its presence in several natural products.16 Various benzofuran derivatives have been isolated from plants kingdom and marine sources.17 Furthermore, they were also provided from bacterial or fungal metabolites.18 Benzofurans occur in numerous natural products, as part of small molecule i.e. benzofury,19 as well as more complex drug such as notorious morphine (as street drug) and macromolecule like rifamycin.20 They also can be assembled in more complex architectures in a wide range of natural products such as fungi, bacteria, etc.Naltrindole (NTI) and its benzofuran derivative (NTB) were proved being antagonist of different opioid receptor agonists in the tail-flick antinociceptive evaluation in mice.21 Amiodarone, (2-{4-[(2-butyl-1-benzofuran-3-yl)carbonyl]-2,diiodophenoxy}ethyl)diethylamine, 1 is an antiarrhythmic agent which nowadays prescribed for treatment of different types of cardiac dysrhythmias, both ventricular and atrial.22 Dronedarone, N-(2-butyl-3-(p-(3-(dibutylamino)propoxy)benzoyl)-5-benzofuranyl) methane sulfonamide, 2 is an efficient drug which stop atrial fibrillation and atrial flutter relapses, which is prescribe for low-risk patient (Fig. 1).23
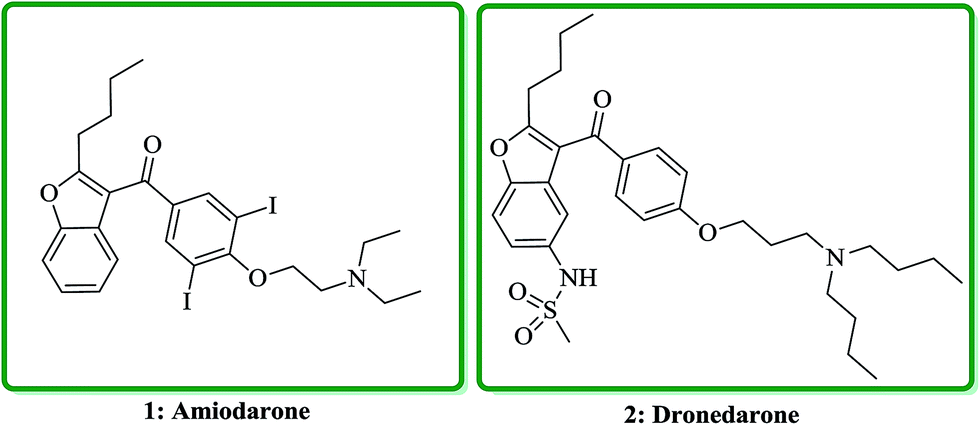 | ||
| Fig. 1 The structure of amiodarone 1 and dronedarone 2. | ||
Psoralen (also called psoralene) (7H-furo[3,2-g]chromen-7-one) 3 is the parent in a family of naturally occurring compounds known as furocoumarins. It is structurally related to coumarin and can be regarded as an umbelliferone derivative (Fig. 2).24
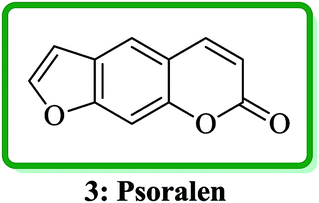 | ||
| Fig. 2 The structure of psoralen 3. | ||
Machicendiol 4, a benzofuran isolated from the extracts of Machilus glaucescens,25 has been long used as traditional medicine in the treatment of asthma, rheumatism, and ulcers for a long period of time.26 It has been found that 2,5-disubstituted benzofurans are particularly active in enhancement of insulin sensitivity.27 The benzofuran-fused benzocarbazol has been found to inhibit the growth of malignant cells and they also showed antibiotic properties (Fig. 3).28,29
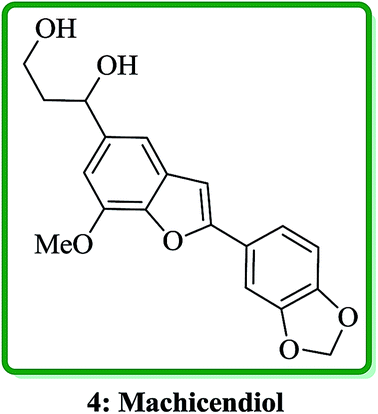 | ||
| Fig. 3 The structure of machicendiol 4. | ||
Ailanthoidol 5 (ref. 30) and XH-14 6 were isolated from the chloroform-soluble fraction of stem woods of Zanthoxylum ailanthoidos.31 Studies on the constituents of plants of Zanthoxylum ailanthoidos, they are used in Chinese traditional herbal medicine. These compounds exhibit different interesting pharmacological activities.32,33 Ailanthoidol 5, a neolignan derivative, demonstrated antiviral, antioxidant and antifungal potencies (Fig. 4).34–39
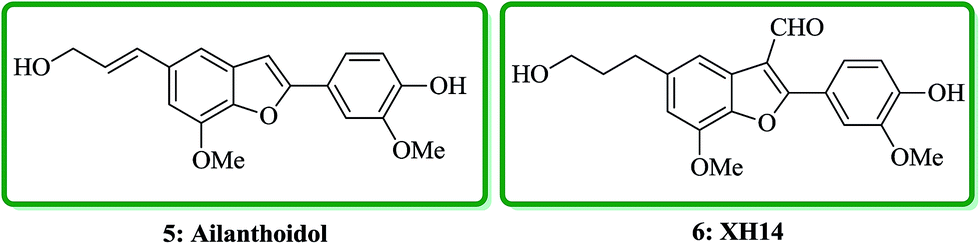 | ||
| Fig. 4 The structure of ailanthoidol 5 and XH-14 6. | ||
Significantly, the benzofuran derivatives containing the pyrazole nucleus were reported to be analgesic, anti-inflammatory, antipyretic, antiarrhythmic, muscle relaxant, psychoanaleptic, anticonvulsant and hypotensive.40–53 Remarkably, a large number of synthetic approaches have been attempted and accomplished for the synthesis of fused benzofurans. The synthesis frequently starting from differently appropriate substituted benzene rings. Most synthetic approaches towards benzofurans are based on the generation of the O–C2 or the C2–C3 bonds, in the vital ring closing step. Nevertheless, those approaches manipulating C3–C3 bond generation, via intramolecular cyclization of an already appropriately functionalized precursor. These approaches are particularly striking and much anticipated. They include: (a) acid-catalyzed cyclization of compounds continuing carbonyl group by dehydration,54,55 (b) palladium56,57 or platinum58-catalyzed59 ring closure by an intramolecular Wittig reaction60–62 or o-(acyloxy)benzyl anions,63 (c) condensation of activated methylene following Dieckmann reaction condtiions64,65 or ketene intermediate involved cyclization,66 (d) acid-catalyzed ring construction of α-aryloxycarbonyls67 or (e) intramolecular Friedel–Crafts reaction,68 (f) photolytic cyclization of α-phenylketones,69 and (g) gold(III)-catalyzed tandem reaction of O-arylhydroxylamines with 1,3-dicarbonyl substrates.70 Moreover, a one-pot reaction for the transformation of allyl aryl ethers to 2-methylbenzofurans via sequential reaction involving Claisen rearrangement/oxidative cyclization has been reported.71 3-Acyl-2-aminobenzofurans give 2-(cycanomethyl) phenyl esters using catalytic quantity of Pd(OAc)2, PCy3, and Zn.72
Recently, the role of benzofuran and its derivatives present in natural products as emerging framework for antimicrobial agents,73 antibreast cancer agents74 and in other natural lead molecules with diverse pharmacological properties have been comprehensively, revealed.75,76 We are especially interested in heterocyclic chemistry77–88 and heterocyclic compounds showing high biological activity.89 In recent years, we have highlighted the applications of several name reactions in the total synthesis of biologically active natural products and applications of asymmetric synthesis in total synthesis of natural products.90–95
In this line very recently, we focused on chemistry of benzofurans and published a chapter in Advances in Heterocyclic Chemistry entitled the recent advances in the synthesis of benzo[b]furans.78 Due to the massive number of pertinent references, coverage of the chemistry of this key heterocycle from different aspects, features and issues were surpassed by the limits in length and pages imposed by the editorial board of this book series. Thus, we had to divide the rest of this vast subject into three reviews. In our two recent reviews, we disclosed the full perspective of reactivity of benzofurans96 and advances in the synthesis of biologically potent compounds bearing at least one benzo[b]furan moiety in their structures, respectively.97 In the present review, we collated the published reports on the total synthesis of natural products containing at least one benzofuran moiety in their complex structures. Noticeably, in spite of brief introduction of the natural products, their sources and the methods used for their characterization, we focused on the key step of construction of benzofuran moiety during the total synthesis of such natural products.
2 Construction of benzofuran as a scaffold in the structures of natural products during their total synthesis
Lignans and neolignans are interesting goals for organic synthetic chemists. The members of this family containing benzofuran showing broad spectrum of biologically activity. Among these compounds, the total synthesis of benzofuran neolignans98,99 have been considered following the biomimetic routes. In this approach, initially an appropriate 4-furyl-3-alkoxy-3-butenoic acid 10 was synthesized. As illustrated in Scheme 1, the aldehyde 7 (ref. 7) was reacted with the lithium salt of protected propynol 8 to afford the corresponding carbinol, which was then transformed into the 3-bromofuran 9 involving sequential reactions including oxidation of propargylic carbinol/selective deprotection of tetrahydropyranyl ether/acid-catalyzed cyclization to afford 3-bromofuran 9 in moderate overall yield. 3-Bromofuran 9 was then converted into the benzofuran 11 via the formation of 4-furyl-3-alkoxy-3-butenoic acid 10. The desired benzofuran 11 converted to 5 in several steps, eventually, ailanthoidol 5 obtained in moderate overall yield.34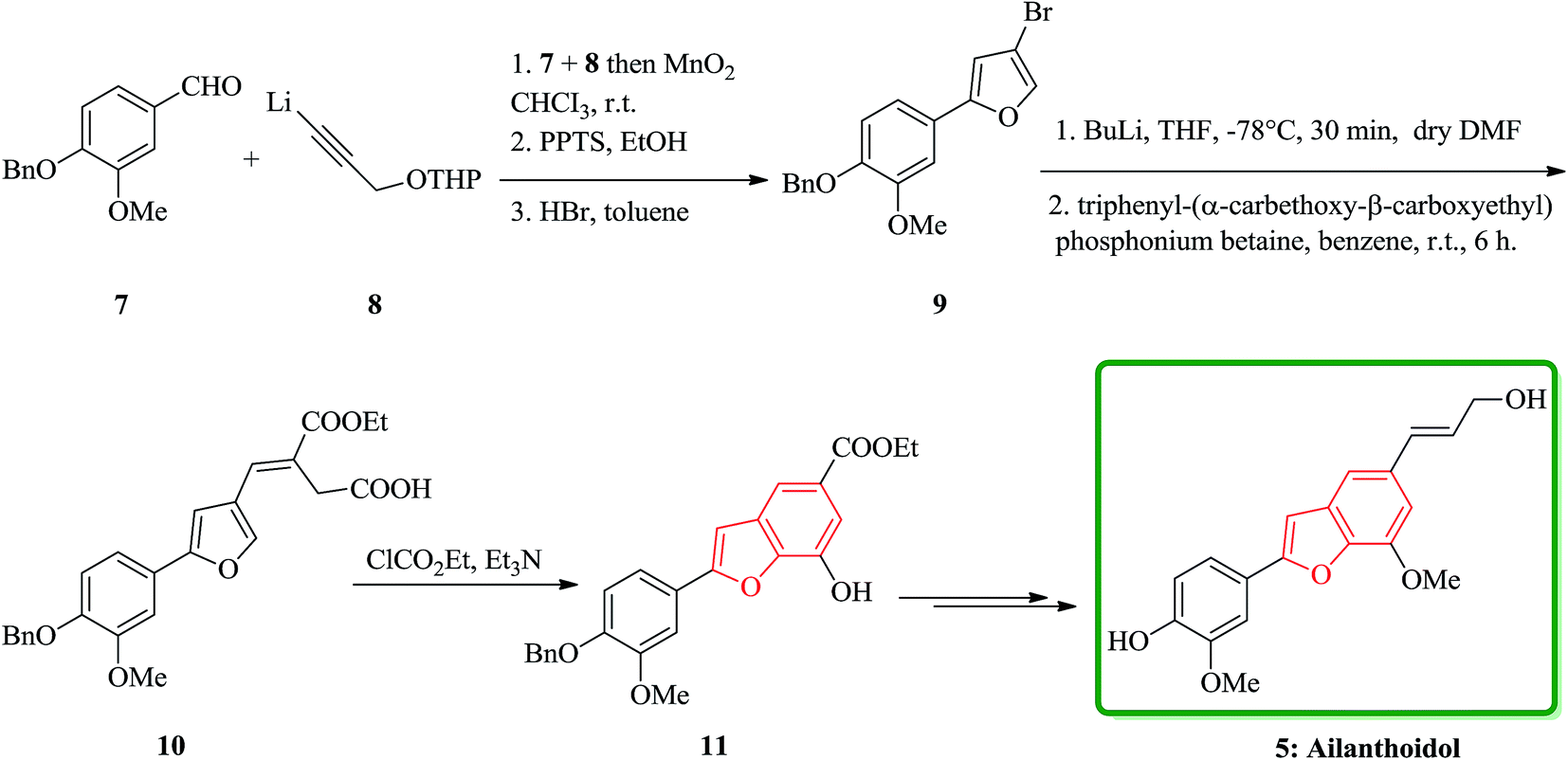 | ||
| Scheme 1 Total synthesis of ailanthoidol 5. | ||
XH-14 6 was initially isolated from the plant so called Salvia miltiorrhiza. Latter on it was found being a potent antagonist against the adenosine receptors.100 Ailanthoidol 5 is also a structurally related compound to 6. Although, there is no report on biological activity of this compound on the adenosine receptor, the extracts of the leaves and bark of this tree had been used as traditional medicine for long period. It has been reported several methods for the synthesis of XH-14 6.100–102 Another method towards the synthesis of XH-14 involved an oxidative dimerization of methyl ferulate (methyl-3-methoxy-4-hydroxycinnamate) to form the benzofuran skeleton has also been reported.103 This strategy gave only low overall yield (34%) and showed no flexibility for the synthesis of other analogs. An improved and efficient strategy for the total synthesis of ailanthoidol 5 has been reported by Lütjens and co-workers in 1998. This approach for the total synthesis found being attractive and practical. In this protocol, the total synthesis of ailanthoidol commenced with the building up of the benzofuran nucleus via coupling of the ortho-halophenol 12 and the alkyne 13 with simultaneous cyclization. The coupling of 12 and 13 was conducted under Sonogashira conditions.104 It affords a better yield and also found being re-producible (Table 1). It was also found that PdC12(PPh3)2 is more efficient catalyst than Pd(PPh3)4 and also iodophenol 12b was proved to react better than bromophenol 12a. The resultant benzofuran 14 was then converted into ailanthoidol in two steps. The first step was involving the removal of the protecting group using TiCl4 and the second step involved the reduction of the ester group using DIBAL to give the desired target 5 in 77% overall yield after crystallization from MeOH (Scheme 2).105
| Entry | Substrate | Conditions | Yield (%) |
|---|---|---|---|
| 1 | 12a | Cu-acetylide of 13, Py, reflux | 65 |
| 2 | 12a | Cu2O, Py, reflux | 62 |
| 3 | 12a | PdCl2(PPh3)2, CuI, NEt3, MeCN | 69 |
| 4 | 12a | Pd(PPh3)4, CuI, NEt3, MeCN | 52 |
| 5 | 12b | Pd(PPh3)2, CuI, NEt3, MeCN | 88 |
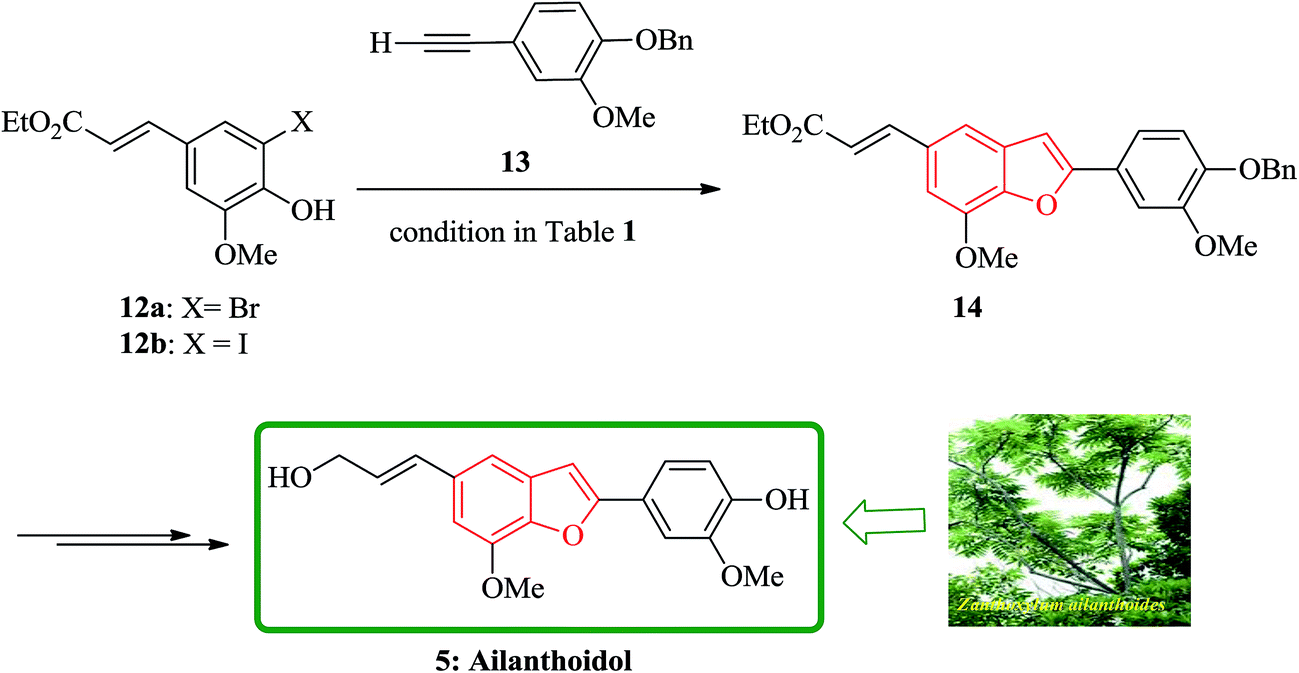 | ||
| Scheme 2 Total synthesis of ailanthoidol 5. | ||
Notably, the synthesis of XH-14 6 was accomplished using a similar protocol, employing the Sonogashira coupling reaction conditions. Nevertheless, in the case of the generation of the intermediate ortho-hydroxytolan, it was isolated preceding to cyclization and then subjected into Pd-catalyzed carbonylative cyclization reaction was to construct the benzofuran ring system with simultaneous acylation at the 3-position. Ethyl-3-methoxy-4-hydroxy-5-iodocinnamate 12b was protected as MOM ether and then coupled with the various substituted alkyne to provide the corresponding MOM protected ortho-hydroxytolan in high yield (92%). For the removal of the MOM protecting group oxalic acid in aqueous methanol was used to afford virtually quantitative yield of the ortho-hydroxytolan 16. Using a catalytic amount of PdCl2 to solution of 16 and NaOAc/MeOH under atmosphere of CO imposed cyclization to a vinyl-palladium(II) species in which after insertion of CO and reaction with methanol afforded the substituted benzofuran 17 in a satisfactory isolated yield. The resulting Pd(0) species were re-oxidized by copper(II)chloride permitting the utilization of a sub-stoichiometric amount of Pd. It was proved that the choice of base is crucial as 16, since a noticeable inclination being subjected to un-catalyzed auto-cyclization under basic conditions were observed. Therefore, the un-functionalized benzofuran 18 was constructed solely when K2CO3 was employed instead of Na2CO3. Conversion of 17 into the desired product 6 was then achieved via straightforward strategy in which after three steps, XH-14 6 was provided. This strategy provides a new gateway for the efficient synthesis of XH-14 6 in which affords the formulated isomer, only. This synthesis is also high yielding and flexible to give a wide variety of differently 2-substituted analogs (Scheme 3).105
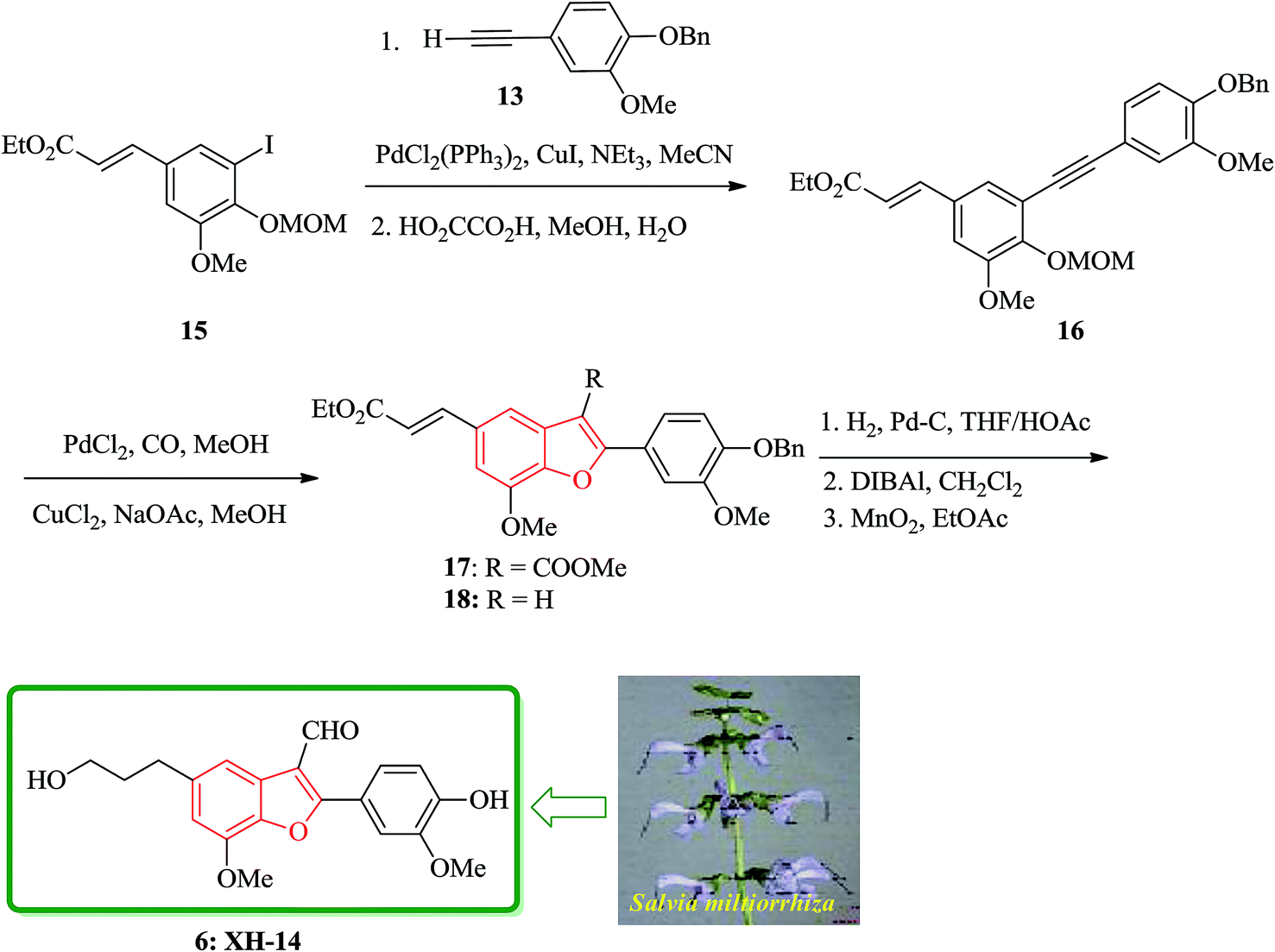 | ||
| Scheme 3 Total synthesis of XH-14 6. | ||
In another attempt, the total synthesis of ailanthoidol was also accomplished in 12 steps manipulating different functional transformations in a 17% overall yield starting from vanillin 19. A convenient method for the synthesis of ailanthoidol from vanillin is established, using trimethylsilyl diazomethane lithium salt to generate diphenyl acetylene which followed by oxymercuration cyclization of the resulting alkyne using mercury acetate in acetic acid as key steps. The mercurial intermediate 21 is found to be a very useful intermediate for the syntheses of analogs by the direct replacement of the mercurial moiety with a variety of functional groups. The desired intermediate 20 upon treatment with mercury acetate in acetic acid and then quenching with saturated sodium chloride solution afforded 2-(p-benzyloxy-m-methoxyphenyl)-3-chloromercurio-5-(5′,5′-dimethyl-1′,3′-dioxan-2′-yl)-7-methoxybenzofuran 21. The chloromercurial intermediate 21 without further purification was isolated and reduced with NaBH4 in THF to afford benzofuran 22 in high yields. After several steps the latter was converted into the desired natural product ailanthoidol 5 (Scheme 4).106
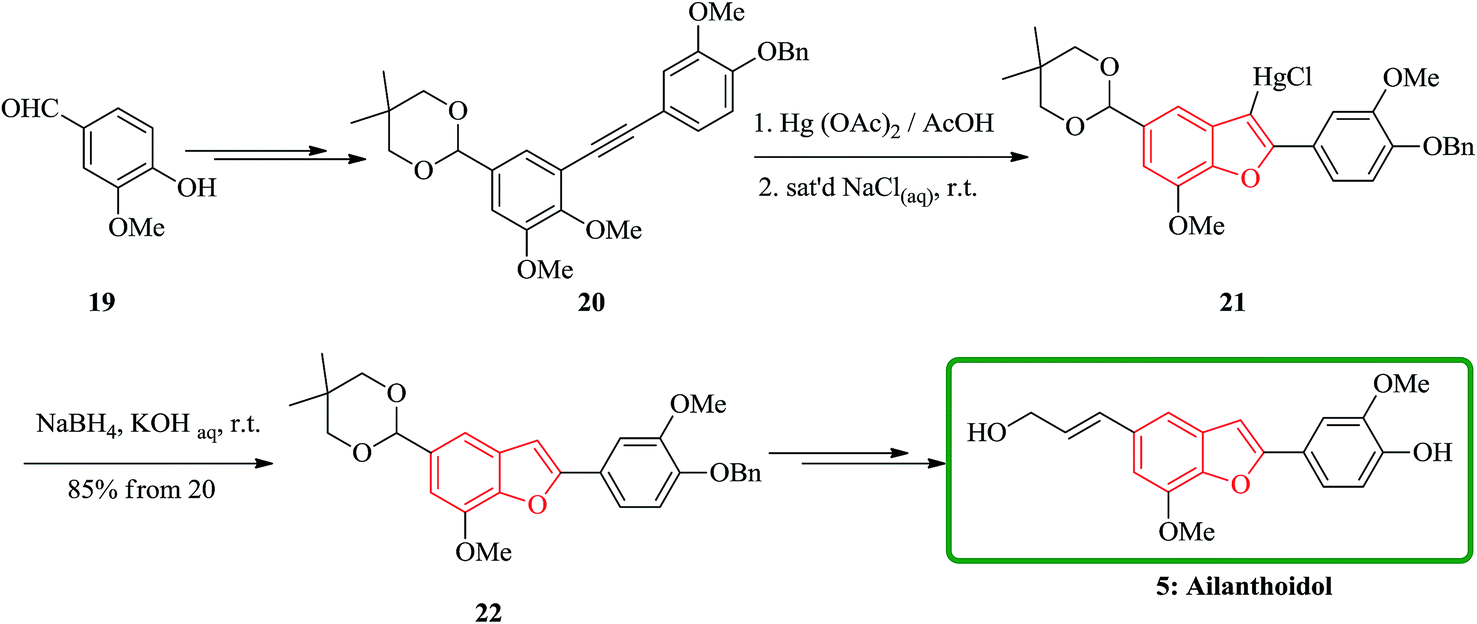 | ||
| Scheme 4 Total synthesis of ailanthoidol 5. | ||
Furthermore, ailanthoidol 5, was also synthesized via a route which is the longest linear sequence is only six steps in 48% overall yield. This pathway started from commercially available 5-bromo-2-hydroxy-3-methoxybenzaldehyde 23. The key transformation in the synthesis is the Stille coupling reaction of benzofuranyl bromide with stannanyl compounds. This synthetic strategy can be modified to give access to a variety of different ailanthoidol analogues. With the aim of developing a successful route to ailanthoidol 5, an alternative method of construction was examined. Accordingly, the removal of the benzyl protecting group with TiCl4 followed by DIBAL or LiAlH4 reduction of the ester to give 5 in the highest 95% yield (over two steps) (Scheme 5).30
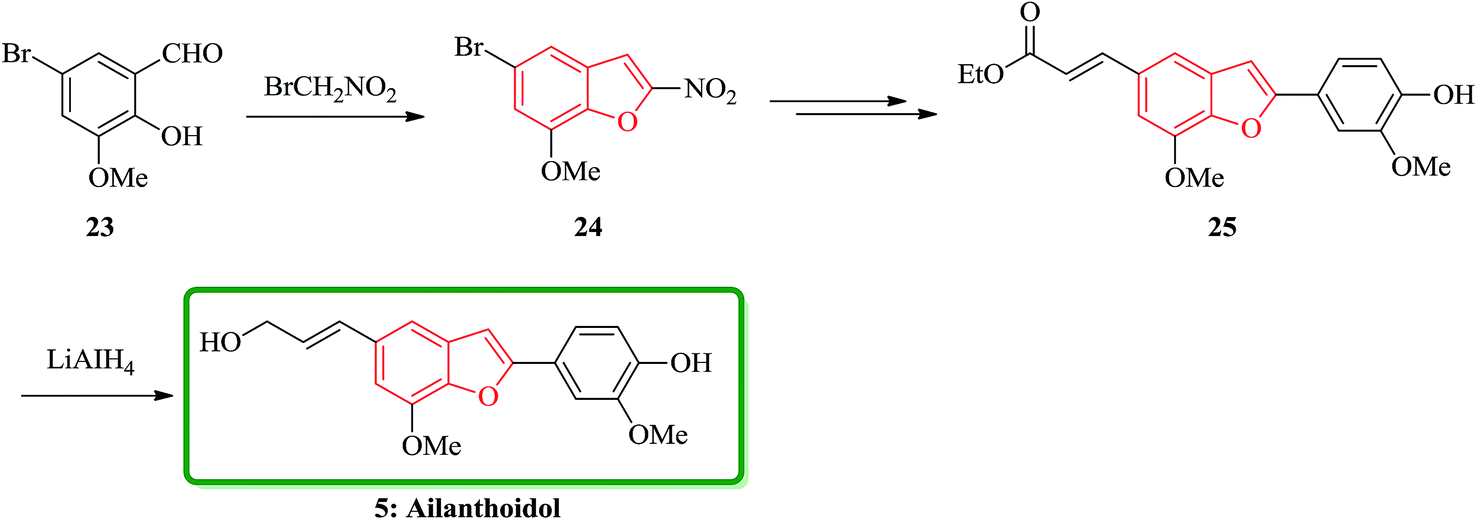 | ||
| Scheme 5 Total synthesis of ailanthoidol 5. | ||
In addition, some other natural products bearing 2-arylbenzofurans moiety in their structures such as, egonol 31a, homoegonol 31b, and demethoxyegonol 31c were also isolated from Styrax japonicum, Styrax officinalis L., and Styrax obassia.107–109 They were found exhibiting cytostatic activity towards human leukemic HL-60 cells.110 The brief total synthesis of all three naturally occurring 31a, 31b, and 31c was achieved only in five steps in overall yields of 40, 40, and 34%, respectively. The bromobenzofuran 28 present in these natural products were all also provided in two steps via selective cross MacMurry coupling in good yields. The introduction of 3-hydroxypropy moiety on the benzofuran rings was accomplished via Sonogashira cross coupling reaction with subsequent hydrogenation followed by hydrolysis. The total synthesis started from readily available 23 and 26 which are coupled by means of selective MacMurry cross coupling reaction110 to afford intermediate 27, which followed by oxidative cyclization to form compound 28. The bromobenzofuran was coupled with propargyl acetate by a palladium-catalyzed Sonogashira reaction to generate 30 as a key intermediate to produce ailanthoidol 5, XH-14 6 and the other three natural products 31a–c (Scheme 6).111
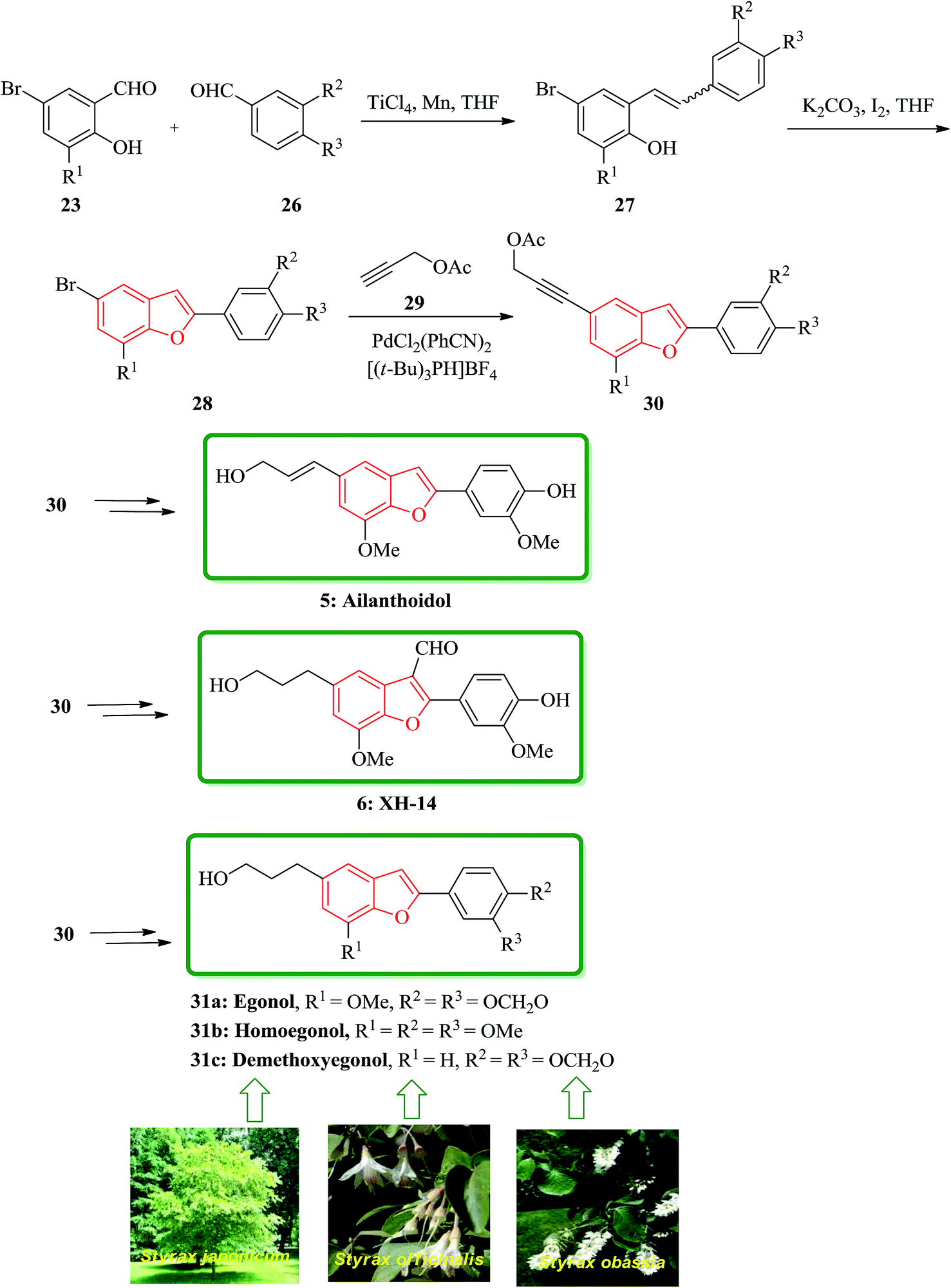 | ||
| Scheme 6 Total synthesis of ailanthoidol 5, XH-14 6 egonol 31a, homoegonol 31b, and demethoxyegonol 31c. | ||
Yang and co-workers have carried out the total synthesis, which could also substantiate unambiguously the structure of XH-14 6. A key feature of this synthetic program was the conventional coupling reaction112 between the copper acetylide 32 (ref. 113) and the aryl bromide 33,114 generating as anticipated the benzofuran 34 with the desired skeleton. Finally, after several steps, hydrolysis of 35 provided the target molecule 6, which was identical in all aspects to the natural XH-14 6 (Scheme 7).115
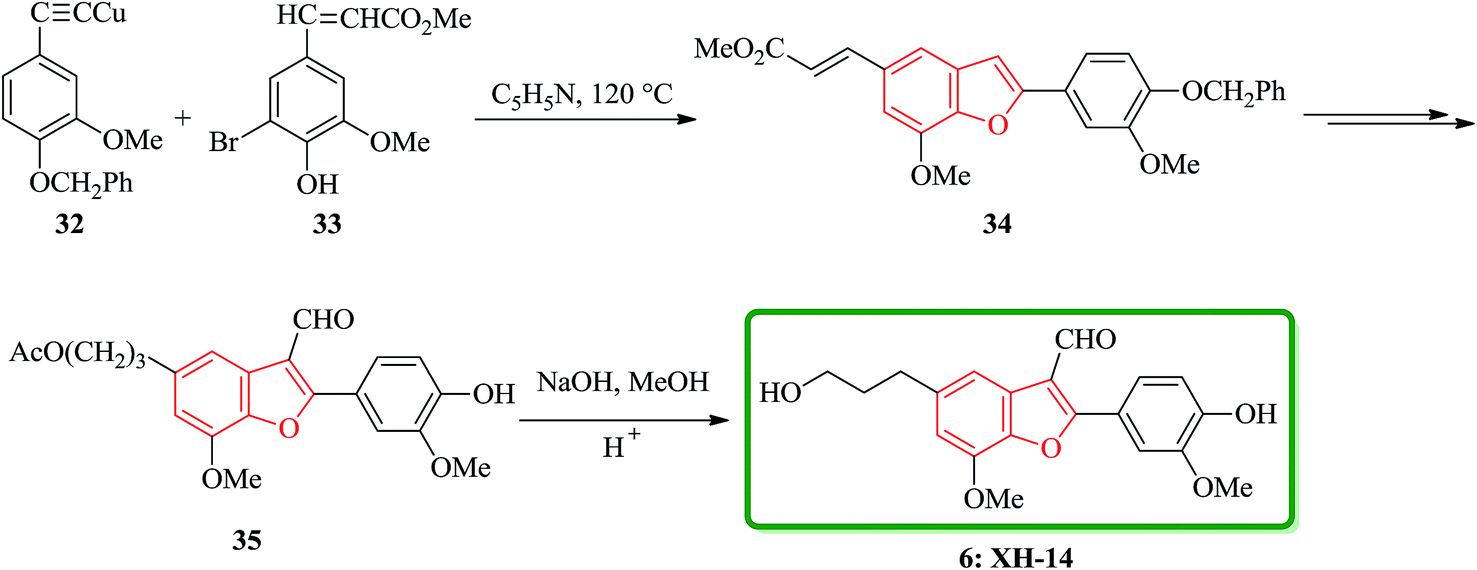 | ||
| Scheme 7 Total synthesis of XH-14 6. | ||
A brief, high yielding practical and highly efficient total synthesis of natural product XH-14 containing benzofuran moiety is achieved in nine steps by Jun and co-workers. In this approach, the key features are Sonogashira coupling, iodine-promoted cyclization, Wittig reaction, and formylation. The total synthesis started from another natural product vanillin 19, which was transformed into diarylyne 36 in three steps. Then, it was subjected into iodine-induced cyclization to afford 3-iodobenzofuranaldehyde 37 containing the benzofuran core. The latter in turn is converted into 2-(4-benzyloxy-3-methoxyphenyl)-3-iodo-5-(3-benzyloxypropyl)-7-methoxybenzofuran 38 in several steps. Upon formylation using n-BuLi/N-formylpiperidine the latter is transformed into 2-(4-benzyloxy-3-methoxyphenyl)-5-(3-benzyloxypropyl)-7-methoxybenzofuran-3-carbaldehyde 39 in 70% yield. Nevertheless, when BCl3 is used for debenzylation of 39 the desired natural product XH-14 6 is obtained in very high yield (90%) (Scheme 8).116
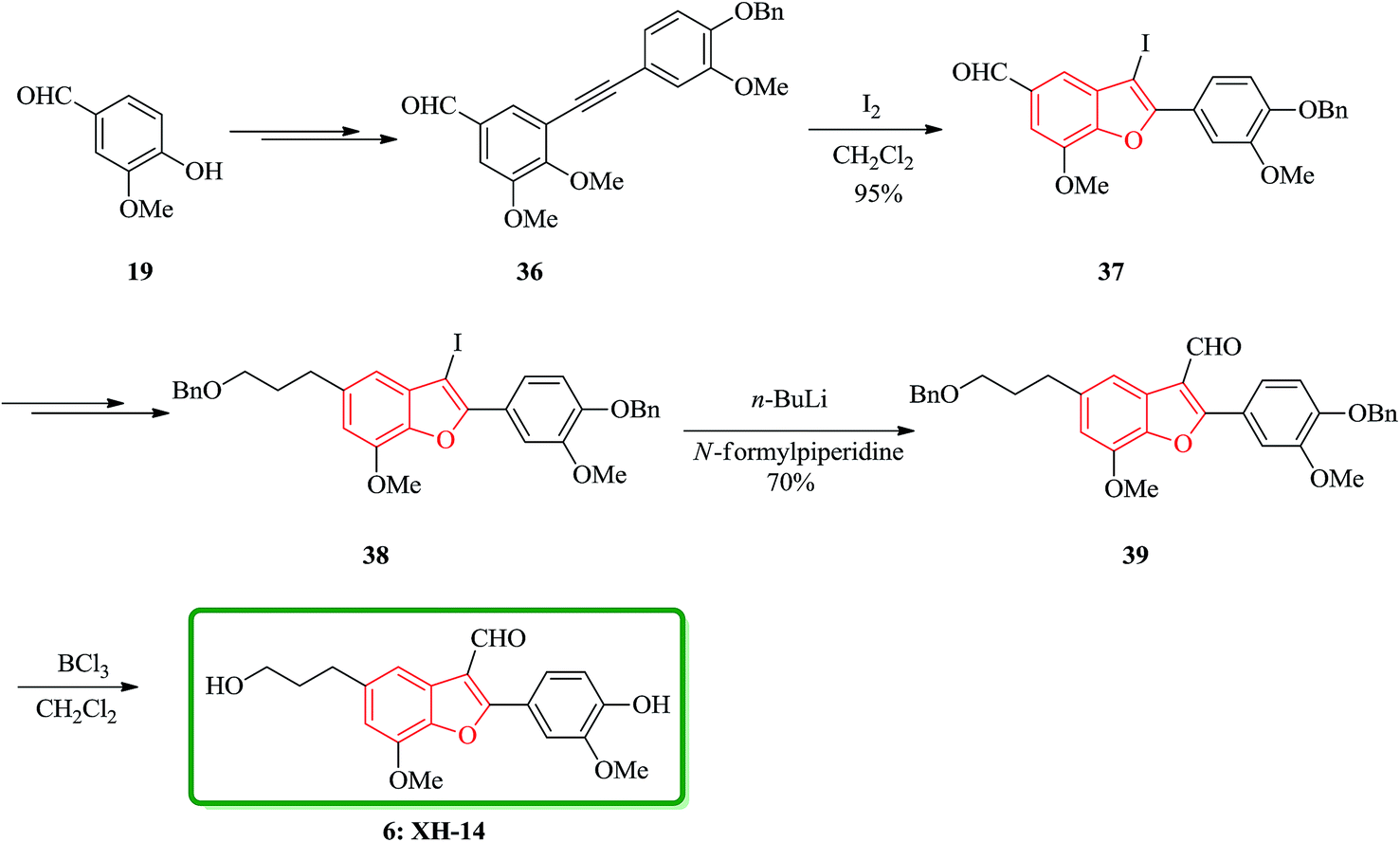 | ||
| Scheme 8 Total synthesis of XH-14 6. | ||
Vibsanol 42, a benzofuran-type lignan isolated from the wood of Viburnum awabuki (Caprifoliaceae), was synthesized by the tandem cyclization of o-tert-butyldimethylsiloxy diaryl alkyne with tetrabutylammonium fluoride and excess paraformaldehyde as the key step. The leaves of Viburnum awabuki (Caprifoliaceae) are known to have been used as a fish poison for the purpose of catching fish around the Okinawa Islands. Vibsanine A, an unprecedented humulene-type diterpene, was isolated from these leaves as a piscicidal compound.117 Recently, vibsanol 42, a natural occurring benzofuran-type lignan showed moderate inhibitory activity toward lipid peroxidation in rat brain homogenates.118 The structure of vibsanol 42 was mainly established on the basis of spectroscopic methods and composed of 2-aryl and 3-hydroxymethyl substituents. It is well known that 2-substituted benzofurans are readily prepared from the o-hydroxyarylalkynes under basic conditions.119 Total synthesis of vibsanol 42 was started from vanillin that after several steps provided the benzofuran precursor 40. The tandem cyclization of 40 gave the desired benzofuran 41 in 67% yields. Finally, the deprotection of 41 smoothly occurred using a catalytic amount of PPTS in MeOH to give vibsanol 42 in 99% yield (Scheme 9).120
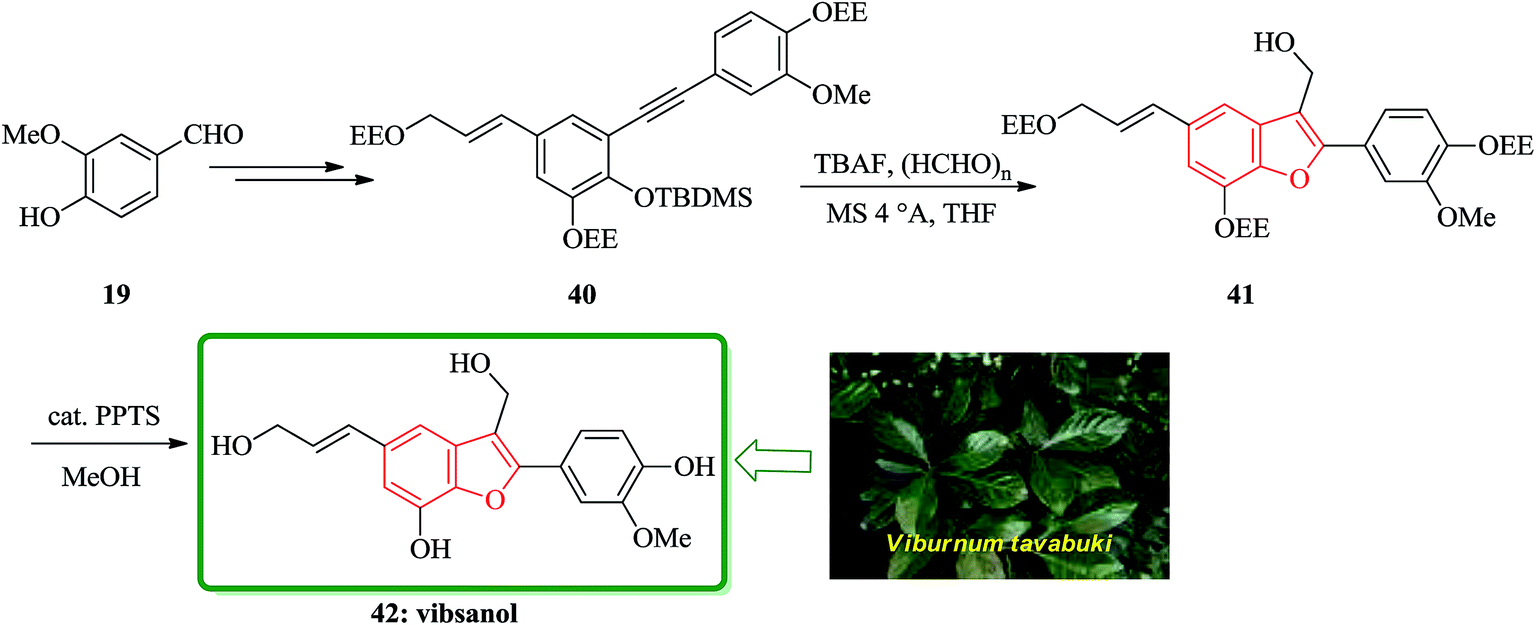 | ||
| Scheme 9 Total synthesis of vibsanol 42. | ||
The first total synthesis of a norneolignan isolated from Ratanhia, 5-(3-hydroxypropyl)-2-(2′-methoxy-4′-hydroxyphenyl)benzofuran 46, is described in 2002. The key steps contain the one-pot reaction for a 2-arylbenzofuran from methyl 3-(4-hydroxyphenyl)propionate 43 with 2-chloro-2-methylthio-(2′-methoxy-4′-acetoxy)acetophenone 44 in the presence of ZnCI2, and reductive desulfurization of the resulting product 45. Significantly, the total synthesis of a norneolignan 46 was accomplished by a one-pot reaction of methyl 3-(4-hydroxyphenyl)propionate and chloride 44 under Friedel–Crafts reaction conditions and reductive desulfurization of the resultant benzofuran 45, as the key steps (Scheme 10).121
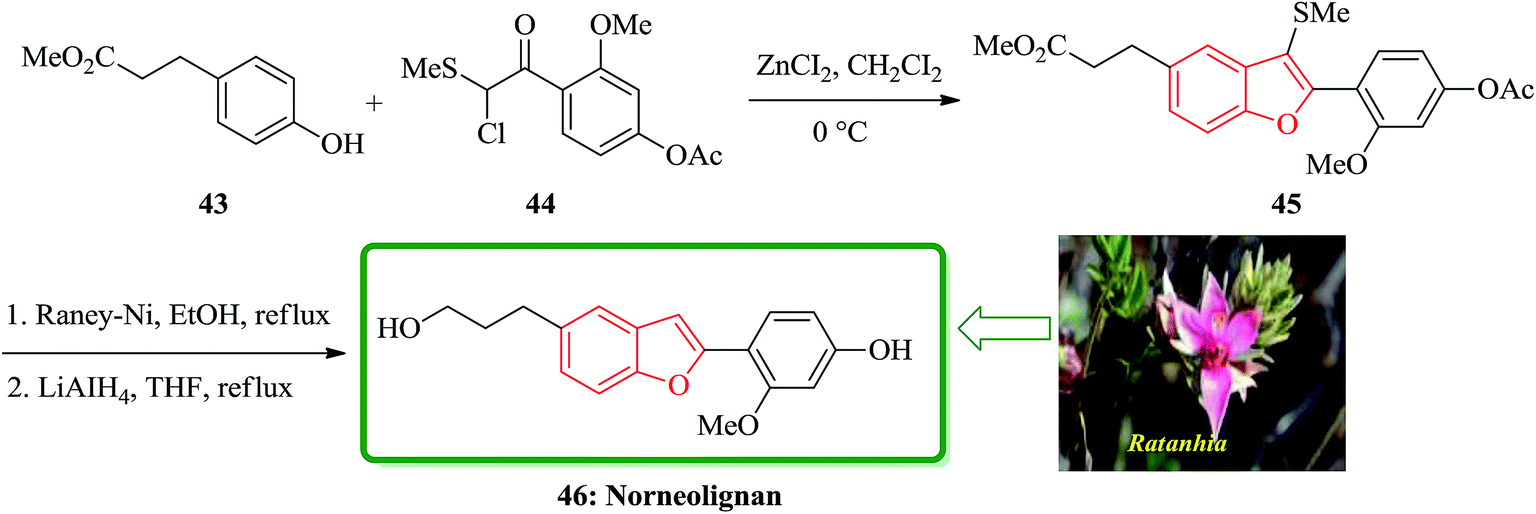 | ||
| Scheme 10 Total synthesis of a norneolignan 46. | ||
Among the natural products bearing benzofuran as scaffold, the eupomatenoids form an expanded class of neolignans,122 are worthy being considered. These compounds initially were isolated from two plant species, which were placed in the archaic angiosperm family eupomatiaceae. Structurally, the eupomatenoids 50 are identified by a 2,3,5-substitution pattern. In this pattern an aryl group is placed as a substituent at the 2-position, a methyl group positioned at 3 and a C3-substituent R stands at position 5. Different eupomatenoids 50a–c, 50f–h were synthesized starting from 2,3,5-tribromobenzofuran 48 via a short and high-yielding synthetic strategy. The total synthesis commenced from tribromobenzofuran 48 and commercially purchasable bromide 47a. The overall yields diverge between 29 and 60% over four to six steps. Remarkably the important and key step of this strategy is to achieve the high regioselectivity from three Pd(0)- and Ni(0)-catalyzed cross-coupling reactions which are performed, sequentially. The order of substitution at the benzofuran nucleus is C-2, C-5 and C-3. In this way, the introduction of the third substituent onto the benzofuran nucleus was possible. Upon double bond equilibration via treatment with iodine,123 (E)-configured eupomatenoid-15 was obtained 50a in 46% overall yield. In a similar manner, eupomatenoids-3 50b and -4 50c were synthesized from the respective aryl bromides 47b124 and 47c. In this way a concise and effective synthesis of 2,3,5-trisubstituted benzofurans via three successive cross-coupling reactions were accomplished. The applicability of this strategy was successfully attempted for the synthesis of a variety of naturally occurring compounds continuing benzofuran moiety such as eupomatenoids but it is also anticipated to be also functional for the synthesis of some other benzofurans (Scheme 11).125
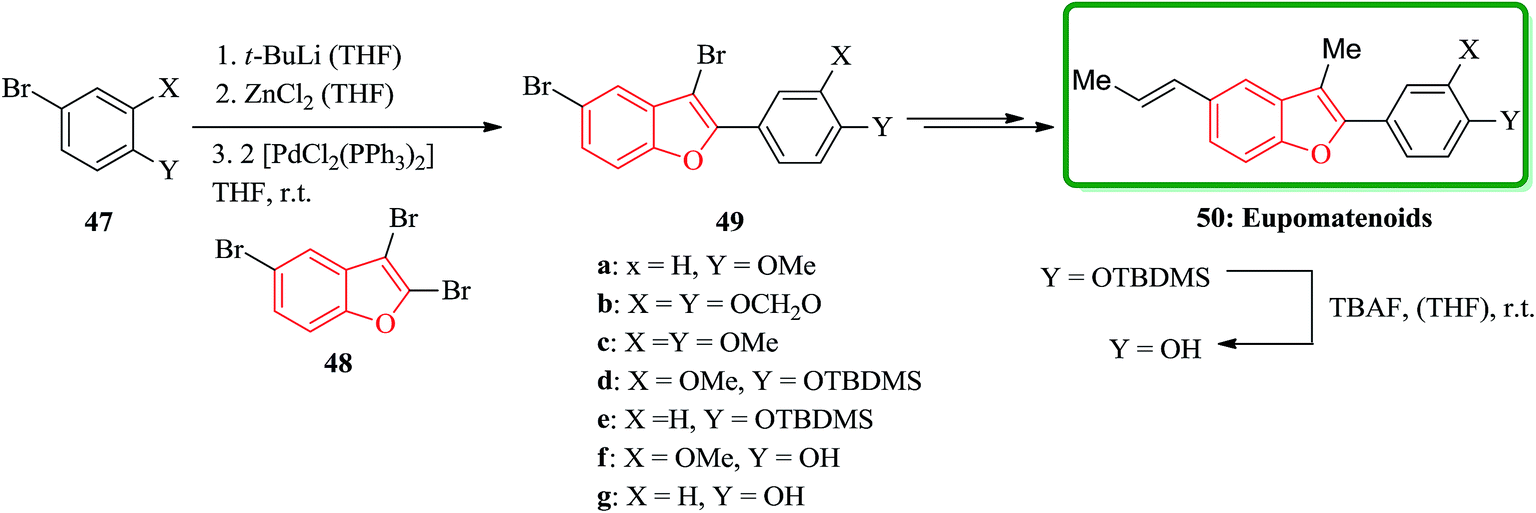 | ||
| Scheme 11 Total synthesis of eupomatenoids 50. | ||
Eupomatenoids, neolignans isolated from Eupomatia laurina and Eupomatia bennettii126 represent naturally occurring 2,3,5-trisubstituted benzofurans 50a and 50b which are interesting targets for total synthesis. Initially, the required precursor 51 was synthesized by a direct bromination of benzofuran in the presence of a base (e.g. KOAc).127 Compound 52 is the product of regioselective cross-coupling reaction between 2,3,5-tribromobenzofuran 48 and the corresponding arylzinc, under optimized conditions. Compound 52 was converted via selective bromine–lithium exchange/methylation to the 2,3-disubstituted 5-bromobenzofurans 53. Ni-catalyzed reaction of compound 53 with alyl magnesium bromide 54 led to the synthesis of desired natural product, eupomatenoids 50 in overall yields of up to 60% (Scheme 12).128
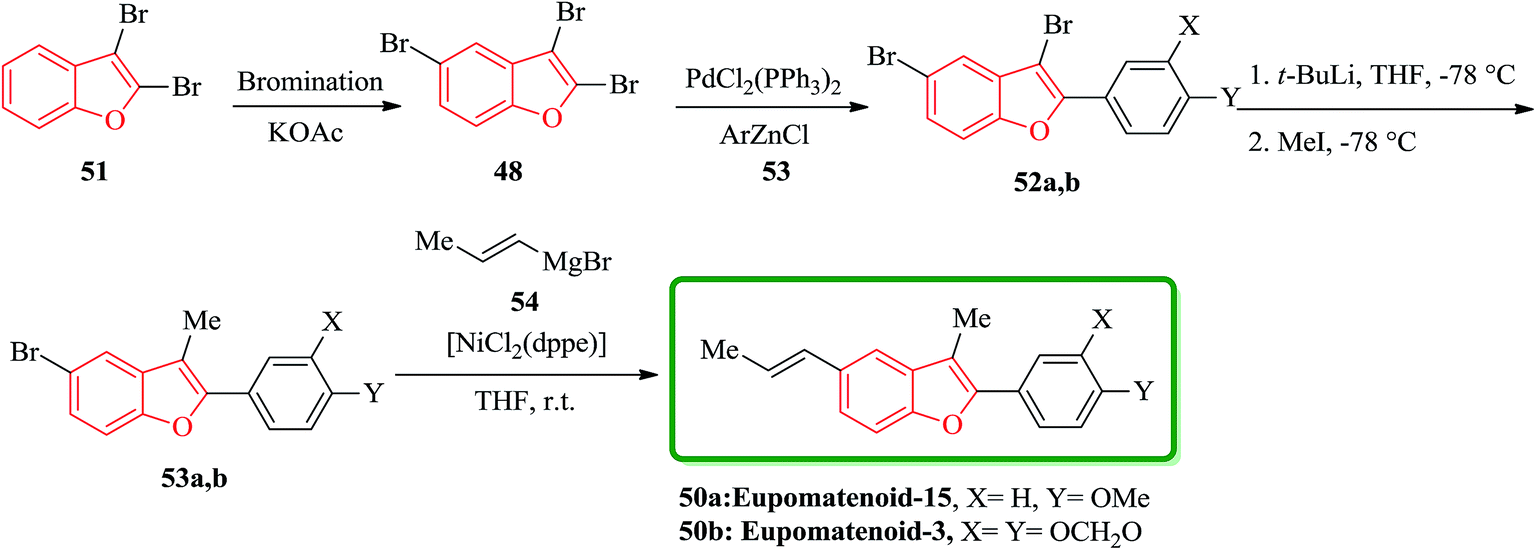 | ||
| Scheme 12 Total synthesis of eupomatenoids 50. | ||
Egonol 31a is a natural benzofuran glycoside occurring widely in Styrax officinalis.129 Primarily, nor-neolignan egonol was isolated by Okada from the seed-oil of Styrax japonicum.130 It has attracted enormous attention due to its versatile biological activities.131 The synthesis of nor-neolignan egonol 59 has been achieved in five steps starting from easily accessible staring materials.132 The total synthesis of nor-neolignan natural product egonol has been anticipated. The benzofuran derivative 59 is actually a known egonol precursor,132 that is itself a natural product. Noticeably, compound 59 was initially isolated from the wood of Anaxagorea clavata.133
The suggested synthetic pathway has three main problems: (a) highly conjugated enol ether derivative 55 is not stable, (b) the Dötz reaction134,135 is competitive due to the formation of naphthol 58 (Scheme 13) with the desired benzofuran-formation135 and finally (c) the enol ether can be subjected to cyclization at the ketene carbon present in intermediate 57 leading to the formation of compound 60. This phenomenon has previously been observed in related enamine intermediates.136 Nevertheless, in some related systems the completion of Dötz reaction has not been reported. Thus, the selective cyclization is considered being done due to the strong complexation followed by annulation at the non-oxygenated vinylketene ligand. For the total synthesis of 59, both dienyne 55 and an enediyne 56 can be used as starting materials.137,138
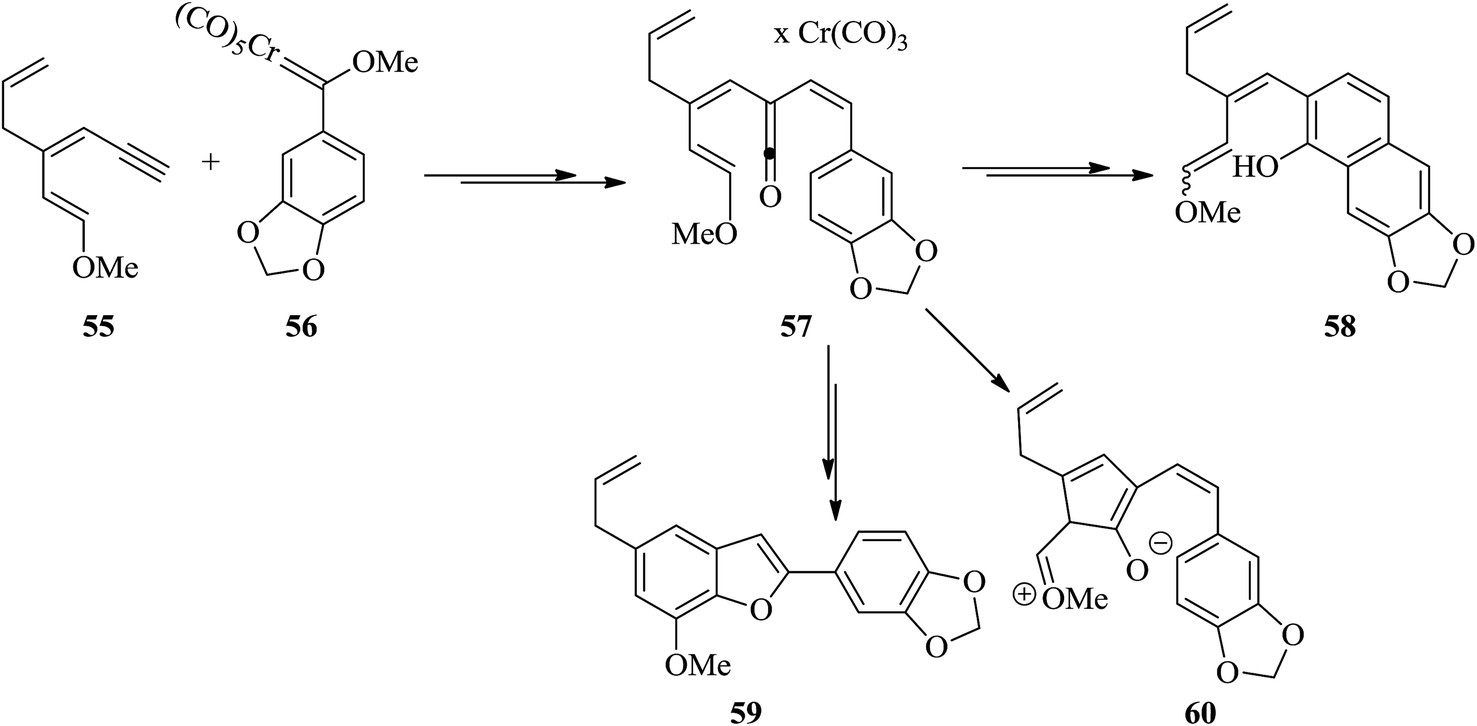 | ||
| Scheme 13 Proposed pathway for the synthesis of compound 59. | ||
The use of silylated methoxydienyne 61 produces egonol precursor 59 in 47% yields along with compound 63 in 15% yields, which is considered as the result of carbene oxidation. After several steps, intermediate 59 is converted into the desired natural product egonol 31a. Noticeably, higher temperatures or/and longer reaction times resulted in the formation of the conjugated alkene moiety. Worthy to mention that the hydroboration of 63 has been reported to give egonol 31a (Scheme 14).132,138
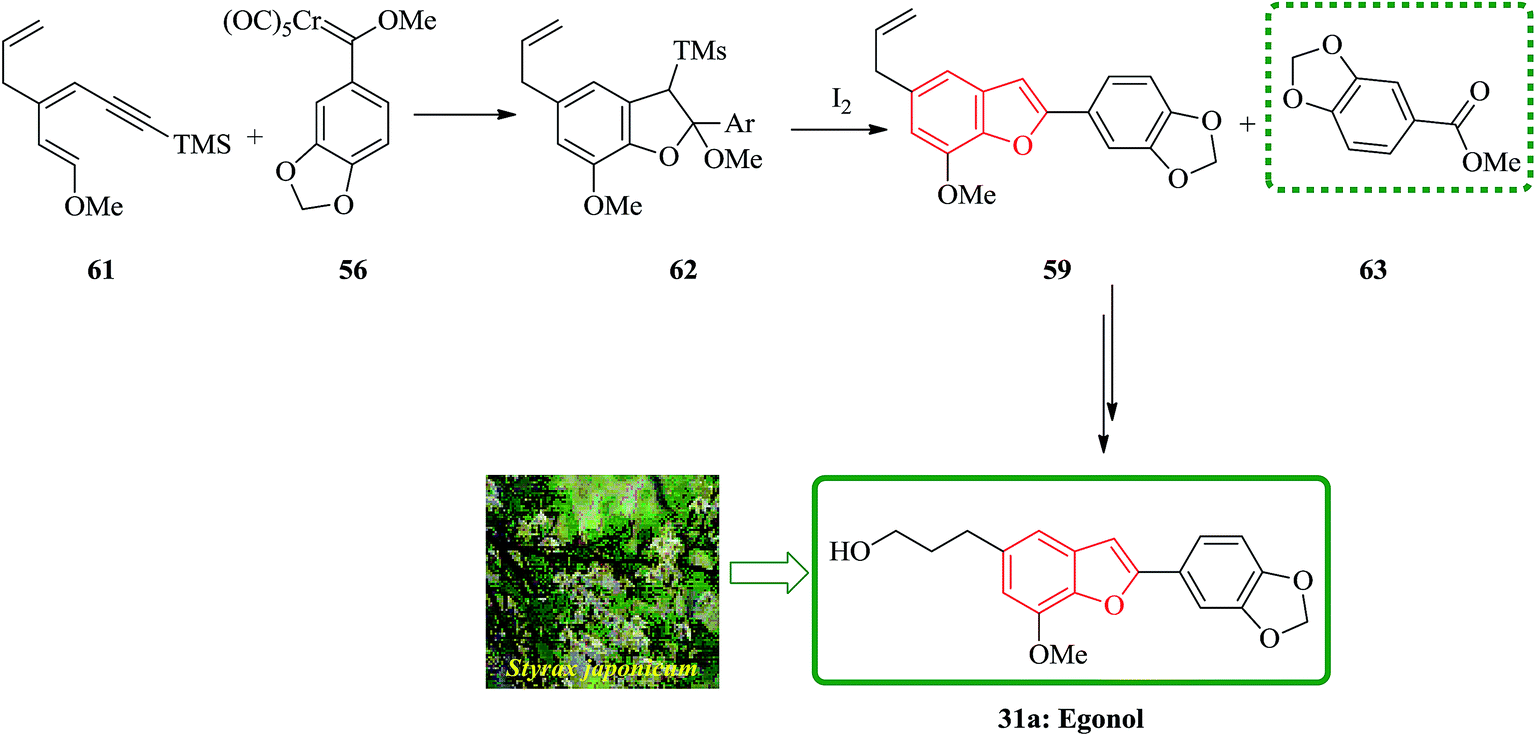 | ||
| Scheme 14 Total synthesis of egonol 31a. | ||
Salvia miltiorrhiza bunge (dan-shen) was extensively utilized as a Chinese customary medicine for the cure of atherosclerosis.139 Hydrosoluble salvianolic acids, which have initially been isolated from water-soluble part of dan-shen are found being the showed several biological activities. They showed anti-tumor, antithrombotic, anti-oxidative, anticoagulant and anti-HIV activities.140 Salvianolic acid C 70, is actually one of the salvianolic acids, which are present in the structure of 2-phenyl-benzofuran neolignan tournefolic acid A 68.141 The total synthesis of the naturally occurring compounds salvianolic acid C 70, tournefolal 69 and tournefolic acid A 68 have been achieved and reported in 2012. Noticeably, the key benzofuran framework were synthesized via selective iodination to obtain 64 followed by Sonogashira coupling142 in which 3-hydroxy-2-iodobenzaldehyde 64 was coupled to the ethynylbenzene analogues 65 in a catalyzed-Pd(Ph3P)2Cl2 and co-catalyzed-CuI reaction to give benzofuran aldehyde 66 in satisfactory yield. The latter can be converted to (E)-3-(7-(benzyloxy)-2-(3,4-bis(benzyloxy)phenyl)benzo[b]furan-4-yl) acrylic acid 67 by Knoevenagel condensation, and then the benzofuran aldehyde 66, which can be transformed into the desired natural product 70 in several steps in overall yields of 40%. On the other hand, upon the debenzylation of 67 and 66, are converted into tournefolic acid A 68 and tournefolal 69 respectively (Scheme 15).143
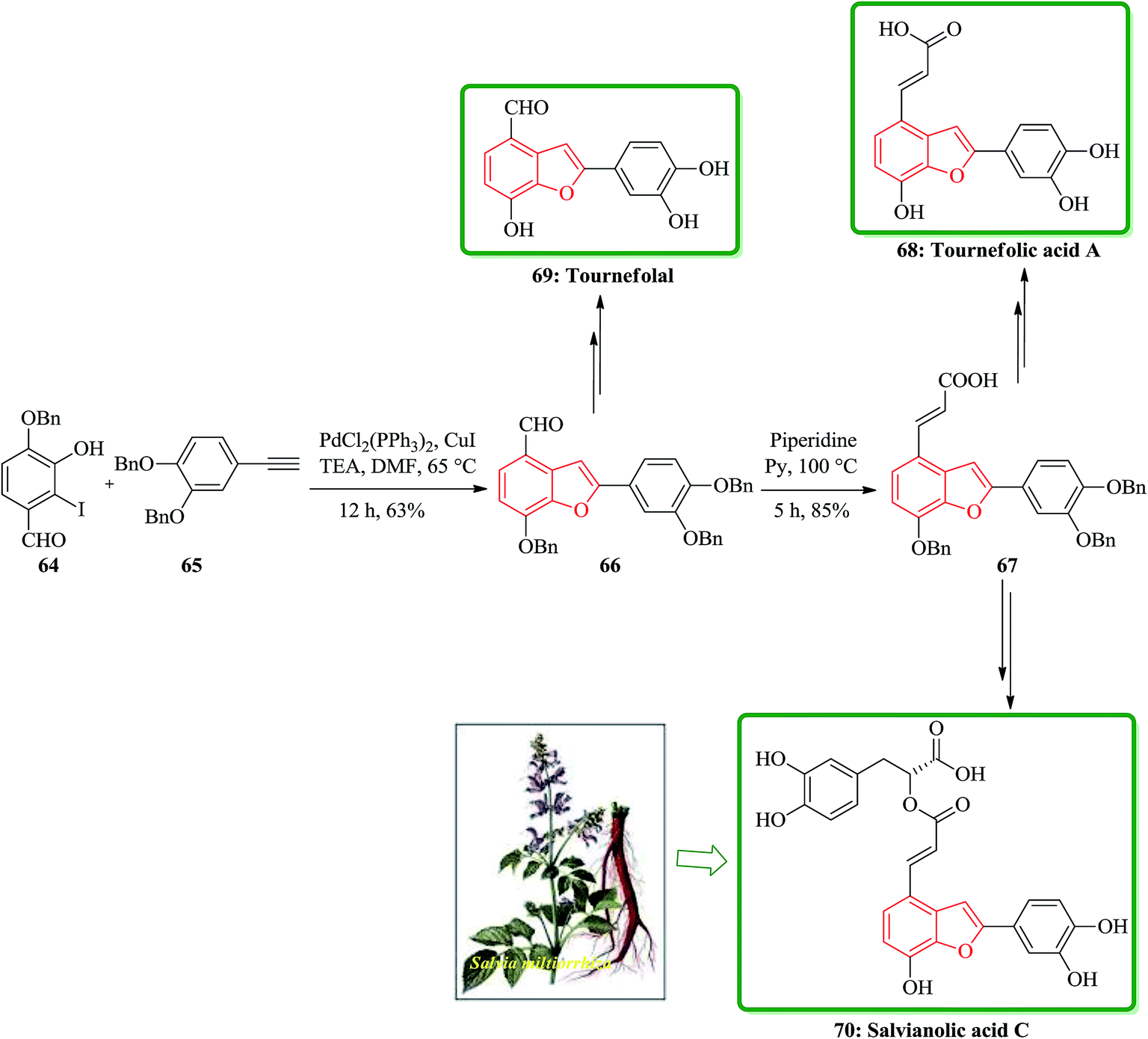 | ||
| Scheme 15 Total synthesis of tournefolic acid A 68, tournefolal 69 and salvianolic acid C 70. | ||
A novel and efficient synthetic approach for the synthesis of biologically potent natural benzofurans is reported in 2007 by Naito and co-workers.144 The important step of this protocol is the well-known [3,3]-sigmatropic rearrangement. TFAA has been established as the best reagent to promote [3,3]-sigmatropic rearrangement for the preparation of cyclic or acyclic dihydrobenzofurans. Alternatively, the TFAT-DMAP system was proved as the most efficient system for the synthesis of different benzofurans. This method is particularly practical since the protection of the phenolic hydroxy groups in the synthesis of hydroxylated 2-arylbenzofurans is non-required.
In accordance with Naito and co-workers protocol144 the synthesis of naturally occurring compounds containing benzofuran moiety such as stemofuran A 75 (ref. 145) eupomatenoid 6 50g,146 and coumestan 83 were achieved.147 These compounds showed various biological activities. For the synthesis of compounds 75, 50g and 83 with no hydroxy group, this synthetic approach was especially remarkable since they can be accomplished without any protection of the phenolic hydroxy groups (Scheme 16). Initially, the synthesis of stemofuran A 75, which had been isolated from Stemona collinsae,145 was attempted. The synthesis of stemofuran A was achieved through condensation of ketones with aryloxyamine followed by reaction with TFAT-DMAP in sequential reactions involving four steps giving the desired products in 72% yield. This reported synthesis of stemofuran A by Pasturel and co-workers148 involved several steps including the required protection/deprotection of the hydroxy group. In the new synthetic route, O-phenylhydroxylamine 72, easily synthesized from phenylboronic acid 71, which was subsequently condensed with dihydroxyacetophenone to furnish the oxime ether 74 in good yield. The oxime ether 74 upon treatment with TFAT mediated by DMAP at ambient temperature gave the desired benzofuran 75 in excellent yield. It was found being identical with stemofuran A 75 by comparison of their spectroscopic and physical data with those of the natural product reported in the literature, previously.145
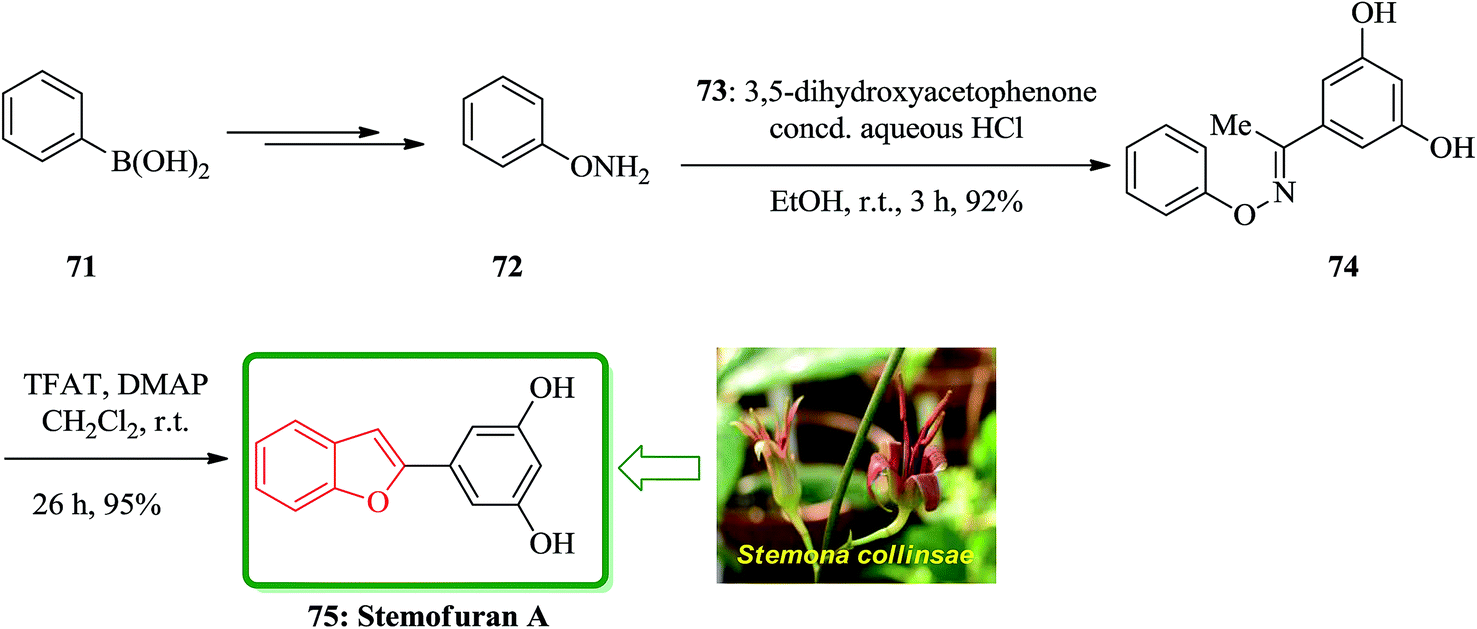 | ||
| Scheme 16 Total synthesis of stemofuran A 75. | ||
In a similar way, eupomatenoid 6 50g were also synthesized via the treatment of oxime ether with TFAT-DMAP. Condensation of O-phenylhydroxylamine 77 bearing the p-bromo group with p-hydroxypropiophenone afforded the oxime ether 79, which upon reaction with TFAT-DMAP in dicloromethane at room temperature gave the 5-bromobenzofuran 80 in 95% yields. Finally, the latter underwent Suzuki coupling reaction with (E)-propenyl boronic acid to furnish eupomatenoid 6 50g in excellent yield. Thus, the total synthesis of eupomatenoid 6 50g in 52% overall yield from (4-bromophenyl)boronic acid 76 in five steps was accomplished and found to be identical with natural eupomatenoid 6 by comparison of its spectroscopic data reported in the literature for the naturally occurring compound (Scheme 17).146
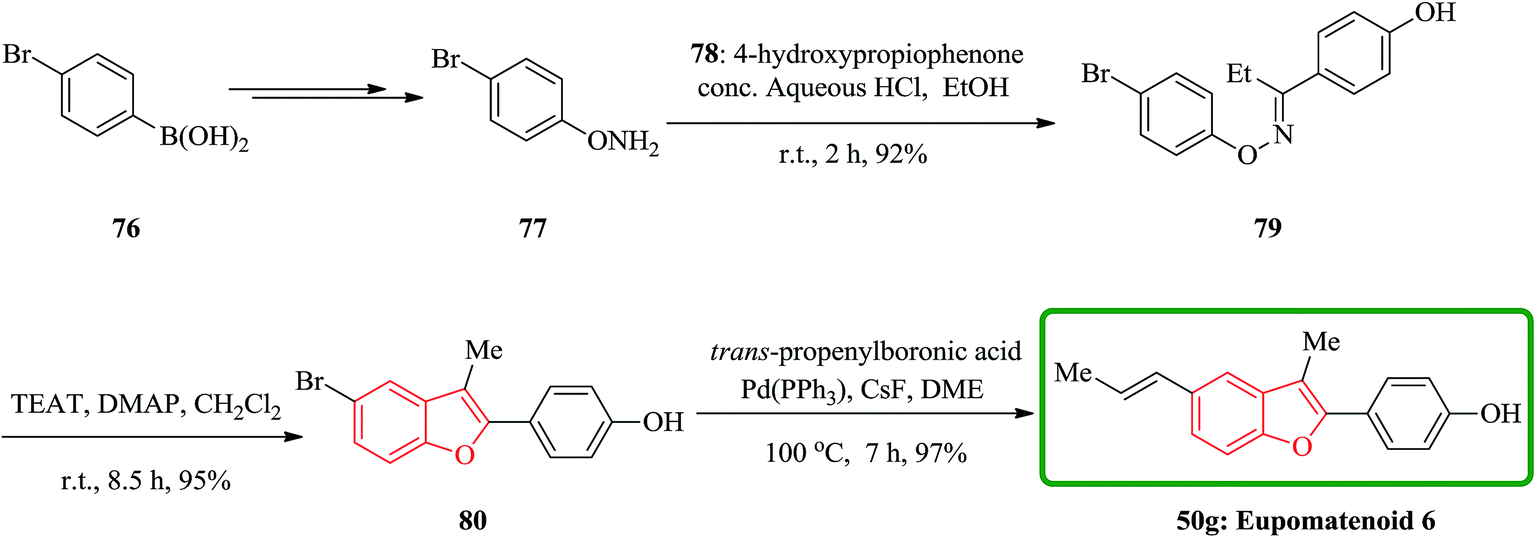 | ||
| Scheme 17 Total synthesis of eupomatenoid 6 50g. | ||
The third desired target was coumestan 83.147 That is a basic pharmacophore having coumestanes such as coumestrol,149 which exhibits estrogenic potency. Due to its unique and remarkable structure, coumestan 83 has attracted the attention of several organic chemists who were attempting independently different approaches.150,151 One of successful reported synthetic strategies involved the synthesis of the benzofuran moiety in the second step. Initial condensation of readily available O-phenylhydroxylamine 72 with 4-chromanone via sequential acylation/rearrangement of the resulting oxime ether 81 gave the desired tricyclic benzofuran 82 in 73% yield in only two steps. Finally, the carbonyl group was introduced upon the treatment of tricyclic benzofuran 82 with PCC to furnish coumestan 83 in good yield (Scheme 18).143
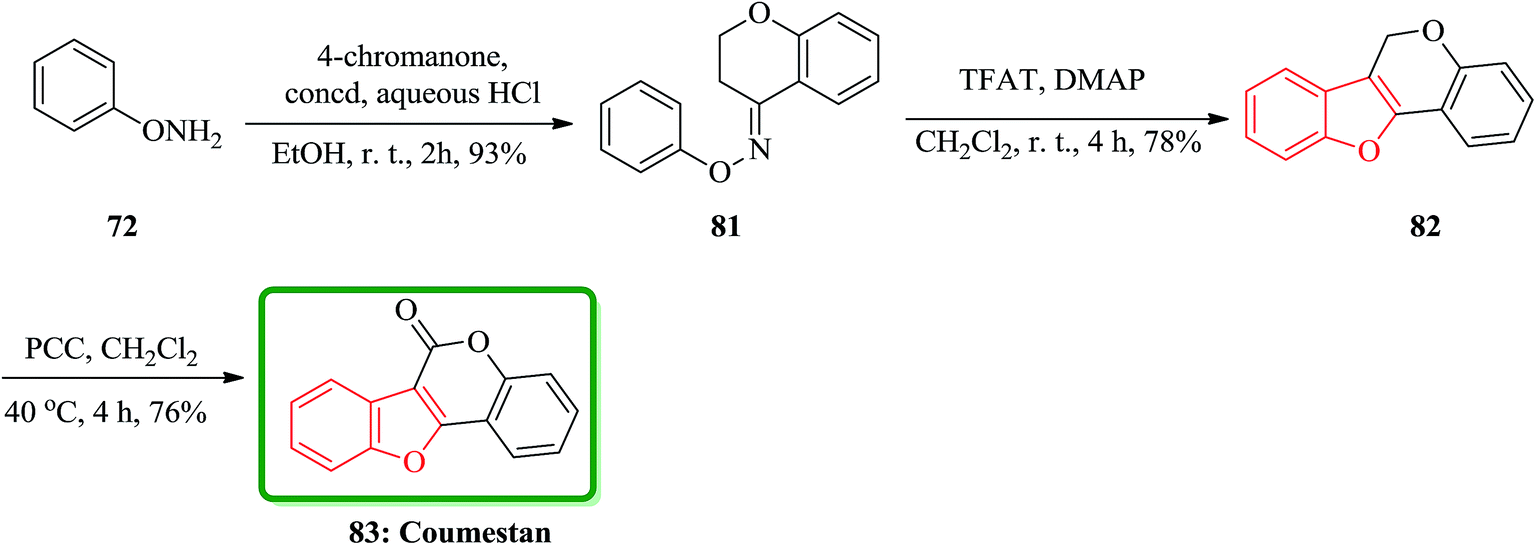 | ||
| Scheme 18 Total synthesis of coumestan 83. | ||
(−)-Machaeriols A, B, C, and D bearing the cannabinoid structure were recently isolated from the bark of the Machaerium multiflorum spruce located in Loreto and Peru.152 They have been reported to have potential in vitro antimicrobial activity against Staphylococcus aureus and methicillin-resistant S. aureus.152 They showed potent in vitro antimalarial activity against Plasmodium falciparum D6 and W2 clones.152 These important biological activities have led to the development of a variety of synthetic approaches to these natural products. An efficient and concise synthesis of the biologically interesting (+)-machaeriol B 89 and its enantiomer 90 was accomplished from O-phenylhydroxylamine 72 in four steps. The key strategies in the synthesis of 89 and 90 involved benzofuran formation through a [3,3]-sigmatropic rearrangement and trans-hexahydrodibenzopyran formation by a domino aldol-type/hetero-Diels–Alder reaction.
Scheme 19 shows a concise synthetic approach to natural (−)-machaeriol B 89 and its unnatural enantiomer 90. The precursor 87 for the total synthesis of 89 and 90 was obtained by a known method.153 Thus, the condensation of O-phenylhydroxylamine 73 with 3,5-bis(dibenzoyloxy)acetophenone 84 in the presence of conc. HCl in EtOH gave the oxime ether 85 in 93% yield. The latter was treated with trifluoroacetyl triflate and N,N-dimethylpyridin-4-amine (DMAP) in CH2Cl2 at room temperature to afford the desired cycloadduct 86 in 95% yield as the sole product via a [3,3]-sigmatropic rearrangement which is the well-known oxa-variant of Fischer's indole synthesis. Removal of the two benzoyl groups from 5-(benzofuran-2-yl)benzene-1,3-diol 1,3-dibenzoate 86 with LiAlH4 in ether at room temperature afforded stemofuran 87 in 90% yield.145 Treatment of benzofuranylbenzenediol 87 with (−)-(S)-citronellal 88a in the presence of EDDA/Et3N in refluxing xylene gave (−)-machaeriol B 89 in 65% yield. The spectroscopic data of the synthetic 89 are in good agreement with the reported data. Conversely, the corresponding treatment of 87 with (−)-(R)-citronellal 88b gave (−)-machaeriol B 90 in 63% yield.154
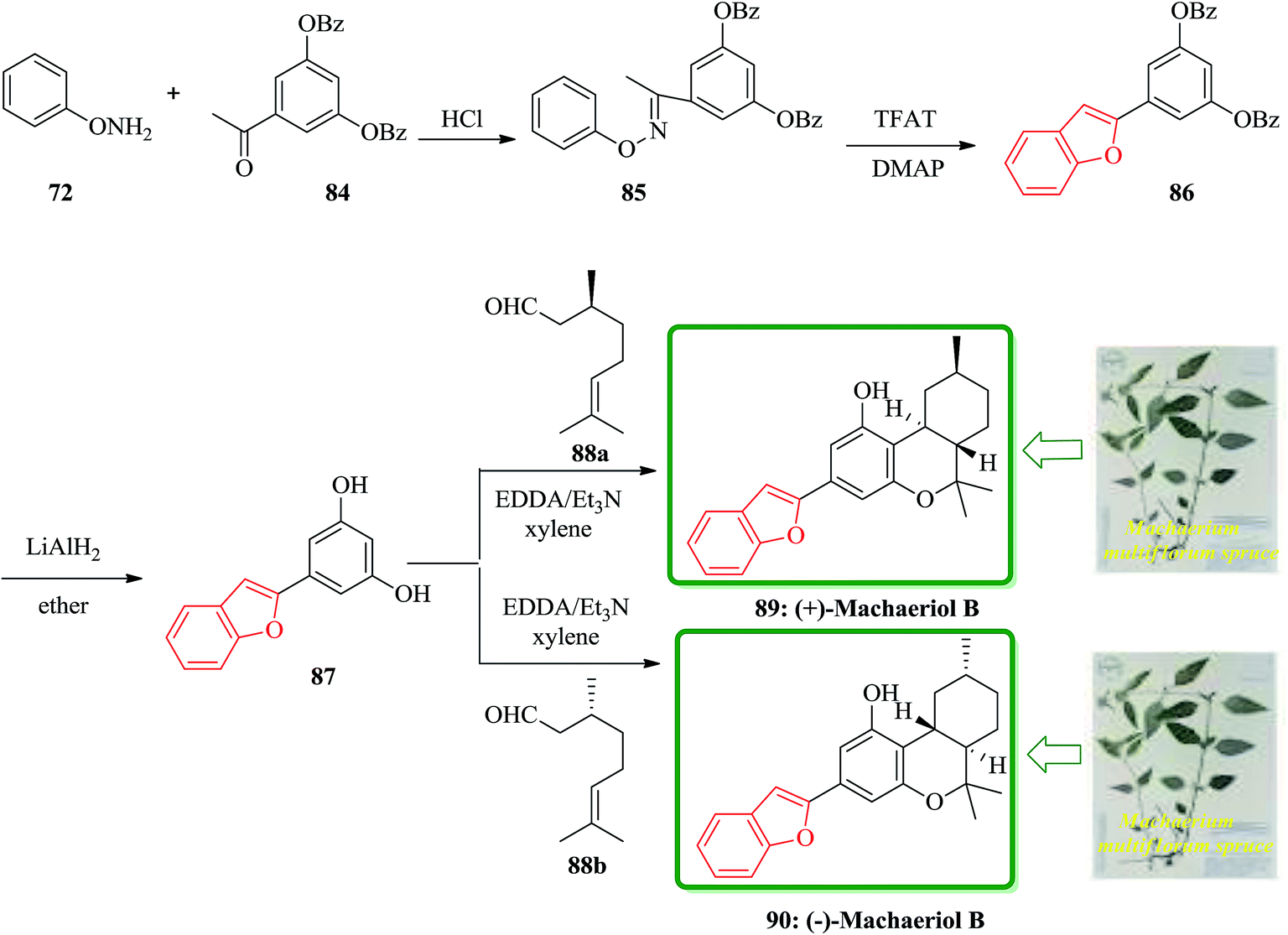 | ||
| Scheme 19 Total synthesis of (+)-machaeriol B 89 and its enantiomer 90. | ||
Also, stemofuran A 75 exhibited a wide range of biological potencies.155,156 A highly effective and facile strategy for the construction of 2-arylbenzo[b]furans has been reported by Ruan and co-workers in 2014.157 As depicted in Scheme 20, stemofuran A 75 was synthesized by a method starting from 2-methoxychalcone epoxide 91 which upon treatment with BF3·Et2O (2 mol%) with subsequent deformylation gave the intermediate 93 in 76% overall yield. Compound 93 underwent demethylation and cyclodehydration reactions in the presence of 48% HBr in acetic acid to give stemofuran A 75 in excellent yield (94%).157
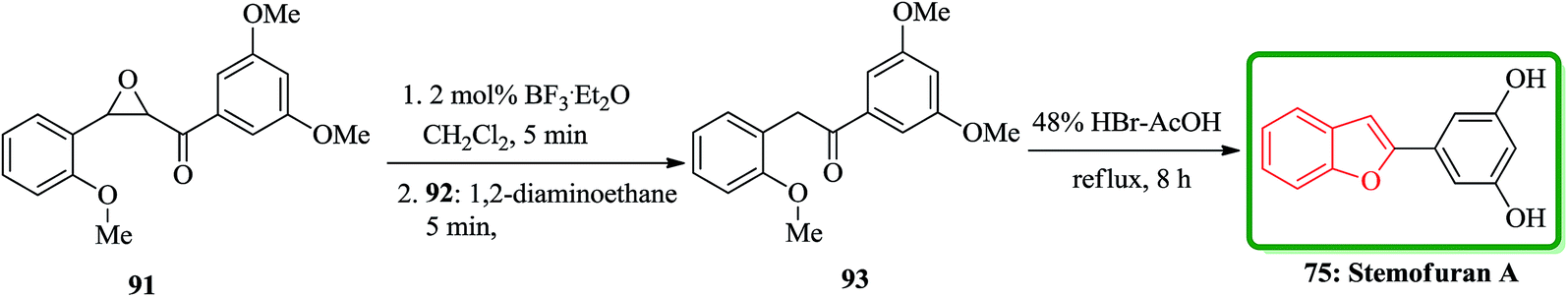 | ||
| Scheme 20 Total synthesis of stemofuran A 75. | ||
The total synthesis of the naturally occurring demethoxy-egonol 31c [5-(3-hydroxypropyl)-2-(3′,4′-methylenedioxyphenyl)benzofuran], a congener of which is used in the treatment of asthma and rheumatism. The key steps involve the construction of a 2-arylbenzofuran skeleton 99 from methyl 3-(4-hydroxyphenyl)propionate with 2-chloro-2-methylthio-(3′,4′-methylenedioxy)acetophenone 97 in the presence of ZnCl2 and successive desulfurization of the resulting product 99 (Scheme 21).158
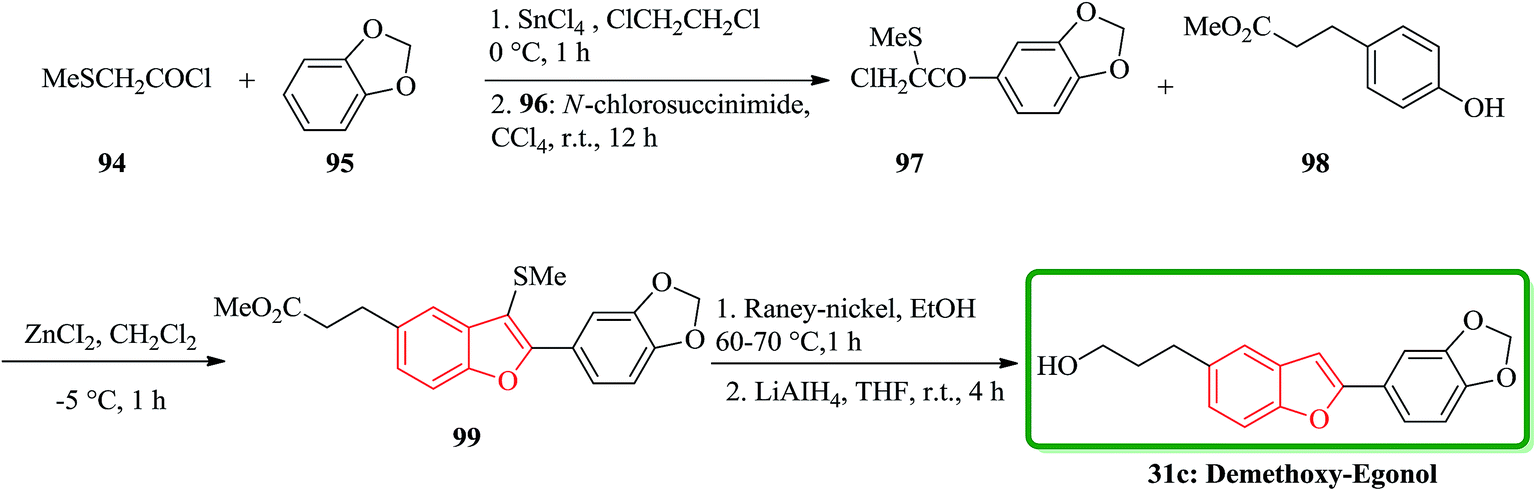 | ||
| Scheme 21 Total synthesis of demethoxy-egonol 31c. | ||
Benzo[b]furan natural product 31a was initially isolated from the Styracaceae family such as Styrax japonicum,159 S. formosanus,160 S. obassia,108 S. macranthus161 and S. officinalis,107 which showed a variety of biological activities including insecticidal, fungicidal, antimicrobial, antiproliferative, cytotoxic and antioxidant properties.131 Egonol, 5-(3-hydroxypropyl)-7-methoxy-2-(3,4-methylenedioxyphenyl) benzofuran was first isolated in 1915 from the seed oil of Styrax japonicum159 and first total synthesized by Kawai162 condensing an o-hydroxybenzaldehyde with an α-chlorophenylacetic acid, which known to be an effective pyrethrum synergist.163 It was reported the most effective total synthesis of egonol 31a in 5 steps with 74% overall yield from vanillin by using Sonogashira coupling reaction. Vanillin 19 reacted with I2/Ag2SO4 in EtOH at room temperature to give iodovanillin 100 in 80% yields. Sonogashira coupling of 100 with 3,4-methylenedioxyphenylacetylene 101 which was easily prepared from piperonal via Colvin rearrangement,164 by using Pd(PPh3)4/CuI/Et3N in DMF yielded benzofuran 102 in 95% yield through successive coupling and cyclization in one-step. Noticeably the latter was very sensitive to the haloaryl substituents as shown in Scheme 22.
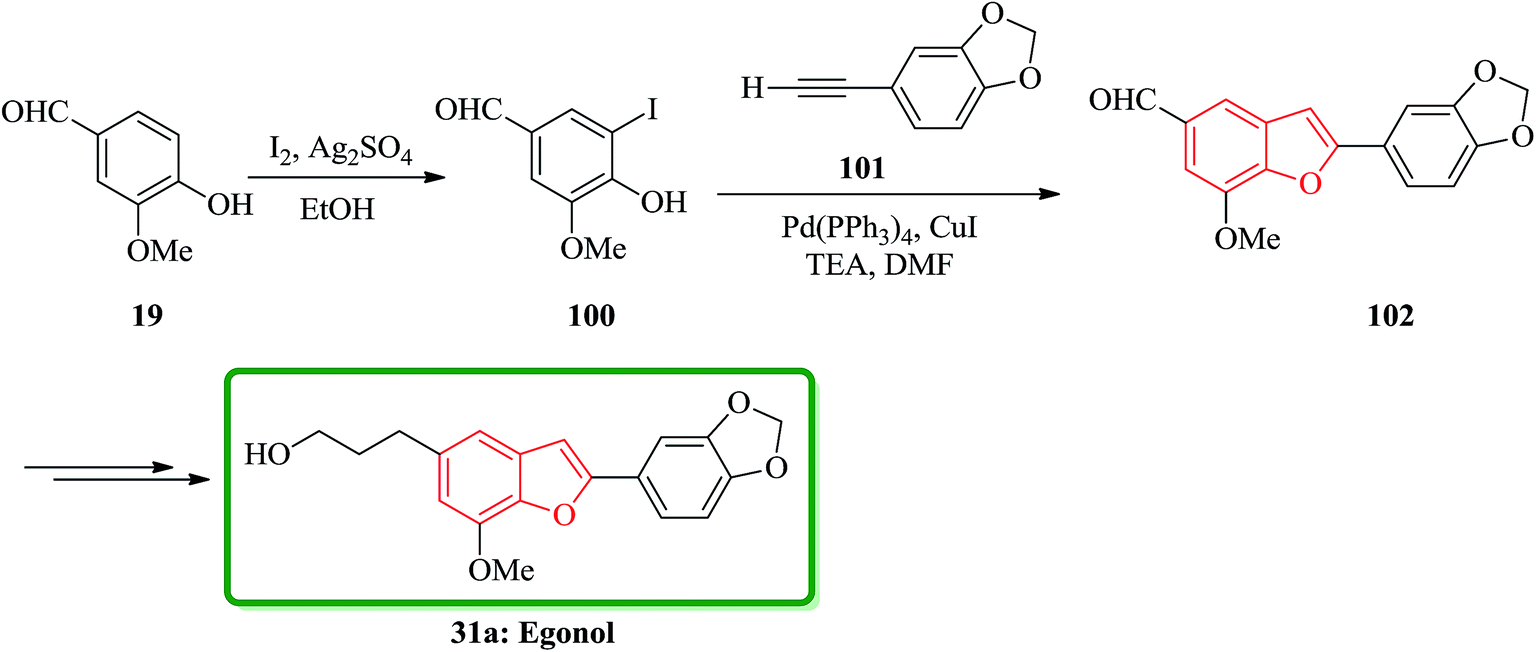 | ||
| Scheme 22 Total synthesis of egonol 31a. | ||
The highly efficient total synthesis of homoegonol 31b was achieved and reported in 2005.165 For the construction of benzofuran moiety present in 31b a facile two-step synthesis of 2-arylbenzofurans was implemented, proceeding via a selective cross-pinacol sort coupling between a salicylaldehyde and an aromatic aldehyde, with subsequent acid-induced cyclization. Therefore, bromobenzofuran 104 was synthesized from salicylaldehyde 23 and aromatic aldehyde 19 in two steps overall yield 61%. Subsequently, bromobenzofuran 104 was subjected into Sonogashira coupling with propargyl acetate to yield alkyne 105, which was subsequently hydrogenated and hydrolyzed to generate homoegonol 31b in satisfactory overall yield (38%) (Scheme 23).166
 | ||
| Scheme 23 Total synthesis of homoegonol 31b. | ||
Recently, Fukuyama and co-workers disclosed167 the results of their study on biological activity related to Phellinus ribis (Schmach) a fungus grown in East Asia which has been used as folk medicines for keeping immunity and for the treatment of gastrointestinal cancer.168 Ribisins A–D were recognized to increase neurite outgrowth in nerve growth factor (NGF). Total synthesis of the desired products 110, 113 and 118, which were found being the biologically potent part of naturally occurring compounds, ribisins A, B and D, have been accomplished. The total synthesis started from optically active pure cis-1,2-dihydrocatechol 107. The key features involve Suzuki–Miyaura cross-coupling reaction, intramolecular Mitsunobu and tandem epoxidation/rearrangement reactions. For the synthesis of ribisins A 110, initially, cis-1,2-dihydrocatechol 107 is transformed into the expected product 108 in several steps. Upon treatment of the latter with diethyl azodicarboxylate (DEAD) mediated by triphenylphosphine, an intramolecular Mitsunobu reaction occurs with the phenolic OH group acting as the internal nucleophile. In this way, the corresponding benzofuran 109 is obtained in high yield, resulted in construction of the tricyclic scaffold of the natural product 110 (Scheme 24).169
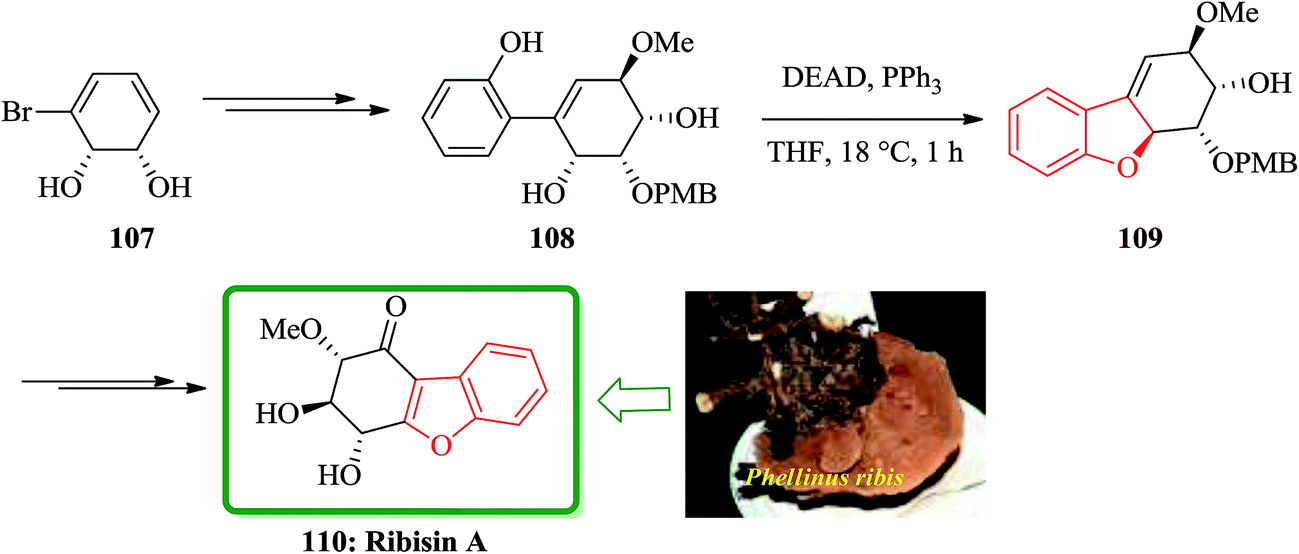 | ||
| Scheme 24 Total synthesis of ribisin A 110. | ||
For the synthesis of ribisins B 113, diol 107 can also be transformed into the expected product 111 in several steps. The latter then can be subjected into a sequential reaction involving an intramolecular Mitsunobu reaction, which resulted in the formation of tricycle 112 in 89% yields. The latter has benzofuran moiety in its structure. Finally, compound 112 can be converted in several steps to the desired ribisins 113 in high yield (Scheme 25).169
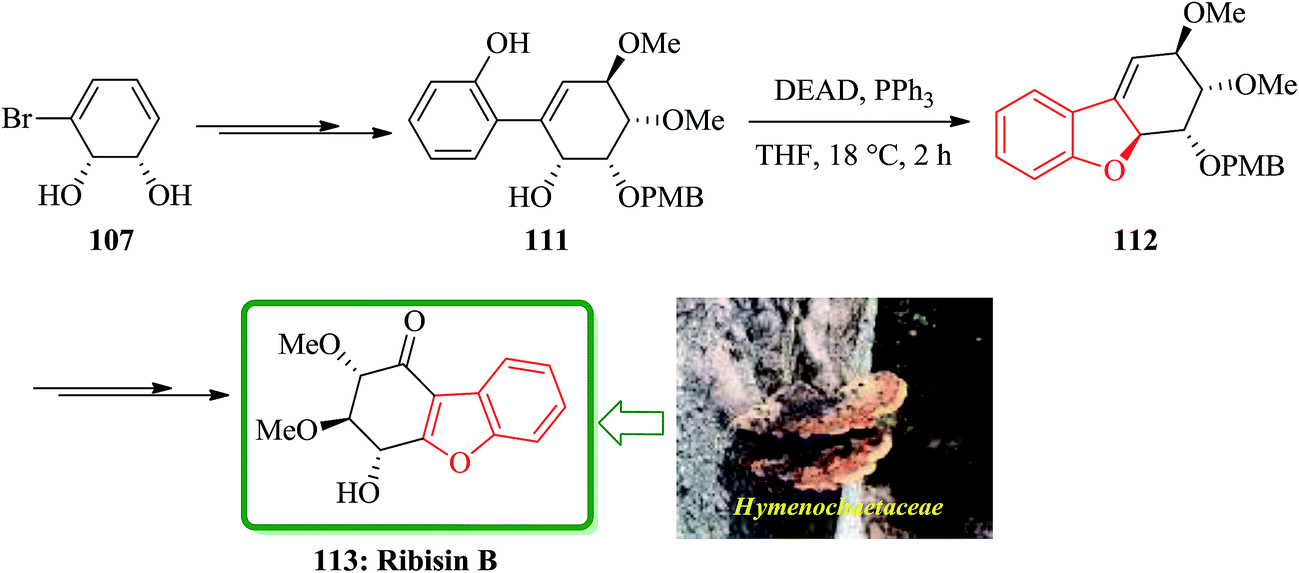 | ||
| Scheme 25 Total synthesis of ribisin B 113. | ||
In continuation of developing a synthetic strategy to obtain 118, Fukuyama and co-workers designed a convenient strategy for the total synthesis of natural product ribisin D. For the purpose, the boronate ester 114 was recognized being capable to cleave aryl isopropyl ethers under mild reaction conditions.170 It was also found that a phenolic hydroxyl group is also needed being present at C6 in the target 118. The reaction of compounds 114 and 115 gave the arylated cyclohexene 116, which was easily subjected into an intramolecular Mitsunobu reaction to afford the cyclodehydration product 117 in excellent yield (94%). The latter that bears the benzofuran moiety was converted into the desired natural product ribisin D 118 in several steps (Scheme 26).169
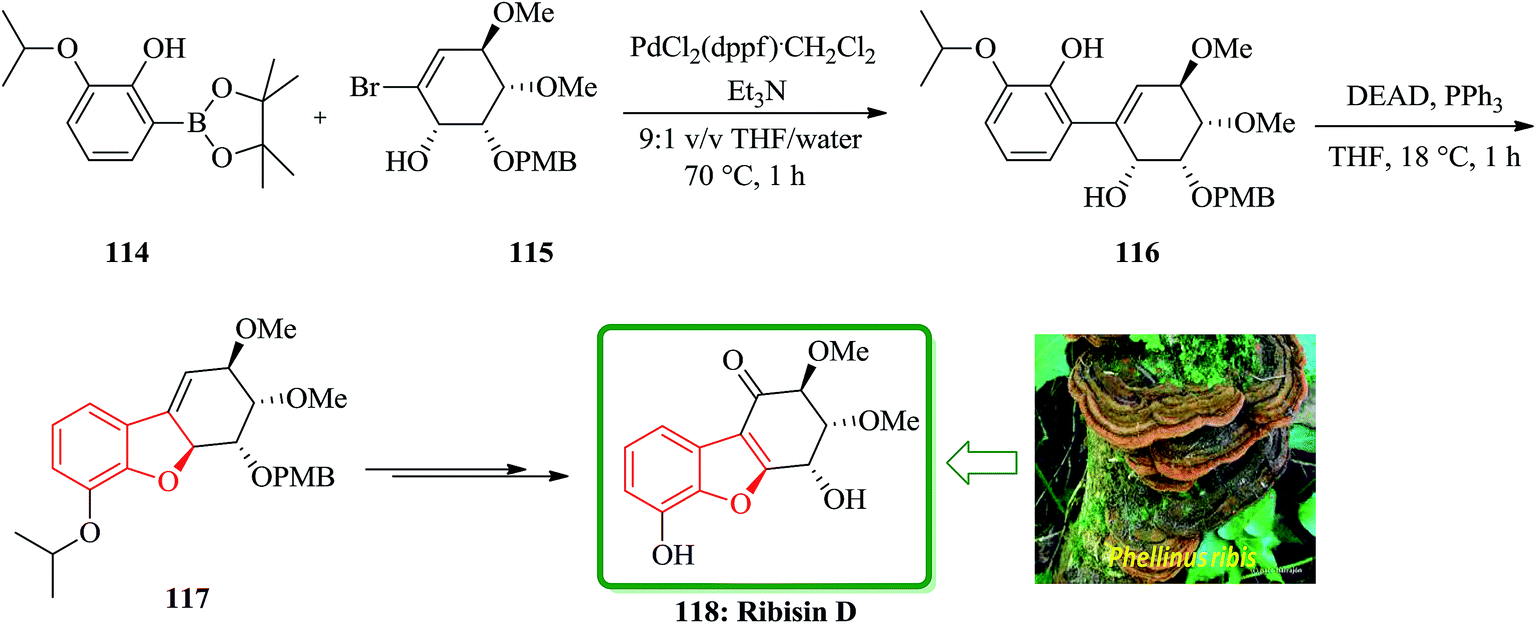 | ||
| Scheme 26 Total synthesis of ribisin D 118. | ||
In 2014, the isolation of four novel naturally occurring compounds as ribisin A–D was achieved and reported. They were isolated from the methanol extraction of the fruiting bodies of P. ribis.167 A concise total synthesis of natural product ribisin A has been accomplished in 11 steps.171 This approach started from market purchasable methyl α-D-glucopyranoside. Ribisin A has a highly oxygenated benzofuran scaffold, thus for its total synthesis, it was taken advantages of the intrinsic chirality of D-glucose. The important features of this total synthesis are applying some name reactions. It involved the Ferrier carbocyclization, Johnson iodination, Suzuki cross-coupling reaction, and Wacker oxidative cyclization. In this total synthesis, initially the commercially available methyl α-D-glucopyranoside 119 was converted to benzofuran precursor 120 in several steps. For the synthesis of the core benzofuran structure, the authors designed a route involving conversion of benzofuran precursor 120 to 121. To oxidize 120, m-CPBA and H2O2 were used which resulted in generation of a complex mixture containing, some unidentified products. Pd(II)-catalyzed Wacker reaction is an efficient protocol for olefin conversation and heterocyclic synthesis via antioxypalladation.172 It was successfully applied to oxidative cyclization of 120 by using PdCl2/CuCl/O2. This oxidation proceeds smoothly at 50 °C in dioxane to give the expected product 121 in satisfactory yield. Upon conventional deprotection of 121 by using TBAF in THF gave the desired compound ribisin A 110. The spectroscopic data for this synthetic product was found being identical to those obtained from the product isolated from natural source (Scheme 27).171
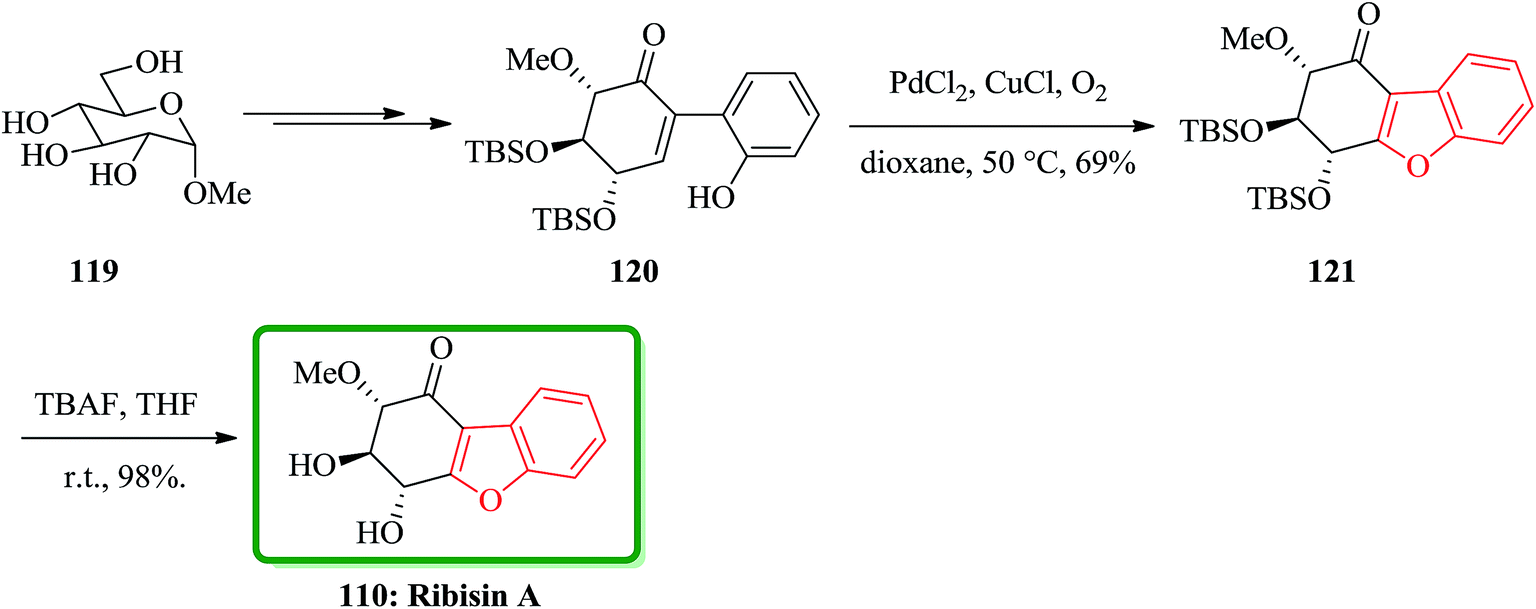 | ||
| Scheme 27 Total synthesis of ribisin A 110. | ||
Cicerfuran 126a, with antifungal potency was isolated from roots of wild chickpea.173 It has been synthesized from sesamol (3,4-methylenedioxyphenol) 122 in seven steps and 37% overall yield. Benzofurans 126a–f and the respective stilbene intermediates were synthesized. They exhibited antifungal and antibacterial potencies. Novak and co-workers accomplished and reported the synthesis of cicerfuran 126a.174 It involves palladium-catalyzed coupling of a styrene and 2-hydroxyaryl halide to form a stilbene, followed by epoxidation, subsequent cyclization and dehydration. Two analogues 126c, 126d of cicerfuran 126a were also synthesized effectively via this method, however the palladium coupling step did not occur with the dioxygenated aryl halides which are required for preparation of cicerfuran itself (Scheme 28, R2 = OH). Palladium-catalyzed coupling of the more reactive aryl acetylenes175–177 with 2-iodophenol afforded two analogues 126b and 126c of cicerfuran, albeit in low yields. In the original synthetic plan, the required stilbene was synthesized by using a Wittig reaction between 2-methoxy-4,5-methylenedioxybenzyltriphenylphosphonium bromide and 2,4-di-tert-butyldimethylsiloxy-benzaldehyde. An alternative pathway to cicerfuran 126a involves epoxidation and cyclization, which affords quantities sufficient for further biological studies. Two other analogues 126e, 126f of cicerfuran were synthesized by this route but were only characterized partially due to decomposition during their purification (Scheme 28).178
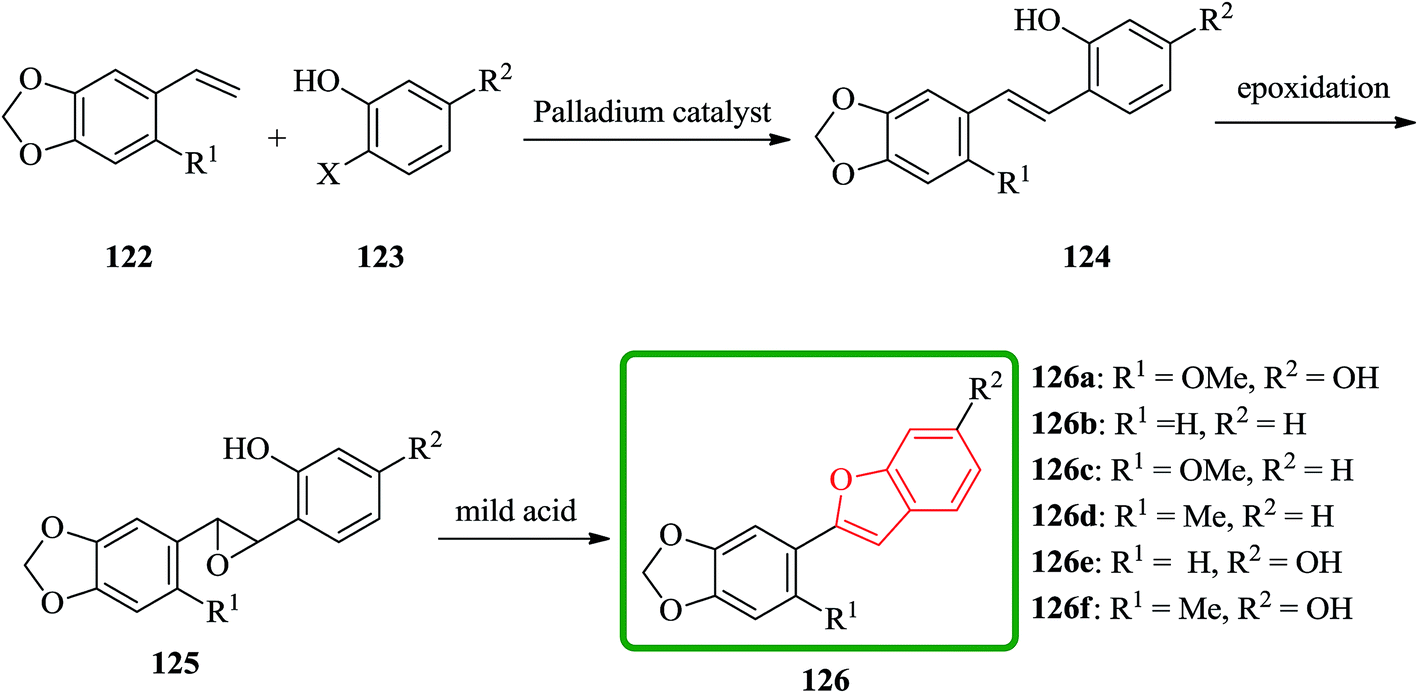 | ||
| Scheme 28 Total synthesis of cicerfuran 126. | ||
Stilbenes 124i and 124j were epoxidized with MCPBA. Stilbene 124j were subjected to sequential epoxidation and cyclization under these conditions to yield 2-(2-methyl-4,5-methylenedioxyphenyl)benzofuran 126d in moderate yield and relatively long reaction time. Notably, when the same process applied to 124i complete decomposition occurred thus, the isolated epoxide 125a underwent acid-catalyzed ring-opening, cyclization and dehydration in the presence of p-toluenesulphonic acid in chloroform to provide 2-(2-methoxy-4,5-methylenedioxyphenyl) benzofuran 126c. The 2-methoxy group in 126c makes the benzofuran moiety much less stable in the presence of acid than that of in 126d, which bears methyl group (Scheme 29).178
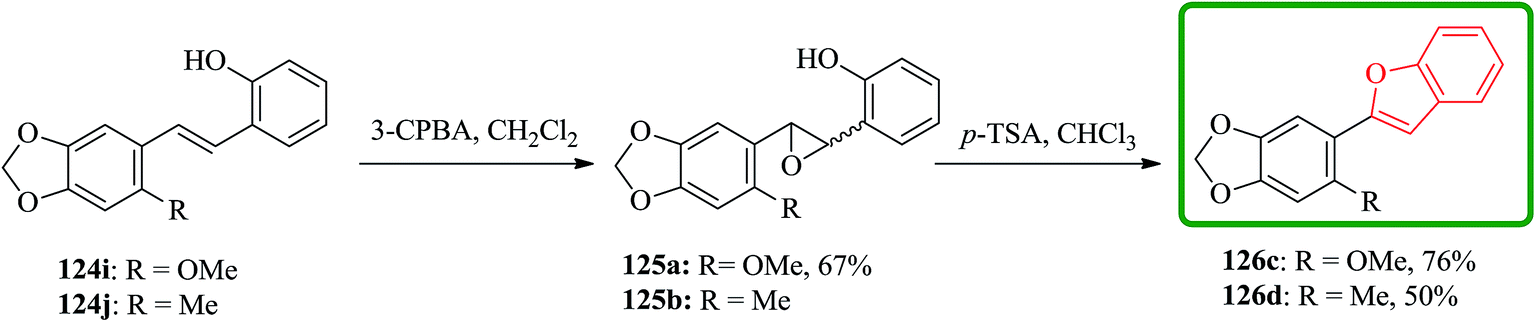 | ||
| Scheme 29 Total synthesis of cicerfuran 126c, d. | ||
Palladium catalyzed coupling of terminal acetylenes with o-hydroxy aryl halides gave corresponding benzofurans in a single step reaction. Aryl acetylenes, which are usually more reactive in palladium-catalyzed coupling reaction were reacted with multioxygenated aryl halides for the synthesis of cicerfuran and its analogues. Three arylbenzofurans, 128, 126b and 126c were prepared via palladium-catalyzed coupling of acetylenes 127a–c with 2-iodophenol 123a as illustrated in Scheme 30.178
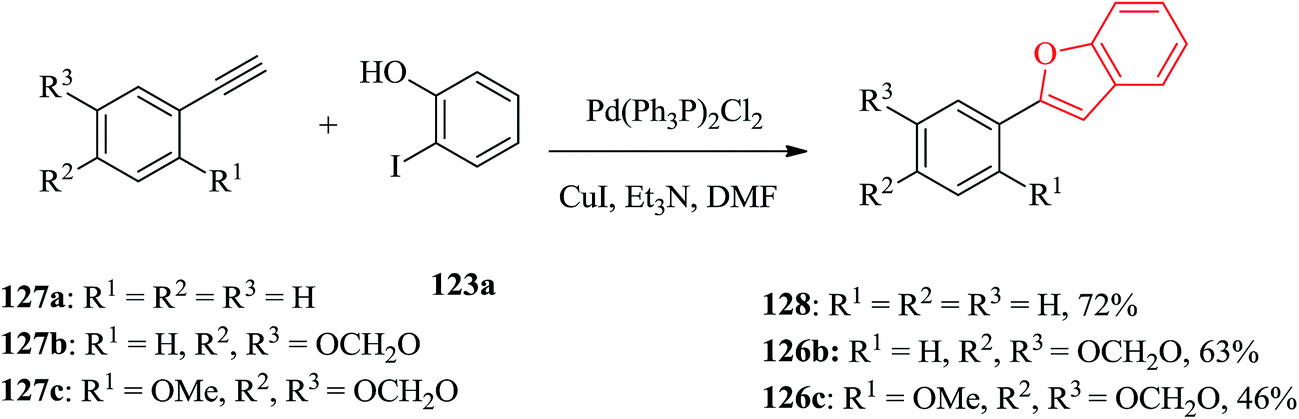 | ||
| Scheme 30 Synthesis of arylbenzofurans 128, 126b and 126c. | ||
Remarkably, acetylation of the hydroxyl groups usually makes the aryl halide more reactive to nucleophilic attack. Therefore, the synthesis of cicerfuran was studied via palladium-catalyzed coupling179 of acetylene 127c with the diacetate of iodoresorcinol 123b, as depicted in Scheme 31.178
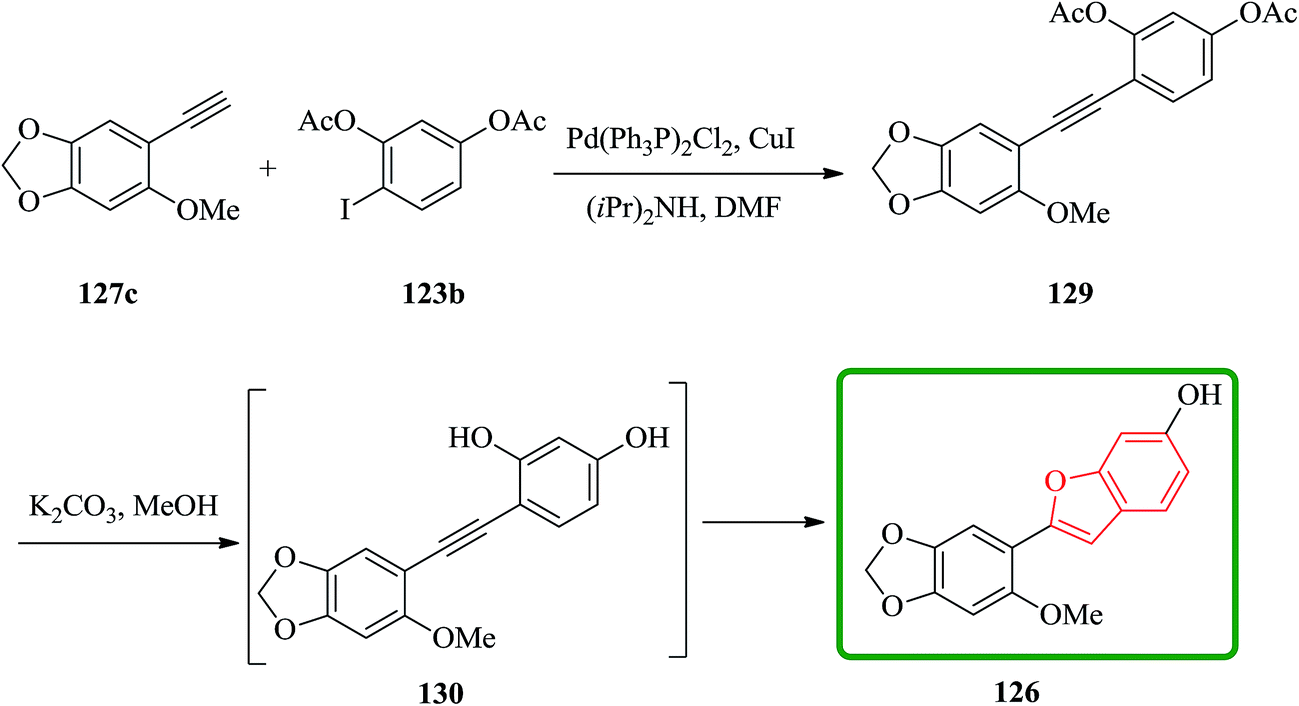 | ||
| Scheme 31 Synthesis of the natural product 126. | ||
In another route, the desired stilbenes 131a–c (prepared via Wittig reactions of phosphonium bromides and benzaldehyde) which obtained approximately as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of the E and Z isomers were epoxidized by using MCPBA. Yields were relatively low apparently because the instability of the OTBDMS protected epoxides 132a–c. These epoxides can be easily converted to the desired compound by using a few crystals of p-toluenesulphonic acid in chloroform (Scheme 32).178
:1 mixtures of the E and Z isomers were epoxidized by using MCPBA. Yields were relatively low apparently because the instability of the OTBDMS protected epoxides 132a–c. These epoxides can be easily converted to the desired compound by using a few crystals of p-toluenesulphonic acid in chloroform (Scheme 32).178
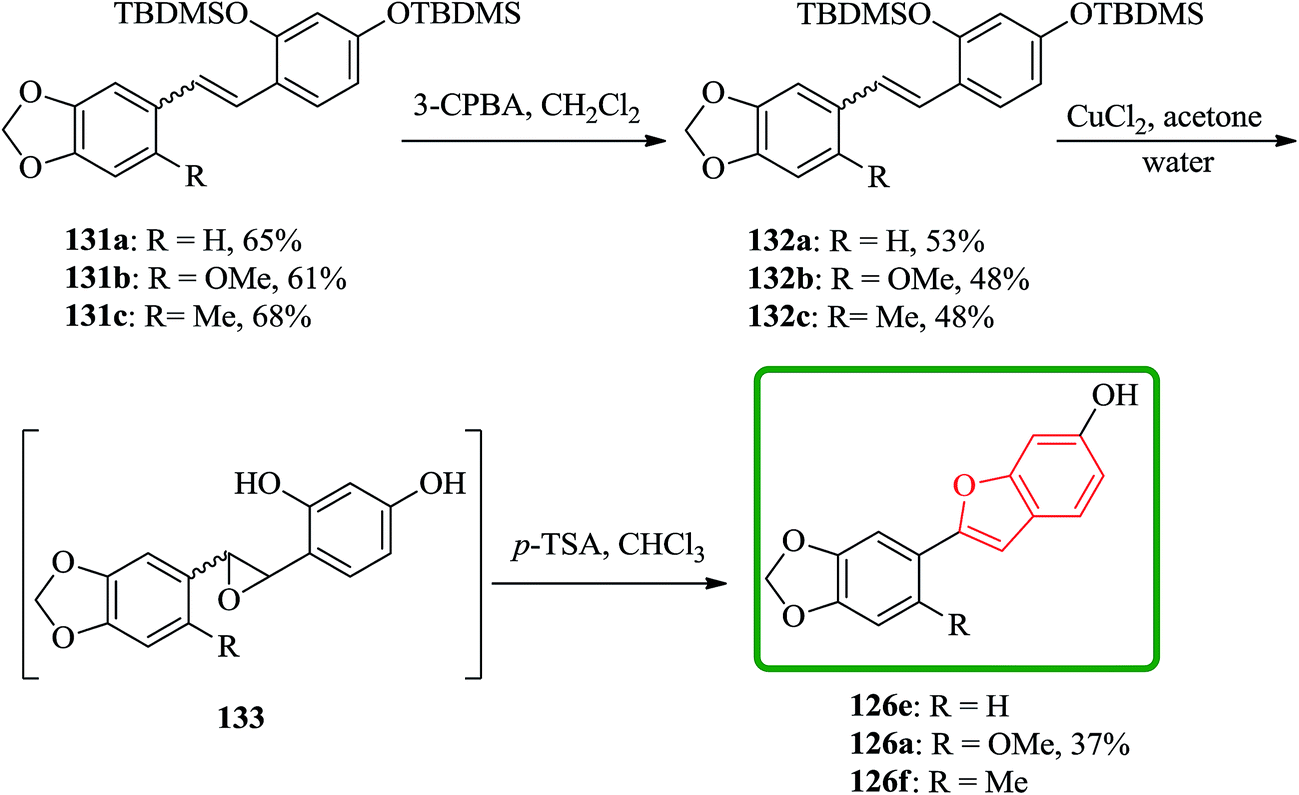 | ||
| Scheme 32 Synthesis of the natural products 126. | ||
Sonogashira coupling/cyclization reaction of aryl iodide 134 with 2-methyl-3-butyn-2-ol 135 was achieved in the presence of Pd(PPh3)2Cl2 and CuI. Deprotection of the acetylene moiety in the same pot using a strong base and the second Sonogashira coupling/cyclization of substituted o-iodophenols led to the formation of the appropriate benzo[b]furans. This protocol was used in the synthesis of natural product cicerfuran 126 (Scheme 33).180
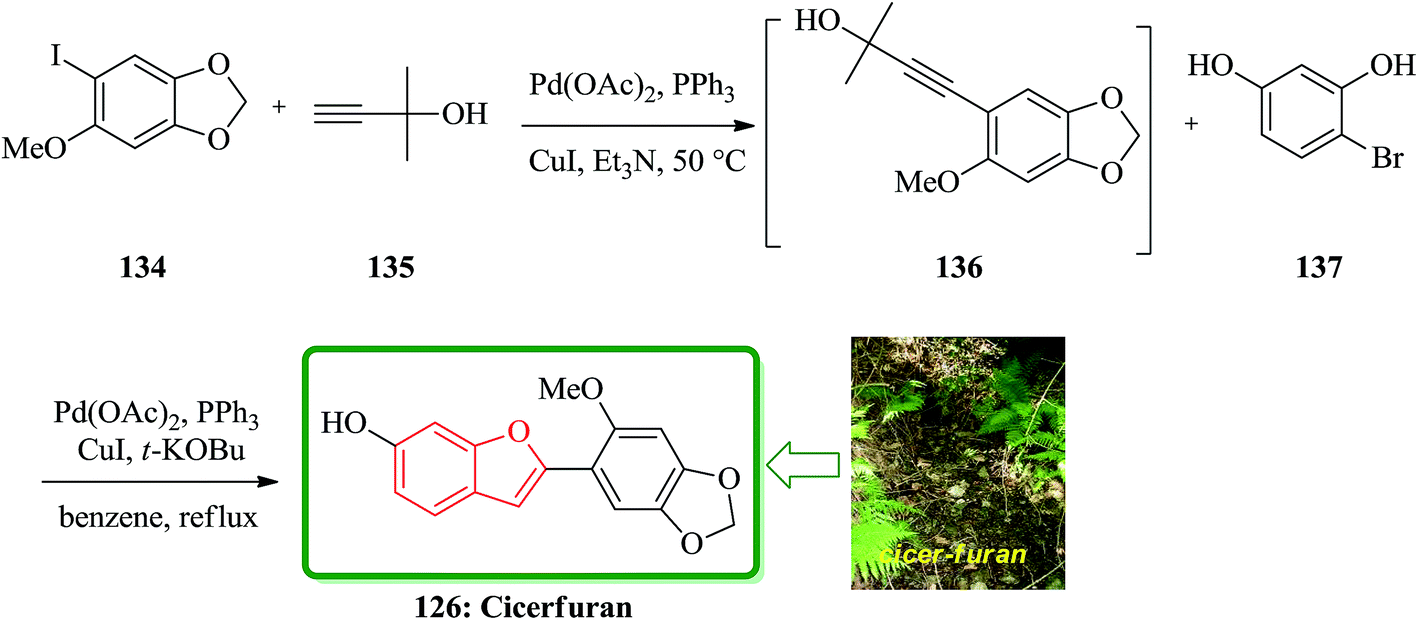 | ||
| Scheme 33 Total synthesis of cicerfuran 126. | ||
A concise total synthesis of eupomatenoid 6 50g was reported by Stevenson research group in seven steps.181 After that, two other five-step synthesis were reported by Bach and co-workers (25% overall yield).182 Eidamshaus and Burch in 2008 accomplished a four-step total synthesis of 50g, in a three-pot approach. Initially, 2-bromo-4-chlorophenol 138 was coupled with 4′-methoxypropiophenone 139, which was followed by concurrent cyclization under optimized reaction conditions to afford compound 140 containing a benzofuran moiety in 45% yield. The latter was then transformed to the desired natural product 50g via a cascade reaction involving Stille reaction and demethylation with ethanethiolate in one-pot fashion (Scheme 34).183
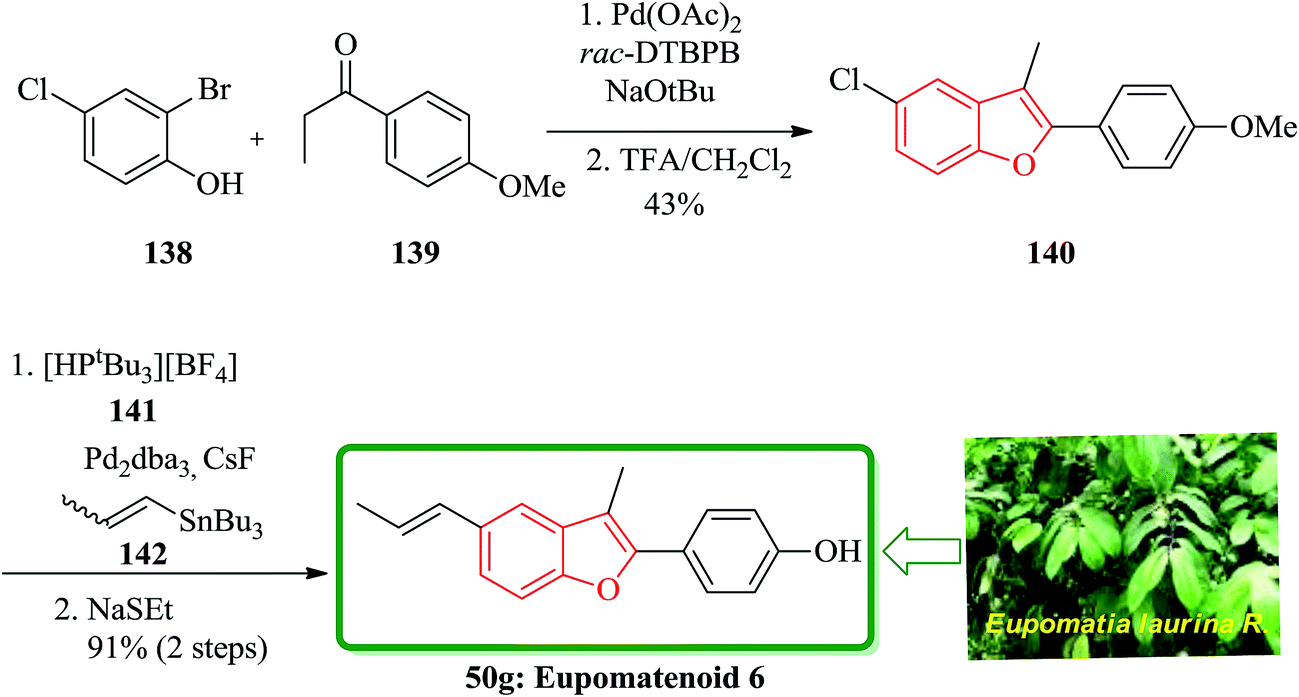 | ||
| Scheme 34 Total synthesis of eupomatenoid 6 50g. | ||
The aglyconic part, which is also called eupomatenoid-6 50g, is a naturally occurring compound. It was initially isolated from extract of the leaves of Piper fulvescens. Compound 146 can be subjected into glycodiversification,184–186 thus can create a set of diverse modulators of Hsp90 activity.187–189 For glycol diversification, eupomatenoid-2 of the 2-(4′-hydroxyphenyl)benzofuran aglycon (a.k.a. eupomatenoid-6) was subjected into glycosylation. Glycosylation of the phenol by glycosylbromides under basic conditions afforded the desired products in the gluco-, galacto- and fuco- series. This procedure failed in the manno- and rhamno-series. However, mannosylation and rhamnosylation of eupomatenoid-6 could be obtained under carefully controlled acidic conditions using O-benzoxazolyl imidate (OBox) donors. Eupomatenoid-6 50g was provided following the previously reported procedure, which is depicted in Scheme 35. This protocol began from 2-bromo-4-chlorophenol 144, which reacted with 1-(4-methoxyphenyl)propan-1-one 143 to afford the intermediate 5-chlorobenzofuran 145. Finally, the latter was transformed to the desired natural product 146 in several steps.190
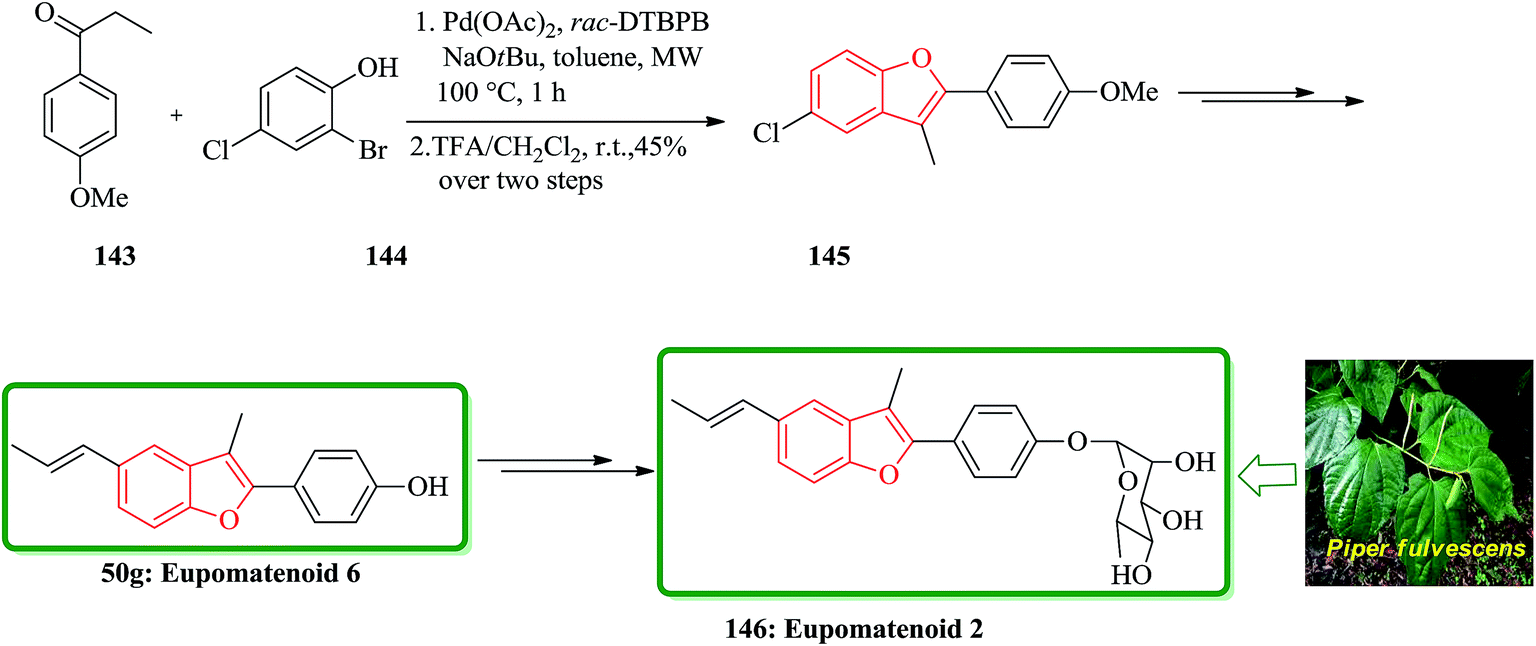 | ||
| Scheme 35 Total synthesis of eupomatenoid 6 50g and eupomatenoid 2 146. | ||
Kendomycin [151, (−)-TAN2162], an ansamycin isolated from different Streptomyces species has been frequently studied over the last decade. It was found being a potent endothel in receptor antagonist and antiosteoperotic with remarkable antibacterial and cytostatic activity.191 The synthesis of the benzofuran fragment 150 started from the known aldehyde 147,192 which is easily available from citronellene. Compound 147 is transformed into ketone 148 in several steps including palladium(0)-mediated rearrangement. The latter was then subjected to acid-catalyzed formation of the furan ring which concomitantly removes the 3-OMOM group to give 149 which was oxidized to carboxylic acid 150 (Scheme 36). After several steps, involving functional groups transformations compound 50g was converted into the desired natural product 151.193
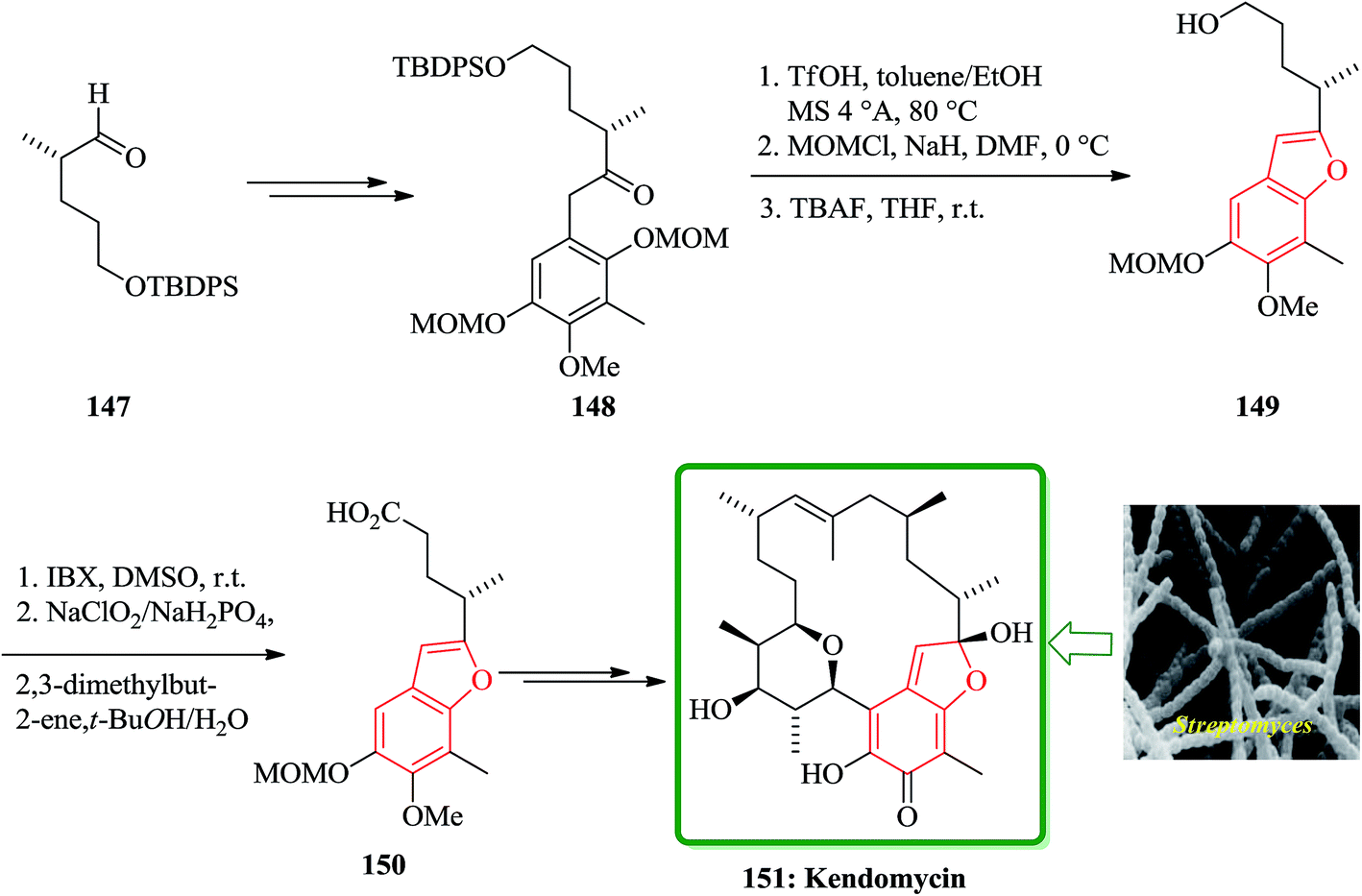 | ||
| Scheme 36 Total synthesis of kendomycin 151. | ||
A pathway for the total synthesis of the bacterial metabolite kendomycin 151 was reported in 2014. Furthermore, an efficient strategy for the total synthesis of 151 was achieved starting from readily available 2-methoxy-3-methylbenzene-1,4-diol 152, which was initially transformed into cycloalkyne 153. The latter was then underwent to a gold-catalyzed hydroalkoxylation resulting in benzofuran 155, which contains benzofuran moiety in its structure. Worthy to mention that benzofuran 155 had been utilized as an intermediate en route to 151. In this strategy, cycloalkyne 153 was submitted to saponification of the remaining acetate. Noticeably, upon treatment of cycloalkyne 153 with PtCl2 the cyclization was not achieved. However, in the presence of electrophilic cationic gold complexes 154, the cyclization of 153 was smoothly proceeded to give the benzofuran derivative 155. The latter was then transformed in several steps to the desired natural product kendomycin 151. The total synthesis was interrupted through the route reported by Mulzer and co-workers.194 However, the subsequent ring contraction reported by these authors via a photo-Fries rearrangement195 could be also occurred (Scheme 37).196
 | ||
| Scheme 37 Total synthesis of kendomycin 151. | ||
Liphagal 160 was isolated from the sponge Aka coralliphaga, collected from reefs in Prince Rupert Bay, Portsmouth, Dominica.197 Liphagal 160 showed significant biological activity involving inhibitory activity against PI3K α (phosphoinositide-3-kinase α).197 Due to its importance, three approaches have been reported for its total synthesis including (A) a relatively short synthesis (nine linear steps) that follows a biomimetic route to the bioactive marine natural product liphagal, from a commercially available starting materials, was described by Mehta and co-workers. Liphagal 160 is the first member of a new ‘liphagane’ type of meroterpenoid carbon skeleton. A mixed biogenetic route for liphagal 160 was suggested197 in which forms the AB rings of this natural product showing a typical sesquiterpene-like structure. For the synthesis of liphagal 160, the key furan precursor was synthesized from an easily available aromatic starting materials. Regioselective mono demethylation of commercially accessible aldehyde 156 after several steps provided 157. One-pot furan annulation198 of 157 went smoothly and furnished the required bromobenzofuran 159 in moderate yield. At the end bromobenzofuran 159 was converted into liphagal 160 after several steps (Scheme 38).199
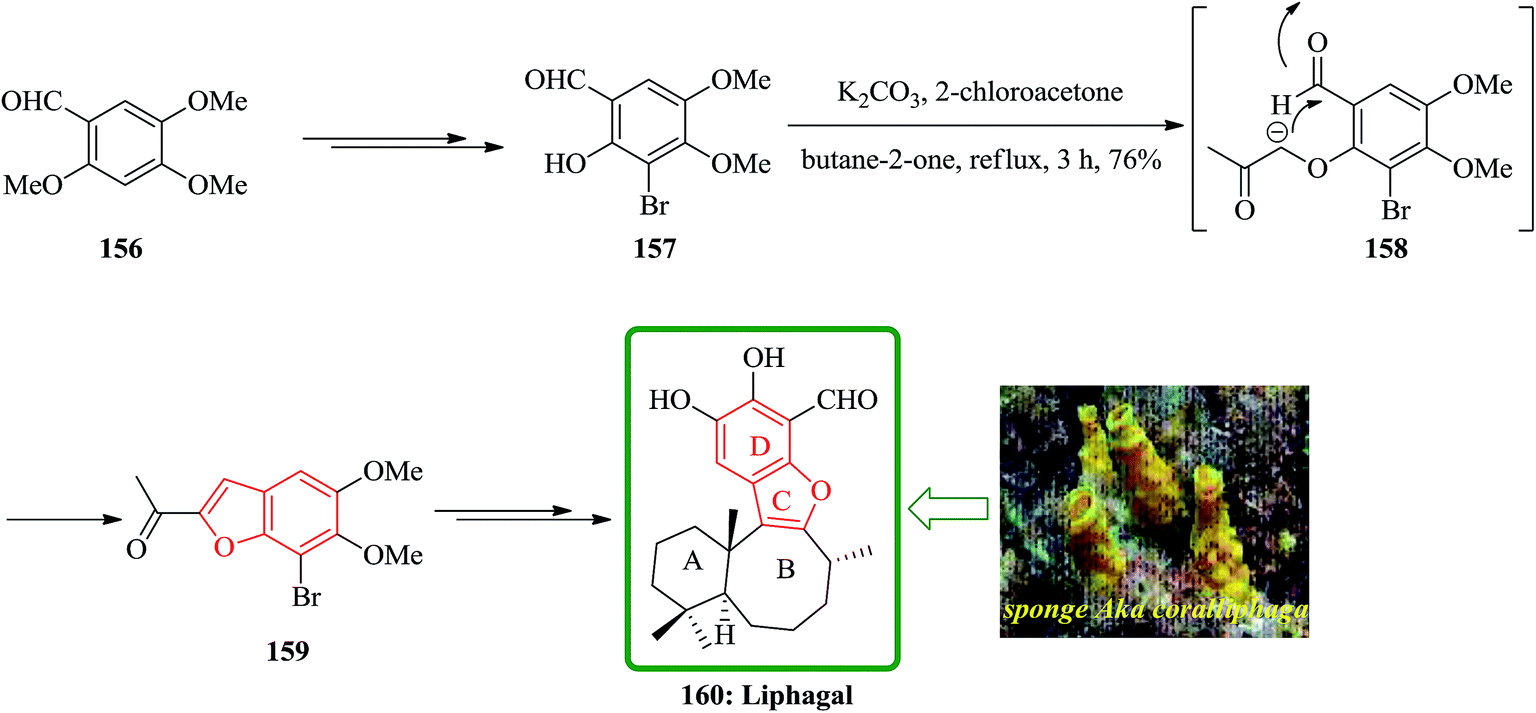 | ||
| Scheme 38 Total synthesis of liphagal 160. | ||
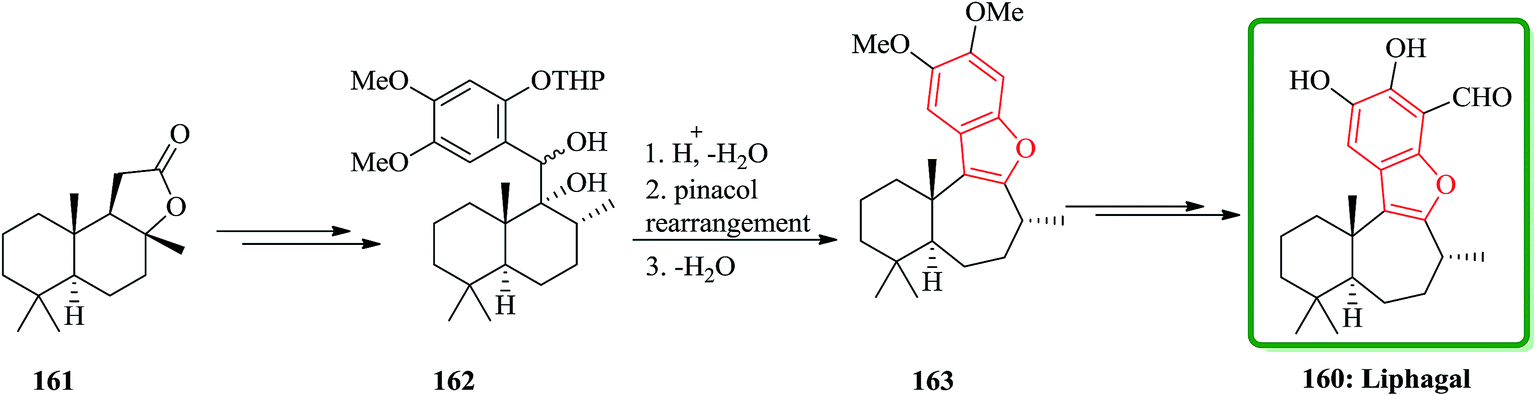
(B) The total synthesis of (+)-liphagal200 has also been accomplished in 13 steps with 9% overall yield and reported. The total synthesis was started from a natural product (+)-sclareolide 161. In this approach, the key step is a ring expansion involving the generation of a highly stabilized benzylic carbocation, which is converted into the seven-membered ring and the benzofuran moiety of the natural product in a single cascade reaction. Compound 161 was converted into 162 in several steps. Having 162 available, the biomimetic step involving ring-expansion reaction was examined. Upon treatment of compound 162 with TFA/CH2Cl2 at −78 °C and then gradual warming to ambient temperature the ring-expanded product 163 was obtained in two steps via pinacol rearmament in 74% overall yield. Then, the synthesis of (+)-liphagal, the desired natural product 160 was accomplished after two steps (Scheme 39).201
 | ||
| Scheme 39 Total synthesis of liphagal 160. | ||
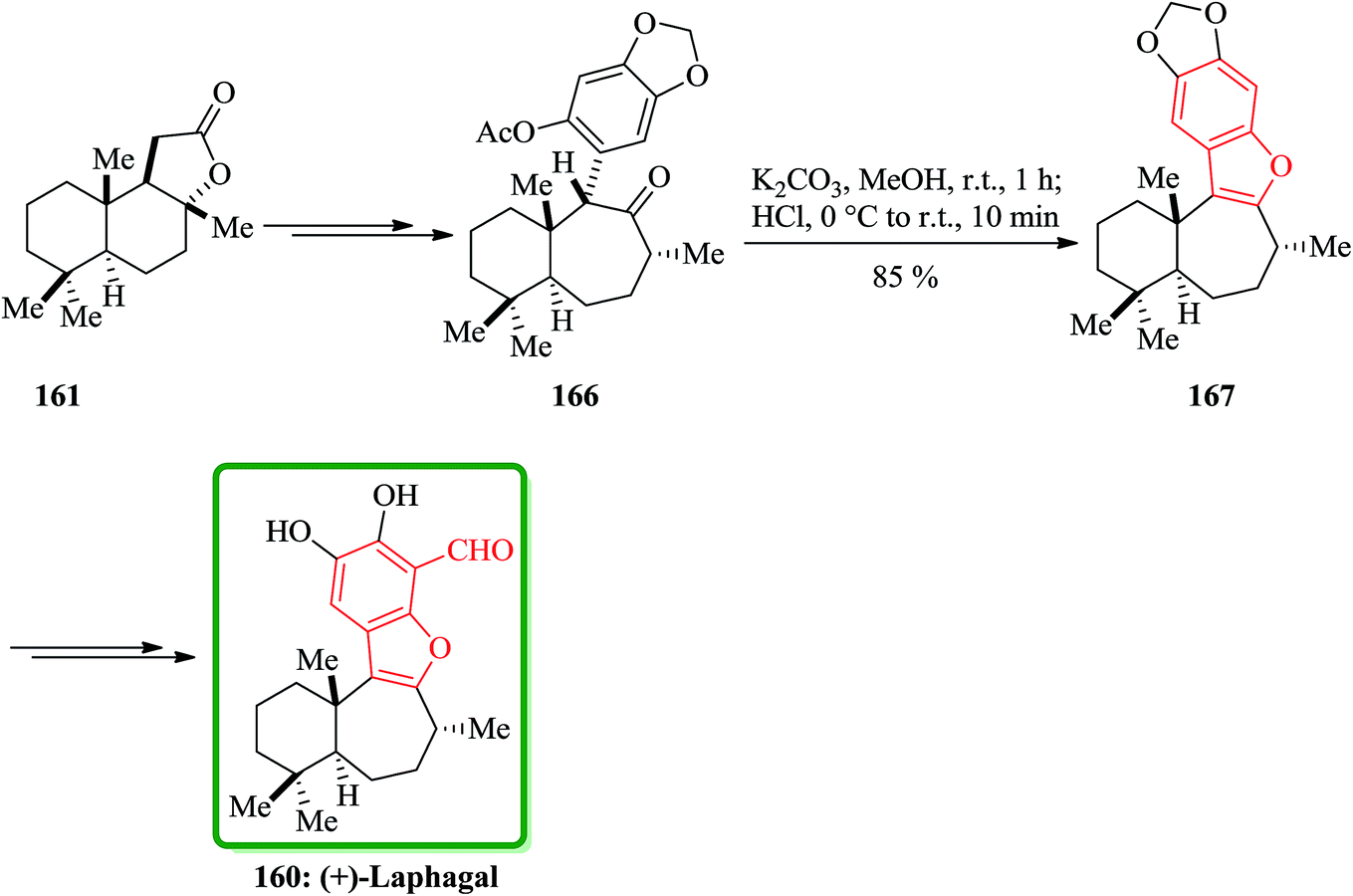
(C) The total synthesis of liphagal202 was started from market purchasable (+)-sclareolide 161. Compound 160 as a structurally outstanding marine natural product, with characteristic tetracyclic core structure was prepared in 29% overall yield in 13 steps modeled biosynthesis. In this total synthesis, starting from 161 and after several steps, the intermediate 166 was provided and transformed into the intermediate 167, which bears the benzofuran moiety, via conventional conditions. Then the latter was converted into the desired natural product (+)-liphagal 160 in several steps (Scheme 40).203
 | ||
| Scheme 40 Total synthesis of liphagal 160. | ||
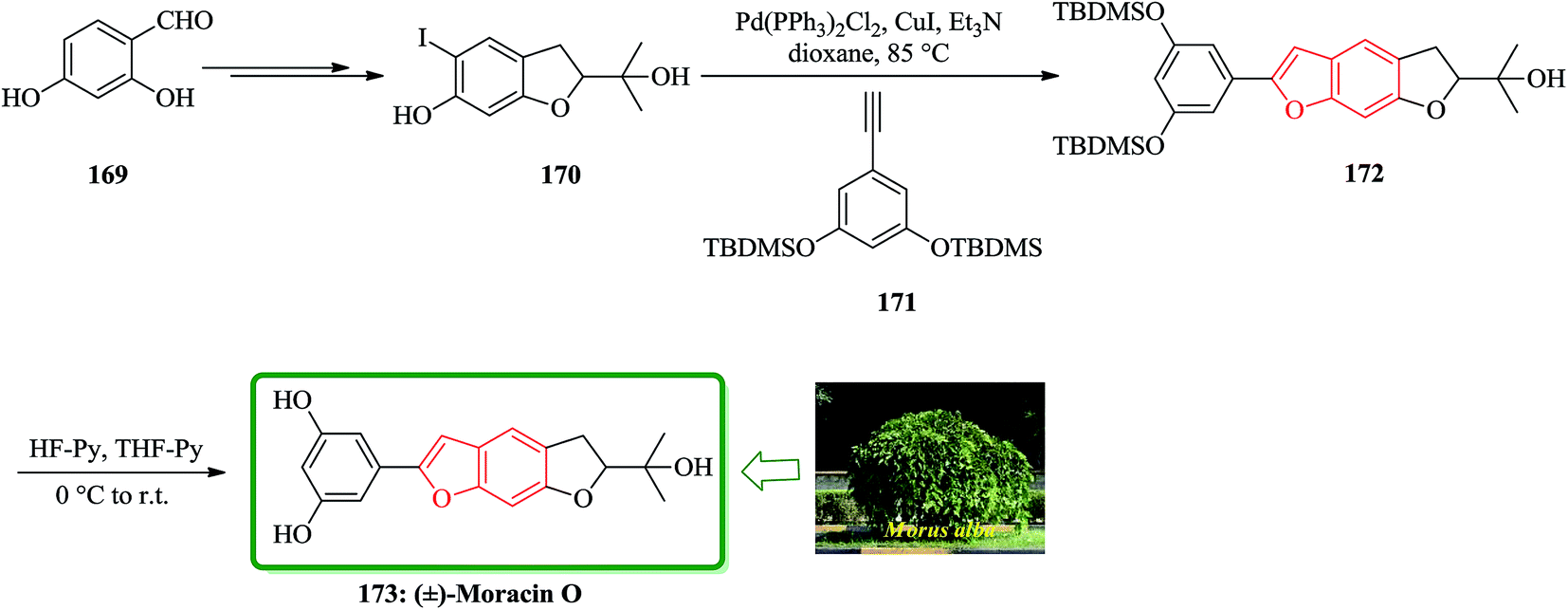
Moracins O and P were first isolated in 1998 from an acetone extract of cortex and phloem tissues of Morus alba shoots infected with Fusarium solani f. sp. Mori. Their structures were determined by their IR, 1H-NMR and 13C-NMR spectral data.204 The first total synthesis of the naturally occurring benzofurans, moracins O and P was achieved using a Sonogashira cross coupling reaction followed by in situ cyclization. In this route, the total synthesis of 173 was started from 2,4-dihydroxybenzaldehyde 169. The reaction of benzohydrofuran nucleus 170 with the substituted acetylene, 1,3-bis-(tert-butyldi-methylsilanyloxy)-5-ethynylbenzene 171 employing Sonogashira cross coupling under basic conditions and in situ cyclization afforded 172 which upon final deprotection with HF-pyridine provided (−)-moracin O 173 in a 75% yield. The NMR spectra of synthetic (−)-173 were identical to the spectra of the corresponding natural products (Scheme 41).204
 | ||
| Scheme 41 Total synthesis of (−)-moracin O 173. | ||
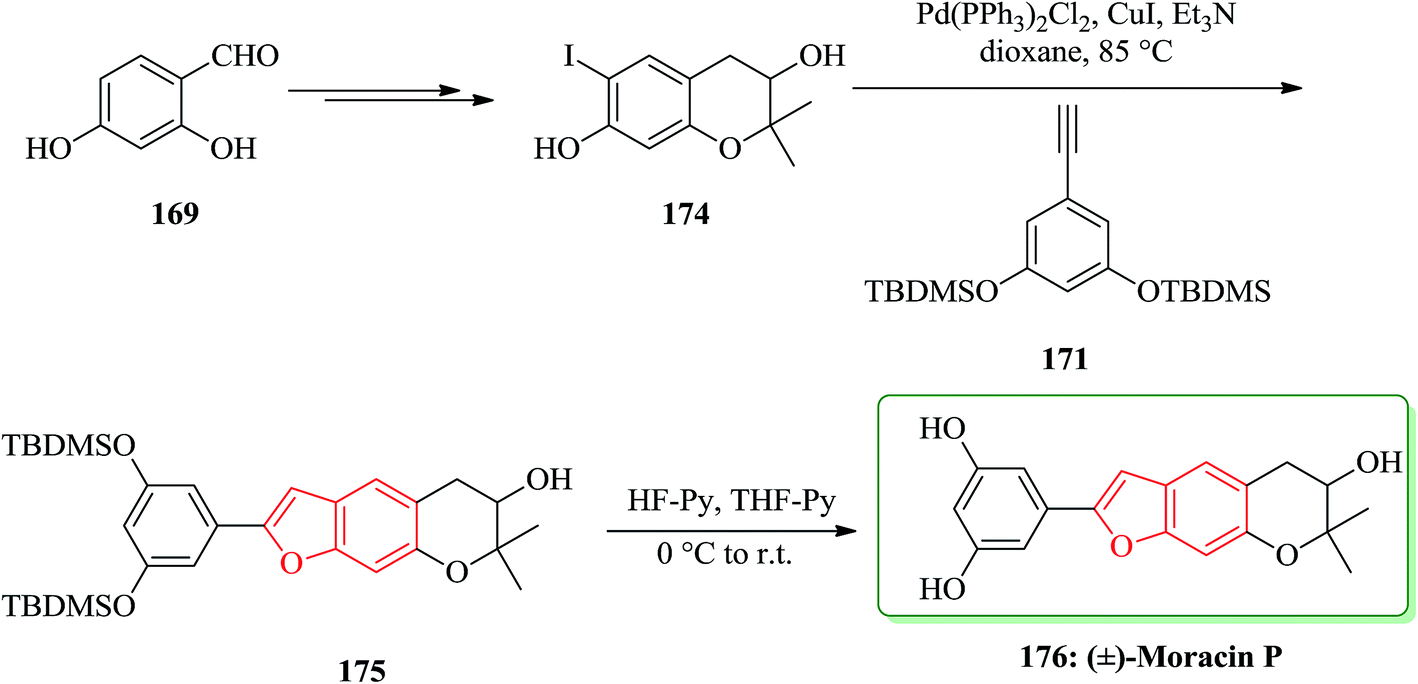
Then, the synthesis of (−)-176 was started from 2,4-dihydroxybenzaldehyde 169 is converted into dihydrochomarine. 174 The latter was reacted with alkyne 171 under Sonogashira cross coupling conditions followed by in situ cyclization in dioxane to afford benzo[b]furan intermediate 175 in a 36% yield. Early attempts to remove the TBDMS groups from benzofuran derivative 175 using TBAF yielded a mixture of products, possibly due to the strong basic conditions and/or the long reaction time which either permitted group migration205 or opening of the pyran ring. The same deprotection reaction with HF–pyridine complex afforded clean removal of the TBDMS protective groups and provided the desired racemic moracin P 176 in a 75% yield (Scheme 42).206
 | ||
| Scheme 42 Total synthesis of (−)-moracin P 176. | ||
The natural products moracins O and P showed being active in vitro inhibitory against hypoxiainducible factor (HIF-1), a mediator, which is a key important during adaptation of cancer cells to tumor hypoxia. Systematic studies revealed the significance of presence of the 2-arylbenzofuran ring and particularly the core framework should have (R)-configuration. The 2-arylbenzofuran is a common unit, consisting of B, C, and D rings. All the benzofuran derivatives 179–191 were synthesized as outlined in Scheme 43. The key and important intermediates for the synthesis of moracin O or P derivatives as shown in Scheme 43 is dihydrobenzofuran 170 which can be synthesized from 2,4-dihydroxybenzaldehyde in several steps. The terminal acetynyl derivatives either were purchased from commercial sources 177e–l or synthesized 177a–d. The acetynyl compounds can be provided via a procedure developed by Ramirez–Corey–Fuchs, in which compounds 177a–d were synthesize in three steps.207 The Sonogashira catalyzed coupling of terminal acetylenes 177a–l with substituted o-iodophenol 170 afforded the moracin O analogues 178a, b and 179–191. Compounds 178a, b were deprotected using HF/pyridine to yield the corresponding phenol analogues 180 and 181 in satisfactory yields. Treatment of compound 181 with ethyl chloroacetate gave the alkylated product 182.208
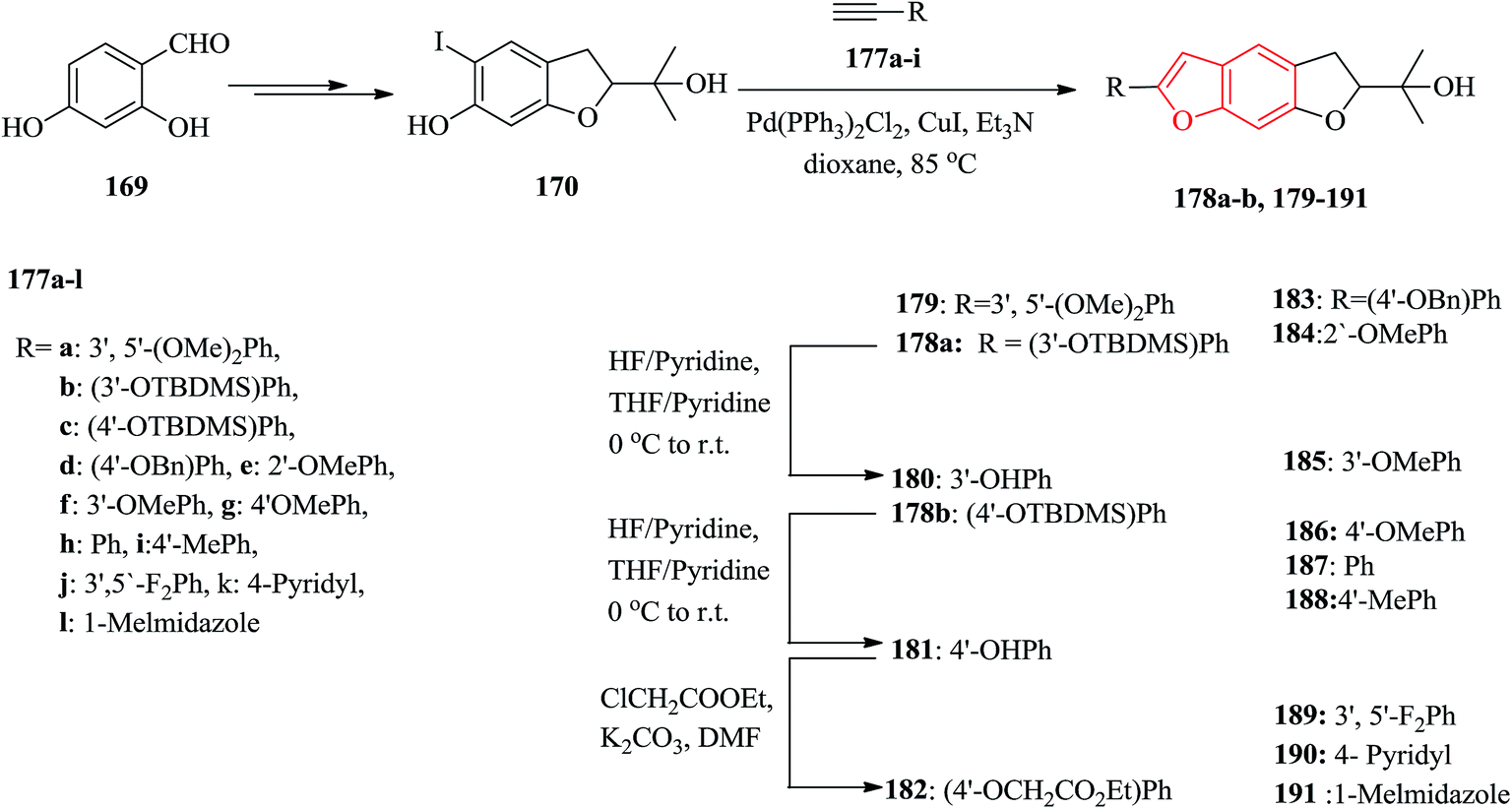 | ||
| Scheme 43 Total synthesis of compounds 179–191. | ||
It has been reported that the (R)-isomer of moracin O was more active than its (S)-isomer. Unpleasantly, the stereogenic center of the synthesized analogues was generated in a nonstereospecific fashion. Thus, it was desirable to synthesize the corresponding (R)-stereoisomer of the analogues 181 asymmetrically in optically pure form for further biological screening. The asymmetric synthetic approach was outlined in Scheme 44. In this pathway, the key intermediate is an optically pure iodobenzofuran derivative (R)-(−)-170 which can be obtained from the prenylated derivative 192 in five steps including a stereoselective synthesis. (R)-(−)-170 reacted with the protected ethynyl benzene compound 177c via Sonogashira reaction to provide (R)-(−)-178 with subsequent deprotection with HF/pyridine, which gave the desired target (R)-(−)-181.208
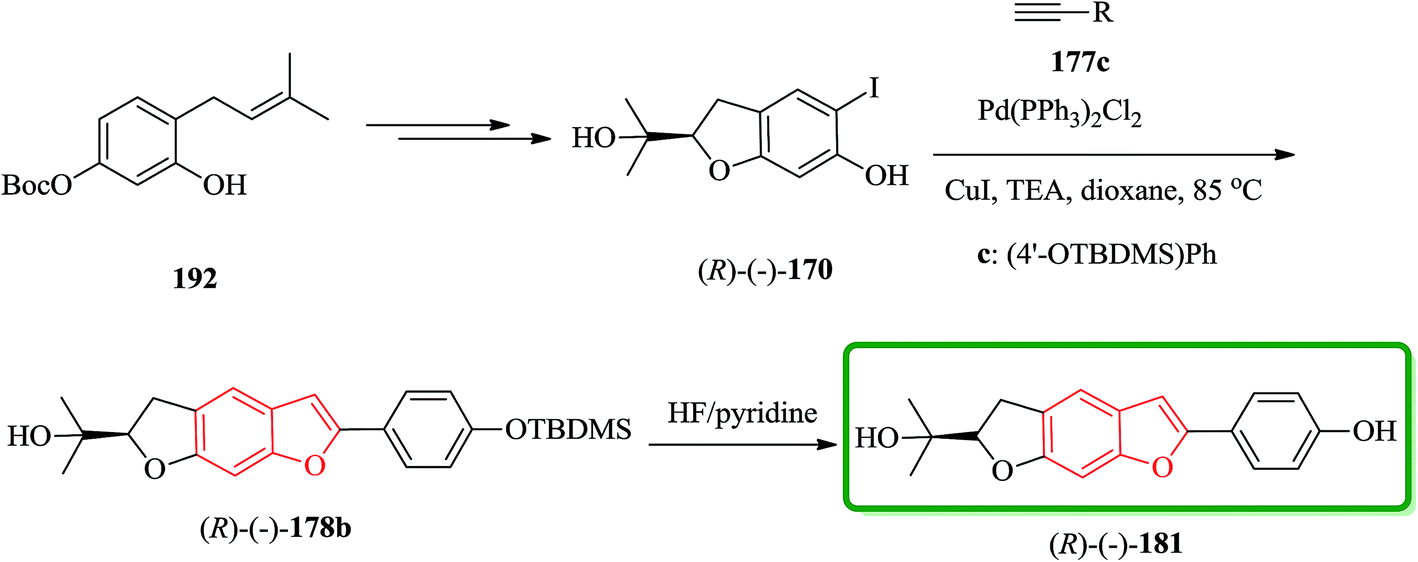 | ||
| Scheme 44 Total synthesis of (R)-(−)-181. | ||
Furoventalene 200 is an irregular isoprenoid benzofuran, which has initially been isolated from the sea fan Gorgonia ventalina.209 Natural product 200 was first synthesized by Weinheimer and Washecheck in a non-regioselective fashion.209 The scaffold of furoventalene 200 was regioselectively build up from methyl 2-fomy1-6-methyl-heptenoate 194 and 2,5-dihydro-3-methyl-4-vinyl-2-furanone 195 via successive 1,6-conjugate addition/aldol-type cyclization to provide a diastereomeric mixture of bicyclic butenolide 196a and 196b. Both of the annulated species can be converted into 200 by a sequential reactions involving, reduction/hydrolysis/dehydrative decarboxylation and dehydrogenation through intermediates 197–199.209 In the total synthesis of 200 dicarbonyl compound 194 is a key compound, which was readily synthesized and provided as the enol form by formylation of the methyl ester of 6-methyl-5-heptenoic acid 193 with ethyl formate in the presence of LDA in THF. The formyl ester 194 upon treatment with the butenolide 195 in Me2SO and KF at ambient temperature annulation product gives a mixture of diastereomer in excellent yield. This mixture can be cleanly separated by column chromatography to afford 196a and 196b (79:21) as crystalline products. Compound 199 was dehydrogenated at ambient temperature using DDQ. The latter was then transformed in several steps to compound, which was identified as furoventalene 200 by comparison of its spectroscopic data with those obtained from the original natural product (Scheme 45).210
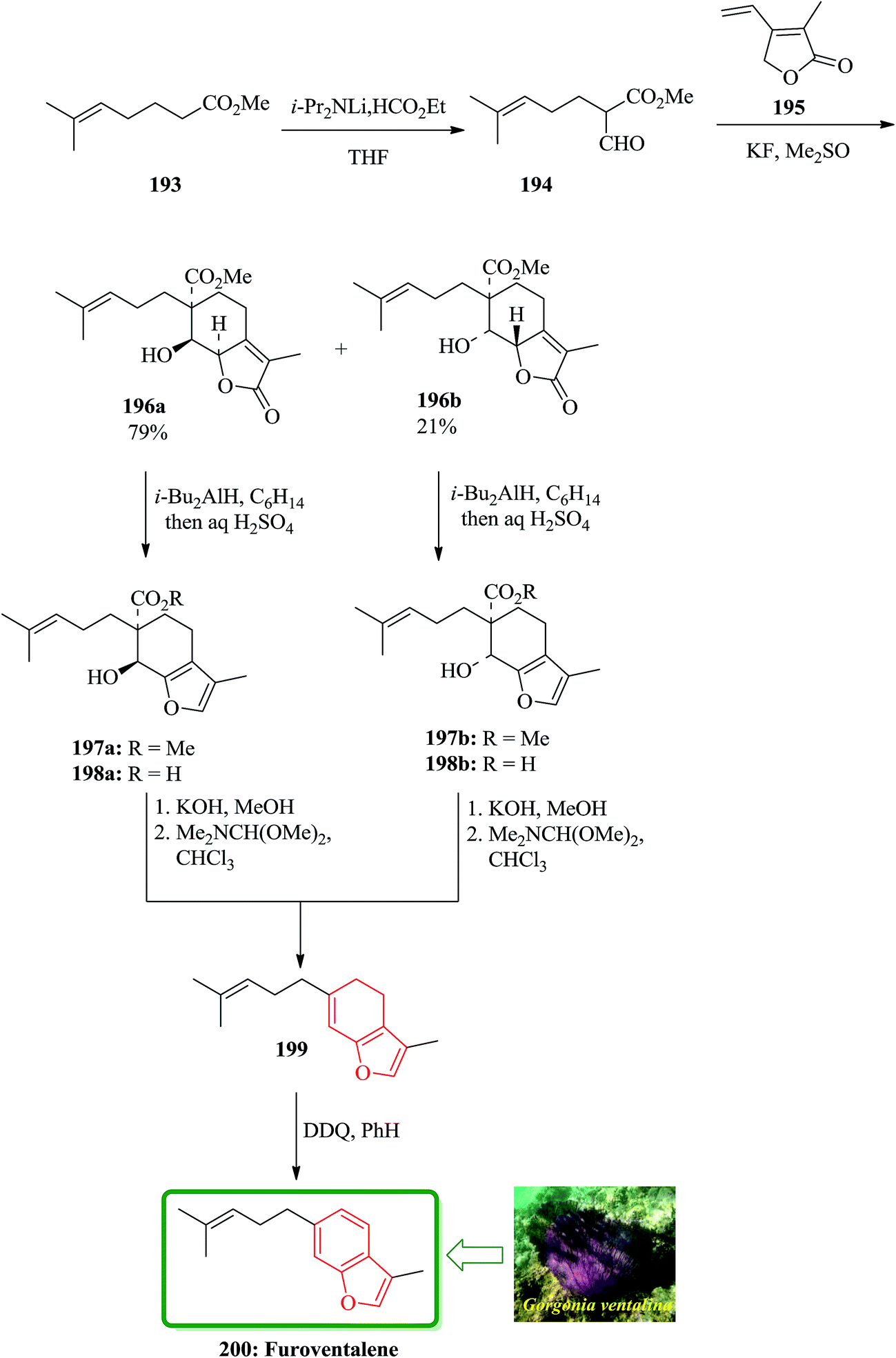 | ||
| Scheme 45 Total synthesis of furoventalene 200. | ||
Khellin 207 is one of several furochromones that was isolated from Ammi visnaga L., a perennial herbaceous plant that cultivates desolate in several Eastern Mediterranean countries.211,212 The total synthesis of 207 was started from 3-furoic acid 201. Regiospecific introduction of the (dimethylamino)-methylene unit adjacent to the ketone was achieved via reaction of a neat mixture of 202 and N,N-dimethyformamide dimethyl acetal (DMF-DMA) (1:1.1) in the presence of TsOH at ambient temperature in couple of days. The desired acyclic precursor 203 (80%) as yellow oil was obtained after chromatography. The latter was subjected to Dieckmann cyclization (potassium tert-butoxide/THF/-78 °C) followed by acid treatment (HCl/THF/4 h) to give the fully substituted benzofuran 204 in 75% yield. Methylation (CH3I/K2CO3/18-crown-6/PhH/A) of 204 yielded the highly versatile benzofuran intermediate 205 (90%). The latter was converted to compound 206 in two steps. Compound 206 is an intermediate which, is converted to the desired natural product khellin 207 (Scheme 46).213
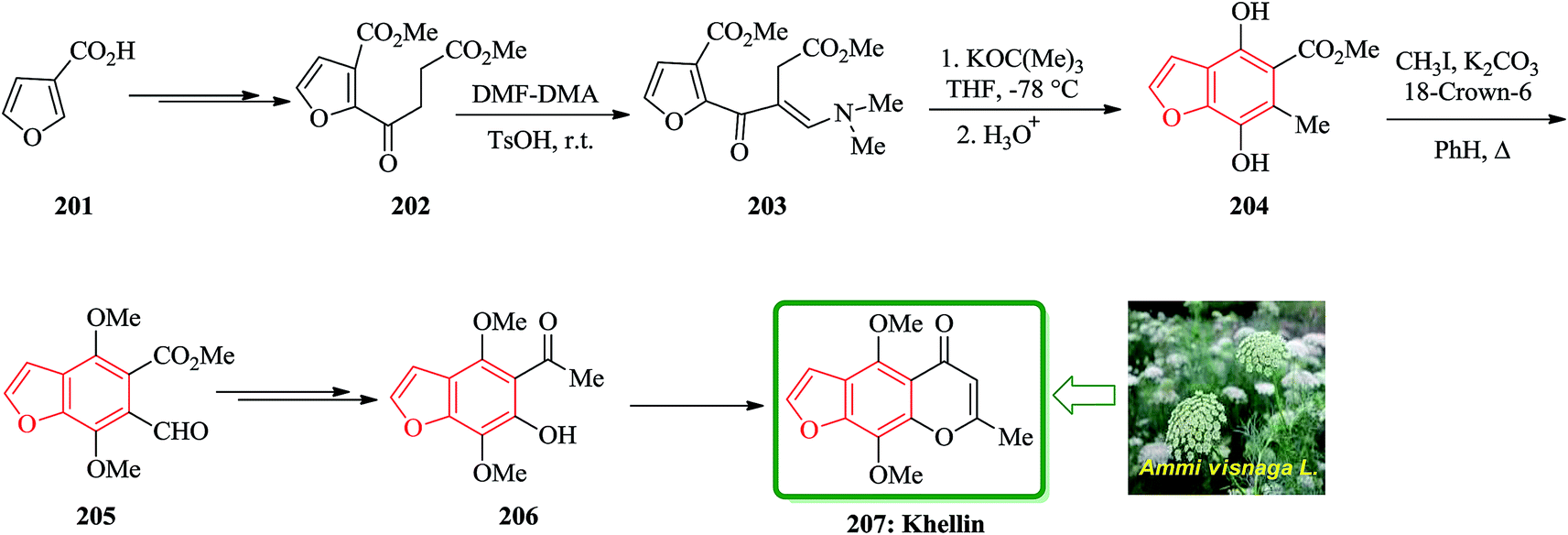 | ||
| Scheme 46 Total synthesis of khellin 207. | ||
Pongamol has been isolated from Pongamia glabra,214 Tephrosiapurpurea.215 T. IanceolotaPongamia glabra216 and T. hamiltonii.217 The structure of pongamol was established as the enol by X-ray crystallography.218 Lanceolatin B was isolated from P. pinnata219 and T. purpurea.220 A new method for dipolar cycloaddition of diazocyclohexane-1,3-diones, leading to benzofuran derivatives has been applied to the tota1 synthesis of natural products from Tephrosia and Pongamia. Total synthesis of pongamol 211 and lanceolatin B 212 started from 6,7-dihydrobenzofuran-4(5H)-one 208, which initially reacted with acetone, DME in the presence of NaH or KH to give compound 209 upon carboxylation and then subjected to dehydrogenation to be converted into methoxy derivative 210. The latter was converted into the desired natural products 211 and 212 via two different reaction routes. The spectroscopic properties of this synthetic materials agreed well with those obtained from natural products reported in the literature (Scheme 47).221
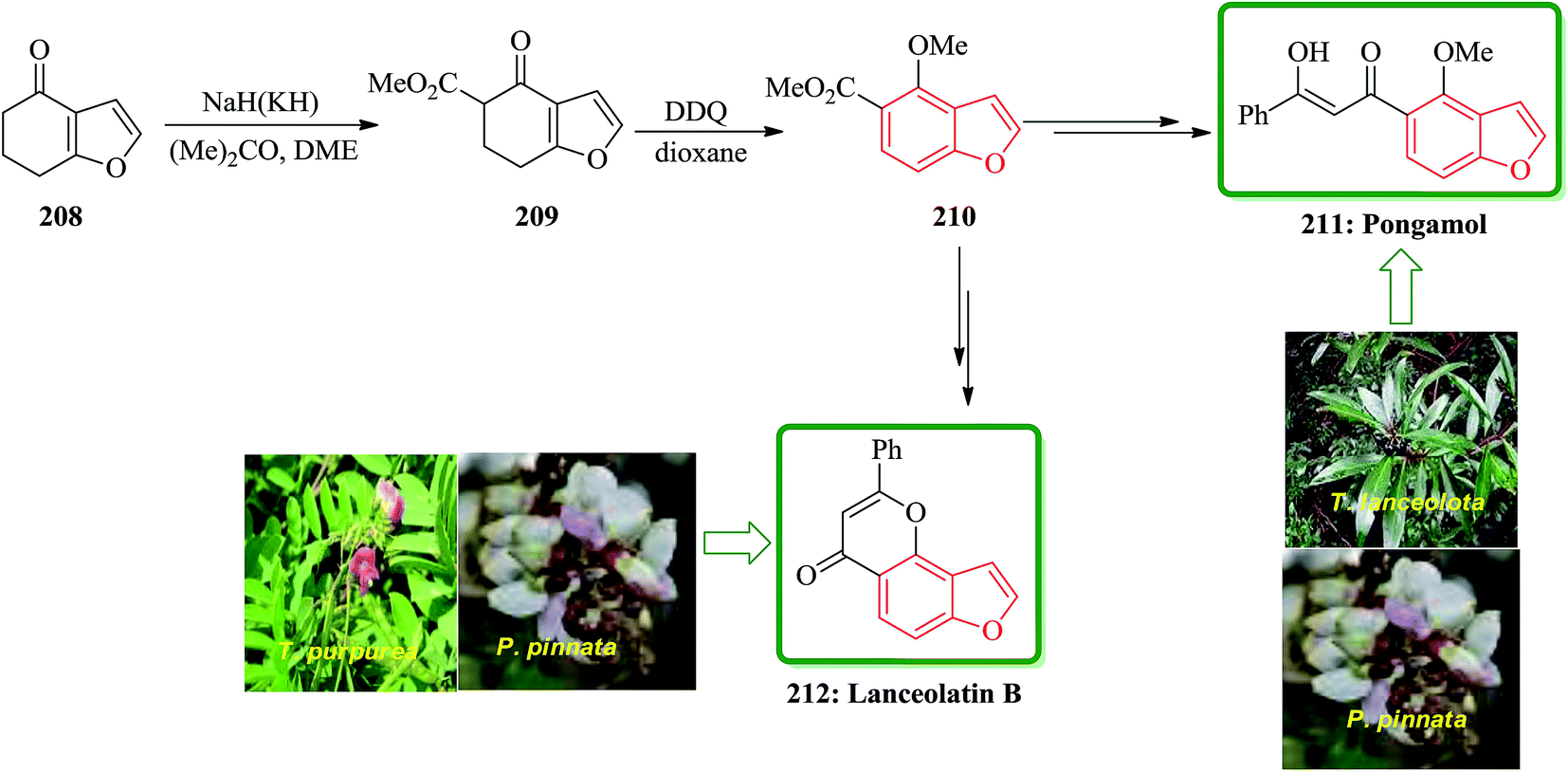 | ||
| Scheme 47 Total synthesis of pongamol 211 and lanceolatin B 212. | ||
Total synthesis of garcifuran B 217, which is the constituents of plants of the Garcinia genus (Guttiferae) was achieved and reported. This plant has been used in traditional herbalmedicines in areas of southeastern Asia, shown later to contain a number of toxic components.222 Garcifurans A (also known as garcinol) and B were isolated from the roots of Garcinia kola Heckel collected in Nigeria by Niwa and co-workers in 1994.223 The total synthesis of garcifuran B 217 started with 5-bromo-2-hydroxybenzaldehyde 213 which was reacted with BrCH(CO2Et)2 in the presence of K2CO3 to provide benzofuran 214 and after 2 steps is converted into 5-bromobenzofuran 216. The reactive trimethylstannyl 215 reacted smoothly with 5-bromobenzofuran 216 to give the desired benzofuran in 44% yield, which was then deprotected by heating under reflux in AcOH/H2O to afford the natural product, garcifuran B 217 (Scheme 48).224
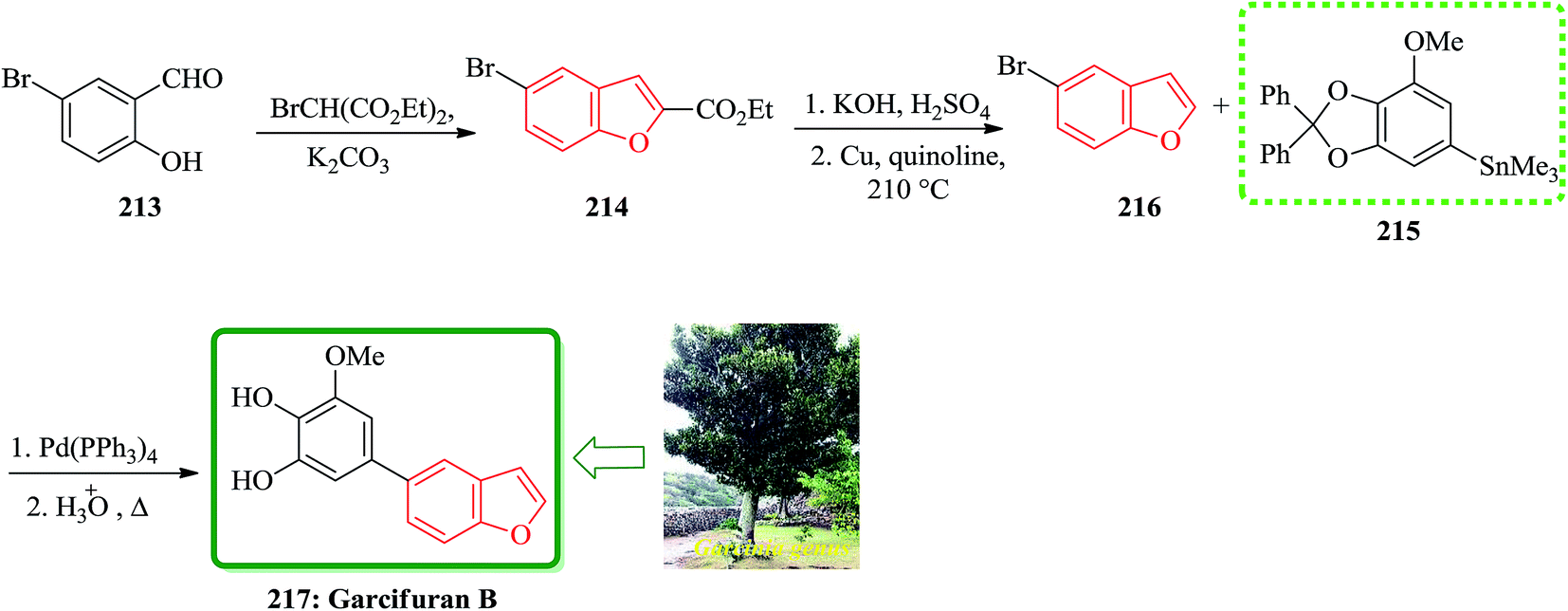 | ||
| Scheme 48 Total synthesis of garcifuran B 217. | ||
Benzofuran derivative 220 was isolated from various yeasts as an antioxidant225 and its structure was determined by degradation studies.226,227 Total synthesis of an antioxidant 220 having a benzofuran skeleton was achieved in four steps via the palladium(0)-catalyzed cross-coupling reaction. Some derivatives of 220 demonstrate antioxidative activity. Scheme 49 illustrates the synthetic route for the synthesis of 220. Regioselective bromination of the known benzodioxole derivative 218 (ref. 228) along with several other steps afforded arylbenzofuran 219 in an excellent yield. The latter is hydrogenated and upon deprotection under the normal conditions gave the desired benzofuran 220 in a 62% overall yield. Remarkably, physicochemical data of the synthetic product were in good agreement with those reported values (Scheme 49).229
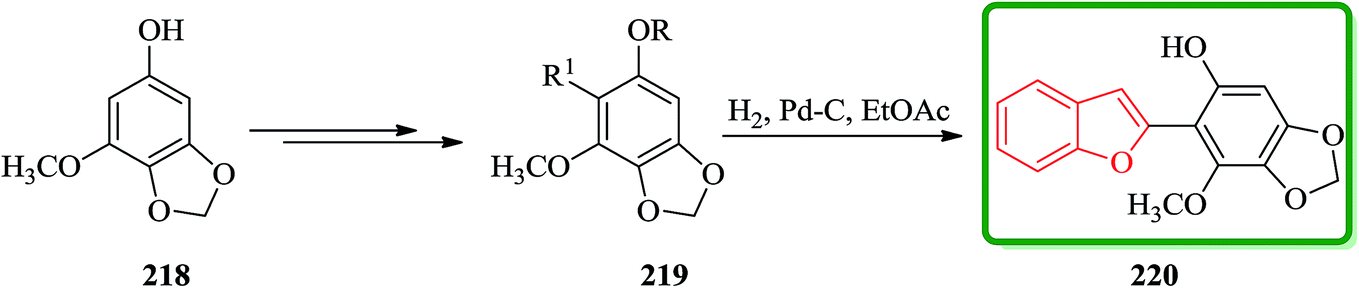 | ||
| Scheme 49 Total synthesis of benzofuran derivative 220. | ||
Novel antibacterial substance, AB0022A, was isolated from the cellular slime mold Dictyostelium purpureum K1001, that it inhibited the growth of Gram-positive bacteria. Because AB0022A was a highly substituted aromatic compound, its structure could not be determined based on only physicochemical and spectral data. Therefore, a dehalogenated derivative from AB0022A was prepared and deduced that its structure is actually l,9-dihydroxy-3,7-dimethoxy-2-hexanoyl-4,6,8-trichlorodibenzofuran. The synthetic product was identical to naturally occurring AB0022A. The strategy for synthesizing AB0022A 224 was as follows. It was selected 1,3,7,9-tetramethoxydibenzofuran 223, which is known to be synthesized from 1,3,5-trimethoxybenzene 221 in three steps.230 At first, they tried to synthesize 1,3,7,9-tetramethoxydibenzofuran 223. Iodination of 1,3,5-trimethoxybenzene 221 and Ullmann coupling gave 2,2′,4,4′,6,6′-hexamethoxybiphenyl 222. Cyclization of this biphenyl under the reported reaction conditions (57% HI aq., reflux) gave a complex mixture, which was methylated with iodomethane to give 1,3,7,9-tetramethoxydibenzofuran 223 in low yield. Finally, the latter was converted after several steps to natural product AB0022A 224 (Scheme 50).231
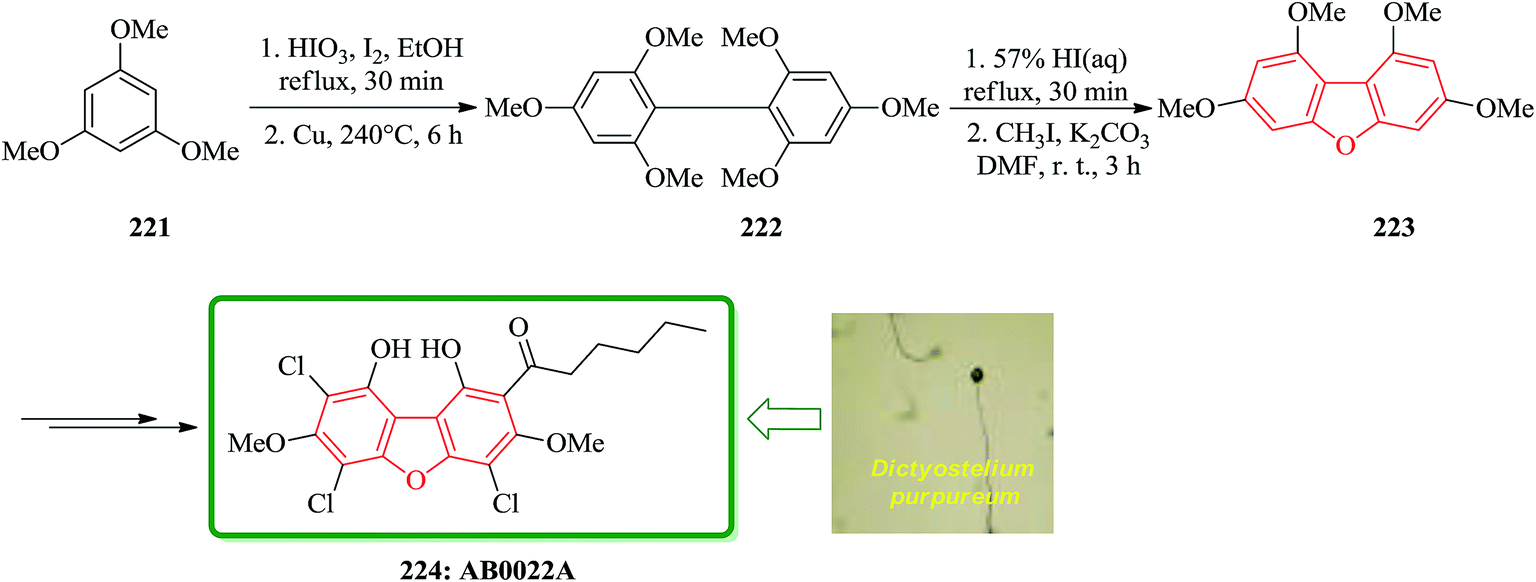 | ||
| Scheme 50 Total synthesis of AB0022A 224. | ||
Frondosins A–E were recently isolated from the sponge Dysidea frondosa. These derivatives, which bear a causal relationship to one another, inhibit the binding of IL-8 to its receptor in the low micromolar range.232 IL-8 promotes the accumulation and activation of neutrophils and has been implicated in a wide range of acute and chronic inflammatory disorders.233 Commercially available 5-methoxysalicylaldehyde 19 was converted into 225. After several steps and under basic conditions frondosin B 226 was produced in pure form and free of double bond isomers (Scheme 51).234
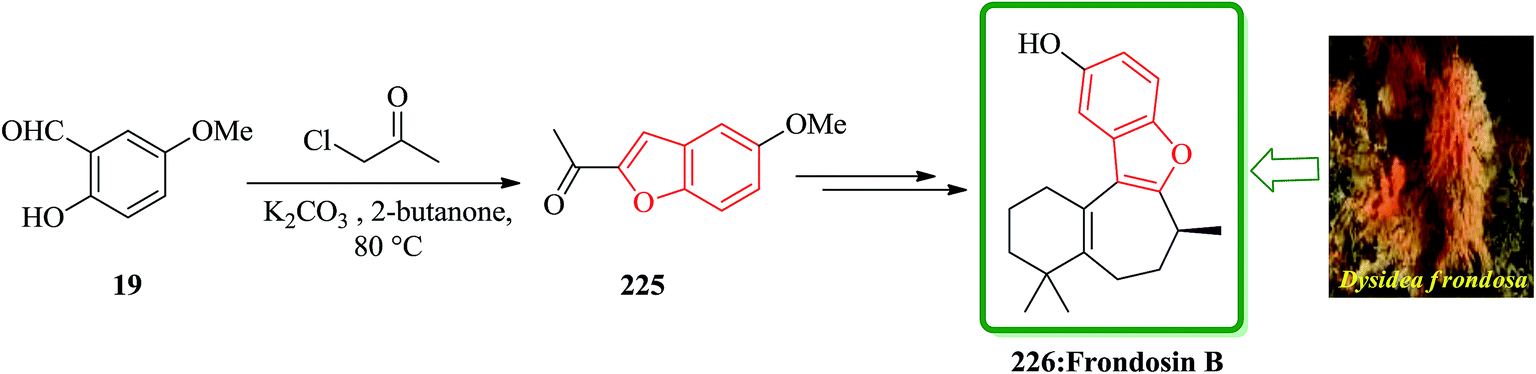 | ||
| Scheme 51 Total synthesis of frondosin B 226. | ||
Chemical examination of the diethyl ether extract from the liverwort Corsinia coriandrina led to the isolation and characterization of a new 2-arylbenzofuran product so-called corsifuran A. Cycloaddition between 4-methoxystyrene 227 and p-quinone 228 catalyzed by ferric(III)chloride hexahydrate in acetonitrile gave 5-hydroxy-2-(4-methoxyphenyl)-2,3-dihydrobenzofuran 229 in moderate yield, which was proved being identical to corsifuran B. Methylation of 229 afforded corsifuran A 230, which showed MS and 1H-NMR data as same as to the natural product isolated from C. coriandrina. Upon to dehydrogenation of 230 using 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) in dioxane corsifuran C 231 was obtained (Scheme 52).235
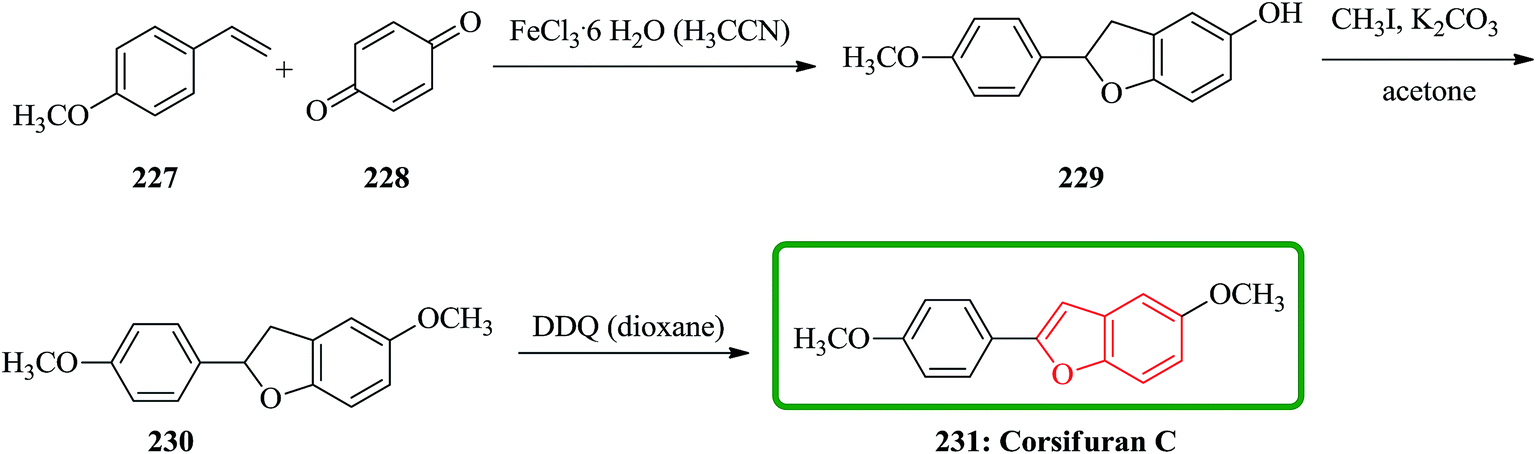 | ||
| Scheme 52 Total synthesis of corsifuran C 231. | ||
Natural 2-acetylbenzofurans calebertin 235a, caleprunin A 235b, and caleprunin B 235c have been isolated from Calea species.236 Caleprunin B 235c had been previously isolated from Eupatorium sternbergianum and called eupatarone.237 These naturally occurring compounds 235a–c were synthesized in an acceptable overall yields. These benzofurans were also provided by direct treatment under MW irradiation of the precursor 1-aryloxypropan-2-ones 233a–c with DMFDMA, with subsequent addition of the catalyst, providing a route that was literally one-step shorter. For the synthesis of 2-acetylbenzofurans 235, first the corresponding 1-aryloxypropan-2-ones 233 were prepared via a base promoting Williamson reaction between the substituted phenols 232, and chloroacetone in refluxing acetone. Then, a series of compounds 234a–f was synthesized in high yields by the reaction of the corresponding 1-aryloxypropan-2-ones 233a–f with DMFDMA. The intramolecular cyclization of 3-aryloxy-4-dimethylamino-3-buten-2-ones 234 gave compound 235a–c. In this way, natural benzofurans 235a–c were provided in good overall yields using phenols 232a–c in a three-step syntheses in which calebertin 235a was obtained in 35%, caleprunin A 235b in 37%, and caleprunin B 235c in 48% yield (Scheme 53).238
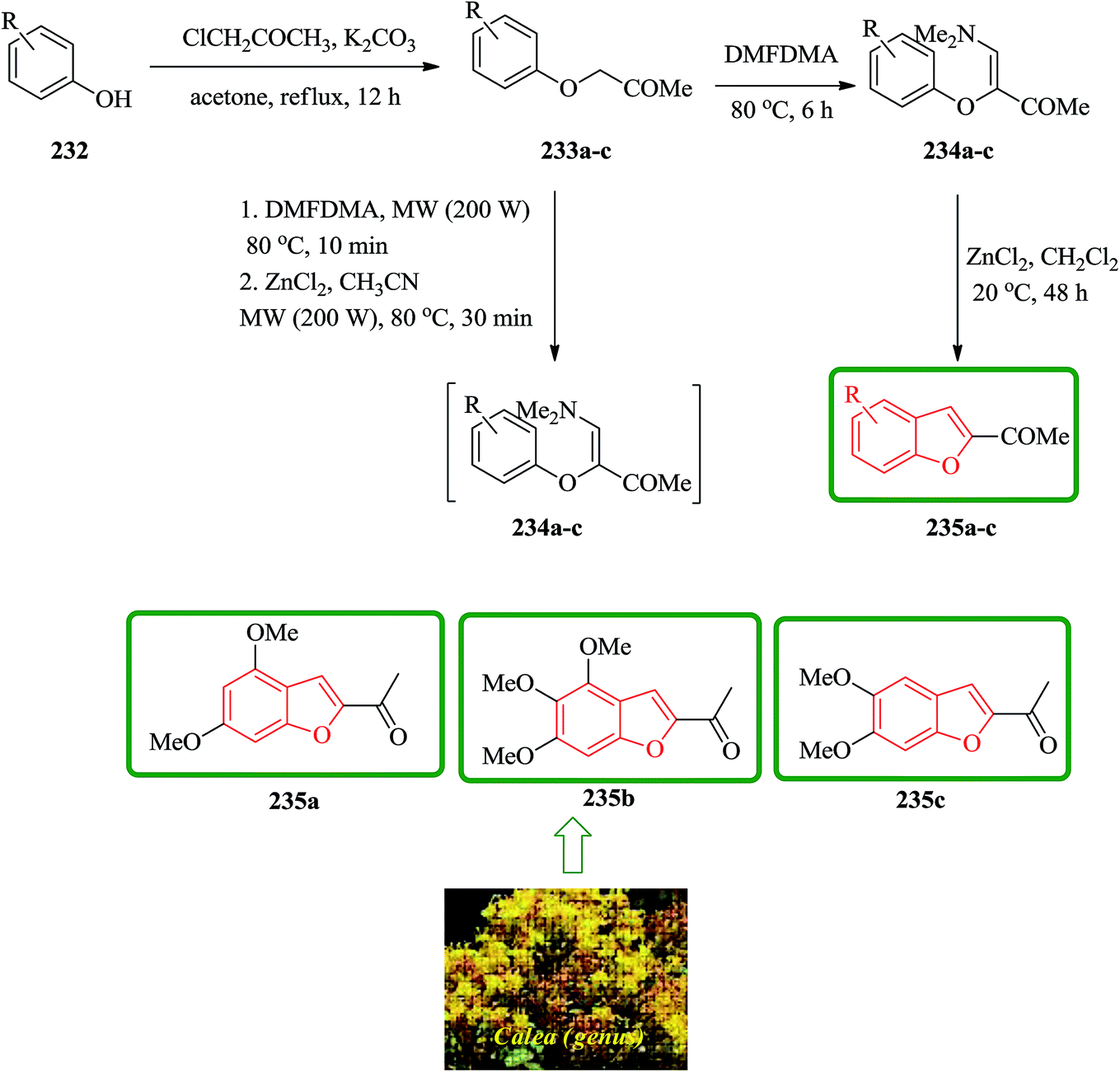 | ||
| Scheme 53 Total synthesis of calebertin 235a, caleprunin A 235b, and caleprunin B 235c. | ||
Furocoumarins 240a are natural tricyclic compounds exhibiting a wide range of biological properties.239 Linear furocoumarins are well-known photosensitizing drugs for the treatment of a number of skin diseases such as psoriasis, vitiligo, mycosis, and eczema,240,241 as well as fungal, viral, and bacterial infections.242,243 Recently, it was reported that some linear furocoumarins were applied to the treatment of cutaneous T-cell lymphoma.244 More notably, they were found to have potential utility in the treatment of human immunodeficiency disease245 and in the prevention of organ transplant rejection.246 A new and efficient method for the synthesis of linear furocoumarins was reported by the Nef reaction.247 This strategy has also been applied to the preparation of four additional benzofuran derivatives. A mixture of 2,4-dihydroxybenzaldehyde and nitromethane was stirred in AcOH in the presence of NH4OAc to give 5-hydroxy-2-(2-nitroethenyl)phenol 236 (ref. 248) in 84% yield. The unsaturated compound 236 was then converted into the desired product 237 (ref. 248) in 87% yields by treatment with NaBH4 in i-PrOH-THF (1:4) at room temperature. It is well known that a nitro group can be easily converted to a carbonyl by the Nef reaction. 4-(2-Nitroethyl)benzene-1,3-diol 237 was thus subjected to the Nef reaction. Interestingly, the predicted aldehyde 238 was not obtained, while the required benzofuran-6-ol 239,249 was produced directly in a one-pot reaction under the reaction conditions. It is visualized that benzofuran-6-ol 239 could produce the intermediate 238 via an intramolecular cyclocondensation under Nef conditions. Based on this finding, a major attempt was there after made to modify the Nef reaction conditions aiming to improve the yield of benzofuran-6-ol 239, which is the key intermediate for the synthesis of diversified furocoumarins 240a (Scheme 54).250
 | ||
| Scheme 54 Total synthesis of diversified furocoumarins 240. | ||
The dibenzofuran-1,4-dione core is found being present in many naturally occurring compounds, some showing interesting biological activities. Some of them are cytotoxic popolohuanone E,251 antipruritic balsaminone A252 and violet-quinone.253 An oxidative cyclization of quinone-arenols 242 resulted in the construction of benzofuran derivatives 243 containing 1,4-dibenzofuran core. The oxidative cyclization was employed as a part of the total synthesis of violet-quinone 244. The quinonearenol 242 was easily synthesized from 4,5-dimethoxy-7-methylnaphthalen-1-ol 241 via a two-step sequential reaction. Relied on, these back grounds, the oxidative cyclization of quinone-arenols 242 was conducted by using benzoquinone 228 as an efficient oxidant in the presence of molecular oxygen, giving raise in 243 in satisfactory yield. Ultimately, MgBr2-iodide-catalyzed selective demethylation of the C4– and C11–OMe motives of 243 gave the desired target violet-quinone 244 in high yield (Scheme 55).254
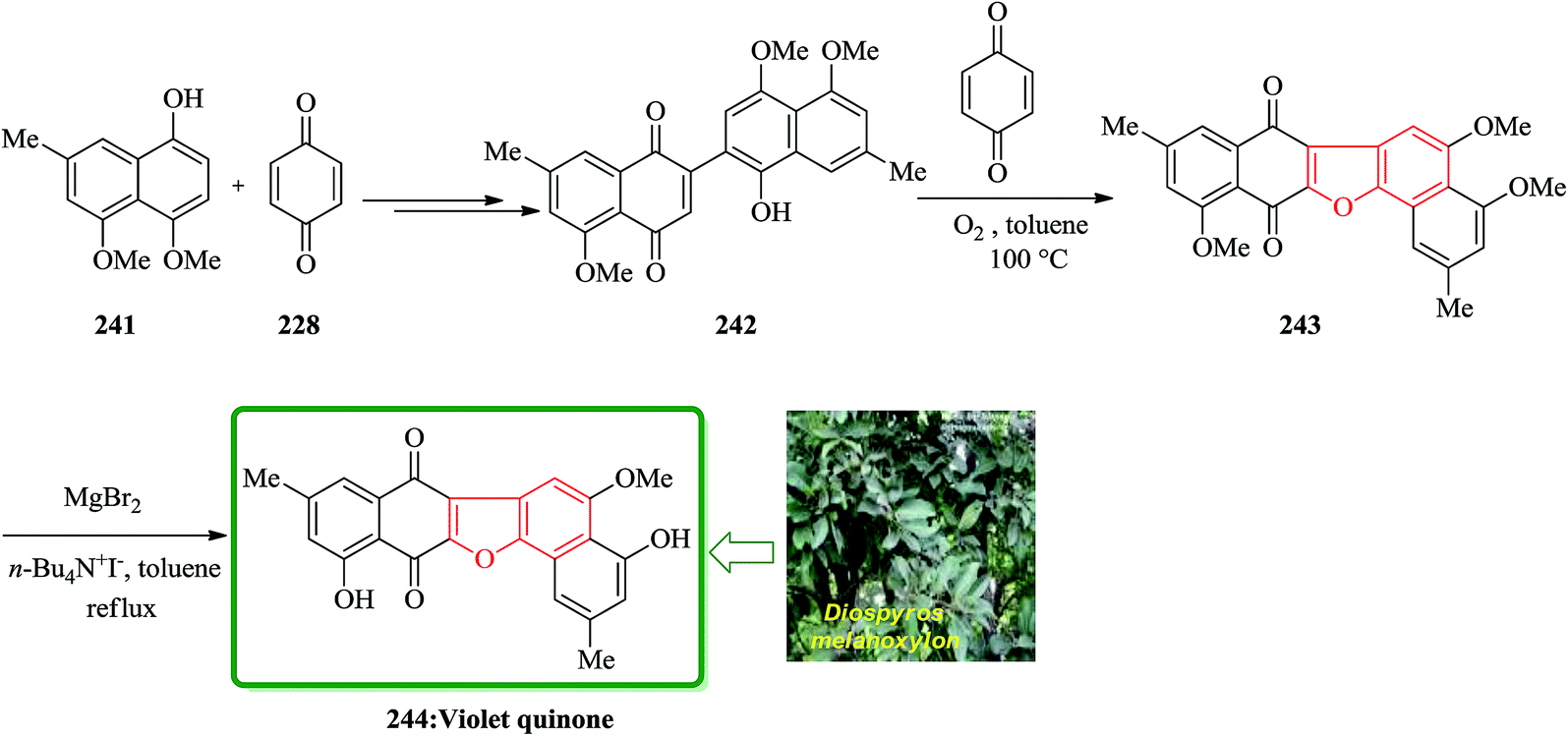 | ||
| Scheme 55 Total synthesis of violet-quinone 244. | ||
Erypoegin H 251 is the most active of lavonoid isolated from the roots of this ornamental plant. It is not only exhibits a broad spectrum of activity against Gram positive bacteria in general, but also exhibits a significant and uniform activity against a panel of 249 different MRSA strains and vancomycin-resistant enterococci.255 The synthetic venture commenced with the di-iodination of resorcinol 247 (ref. 256) followed by consecutive attachment of a pivaloyl and a trimethylsilylethoxymethyl group. The resulting crude product from 250 was subjected to an intramolecular etherification under standard conditions to complete the construction of tetracyclic framework of erypoegin H 251, which was obtained in a respectable 28% yield over the nine steps of the longest linear sequence. The resulting compound 249, upon exposure to catalytic amounts of PtCl2 in toluene under a CO atmosphere,257,258 underwent a clean cycloisomerization with the formation of the desired benzofuran derivative 250. This reaction was best performed in the presence of powdered molecular sieves to sequester traces of water that might protonate the putative organo platinum intermediate of type C and/or D and hence reduce the efficiency of the O → C shift. Under these optimized conditions, the cycloisomerization of 249 proceeded exceedingly well and afforded 250 in 84% yield on a multi-gram scale (Scheme 56).259
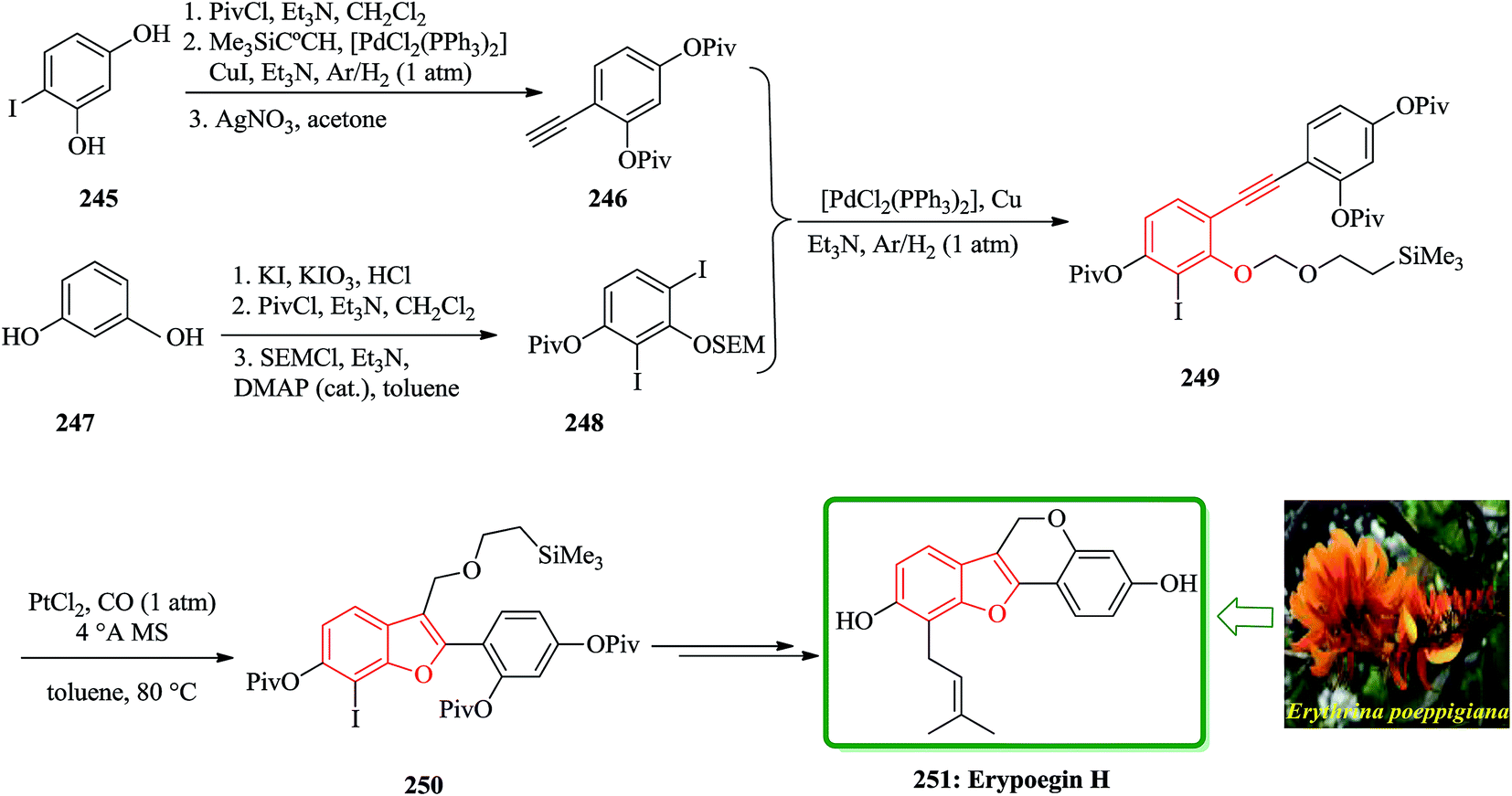 | ||
| Scheme 56 Total synthesis of erypoegin H 251. | ||
The study on the phytoalexins of cowpea, Vigna unguiculata (L.) Walp, showed that a natural product antifungal so called vignafuran 255 which has benzofuran moiety in its structure.260 Interestingly, the total synthesis of this naturally occurring compound was accomplished via an efficient one-pot manner. In this sequential approach for the formation of the benzofuran moiety, aryl halides protected iodophenols and carbinol-based acetylene sources were employed. The sequence involved alternating palladium-catalyzed Sonogashira couplings/deprotection and ring closing step. Initially, a suitable O-methyl-iodoresorcinol was silylated to prepare the required corresponding aryl halides 252.261 In a suitable vessel 252, reacted with 1-ethynyl-cyclohexanol 253 and catalytic amounts of suitable Pd catalyst under optimized reaction conditions. The progress of this reaction was monitored which upon its completion, potassium hydroxide, compound 254 and small amount of catalyst were added to the reaction mixture. It is presumed that the reaction gives the intermediate diarylacetylene, which was transformed to vignafuran 255 upon treatment with tetrabutylammonium fluoride. This achievement is a unique example of total synthesis of natural products via one pot manner, attractively showing the value of the ‘one-pot’ cascade Sonogashira coupling based strategy (Scheme 57).262
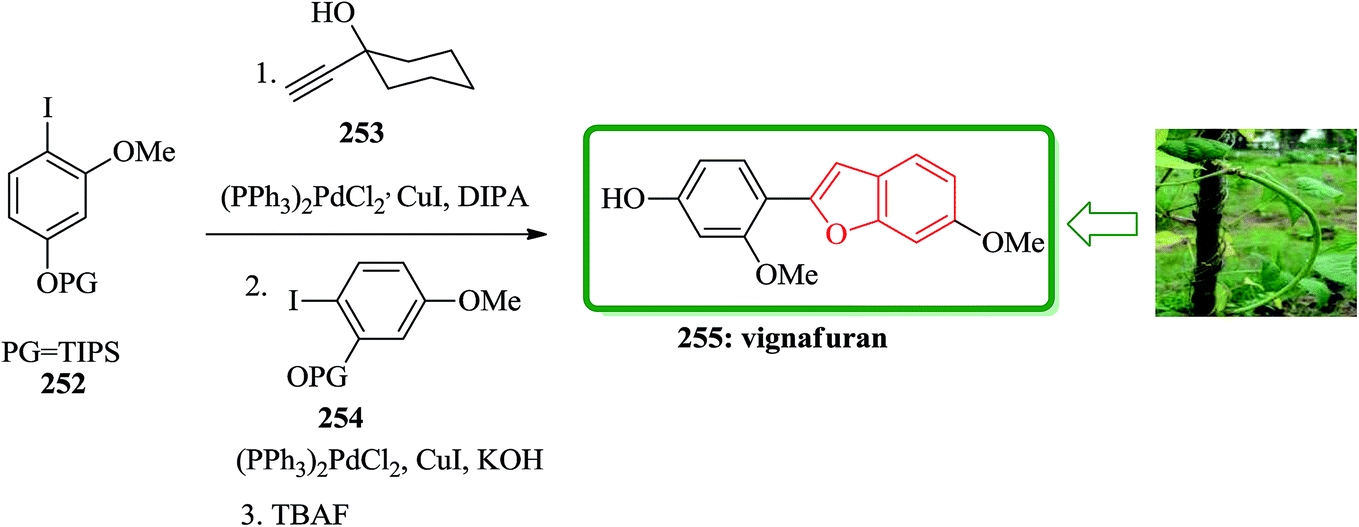 | ||
| Scheme 57 Total synthesis of vignafuran 255. | ||
The first total synthesis of glycyrol, isolated from glycyrrhizae radix, with a unique skeleton of a benzofuran coumarin was reported in 2008. Glycyrrhizae radix is a traditional medicine in the East Asia, and contains biologically active natural products such as glycyrrhizin, glycyrol, glycycoumarin, and liquoric acid.263 Glycyrol has antibacterial activity against upper airway respiratory tract pathogens.264 The key steps are Smiles rearrangement and selective introduction of prenyl and O-methyl groups. Preparation of O-benzyl-(diacetoxyiodo)arene 257 as a Smiles rearrangement precursor for the construction of benzofuran coumarin had unexpected difficulties. Benzylation of commercially available 2-iodophenol was achieved and after several steps, a crude 1-benzyloxy-3-(diacetoxyiodo)benzene 257 was provided. However, 1-benzyloxy-3-(diacetoxyiodo)benzene 257 was more unstable than commercially available 3-methoxy-1-(diacetoxyiodo)benzene and decomposed within one day, even with refrigeration. It was guessed that the (diacetoxyiodo)benzene is likely to be an oxidizing agent and the benzylic position could be susceptible to this reagent, although the reactivity of (diacetoxyiodo)benzene is not so powerful as common oxidizing agents. Fortunately, a base-catalyzed condensation of 4-hydroxycoumarin 259 with freshly prepared 1-benzyloxy-3-(diacetoxyiodo)benzene 257 successfully yielded an iodiumacetate salt 260, which was directly converted to 2-iodo-4-phenoxycoumarin 261 in 87% yield by refluxing in DMF via Smiles rearrangement. The palladium-mediated intramolecular coupling reaction of vinyl iodide with the phenyl group in 261 was readily achieved by using palladium(II) acetate and triethylamine in refluxing toluene to provide the crude benzofuran 262. Finally, simultaneous deprotection of the MOM and benzyl groups with N,N-dimethylaniline and aluminum chloride in refluxing methylene chloride, followed by careful purification on a silica gel, furnished the desired target material glycyrol 263 in 68% yield in two steps (Scheme 58).265
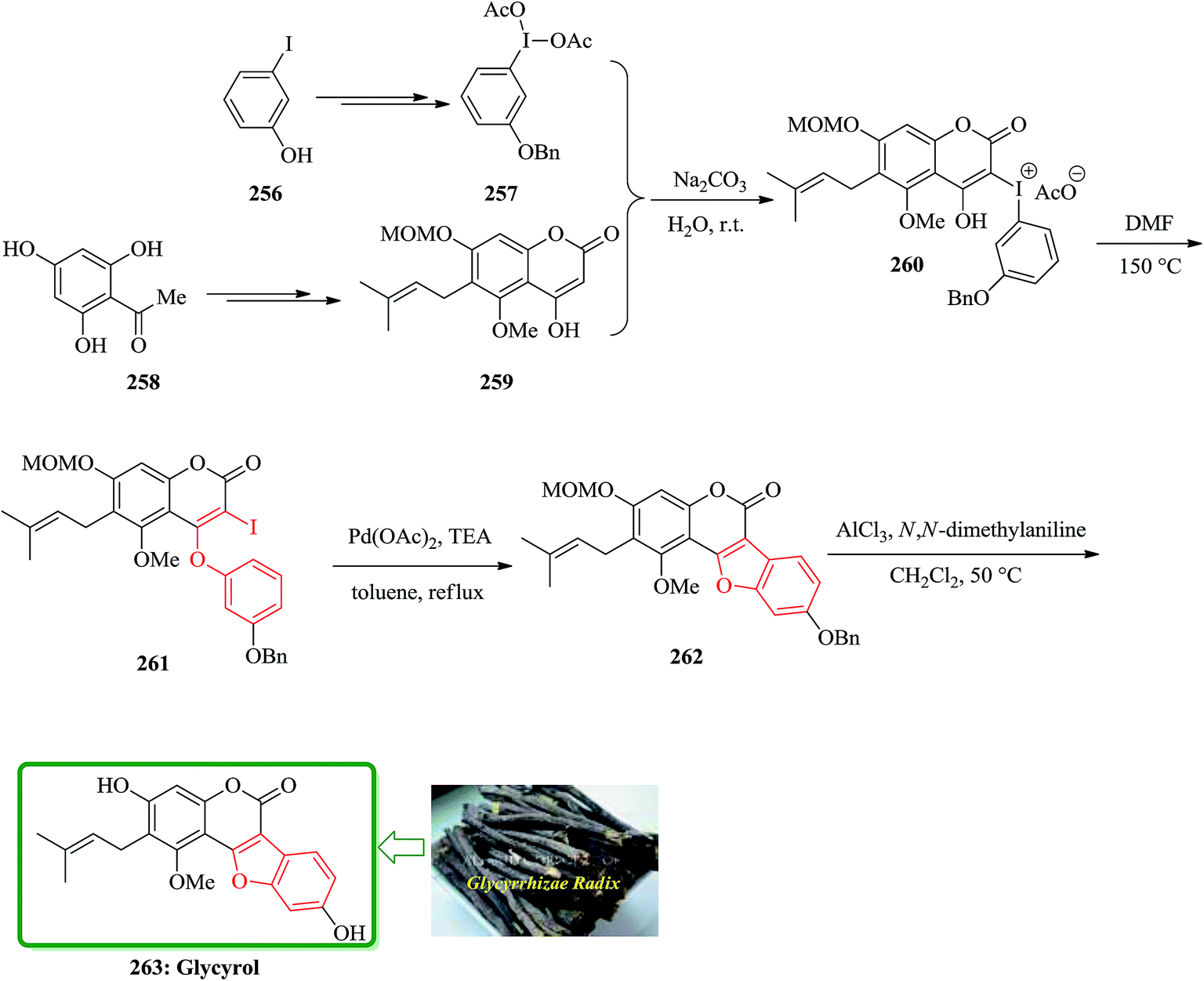 | ||
| Scheme 58 Total synthesis of glycyrol 263. | ||
Gnetuhainin B 272 was initially isolated from the lianas of Gnetum hainanense by Lin and co-workers.266 The structure of viniferifuran as the congerer of gnetuhainin, extracted from Vitis vinifera ‘Kyohou’ was fully characterized based on the widespread 1H-NMR and 13C-NMR data and elemental analysis and reported by Niwa.267 On the other hand, in 1998, Boyd research group based on extensive spectroscopic data revealed the structures of two novel oligostilbenes, malibatols A and B, which were long ago isolated from the extract of the leaves of Hopea malibato.268 Malibatols A and B were found showing cytotoxicity to the host cells (CEM SS) in an extensively antiviral test. Significantly, an oxidized analogue of malibatol A, has an oxidized analogue so-called shoreaphenol or hopeafuran. It was initially isolated from the bark of Shorea robusta and the stem wood of Hopea utilis.269 Oligostilbenes270 are a typical of highly oxygenated naturally occurring compounds, which bear more than two stilbene units. In the total synthesis of these compounds a region selectively Bi(OTf)3-catalyzed cyclodehydration was performed for the facile access to 3-arylbenzofuran moiety. Consequently, for the introduction of aryl group at the C-2 position of benzofuran a Pd-catalyzed direct C–H activation of benzofuran followed by cross-coupling with aryl halide is a key reaction. In an approach towards the total synthesis of these analogues, Chakraborty and co-workers synthesized aryloxyketone 266, which was in turn can be readily synthesized from the treatment of phenol 264 (ref. 271) with α-bromoketone 265 (ref. 272) mediated by K2CO3. Upon the treatment of ketone 266 with BCl3 the desired benzofuran 267 was obtained in satisfactory yield. On the other hand, the ester group in 268 was transformed into formyl group through a two-step sequential reaction including DIBAL reduction/Dess–Martin oxidation273 in excellent overall yield. Upon Horner–Wadsworth–Emmons type olefination of 269 using diethyl 4-methoxybenzylphosphonate gave 272 in virtually quantitative yield. For the construction of the seven-membered ring implanted in malibatol A 270 and shoreaphenol or hopeafuran 271, the epoxide ring opening by nucleophilic attack of the neighboring aromatic moiety was successfully conducted (Scheme 59).274
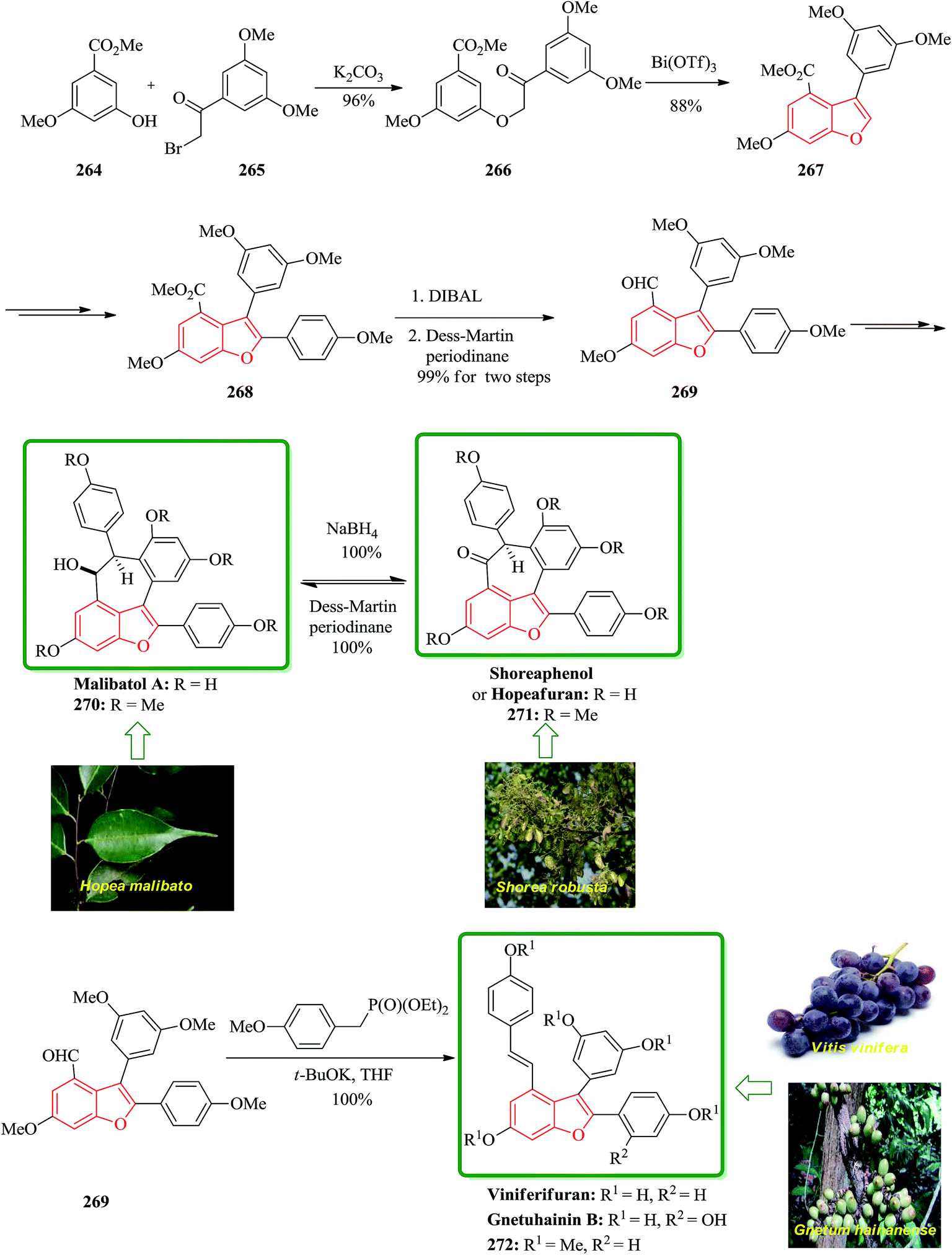 | ||
| Scheme 59 Total synthesis of natural products 270–272. | ||
The total synthesis of kynapcin-24, 279 was achieved in 12% overall yield from commercially available 3,4-dihydroxybenzaldehyde by a route in which the longest linear sequence is only 14 steps. Compound 279 was initially isolated from the Korean mushroom Polyozellus multiflex Murr Prolyl endopeptidase (PEP), a serine protease, is known to cleave a peptide substrate on the C-terminal side of a proline residue.275 Additionally, the PEP activity of Alzheimer's patients has been found to be significantly higher than that of the normal person.276 Recently Song and co-workers reported the synthesis of two novel PEP inhibitors, one of them is the benzofuran dimer kynapcin-24 279. Propeptin has inhibition similar to 279 that is a hydrophilic and large-molecular weight peptide, which may make it difficult to penetrate into the blood–brain barrier.
The key transformations in the total synthesis are copper-mediated and palladium-catalyzed coupling reactions of the iodide 3-iodo-5,6-diisopropoxy-2-[(tetrahydropyran-2-yloxy)methyl]benzofuran with the corresponding stannane 5,6-diisopropoxy-2-[(tetrahydropyran-2-yloxy)methyl]-3-(tributylstannyl)benzofuran, and a 5-endo-dig iodocyclization of a (hydroxyphenyl)propargyl ether. For the total synthesis of kynapcin-24 279, coupling of phenyl iodide 273 with protected propargyl alcohol 274 instead of methyl propynoate proceeded smoothly in dioxane under the copper-mediated palladium catalysis to give the desired 275 in excellent 96% yield. The latter is reacted with NIS in the presence of hydrazine hydrate to give benzofuran 276. The latter upon lithiation and quenching with tributylstannyl chloride provided stannane benzofuran 277 in 73% yields. Then 277 reacted with iodide 276 reacted under the copper-mediated palladium-catalyzed coupling to give dibenzofuran 278 in 72% yield. The latter was subjected to sequential deprotection, oxidation, and oxidation-esterification using pyridinium p-toluene-sulfonate, 2-iodoxybenzoic acid, and silver(I)oxide-thionyl chloride, providing the desired target 279 in 98% yield (Scheme 60).277
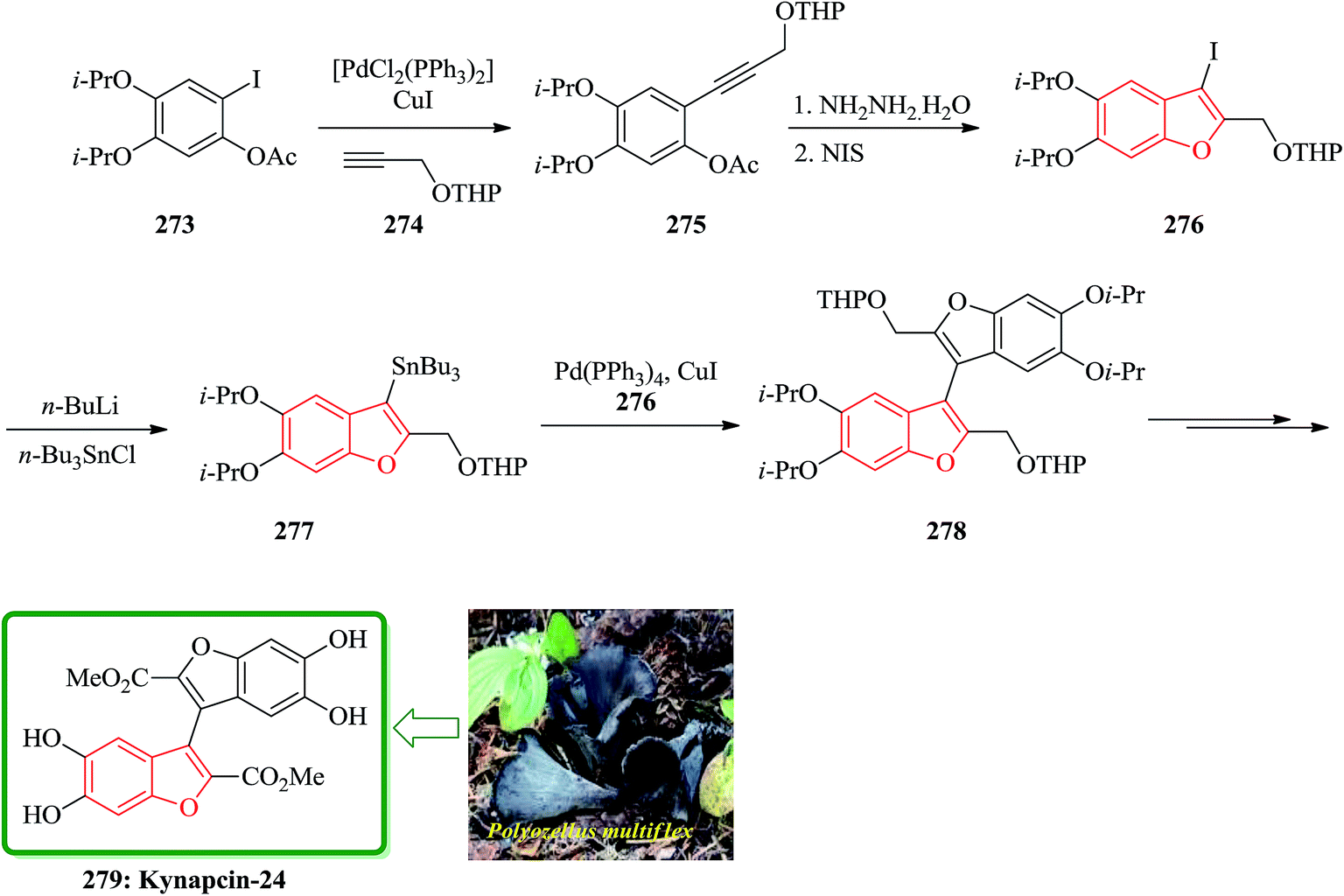 | ||
| Scheme 60 Total synthesis of kynapcin-24 279. | ||
(±)-Laetirobin 285 as a new cytostatic agent was isolated from the fruiting bodies of the fungus Laetiporus sulphureus and its structure was fully characterized.278 It was found, laetirobin has the potency to prevent tumor cell division (mitosis) and appealing automatic cell death (apoptosis). A brief and efficient total synthesis of laetirobin was achieved in 12% overall yield in six steps. In this approach, the total synthesis started from market purchasable 2,4-dihydroxyacetophenone 280. The latter was converted to 281 in several steps compound 281 was the reacted with protected dipropargyl alcohol to give the tosylate 283 sequential reactions involving (a) the double Sonogashira reaction of a bis(alkyne), (b) a highly efficient copper(I)-catalyzed construction of a bis(benzo[b]furan), and (c) the biomimetic [4 + 2] dimerization. The phenol 284 was synthesized by treatment of tosylate 283 with newly activated Mg in MeOH.279 The optimal conditions for such conversion is using of 25 mol% of copper(I) iodide under the conditions of a modified Stephens–Castro reaction. After several steps, phenol 284, was transformed into the desired natural product (±)-285 (Scheme 61).280
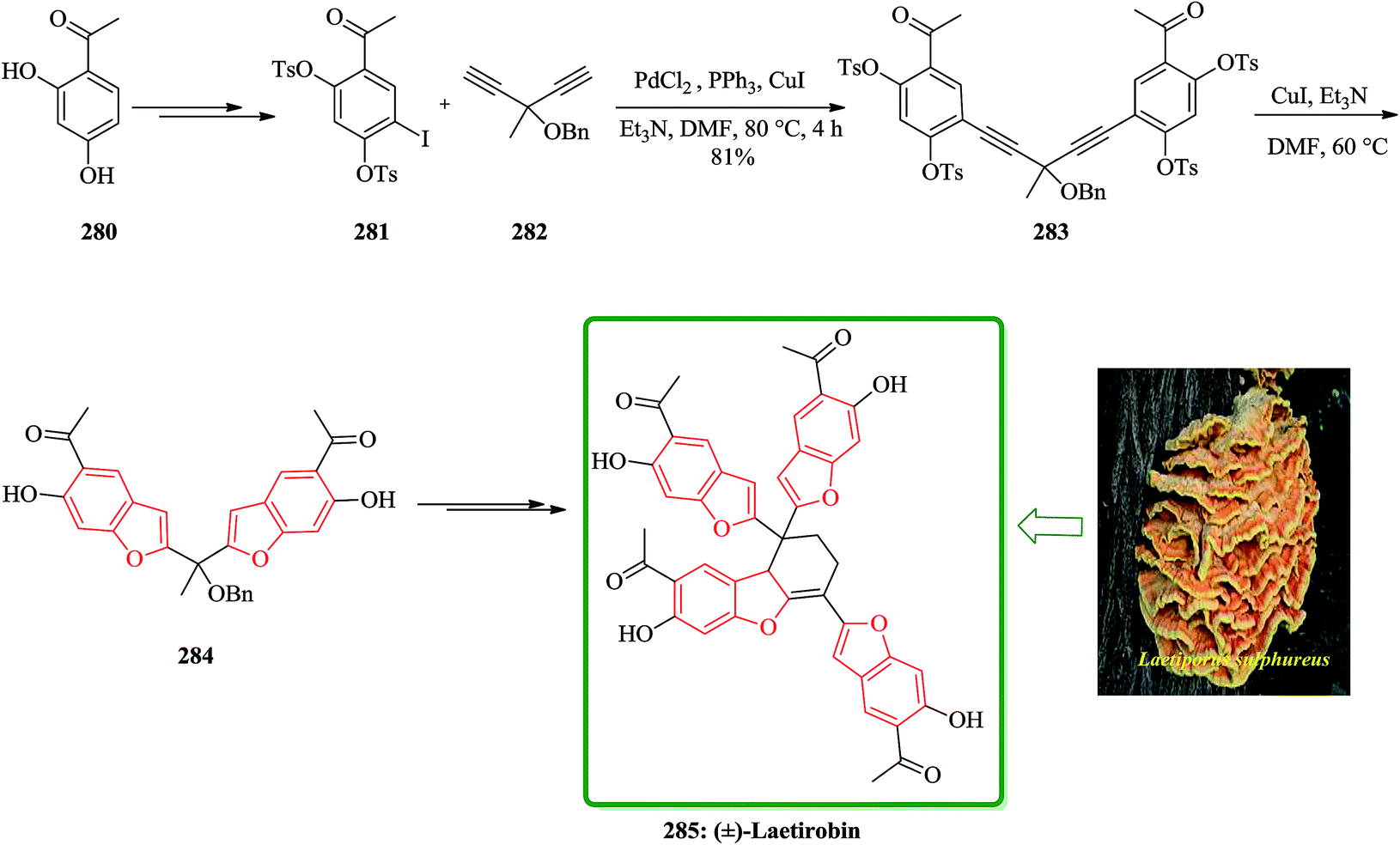 | ||
| Scheme 61 Total synthesis of (±)-laetirobin 285. | ||
Malibatol A 270 and shoreaphenol 271, are two dimeric resveratrol polyphenolic benzofurans which isolated initially from Hopea malibato and Shorea robusta, respectively.268,269 A flexible protocol for the synthesis of hexacyclic dimeric resveratrol polyphenolic benzofurans has been achieved and revealed in 2010. In this approach, firstly benzyl ethers 287 were synthesized from appropriate 286 in high yield. Then, benzofuran formed from keto benzyl ethers 287 was converted into a compound bearing benzofuran moiety 288 via a two-step reaction, which in general gives a satisfactory yields of the products (71–85% yield). In this procedure, when pentacyclic benzofuran 288 is used, the oxygen-substituted, seven-membered ring in the malibatol A 270 and shoreaphenol 271 are constructed. Therefore, in a one pot reaction, upon epoxidation of stilbene 288 using bromohydrin (NBS, NaOH), and subsequent treatment of the epoxide with BBr3 led to cyclization and inclusive demethylation gave racemic malibatol A 270 as a sole diastereoisomer in acceptable yield. Upon oxidation of malibatol A 270 in the presence of PDC shoreaphenol 271, is obtained, albeit in the moderate yield (Scheme 62).281
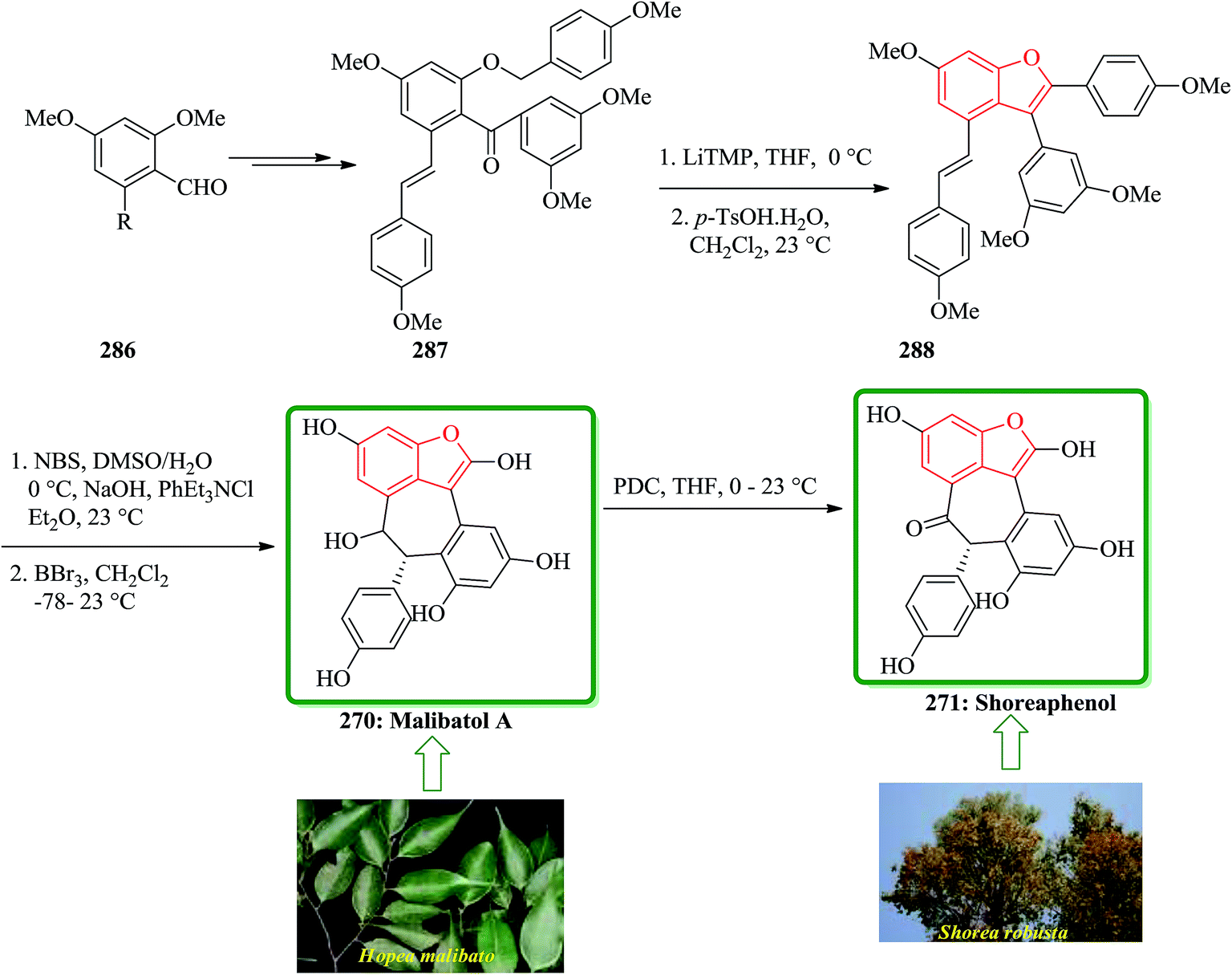 | ||
| Scheme 62 Total synthesis of malibatol A 270 and shoreaphenol 271. | ||
Syah and co-workers reported the isolation and characterization of a novel oligostilbenoid from the tree bark of Hopea mengarawan.282 This natural product exhibited potent immunosuppressive activity.283 As a matter of fact, several of oligomeric stilbenes have been isolated and recognized having divergent means of connectivity of their basic 1,2-diphenylethylene scaffold. Several remarkable biological functions of this family have been acknowledged comprising antibacterial antifungal, anti-inflammatory, and anticancer activities.284 A total synthesis of diptoindonesin G starts from readily available aryloxyketone 289 including one pot sequential cyclization/intramolecular Friedel–Crafts acylation reaction of aryloxyketone in cascade manner which gives compound 290 and 292 bearing benzofuran framework respectively. The latter upon treatment with BCl3 in CH2Cl2 undergoes regioselective demethylation to give the tetracyclic 6H-anthra[1,9-bc]furan-6-one G. In fact, treatment of 289 with BCl3, resulted in benzofuran 290 in excellent yield. The latter was subjected to Pd-catalyzed direct arylation285 to assemble an aryl group at the C2 position of the benzofuran286 unit of 290. Reaction of 290 under the conditions, previously reported for the synthesis of oligostilbenoids gave diptoindonesin G 293 in 18–22% yield (Scheme 63).287
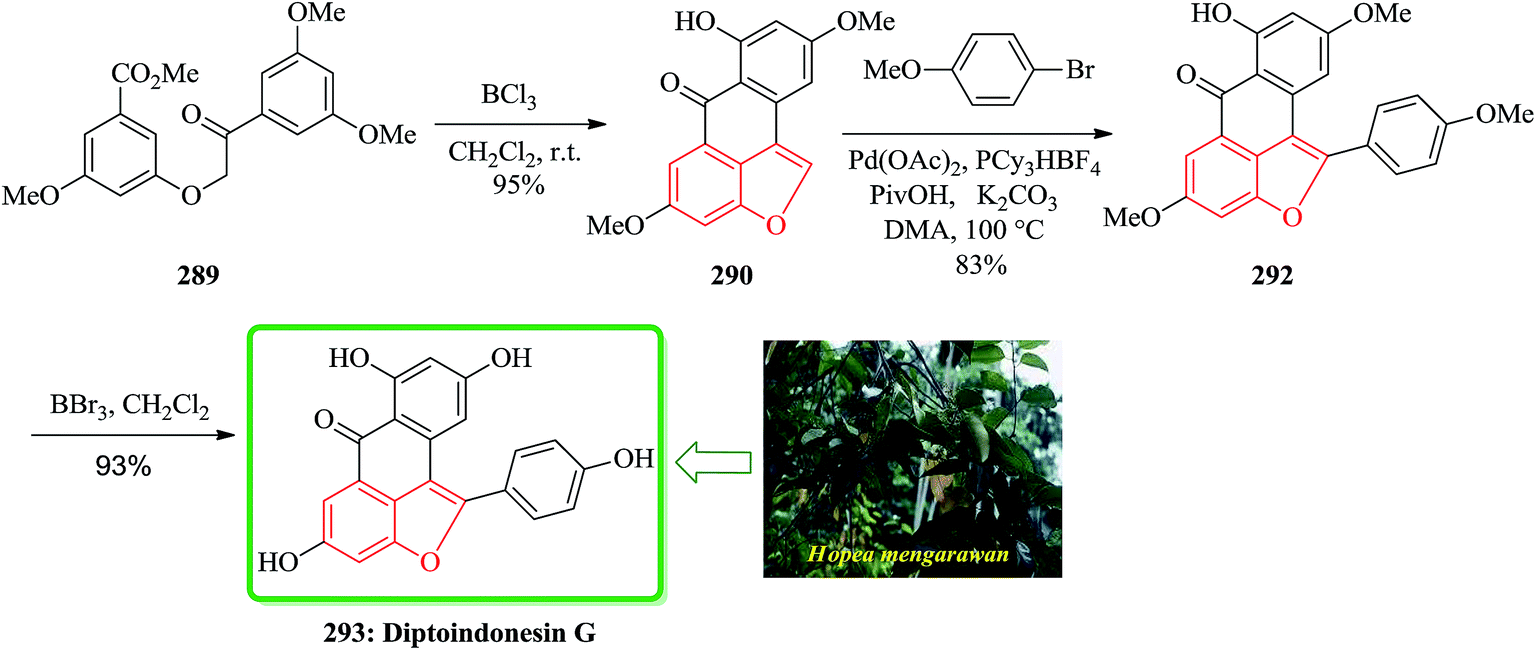 | ||
| Scheme 63 Total synthesis of diptoindonesin G 293. | ||
(+)-(R)-Concentricolide (+)-297, is the enantiomer of an anti-HIV-1 agent which was initially isolated from Daldinia concentrica. The concise total synthesis of (+)-297 was achieved in 7 steps starting from 2-iodophenol. This total synthesis disclosed the (S)-configuration for the naturally occurring form of the furanophthalide. The key steps in this strategy are an anionic ortho-Fries rearrangement to give 3-iodosalicylamide, easy formation of the benzofuran system using the Sonogashira coupling/cyclization via tandem manner as well as orthometalation to attach a propanoyl group, and CBS reduction, creating the stereogenic center, enantioselectively. This brief total synthesis started with market purchasable 2-iodophenol 294, which after 2 steps provided 3-iodosalicylamide 295. The latter upon treatment with trimethylsilylacetylene mediated by bis(triphenylphosphine)-palladium(II) chloride and in the presence of cuprous iodide under optimized conditions gave benzofuran 296 in high yield. Interestingly, it was found that the elevated temperature decreases the effectiveness of the catalyst system required for the cyclization of Sonogashira intermediate to the corresponding benzofuran 296, thus, much higher catalyst loading as well as portion wise addition is needed for the completion of the reaction via tandem fashion (Scheme 64).288
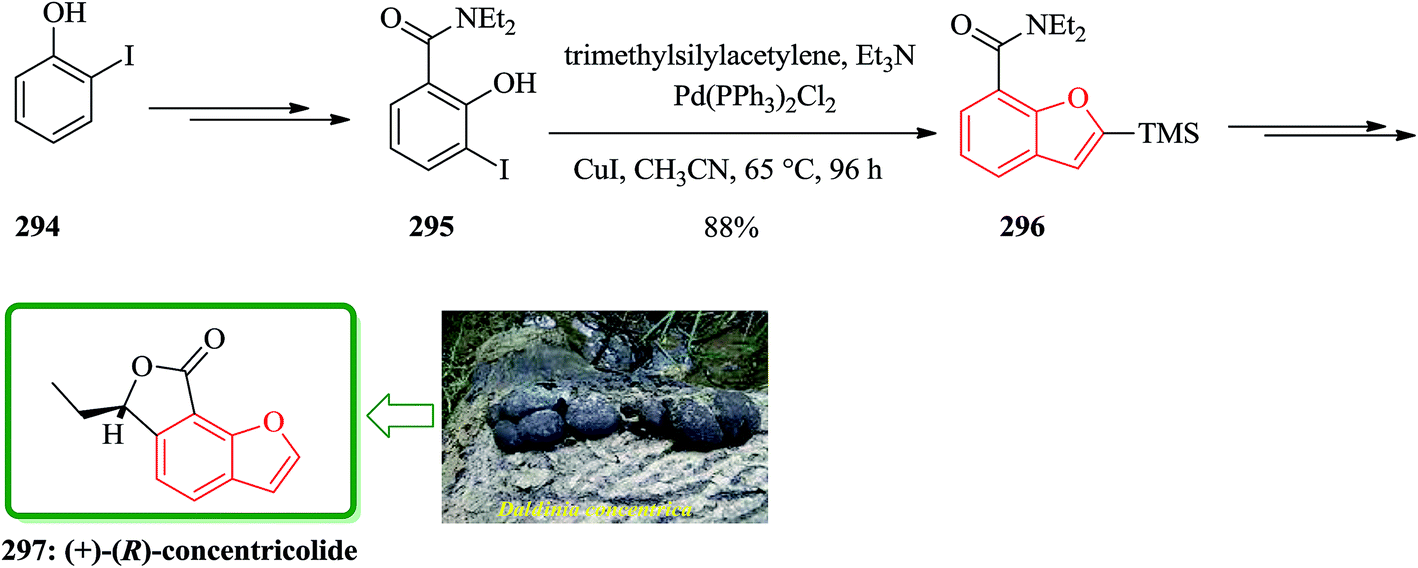 | ||
| Scheme 64 Total synthesis of (+)-(R)-concentricolide (+)-297. | ||
Synthesis of new iboga-analogues, replacing the indole ring with a benzofuran moieties has been reported in 2011. The 3-benzofuranethanol 299 was obtained via Larock's heteroannulation reaction289 between 2-iodophenol and internal alkyne 298, which subsequently treated with tetrabutyl ammonium fluoride in 55% yield in two steps, finally, compounds 300a and 300b were provided. Pd(II)–Ag(I) mixed metal mediated cyclization strategy was first developed by Trost in the synthesis of ibogamine.290 This protocol was applied to 300a and 300b to afford 301a and 301b in 42% and 22% yields, respectively (Scheme 65).291
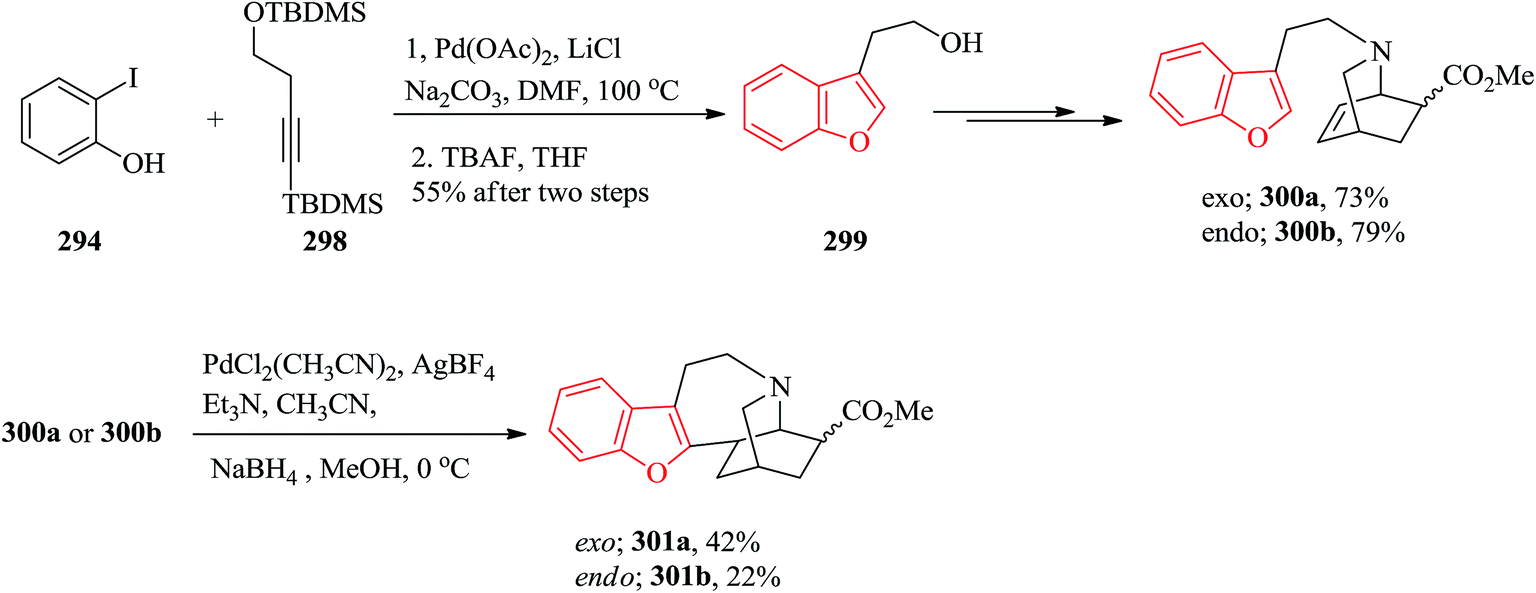 | ||
| Scheme 65 Synthesis of 301a, b. | ||
To synthesize of iboga analogues 304, the requisite benzofuran alcohol 302 was obtained in one-pot from 2-iodophenol via Sonogashira coupling with 3-butyn-1-ol at ambient temperature. After two steps, 302 afforded 303a and 303b in high yields. Compound 303a underwent the mixed-metal-mediated cyclization. This reaction proceeded smoothly and nicely to afford the desired product 304a in moderate yield. A similar cyclization of compound 303b also occurred to give the product 304b, although in lower yield (Scheme 66). Unexpectedly, the endo-isomers 300b and 303b found to be more polar than their exo-isomers.291
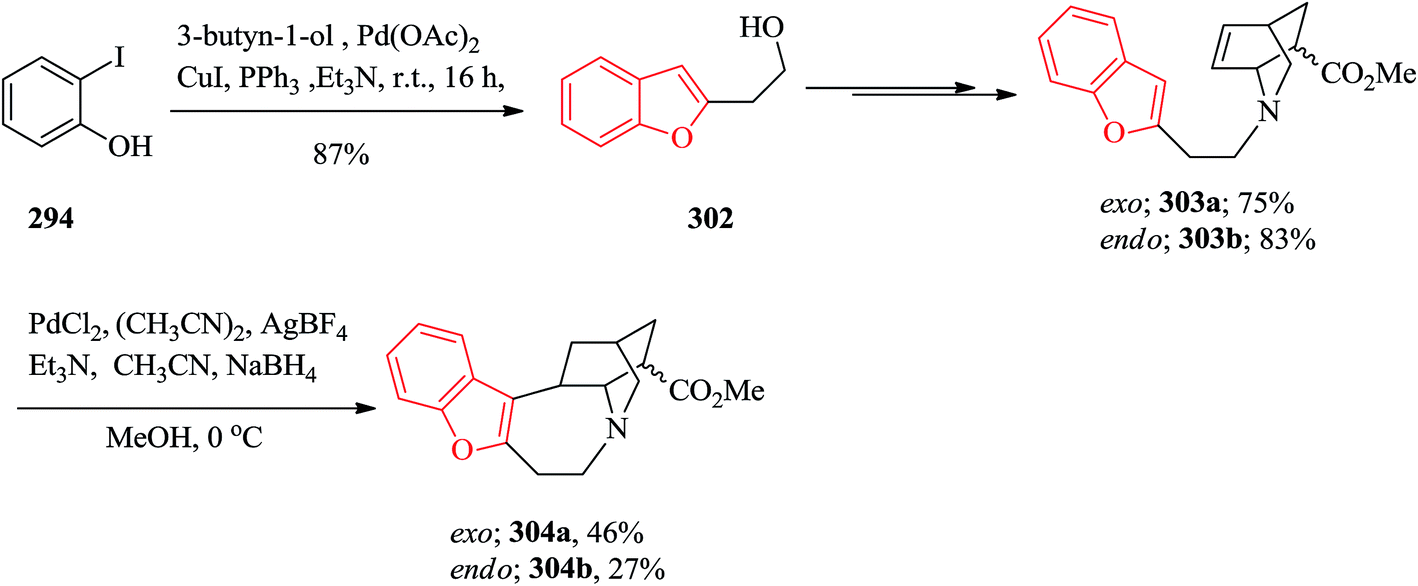 | ||
| Scheme 66 Synthesis of iboga analogues 304. | ||
2-Benzoylbenzo[b]furans and aurones (2-benzylidene-3-(2H)-benzofuran-3-ones) are occurring in nature and bearing the same carbon unit scaffold (C6–C3–C6). 2-Benzoylbenzo[b]furans were initially isolated from different plants, used traditionally as medicine by native inhabitants.292 Both compounds were screened, showing interesting biological activities.293 In some cases, they were employed as intermediates for the total synthesis of biological active compounds, i.e., aromatase inhibitors.294 The naturally occurring aurones (2-benzylidene-3(2H)-benzofuran-3-ones) can be cleanly transformed to another class of natural products 2-benzoylbenzo[b]furans by an efficient reduction, acid-mediated rearrangement, and oxidation cascade. This facile transform was performed with no purification of intermediates. This simple conversion may be considered as a possible biosynthesis route of 2-benzoylbenzo[b]furans in plants. The aurones were prepared following a previously reported method295 and provided as solely Z-isomers, in respective to the configuration of naturally occurring aurones. The reduction of aurones was conducted with sodium borohydride in methanol at ambient temperature to afford the corresponding allylic alcohols (2,3-dihydrobenzofuran-3-ols) 306. These alcohols are sensitive to high temperature and acidic conditions. The isomerization was taken place at room temperature in a mixture of water and acetonitrile mediated by aqueous HCl to give 309. The plausible mechanism involves a carbocation generation 307 followed by rearrangement of the later to the extracyclic methine carbon 308, stabilized by the B-aryl group. The organic solution of the rearranged alcohol was directly used in the oxidation step using MnO2 as an oxidant in dry conditions. These three steps conversion were applied for the synthesis of a series of aurones 305 in high to excellent yields. The benzoylbenzo[b]furans analogs 310 were obtained with excellent yields (76–86%) (Scheme 67).296
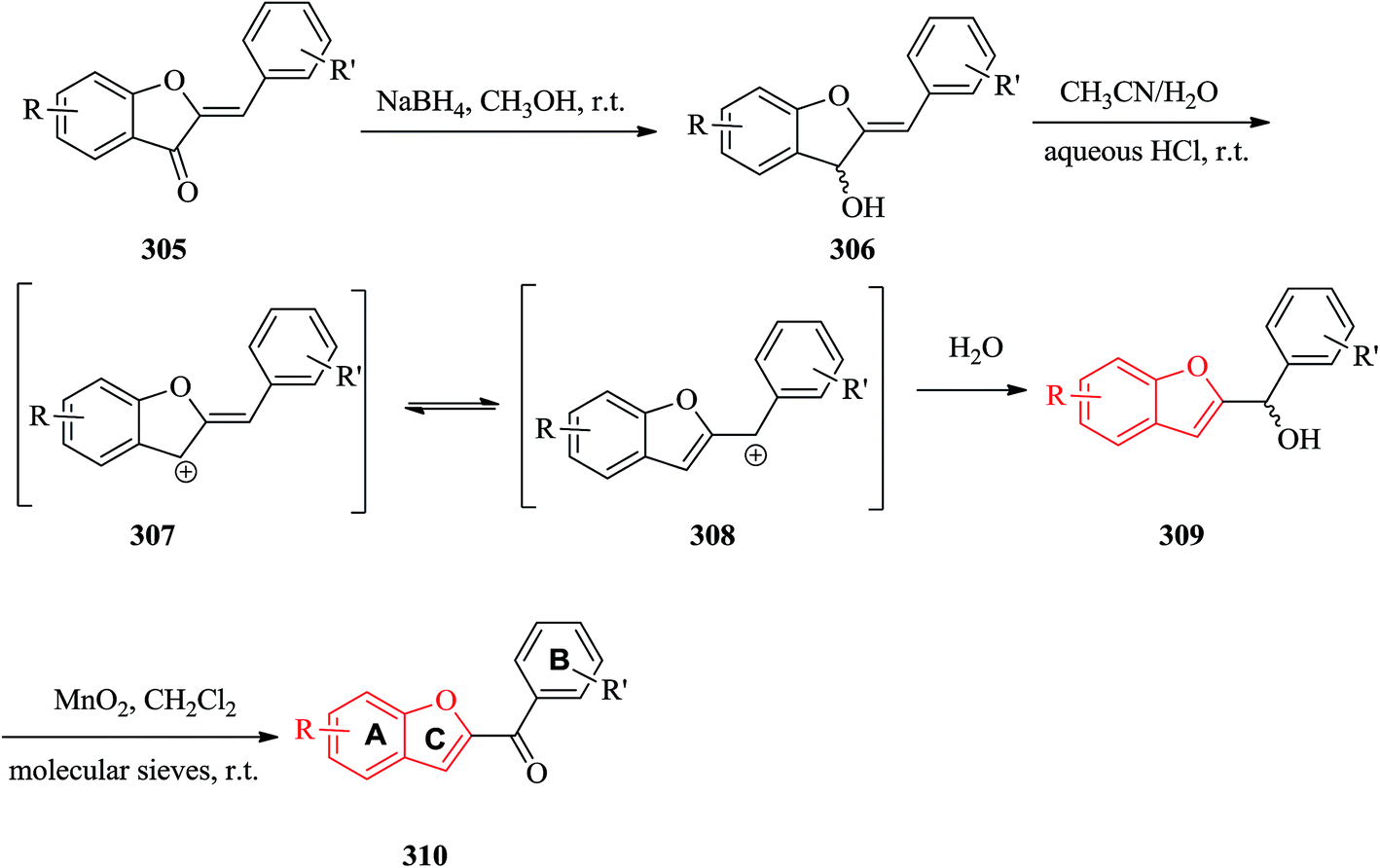 | ||
| Scheme 67 Synthesis of the benzoylbenzo[b]furans analogs 310. | ||
In 2005, the naturally occurring compound (+)-fulicineroside was isolated from the slime mold Fuligo cinerea, the plant was found and collected in the Czech republic.297 The total synthesis of (+)-fulicineroside 315 was accomplished and reported in 2013. The total synthesis was started with the Ullman-type coupling298 commercially available resorcin 312 with readily accessible 1-bromo-3,5-dimethoxybenzene 311 to obtain the corresponding biarylether phenol as an intermediate which without isolation is transformed into the dimethyl carbamate 313 via a one-pot fashion. The dimethyl carbamate 313 under Fagnou's C–H activation conditions299 and Pd-mediated C–H activation provided compound 314 containing the dibenzofuran ring moiety present in the structure of the desired natural product 315.300 Worthy to mention that in this treatment the reaction times were remarkably decreased and the best yields were obtained when AgOAc was used as an oxidant instead of molecular oxygen present in air under ambient conditions. Compound 314 was transformed into (+)-fulicineroside 315 as the desired target via multi-steps reaction through different functional group transformations (Scheme 68).301
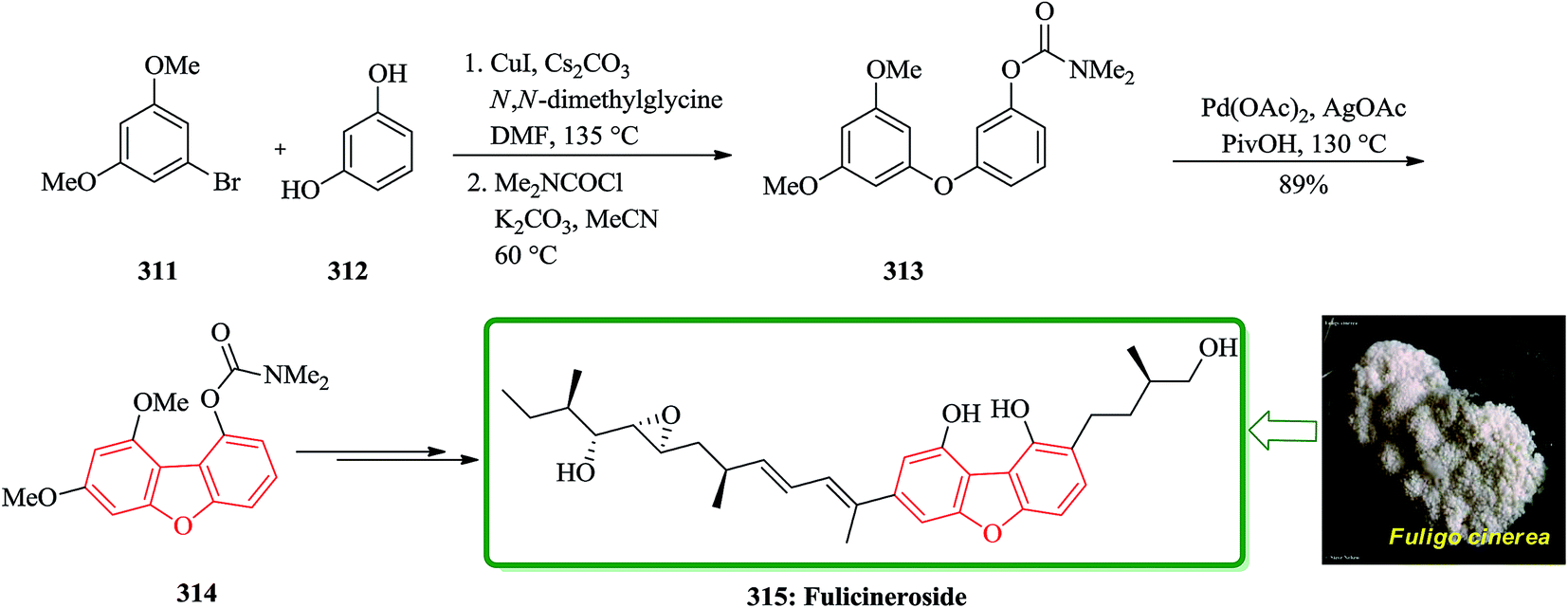 | ||
| Scheme 68 Total synthesis of fulicineroside 315. | ||
Coumestrol 319 is an essential dietary ingredient found in forage plants, cabbages and soybeans.302 Due to its importance in human nutrition, it has been extensively studied.303,304 The total synthesis of 319 based on the iron-catalyzed cross-dehydrogenative coupling (CDC) was achieved and revealed in 2013. In this approach, a modified aerobic oxidative cross-coupling applied for the construction of benzofuran, a moiety present in coumestrol 319. Ethyl 2-(2,4-dimethoxybenzoyl)acetate 317 and 3-methoxy phenol 316, were reacted in DCE as solvent at 70 °C in the presence of FeCl3 as the catalyst and 2,2′-bipyridine as additives to give compound 318. The latter was then submitted to sequential deprotection/lactonization giving the desired natural product in good (59%) overall yield (Scheme 69).305
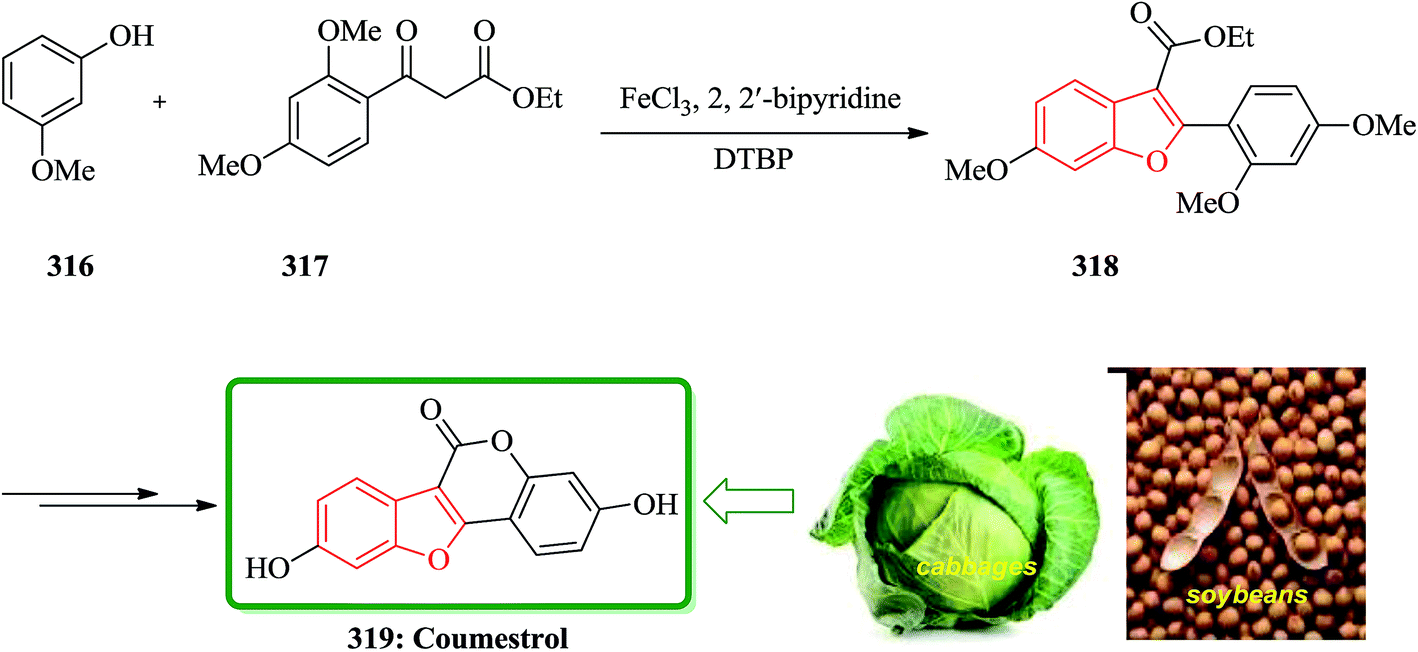 | ||
| Scheme 69 Total synthesis of coumestrol 319. | ||
The dried root of Salvia miltiorrhiza bunge so called danshen, in the Lamiacea family is one of the mostly common used Chinese folk medicines (CFM). This medicine has a history of at least 2000 years in China and has been also used globally, since 1970s. It helps circulation and develop blood thus to provide therapeutic relief from stroke and angina pectoris. Moreover, it shows antiviral, antioxidant and antitumor potencies.306–309 In 2013, the total synthesis of a methylated analogue of (+)-salvianolic acid C has been accomplished and reported. Key features in this synthetic approach are using readily available and inexpensive Cu(I) acetylide, significant carboxyl activation under microwave irradiation (MW), and using kinetic resolution of a racemic mixture of secondary alcohol via lipase catalyzed danshensu. The total synthesis starts from coupling of readily accessible 321 with 320 under the optimal reaction conditions reported by Scammells and co-workers102 to give the 2-arylbenzo[b]furan core 322 (51% yield). Finally, reaction of 323 and 324, in the presence of Et3N gave carboxylic acid 71 in satisfactory yield (Scheme 70).310
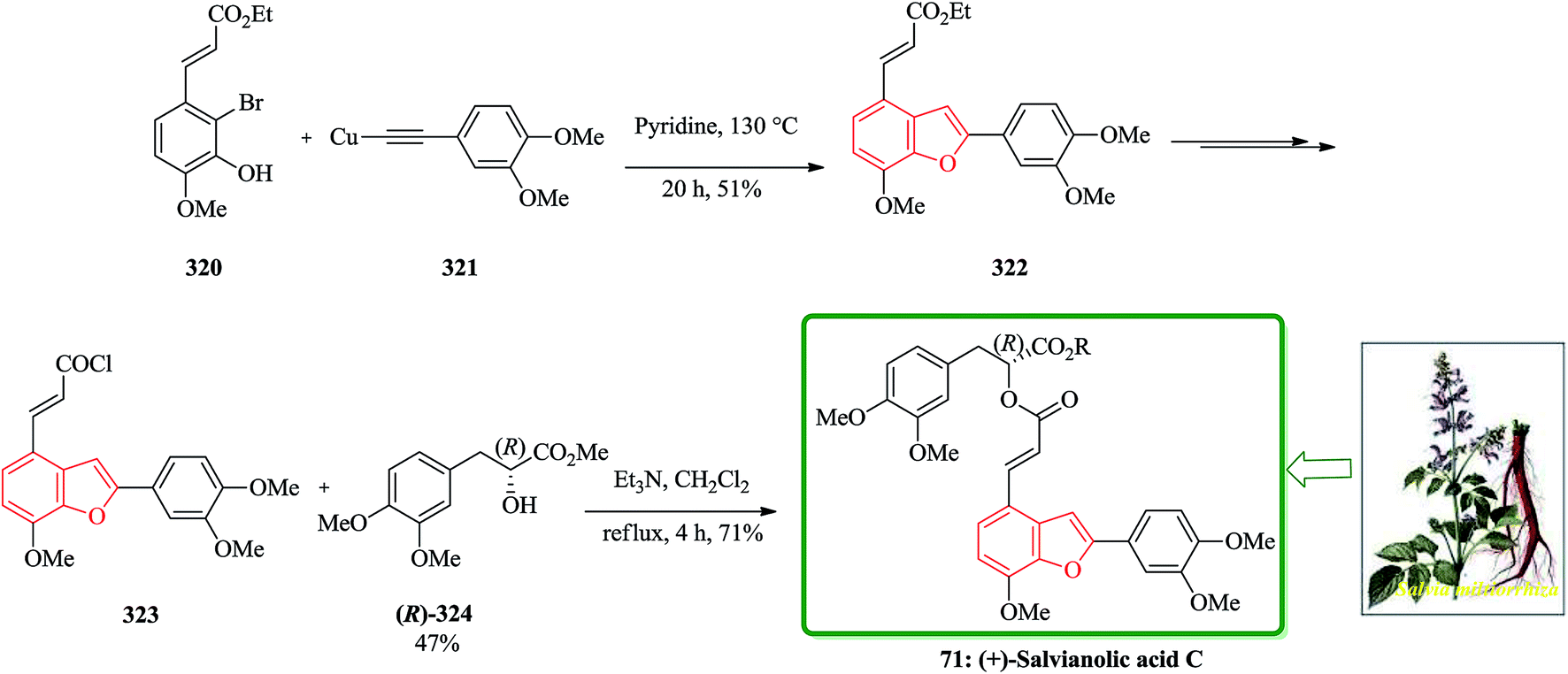 | ||
| Scheme 70 Total synthesis of (+)-salvianolic acid C 71. | ||
Pimpinellin 328 acts as a phytoalexin in parsley and celery. It was found to serve as an inhibitor of trichothecene toxin biosynthesis. It has been isolated from a variety of plant cradles,311 such as Pimpinella saxifraga L.312 The total synthesis of pimpinellin 328 involves the Au(I)-catalyzed intramolecular hydroarylation (IMHA) of the appropriate aryl propiolate esters, which were themselves provided by the reaction of the respective phenols with either 3-(trimethylsilyl)propiolic acid or propiolic acid and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride or dicyclohexylcarbodiimide. The total synthesis of pimpinellin 328 started from vanillin 19, which was transformed to substituted arene 325 in 84% yield. The latter was then submitted to a Sonogashira cross-coupling313 reaction with triisopropylsilylacetylene to afford a 1:8 mixture of acetylene 326 (5%) and the isomeric benzofuran 327 (39%). Delightfully, they could be separated by column chromatographically. The rather moderate yields linked with the transformed 325 → 326 + 327 can be ascribed to possible competitive oxidative coupling of the triisopropylsilylacetylene. It is worthy to mention that such process is expected to generate likely volatile compounds, which actually were not detected in the obtained crude product mixture. Compound 327 is transformed into the desired natural product 328 in several steps including a step required for the assembly of the lactone ring, present in the species isolated from natural products (Scheme 71).314
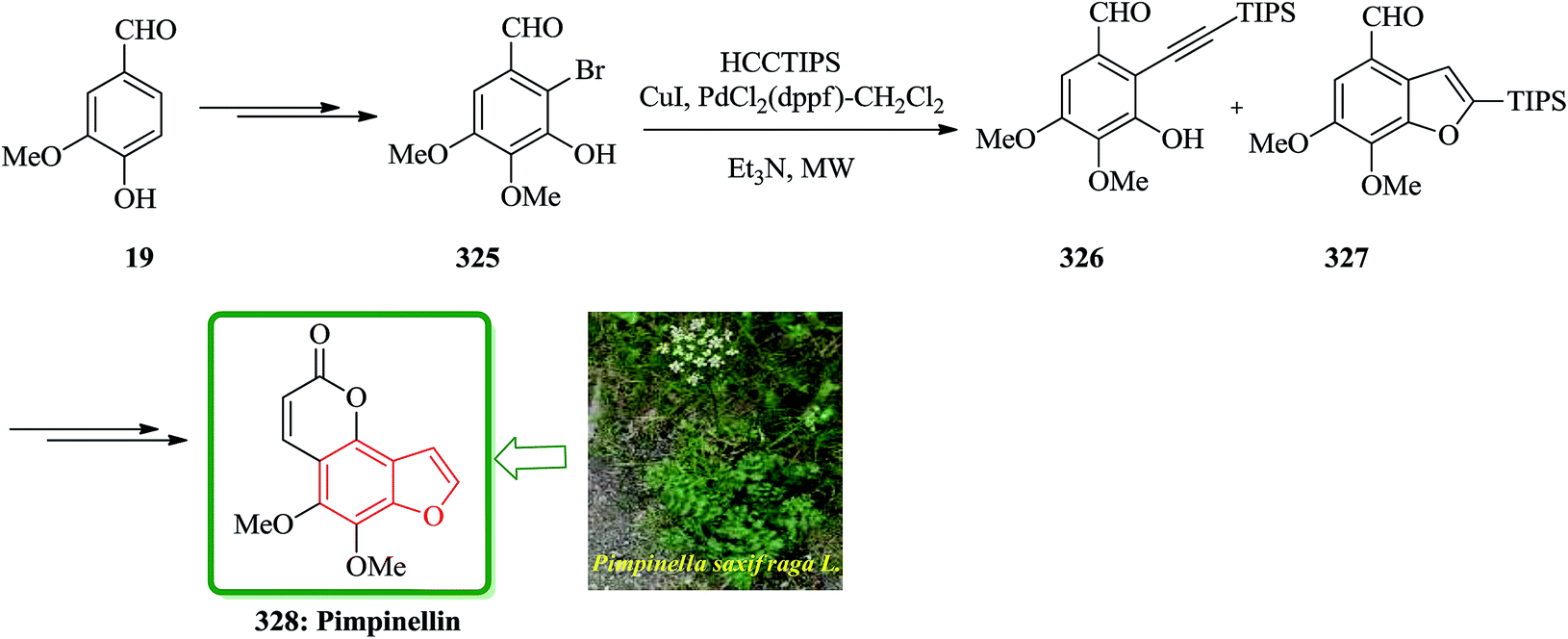 | ||
| Scheme 71 Total synthesis of pimpinellin 328. | ||
Xylarianaphthol-1 336, a dinaphthofuran derivative showing divers biological activities was originally isolated from a marine sponge-derived fungus of order Xylariales on the control of a bioassay employing the transfected human osteosarcoma MG63 cells.315,316 The total synthesis of 336 was achieved as illustrated in Scheme 72. The total synthesis started from coupling of 1,5-naphthalenediol mono-methoxymethyl (MOM) ether 329 with bromobenzoquinone 330 mediated by K2CO3 in DMSO317 to afford a C–O coupling product. Upon reduction of the quinone moiety, present in acetonitrile using aqueous Na2S2O4, the mono-triflation was regioselectively proceeded resulting in the formation of compound 332. The latter was further treated with sodium acetate to produce acetate 333, which can be used as the precursor of the key intramolecular arylation. Among several efforts to find optimal conditions the combination of Pd(PPh3)4 and NaOAc was found most operative to promote Mizoroki–Heck-type intramolecular arylation, which is leading into the formation of the desired pentacyclic product 334 in satisfactory yield.318
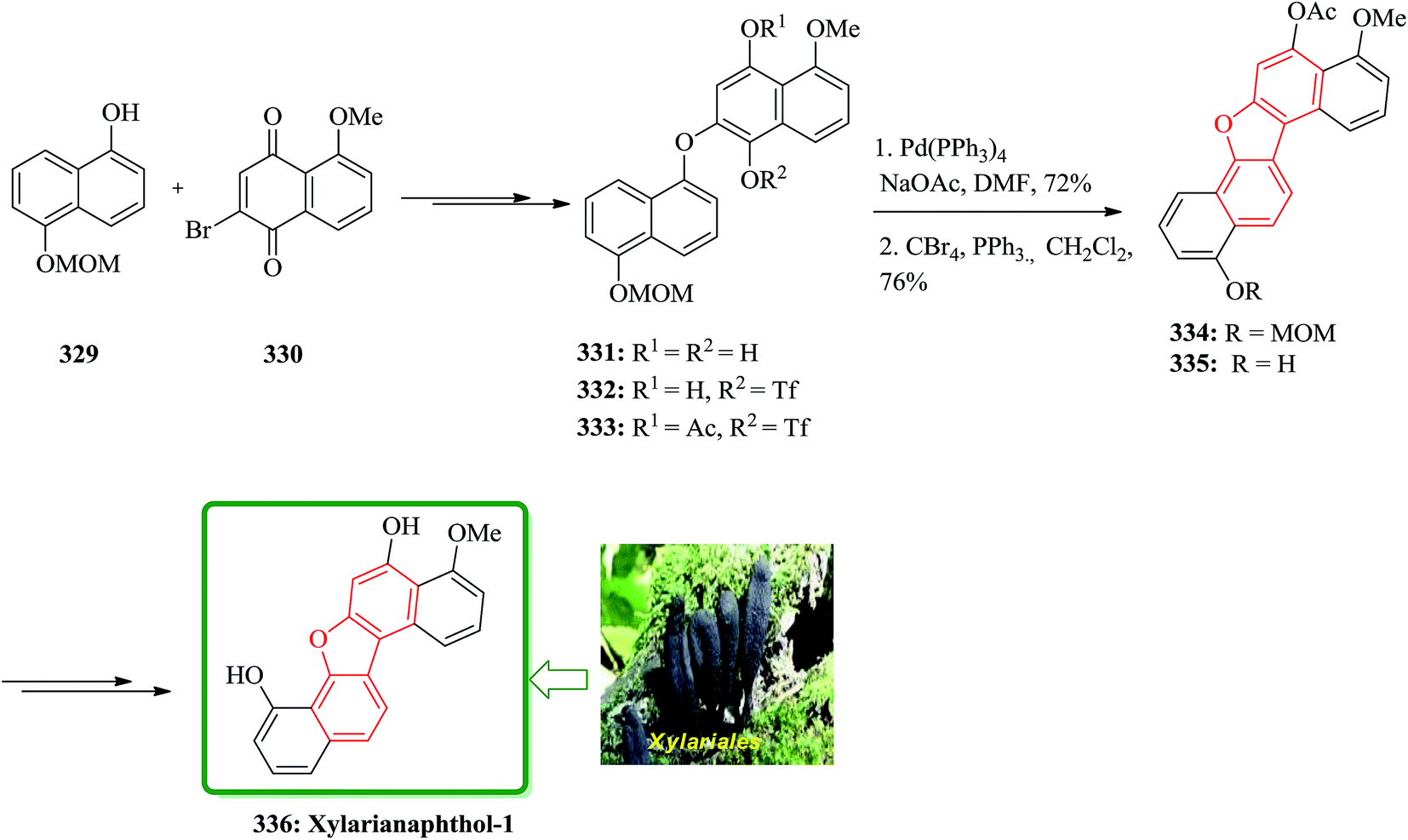 | ||
| Scheme 72 Total synthesis of xylarianaphthol-1 336. | ||
Propolisbenzofuran B 340, is a biologically active naturally occurring compound, which was initially isolated from honeybee propolis resin. The total synthesis of 340 includes a silicon-tether controlled oxidative ketone–ketone cross coupling and a benzofuran construction via cascade manner to provide the core structure of the target. The total synthesis commenced with easily accessible 3-methoxycyclohex-2-enone which in several steps is converted into 1,4-diketone 337 in accordance with a pathway reported by Clift and co-workers previously.319 This 1,4-diketone 337 was then converted into dihydroquinone 338 using PCC on silica in excellent yield. Worthy to mention that initially the Ley oxidation was used but found being unsuccessful since remarkable unchanged starting material was recovered. Even increases in catalyst loading in this case did not work, it could be attributed to the presence of the adjacent ethyl group, which apparently hinders initial formation of the required ruthenate ester. Delightfully, the ethyl substituent could not prevent the construction and aromatization of benzofuran via cascade reaction. Compound 338 was converted into ethyl substituted benzofuran 339 in satisfactory yield. Completion of the synthesis from this point was direct and classical. Upon removal of the silylether using 20% HF followed by acetylation of the resulting primary alcohol, which proceeded clean and smoothly caused to selective deprotection of the isopropyl ethers which were performed in the presence of AlCl3. These three sequential steps gave the desired natural product, propolisbenzofuran B 340 in 77% overall yield. This synthetic product showed identical spectral data with those of obtained and reported for the species isolated from natural sources (Scheme 73).320
 | ||
| Scheme 73 Total synthesis of propolisbenzofuran B 340. | ||
Daphnodorin A 344, is a member of the daphnodorins. The total synthesis of 344 was achieved and reported in 2014. Key aspects of the synthetic protocol involve the assembly of 2-substituted-3-functionalized benzofuran through intramolecular Heck reaction321 and a mild Barton–McCombie deoxygenation process catalyzed by triethylborane. This strategy provided daphnodorin A in 7 steps with overall yield of 19.7% or 15 steps with overall yield of 5.6%. Initially, compound 341 and the desired o-iodophenol 342 were synthesized. Then the corresponding o-iodophenol 342 reacted, subjected into conjugate addition followed by intramolecular Heck reaction with ynone 341 to form an entirely protected daphnodorin B 343. Finally, upon deprotection of the latter daphnodorin A 344 was provided (Scheme 74).322
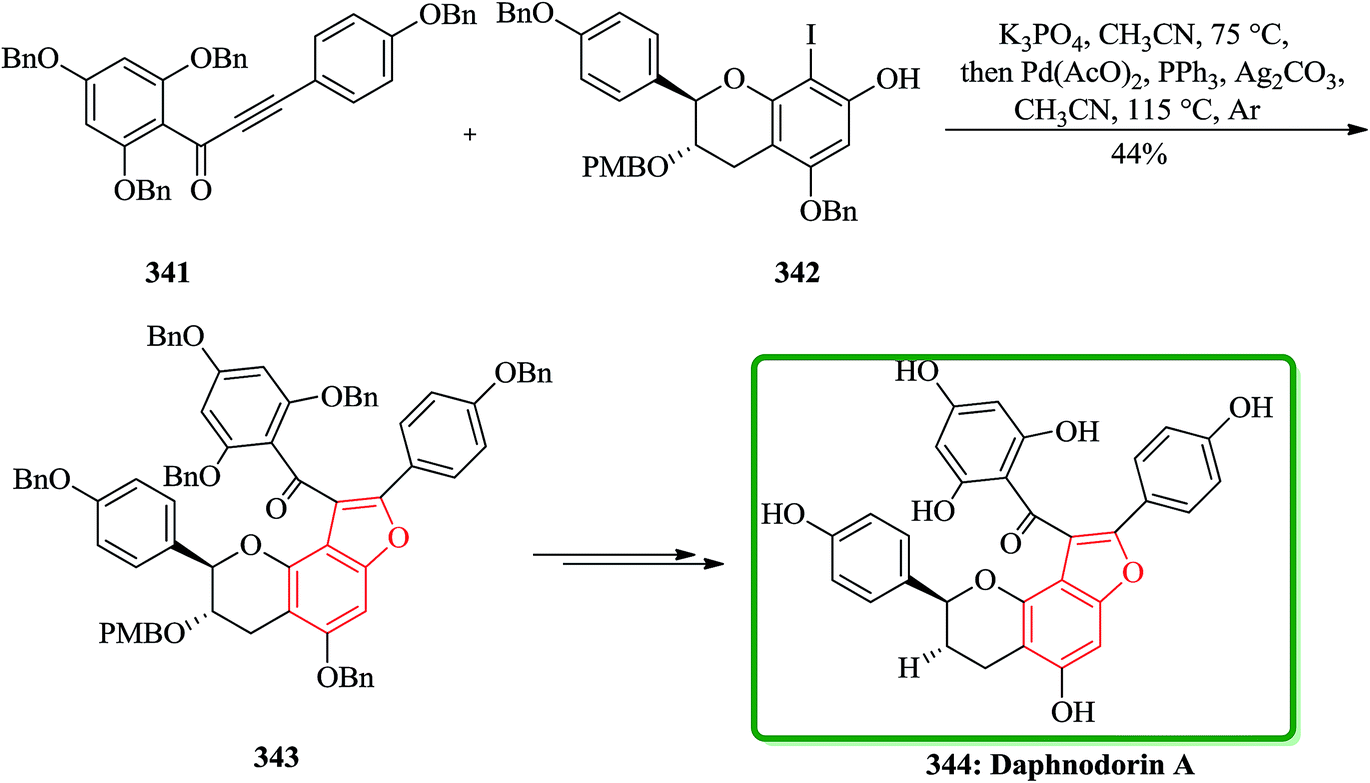 | ||
| Scheme 74 Total synthesis of daphnodorin A 344. | ||
Two new flavones (±)-anastatins A and B, isolated from Anastatica hierochuntica have a benzofuran moiety as scaffold in their structures and their total synthesis was reported very recently. The key features for their synthesis are bromination, Suzuki coupling reaction,323 and an oxidation/oxa-Michael reaction.324 The concise total synthesis of (±)-anastatins A and B were accomplished in eight steps starting from the market purchasable phloroglucinol with acceptable overall yield of 9% and 10%, respectively. The key intermediate 345 was synthesized in accordance with the procedure reported, previously.325 Then, the stage was fixed for the assembly of the benzofuran moiety which was achieved through a one pot oxidation/oxa-Michael reaction using Ag2O in DMF via cascade manner to afford compound 346 in 75% yield. Noticeably, the relatively low yield was probably due to decomposition of product 346 under influence of Ag2O, which is used as oxidant with long reaction time. Upon hydrogenation of 346 in the presence of Pd/C, the total synthesis of (±)-anastatin B was completed. This hydrogenation step provided the natural product virtually in quantitative yield (Scheme 75).326
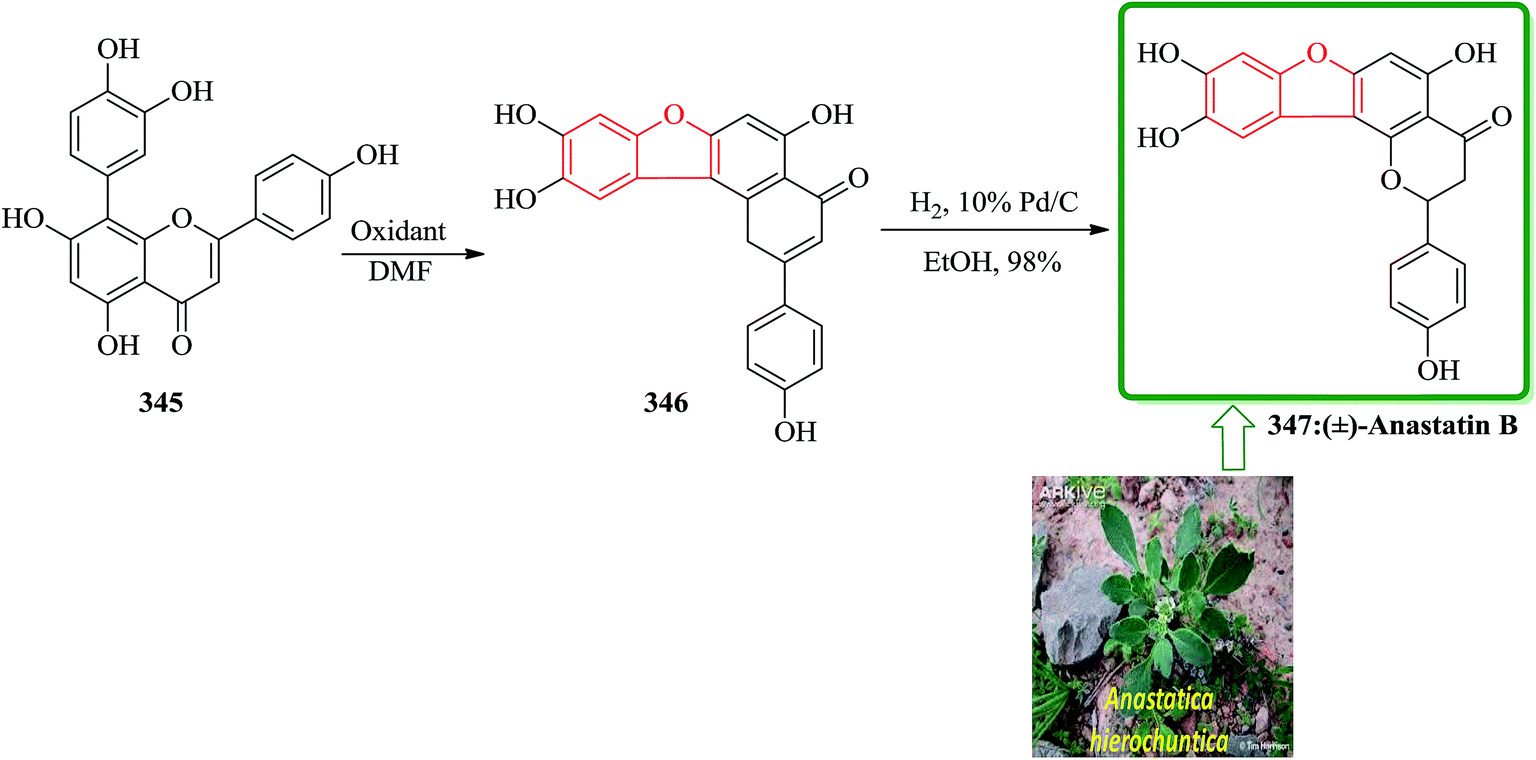 | ||
| Scheme 75 Total synthesis of (±)-anastatins B 347. | ||
Moreover, the synthesis of (±)-anastatin A 350 was achieved in similar way. Intermediate 348 under the same conditions afforded compound 349 in 41% yields, starting from intermediate 348. However, in this case the regioselectivity of the intramolecular Michael addition for the construction of the cyclized product 349 is significant. The possible regioisomer causing from cyclization of the 5-OH group onto the ortho quinone intermediate was not constructed and even detected. Compound 349, upon hydrogenation on 10% Pd/C in ethanol gave the desired (±)-anastatin A 350 (Scheme 76).326
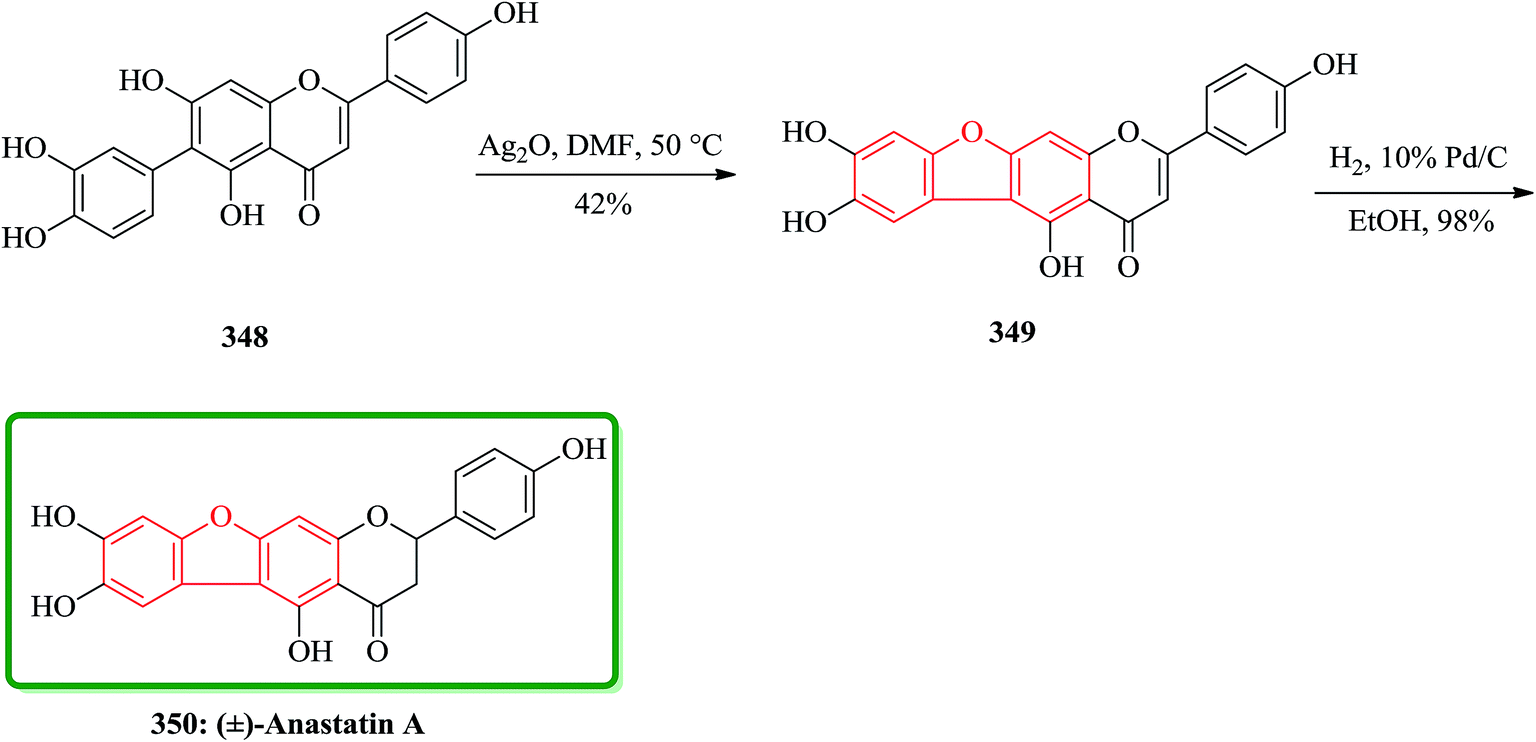 | ||
| Scheme 76 Total synthesis of flavones (±)-anastatins A 350. | ||
Vialinin C 355 was initially isolated from dry fruiting bodies of non-poisonous and eatable Chinese mushroom, Thelephora vialis. Ganbajunin B 356 has the same origin as C 355. The structures of 355 and ganbajunin B 356 were established unambiguously, only after they were synthesized. The total synthesis of compounds 355 and 356 has been achieved and revealed very recently.327 Compound 354, which contains benzofuran moiety was synthesized from the reaction of sequential Suzuki–Miyaura coupling.328 The reaction of 351, 352 and 353 gave 354 which after several steps gave the desired natural product 355 in satisfactory overall yield. In another route, the benzofuran derivative 354 was also used as a precursor for the synthesis of ganbajunin B 356 in 30% overall yield (Scheme 77).329
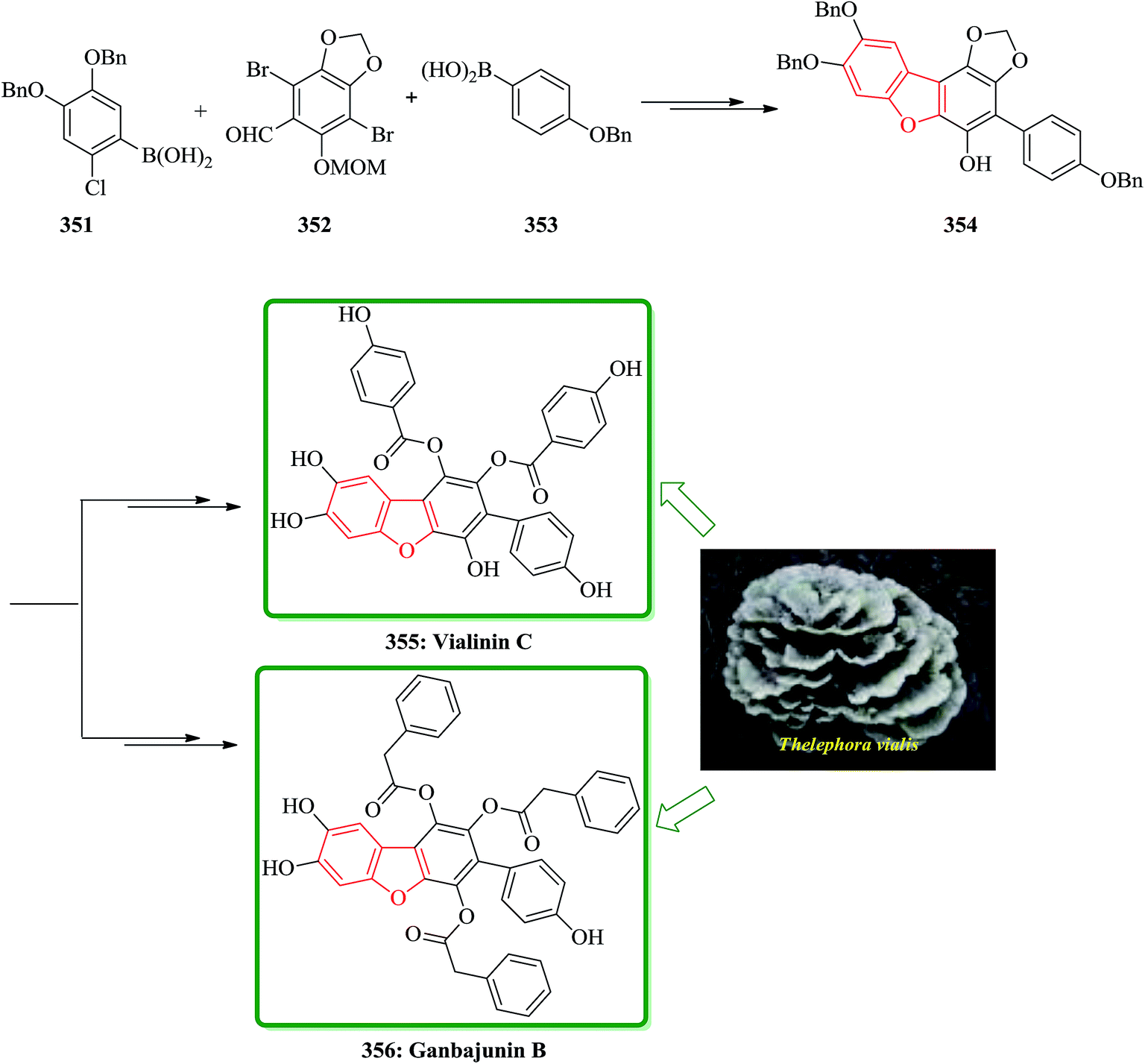 | ||
| Scheme 77 Total synthesis of vialinin C 355 and ganbajunin B 356. | ||
The naturally occurring compound diptoindonesin (Dip) G 293 was initially isolated from tree barks of Hopea mengarawan in Indonesia282 and from Hopea chinensis stem barks in China.283 Dip G has a tetracyclic core with A–D rings bearing a ketone and three phenolic OH groups and also involves an additional E-ring bearing one more phenolic OH group. Dip G 293 exhibited anti-proliferation effect in murine leukemia P-388 cells.282 A convergent synthetic approach for the total synthesis of diptoindonesin G 293 has been achieved and reported by Tang and co-workers in 2009.330 The protocol comprises a regioselective dehydrative cyclization of arylacetals, a regioselective bromination of benzofurans, a sequential cross-coupling of bromo-benzofurans with aryl boronicacids and a BBr3-mediated tandem cyclization and demethylation. This approach started with commercially available mono-protected resorcinol derivative 357. The latter can be converted into the benzofuran core 360 by the sequence of alkylation with bromodimethylacetal and cyclodehydration via an intermediate 359 using Amberlyst-15.331 Notably the cyclization was taken place regioselectively, which is consistent with similar reactions reported previously.331 Compound 360 was transformed into penultimate intermediate 361 in several steps including cross-coupling with 3,5-dimethoxyphenyl boronic acid, which occurred at high temperature. The desired target Dip G 293 was synthesized from 361 via BBr3 mediated tandem cyclization and demethylation in accordance with the procedure reported, previously (Scheme 78).287
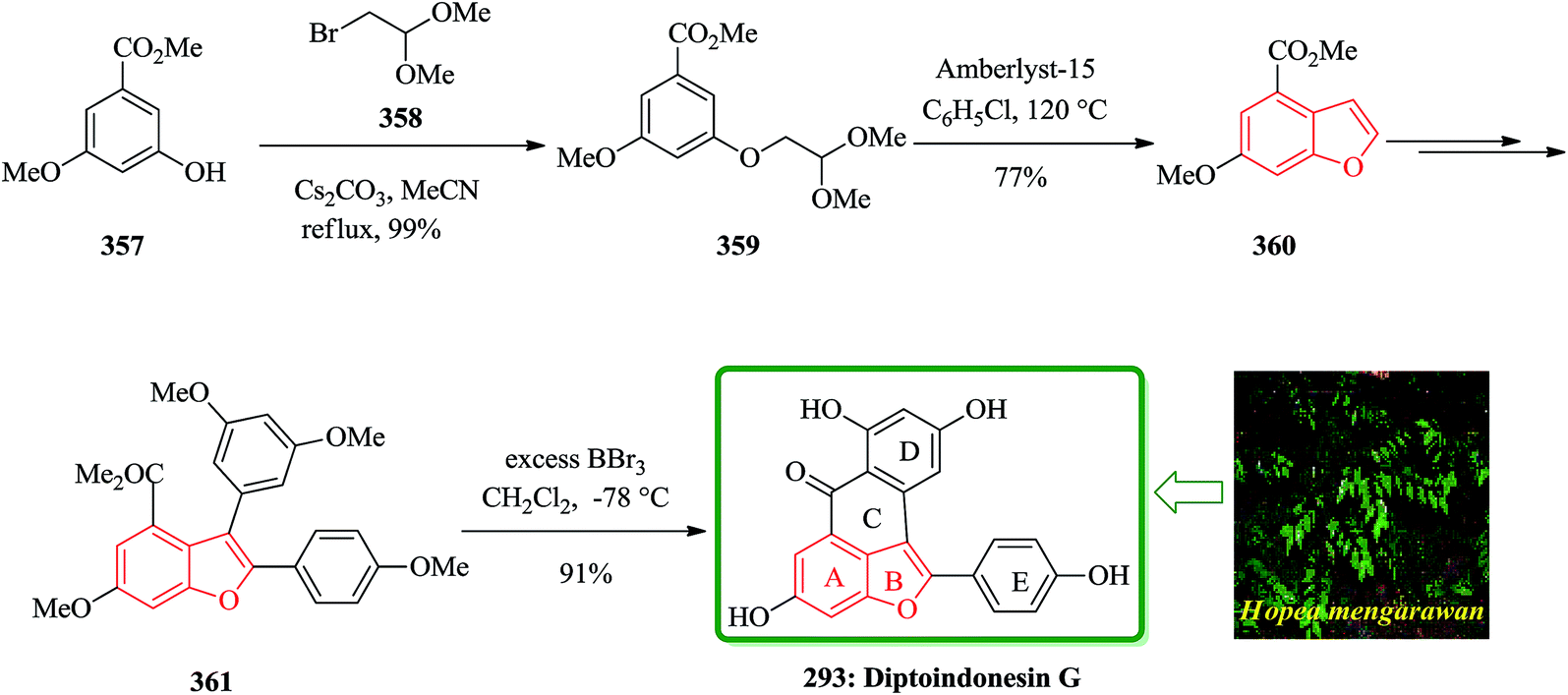 | ||
| Scheme 78 Total synthesis of diptoindonesin (Dip) G 293. | ||
A diverse total synthetic approach for the total synthesis of several natural products containing highly substituted benzofuran starting from furan derivatives has been achieved and reported by Tang and co-workers.332 The key step in their strategy was Rh-catalyzed carbonylative benzannulation methodology, which led to the formation of various highly substituted benzofurans present in natural products. This protocol started with market purchasable or readily accessible 2,3-dibromofuran 362. In this line, Tang and their research group accomplished and reported the first formal total synthesis of natural products amurensin H (or viniferifuran) 368, malibatol A 270 and shoreaphenol (or hopeafuran) 271 containing benzofuran scaffold via Rh-catalyzed benzannulation. Initially, dibromofuran 362 or dibromofurfural 363 was converted to 365 (Scheme 79). This group tried to find that the key benzannulation reaction worked smoothly for substrate 365, which bears a smaller bromine substituent. Then substrate 365 provided the highly substituted benzofuran core 366. After several steps, permethylated precursor 367 was produced, which was then converted to natural products via different routs333,334 to 368, 270 and 271.332
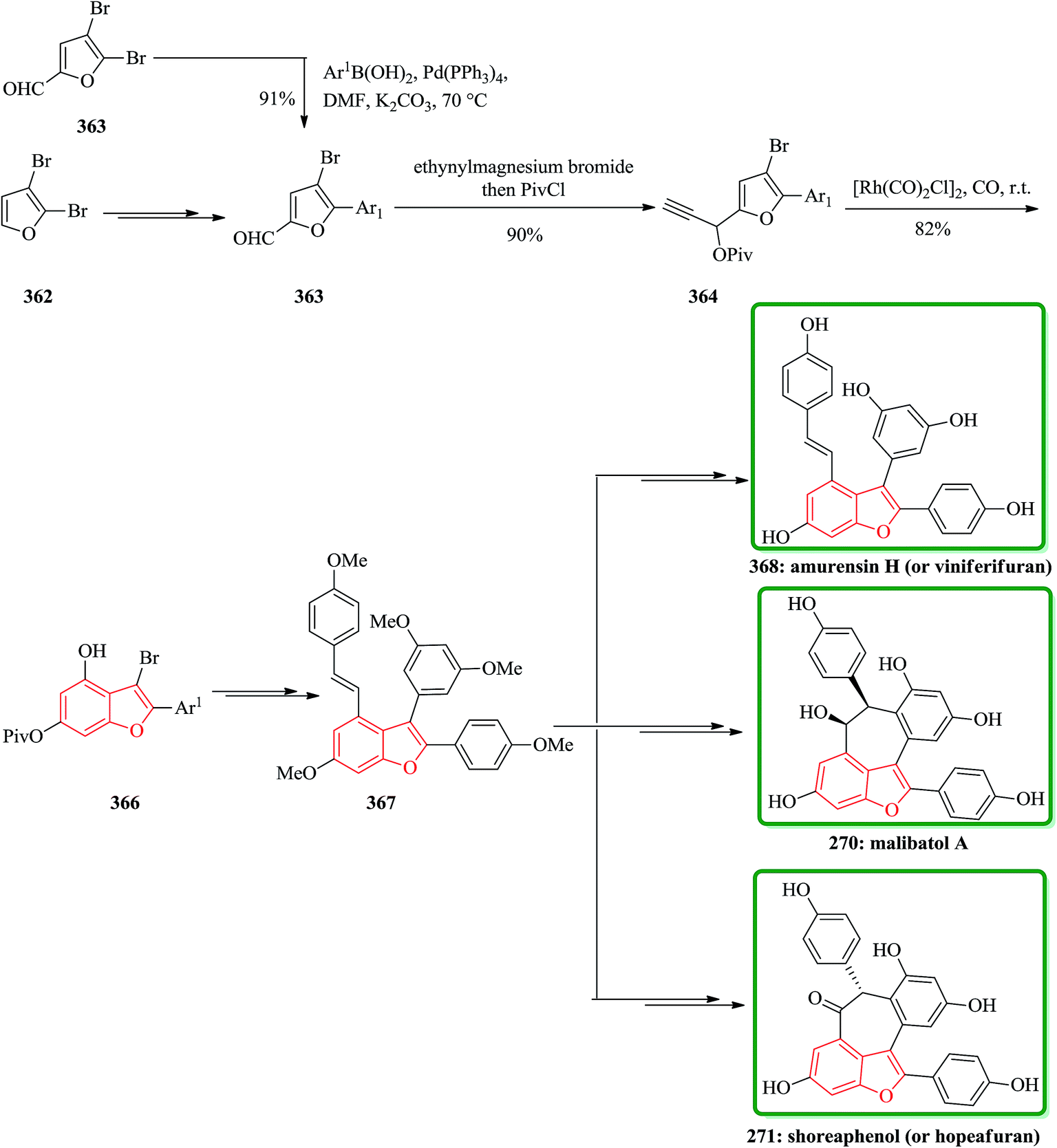 | ||
| Scheme 79 Formal synthesis of benzofuran-containing natural products amurensin H (or viniferifuran) 368, malibatol A 270, shoreaphenol (or hopeafuran) 271 via Rh-catalyzed benzannulation. | ||
Alternatively, compound 366 was converted into intermediate 369, the latter was subjected to methylation with subsequent cross-coupling with Ar2B(OH)2 followed by removal of pivalate gave the desired benzofuran, which could be transformed to anti-proliferation compound PAA 370 and natural product fuliginosin A 371 in two and four steps, respectively, via different functional group transformations. This is the first strategy for the total synthesis of fuliginosin A. It is also an example of the confirmation of structure of a natural product by its total synthesis. The overall yields for 370 and 371 are 24.3% and 3.6%, respectively, starting form 363 (Scheme 80).332
 | ||
| Scheme 80 Synthesis of PPA 370 and fuliginosin A 371. | ||
3 Conclusion
Benzofurans are significant moiety in a wide range of biologically potent naturally occurring compounds as well as synthetic products. Investigation on natural products including benzofuran has extraordinarily improved during the past few decades. New discovered naturally occurring compounds having complex structures have been extracted, well characterized, demonstrated important biological activities, therefore synthesized from commercially accessible or easily available starting precursors. Because of this extensive scope of biological properties, from long time ago, benzofurans have attracted the attentions and stirred up the interests of several research groups. Several of them display antimicrobial, anticancer, antioxidant, immune modulatory and anti-inflammatory activities. Benzo[b]furans have also attracted massive interest because of their existence in natural products, biologically active compounds, and other molecules of medicinal interest. In this review, we tried to highlight the total synthesis of natural product containing benzofuran moiety, since they have been found being a foremost source of drug discovery and drug development for a wide variety of diseases. The benzofuran framework can be labeled a ‘skeleton key’ as it is an unprecedented core in diverse compounds acting at different targets to inspire variety of pharmacological activities having various substitution patterns.Abbreviations
| (PTP-1B) | Protein tyrosine phosphatase inhibitors |
| (NTI) | Naltrindole |
| (DMAP) | N,N-dimethylpyridin-4-amine |
| (DEAD) | Diethyl azodicarboxylate |
| (DMF-DMA) | N,N-Dimethyformamide dimethyl acetal |
| (DDQ) | 2,3-Dichloro-5,6-dicyano-p-benzoquinone |
| (PEP) | Prolyl endopeptidase |
| (CDC) | Cross-dehydrogenative coupling |
| (MW) | Microwave irradiation |
| (IMHA) | Intramolecular hydroarylation |
| (Dip) | Diptoindonesin |
Acknowledgements
The authors are grateful to Department of Chemistry of Alzahra University for the encouragements and Alzahra University Research Council, for partial financial support. MMH is thankful to Iran National Research Foundation (INSF) for the partial financial supports.References
- P. Cagniant and D. Cagniant, Adv. Heterocycl. Chem., 1975, 18, 337–482 CrossRef CAS.
- C. W. Bird and G. W. H. Cheeseman, in Compr. Heterocycl. Chem., ed. Katritzky, A. R., Pergamon, New York, 1984, vol. 4, pp. 89–153 Search PubMed.
- D. M. X. Donelly and M. J. Meegan, in Compr. Heterocycl. Chem., ed. Katritzky, A. R., Pergamon, New York, 1984, vol. 4, pp. 657–712 Search PubMed.
- P. C. Stevenson, M. S. J. Simmonds, M. A. Yule, N. C. Veitch, G. C. Kite, D. Irwin and M. Legg, Phytochemistry, 2003, 63, 41–46 CrossRef CAS PubMed.
- H. Tanaka, T. Oh-Uchi, H. Etoh, M. Sako, M. Sato, T. Fukai and Y. Teteishi, Phytochemistry, 2003, 63, 597–602 CrossRef CAS PubMed.
- Z. Ali, T. Tanaka, I. Iliya, M. Iinuma, M. Furusawa, T. Ito, K.-I. Nakaya, J. Murata and D. Darnaedi, J. Nat. Prod., 2003, 66, 558–560 CrossRef CAS PubMed.
- G. K. Rao, K. N. Venugopala and P. N. S. Pai, J. Pharmacol. Toxicol., 2007, 2, 481–488 CrossRef CAS.
- M. Koca, S. Servi, C. Kirilmis, M. Ahmedzade, C. Kazaz, B. Özbek and G. Ötük, Eur. J. Med. Chem., 2005, 40, 1351–1358 CrossRef CAS PubMed.
- L. Pieters, S. V. Dyck, M. Gao, R. Bai, E. Hamel, A. Vlietinck and G. Lemiére, J. Med. Chem., 1999, 42, 5475–5481 CrossRef CAS PubMed.
- K. M. Dawood, H. Abdel-Gawad, E. A. Rageb, M. Ellithey and H. A. Mohamed, Bioorg. Med. Chem., 2006, 14, 3672–3680 CrossRef CAS PubMed.
- K. Tsujihara, M. Hongu, K. Saito, H. Kawanishi, K. Kuriyama, M. Matsumoto, A. Oku, K. Ueta, M. Tsuda and A. Saito, J. Med. Chem., 1999, 42, 5311–5324 CrossRef CAS PubMed.
- Y. Liao, A. P. Kozikowski, A. Guidotti and E. Costa, Bioorg. Med. Chem. Lett., 1998, 8, 2099–2102 CrossRef CAS PubMed.
- A. H. Banskota, Y. Tezuka, K. Midorikawa, K. Matsushige and S. Kadota, J. Nat. Prod., 2000, 63, 1277–1279 CrossRef CAS.
- M. Ono, H. Kawashima, A. Nonaka, T. Kawai, M. Haratake, H. Mori, M. Kung, H. F. Kung, H. Saji and M. Nakayama, J. Med. Chem., 2006, 49, 2725–2730 CrossRef CAS PubMed.
- D. Manish, B. K. Tripathi, A. K. Tamrakar, A. K. Srivastava, B. Kumar and A. Goel, Bioorg. Med. Chem., 2007, 15, 727–734 CrossRef PubMed.
- D. H. Choi, J. W. Hwang, H. S. Lee, D. M. Yang and J. G. Jun, Bull. Korean Chem. Soc., 2008, 29, 1594–1596 CrossRef CAS.
- Z. Liu, G. Xia, S. Chen, Y. Liu, H. Li and Z. She, Mar. Drugs, 2014, 12, 3669–3680 CrossRef PubMed.
- F. Xu, Y. Zhang, J. Wang, J. Pang, C. Huang, X. Wu, Z. She, L. L. P. Vrijmoed, E. B. Gareth Jones and Y. Lin, J. Nat. Prod., 2008, 71, 1251–1253 CrossRef CAS PubMed.
- P. Dawson, J. Opacka-Juffry, J. D. Moffatt, Y. Daniju, N. Dutta, J. Ramsey and C. Davidson, Biol. Psychiatry, 2014, 48, 57–63 CAS.
- N. Maggi, C. R. Pasqualucci, R. Ballotta and P. Sensi, Chemotherapy, 1966, 11, 285–292 CrossRef CAS PubMed.
- M. Sofuoglu, P. S. Portoghese and A. E. Takemori, J. Pharmacol. Exp. Ther., 1991, 257, 676–680 CAS.
- G. V. Naccarelli, D. L. Wolbrette, H. M. Patel and J. C. Luck, Current Opinion in Cardiology, 2000, 15, 64–72 CrossRef CAS PubMed.
- S. Kathofer, D. Thomas and C. A. Karle, Cardiovasc. Drug Rev., 2005, 23, 217–230 CrossRef CAS PubMed.
- Q. Wu, L. A. Christensen, R. J. Legerski and K. M. Vasquez, EMBO Rep., 2005, 6, 551–557 CrossRef CAS PubMed.
- E. G. Schneiders and R. Stevensonm, J. Org. Chem., 1979, 44, 4710–4711 CrossRef.
- F. G. Schreiber and R. Stevenson, J. Chem. Soc., Perkin Trans. 1, 1976, 1, 1514–1518 RSC.
- R. L. Shriner and P. McCutchan, J. Am. Chem. Soc., 1929, 81, 2193–2195 CrossRef.
- M. W. Rathke, J. Am. Chem. Soc., 1970, 92, 3222–3223 CrossRef CAS.
- M. W. Rathke and D. F. Sullivan, J. Am. Chem. Soc., 1973, 95, 3050–3051 CrossRef CAS.
- S. Y. Lin, C.-L. Chen and Y.-J. Lee, J. Org. Chem., 2003, 68, 2968–2971 CrossRef CAS PubMed.
- W. S. Sheen, I. L. Tsai, C. M. Teng and I. S. Chen, Phytochemistry, 1994, 36, 213–215 CrossRef CAS.
- C. L. Kao and J. W. Chern, Tetrahedron Lett., 2001, 42, 1111–1113 CrossRef CAS.
- C. L. Kao and J. W. Chern, J. Org. Chem., 2002, 67, 6772–6787 CrossRef CAS PubMed.
- C. Fuganti and S. A. Serra, Tetrahedron Lett., 1998, 39, 5609–5610 CrossRef CAS.
- R. Basawaraj, B. Yadav and S. S. Sangapure, Indian J. Heterocycl. Chem., 2001, 11, 31–34 CAS.
- S. M. Rida, S. A. M. El-Hawash, H. T. Y. Fahmy, A. A. Hazzaa and M. M. M. El-Meligy, Arch. Pharmacal Res., 2006, 29, 826–833 CrossRef CAS.
- R. K. Ujjinamatada, R. S. Appala and Y. S. Agasimundin, J. Heterocycl. Chem., 2006, 43, 437–441 CrossRef CAS.
- S. Wachi, K. Takagi, G. Menichi and M. Hubert-Habart, Bull. Soc. Chem., 1978, 56, 230–233 Search PubMed.
- D. V. Singh, A. R. Mishra, R. M. Mishra, A. K. Pandey, C. R. Singh and A. K. Dwivedi, Indian J. Heterocycl. Chem., 2005, 14, 319–322 CAS.
- A. M. Farag, A. S. Mayhoub, S. E. Barakat and A. H. Bayomi, Bioorg. Med. Chem., 2008, 16, 4569–4578 CrossRef CAS PubMed.
- B. Insuasty, A. Tigreros, F. Orozco, J. Quiroga, R. Abonia, M. Nogueras, A. Sanchez and J. Cobo, Bioorg. Med. Chem., 2010, 18, 4965–4974 CrossRef CAS PubMed.
- A. M. Farag, A. S. Mayhoub, S. E. Barakat and A. H. Bayomi, Bioorg. Med. Chem., 2008, 16, 881–889 CrossRef CAS PubMed.
- I. M. El-Deeb and S. H. Lee, Bioorg. Med. Chem., 2010, 18, 3961–3973 CrossRef CAS PubMed.
- B. S. Holla, P. M. Akberali and M. K. Shivananda, Il Farmaco, 2000, 55, 256–263 CrossRef CAS PubMed.
- A. Mustafa, C. A. Hismat and M. M. J. Yannis, J. Prakt. Chem., 1970, 312, 1011–1019 CrossRef CAS.
- M. H. Elnagdi, M. R. H. Elmoghayor, E. A. A. Hafez and H. H. Alnima, J. Org. Chem., 1975, 40, 2604–2607 CrossRef CAS.
- H. G. Garg, J. Med. Chem., 1972, 15, 446–447 CrossRef CAS PubMed.
- F. Manna, F. Chementi, R. Fioravanti and A. Bolasco, Bioorg. Med. Chem. Lett., 2005, 15, 4632–4635 CrossRef CAS PubMed.
- J. H. Ahn, H. M. Kim, S. H. Jung, S. K. Kang, K. R. Kim, S. D. Rhea, S. D. Yong, H. G. Cheon and S. S. Kim, Bioorg. Med. Chem. Lett., 2004, 14, 4461–4465 CrossRef CAS PubMed.
- R. V. Ragavan, V. Vijayakumar and N. S. Kumari, Eur. J. Med. Chem., 2010, 45, 1173–1180 CrossRef CAS PubMed.
- Y. R. Prasad, A. L. Rao, L. Prasoona, K. Murali and K. P. Ravi, Bioorg. Med. Chem. Lett., 2005, 15, 5030–5034 CrossRef CAS PubMed.
- J. C. Jung, E. B. Watkins and M. A. Avery, Heterocycles, 2005, 65, 77–94 CrossRef CAS.
- A. E. Rashad, M. I. Hegab, R. E. Abdel-Megeid, J. A. Micky and F. M. E. Abdel-Megeid, Bioorg. Med. Chem., 2008, 16, 7102–7106 CrossRef CAS PubMed.
- P. D. Seemuth and H. Zimmer, J. Org. Chem., 1978, 43, 3063–3065 CrossRef CAS.
- B. Ledoussal, A. Gorgues and A. Le Coq, J. Chem. Soc., Chem. Commun., 1986, 171–172 RSC.
- L. Abmann and W. Friedrichsen, Heterocycles, 1989, 29, 1003–1004 CrossRef.
- A. Arcadi, S. Cacchi, M. Del Rosario, G. Fabrizi and F. Marinelli, J. Org. Chem., 1996, 61, 9280–9288 CrossRef CAS.
- I. Nakamura, Y. Mizushima and Y. Yamamoto, J. Am. Chem. Soc., 2005, 127, 15022–15023 CrossRef CAS PubMed.
- X. Z. Jiang, W. L. Liu, W. Zhang, F. Q. Jiang, Z. Gao, H. Zhuang and L. Fu, Eur. J. Med. Chem., 2011, 46, 3526–3530 CrossRef CAS PubMed.
- A. Hercouet and M. Le Corre, Tetrahedron, 1981, 37, 2867–2873 CrossRef CAS.
- L. Capuano, S. Drescher, V. Hammerer and M. Hanisch, Chem. Ber., 1988, 121, 2259–2261 CrossRef CAS.
- (a) S. Ghosh and J. Das, Tetrahedron Lett., 2011, 52, 1112–1116 CrossRef CAS; (b) S.-E. Syu, Y.-T. Lee, Y.-J. Jang and W. Lin, Org. Lett., 2011, 13, 2970–2973 CrossRef CAS PubMed.
- Y. Liu, H. K. Jacobs and A. S. Gopalan, Tetrahedron Lett., 2011, 52, 2935–2939 CrossRef CAS PubMed.
- D. C. Schroeder, P. O. Corcoran, C. A. Holden and M. C. Mulligan, J. Org. Chem., 1962, 27, 586–591 CrossRef CAS.
- G. Lamotte, P. Demerseman and R. Royer, Synthesis, 1984, 1068–1070 CrossRef CAS.
- W. T. Brady and Y. F. Giang, J. Org. Chem., 1986, 51, 2145–2147 CrossRef CAS.
- J. K. MacLeod and B. R. Worth, Tetrahedron Lett., 1972, 13, 237–240 CrossRef.
- J. Einhorn, P. Demerseman and R. Royer, J. Heterocycl. Chem., 1985, 22, 1243–1247 CrossRef CAS.
- G. Pandey, A. Krishna and U. T. Bhalerao, Tetrahedron Lett., 1989, 30, 1867–1870 CrossRef CAS.
- Y. Liu, J. Qian, S. Lou and Z. Xu, J. Org. Chem., 2010, 75, 6300–6303 CrossRef CAS PubMed.
- S. W. Youn and J. I. Eom, Org. Lett., 2005, 7, 3355–3358 CrossRef CAS PubMed.
- M. Murai, K. Miki and K. Ohe, Chem. Commun., 2009, 3466–3468 RSC.
- A. Hiremathad, M. R. Patil, K. R. Chethana, K. Chand, M. A. Santos and R. S. Keri, RSC Adv., 2015, 5, 96809–96828 RSC.
- G. Khodarahmi, P. Asadi, F. Hassanzadeh and E. Khodarahmi, J. Res. Med. Sci., 2015, 20, 1094–1104 CrossRef CAS PubMed.
- C. Selvam, B. C. Jordan, S. Prakash, D. Mutisya and R. Thilagavathi, Eur. J. Med. Chem., 2017, 128, 219–236 CrossRef CAS PubMed.
- K. C. Rajeshwari, A. Hiremathad, M. Singh, M. A. Santos and R. S. Keri, Pharmacol. Rep., 2017, 69, 281–295 CrossRef PubMed.
- (a) M. M. Heravi and B. Talaei, Adv. Heterocycl. Chem., 2016, 118, 195–291 CrossRef; (b) M. M. Heravi and B. Talaei, Adv. Heterocycl. Chem., 2015, 114, 147–225 CrossRef CAS; (c) M. M. Heravi and V. Fathi Vavsari, Adv. Heterocycl. Chem., 2015, 114, 77–145 CrossRef CAS; (d) M. M. Heravi and B. Talaei, Adv. Heterocycl. Chem., 2014, 113, 143–244 CrossRef CAS.
- M. M. Heravi and V. Zadsirjan, Adv. Heterocycl. Chem., 2015, 117, 261–376 CrossRef.
- M. M. Heravi, S. Khaghaninejad and N. Nazari, Adv. Heterocycl. Chem., 2014, 112, 183–234 CrossRef CAS.
- M. M. Heravi, S. Khaghaninejad and M. Mostofi, Adv. Heterocycl. Chem., 2014, 112, 1–50 CrossRef CAS.
- S. Khaghaninejad and M. M. Heravi, Adv. Heterocycl. Chem., 2014, 111, 95–146 CrossRef CAS.
- M. M. Heravi and T. Alishiri, Adv. Heterocycl. Chem., 2014, 113, 1–66 CrossRef CAS.
- M. M. Heravi and B. Talaei, Adv. Heterocycl. Chem., 2017, 122, 43–114 CrossRef.
- V. Zadsirjan, M. Shiri, M. M. Heravi, T. Hosseinnejad, S. A. Shintre and N. A. Koorbanally, Res. Chem. Intermed., 2017, 43, 2119–2142 CrossRef CAS.
- (a) R. Mirsafaei, M. M. Heravi and S. Ahmadi, J. Mol. Catal. A: Chem., 2015, 402, 100–108 CrossRef CAS; (b) M. M. Heravi, E. Hashemi, Y. S. Beheshtiha, S. Ahmadi and T. Hosseinnejad, J. Mol. Catal. A: Chem., 2014, 394, 74–82 CrossRef CAS.
- M. M. Heravi, E. Hashemi, Y. S. Beheshtiha, K. Kamjou, M. Toolabi and N. Hosseintash, J. Mol. Catal. A: Chem., 2014, 392, 173–180 CrossRef CAS.
- M. M. Heravi, F. Mousavizadeh, N. Ghobadi and M. Tajbakhs, Tetrahedron Lett., 2104, 55, 1226–1228 CrossRef.
- M. M. Heravi, M. Khorasani and F. Derikvand, Catal. Commun., 2007, 8, 1886–1890 CrossRef CAS.
- M. M. Heravi, M. Daraie and V. Zadsirjan, Mol. Diversity, 2015, 15, 577–623 CrossRef PubMed.
- M. M. Heravi and V. F. Vavsari, RSC Adv., 2015, 5, 50890–50912 RSC.
- M. M. Heravi, T. B. Lashaki and N. Poorahmad, Tetrahedron: Asymmetry, 2015, 26, 405–495 CrossRef CAS.
- M. M. Heravi, E. Hashemi and N. Nazari, Mol. Diversity, 2014, 18, 441–472 CrossRef CAS PubMed.
- M. M. Heravi, E. Hashemi and F. Azimian, Tetrahedron, 2014, 70, 7–21 CrossRef CAS.
- M. M. Heravi, E. Hashemi and N. Ghobadi, Curr. Org. Chem., 2013, 17, 2192–2224 CrossRef CAS.
- M. M. Heravi and E. Hashemi, Tetrahedron, 2012, 68, 9145–9178 CrossRef CAS.
- M. M. Heravi, V. Zadsirjan and M. Dehghani, Curr. Org. Chem., 2016, 20, 1069–1134 CrossRef CAS.
- M. M. Heravi and V. Zadsirjan, Curr. Org. Synth., 2016, 13, 780–833 CrossRef CAS.
- R. S. Ward, Chem. Soc. Rev., 1982, 11, 75–125 RSC.
- R. S. Ward, Tetrahedron, 1990, 46, 5029–5041 CrossRef CAS.
- Z. Yang, H. B. Liu, C. M. Lee, H. M. Chang and H. N. C. Wong, J. Org. Chem., 1992, 57, 7248–7257 CrossRef CAS.
- S. A. Hutchinson, H. Liitjens and P. J. Scammells, Bioorg. Med. Chem. Lett., 1997, 7, 3081–3084 CrossRef CAS.
- P. J. Scammells, S. P. Baker and A. R. Beauglehole, Bioorg. Med. Chem., 1998, 6, 1517–1524 CrossRef CAS PubMed.
- Y. H. Kuo and C. H. Wu, J. Nat. Prod., 1996, 59, 625–628 CrossRef CAS.
- M. M. Heravi and S. Sadjadi, Tetrahedron, 2009, 65, 7761–7775 CrossRef CAS.
- H. Lütjens and P. J. Scammells, Tetrahedron Lett., 1998, 39, 6581–6584 CrossRef.
- M. Inoue, M. W. Carson, A. J. Frontier and S. J. Danishefsky, J. Am. Chem. Soc., 2001, 123, 1878–1889 CrossRef CAS PubMed.
- R. Segal, I. M. Goldzweig, S. Sokolo and D. V. Zaitschek, J. Chem. Soc. C, 1967, 2402–2404 RSC.
- (a) Q. L. Li, B. G. Li, H. Y. Qi, X. P. Gao and G. L. Zhang, Planta Med., 2005, 71, 847–851 CrossRef CAS PubMed; (b) M. Takanashi, Y. Takizawa and T. Mitsuhashi, Chem. Lett., 1974, 869 CrossRef CAS.
- T. Hirano, M. Goto and K. Oka, Life Sci., 1994, 55, 1061–1069 CrossRef CAS PubMed.
- M. M. Heravi and Z. Faghihi, Curr. Org. Chem., 2012, 16, 2097–2123 CrossRef CAS.
- X. F. Duan, G. Shen and Z. B. Zhang, Synthesis, 2010, 7, 1181–1187 CrossRef.
- C. E. Castro, E. J. Gaughan and D. C. Owsley, J. Org. Chem., 1966, 31, 4071 CrossRef CAS.
- R. D. Stephens and C. E. Castro, J. Org. Chem., 1963, 28, 3313–3315 CrossRef CAS.
- (a) A. Galet, J. Am. Chem. Soc., 1946, 68, 376–377 CrossRef; (b) H. Hellmann and W. Elser, Justus Liebigs Annalen der Chemie, 1961, 639, 77 CrossRef CAS.
- Z. Yang, P. M. Hon, K. Y. Chui, Z. L. Xu, H. M. Chang, C. M. Lee, Y. X. Cui, H. N. C. Wong, C. D. Poon and B. M. Fung, Tetrahedron Lett., 1991, 32, 2061–2064 CrossRef CAS.
- H. B. Bang, S. Y. Han, D. H. Choi, D. M. Yang, J. W. Hwang, H. S. Lee and J.-G. Jun, Synth. Commun., 2009, 39, 506–515 CrossRef CAS.
- K. Kawazu, Agric. Biol. Chem., 1980, 44, 1367–1372 CAS.
- Y. Fukuyama, M. Nakahara, H. Minami and M. Kodama, Chem. Pharm. Bull., 1996, 44, 1418–1420 CrossRef.
- (a) F. Toda and M. Nakagawa, Bull. Chem. Soc. Jpn., 1959, 32, 514–516 CrossRef CAS; (b) I. Candiani, S. DeBemardinis, W. Cabri, M. Marchi, A. Bedeschi and S. Penco, Synlett, 1993, 9–69 Search PubMed; (c) K. Hiroya, K. Hashimura and K. Ogasawara, Heterocycles, 1994, 38, 2463–2472 CrossRef CAS.
- A. Sakai, T. Aoyama and T. Shioiri, Tetrahedron Lett., 1999, 40, 4211–4214 CrossRef CAS.
- H.-D. Choi, P. J. Seo and B.-W. Son, Arch. Pharmacal Res., 2002, 25, 786–789 CrossRef CAS.
- (a) B. F. Bowden, E. Ritchie and W. C. Taylor, Aust. J. Chem., 1972, 25, 2659–2669 CrossRef CAS; (b) K. Picker, E. Ritchie and W. C. Taylor, Aust. J. Chem., 1973, 26, 1111–1119 CrossRef CAS; (c) R. W. Read and W. C. Taylor, Aust. J. Chem., 1979, 32, 2317–2321 CrossRef CAS; (d) A. R. Carroll and W. C. Taylor, Aust. J. Chem., 1991, 44, 1615–1626 CrossRef CAS; (e) A. R. Carroll and W. C. Taylor, Aust. J. Chem., 1991, 44, 1627–1633 CrossRef CAS.
- T. A. Engler and W. Chai, Tetrahedron Lett., 1996, 37, 6969–6970 CrossRef CAS.
- W. J. Gensler and J. E. Stouffer, J. Org. Chem., 1958, 23, 908–910 CrossRef CAS.
- T. Bach and M. Bartels, Tetrahedron Lett., 2002, 43, 9125–9127 CrossRef CAS.
- (a) R. Ahmed and R. Stevenson, Phytochemistry, 1975, 14, 2710–2712 CrossRef CAS; (b) B. A. McKittrick and R. Stevenson, J. Chem. Soc., Perkin Trans. 1, 1983, 475–482 RSC; (c) T. A. Engler, W. Chai and K. O. LaTessa, J. Org. Chem., 1996, 61, 9297–9308 CrossRef CAS; (d) T. A. Engler and W. Chai, Tetrahedron Lett., 1996, 37, 6969–6970 CrossRef CAS.
- T. Benincori, E. Brenna, F. Sannicolo, L. Trimarco, P. Antognazza, E. Cesarotti, F. Demartin and T. Pilati, J. Org. Chem., 1996, 61, 6244–6251 CrossRef CAS PubMed.
- T. Bach and M. Bartels, Synthesis, 2003, 6, 925–939 CrossRef.
- (a) T. J. Jackson and J. W. Herndon, Tetrahedron, 2001, 57, 3859–3868 CrossRef CAS; (b) J. W. Herndon, Y. Zhang, H. Wang and K. Wang, Tetrahedron Lett., 2000, 41, 8687–8690 CrossRef CAS; (c) J. W. Herndon and A. Hayford, Organometallics, 1995, 14, 1556–1558 CrossRef CAS; (d) J. W. Herndon and H. Wang, J. Org. Chem., 1998, 63, 4562–4563 CrossRef CAS; (e) Y. Zhang and J. W. Herndon, Tetrahedron, 2000, 56, 2175–2184 CrossRef CAS; (f) M. L. Waters, M. E. Bos and W. D. Wulff, J. Am. Chem. Soc., 1999, 121, 6403–6413 CrossRef CAS.
- A. Rahm and W. D. Wulff, J. Am. Chem. Soc., 1996, 118, 1807–1808 CrossRef CAS.
- R. S. Ward, Nat. Prod. Rep., 1999, 16, 75–96 RSC.
- (a) R. S. Mali and A. P. Massey, J. Chem. Res., Synop., 1998, 230–231 RSC; (b) Y. Aoyagi, T. Mizusaki, A. Hatori, T. Asakura, T. Aihara, S. Inaba, K. Hayatsu and A. Ohta, Heterocycles, 1995, 41, 1077–1084 CrossRef CAS; (c) F. G. Schreiber and R. Stevenson, Chem. Lett., 1975, 1257–1258 CrossRef CAS; (d) E. Ritchie and R. Taylor, Aust. J. Chem., 1969, 1329–1330 CrossRef CAS.
- A. M. Puentes de Diaz, Phytochemistry, 1997, 44, 345–346 CrossRef CAS.
- A. De Meijere, H. Schirmer and M. Duetsch, Angew. Chem., Int. Ed., 2000, 39, 3964–4002 CrossRef CAS PubMed.
- K. H. Dotz and P. Tomuschat, Chem. Soc. Rev., 1999, 28, 187–198 RSC.
- A. De Meijere, Pure Appl. Chem., 1996, 68, 61–72 CrossRef CAS.
- G. B. Stone and L. S. Liebeskind, J. Org. Chem., 1990, 55, 4614–4622 CrossRef CAS.
- J. Zhang, Y. Zhang, Y. Zhang and J. W. Herndon, Tetrahedron, 2003, 59, 5609–5616 CrossRef CAS.
- (a) T. O. Cheng, Int. J. Cardol., 2007, 121, 9–22 CrossRef PubMed; (b) X. B. Wang, S. L. Morris-Natschke and K. H. Lee, Med. Res. Rev., 2007, 27, 133–148 CrossRef CAS PubMed; (c) L. Zhou, Z. Zuo and M. S. Chow, J. Clin. Pharmacol., 2005, 45, 1345–1359 CrossRef CAS PubMed.
- (a) Y. R. Lu and L. Y. Foo, Tetrahedron Lett., 2001, 42, 8223–8225 CrossRef CAS; (b) X. N. Yang, Y. J. Wang, Y. S. Liu and X. Tang, J. Ethnopharmacol., 2008, 117, 408–414 CrossRef CAS PubMed; (c) X. S. Ouyang, K. Takahashi, K. Komatsu, N. Nakamura, A. H. Baba and J. Azuma, Jpn. J. Pharmacol., 2001, 87, 289–296 CrossRef CAS PubMed; (d) Y. T. Wu, Y. F. Chen, Y. J. Hsieh, I. Jaw, M. S. Shiao and T. H. Tsai, Int. J. Pharm., 2006, 326, 25–31 CrossRef CAS PubMed; (e) J. F. Zhao, C. H. Liu, Y. Y. Hu, L. M. Xu, P. Liu and C. Liu, Hepatobiliary Pancreatic Dis. Int., 2004, 3, 102–105 CAS.
- Y. L. Lin, Y. Y. Chang, Y. H. Kuo and M. S. Shiao, J. Nat. Prod., 2002, 65, 745–747 CrossRef CAS.
- (a) C. H. Cho, B. Neuenswander, G. H. Lushington and R. C. Larock, J. Comb. Chem., 2008, 10, 941–947 CrossRef CAS PubMed; (b) H. B. Bang, S. Y. Han, D. H. Choi, J. W. Hwang and J. G. Jun, ARKIVOC, 2009, 2, 112–125 Search PubMed; (c) J. R. Hwu, K. S. Chuang, S. H. Chuang and S. C. Tsay, Org. Lett., 2005, 7, 1545–1548 CrossRef CAS PubMed.
- S. D. Shen, G. P. Zhang, M. Leib and L. H. Hu, ARKIVOC, 2012, 4, 204–213 Search PubMed.
- N. Takeda, O. Miyata and T. Naito, Eur. J. Org. Chem., 2007, 1491–1509 CrossRef CAS.
- T. Pacher, C. Seger, D. Engelmeier, S. Vajrodaya, O. Hofer and H. Greger, J. Nat. Prod., 2002, 65, 820–827 CrossRef CAS.
- D. C. Chauret, C. B. Berrnard, J. T. Arnason and T. J. Durst, J. Nat. Prod., 1996, 59, 152–155 CrossRef CAS PubMed.
- S. B. Pandit and S. Y. Gadre, Synth. Commun., 1988, 18, 157–166 CrossRef CAS.
- J. Y. Pasturel, G. Solladie and J. Maignan, Chem. Abstr., 2003, 139, 36375–36377 Search PubMed.
- E. M. Bickoff, R. L. Lyman, A. L. Livingston and A. N. Booth, J. Am. Chem. Soc., 1958, 80, 3969–3971 CrossRef CAS.
- G. A. Kraus and N. Zhang, J. Org. Chem., 2000, 65, 5644–5646 CrossRef CAS PubMed.
- Y. R. Lee, J. Y. Suk and B. S. Kim, Org. Lett., 2000, 2, 1387–1389 CrossRef CAS PubMed.
- (a) I. Muhammad, X.-C. Li, M. R. Jacob, B. L. Tekwani, D. C. Dunbar and D. Ferreira, J. Nat. Prod., 2003, 66, 804–809 CrossRef CAS PubMed; (b) I. Muhammad, X.-C. Li, D. C. Dunbar, M. A. ElSohly and I. A. Khan, J. Nat. Prod., 2001, 64, 1322–1325 CrossRef CAS PubMed.
- (a) O. Miyata, N. Takeda and T. Naito, Org. Lett., 2004, 6, 1761–1763 CrossRef CAS PubMed; (b) N. Takeda, O. Miyata and T. Naito, Eur. J. Org. Chem., 2007, 1491–9150 CrossRef CAS.
- H. J. Lee, Y. R. Lee and S. H. Kim, Helv. Chim. Acta, 2009, 92, 1404–1412 CrossRef CAS.
- J. D. Lambert, R. O. Meyers, B. N. Timmermann and R. T. Dorr, Cancer Lett., 2001, 171, 47–56 CrossRef CAS PubMed.
- (a) O. Dann, J. Lang and H. Vohl, Justus Liebigs Ann. Chem., 1960, 631, 116–128 CrossRef CAS; (b) S. Johann, B. B. Cota, E. M. Souza-Fagundes, M. G. Pizzolatti, M. A. Resende and C. L. Zani, Mycoses, 2009, 52, 499–506 CrossRef CAS PubMed.
- L. Ruan, M. Shi, S. Mao, L. Yu, F. Yang and J. Tang, Tetrahedron, 2014, 70, 1065–1070 CrossRef CAS.
- H. D. Choi, M. C. Ha, P. J. Seo, B. W. Son and J. C. Song, Arch. Pharmacal Res., 2000, 23, 438–440 CrossRef CAS.
- H. Okada, J. Pharm. Soc. Jpn., 1915, 657–666 CAS.
- C. Y. Hopkins, D. F. Ewing and M. J. Chisholm, Can. J. Chem., 1967, 45, 1425–1429 CrossRef CAS.
- Y. Luo, Z. He and H. Li, Fitoterapia, 2007, 78, 211–214 CrossRef CAS PubMed.
- S. Kawai, T. Nakamura and N. Sugiyama, Ber. Dtsch. Chem. Ges. A, 1939, 72, 1146–1149 CrossRef.
- M. Takanashi and Y. Takizawa, Phytochemistry, 1988, 27, 1224–1226 CrossRef CAS.
- E. W. Colvin and B. J. Hamill, J. Chem. Soc., Perkin Trans. 1, 1977, 869–874 RSC.
- H. L. Teles, J. P. Hemerly, P. M. Pauletti, J. R. C. Pandolfi, A. R. Araujo, S. R. Valentini, M. C. M. Young, V. S. Bolzani and D. H. S. Silva, Nat. Prod. Res., 2005, 19, 319–323 CrossRef CAS PubMed.
- X. F. Duan, J. X. Feng and Z. B. Zhang, Synthesis, 2010, 3, 0515–0519 CrossRef.
- Y. Liu, M. Kubo and Y. Fukuyama, J. Nat. Prod., 2012, 75, 2152–2157 CrossRef CAS PubMed.
- (a) I.-K. Lee, J.-H. Lee and B.-S. Yun, Bioorg. Med. Chem. Lett., 2008, 18, 4566–4568 CrossRef CAS PubMed; (b) Y. Liu and F. Wang, Carbohydr. Polym., 2007, 70, 386–392 CrossRef CAS.
- P. Lan, M. G. Banwell and A. C. Willis, J. Org. Chem., 2014, 79, 2829–2842 CrossRef CAS PubMed.
- (a) M. G. Banwell, B. L. Flynn and S. G. Stewart, J. Org. Chem., 1998, 63, 9139–9144 CrossRef CAS; (b) B. L. Flynn and M. G. Banwell, Heterocycles, 2012, 84, 1141–1170 CrossRef CAS.
- C. Zhang, J. Liu and Y. Du, Tetrahedron Lett., 2014, 55, 959–961 CrossRef CAS.
- (a) M. Reiter, S. Ropp and V. Gouverneur, Org. Lett., 2004, 6, 91–94 CrossRef CAS PubMed; (b) M. Reiter, H. Turner, R. Mills-Webb and V. Gouverneur, J. Org. Chem., 2005, 70, 8478–8485 CrossRef CAS PubMed; (c) X. Liao, H. Zhou, X. Z. Wearing, J. Ma and J. M. Cook, Org. Lett., 2005, 7, 3501–3504 CrossRef CAS PubMed; (d) T. Hayashi, K. Yamasaki, M. Mimura and Y. Uozumi, J. Am. Chem. Soc., 2004, 126, 3036–3037 CrossRef CAS PubMed.
- P. C. Stevenson and N. C. Veitch, Phytochemistry, 1998, 48, 947–951 CrossRef CAS.
- Z. Novak, G. Timari and A. Kotschy, Tetrahedron, 2003, 59, 7509–7513 CrossRef CAS.
- N. G. Kundu, M. Pal, J. S. Mahanty and M. De, J. Chem. Soc., Perkin Trans. 1, 1997, 2815–2820 RSC.
- A. Arcadi and E. Marinelli, Synthesis, 1986, 749–751 CrossRef CAS.
- S. Torii, L. H. Xu and H. Okumoto, Synlett, 1992, 515–516 CrossRef CAS.
- S. N. Aslam, P. C. Stevenson, S. J. Phythian, N. C. Veitch and D. R. Hall, Tetrahedron, 2006, 62, 4214–4226 CrossRef CAS.
- R. T. Scannell and R. Stevenson, J. Heterocycl. Chem., 1980, 17, 1727–1728 CrossRef CAS.
- Y. Jiang, B. Gao, W. Huang, Y. Liang, G. Huang and Y. Ma, Synth. Commun., 2009, 39, 197–204 CrossRef CAS.
- R. Stevenson and B. A. McKittrick, J. Chem. Soc., Perkin Trans. 1, 1983, 475–482 Search PubMed.
- T. Bach and M. Bartels, Synthesis, 2003, 925–939 CrossRef CAS.
- C. Eidamshaus and J. D. Burch, Org. Lett., 2008, 10, 4211–4214 CrossRef CAS PubMed.
- J. Jiang, J. B. Biggins and J. S. Thorson, J. Am. Chem. Soc., 2000, 122, 6803–6804 CrossRef CAS.
- J. M. Langenhan, B. R. Griffith and J. S. Thorson, J. Nat. Prod., 2005, 68, 1696–1711 CrossRef CAS PubMed.
- C. J. Thibodeaux, C. E. Melancon and H. W. Liu, Angew. Chem., Int. Ed., 2008, 47, 9814–9859 CrossRef CAS PubMed.
- S. M. Patel, M. d. l. Fuente, S. Ke and A. M. R. Guimaraes, Chem. Commun., 2011, 47, 10569–10571 RSC.
- C. Albermann, A. Soriano, J. Jiang and H. Vollmer, Org. Lett., 2003, 5, 933–936 CrossRef CAS PubMed.
- H. Cheng, X. Cao, M. Xian and L. Fang, J. Med. Chem., 2004, 48, 645–652 CrossRef PubMed.
- L. Morelli, A. Bernardi and S. Sattin, Carbohydr. Res., 2014, 390, 33–41 CrossRef CAS PubMed.
- (a) H. B. Bode and A. Zeeck, J. Chem. Soc., Perkin Trans. 1, 2000, 323–328 RSC; (b) H. B. Bode and A. Zeeck, J. Chem. Soc., Perkin Trans. 1, 2000, 2665–2670 RSC.
- Y. Yu, H. Men and C. Lee, J. Am. Chem. Soc., 2004, 126, 14720–14721 CrossRef PubMed.
- T. Magauer, H. J. Martin and J. Mulzer, Angew. Chem., Int. Ed., 2009, 48, 6032–6036 CrossRef CAS PubMed.
- T. Magauer, H. J. Martin and J. Mulzer, Chem.–Eur. J., 2010, 16, 507–519 CrossRef CAS PubMed.
- P. Magnus and C. Lescop, Tetrahedron Lett., 2001, 42, 7193–7196 CrossRef CAS.
- L. Hoffmeister, P. Persich and A. Fürstner, Chem.–Eur. J., 2014, 20, 4396–4402 CrossRef CAS PubMed.
- F. Marion, D. E. Williams, D. O. Patrick, I. Hollander, R. Mallon, S. C. Kim, D. M. Roll, L. Feldberg, R. V. Soest and R. J. Andersen, Org. Lett., 2006, 8, 321–324 CrossRef CAS PubMed.
- H. Landelle, A. M. Godard, D. Laduree, E. Chenu and M. Robba, Chem. Pharm. Bull., 1991, 39, 3057–3060 CrossRef CAS.
- G. Mehta, N. S. Likhite and C. S. A. Kumar, Tetrahedron Lett., 2009, 50, 5260–5262 CrossRef CAS.
- A. C. Anderson and D. L. Wright, Org. Biomol. Chem., 2009, 7, 840–850 Search PubMed.
- J. H. George, J. E. Baldwin and R. M. Adington, Org. Lett., 2010, 12, 2394–2397 CrossRef CAS PubMed.
- (a) B. Vanhaesebroeck, L. Stephens and P. Hawkins, Nat. Rev. Mol. Cell Biol., 2012, 13, 195–203 CrossRef CAS PubMed; (b) M. P. Wymann and C. Schultz, ChemBioChem, 2012, 13, 2022–2035 CrossRef CAS PubMed.
- (a) V. Deore, M. Kumar Lohar, R. Mundada, A. Roychowdhury, R. Vishwakarma and S. Kumar, Synth. Commun., 2011, 41, 177–183 CrossRef CAS; (b) T. Kamishima, T. Kikuchi, K. Narita and T. Katoh, Eur. J. Org. Chem., 2014, 3443–3450 CrossRef CAS.
- (a) A. Shirata, K. Takahashi, M. Taksugi, S. Nago, S. Ishikawa, S. Ueno, L. Munoz and T. Masamune, Bull. Seric. Exp. Stn., 1983, 28, 793 CAS; (b) F. Ferrari, F. D. Monache, R. S. Compagnone, A. I. Suarez and S. Tillett, Fitoterapia, 1998, 69, 554–555 CAS; (c) L. Cui, M. Na, H. Oh, E. Y. Bae, D. G. Jeong, S. E. Ryu, S. Kim, B. Y. Kim, W. K. Oh and J. S. Ahn, Bioorg. Med. Chem. Lett., 2006, 16, 1426–1429 CrossRef CAS PubMed.
- R. Kakarla, M. Ghosh, J. A. Anderson, R. G. Dulina and M. J. Sofia, Tetrahedron Lett., 1999, 40, 5–8 CrossRef CAS.
- N. Kaur, Y. Xia, Y. Jin, N. T. Dat, K. Gajulapati, Y. Choi, Y. S. Hong, J. J. Lee and K. Lee, Chem. Commun., 2009, 1879–1881 RSC.
- T. Gibtner, F. Hampel, J. P. Gisselbrecht and A. Hirsch, Chem.–Eur. J., 2002, 8, 408–432 CrossRef CAS.
- Y. Xia, Y. Jin, N. Kaur, Y. Choi and K. Lee, Eur. J. Med. Chem., 2011, 46, 2386–2396 CrossRef CAS PubMed.
- A. J. Weinheimer and P. H. Washecheck, Tetrahedron Lett., 1969, 3315–3318 CrossRef CAS.
- F. Kido, Y. Noda, T. Maruyama, C. Kabuto and A. Yoshikoshi, J. Org. Chem., 1981, 46, 4264–4266 CrossRef CAS.
- S. Danishefsky, J. F. Kerwin and S. Kobayashi, J. Am. Chem. Soc., 1982, 104, 358–360 CrossRef CAS.
- S. Danishefsky, E. R. Larson and D. Askin, J. Am. Chem. Soc., 1982, 104, 6457–6458 CrossRef CAS.
- R. B. Gammill and B. R. Hyde, J. Org. Chem., 1983, 48, 3863–3865 CrossRef CAS.
- S. Narayanaswamy, S. Rangaswami and T. R. Scshadri, J. Chem. Soc. C, 1954, 187l–l873 Search PubMed.
- A. Pelter, R. S. Ward, E. V. Rao and N. R. Raju, J. Chem. Soc., Perkin Trans. 1, 1981, 2491–2498 RSC.
- S. Rangaswami and V. Sasuy, Curr. Sci., 1955, 24, 13–14 CAS.
- P. Rajani and P. N. Sarma, Phytochemistry, 1988, 27, 648–649 CrossRef CAS.
- V. S. Parmar, J. S. Ratho, R. Jain, D. A. Henderson and J. F. Malone, Phytochemistry, 1989, 28, 59l–593 CrossRef.
- T. Tanaka, M. Iinuma, K. Yuki, Y. Fujii and M. Mizuno, Phytochemistry, 1992, 31, 993–998 CrossRef CAS.
- S. Rangaswti and B. V. Ramasastry, Bull Inst. Nat. Sci., 1955, 4, 149 Search PubMed.
- M. C. Pirrung and Y. R. Lee, Tetrahedron Lett., 1994, 35, 6231–6234 CrossRef CAS.
- M. Niwa, K. Terashima and M. Aqil, Heterocycles, 1993, 36, 671–673 CrossRef CAS.
- M. Niwa, K. Terashima, J. Ito and M. Aqil, Heterocycles, 1994, 38, 1071–1076 CrossRef CAS.
- T. R. Kelly, A. Szabados and Y.-J. Lee, J. Org. Chem., 1997, 62, 428–429 CrossRef CAS PubMed.
- M. Forbes, F. Zilliken, G. Roberts and P. Gyrgy, J. Am. Chem. Soc., 1958, 80, 385–389 CrossRef CAS.
- M. A. P. Meisinger, F. A. Kuehl, E. L. Rickes, N. G. Brink, K. Folkers, M. Forbes, F. Zilliken, G. Roberts and P. Gyorgy, J. Am. Chem. Soc., 1959, 81, 4979–4982 CrossRef CAS.
- A. F. Wagner, E. Walton, A. N. Wilson, J. O. Rodin, F. W. Holly, N. G. Brink and K. Folkers, J. Am. Chem. Soc., 1959, 81, 4983–4989 CrossRef CAS.
- B. A. McKittrick and R. Stevenson, J. Chem. Soc., Perkin Trans. 1, 1984, 709–712 RSC.
- S. Jinno, T. Okita and K. Inouye, Bioorg. Med. Chem. Lett., 1999, 9, 1029–1032 CrossRef CAS PubMed.
- M. V. Sargent, P. O. Stransky, V. A. Patrick and A. H. White, J. Chem. Soc., Perkin Trans. 1, 1983, 231–239 RSC.
- T. Sawada, M. Aono, S. Asakawa, A. Ito and K. Awano, J. Antibiot., 2000, 53, 959–966 CrossRef CAS PubMed.
- A. D. Patil, A. J. Freyer, L. Killmer, P. Offen, B. Carte, A. J. Jurewicz and R. K. Johnson, Tetrahedron, 1997, 53, 5047–5060 CrossRef CAS.
- R. C. Hock, I. U. Schraustätter and C. G. Cochrane, J. Lab. Clin. Med., 1996, 128, 134–145 CrossRef.
- M. Inoue, A. J. Frontier and S. J. Danishefsky, Angew. Chem., 2000, 112, 777–780 CrossRef.
- S. H. von Reuß and W. A. König, Phytochemistry, 2004, 65, 3113–3118 CrossRef PubMed.
- A. G. Ober, F. R. Fronczek and N. H. Fischer, J. Nat. Prod., 1985, 48, 242–248 CrossRef CAS.
- A. G. González, B. M. Fraga, M. G. Hernández and V. P. García, Phytochemistry, 1982, 21, 1826–1827 CrossRef.
- M. d. Carmen Cruz and J. Tamariz, Tetrahedron, 2005, 61, 10061–10072 CrossRef.
- D. Bethea, B. Fullmer, S. Syed, G. Seltzer, J. Tiano, C. Rischko, L. Gillespie, D. Brown and F. P. Gasparro, J. Dermatol. Sci., 1999, 19, 78–88 CrossRef CAS PubMed.
- K. M. Grundmann, R. Ludwig, T. M. Zollner, F. Ochsendorf, D. Thaci, W. H. Boehncke, J. Krutmann, R. Kaufmann and M. Podda, J. Am. Acad. Dermatol., 2004, 50, 734–739 CrossRef.
- (a) P. E. Grimes, Clin. Dermatol., 1997, 15, 921–926 CrossRef CAS PubMed; (b) H. Petering, C. Breuer, R. Herbst, A. Kapp and T. Werfel, J. Am. Acad. Dermatol., 2004, 50, 68–72 CrossRef PubMed.
- (a) P. V. M. M. Diederen, H. Weelden, C. J. G. Sanders, J. Toonstra and W. A. Vloten, J. Am. Acad. Dermatol., 2003, 48, 215–219 CrossRef PubMed; (b) G. Miolo, R. Tomanin, A. D. Rossi, A. F. Dall, F. Zacchello and M. Scarpa, J. Photochem. Photobiol., B, 1994, 26, 241–247 CrossRef CAS; (c) C. Lage, M. Pádula, T. A. M. Alencar, S. R. F. Gonçalves, L. S. Vidal, J. Cabral-Neto and A. C. Leitão, Mutat. Res., Rev. Mutat. Res., 2003, 544, 143–157 CrossRef CAS.
- G. D. Cimino, H. B. Gamper, S. T. Isaacs and J. E. Hearst, Annu. Rev. Biochem., 1985, 54, 1151–1193 CrossRef CAS PubMed.
- R. L. Edelson, Arch. Dermatol., 1999, 135, 600–601 CrossRef CAS.
- (a) J. J. Goupil, French Patent, 2, 698, 270–319, 1994; (b) J. L. Goaster, French Patent, 691, 629–691, 1993.
- A. Wiesmann, A. Weller, G. Lischka, T. Klingebiel, L. Kanz and H. Einsele, Bone Marrow Transplant., 1999, 23, 151–155 CAS.
- P. Ceccherelli, M. Curini, M. C. Marcotullio, F. Epifano and O. Rosati, Synth. Commun., 1998, 28, 3057–3064 CrossRef CAS.
- D. Dauzonne and R. Royer, Synthesis, 1984, 1054–1057 CrossRef CAS.
- P. J. Coleman, K. M. Brashear, B. C. Askew, J. H. Hutchinson, C. A. McVean, L. T. Duong, B. P. Feuston, C. Fernandez-Metzler, M. A. Gentile, G. D. Hartman, D. B. Kimmel, C. T. Leu, L. Lipfert, K. Merkle, B. Pennypacker, T. Prueksaritanont, G. A. Rodan, G. A. Wesolowski, S. B. Rodan and M. E. Duggan, J. Med. Chem., 2004, 47, 4829–4837 CrossRef CAS PubMed.
- B. L. Zhang, F. D. Wang and J. M. Yue, Synlett, 2006, 4, 567–570 Search PubMed.
- J. R. Carney and P. J. Scheuer, Tetrahedron Lett., 1993, 34, 3727–3730 CrossRef CAS.
- K. Ishiguro, Y. Ohira and H. Oku, J. Nat. Prod., 1998, 61, 1126–1129 CrossRef CAS PubMed.
- A. V. B. Sankaram, V. V. N. Reddy and G. S. Sidhu, Phytochemistry, 1981, 20, 1093–1096 CrossRef CAS.
- T. Takeya, H. Kondo, T. Otsuka, K. Tomita, I. Okamoto and O. Tamura, Org. Lett., 2007, 9, 2807–2810 CrossRef CAS PubMed.
- (a) H. Tanaka, M. Sato, T. Oh-Uchi, R. Yamaguchi, H. Etoh, H. Shimizu, M. Sako and H. Takeuchi, Phytomedicine, 2004, 11, 331–337 CrossRef CAS PubMed; (b) H. Tanaka, M. Sato, S. Fujiwara, M. Hirata, H. Etoh and H. Takeuchi, Lett. Appl. Microbiol., 2002, 35, 494–498 CrossRef CAS PubMed.
- F. L. Weitl, J. Org. Chem., 1976, 41, 2044–2045 CrossRef CAS PubMed.
- A. Fürstner and P. W. Davies, J. Am. Chem. Soc., 2005, 127, 15024–15025 CrossRef PubMed.
- (a) A. Fürstner, P. W. Davies and T. Gress, J. Am. Chem. Soc., 2005, 127, 8244–8245 CrossRef PubMed; (b) A. FPrstner and C. A. Wssa, J. Am. Chem. Soc., 2006, 128, 6306–6307 CrossRef PubMed.
- A. Furstner, E. K. Heilmann and P. W. Davies, Angew. Chem., Int. Ed, 2007, 46, 4760–4763 CrossRef PubMed.
- (a) N. W. Preston, K. Chamberlain and R. A. Skipp, Phytochemistry, 1975, 14, 1875–1876 CrossRef; (b) R. P. Duffley and R. Stevenson, J. Chem. Soc., Perkin Trans. 1, 1977, 802–804 RSC; (c) M. Meyer, C. Deschamps and D. Molho, Bull. Soc. Chim. Fr., 1991, 91–99 CAS; (d) M. Na, D. M. Hoang, D. Njamen, J. T. Mbafor, Z. T. Fomun, P. T. Thuong, J. S. Ahn and W. K. Oh, Bioorg. Med. Chem. Lett., 2007, 17, 3868–3871 CrossRef CAS PubMed; (e) T. Kinoshita and K. Ichinose, Heterocycles, 2005, 65, 1641–1654 CrossRef CAS.
- M. A. Khalizadeh, A. Hosseini, M. Shokrollahzadeh, M. R. Halvagar, D. Ahmadi, F. Mohannazadeh and M. Tajbakhsh, Tetrahedron Lett., 2006, 47, 3525–3528 CrossRef.
- M. Csekei, Z. Novak and A. Kotschy, Tetrahedron, 2008, 64, 8992–8996 CrossRef CAS.
- (a) E. E. Shul’ts, T. N. Petrova, M. M. Shakirov, E. I. Chernyak and G. A. Tolstikov, Chem. Nat. Compd., 2000, 36, 362–368 CrossRef; (b) T. Shiozawa, S. Urata, T. Kinoshita and T. Saitoh, Chem. Pharm. Bull., 1989, 37, 2239–2240 CrossRef CAS.
- Y. Tanaka, H. Kikuzaki, S. Fukuda and N. Nakatani, J. Nutr. Sci. Vitaminol., 2001, 47, 270–273 CrossRef CAS PubMed.
- Y. L. Jin, S. Kim, Y. S. Kim, S. A. Kim and H. S. Kim, Tetrahedron Lett., 2008, 49, 6835–6837 CrossRef CAS.
- K.-S. Huang, Y.-H. Wang, R.-L. Li and M. Lin, J. Nat. Prod., 2000, 63, 86–89 CrossRef CAS.
- (a) J. Ito, Y. Takaya, Y. Oshima and M. Niwa, Tetrahedron, 1999, 55, 2529–2544 CrossRef CAS; (b) I. Kim and J. Choi, Org. Biomol. Chem., 2009, 7, 2788–2795 RSC.
- J.-R. Dai, Y. F. Hallock, J. H. Cardellina and M. R. Boyd, J. Nat. Prod., 1998, 61, 351–353 CrossRef CAS PubMed.
- (a) A. Saraswathy, K. K. Purushothaman, A. Patra, A. K. Dey and A. B. Kundu, Phytochemistry, 1992, 31, 2561–2562 CrossRef CAS; (b) T. Tanaka, T. Ito, Y. Ido, K. Nakaya, M. Iinuma and V. Chelladurai, Chem. Pharm. Bull., 2001, 49, 785–787 CrossRef CAS PubMed.
- (a) Y. Takaya and M. Niwa, Trends Heterocycl. Chem., 2001, 7, 41–54 Search PubMed; (b) E.-K. Seo and A. D. Kinghorn, Stud. Nat. Prod. Chem., 2000, 23, 531 CAS.
- (a) T. K. Chakraborty and G. V. Reddy, J. Org. Chem., 1992, 57, 5462–5469 CrossRef CAS; (b) N. Hoffmann and J.-P. Pete, Synthesis, 2001, 1236–1242 CAS; (c) S. Inoue, C. Nakagawa, H. Hayakawa, F. Iwasaki, Y. Hoshino and K. Honda, Synlett, 2006, 1363–1366 CrossRef CAS.
- L. Chen, Q. Ding, P. Gillespie, K. Kim, A. J. Lovey, W. W. McComas, J. G. Mullin and A. Perrota, World Pat.WO 02057261 A2, 2005.
- M. M. Heravi, F. Dirkwand, H. A. Oskooie and M. Ghassemzadeh, Heterocycl. Commun., 2005, 11, 75–78 CAS.
- I. Kim and J. Choi, Org. Biomol. Chem., 2009, 7, 2788–2795 CAS.
- A. Yaron and F. Naider, Crit. Rev. Biochem. Mol. Biol., 1993, 28, 31–81 CrossRef CAS PubMed.
- T. Aoyagi, T. Wada, M. Nagai, F. Kojima, S. Harada, T. Takeuchi, H. Takahashi, K. Hirokawa and T. Tsumita, Experientia, 1990, 46, 94–97 CrossRef CAS PubMed.
- L. Y. Yang, C. F. Chang, Y. C. Huang, Y. J. Lee, C. C. Hu and T. H. Tseng, Synthesis, 2009, 7, 1175–1179 Search PubMed.
- M. J. Lear, O. Simon, T. L. Foley, M. D. Burkart, T. J. Baiga, J. P. Noel, A. G. DiPasquale, A. L. Rheingold and J. J. La Clair, J. Nat. Prod., 2009, 72, 1980–1987 CrossRef CAS PubMed.
- M. Sridhar, B. A. Kumar and R. Narender, Tetrahedron Lett., 1998, 39, 2847–2850 CrossRef CAS.
- O. Simon, B. Reux, J. J. La Clair and M. J. Lear, Chem.–Asian J., 2010, 5, 342–351 CrossRef CAS PubMed.
- D. Y. K. Chen, Q. Kang and T. R. Wu, Molecules, 2010, 15, 5909–5927 CrossRef CAS PubMed.
- L. D. Juliawaty, E. H. Hakim, S. A. Achmad, Y. M. Syah, J. Latip and I. M. Said, Nat. Prod. Commun., 2009, 4, 947–950 CAS.
- H. M. Ge, W. H. Yang, Y. Shen, N. Jiang, Z. K. Guo, Q. Luo, Q. Xu, J. Ma and R. X. Tan, Chem.–Eur. J., 2010, 16, 6338–6345 CrossRef CAS PubMed.
- (a) K.-S. Huang, M. Lin and G.-F. Cheng, Phytochemistry, 2001, 58, 357–362 CrossRef CAS PubMed; (b) Y. Meng, P. C. Bourne, P. Whiting, V. Sik and L. Dinan, Phytochemistry, 2001, 57, 393–400 CrossRef CAS PubMed; (c) H.-F. Luo, L.-P. Zhang and C.-Q. Hu, Tetrahedron, 2001, 57, 4849–4854 CrossRef CAS; (d) C. Privat, J. P. Telo, V. Bernades-Genisson, A. Vieira, J. P. Souchard and F. Nepveu, J. Agric. Food Chem., 2002, 50, 1213–1217 CrossRef CAS PubMed; (e) S. Wang, D. Ma and C. Hu, Helv. Chim. Acta, 2005, 88, 2315–2321 CrossRef CAS.
- (a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC; (b) B.-J. Li, S.-D. Yang and Z.-J. Shi, Synlett, 2008, 949–957 CAS; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (d) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (e) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677–685 RSC.
- (a) N. S. Nandurkar, M. J. Bhanushali, M. D. Bhor and B. M. Bhanage, Tetrahedron Lett., 2008, 49, 1045–1048 CrossRef CAS; (b) S.-D. Yang, C.-L. Sun, Z. Fang, B.-J. Li, Y.-Z. Li and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1473–1476 CrossRef CAS PubMed; (c) H.-Q. Do, R. M. K. Khan and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 15185–15192 CrossRef CAS PubMed; (d) S. Matsuda, M. Takahashi, D. Monguchi and A. Mori, Synlett, 2009, 1941–1944 CAS; (e) M. Ionita, J. Roger and H. Doucet, ChemSusChem, 2010, 3, 367–376 CrossRef CAS PubMed.
- K. Kim and I. Kim, Org. Lett., 2010, 12, 5314–5317 CrossRef CAS PubMed.
- C. W. Chang and R. J. Chein, J. Org. Chem., 2011, 76, 4154–4157 CrossRef CAS PubMed.
- (a) R. C. Larock, E. K. Yum, M. J. Doty and K. K. C. Sham, J. Org. Chem., 1995, 60, 3270–3271 CrossRef CAS; (b) B. C. Bishop, I. F. Cottrell and D. Hands, Synthesis, 1997, 1315–1320 CrossRef CAS.
- B. M. Trost, S. A. Godleski and J. P. Genet, J. Am. Chem. Soc., 1978, 100, 3930–3931 CrossRef CAS.
- S. Paul, S. Pattanayak and S. Sinha, Tetrahedron Lett., 2011, 52, 6166–6169 CrossRef CAS.
- (a) R. Hänsel, H. Rimpler and R. Schwar, Tetrahedron Lett., 1965, 6, 1545–1548 CrossRef; (b) Z.-F. Hu, L.-L. Chen, J. Qi, Y.-H. Wang, H. Zhang and B.-Y. Yu, Fitoterapia, 2011, 82, 190–192 CrossRef CAS PubMed.
- (a) M. W. Khan, M. J. Alam, M. A. Rashid and R. Chowdhury, Bioorg. Med. Chem., 2005, 13, 4796–4805 CrossRef CAS PubMed; (b) I. Hayakawa, R. Shioya, T. Agatsuma, H. Furukawa, S. Naruto and Y. Sugano, Bioorg. Med. Chem. Lett., 2004, 14, 455–458 CrossRef CAS PubMed.
- M. R. Saberi, T. K. Vinh, S. W. Yee, B. J. Griffiths, P. J. Evans and C. Simons, J. Med. Chem., 2006, 49, 1016–1022 CrossRef CAS PubMed.
- (a) C. Beney, A.-M. Mariotte and A. Boumendjel, Heterocycles, 2001, 55, 967–972 CrossRef CAS; (b) V. Wallez, S. Durieux-Poissonnier, P. Chavatte, J. A. Boutin, V. Audinot, J.-P. Nicolas, C. Bennejean, P. Delagrange, P. Renard and D. Lesieur, J. Med. Chem., 2002, 45, 2788–2800 CrossRef CAS PubMed.
- S. Yahiaoui, M. Peuchmaur and A. Boumendjel, Tetrahedron, 2011, 67, 7703–7707 CrossRef CAS.
- T. Rezanka, L. O. Hanus, P. Kujan and V. M. Dembitsky, Eur. J. Org. Chem., 2005, 2708–2714 CrossRef CAS.
- D. Ma and Q. Cai, Org. Lett., 2003, 5, 3799–3802 CrossRef CAS PubMed.
- B. Liégault, D. Lee, M. P. Huestis, D. R. Stuart and K. Fagnou, J. Org. Chem., 2008, 73, 5022–5028 CrossRef PubMed.
- (a) A. Shiotani and H. Itatani, J. Chem. Soc., Perkin Trans. 1, 1976, 1236–1241 RSC; (b) H. Hagelin, J. D. Oslob and B. kermark, Chem.–Eur. J., 1999, 5, 2413–2416 CrossRef CAS; (c) T. Gensch, M. Rçnnefahrt, R. Czerwonka, A. Jäger, O. Kataeva, I. Bauer and H.-J. Knçlker, Chem.–Eur. J., 2012, 18, 770–776 CrossRef CAS PubMed.
- R. Bartholomaus, F. Dommershausen, M. Thiele, N. S. Karanjule, K. Harms and U. Koert, Chem.–Eur. J., 2013, 19, 7423–7436 CrossRef PubMed.
- E. M. Bickoff, A. N. Booth, R. L. Lyman, A. L. Livingston, C. R. Thompson and F. Deeds, Science, 1957, 126, 969–970 CAS.
- M. S. Kurzer and X. Xu, Annu. Rev. Nutr., 1997, 17, 353–381 CrossRef CAS PubMed.
- M. S. Kurzer, J. Nutr., 2003, 133, 1983S–1986S Search PubMed.
- U. A. Kshirsagar, R. Parnes, H. Goldshtein, R. Ofir, R. Zarivach and D. Pappo, Chem.–Eur. J., 2013, 19, 13575–13583 CrossRef CAS PubMed.
- R. J. Y. Chen, T. Jinn, Y. Chen, T. Chung, W. Yang and J. T. C. Tzen, Acta Pharmacol. Sin., 2011, 32, 141–151 CrossRef CAS PubMed.
- Y. Lu and L. Foo, Phytochemistry, 2002, 59, 117–140 CrossRef CAS PubMed.
- R. Jiang, K. Lau, P. Hon, T. C. W. Mak, K. Woo and K. Fung, Curr. Med. Chem., 2005, 12, 237–246 CrossRef CAS PubMed.
- T. o. Cheng, Int. J. Cardiol., 2007, 121, 9–22 CrossRef PubMed.
- B. L. Alford and H. M. Hügel, Org. Biomol. Chem., 2013, 11, 2724–2727 CAS.
- (a) W. Steck, Phytochemistry, 1970, 9, 1145–1146 CrossRef CAS; (b) M. Vanhaelen and R. Vanhaelen-Fastré, Phytochemistry, 1974, 13, 306–307 CrossRef CAS; (c) J. Reisch, A. Wickramasinghe and V. Kumar, J. Nat. Prod., 1989, 52, 1379–1382 CrossRef CAS; (d) A. H. Muller, L. R. O. Degáspari, P. C. Vieira, M. F. da Silva, J. B. Fernandes and J. R. Pirani, Phytochemistry, 1993, 34, 585–586 CrossRef CAS; (e) F. M. Oliveira, A. E. G. Sant’ana, L. M. Conserva, J. G. S. Maia and G. M. P. Guilhon, Phytochemistry, 1996, 41, 647–649 CrossRef CAS.
- (a) A. E. Desjardins, G. F. Spencer and R. D. Plattner, Phytochemistry, 1989, 28, 2963–2969 CrossRef CAS; (b) C. C. Wang, J. E. Lai, L. G. Chen, K. Y. Yen and L. L. Yang, Bioorg. Med. Chem., 2000, 8, 2701–2707 CrossRef CAS PubMed; (c) J. Singhuber, I. Baburin, G. F. Ecker, B. Kopp and S. Hering, Eur. J. Pharmacol., 2011, 668, 57–64 CrossRef CAS PubMed.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- A. Cervi, P. Aillard, N. Hazeri, L. Petit, C. L. L. Chai, A. C. Willis and M. G. Banwell, J. Org. Chem., 2013, 78, 9876–9882 CrossRef CAS PubMed.
- E. Gellert, R. Hamet and E. Schlittler, Helv. Chim. Acta, 1951, 34, 642–651 CrossRef CAS.
- T. Matsui, Y. Sowa, H. Murata, K. Takagi, R. Nakanishi, S. Aoki, M. Yoshikawa, M. Kobayashi, T. Sakabe, T. Kubo and T. Sakai, Int. J. Oncol., 2007, 31, 915–922 CAS.
- M. L. Bolognesi, F. Lizzi, R. Perozzo, R. Brun and A. Cavalli, Bioorg. Med. Chem. Lett., 2008, 18, 2272–2276 CrossRef CAS PubMed.
- N. Kotoku, K. Higashimoto, M. Kuriok, M. Arai, A. Fukud, Y. Sumii, Y. Sowa, T. Sakai and M. Kobayashi, Bioorg. Med. Chem. Lett., 2014, 24, 3389–3391 CrossRef CAS PubMed.
- (a) M. D. Clift, C. N. Taylor and R. J. Thomson, Org. Lett., 2007, 9, 4667–4669 CrossRef CAS PubMed; (b) C. T. Avetta, L. C. Konkol, C. N. Taylor, K. C. Dugan, C. L. Stern and R. J. Thomson, Org. Lett., 2008, 10, 5621–5624 CrossRef CAS PubMed; (c) M. D. Clift and R. J. Thomson, J. Am. Chem. Soc., 2009, 131, 14579–14593 CrossRef CAS PubMed.
- B. T. Jones, C. T. Avetta and R. J. Thomson, Chem. Sci., 2014, 5, 1794–1798 RSC.
- M. M. Heravi and A. Fazeli, Heterocycles, 2010, 81, 1979–2026 CrossRef CAS.
- H. Yuan, K. Bi, W. Chang, R. Yue, B. Li, J. Yea, Q. Sun, H. Jin, L. Shan and W. Zhang, Tetrahedron, 2014, 70, 9084–9092 CrossRef CAS.
- M. M. Heravi and E. Hashemi, Monatsh. Chem., 2012, 143, 861–880 CrossRef CAS.
- M. M. Heravi and P. Hajiabbasi, Mol. Diversity, 2014, 18, 411–439 CrossRef CAS PubMed.
- X. Zheng, W. D. Meng and F. L. Qing, Tetrahedron Lett., 2004, 45, 8083–8085 CrossRef CAS.
- G. Pan, Y. Ma, K. Yang, X. Zhao, H. Yang, Q. Yao, K. Lu, T. Zhu and P. Yu, Tetrahedron Lett., 2015, 56, 4472–4475 CrossRef CAS.
- Y. Q. Ye, C. Negishi, Y. Hongo, H. Koshino, J.-i. Onose, N. Abe and S. Takahashi, Bioorg. Med. Chem., 2014, 22, 2442–2446 CrossRef CAS PubMed.
- Y.-Q. Ye, H. Koshino, J. Onose, K. Yoshikawa, N. Abe and S. Takahashi, Org. Lett., 2009, 11, 5074–5077 CrossRef CAS PubMed.
- N. Radulovic, D. N. Quang, T. Hashimoto, M. Nukada and Y. Asakawa, Phytochemistry, 2005, 66, 1052–1059 CrossRef CAS PubMed.
- J.-T. Liu, T. J. Do, C. J. Simmons, J. C. Lynch, W. Gu, Z.-X. Ma, W. Xu and W. Tang, Org. Biomol. Chem., 2016, 14, 8927–8930 CAS.
- I. Kim, S.-H. Lee and S. Lee, Tetrahedron Lett., 2008, 49, 6579–6584 CrossRef CAS; J. H. Lee, M. Kim and I. Kim, J. Org. Chem., 2014, 79, 6153–6163 CrossRef PubMed.
- J.-t. Liu, C. J. Simmons, H. Xie, F. Yang, X. Zhao, Y. Tang and W. Tang, Adv. Synth. Catal., 2016, 358, 1–6 CrossRef.
- G. A. Kraus and V. Gupta, Tetrahedron Lett., 2009, 50, 7180–7182 CrossRef CAS.
- D. Y. K. Chen, Q. Kang and T. R. Wu, Molecules, 2010, 15, 5909–5927 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |