
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal organic frameworks as precursors for the manufacture of advanced catalytic materials
Lide
Oar-Arteta
,
Tim
Wezendonk
,
Xiaohui
Sun
,
Freek
Kapteijn
 and
Jorge
Gascon
*
and
Jorge
Gascon
*
Catalysis Engineering, Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: j.gascon@tudelft.nl
First published on 22nd February 2017
Abstract
The use of metal organic frameworks as hard templates for the preparation of heterogeneous catalysts is thoroughly reviewed. In this critical article, the main factors to consider when using a MOF as a sacrificial template are first discussed. Then, the existing literature on the topic is reviewed, classifying the different examples according to the MOF metal. Finally, the main advantages, limitations and perspectives of the so-called MOF mediated synthesis are outlined.
 Lide Oar-Arteta | Lide Oar-Arteta (1986) received her PhD (2014) from the University of The Basque Country (UPV/EHU) after a secondment in the Institut de Chimie des Milieux et Matériaux de Poitiers (IC2MP) in 2012. In 2015, she joined the Catalysis Engineering group of Delft University of Technology (The Netherlands) where she is now a Marie-Slodowska Curie postdoctoral researcher in collaboration with Haldor Topsøe A/S (Denmark). Her current research interests focus on the preparation of metal–organic framework derived catalysts for their application in different catalytic processes and with special emphasis on the Fischer–Tropsch synthesis. |
 Tim Wezendonk | Tim A. Wezendonk (1986) obtained his MSc in Chemical Engineering at the Delft University of Technology in 2013. For his thesis work on MOF-based catalyst synthesis for Fischer–Tropsch to Olefins, he was awarded the Netherlands Process Technology best research thesis prize. He continued working in this topic at the TU Delft as a PhD student under the supervision of Prof. Gascon and Prof. Kapteijn, elaborating on the mechanisms at play in this novel synthesis method, investigating the structural and elemental influence of various MOFs, and elucidating the evolution of the active phase of MOF-derived Fe catalysts during Fischer–Tropsch operation. |
 Xiaohui Sun | Xiaohui Sun received his BS degree (2010) and MS degree (2013) from the Department of Chemical Engineering at China University of Petroleum (Huadong). The same year he started his PhD at the department of Chemical Engineering at Delft University of Technology. His research now is focused on the application of zeolite-based and MOF-mediated catalysts in heterogenous hydrogenation reactions. |
 Freek Kapteijn | Freek Kapteijn (1952), MSc in Chemistry and Mathematics, received his PhD in 1980 at the University of Amsterdam. After post-doc positions (Coal Science) in Amsterdam and Nancy (ENSIC), he became an associate professor in Amsterdam. He moved to Delft University of Technology in 1992, became ‘Anthonie van Leeuwenhoek professor’ in 1999, and since 2008 has been chair of Catalysis Engineering, with visiting professorships at ETH Zürich, Tianjin and Zhejiang Normal University. His research interest focuses on the interplay of catalysis and engineering, comprising structured and multifunctional catalysts, adsorption, separation and (catalytic) membranes. He has co-authored over 600 publications in peer-reviewed journals and book chapters. |
 Jorge Gascon | Jorge Gascon (1977) received his MSc in Chemistry in 2002 and his PhD in Chemical Engineering in 2006, both at the University of Zaragoza (Spain). Since 2013 he has been ‘Anthonie van Leeuwenhoek Professor’ of Catalysis Engineering at TUDelft. His research interests include fundamental aspects and applications of new nano-structured materials and composites. He has co-authored over 170 publications, several patents and book chapters. He has been the recipient of several fellowships and awards, including VENI (2010), VIDI (2013) and ERC Starting (2013) personal grants and the 2013 ExxonMobil Chemical European Science and Engineering Award. |
1. Introduction
Translating material design into an actual catalyst through controlled synthesis is one of the most important challenges heterogeneous catalysis faces nowadays. Despite many advances, conventional synthetic techniques usually fall short in delivering materials with the desired properties. The preparation of metal nanoparticle based catalysts, where it is not always possible to achieve desired metal loadings and controlled particle size, is an outstanding example of this challenge. Recent advances in this field include (i) the use of colloidal,1 reverse micelle,2 and dendrimer chemistry3 in the production of metal and metal oxide nanoparticles with well-defined sizes, shapes, and compositions, (ii) the introduction of sol–gel4 and atomic layer deposition5 chemistry for the production and modification of high-surface-area supports and active phases, (iii) the design of dumbbells, core@shell, and other complex nanostructures which lead to multiple functionality in catalysis6 and (iv) the mixed molecular-nanostructure approach that can be used to develop more demanding catalytic sites, by derivatization of the surface of solids or tethering or immobilizing other chemical functionalities.7Metal organic frameworks (MOFs) are crystalline solids consisting of infinite lattices built up from inorganic secondary building units (SBUs, metal ions or clusters) and organic linkers, connected by coordination bonds of moderate strength. MOFs are among the materials with the highest porosity, and their pore size, shape, dimensionality, and chemical environment can be finely tuned.8 As a consequence, MOFs have been extensively studied not only in catalysis but also in different fields such as fuel storage, batteries, and supercapacitors.9 Specially in catalysis, the high versatility of MOF design provides clear advantages, since in principle, it should be possible to rationally design not only the active site but also its environment with an unprecedented degree of precision.10 However, the main constraint for MOF application in catalysis is their relatively low thermal and chemical stability, especially when compared with inorganic porous solids. Although a handful of these materials are particularly robust,11–14 it is very difficult to anticipate the stability of a given MOF under certain reaction conditions, even when the thermal stability is very high.
As Billy Ocean once sang “when the going gets tough, the tough get going” and so it does apply to MOFs in catalysis. MOFs may not only be envisioned as catalytic solids themselves, but also as precursors for developing highly stable materials with special catalytic properties through the so-called MOF mediated synthesis (MOFMS). This synthetic technique makes use of the MOF as a sacrificial template. Under a certain atmosphere and at high temperature the framework collapses, the organic linker is completely or partially burnt away or carbonized (converted into a carbon matrix)15 and the metal ions/clusters become mobile within the newly created matrix or just evaporate. The essence of MOFMS lies not only in its simplicity but also in its versatility. Its variants embrace a whole range of approaches giving place to different types of materials. Depending upon the heat treatment and, in some cases further post-treatment(s), MOF precursors may lead to the formation of bulk and supported catalytic materials bearing unprecedented properties (Fig. 1).
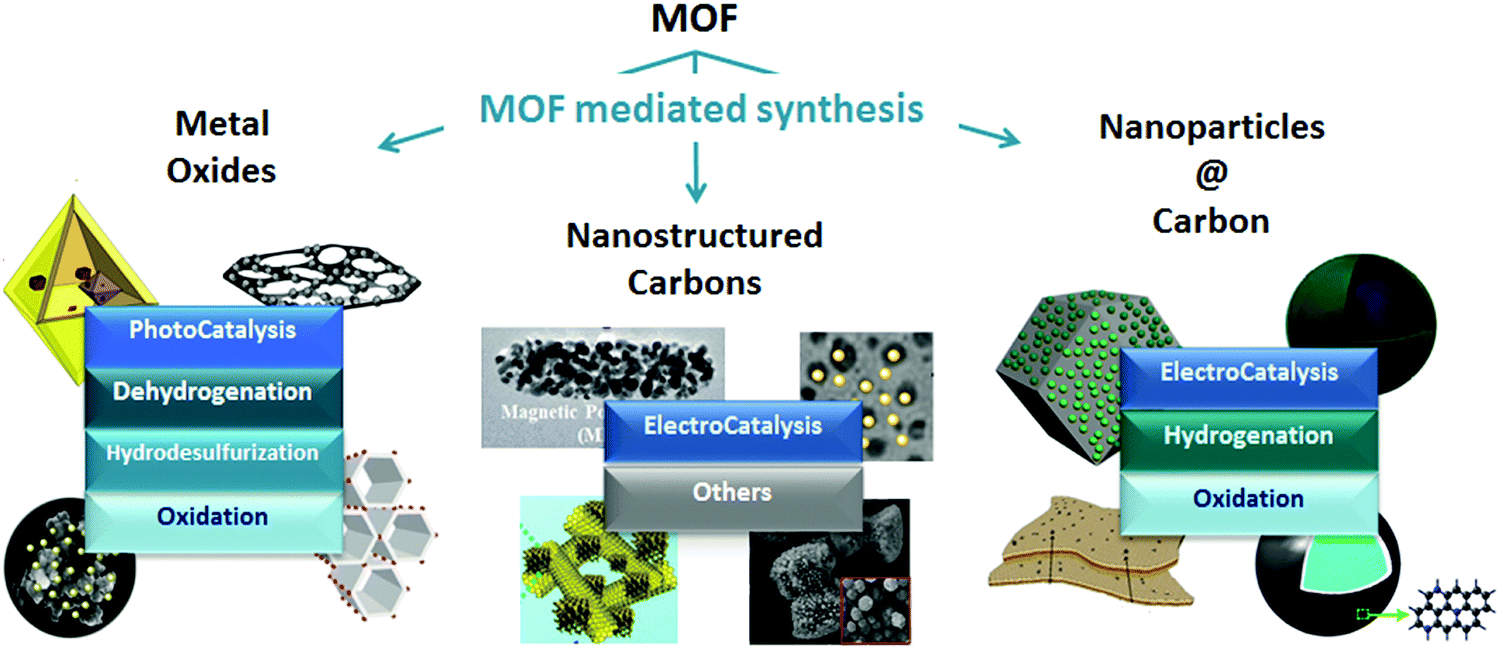 | ||
| Fig. 1 Examples of nanostructures obtained through MOF mediated synthesis and their application in catalysis.16–26 | ||
In recent years, the synthesis of nanostructured metal oxides (MOs) has been a matter of intense investigation, with special emphasis on controlling the shape, composition and porosity of the resulting MOs.27 The MOFMS has come to light as a promising technique for tailoring metal MOs in a simple manner. Calcination in air is the usual approach for the preparation of metal oxides from MOFs, but acid leaching and other heat treatments are also applied. Carbon materials may act as catalysts and catalyst supports. The main hurdle in the case of carbons as both support and catalyst lies in the development of novel methods able to tailor the porosity and morphology of carbonaceous nanostructures.28 MOFMS yields well-structured hierarchical carbons by removing the metal phase during or after the carbonization of the MOF by volatilization at high temperature or acid leaching, respectively. When it comes to supported nanoparticles, the tendency of nanoparticles to grow into larger crystallites is an impediment for stable catalytic performance.29 Carbonizing the MOF in an inert atmosphere leads to a highly dispersed metal phase encapsulated in a porous carbon matrix, the spatial restriction created by encapsulation minimizing the sintering of the metal nanoparticles. As in the case of nanostructured carbons, the conventional strategy to obtain nanoparticles@carbon (NP@C) consists of carbonizing the MOF in an inert atmosphere at high temperature. However, contrary to the approach for carbons, in this case the metal phase remains retained in the resulting material. Therefore, during the pyrolysis, a part of the organic ligand decomposes and evaporates to the gas phase, whereas the other part becomes carbonized in the framework.
As recently highlighted by Sun et al.30 and Xia et al.,31 MOFMS has been extensively applied during the last few years in the field of electrochemistry in both electrochemical energy storage and conversion. MOFMS has been thoroughly applied in supercapacitors, batteries and fuel cells. Moreover, over the past couple of years, the MOFMS technique has also been applied in the field of heterogeneous catalysis, the main topic of this review article.
Herein we highlight the main properties of MOFMS as a novel technique for the preparation of heterogeneous catalysts. Before reviewing the different examples from the literature, we first bring to the attention of the reader important considerations to be taken into account in the design of new solids via MOFMS. After the scene has been set, we thoroughly review the different examples from the literature. We do so by classifying these materials according to the metal in the framework of the pristine MOF. Last but not least, we share our personal assessment of the most promising catalytic materials and catalytic applications and the most important issues that still need to be addressed. This review aims at giving a critical overview of the current situation for the MOFMS in heterogeneous catalysis thus providing guidelines for the design of new generations of catalytic materials using MOFs as sacrificial agents.
2. General considerations about MOF mediated synthesis
Although the organic linker plays an essential role in the outcome of the final catalyst, it is the nature of the metal that governs the MOF mediated synthesis and determines the catalytic process the MOF mediated catalyst will be applied in. In short, the parent MOF is selected according to the metal(s) in its framework, whereas the organic linker and the synthesis conditions are envisioned as the tools that allow optimizing the final properties of the MOF mediated catalyst. Accordingly, once the MOF decomposes and envisaging the MOFMS as an essential heat treatment, the following parameters are key in determining the final properties of the resulting MOF derived catalyst:(a) The Gibbs free energy (ΔG) of a reaction, such as oxidation of metals, quantifies the thermodynamic driving force necessary to make a reaction “work” under given conditions. A negative value for ΔG indicates that oxidation can proceed spontaneously without external inputs, while a positive value indicates that it will not. Widely used in extractive metallurgy, the Ellingham diagram (Fig. 2) plots the formation free energy of oxides versus temperature.32 The lower the position of a metal in the Ellingham diagram, the greater is the stability of its oxide and besides, the intersection of two lines in this diagram implies an oxidation–reduction equilibrium. Therefore, reduction using a given reductant is possible at temperatures above the intersection point where the ΔG line of that reductant is lower on the diagram than that of the metallic oxide to be reduced. In MOFMS, the carbon formed during the heat treatment acts as the reducing agent and reduction of metal oxides is carried out by a carbothermic reaction. Likewise, when the number of electrons involved is the same, the Gibbs free energy of oxidation follows exactly the opposite trend to the reduction potential of the corresponding metal ion for different metals. Accordingly, Das et al. quantified for the first time that metal ions with a standard reduction potential of −0.27 V or higher, such as Co, Ni and Cu, present in MOFs always form pure metal nanoparticles during thermolysis in N2 up to 900 °C, whereas metal ions with a reduction potential lower than −0.27 V, such as Mg, Al, Mn, Zn or Cr tend to combine with the oxygen present in the organic part of the MOF and form metal oxide nanoparticles during the same thermolysis treatment.33
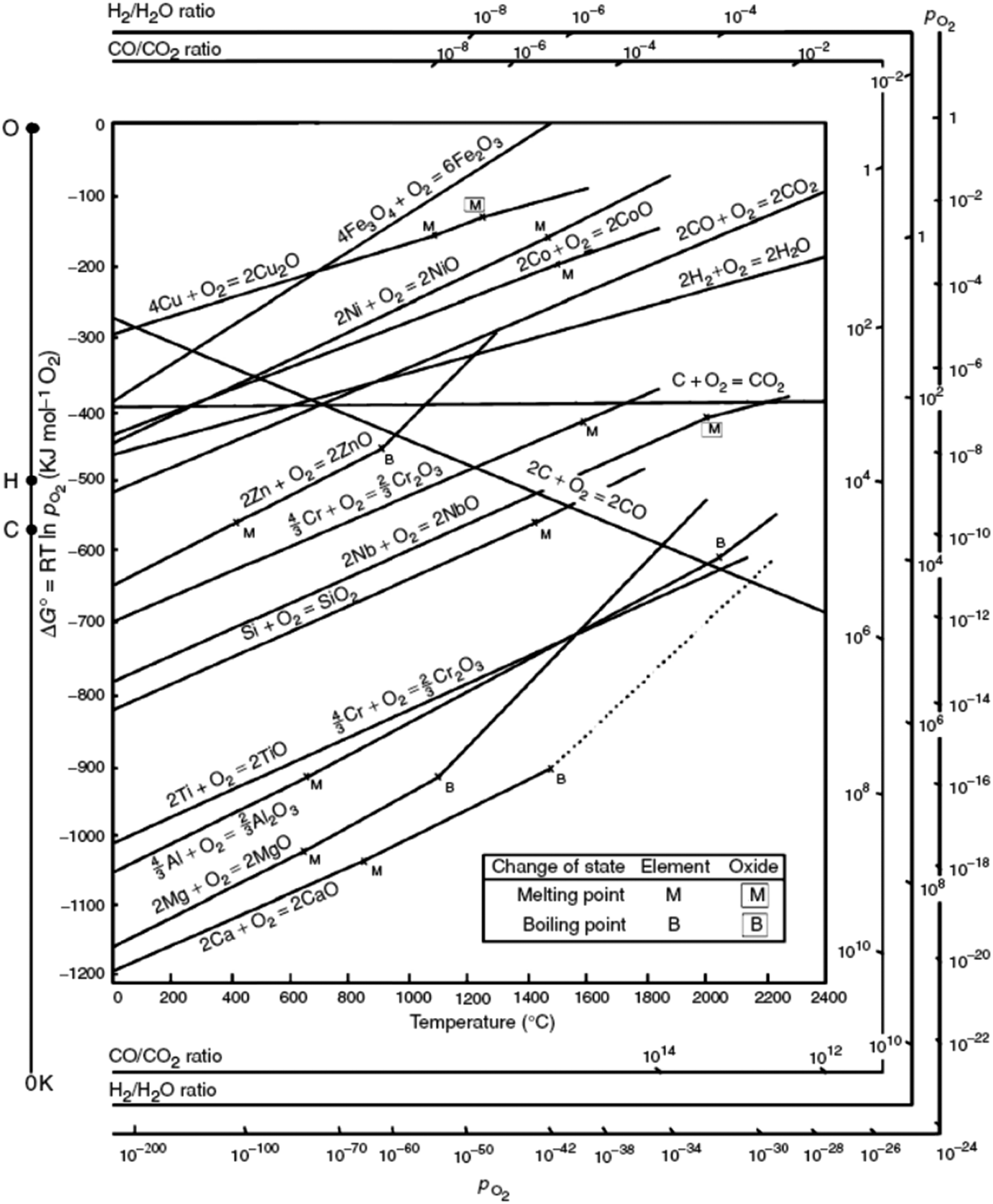 | ||
| Fig. 2 Ellingham-diagram for the formation of oxides based on their standard free energy of formation over temperature.34 | ||
(b) Another property of utmost importance for MOFMS is the Tamman temperature of the actual metal species, which is approximately half the melting point of the metal species itself (Table 1). The exact definition of the Tamman temperature is that at which the atoms or molecules of the solid acquire sufficient energy for their bulk diffusion. Otherwise, the Huttig temperature which is approximately one third of the melting point of the metal species refers to surface species having sufficient mobility to begin to agglomerate and sinter. In MOFMS, the Tamman temperature of the metal species is typically above the temperature required to make the organic framework collapse. Therefore, once the framework is decomposed it is according to their Tamman temperature that atoms start colliding and forming the corresponding nanoparticles, which, in line with thermodynamics, consist of metals, metal oxides, metal carbides, metal oxocarbides and/or a mixture of them. Increasing the temperature boosts the buildup of larger particles at the expense of smaller ones which is better known as the sintering process. In short, the Tamman temperature of different metal species is responsible for delimiting the temperature range in which the MOFMS must proceed if control over nanoparticle size is to be achieved. Likewise, the vapour pressure of the formed metallic species or mixture of them needs also to be taken into consideration in order to get rid of or retain the metal species in the final catalytic material.
| Metal | Melting point, °C | Boiling point, °C |
|---|---|---|
| Al | 660 | 2519 |
| Cr | 1907 | 2671 |
| Co | 1495 | 2927 |
| Cu | 1085 | 2560 |
| Fe | 1538 | 2861 |
| Mg | 650 | 1090 |
| Mn | 1246 | 2061 |
| Ni | 1455 | 2913 |
| Ru | 2334 | 4150 |
| Ti | 1668 | 3287 |
| V | 1910 | 3407 |
| Zn | 420 | 907 |
| Zr | 1855 | 4409 |
3. MOF mediated synthesis of heterogeneous catalysts
(a) Aluminium (Al)-MOFMS
Aluminium is the third most abundant element in the Earth's crust (after oxygen and silicon) and the most abundant metal.36 Among the different transition aluminas known, γ-alumina (γ-Al2O3) is perhaps the most important with direct applications as a catalyst and catalyst support in the automotive and petrochemical industries.37 The usefulness of this oxide is due to a favourable combination of its textural properties, such as surface area, pore volume, and pore size distribution and its acid/base characteristics, which are mainly related to surface chemical composition, local microstructure, and phase composition.The low position of Al in the Ellingham diagram indicates the high stability of its oxide even at high temperature and in the presence of carbon. The melting point of Al2O3 is 2072 °C, its boiling point is 2977 °C and the reduction potential of Al3+ is −1.66 V. Accordingly, bare pyrolysis of Al-MOFs should yield basically Al2O3 (reduction potential below −0.27 V33) in a carbon matrix (high boiling point) at least up to 2977 °C, when it starts to evaporate. Therefore, if required, acid leaching applies for the removal of this metal oxide, leading to Al free carbons.
Due to their thermal and chemical stability, Al-containing MOFs, such as the well-known Al-MIL-53, Al-MIL-101-NH2 and CAU-1 are subjects of intensive research.38 Al-MOFs used in the synthesis of carbon supports for catalysis include CAU-1 and MOF-253. Prepared for the first time by Ahnfeldt et al.,38 the 3D microporous framework Al-CAU-1 ([Al4(OH)2-(OCH3)4(H2N-bdc)3]·xH2O) (bdc = 1,4-benzenedicarboxylate) is formed with two types of cages, distorted octahedral and distorted tetrahedral, with effective accessible diameters of approximately 1 and 0.45 nm, respectively. CAU-1 exhibits high porosity and thermal stability up to 360 °C. In this manner, reaction of AlCl3·6H2O with 2,2′-bipyridine-5,5′-dicarboxylic acid (H2bpydc) affords Al(OH)(bpydc) (MOF-253) which displays a BET surface area of 2160 m2 g−1 being stable up to 400 °C.39 The Al-MOF mediated synthesis reported so far includes the preparation of carbon supports for electrocatalysis and coupling reactions.
In electrocatalysis, Afsahi et al. confirmed the potential of Al-MOFs as templates to prepare efficient fuel cell electrocatalysts.40 Firstly, they synthesized desolvated Al-MOF-253 and subsequently they introduced PtCl2(CH3CN)2 together with acetonitrile. The mixture was heated at 85 °C for 72 h and the resulting solid was collected by filtration and immersed in acetonitrile for 3 days. Finally, the product was collected by filtration and dried at 150 °C for 12 h under vacuum. To prepare PEMFC electrocatalysts, the obtained Pt-MOF materials were heat treated at temperatures ranging between 700 and 1050 °C at 1 °C min−1 for 4 h under Ar. The membrane–electrode assemblies (MEAs) made from the obtained Pt nanoparticles (7–10 nm) in carbon were tested as both an anode and a cathode in a H2/air single cell fuel cell. As the anode, the most promising electrocatalyst (carbonized at 950 °C) demonstrated an open circuit voltage of 970 mV and a power density of 0.58 W mgPt−1 comparable to the commercial electrode power density (0.64 W mgPt−1) at 0.6 V in a single cell test. At the cathode, a power density of 0.38 W mgPt−1 at 0.6 V was achieved. Although the results show that this rather simple technique allows the production of electrocatalysts with performances comparable to the commercial materials, the authors propose further improvements by (i) changing the heating inert atmosphere to a reactive gas such as ethylene or acetylene, possibly allowing better preservation of the original atomic dispersion of Pt in the MOF and (ii) starting with MOFs containing other metals such as Fe or Co in order to replace the highly expensive Pt and Pt groups metals (PGMs).
Quantum chemistry calculations predict an electrocatalytic behaviour of Co9S8 similar to that observed for a Pt electrode. In view of this, Zhu et al. prepared highly active Co9S8/CNS catalysts for ORR from aluminum-based MIL-101-NH2 and Co/S precursors.41 MIL-101-NH2 was selected as a prototypical host for the encapsulation of thiourea (TU) and cobalt chloride to form the catalyst precursor, due to its facile, economical and scalable synthesis. The as-obtained Co(II)TU@MIL-101-NH2 sample was finally heated in Ar at 600 °C (or 700, 800, 900, 1000 °C) for 5 h with a heating ramp of 10 °C min−1. The residual Al component and other impurities were removed by immersing the samples in a HF (20 wt%) solution for 24 h. The authors found that the resulting Co9S8@CNS material carbonized at 900 °C exhibited remarkably comparable ORR catalytic activity, and superior long-term stability and methanol tolerance compared with commercial 20 wt% Pt/C catalyst under alkaline conditions. They associated the outstanding electrocatalytic performance of the catalyst for the ORR with the unique honeycomb-like open structure with high surface area and pore volume, proper degree of graphitization, and high content of the active species combined with their synergetic interactions.
The palladium-catalyzed Suzuki–Miyaura coupling of aryl halides with aryl boronic acids is one of the most powerful methods for constructing biaryl structures, which are important units in pharmaceuticals, herbicides, and natural products.42 On the other side, N-decorated carbon materials enhance the mechanical and energy-storage properties, and show high stability as supports for noble metal nanoparticles.43 In view of this, Zhang et al. successfully fabricated a novel catalyst based on Pd nanoparticles supported on N-doped nanoporous carbon from Al-based MOFs with high catalytic activity for the Suzuki–Miyaura coupling reactions at room temperature.16 The as-prepared CAU-1 was carbonized at 800 °C for 10 h in N2 giving N-doped nanoporous carbon (NPC) and subsequently Pd nanoparticles were immobilized on the N-doped NPC by impregnation. Additionally, for the preparation of aluminium-free N-doped NPC-Pd, prior to Pd impregnation, N-doped NPC was treated with 10% HCl for 5 h, giving Al free N-doped NPC-Pd. Catalytic results indicated that N-doped NPC-Pd could catalyze the reaction efficiently even at room temperature within 1 h to afford the corresponding biphenyl in 97% yield with EtOH/H2O as the solvent, compared to 78% yield obtained with the commercially available 5% Pd/C catalyst under the same conditions. The reaction did not take place in the absence of a catalyst nor using N-doped NPC as a catalyst, which indicated that aluminium oxide has no catalytic activity for the coupling reaction. However, for the aluminium-free N-doped NPC-Pd only 69% yield of biphenyl was obtained which demonstrated that the presence of aluminium oxide in N-doped NPC might be helpful to improve the dispersion of Pd nanoparticles.
In the aforementioned examples, although for different catalytic purposes, the authors successfully prepared carbon materials for supporting noble and non-noble metal particles from Al-MOFs. Although the presence of aluminium and aluminium oxide are thought to improve the dispersion of Pd nanoparticles for coupling reactions, no further explanation on the mechanism is given. Besides, no clear effect of Al is reported in the electrocatalytic performance of MOF derived catalysts. In this sense, there is a gap in Al-MOF derived catalysis. Filling this gap would imply (i) deeper analysis on the role of Al2O3 in improving the metal dispersion of noble metal nanoparticles and (ii) further research on carbon free nanostructured Al2O3 synthesis from MOFs through calcination in air.44 However, for the latter and based on the wide availability of mesoporous aluminas the next question must be posed: is it really worth applying MOF mediated synthesis to this end?
(b) Chromium (Cr)-MOFMS
Chromium is found mainly in nature as chromite. The melting points of Cr and Cr2O3 are 1907 °C and 2435 °C, respectively, and their boiling points are 2671 °C and 4000 °C. Besides, the reduction potentials of Cr3+ to Cr2+ and of Cr3+ to Cr are −0.40 V and −0.74 V, respectively. Although Cr oxidation states range from +6 to +2, its most stable states found in the environment are +6 and +3. Whereas Cr(III) is believed to be an essential element, hexavalent chromium (Cr(VI)) is acutely toxic, a proven mutagen and carcinogenic heavy metal pollutant,45 and is believed to be the second most common inorganic contaminant after lead.46Probably the best-known application of chromium in catalysis applies to ethylene polymerization or the Phillips polymerization process. The Cr(VI)/SiO2 Phillips catalyst, patented in 1958 by Hogan and Banks, is a highly versatile system accounting for the production of about half of the world's market47 of high density (HDPE) and also linear low density (LLDPE) polyethylene, making it one of the world's most important industrial catalysts for over half a century.48 In addition, Cr based catalysts are used in high temperature water gas shift,49 dehydrogenation of propane and isobutane50 and organic transformations.51
Regarding Cr-MOFs and firstly reported in 2005 by Férey et al.,11 MIL-101(Cr) is a chromium terephthalate MOF with the molecular formula Cr3F(H2O)2O[(O2C)C6H4(CO2)]3·nH2O (where n is ∼25) and stable up to 275 °C in air. The large pores (2.9 and 3.4 nm) and high BET area (>3000 m2 g−1) with a huge cell volume together with the coordinatively unsaturated open metal sites that can be subjected to diverse post-synthesis functionalization or guest encapsulation, and excellent hydrothermal/chemical stability, make MIL-101 particularly attractive for different applications including selective gas adsorption/separation, energy storage and heterogeneous catalysis for oxidation, hydrogenation, condensation, degradation and coupling reactions.52
Consequently, Cr-MOFMS has been applied in the catalytic dehydrogenation of isobutane. In this industrially important route for producing isobutene, a vital component for the synthesis of octane number boosters for unleaded gasoline, the Cr2O3/Al2O3 catalyst has been applied on a commercial scale for years.53 A few years ago, Zhao et al. studied the reactivity of isobutane dehydrogenation over a series of non-ordered mesoporous chromia/alumina catalysts promoted with K2O using the MIL-101(Cr) metal–organic framework as a molecular host and chromium precursor.50 They impregnated MIL-101 with aluminium isopropoxide (Al(i-OC3H7)3) and KOH as the aluminium and potassium sources, respectively, and calcined the resulting material in air at different temperatures ranging from 600 to 900 °C for 4 h obtaining K2O–Cr2O3/Al2O3 catalysts with large specific surface areas and high pore volumes. They found that the pore texture, structural phase, reducibility and surface concentrations of Cr3+ and Cr6+ species over the catalysts depended on the chromia loadings and calcination temperature. They proposed that the Cr3+ species were mainly the active sites and the catalytic selectivity depended on the surface Cr3+/Cr6+ value over the catalyst. The K2O addition to the catalyst slightly decreased the specific surface area and the surface Cr3+/Cr6+ value but it greatly improved the isobutene selectivity and reduced the deactivation rate. Accordingly, the catalyst with 1.5 wt% K2O and 10 wt% Cr2O3 loadings calcined in air at 800 °C for 4 h was found to exhibit the highest isobutane conversion activity with high isobutene selectivity and a more stable dehydrogenation activity than the conventional supported chromia catalyst prepared by the conventional impregnation method.
Another noteworthy example of MOF derived Cr catalysts is given by Qiu et al. who worked on the controlled growth of dense and ordered MOF derived nanoparticles on graphene oxide (GO) for organic transformations.51 In their synthesis procedure, GO was mixed with metal ions for a long time before the organic ligand was added enabling a sufficient coordination between the surface groups of GO with metal ions, providing dense and homogeneous nucleation nods for the formation of MOFs. Thus, MIL-101–GO hybrids with high-density and ordered MOF particles were achieved, which featured different sizes and morphologies as compared to the parent MOFs. Cr-containing/rGO nanomaterials were obtained after pyrolysis of a MIL-101–GO hybrid at 900 °C under Ar for 8 h. The higher activity of this material in ethylbenzene oxidation compared to the merely carbonized MIL-101 and MIL-101–GO prepared by other reported method was attributed to the smaller size of Cr3C2 particles dispersed by rGO, leading to more exposed active sites for organic transformations. The authors postulate that this method might bring new opportunities for the synthesis of growth-controllable MOF–GO and metal-containing/rGO nanohybrids for advanced functional applications by combining both fascinating materials.
All in all, both the high number of Cr catalysed processes and the versatility of MIL-101(Cr) regarding synthesis, design and applications52 offer a large amount of possibilities for MOFMS. Regarding the preparation of CrOx, it is worth re-emphasizing the potential hazard that handling chromium(VI) oxide (CrO3) constitutes during the synthesis, loading, and unloading of the catalyst. Keeping this in mind and proceeding accordingly, the stability of MIL-101 up to 275 °C in air establishes the lower limit of temperature for its calcination in air, whereas the upper one needs to be established according to the targeted porosity of the final metal oxide depending on its catalytic application. Otherwise, for the preparation of supported nanoparticles and back to Cr's properties, the reduction potentials of Cr3+ to both Cr2+ and metallic Cr predict that the heat treatment in an inert atmosphere of Cr(III)-MIL-101 leads mainly to Cr(II), in the form of Cr3C2 at temperatures close to 1200 °C according to the Ellingham diagram.
(c) Cobalt (Co)-MOFMS
In nature cobalt is found in cobaltite, skutterudite and erythrite whereas most cobalt is obtained as a by-product of nickel refining. The high position of Co in the Ellingham diagram evidences the low stability of its oxide. This means that the pyrolysis of Co-MOFs will lead to metallic Co by carbothermal reduction at low temperature close to 200 °C. The melting point of metallic Co is 1495 °C, its boiling point is 2927 °C and the reduction potential of Co2+ is −0.27 V, thus confirming the theory by Das et al.33The use of Co in different catalytic applications mainly depends on the following facts: (i) the oxidation–reduction properties of cobalt and its ability to demonstrate several valencies I + II + III with easy electron transfer between these states, (ii) cobalt's ability to form complexes by accepting atoms from other molecules, (iii) cobalt chemicals in solution and in polymerisation systems can decompose to give more than one ion to take part in catalysis, and (iv) solid cobalt compounds have vacancies in their crystal lattices which can take part in catalysis. Therefore, cobalt is used as a catalyst in oxidation reactions such as the conversion of xylene to terephthalic acid; the steam reforming of ethanol for hydrogen production proceeds over cobalt oxide-base catalysts;54 cobalt-based catalysts are used in reactions involving carbon monoxide being the main catalyst in the low-temperature Fischer–Tropsch process for the hydrogenation of carbon monoxide into liquid fuels;55 hydroformylation of alkenes often uses cobalt octacarbonyl as a catalyst,56 besides, the hydrodesulfurization of petroleum uses a catalyst derived from cobalt and molybdenum, and furthermore, cobalt is also used as a catalyst in electro- and photocatalytic processes.
ZIF-67 is the Co-MOF preferred choice when it comes to Co-MOFMS. ZIF-67 (Co(HmIM)2) is isostructural to ZIF-8, and is formed by bridging 2-methylimidazolate anions and cobalt cations resulting in a sodalite (SOD) topology with a pore size of about 0.34 nm.57 ZIF-67 nanocrystals can be synthesized in aqueous solutions at room temperature, thus not only prohibiting the usage of toxic organic solvent, but also decreasing the cost of preparation of MOF materials. Furthermore, the concentration of reagents is proved to play an important role in controlling particle size, and diluting the synthesis solution can increase the particle size of ZIF-67.58
In photocatalysis, work done so far on Co-MOFMS includes dye-sensitized solar cells (DSCs), which have the ability to convert solar energy into electricity. DSCs are generally composed of a dye-sensitized photoanode, an electrolyte, and a counter electrode (CE), the latter being crucial to the photovoltaic performance of DSCs.1 Typically, a Pt CE enables DSCs to achieve high power conversion efficiency (PCE), but the high cost of Pt severely limits the overall development of the cells. Against this background, Jing et al. developed a facile one-step approach using ZIF-67 as a sacrificial template in the synthesis of a CE catalyst for DSCs.59 ZIF-67 was pyrolyzed in N2 following a heating rate of 5 °C min−1 and maintained at 850 °C for 2 h, yielding ZIF-67-850 which was found to be the optimum calcination temperature, producing the highest degree of graphitization with ameliorated surface wettability. ZIF-67-850 exhibited good electrochemical performance and the PCE attained was very close to that observed for the Pt CE in the liquid I3−/I− redox couple electrolyte. Characterizing the structure of the products indicated that cobalt nanoparticles were embedded in an N-doped graphitic carbon matrix while cobalt and cobalt oxide nanoparticles were exposed on the external surface of the carbon. The authors attributed the excellent performance of ZIF-67-850 to the synergetic effects between the Co and CoO coupled with the nitrogen doped graphitic carbon. They finally concluded that the electrocatalytic performance, cycle stability and low-cost of the preparation make this novel catalyst a promising candidate to replace Pt-based materials in solar cell applications.
Pt nanoparticles supported on Vulcan™ XC-72R microporous carbon are the most common catalysts used to accelerate the sluggish oxygen reduction reaction (ORR) at the cathode of a proton exchange membrane fuel cell (PEMFC). There are several examples of Co-MOFMS in electrocatalysis, with special emphasis on the ORR. As a matter of fact, Ma et al. identified a Co-imidazolate framework Co(Im)2 (Im = imidazolate) described elsewhere60 as an efficient precursor for this reaction.61 The sample started to demonstrate ORR activity after heating it at 600 °C and its optimal performance was achieved when the sample was pyrolyzed at 750 °C with an onset potential comparable to the best cobalt-based non-PGM catalysts.62 The combined analysis of TEM, XPS and XAFS led the authors to believe that upon thermal activation, the catalytic sites were constructed by Co–N interactions through pyridinic or pyrrolic N species retained in the carbon.63 This model explains why the ORR activity decreases with higher pyrolysis temperatures, as more N is lost with increasing carbonization, and comprises the starting point for prospective work on the topic.
Xia et al. obtained similar results by pyrolyzing Co-based ZIF-67 in an Ar atmosphere with temperatures in the range of 600–900 °C.64 The current density of the ORR was also maximized in this case for the sample carbonized at 750 °C, explained by the optimum graphitic N atoms witnessed by XPS.63 Moreover, the authors studied the effect of the crystallite size of the MOF precursor and showed that as the size decreased, the onset potential and kinetic current density increased. Finally, they concluded that the high activity of the nano-electrocatalyst is attributed to the fully exposed nano-structure together with good mass- and electron-transport properties.
In other work, Xia et al. proved that MOFs can be coated on highly ordered mesoporous carbon prior to pyrolysis to provide a final catalyst structure with improved mass and electron transfer characteristics.65 This procedure comprised the coating of a Co benzimidazole MOF on a CMK-3 mesoporous carbon and subsequently pyrolyzing it at 700 °C, and optionally oxidizing the Co phase in air at 150 °C. The heat treatment resulted in finely dispersed Co agglomerates over the 3D pore structure with sizes around 15–30 nm, without causing pore blockage due to the interconnected carbon matrix (Fig. 3). The different heat treatment procedures and additional acid leaching of Co generates a tunable set of ORR catalysts that gives highly relevant insight into the Co core and carbon shell, next to very efficient ORR catalysts in terms of activity and stability.
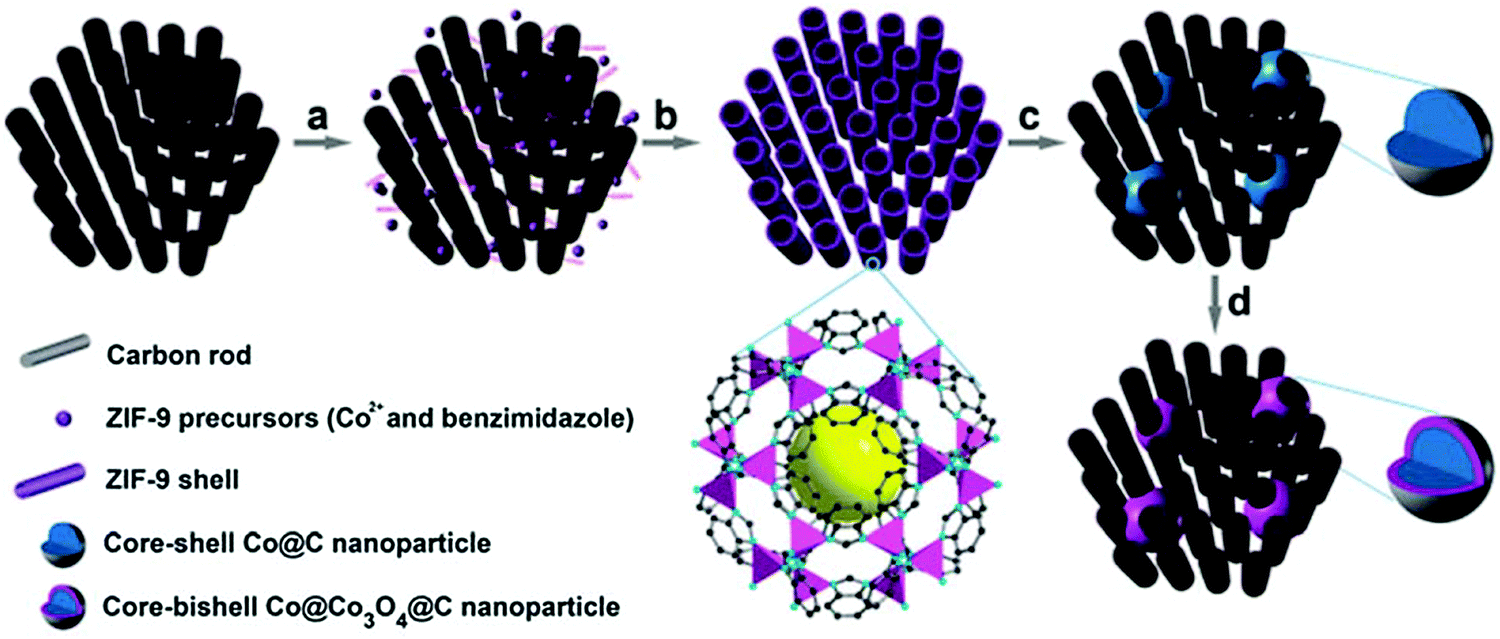 | ||
| Fig. 3 Schematic illustration of the synthesis of Co@Co3O4@C–CM: (a) functionalization of the CM with carboxylate groups, (b) growth of the MOF on the CM, (c) heat treatment under Ar flow and (d) oxidation in air at 90 °C. Reproduced from ref. 65 with permission from The Royal Society of Chemistry. | ||
With their work Wang et al. provided further insight into Co-MOFMS for electrocatalysis by analysing three representative MOF structures (ZIF-67, ZIF-8 and Co2(bdc)2(dabco) (dabco = 1,4-diazabicyclo[2.2.2]-octane)) as the catalyst precursors for the ORR.66 Such choices allowed them to study how the metal/ligand combination affected the ORR performance. The ORR catalysts were prepared via pyrolysis in Ar of the MOFs at different temperatures ranging from 600–1000 °C for 10 min and with 10 °C min−1 heating ramp. They found that among the three MOF precursors, the ZIF-67 derived catalyst exhibited the best ORR activity in both alkaline and acidic electrolytes. They suggested that Co atoms with stronger interactions with the carbon matrix, for instance, those directly bonding with the pyrrolic/pyridinic N, were retained in the nanosized voids and these N-coordinated Co sites inside the nanoscale pores served as the active ORR sites. Furthermore, the best ORR performance was obtained when the porosity and the graphitic structure of ZIF-67 were optimized at 900 °C pyrolysis temperature, and the Co content was reduced from 30 wt% Co to 4.7 wt% by acid leaching in HCl due to the removal of Co particles in the nanopores which were blocking the CoNx active sites. Notably, the acid leached sample exhibited even more positive onset and half-wave potentials than those of Pt/C in alkaline electrolytes, as well as better stability and tolerance to methanol crossover. The overall performance of the acid leached sample is among the best of the contemporary noble metal free ORR catalysts in both alkaline and acidic electrolytes which the authors ascribed to the highly dispersed active CoNx sites in the nanoporous conductive system.
Kong et al. worked on the one-step pyrolysis of a unique “cage-in-cage” Co-MOF to prepare nitrogen-doped carbon cubes with numerous embedded metallic Co nanoparticles and tested them for the ORR.67 In their case, CPM-24 Co-MOF was selected since (i) its highly symmetric isotropic cubic symmetry was expected to contribute to the uniformity in the distribution of heteroatoms in a carbon matrix, (ii) it consists of a unique nested Co24@Co48 nearly spherical cage-within-cage configuration which best meets the desired structural features that could facilitate the creation of a core–shell configuration and (iii) it also consists of the Co–N linkage which allows the entrapment of nitrogen sites for an enhanced activity. CPM-24 Co-MOF was synthesized via a modified indium assisted multicomponent self-assembling method previously reported by the same authors.68 CPM-24 Co-MOF cubic crystals were heated for 2 h under Ar, with a heating rate of 3 °C min−1 and the final temperature reached between 550 and 900 °C depending on the sample. As a result, ferromagnetic Co nanoparticle-embedded and nitrogen-doped carbon composites were obtained. The sample prepared at 700 °C demonstrated excellent methanol-tolerance and showed high ORR activity comparable to the commercial Pt/C (10 wt%) catalyst in alkaline medium and also showed higher durability than Pt/C. The authors proved through long-time etching by HF that Co nanoparticles in the composites were essential in enhancing the ORR electrocatalytic performance which in principle contradicts what Wang et al.66 previously suggested when attributing the catalytic activity mainly to Co–Nx moieties. In contrast, Kong et al. believe that nitrogen-doped carbon activated by Co nanoparticles played the important role in ORR performance of their MOF derived Co electrocatalysts.
To rationally design the reaction interface for an oxygen evolution reaction (OER), an optimal electrode structure involving high-performance catalysts is essential in reducing the overpotential, promoting the reaction kinetics, and enhancing the specific activity for the OER, thus improving the energy efficiency.69 To this end, a lamellar Co naphthalenedicarboxylate MOF was used by Ma et al. to grow nanowire arrays (NAs) on a Cu substrate plate, followed by carbonization at 600 °C.70 Specifically, the material was first heated from room temperature to 400 °C with a ramp rate of 1 °C min−1, and the temperature was kept at 400 °C for 2 h, which was further increased to 600 °C and stabilized for another 4 h. Pyrolysis of the layered crystalline MOF structure created slit-like pores and the array of thick nanowires provided a large accessible surface area. Noticeably, the OER current of Co3O4C–NA largely exceeded that of a similarly loaded IrO2/C coated on Cu despite the slightly lower onset potential of IrO2/C, featuring a much better catalytic performance of Co3O4C–NA and also in terms of durability. Moreover, the carbonized MOF on Cu proved to be a better electrode configuration than calcined MOF on Cu, evidenced by electrochemical impedance spectra and polarization curves.
Leaving electrocatalysis behind, there are a large number of examples that endorse the application of MOF derived cobalt catalysts in conventional heterogeneous catalysis in oxidation and hydrogenation reactions.
The low-temperature oxidation of CO has drawn tremendous interest due to its wide applications such as cleaning air, lowering automotive emission, and detecting trace amounts of CO. Bao et al. targeted the preparation of Co oxides from MOFs and demonstrated a novel “in situ self-reduction” route for the fabrication of a nanocomposite consisting of Ag/Co3O4 from Prussian blue analogues (PBAs) without using any reductant.71 After calcining the uniform Ag3[Co(CN)6] nanocubes at different temperatures in air, porous Ag/Co3O4 nanocomposites with abundant well-knit Ag/Co3O4 nanojunctions were obtained. The prepared samples exhibited excellent catalytic activity and stability for catalytic CO oxidation and the activity was maintained for more than 18 h, even after the catalyst was exposed to air for two months. Without any pretreatment, Ag/Co3O4 catalysts prepared by these authors showed outstanding catalytic performance compared with other oxide supported-Ag catalysts reported in recent years. The presence of abundant and highly dispersed Ag/Co3O4 junctions in the nanocomposites was suggested to be responsible for the excellent catalytic performance. On the junctions, O2 and CO adsorb on Ag and Co3O4, respectively, and react with each other more easily and besides, Ag particle aggregation could be avoided during a catalytic reaction. Coming back to ZIF-67 mediated synthesis for supported Co nanoparticle preparation, Wang et al. recently disclosed the preparation of an efficient metallic Co-based catalyst for low-temperature CO oxidation, where the Co was embedded in porous carbon.72 ZIF-67 was pyrolyzed at 600 °C for 10 h in Ar and the obtained Co/C-600 catalyst was used for CO oxidation under dry conditions yielding 100% CO conversion at 0 °C (T100). The apparent activation energy of CO oxidation over the Co/C-600 catalyst was comparable to those of the Co3O4,73 Pd74 and Au75 catalysts. When comparing the activity of Co/C-600 to Co supported on activated carbon, it was found that the light-off temperature decreased by 78 °C for Co/C-600. The catalytic activity over the Co/C-600 catalyst remained unchanged after 24 h of time on stream at room temperature, thus indicating that Co/C-600 was highly stable. However, in the presence of small amounts of moisture, Co3O4 deactivates quickly even under ambient temperatures. Interestingly, catalytic stability tests for Co/C-600 under wet gas conditions showed improved moisture tolerance as compared to other Co-based materials and moreover, the activity of the catalyst could be fully restored upon a mild heating treatment in an oxidative atmosphere.
Air pollution by volatile organic compounds (VOCs) has been a serious environmental problem in past decades. Among the VOCs, benzene is an important chemical feedstock and a gasoline ingredient.76 For complete oxidation of benzene to CO2 and H2O in this case, Li et al. studied tricobalt tetraoxide-supported palladium catalysts derived from MOFs.77 Porous polyhedron (PP) Co3O4 catalysts were prepared through one-step calcination of ZIF-67 in a muffle oven at an elevated temperature (250, 350, or 550 °C) at a ramping rate of 1 °C min−1 and held for 1 h. Finally, Pd/Co3O4-PP-X (X = 250, 350, or 550 °C) were prepared by a conventional impregnation method and subjected to calcination at 200 °C for 4 h. Among the MOF derived catalysts obtained at different temperatures, Co3O4-PP-350 exhibited the highest activity for the complete catalytic conversion of benzene. This catalyst also showed better performance than that of Pd supported on Co3O4 nanoparticles (NPs) prepared by a hydrothermal method and calcined at the same temperature. They resolved that (i) the proper porosity and nanoparticle size of the catalyst could be controlled by adjusting the calcination temperature, (ii) the surface adsorbed oxygen, which is associated with the PdOx species, is crucial for improved catalytic performance and (iii) the reducibility of the catalyst has a significant effect on the catalytic activity for the oxidation of benzene. As a consequence, the porous structure could expose more PdOx species on the surface of the catalyst, and the electron transfer from Pd to O for Pd/Co3O4-PP-350 might make the surface adsorbed oxygen more active, rendering Pd/Co3O4-PP-350 as the best catalyst for the complete oxidation of benzene.
Direct oxidation of alcohols to esters with molecular oxygen is an attractive and crucial process for the synthesis of fine chemicals. The heterogeneous catalysts used so far are based on noble metals or require the addition of base additives. Therefore, the development of reusable non-noble metal based catalysts for the oxidative direct esterification of alcohols under mild conditions is an attractive and challenging subject in both green chemistry and organic synthesis. To this purpose, Zhong et al. developed a simple, cost effective, and environmentally friendly protocol for direct aerobic oxidative esterification of alcohols, using a novel nitrogen doped graphite enclosed cobalt material from ZIF-67 as a catalyst.17 The conversion of ZIF-67 into Co@C–N was performed by direct thermal treatment under a flow of Ar at 800 °C for 8 h with a heating rate of 1 °C min−1. The strong coordination interaction between Co and N atoms in the parent MOF allowed a stepwise collapse of the MOF structure during slow-heating procedures to prevent a serious aggregation of Co. The resulting carbon–nitrogen composite played an important role in isolating the Co nanoparticles, which were surrounded tightly by graphitized carbon. The catalytic system featured a broad substrate scope for aromatic and aliphatic alcohols as well as diols, giving their corresponding esters in good to excellent yields at room temperature and under atmospheric conditions without the assistance of any base additives. Moreover, the catalyst was easily recovered due to its good magnetic properties and the reactivity could be fully restored for up to five runs when the reused catalyst was treated in H2 at 400 °C for 1 h. In a similar manner, Zhou et al. recently reported a similar route to generate Co NPs with surface-oxidized CoO species uniformly incorporated into N-doped porous carbon by one-step pyrolysis of ZIF-67, as a hard template.78 The pyrolysis was carried out at different temperatures and times under N2 and it was found that the MOF pyrolized at 700 °C for 3 h showed the best properties. Thus, the resultant Co–CoO@N-doped porous carbon nanocomposite as an inexpensive, stable and magnetically recyclable catalyst exhibited excellent catalytic performance for direct homo-and cross-coupling esterifications of primary alcohols under mild conditions, with 1 bar of O2 as an environmentally friendly oxidant. The special structure of the nanocomposite and the synergistic effect between Co and CoO NPs were proposed by these authors to be responsible for the superior activity compared to other related catalysts. In addition, the N-doped porous carbon scaffold not only stabilized the NPs but also greatly facilitated the accessibility and adsorption of substrates to the active sites and the diffusion of products.
The development of a highly efficient and environmentally sound methodology for the synthesis of amides from inexpensive and abundant feedstocks is highly desirable in both academic research and industrial applications. In this regard, Bai et al. addressed a novel oxidative amidation of aldehydes using MOF mediated heterogeneous Co-based catalysts.79 The as-prepared CoIIMOF (Co9(btc)6(tpt)2(H2O)15, tpt = 2,4,6-tris(4-pyridyl)-1,3,5-triazine) was heated at 600 °C for 8 h with a heating rate of 1 °C min−1 in Ar. With 4-methylbenzaldehyde as a model substrate and DMF as the solvent, Co@CN600 gave N,N-4-trimethylbenzamide in 90% yield at 80 °C. Moreover Co@CN600 could be easily separated by using an external magnetic field and could be reused at least five times with only a slight loss of activity. Control experiments using (i) homogeneous Co(NO3)2, (ii) the parent CoIIMOF, (iii) CoO or Co3O4 nanoparticles, (iv) metallic Co nanoparticles (20–30 nm), (v) activated carbon and (vi) a physical mixture of Co and carbon as the catalysts highlighted the importance of synergic interactions between the C–N composite and Co nanoparticles in determining the activity of the Co@C–N materials in the oxidative amidation reaction. With the optimized reaction conditions in hand, the authors also proved that the MOF derived catalysts were highly active in oxidative amidation of a wide range of aldehydes with formamides, affording the corresponding amides in good to excellent yields under mild reaction conditions. The performance was mainly attributed to the resulting small Co nanoparticles embedded in the N-doped carbons highly dispersed with an average size of ca. 7 nm. Three distinct peaks were observed in the N1s spectra of Co@C–N600, suggesting the presence of three kinds of coordination environments for N atoms: pyridine type nitrogen bonded to a metal (advantageous to prevent a serious aggregation of Co during pyrolysis of the MOF), pyrrole-type nitrogen (usually found in the carbonized nitrogen-containing organic materials and which might confer the porous N-doped carbons with a remarkably enhanced chemical reactivity due to their extended electronic structures) and N typical for ammonium species.
The ability to transport electrons has been identified as the key property of the resultant catalyst in advanced oxidation reactions. Andrew et al. adopted MOFMS, and combined ZIF-67 with graphene oxide (GO), in order to enhance electron transport and use GO as a co-catalyst to activate peroxymonosulfate (PMS).80 The ZIF-67/GO nanocomposite was carbonized in N2 at 600 °C for 6 hours, and subsequently washed with water and ethanol thoroughly to obtain the final product, magnetic cobalt–graphene (MCG). MCG was used as a heterogeneous catalyst to activate PMS for the decolorization of acid yellow in water. MCG exhibited a unique morphology and porous structure, and sufficient saturation magnetization also allowed it to be readily collected from water after the decolorization reaction. Moreover, this material exhibited an enhanced catalytic activity in the activation of PMS compared to the merely carbonized ZIF-67. To evaluate the long-term catalytic activity of MCG, a 50-cycle decolorization test was performed and the regeneration efficiency remained the same over 50 cycles, showing its stable and effective catalytic activity.
Hydrogen as a clean energy has attracted wide attention. Xing et al. focused their efforts on the preparation of multilayer core–shell composites from Co-MOFs loaded on reduced graphene oxide (rGO) sheets for hydrogen generation from NaBH4.81 The as-obtained Co-MOF–poly vinylpyrrolidone (PVP)–GO black powder was further heated with a ramp of 5 °C min−1 to 700 °C in N2 for 1 h yielding Co@N–CG and further heat treated at 200 °C for 24 h in air yielding Co@CoOx@N–carbon–rGO (Co@CoOx@N–CG). Additionally, the authors prepared Co@CoOx@CG, Co@CoOx@N–C and Co@CoOx@C, in the absence of PVP, GO and both of them, respectively. The Co@carbon and Co@carbon–rGO core–shell structures were obtained with the formation of cores from metal ions and carbon shells from carbonization of ligand molecules. Controllable oxidation of Co cores to Co shells generated multilayer core–shell structures, namely Co@CoOx@carbon. In hydrogen generation through hydrolysis of NaBH4, multilayer core–shells, Co@CoOx@carbon possessed higher catalytic activity with lower activation energy compared to single core–shells. The authors explained the improved catalytic activity by (i) the synergy effects between Co and CoOx on the interface region and (ii) the carbon shells or N-doped carbon shells protected the Co or Co@CoOx cores from growing, aggregating, or breaking up for excellent stability. Moreover, the rGO sheets facilitated the magnetic momentum transfer and improved the flexibility of these core–shell catalysts. The authors deem these core–shell structures anchored on rGO sheets to be an excellent alternative for the design of effective heterogeneous catalysts involving energy industrial routes.
Likewise, transition metal-catalyzed transfer hydrogenation protocols are convenient and alternative green chemical methods to traditional energy-consuming hydrogenation processes, besides being able to provide high atom efficiency and generating advantageous economics.82,83 The use of hydrogen donor reagents such as alcohols in transfer hydrogenation reactions can avoid the use of autoclaves and high-pressure hydrogen, being highly relevant for industrial applications. Not long ago, Long et al. hinted at the transfer hydrogenation of unsaturated bonds in the absence of base additives catalyzed by MOF derived cobalt heterogeneous catalysts.84 They selected [Co(bdc)(ted)0.5] MOF as a template due to the presence of triethylenediamine basic sites which are deemed to be favorable for transfer hydrogenation reactions. In the transfer hydrogenation of acetophenone to phenethanol as a model process using isopropanol as both reductant and solvent, the Co@C–N materials prepared under longer thermolysis times and higher temperatures exhibited higher activity and selectivity, thus the final selected catalyst was obtained by pyrolysis in argon of the Co MOF at 900 °C for 15 h. Multiple advantages of the proposed catalytic system included a remarkable versatility not only for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrogenation but also for CC, C
O hydrogenation but also for CC, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, and NO bonds, an environmental-benign protocol and the use of a magnetically separable non-noble metal catalyst in a simple and safe reaction setup. Both mechanisms proposed in the literature for the transfer hydrogenation with base additives suggest that the presence of bases facilitates the formation of metal hydrides in the transition metal.85 Therefore, the authors assumed that basic sites on Co@C–N might play a similar role to that of base additives to facilitate the transfer of protons to Co nanoparticles to form Co metal hydride species, thus promoting the production of the desired hydrogenation products. Combination of high efficiency, versatility and recyclability as well as mild reaction conditions in the absence of bases and gaseous hydrogen makes this system an attractive alternative pathway for various hydrogenation processes.
N, and NO bonds, an environmental-benign protocol and the use of a magnetically separable non-noble metal catalyst in a simple and safe reaction setup. Both mechanisms proposed in the literature for the transfer hydrogenation with base additives suggest that the presence of bases facilitates the formation of metal hydrides in the transition metal.85 Therefore, the authors assumed that basic sites on Co@C–N might play a similar role to that of base additives to facilitate the transfer of protons to Co nanoparticles to form Co metal hydride species, thus promoting the production of the desired hydrogenation products. Combination of high efficiency, versatility and recyclability as well as mild reaction conditions in the absence of bases and gaseous hydrogen makes this system an attractive alternative pathway for various hydrogenation processes.
Generally, different anilines are prepared by reduction of their corresponding nitro substrates. Nevertheless, selective hydrogenation of a nitro group in the presence of other reducible substituents is challenging. Wang et al. synthesized chemoselective nitrogen-doped carbon supported Co MOF derived catalysts for the selective hydrogenation of nitroarenes starting from ZIF-67.86 Both conversion and selectivity decreased with an increase in pyrolysis temperature of the MOF, suggesting that the presence of nitrogen, which decreases with increasing temperature, might play an important role in this catalytic system. Shen et al. focused on the hydrogenation of nitrobenzene and proposed a facile and efficient approach to fabricate non-noble metal@noble metal core–shell catalysts by using Co-MOF as a sacrificial template.18 Co@Pd core–shell NPs embedded in a N-doped carbon matrix (Co@Pd/CN) were prepared using ZIF-67 and Pd(NO3)2 as the precursor and Pd source, respectively. First, ZIF-67 was pyrolyzed at 600 °C under Ar for 8 h and the obtained Co NPs were used as a sacrificial template to reduce an aqueous Pd(NO3)2 solution via galvanic replacement reaction resulting in the coverage of the MOF-derived Co NPs by an ultrathin Pd shell. The highly exposed Pd atoms on Co nanoparticles resulted in a highly active, extremely stable catalyst for nitrobenzene hydrogenation. The Co@Pd/NC showed much higher hydrogenation activity than MOFs supporting noble NPs, thus opening a new avenue for MOF-templated non-noble@noble metal core–shell catalysts that could far surpass the traditional MOFs supporting noble NPs in catalytic properties. Moreover, as a novel approach, the one-step pyrolysis of ZIF-67 carried out by Ma et al. produced an N-doped porous carbon for the tandem dehydrogenation of ammonia borane and hydrogenation of nitro compounds at room temperature.87
Most of the examples dealing with Co-MOFMS for heterogeneous catalyst preparation refer to metal nanoparticles in carbon where pyrolysis under an inert atmosphere is applied. Ample data are available on this topic when compared with other metal based MOFs. Besides, due to the ease of synthesis and low price, ZIF-67 has been the most studied MOF in MOF derived catalyst preparation. ZIF stands for a zeolitic imidazolate framework and refers to a sub-family of MOFs with imidazolate as the linker. The structures of ZIFs are similar to conventional aluminosilicate zeolites and thus, generally display properties that combine the advantages of both zeolites and MOFs, such as ultrahigh surface areas, unimodal micropores, high crystallinities, abundant functionalities and exceptional thermal and chemical stabilities.88 In MOFMS, the main feature of ZIF-67 derived catalysts is the content of N which results in advantageous properties in different catalytic processes due to the resulting (i) nitrogen-doped carbon, (ii) Co–Nx moieties and (iii) synergetic interactions between the C–N composite and Co nanoparticles. Thermodynamics predict that the carbothermal reduction of Co ions in the framework of the MOF leads mainly to metallic cobalt. Metallic cobalt catalyses the graphitization of surrounding carbon which in some processes comprises the main limitation for the use of these catalysts, since the hardly accessible nanoparticles might lead to diffusion limitations of the reactants to the active Co sites during the process.
All things considered and since in some processes the nature of the active sites still remains under debate, full understanding and control of the synthesis process calls for further research on Co-MOFMS, including ZIF-67-MOFMS.
(d) Copper (Cu)-MOFMS
The applications of copper (Cu) and Cu-based nanoparticles, which are based on this earth-abundant and inexpensive metal, have generated a great deal of interest in recent years, especially in the field of catalysis.89 Higher in the Ellingham diagram than Co, the reduction potential of Cu2+ is +0.34 V and the melting and boiling points of Cu are 1085 °C, and 2560 °C, respectively. The catalytic application of Cu is broad, including electrocatalysis, photocatalysis, and gas-phase thermal catalysis.HKUST-1 (MOF-199) is one of the most cited and employed Cu-MOFs because of its large surface area, high pore volume, high chemical stability and easy synthesis. First reported in 1999 by Chui et al.,90 HKUST-1 is made up of copper nodes with 1,3,5-benzenetricarboxylic acid (btc) struts between them. In the framework of HKUST-1, Cu(II) ions form dimers, where each copper atom is coordinated by four oxygens from btc linkers and water molecules,91 forming face-centered-cubic crystals that contain an intersecting three-dimensional (3D) system of large square-shaped pores (9 Å by 9 Å). Cu-bdc's such as MOF-2 are other examples of Cu-MOFs with markedly high surface area, which makes them superior candidates for gas separation and sieving applications.92 The coordination mode in MOF-2 is the same as the coordination mode in HKUST-1. However, in contrast to HKUST-1, the architecture of MOF-2 is based on stacking lamellar phase sheets of Cu2(bdc)2 in two dimensions.93 MOFs with open coordination sites, like HKUST-1 and MOF-2, will likely be scrutinized for possessing both homogeneous and heterogeneous catalytic behaviour.92
Both metal oxides and nanoparticles@carbon have been reported using mainly HKUST-1 as the precursor of electro- and photocatalysts as well as for CO oxidation and the transformation of alcohols and aldehydes.
In photocatalysis, Li et al. described a facile synthetic method for the preparation of Cu/TiO2 photocatalysts with hollow structures by treating HKUST-1 at 140 °C in the presence of ascorbic acid (AA) and ethylene glycol (EG) as solvent and TiO2 hollow particles.20 The released Cu2+ cations were simultaneously reduced by AA to produce Cu nanoparticles when they penetrated through the TiO2 shells. The AA-reduced Cu/TiO2 hybrid structures exhibited enhanced charge separation and hydrogen production performance. Likewise, Mondal et al. constructed MOF-derived Cu/CuO@TiO2 photocatalysts where the copper species were adsorbed on anatase.19 Titanium isopropoxide (Ti(ipro)4) was added to the Cu-bdc MOF in an ethanol/water mixture and then the amorphous material was calcined at 350 °C for 5 h in a N2 atmosphere. The obtained mesoporous Cu/CuO@TiO2 showed a considerably higher rate of H2 production than conventional CuO loaded TiO2, attributed to the presence of surface deposited Cu0 species and the small size of the heterojunction between CuO and TiO2, which facilitated interfacial charge carrier transfer from TiO2 nanoparticles.
In the field of electrocatalysis, palladium is a potential candidate to substitute platinum as a catalyst for the ORR since on the one hand, Pd is a member of the Pt group metals and thus its catalytic ability is close to that of Pt and on the other hand, the Pd abundance in the earth crust is higher than that of Pt. Further, addition of transition metals such as Cu94 to the catalytic system enhances the catalytic performance and decreases precious-metal loading. In this line, Luo et al. successfully synthesized a PdCu alloy on carbon (PdCu/C) using HKUST-1 as the catalyst precursor.95 The synthesis procedure consisted of adding K2PdCl6, sodium acetate, HKUST-1 (Basolite C300, Sigma-Aldrich) and carbon black (heat treated Vulcan XC-72) into methanol and purging by nitrogen in an ice-water bath (ca. 2–4 °C) for 20 min under continuous stirring. Subsequently, the system was sealed under CO in an ice-water bath for 2 h and then sealed under N2 at room temperature for 12 h under stirring. Finally, the product was filtered and rinsed with ultra-pure water and the obtained black powder was dried under air at 60 °C overnight. The improved ORR activity obtained on the MOF derived PdCu/C compared to PdCu/C synthesized from a metal chloride precursor (CuCl2) was correlated with Pd surface modification induced by alloying, further proved by CO-stripping voltammograms. Compared with the commercial Pt/C Johnson Matthey (JM) catalyst, the ORR mass activity of PdCu/C was lower in acid medium, whereas it was around 3-fold higher in alkaline solution. In view of these results, the authors claim that using HKUST-1 precursors for the synthesis of PdCu nanoalloyed NPs, not only extends the application of MOFs in the field of electrocatalysis, but also brings a new methodology in advanced non-Pt active centers for the ORR. Besides, work done by Raoof et al. highlights the good behaviour of HKUST-1 derived Cu/nanoporous carbon composites for the hydrogen evolution reaction (HER).96 The Cu/NPC composite catalyst was synthesized by direct carbonization of HKUST-1, the latter prepared according to a previously reported procedure.97 HKUST-1 was then heat treated in N2 at 550 °C, followed by 6 h at 900 °C yielding the Cu/NPC composite. Electrochemical measurements of the Cu/NPC on a glassy carbon electrode indicate improvement of HER catalysis which they attribute to the existence of Cu, CuO, and Cu2O species together with improved surface area, and the expected hydrogen adsorption properties of the Cu/NPC composite.
Copper based catalysts look also promising for substituting the limited available noble metals in the preferential oxidation of CO (PROX) in the presence of hydrogen, which is one of the most promising ways to reduce the CO content of effluents from reforming units.98 Not yet envisioned as a synthesis method itself, Zamaro et al. realized that HKUST-1 in CO oxidation was not active by itself, but after ‘in situ’ activation in the reaction atmosphere at temperatures above 230 °C, highly dispersed and highly active CuO nanoparticles were produced.99 The transformation involved a gradual segregation of CuO nanoparticles from the MOF, simultaneously with a loss of MOF crystallinity. Moreover, the incorporation of a Ce precursor in the network of HKUST-1 with subsequent activation yielded a highly dispersed mixture of CuO and CeO2 nanoparticles with a high degree of interaction that was highly active in CO oxidation above 230 °C. Later, as a proof of concept, Liu et al. used the same MOF and TiO2 whiskers100 as supports to prepare a well-dispersed and size-controllable CuO/TiO2 catalyst derived from the thermolysis of the Cu-btc/TiO2 composite through calcination in air.101 The CuO/TiO2 catalyst behaved excellently in CO oxidation and the strategy could be translated to other supports, with CuO/Al2O3 showing excellent activity.
With focus on the transformation of alcohols, Niu et al. synthesized a hybrid catalyst for the catalytic reduction of 4-nitrophenol consisting of non-noble Cu/Cu2O NPs supported on porous carbon using HKUST-1 as a sacrificial template.102 After thoroughly mixing HKUST-1 with a phenol formaldehyde resin as an additional carbon source, the resulting solid product was carbonized under a N2 atmosphere at 600 °C for 4 h. The Cu/Cu2O/C composite demonstrated excellent catalytic activity for the reduction of 4-nitrophenol in the presence of NaBH4, comparable with noble metal based catalysts, such as SiO2/Fe3O4–C/Au,103 Au–Ag/GO,104 and Au/CuO.105 Moreover, the synthesized catalyst could be reused for at least five cycles due to its good stability. The authors claimed that Cu/Cu2O NPs were responsible for the rapid reduction of 4-nitrophenol while the carbon matrix could prevent aggregation of Cu/Cu2O NPs providing high surface-to-volume ratio and chemical stability. In another recent work, a new catalytic system based on HKUST-1, TEMPO and NMI (TEMPO: 2,2,6,6-tetramethylpiperidine-N-oxyl; NMI: N-methylimidazole) was developed by Kim et al. for aerobic alcohol oxidation.106 As-prepared HKUST-1 was calcined under Ar at 800 °C for 6 h with a ramping rate of 5 °C min−1. For the reaction, a test tube was loaded with equimolar amounts of the catalyst and TEMPO and evacuated and backfilled with oxygen. The alcohol and NMI with acetonitrile were then added to the mixture and kept under stirring at 70 °C under an O2 balloon. Simple pyrolysis of HKUST-1 afforded a robust and chemically inert copper–carbon nanocomposite (Cu@C) to form the Cu@C/TEMPO/NMI system which exhibited outstanding performance for the aerobic oxidation of alcohols to their corresponding aldehydes. Furthermore, this catalyst system revealed a broad scope for the substrate and excellent reusability for the aerobic oxidation of alcohols compared to the non-carbonized HKUST-1/TEMPO/NMI system.
In the catalytic hydrotreatment of furfural, Cu catalysts have strong ability to polarize the C–O bond while leaving the furan ring unaffected and furthermore, the appropriate addition of nickel might enhance their performance due to the synergistic effect of bimetal.107 Accordingly, Wang et al. addressed the preparation of CuNi bimetallic nanoparticles embedded in a carbon matrix (CuNi@C) from MOFs and applied them in the selective conversion of biomass-derived furfural to cyclopentanone.108 For the preparation of CuNi@C bimetallic catalysts with the Ni/Cu molar ratio from 0 to 2.5, a predetermined amount of Ni(NO3)2·6H2O ethanol solution was added to as-prepared HKUST-1. The mixture was kept under stirring for 24 h and then dried at 80 °C overnight. The resulting solid was calcined in N2 for 2 h at 500 °C to obtain the CuNi@C catalyst. The authors found that CuNi0.5@C showed the best catalytic performance when compared to other Cu/Ni ratios and also when compared to CuNi0.5 prepared by the classical precipitation method. In addition, CuNi0.5@C could be reused four times with good activity and stability. The enhanced structure of the composite, the nano-scaled metallic copper and nickel without oxide impurity highly dispersed in a porous carbon matrix are thought to be responsible for the outstanding catalytic performance in furfural conversion to cyclopentanone.
In summary, despite the strong background on the applications of bulk Cu in various fields the use of Cu NPs is restricted by their inherent instability under atmospheric conditions, i.e. their sensitivity for oxidation. The possible modification of the chemical and physical properties of these nanoparticles using different synthetic strategies and conditions and/or via post-synthetic chemical treatments has been largely responsible for the rapid growth of interest in these nanomaterials and their applications in catalysis.89 In this sense MOFMS reinforces the idea of creating a chemically inert metal or metal oxide–carbon composite by pyrolysis of MOFs as a powerful general strategy for preparing stable and reusable copper heterogeneous catalysts. Nevertheless, the high reduction potential for Cu2+ means that when calcined in an inert atmosphere the vast majority of copper appears in its metallic form. Metallic copper strongly promotes the graphitization of the surrounding carbon which leads to poor accessibility of reactants to active Cu sites during the catalytic process.
(e) Iron (Fe)-MOFMS
Iron minerals are ubiquitous in nature and there is abundant availability of them at minimal cost.109 Haematite is the most common iron containing ore, but iron is widely distributed in other minerals such as magnetite and taconite. The melting point of metallic Fe is 1538 °C, its boiling point is 2861 °C and the reduction potential of Fe3+ and Fe2+ are −0.04 and −0.41 V, respectively. The magnetic nature of iron enables its easy separation from aqueous media; its high boiling point ensures high iron loading after pyrolysis and the formation of carbides due to C incorporation in the Fe lattice and prompts the metallic, carbidic, oxidic dynamic system in the pyrolysis. As in the case of Co and Cu, Fe also catalyzes graphite formation in the carbons seen as graphite layers.Iron catalysis embraces Fe/N/C systems for ORR catalysis for fuel cell applications, FeC for FTS, Fe-mixed-C systems for nitro/chloro conversion and separation from waste water (dye and oil experiments), among others.
Recently, de Krafft et al. reported a simple MOF-templated strategy for the synthesis of crystalline octahedral nanoshells combining the desired photocatalytic properties from both types of oxides (Fig. 4).110 In their work, they coated MIL-101(Fe) particles with an amorphous shell of titania by acid-catalyzed hydrolysis and condensation of titanium(IV) bis(ammonium lactato)dihydroxide (TALH) in water,111 followed by calcination in air at 550 °C for 16 h. They found the resulting crystalline Fe2O3@TiO2 nanoparticles to have interesting photophysical properties as they exhibited photocatalytic hydrogen production from water using visible light, while neither component alone was able to do so. Kim and Yoon112 reported the pyrolysis of MIL-100(Fe) and the effects of furfuryl alcohol (FA) added to the MOF-pores acting as a carbon precursor. They displayed the ferromagnetic properties of the polycrystalline Fe2O3@C system, allowing for facile particle separation from a slurry phase. Elemental analysis revealed very minor hydrogen content, clearly indicating the conversion of the organic linkers toward carbonaceous materials upon pyrolysis. Indeed, the highest carbon loading of the composite material was obtained when fully soaking the MOF in FA, filling the interstitial sites of the MOF crystals in addition to the pores. Banerjee et al.113 pyrolyzed MIL-53(Fe) at 500 and 600 °C to produce Fe3O4–carbon nanocomposites as recyclable adsorbent materials for removal and recovery of environmental pollutants. The sample pyrolyzed at 500 °C was highly hydrophobic due to the presence of small micropores around 0.8 nm in diameter and the absence of hydrophilic surface groups, much contrary to the sample pyrolyzed at 600 °C comprising larger mesopores with an average of 2 nm. This allowed for effective separation of oil/water mixtures using the super hydrophobic sample, achieving separation factor values over 40. The recyclability of the material is excellent, due to its magnetic character, retaining over 75% of the separation capacity after 9 cycles. Quin et al. reported the synthesis of CoFe2O4 nanocomposites from Fe-containing MOF (MIL-100-Fe) for phenol oxidation in aqueous solution using peroxymonosulfate as an oxidant.114 They immobilized cobalt nitrate hexahydrate into the pores of MIL-100 by incipient wetness impregnation and calcined it at different temperatures (400–600 °C). The resulting magnetic nanoparticles reduced phenol concentration by more than 95% after 120 min and the regenerated catalysts exhibited good performance and stability in phenol degradation. Likewise, Dong et al. reported the preparation of magnetic porous carbons (MPCs) as supports for Au and Pd noble metal NPs.21 They obtained the MPC through calcination of Fe-MIL-88A up to 700 °C under N2 for 1 h. Accordingly, both Au/MPC and Pd/MPC nanocatalysts exhibited excellent catalytic activity for the reduction of 4-nitrophenol (4-NP) in comparison with most other reported catalysts.115 Moreover, the Pd/MPC catalyst showed high efficiency toward hydrodechlorination of chlorophenols. In both the synthesized catalysts, the MPC provided a large surface area and mesopores on which the Au and Pd NPs were finely dispersed. The catalysts could be easily recovered from the reaction mixture by using a magnet because of the magnetic NPs encapsulated in the porous carbon, and could be conveniently reused. These methods are presented by the authors as an eco-friendly strategy to convert nitro- and chloro-compounds. A short while ago, Li et al. implemented MOFMS in the hydrogenation of nitro compounds by developing a highly efficient, low-cost, and magnetically recyclable γ-Fe2O3@porous carbon catalyst.116 In their study, Fe-MIL-88A samples were thermally treated at different temperatures and times in N2 to afford Fe-based NPs encapsulated inside porous carbon. The reduction of nitrobenzene to aniline in the presence of hydrazine hydrate (N2H4·H2O) was first used to explore the catalytic activity of different catalysts. As a result, the catalyst pyrolyzed at 500 °C for 1 h gave the best activity and could be readily recycled with an external magnet being reused at least 10 times without any loss of activity. They concluded that at this temperature the small γ-Fe2O3 NPs were well dispersed inside the porous carbon, which effectively limited the aggregation and growth of the high-density NPs and still facilitated the transportation of substrates, intermediates and products. Encouraged by the excellent performance of Fe-500-1 h in the reduction of nitrobenzene, a wide variety of functionalized nitro compounds were also investigated. As a result, most of the corresponding substituted anilines were obtained with high conversion and selectivity. Notably, halogen-substituted nitroarenes were effectively reduced to the corresponding haloaromatic amines without obvious dehalogenation. This catalyst also showed absolute selectivity in the reduction of nitroarenes in the presence of other functional groups, including amino, phenolic hydroxyl and alcoholic hydroxyl. Last but not least, it was also able to offer a high chemoselectivity in the reduction of substituted nitroarenes with quite challenging reducible functional groups, including nitrile, aldehyde and ketone, giving the only product of the corresponding anilines while the reducible functional groups were unchanged. Lee et al.117 pyrolyzed Fe-MIL-88-A and Fe-MIL-88-B in a range of temperatures between 600–1000 °C under nitrogen flow, creating magnetic nanoparticle-embedded porous carbon materials. The XRD results showed that the Fe phase shifted from a mix of Fe2O3 and Fe3C at 600 °C to fully metallic Fe at 1000 °C. The porosity of the carbon materials went through an optimum at 800 °C, indicating pore collapse and decreasing surface areas at elevated temperatures. The two Fe-MOFs pyrolyzed at 800 °C were used for water treatment using methylene blue as a model compound, obtaining adsorption capacity values of 60.2 and 83.9 mg g−1 for pyrolyzed MIL-88-A and MIL-88-B, respectively.
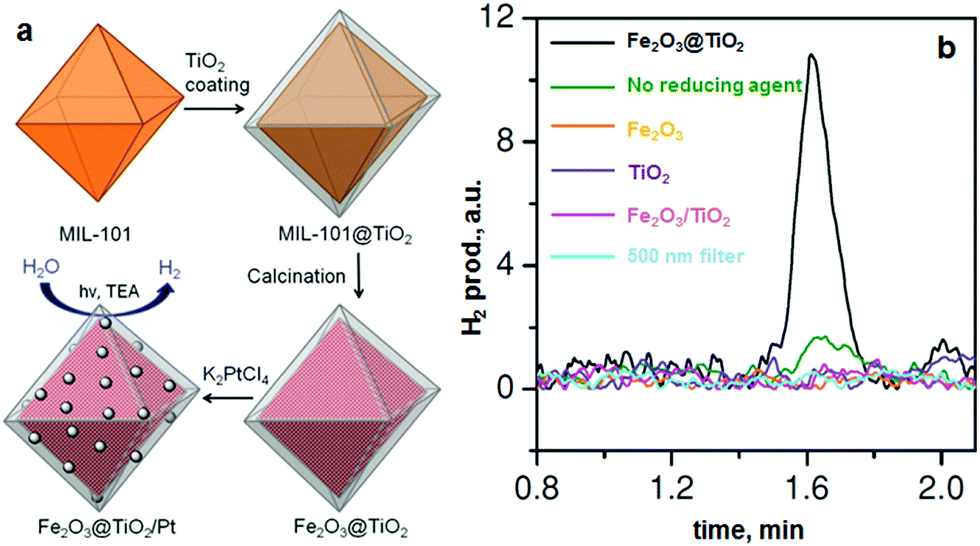 | ||
| Fig. 4 (a) MOF-templated synthesis of Fe2O3@TiO2 by coating MIL-101(Fe) with TiO2 followed by calcination, and its use for photocatalytic hydrogen production after depositing Pt particles; (b) the H2 peaks in GC analysis of the headspace using 0.5 mg of different catalyst samples. From de Krafft et al. 2012.110 Copyright © 2012 by John Wiley Sons, Inc. Reprinted by permission of John Wiley & Sons, Inc. | ||
Additionally, MOFMS has been applied in various attempts to improve the catalysis of the oxygen reduction reaction for fuel cell applications. In order to obtain the desired Fe/N/C composites, different synthesis procedures have been followed. For example, pyrolysis of low-boiling temperature Zn-based MOFs produce highly porous carbons that provide excellent support surface for Fe nanoparticles. Similarly, Fe-based MOFs, often with amino-functionalized linkers, are pyrolyzed to produce Fe/N/C in a one-step synthesis. The main difference between the two synthetic pathways is the location of the Fe species. Lower surface loadings can be achieved through impregnation of the pyrolyzed MOF, however; only Fe-MOF pyrolysis results in the formation of encapsulated Fe nanoparticles in the carbon matrix. Morozan et al.118 used the most complex MOFMS strategy comprising a first pyrolysis of FA impregnated ZIF-8 at 1100 °C, subsequent Fe and N functionalization by impregnation and ball milling, followed by secondary Ar pyrolysis at 1050 °C and even a third NH3 pyrolysis at 950 °C. The various approaches generated metallic and carbidic iron next to large fractions of FeN4 species in a micro- and mesoporous carbonaceous matrix, of which larger mesopores did not further enhance the catalytic ORR activity. The boost in PEMFC performance after ammonia pyrolysis was ascribed to the increase in the surface area, interconnecting existing active sites for catalysis instead of increasing the active site density. Zhang et al.119 concluded that Fe species indeed are crucial to the catalytic performance, ruling out that the initial activity loss of the Fe/N/C catalysts is due to the Fenton reaction of Fe with H2O2. Furthermore, the kinetics of Fe/N/C and Fe-free N/C catalysts during the ORR were similar, revealing that micropore flooding through electro-oxidation of the carbon surface was indeed the main reason for the activity drop, in agreement with their previous study.120
Mao et al.121 mixed MIL-100(Fe) with aminotetrazole and pyrolyzed the materials at temperatures between 600–900 °C to produce Fe–N–C composites. The products were washed with HCl to produce N-doped carbon shells encapsulating the metallic Fe nanoparticles, removing Fe3C and Fe2O3 particles on the carbon surface. The treatment resulted in doubling of the micropore area, liberating cavities to trap oxygen while exposing active sites. The electrocatalytic properties of the materials showed that increasing pyrolysis temperatures resulted in increased current density and a positive shift of the onset potential during linear sweep voltammetry. The trend can be related to the facilitated electron transfer by the enhanced graphitic crystallinity witnessed in XRD. The highest catalytic activity can be achieved with the sample pyrolyzed at 800 °C (NC-800); the authors argue that higher pyrolysis temperatures decrease the N-doping and thus the number of active sites, in agreement with lowered N-content from EDS results. Cyclic voltammetry showed that NC-800 outperformed metal-free N-doped carbons, confirming the involvement of Fe in catalysis. Furthermore, the MOF-based catalyst showed improved stability during continuous ORR compared to the commercial Pt–C catalyst and comprised a higher methanol tolerance. In another study, Afsahi et al.122 used a sodalite-like Fe MOF consisting of benzene tris tetrazole linkers to obtain hybrid Fe–N–C after pyrolysis in the range of 700–1000 °C. The products were subsequently acid leached in 1 M H2SO4 and heat treated at the pyrolysis temperature under a NH3 atmosphere. XRD of the pyrolyzed samples shows multi-crystalline Fe phases including Fe carbides, oxides, sulphides and nitrides, with all patterns showing much resemblance. Acid leaching removed the unstable Fe oxides and sulphides, and after NH3 heat treatment Fe–N–C catalysts were produced. The particle size of the Fe spheres varies between 10–60 nm, which are dispersed in a highly porous cube-shaped carbon material. After acid washing, the Fe content drastically reduced from 40 wt% to around 5–10%, an increase in pyrolysis temperature was found to further carbonize the structure, removing N species. The methodological approach allowed the authors to conclude that the ORR catalytic activity originates from the pyridinic and quaternary nitrogen species. Additionally, the performance of the catalysts in a fuel cell did not exceed the one from Dodelet's group,123 and it is believed that the higher Fe content plays a prominent role in the reduced activity. Li et al.124 pyrolyzed ball-milled MIL-101(Fe) mixed with melamine in a range of temperatures between 500–1000 °C to obtain Fe/Fe3C embedded in graphite layers and carbon nanotubes. High temperatures above 700 °C and a melamine carbon precursor were necessary to produce the graphitic nature and to form the carbon nanotubes. Raman spectra confirmed the more graphitic nature of high temperature samples, nearly doubling the G-band over I-band intensity values. The ORR activity in terms of onset potential was very close to commercial Pt–C, and the electron transfer number from the Koutecky–Levich plots was estimated to be 3.6 compared to 3.7 for Pt–C. Zhao et al.125 pyrolyzed nanocrystalline spindles of MIL-88B-NH2 in Ar at 900 °C to produce very active ORR catalysts. The XPS results indicated that the amino-type nitrogen was completely converted into pyridine- and quaternary-type nitrogen species, forming a well-integrated Fe/N/C catalyst. The micro- and mesoporous matrix of the catalyst was found to consist predominantly of amorphous carbon, with layered graphite species surrounding the Fe nanoparticles. The power density of an alkaline direct methanol fuel cell increased 1.7 times when using the pyrolyzed MOF as compared to the commercial Pt/C catalyst. Furthermore, the MOF-derived catalyst showed superior stability toward methanol poisoning, whereas the current of the commercial Pt/C decreased by 80% upon introduction of 2 M methanol during chronoamperometric response measurements at 0.84 V in oxygen saturated 0.1 M KOH.
Recently, MOFs have been used as a precursor to produce high-potential Fe catalysts for the Fischer–Tropsch synthesis reaction (Fig. 5).126 The Fischer–Tropsch synthesis (FTS) is a process for flexible production of key chemicals from synthesis gas; the main reaction being the hydrogenation of CO. Santos et al. employed the commercially available Fe–BTC as a precursor in MOFMS. Heat treatment of the MOF at 500 °C in an inert atmosphere resulted in the formation of a Fe@C composite that showed exceptional activity and stability in FTS. The key factor in catalytic performance consisted of maintaining the dispersion of the active phase. The pyrolysis of the MOF at low heating rates gently carbonized the framework while capturing the Fe in a carbonaceous matrix formed by the decomposition of the linker. With a pyrolysis temperature that is a factor of three lower than the melting point of the metal, sintering could be effectively reduced to synthesize highly dispersed Fe@C systems. Furthermore, the addition of a carbon source allowed for further tuning the porosity and Fe loading of the materials. Wezendonk et al. investigated the conversion of the Fe–BTC towards Fe@C by in situ XAFS and Mossbauer that revealed many intermediate Fe phases.127 Starting from the Fe(III) oxo-clusters, the pyrolysis formed mixtures of Fe carbides and a Wüstite phase. After reduction of the catalyst, the Wüstite was fully converted to metallic Fe and thus, upon syngas exposure creates highly active Hägg carbides. The system showed no deactivation, most probably due to the unique spatial confinement, witnessed with XPS and TEM. Hence, the catalyst combines a high loading of 40 wt% Fe with a high dispersion of which over 95% can be transformed into the active phase, resulting in the highest reported values for Fe-based FTS activity. Hence, the spatial confinement created by the porous carbon matrix on one hand allows for near-full transformation into an active phase and on the other hand prevents the metal NPs from sintering even under high-temperature FTS. Bing An et al.128 used a MOF with dicarboxylic linkers instead of tricarboxylic MOFs to obtain larger external surface Fe particles to properly assess the active phase with microscopy. They found that the pyrolysis additionally leaves behind carbonates on the Fe surface, which is liberated from carbonate coverage during FTS. They confirm that the Hagg carbide phase is the active phase in HTFTS, and show microscope results of spent catalysts evidencing carbide surface growth on the large Fe oxide particles. Because of the slower active phase evolution, steady state conversion is achieved only over 60 h instead of 3 h for the system with much smaller particles.22 High activities were achieved at 300 °C with the activity correlating well with the thickness of the carbide shell on the large particles. Interestingly, a control experiment employing pyrolyzed molecular Fe-oxo clusters showed little activity, underlining the importance of a framework-type precursor for pyrolysis and generating dispersed Fe particles. Wezendonk et al.129 further investigated the effect of the MOF precursor using different MOFs while employing fixed pyrolysis conditions. It was found that under the same temperature, the different MOFs undergo a similar degree of carbonization of the framework by decarboxylation. The smallest Fe nanoparticles were obtained when pyrolyzing the most porous MOFs, confirming that the spatial distribution of the MOF is the key to produce catalysts with high dispersion and loading. Indeed, the performance could be related to the surface area of Fe, confirming the dispersion-activity relationship,22 while optimal hydrocarbon selectivity could be obtained with promoters such as K, Cu, Mn and S.
 | ||
| Fig. 5 (a) Time-on-stream evolution of CO conversion for unpromoted Fe@C catalysts with different Fe loadings; (b) time-on-stream evolution of CO conversion for K-promoted 38-Fe@C catalysts; and (c) Fe phase transformation during pyrolysis of Fe–BTC towards Fe@C. Reprinted by permission from Macmillan Publishers Ltd: Nature Communications,126,127 copyright 2015 Reprinted with permission from Wezendonk et al. 2016.126,127 Copyright 2016, American Chemical Society. | ||
(f) Magnesium (Mg)-MOFMS
Magnesium is the eighth most abundant element in the Earth's crust and it occurs naturally only in combination with other elements, where it invariably has a +2 oxidation state.36 The melting point of Mg is 650 °C, its boiling point is 1090 °C and the reduction potential of Mg2+ is −2.37 V. MgO is popular for its broad application as an industrial base catalyst and a catalyst support employed in aldol additions, Claisen–Schmidt condensations, transesterifications, and other basic catalysed reactions.130To the best of our knowledge, there is only one study reported to date dealing with Mg-MOF derived catalytic materials. In this example, a three dimensional framework with parallel hexagonal channels constructed from Mg2+ ions and 3,5-pyridine dicarboxylic acid was employed.131 In their work, Salehifar et al. developed a low-temperature synthesis procedure to obtain MgO nanorods via thermal decomposition of this Mg-MOF and evaluated its photocatalytic activity.132 The as-prepared Mg-MOF was treated in an autoclave under hydrothermal conditions at 200 °C for 6 h. The obtained product was washed with distilled water and absolute methanol several times and dried at 70 °C for 5 h to yield MOF derived MgO nanorods. In order to evaluate the photocatalytic activity of the MgO nanorods, the decolorization of methylene blue (MB) was studied. The authors found that in the presence of MOF derived MgO, the MgO nanorods obtained from MOFs have remarkable photocatalytic activity compared to other studies in the literature.133 Here, MOF mediated synthesis brings not only simplicity but also decreases the costs and the temperature required in comparison with other similar techniques for synthesizing MgO.133–135
To sum up, Mg-MOFs are still practically unexplored as catalyst precursors. However, the fact that one of the most fruitful methods for preparing mesoporous MgO was the “template” method, whose synthesis procedures require expensive templates and multiple steps besides being time-consuming130 makes MOFMS an auspicious alternative technique for the synthesis of MgO. As in the case of Zn, since Mg2+ has one of the lowest reduction potentials, the preparation of nanostructured MgO from Mg-MOFs can be achieved by both direct pyrolysis in an inert atmosphere (MgO@C) and calcination in air (MgO) of the pristine MOF. Similar to that of Zn, the boiling point of Mg is considerably low, therefore one might think of using Mg-MOFs as templates for the preparation of hierarchical carbons by direct pyrolysis at around 1000 °C. However, looking at the Ellingham diagram, MgO with a boiling point of 3600 °C is only reduced by carbon at around 1850 °C thus making this preparation route of carbons far from energy efficient and thus non-cost effective.
(g) Manganese (Mn)-MOFMS
The chemistry of manganese is quite complex. Compounds containing manganese with formal oxidation states from −3 to +7 are known, however, under normal conditions, the +2 state is the most stable.136 The melting point of Mn is 1246 °C, its boiling point is 2061 °C and the reduction potential of Mn2+ is −1.18 V. Therefore, in this case as well, pyrolysis in an inert atmosphere of the pristine Mn-MOF will lead to a mixture of Mn oxides in carbon whereas the calcination in air will generate the corresponding carbon-free metal oxide.Due to their unique redox properties, low cost, and environment compatibility, manganese oxides are well known candidates for numerous catalytic applications137 including decomposition of hydrogen peroxide, partial oxidation with hydrogen peroxide, oxidation of CO, methane, aromatic compounds, aqueous phenol in aqueous solution, NO–CO reaction, and chemical-looping combustion processes.138
The only MOF derived Mn based catalyst reported so far in the literature appears in the work carried out by Peng et al.139 These authors developed a novel methodology for the formation of large-pore mesoporous mixed valence Mn3O4via the conversion of a Mn-MOF fabricated in ionic liquid (IL)–water mixtures for the degradation of MB. After IL and water removal, the Mn–BDC underwent thermolysis under N2 with a heating rate of 5 °C min−1 from room temperature to 400 °C. After reaching the target temperature, the material was calcined for 2 h and then naturally allowed to cool down to room temperature. In the presence of Mn3O4, more than 99.7% MB degraded after only 1.5 h at 75 °C, compared to the reported behaviour for Mn3O4 crystals,140 which needed 3 h to achieve a MB degradation >99.7% at 80 °C under similar concentration conditions.
The catalytic properties of manganese oxides depend on their structures, oxidation states and surface areas. To improve the catalytic properties of manganese oxides, therefore, these properties should be controlled. Manganese oxide structures are generally controlled through the synthesis conditions and they are usually supported on materials with high surface areas.138 MOF mediated synthesis of Mn-MOFs to obtain mesoporous Mn oxides looks the most encouraging and straightforward approach for its application in catalysis, mainly in oxidation reactions and photocatalytic degradation of dyes. Here, the oxidation state of Mn in the pristine MOF, the atmosphere employed during the decomposition of the MOF together with the temperature of the heat treatment will play an essential role for controlling the size and the oxidation species present in the final catalyst due to the broad spectrum of Mn oxides available.
(h) Nickel (Ni)-MOFMS
Nickel is cheap, sufficiently active for certain reactions, and allows suitable catalysts to be economically produced.141 The melting point of Ni is 1455 °C, its boiling point is 2913 °C and the reduction potential of Ni2+ is −0.25 V. Nickel's position is just above palladium in the periodic table, and as a group 10 metal, it can readily catalyse many elementary reactions for which palladium or platinum are active as well. Because of these commonalities, nickel is often viewed solely as a low-cost replacement catalyst for cross-coupling reactions. However, this common misconception is clearly refuted by the numerous and diverse nickel catalysed reactions reported in the literature,142 such as, oxidative dehydrogenation of ethane,143 partial oxidation144 and both dry145 and steam reforming of methane,146 methanation of CO147 and hydrotreating processes148 among others. Besides RANEY® nickel is used in a large number of industrial processes mainly focused on hydrogenation.149,150 Furthermore, particularly remarkable is the recent development of nickel based homogeneous or organo-nickel chemistry.151p-Aminophenol is an important intermediate in the preparation of several analgesic and antipyretic drugs. It is also used as a corrosion inhibitor in paints and as an anticorrosion-lubricating agent in fuels for two-stroke engines and in the dye industry it is used as a wood stain and as a dyeing agent for fur and feathers. A major process for the preparation of p-aminophenol is via the hydrogenation of nitrobenzene in the presence of strong acids.152 Mesoporous carbon nitrides (m-CN) have pendant amino groups153 intrinsically present on their mesoporous walls, which can serve as nucleation centers for the grafting of metal NPs, thereby leading to m-CN-material-based catalysts containing well-dispersed catalytically active sites.154 Very recently, Zuo et al. reported the preparation of a N-doped mesoporous carbon supported Ni nanoparticle catalyst (Ni/m-CN) via carbonization of Ni-MOF [Ni(Hbtc)(4,4′-bipyridine)] at 700 °C.155 When the resulting Ni/m-CN catalyst was tested in the catalytic reduction of 4-nitrophenol using NaBH4 as a stoichiometric reductant, the reaction proceeded very rapidly. The catalytic performance was preserved without any loss of efficiency after six cycles, highlighting its excellent activity and stability upon reuse besides being easily recovered due to the superparamagnetism of the catalyst. Compared to other catalysts in the literature, the activity of the Ni/m-CN catalyst was approximately the same as that of Ag-doped carbon spheres and Fe3O4@SiO2–Ag catalysts156,157 and considerably higher than other Ni and Co based catalysts.158,159
Hydrodesulfurization (HDS) is the oil refinery process for removal of sulfur from sulfur-containing organics present in crude oil fractions. The process typically relies on alumina supported heterogeneous catalysts with molybdenum–cobalt or molybdenum–nickel sulfides as active components.160 Not long ago, in a more sophisticated version of MOFMS, Larabi et al. reported the application of the well-defined MOF [Ni2(dhtp)] (H4dhtp = 2,5-dihydroxyterephthalic acid),161 as a precursor to bimetallic nickel containing molybdenum sulfide nanoparticles, for HDS reactions.162 The active material was prepared by subliming Mo(CO)6 on activated [Ni2(dhtp)], which has the capacity of grafting metal carbonyls on the organic linker. The porous, highly dispersed, bimetallic Ni–Mo material was sulfided for 4 h at 350 °C by reaction with H2S in the presence of 42 atm H2. The HDS activity of the activated catalyst was measured for the conversion of dibenzothiophene to biphenyl or cyclohexylbenzene at 350 °C in the presence of 38 atm H2. The MOF-derived sulfided material resulted in an active bulk HDS catalyst, stable over several cycles and yielded almost an order of magnitude improvement in the catalytic activity per molybdenum center when compared to the reference unsupported bulk NiMoO4 material. Furthermore, hydrodenitrogenation (HDN) and hydrogenation activities were also relatively high for this material.
As in the case of Co and Cu, the reduction potential of Ni2+ suggests that pyrolysis of Ni(II)-MOFs tends to form metallic nickel embedded in carbon at a relatively low temperature (250 °C) according to the Ellingham diagram. Metallic Ni is also a well-known catalyst for graphitization of carbon.163 Therefore pyrolysis parameters such as temperature during Ni-MOFMS require special attention in order to control graphite layer formation around Ni nanoparticles, which might lead to internal diffusion limitations or even to total isolation of the active phase. It should not escape our attention that preparation of NiO catalysts from Ni-MOF leads to highly porous structured oxides, comprising another appealing, unexplored and simple application of Ni-MOFMS. On the other hand, grafting on Ni-MOFs has already given way to superior catalytic materials for hydrotreating processes, thus paving the way for the application of these or similar materials in other catalytic processes. In any case, there is still a long way to go regarding MOFMS for this metal.
(i) Ruthenium (Ru)-MOFMS
Ruthenium is one of the rarest metals on Earth. Although it is found uncombined in nature, it is more commonly found associated with other metals like palladium in minerals like pentlandite and pyroxinite and commercially it is obtained from the wastes of nickel refining. The melting point of Ru is 2334 °C, its boiling point is 4150 °C and the reduction potential of Ru2+ is −0.45 V.Ruthenium is used in catalysts for ammonia and acetic acid production. It is also an excellent catalyst for converting deNOx to N2, but the oxide is relatively highly volatile, so unsuited for use in three way catalytic converters (TWCs). Ruthenium catalysts are widely known for dissociating the CO bond easily and for being effective for methanation or Fischer–Tropsch synthesis.164 By and large, molecular ruthenium catalysts are used to perform selective carbon–carbon bond formation by combination of simple substrates and their tolerance toward functional groups has allowed access to high value, multifunctional molecules. Ru catalysts allow the coupling of functional alkenes or alkynes with a variety of unsaturated molecules such as alkenes, dienes, alkynes and diynes. A large range of electron-rich ruthenium or hydridoruthenium complexes are currently used for the formation of cyclic and polycyclic compounds on reaction with substrates containing several pathways, such as C–H bond activation, the distribution of carbine from diazoalkanes and especially their versatility in making a large variety of ruthenacycle intermediates.165 New ruthenium catalysts enable carbon–carbon, carbon–hydrogen, and carbon–heteroatom bond formation and cleavage, and are able to provide non-classical activation modes.
Very recently, Chen et al. provided a novel concept for MOF mediated synthesis by hybridization of clay minerals with metal–organic frameworks as efficient catalysts for benzene hydrogenation.23 To this end, they grew a ruthenium MOF [RuII,III2(btc)2XxY1.5−x] (X, Y = Cl− or OH−) on montmorillonite (Mt) via a solvothermal reaction, fabricating a composite material (Ru-MOF_Mt). In order to obtain stable Ru NCs, the prepared hybrids were annealed in H2 at 300 °C, thus partially decomposing the frameworks and producing Ru particles that act as active centers for hydrogenation. In the catalytic tests, benzene was proven to be completely converted into cyclohexane within 2.5 h at 160 °C and a H2 pressure of 6.0 MPa. The turnover frequency (TOF) of benzene reached 3478 h−1, which is higher than that reported for Ru/C or other heterogeneous catalysts.166,167 Moreover, the Ru-MOF_Mt catalyst was reused 5 times and no obvious degradation was detected, which demonstrated the good stability of the Ru particles protected by both the partly decomposed btc oligomers and the clay layers.
To sum up, the low availability of Ru makes this metal far expensive. Since in principle MOFMS targets high loadings of highly dispersed metals in supported catalysts, applying this technique in this case may seem quite pointless. Alternatively, diluting Ru-MOFs with other solids comprises a more suitable option for this metal. Both organic and inorganic materials might be used for this purpose but always keeping in mind this will directly affect both the porosity of the carbon matrix and the final state of the active metal phase. In any case, the reduction potential of Ru2+ predicts a strong tendency during the MOFMS in inert atmosphere to form metallic Ru.
(j) Titanium (Ti)-MOFMS
Titanium is the second most abundant transition metal after iron, its melting and boiling points being 1668 °C and 3287 °C, respectively. For TiO2, the melting point ranges from 1830 to 1855 °C and its boiling point from 2500 to 3000 °C. The reduction potentials of Ti3+ and Ti2+ to metallic Ti are −1.21 V and −1.63 V, respectively, and in the case of Ti3+ to Ti2+ is −0.37 V. Probably the best-known applications of Ti in catalysis are as TiCl3 and TiCl4 Ziegler–Natta catalysts for the production of polypropylene and as TiO2 catalytic support for different processes and as a photocatalyst. Indeed, during the most recent decades TiO2 based photocatalysts have attracted great interest due to their excellent chemical stability, nontoxicity, and low price.168 Its low cost and simple nature of processing make the sol–gel route the common technique for the synthesis of TiO2. Nevertheless, the control of TiO2 in terms of phase, shape and morphology, and especially the nanoscale synthesis of TiO2 particles through a simple and controllable method remains a challenge.169Among Ti based MOFs, MIL-125 is the first example of a highly porous and crystalline titanium(IV) dicarboxylate with high thermal stability and photochemical properties.170 Its structure is built up from a pseudo cubic arrangement of octameric wheels, built up from edge- or corner-sharing TiO5(OH) octahedra, and terephthalate dianions leading to a three-dimensional periodic array of two types of hybrid cages with accessible pore diameters of 6.13 and 12.55 Å.
TiO2 is widely used as a photocatalyst for CO2 reduction;171 however, this material often exhibits low efficiency.172 Noble metal co-catalyst species favour electron–hole separation and induce interfacial electron transfer or allow visible light absorption. In this regard, the potential of gold nanoparticles (GNPs) has received recently considerable attention.173 For instance, Khaletskaya et al. targeted gold/titania nanocomposites, GNP/TiO2 through the pyrolysis of GNP/NH2-MIL-125 nanocrystals and tested them for the UV light promoted reduction of CO2 to methane.169 Preformed GNPs, aminoterephthalic acid and titanium(IV) isopropoxide were dissolved in a mixture of N,N-dimethylformamide (DMF) and methanol (MeOH) (DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH = 1:1 (v/v)). The mixture was heated to 423 K, and kept at this temperature for 15 h which yielded GNP/NH2-MIL-125. GNP/NH2-MIL-125 was calcined in O2 at 450 °C for 2 h resulting in the selective rutile-phase formation of GNP/TiO2 with the retained size and morphology of the original MOF crystals which enabled the controlled nanoscale synthesis of TiO2 in terms of phase, shape, and morphology. In catalytic terms, the aforementioned properties translated into high activity of the GNP/TiO2 photocatalyst for CO2 reduction compared to P-25 and AUROlite (commercial Au/TiO2) besides not showing any signs of deactivation in the second reaction run. The authors do however propose further improvement of GNP/TiO2 materials by (i) directing toward a more uniform distribution of the GNPs, (ii) further downscaling and tailoring of the primary particle size of TiO2, (iii) elucidating the effects of light heteroelement doping (C, N) together with GNP loading and (iv) addressing CO2 reduction under visible light conditions.
:MeOH = 1:1 (v/v)). The mixture was heated to 423 K, and kept at this temperature for 15 h which yielded GNP/NH2-MIL-125. GNP/NH2-MIL-125 was calcined in O2 at 450 °C for 2 h resulting in the selective rutile-phase formation of GNP/TiO2 with the retained size and morphology of the original MOF crystals which enabled the controlled nanoscale synthesis of TiO2 in terms of phase, shape, and morphology. In catalytic terms, the aforementioned properties translated into high activity of the GNP/TiO2 photocatalyst for CO2 reduction compared to P-25 and AUROlite (commercial Au/TiO2) besides not showing any signs of deactivation in the second reaction run. The authors do however propose further improvement of GNP/TiO2 materials by (i) directing toward a more uniform distribution of the GNPs, (ii) further downscaling and tailoring of the primary particle size of TiO2, (iii) elucidating the effects of light heteroelement doping (C, N) together with GNP loading and (iv) addressing CO2 reduction under visible light conditions.
Oxidative desulfurization (ODS) has been extensively explored in the recent literature as a means to attain deep desulfurization of petroleum streams.174 In ODS, sulfur compounds are oxidized to their corresponding sulfoxides and sulfones which can be more easily extracted due to increased polarity. On this subject, McNamara et al. not only addressed the performance of Ti-MOF derived catalysts for the oxidation of dibenzothiophene but also focused on the effect of the porosity of the MOF precursor on the final morphology of the resulting MOF-templated material.175 As well as the microporous MIL-125(Ti), they prepared a hierarchically microporous/mesoporous analogue of MIL-125(Ti) made up of agglomerations of microporous MOF nanoparticles exhibiting interparticle mesoporosity. The main differences in the synthesis of this mesoporous MOF compared to the microporous analogue are the preparation of a dry precursor powder for use in a vapor-assisted crystallization setup and the addition of cetyltrimethylammonium bromide (CTAB) as a templating agent. Both microporous and microporous/mesoporous MOF materials were carbonized in Ar at different temperatures ranging from 600 to 1100 °C at a ramp rate of 10 °C min−1 for 6 h and were passivated under 1% O2 in He for 1 h afterwards. The results showed that altering the MIL-125 morphology to obtain interparticle mesoporosity yielded materials with significantly enhanced mesoporosity when pyrolyzed compared to pyrolysis of the strictly microporous MIL-125 material. Furthermore, pyrolysis of the hierarchically micro-porous/mesoporous MIL-125 yielded a carbonaceous material with smaller Ti nanoparticles compared to pyrolysis of the microporous MIL-125, which was attributed to the confined environment of the Ti particles embedded in smaller carbon particles. In addition, increasing the pyrolysis temperature led to higher Ti content (>40 wt%) in the final materials and reduced Ti oxide phases including a Ti oxycarbide phase. Oxidation of sulfur-containing dibenzothiophene using tert-butyl hydroperoxide revealed that the meso-materials exhibited enhanced activities over the micro-materials pyrolized at the same temperature due to their increased mesoporosity and smaller Ti nanoparticle sizes. The above results demonstrated that, by effectively tuning the morphology of the precursor MOFs, the properties of the pyrolyzed materials could be tailored to suit specific applications.
Presumably, the most outstanding feature of Ti-MOFMS is the strong temperature dependence on the carbothermal reduction of Ti species under pyrolysis conditions which prompts the generation of oxides, carbides, and oxycarbides supported on nanoporous carbon.176 Thus, as well as the metal loading and the size of the nanoparticles, the carbonization temperature used during the pyrolysis allows the final phase of Ti to be tuned, from Ti4+ species to more reduced oxidation states of Ti (Ti2+ and Ti3+) via carbonization and carbothermal reduction. Otherwise, the synthesis of pure TiO2 needs to be conducted though calcination in air above 300 °C in the case of NH2-MIL-125 and 400 °C for MIL-125.177
(k) Vanadium (V)-MOFMS
Vanadium is the 21st most abundant element in the Earth's crust and the 2nd most abundant transition metal in sea water.178 The melting point of V is 1910 °C, its boiling point being 3407 °C. Vanadium oxide standards include V2O5, VO2, V2O3, and VO, where vanadium is present in oxidation states V5+, V4+, V3+, and V2+, respectively,179 the prevailing valence states of V in nature being V(III), V(IV), and V(V). Furthermore, the V–C system is a typical binary system which has many different stoichiometries. V2C, V4C3, V6C5, V8C7, and VC have been synthesized and investigated for many years.180 Boiling points of V2O5 and VC are 2030 and 3900 °C, respectively. The reduction potentials of V3+ to V2+ and V2+ to V are −0.26 and −1.18 V, respectively.Vanadium oxide or supported vanadium oxide catalysts which contain a vanadium oxide phase deposited on a high surface area oxide support (e.g., Al2O3, SiO2, TiO2) have found extensive applications as oxidation catalysts in the chemical, petroleum and environmental industries. These reactions include the oxidation of sulfur dioxide to sulfur trioxide in the production of sulfuric acid, the oxidative dehydrogenation of propane to propene, the selective oxidation of methane to formaldehyde, the oxidative dehydrogenation of alkanes and the selective photo-oxidation of alcohols.181,182 Moreover, transition metal carbides, pure or modified by oxygen, have been extensively studied as catalysts, due to their attractive advantages in terms of activity, selectivity, stability, and resistance to poisoning in hydrocarbon reactions.183 Among these, vanadium carbide is of importance for industrial applications due to its excellent high temperature strength, and high chemical and thermal stability even at high temperatures, that make it more resistant to sintering and attrition than other solids.184
MIL-47 is a vanadium benzenedicarboxylate MOF with chemical formula VIVO(O2C-C6H4-CO2), composed of VIVO6 octahedra linked by a 1,4-benzenedicarboxylate group forming a three-dimensional, orthorhombic, porous solid and it degrades below 400 °C under both nitrogen and air flow.185
Substantial leaching of active V species often limits the reusability of V-based solid catalysts in liquid-phase oxidation reactions thus calling for the development of more stable, novel materials. Kim et al. worked on the synthesis of active vanadium oxide and carbide species dispersed on a carbon support from a V-based MOF template and evaluated its catalytic performance in the liquid-phase oxidation of dibenzothiophene (DBT) with tert-butyl hydroperoxide (TBHP).186 The activated MIL-47(V) was carbonized under Ar with a ramp rate of 10 °C min−1 at temperatures ranging from 600 °C to 1100 °C for 6 h. After cooling to 25 °C under Ar, the resulting catalyst was passivated under 1% of O2 in He for 1.5 h prior to exposure to ambient atmosphere. Notably, the pyrolysis of MIL-47(V) provided carbon supports with high surface areas (∼300–400 m2 g−1), high mesoporosity (Vmeso/Vpore ∼ 0.9), high V loadings (35–70 wt%), and small (∼18 nm) V crystallites dispersed on the surface. The effect of the pyrolysis temperature was clear-cut on different surfaces and bulk phases of V achieved, higher temperatures leading to increased amounts of V carbide at the surface, even reaching 100% V carbide composition at the surface of the sample carbonized at 1100 °C. The MOF derived V carbide catalysts carbonized at high temperature (1000 and 1100 °C) exhibited good activities and enhanced stabilities compared to the conventional V on activated carbon synthesized via a conventional impregnation and to MOF derived V catalysts carbonized at lower temperatures (600 °C). The authors claimed this may likely be attributed to the advantages of the V carbide over the oxide counterparts, which can provide enhanced chemical stability and enhanced anchoring on the carbon support during this reaction and which could not be obtained for the conventional V supported by impregnation on activated carbon even when carbonized at 1100 °C.
Traditionally, vanadium carbide has been prepared by various high temperature reactions. Recently, other synthesis methods have been developed including mechanical alloying, temperature programmed reaction and gas reduction–carburization, aluminothermic reduction of vanadium oxide and carburization of vanadium oxide with an organic reagent such as cyanamide.184 However, industrial applications of the methods are still limited due to agglomeration problems, wide size distributions, low yields, complex monitoring and high costs.187 Based on these terms, MOFMS comprises a novel route to obtain transition metal carbides by one step pyrolysis above 400 °C when MIL-47 is being used as the parent MOF.
(l) Zinc (Zn)-MOFMS
Its high availability, low toxicity and environmental benignity188 make Zn a highly attractive metal for application in catalysis. Zn has a melting point of 419.5 °C and a boiling point of 907 °C. The reduction potential of the zinc ion is −0.76 V and the melting point of its oxide, ZnO, is 2100 °C, its Tamman temperature being 900 °C.189 Due to its electronic properties, ZnO is mainly used as a photocatalyst and also as a promoter/support in heterogeneous catalysis.Among Zn-MOFs, MOF-5 and ZIF-8 are the most obvious candidates to be used as templates in MOFMS. Consisting of Zn4O building blocks linked together by terephthalate bridges to form a zeolite-like, cubic framework, the group of O. M. Yaghi published the structure of MOF-5 in late 1999.190 Since then, MOF-5 has been subjected to numerous studies, especially in gas storage191–194 and separation195 as well as in catalysis.196–198 ZIF-8 (Zn(mIM)2, mIM = 2-methylimidazolate) exhibits a SOD topology comprised of cages connected through six-membered windows, and is currently the most widely investigated zeolitic imidazolate framework (ZIF) material for a wide range of applications.199 The unique properties of ZIF-8 such as pressure-dependent pore size, reversible guest trapping and amorphization200 have boosted its use for gas separation,201 sensing202 and catalysis.203
Depending upon the heat treatment Zn-MOFs give way to two different MOF derived catalytic materials: (i) nanostructured metal oxides and (ii) hierarchical nanoporous carbons.
In order to prepare nanostructured ZnO from Zn-MOFs, the typical approach consists of calcining the MOF under air at a temperature above the decomposition temperature of the framework, around 300 °C for both MOF-5204 and ZIF-8.205 However, as the reduction potential of Zn2+ is lower than −0.27 V and thus below 900 °C zinc ions are unable to reduce to a zero oxidation state, the calcination under inert atmosphere of Zn-MOFs also gives the corresponding metal oxide. The main advantage of MOFMS compared to other preparation methods lies in the high porosity and structure/morphology control of the resulting MOF derived ZnO nanoparticles206 which results in improved catalytic performance.
The major limitation for achieving high photocatalytic efficiency in the ZnO nanostructure systems is the fast recombination of charge carriers.207 Therefore, the design and modification of ZnO photocatalysts with high sensitivity and reactivity has attracted much attention.208 In this field, Yang et al. demonstrated that ZnO nanoparticles prepared by calcination of MOF-5 in air at 600 °C, formed hierarchical aggregates with a three-dimensional cubic morphology. These aggregates exhibited high photocatalytic degradation of Rhodamine-B (RhB) under UV irradiation, comparable to degradation by the commercial P-25 TiO2 catalyst.209 The authors investigated the outstanding photocatalytic activity by measuring the room temperature photoluminescence (PL) of the sample. The PL spectrum indicated that the rate of recombination between the photogenerated holes and electrons was lowered on the surface of the prepared ZnO crystals thus resulting in a superior photocatalytic activity. In other work, Cao et al. developed a simple method to prepare metal oxide/3D graphene network (3DGN) composites using ZIF-8 as the precursor of the metal oxide and 3DGN as the backbone. After the preparation of the ZIF-8/3DGN composite, the synthesis consisted of a facile two-step annealing process under Ar at 450 °C for 1 h, which conferred the ZIF-8/3DGN material with a more textured and rougher surface, followed by calcination in air at 380 °C for 1 h to give the final ZnO/3DGN material.24 In their case, the improved performance of the ZnO/3DGN composite in the photodegradation of the dye methylene blue (MB) under irradiation with UV light was ascribed to the high specific surface area of the composite whereas the prevention of the recombination of photoinduced holes and electrons was attributed to good contact and effective interaction between 3DGN and ZnO. Similarly to Cao et al., Pan et al. also selected nonmetal doping in order to improve the low efficiency of charge separation in ZnO nanoparticles, using in this case the organic linker of the pristine MOF as the carbon source.210 The authors prepared C-doped ZnO via two-step pyrolysis under N2 of ZIF-8 and applied it in the photodegradation of RhB and phenol and photo electrochemical (PEC) water splitting. To this end, ZIF-8 was first calcined in air at 350 °C for 2 h (at a heating rate of 5 °C min−1) to partially remove the organic groups resulting in a porous structure, then the temperature was increased to 400 °C for 1 h (at a heating rate of 2 °C min−1) to remove the organic residues and increase the crystallization of ZnO. Compared to a commercial C-doped ZnO catalyst, the results showed that the ZIF-8 derived catalyst exhibited around 2-fold higher photoactivity in both the photodegradation of RhB and phenol and 4-fold higher in PEC water splitting. The authors claimed that the inter-connected porous structure of ZnO nanoparticles played an important role in mass transfer, charge transfer and light absorption during the photoreactions.211
Besides its use in photocatalysis, MOF derived ZnO is also advantageous in some other catalytic applications. During the catalytic oxidation of CO, creating a high density of noble metal nanoparticles and a high support interface is the key to maximize catalytic performance.212–214 For this purpose, Liu et al. employed MOF-5 as a precursor to prepare ZnO-supported Pt nanoparticles which exhibited excellent catalytic activity for CO oxidation.215 Nanocrystalline ZnO-supported platinum nanoparticles were synthesized by introducing inorganic platinum salt into the pores of MOF-5 followed by heating in air at 600 °C for 1 h, which allowed the platinum nanoparticle size to be tuned by changing the concentration of the platinum precursor. The Pt/ZnO obtained from the MOF resulted in a catalytic activity twice as high as that of Pt/ZnO obtained by conventional methods. The authors suggested that the good catalytic activity of the Pt/ZnO samples may result from metal (Pt)–support (ZnO) interaction and depended on the Pt particle size.
Moving forward in this topic, the removal of Zn from the framework during MOFMS results in highly porous hierarchical carbons. As shown in the Ellingham diagram, above 950 °C the Gibbs free energy of formation for C is lower than that for ZnO meaning that carbon is able to reduce zinc oxide, the carbon itself being oxidized to CO, and to CO2 above 1050 °C. Accordingly, in the vast majority of carbons from Zn-MOFs the pyrolysis of the pristine MOF takes place around 900–950 °C for several hours in order to completely evaporate the metal. An alternative and far less employed approach consists of pyrolysis at lower temperature decomposing the framework to form the carbon, and subsequently removing the ZnO by acid leaching.
These MOF mediated porous carbons find their main application in electrocatalysis. Two main strategies exist in order to optimize the design of ORR catalysts thus boosting their performance, which consist of (i) enhancing Pt dispersion in the catalyst and/or (ii) replacing the expensive Pt with non-precious metals. In order to maintain good Pt nanoparticle dispersion, recent efforts have been focused on the development of new carbon support nanostructures in an attempt to further improve the activity and stability of PEMFCs.216 In their work, Khan et al. developed highly efficient ORR electrocatalysts based on Pt/Ni bimetallic nanoparticles dispersed on highly porous carbon obtained via pyrolysis of MOF-5.217 Firstly, MOF-5 was heated in an argon atmosphere at 950 °C for 9 h for improved graphitization and removal of all impurities including Zn/ZnO. Subsequently, the obtained carbon material (PC 950) was sonicated in ethylene glycol, heated up to 100 °C and H2PtCl6·6H2O and Ni(NO3)2·6H2O in ethylene glycol were separately added dropwise. Finally, the resultant mixture was heated up to 180 °C for 4 h to complete the polyol reduction synthesis method.218 As a result, Pt–Ni/PC 950 (15:15%) demonstrated superior ORR activity over a Pt/C (20%) commercial catalyst in terms of both Eonset and E1/2, attributed to the synergistic effect between Pt–Ni nanoparticles and the carbon encapsulating effect of the MOF derived material. Previously these authors applied MOF-5 as a template for the preparation of porous carbon as an electrode material in ethanol fuel cells (DEFCs).219 In this case, MOF-5 was carbonized at 900 °C in Ar for 6 h and the carbon material (PC-900) was deposited with PtFe (20%) nanoparticles using the same polyol reduction method. The electrocatalytic capability of the catalyst for ethanol electro-oxidation was investigated and compared with those of PtFe and Pt supported on Vulcan XC72 carbon catalysts (PFe/CX-72 and Pt/XC-72) prepared via the same method. They observed that the catalyst PtFe/PC-900 showed an outstanding normalized activity per gram of Pt and superior power density compared to the commercially available carbon-supported catalysts.
The second course of action involves reducing the cost and promoting a wider use of PEMFCs replacing the expensive Pt based electrocatalysts with a lower-cost alternative.220,221 Fe-Based cathode catalysts are promising contenders, but their power density is low compared with Pt-based cathodes, largely due to poor mass-transport properties. In view of this, Proietti et al. used ZIF-8 as a host for Fe and N precursors by means of mixing iron acetate and phenantroline together with ZIF-8 using low-energy ballmilling.123 The mixture was pyrolyzed in Ar at increasing temperatures and optionally in NH3 at 950 °C, creating highly porous Fe/N/C composites. With an increase in the Ar pyrolysis temperature to 1050 °C, Zn could be removed completely to get purely microporous carbons. NH3 pyrolysis resulted in an additional 17 wt% loss by hydrogasification, doubling the BET area and introducing substantial mesoporosity besides increasing the nitrogen content in the sample. The introduction of mesopores together with the general increase in the microporous surface area and the nitrogen content resulted in an over four times increased ORR activity compared to the microporous carbon.
In 2009, a new class of catalysts based on earth-abundant carbon materials was discovered as an efficient, low-cost, metal-free alternative to platinum for the ORR in fuel cells. Since then, tremendous progress has been made, and carbon-based metal-free catalysts have been demonstrated to be effective for an increasing number of catalytic processes.222 A good example of this is work done by Aijaz et al. who designed high-surface-area N-decorated nanoporous carbons using ZIF-8 along with furfuryl alcohol (FA) and NH4OH as the secondary carbon and nitrogen sources, respectively.223 FA and NH4OH were first introduced into the cavities of activated ZIF-8 at room temperature. After filtration and washing with ethanol, the FA–NH4OH/ZIF-8 composite was heated in Ar at 80 °C for 24 h, then at 150 °C for 7 h for FA polymerization, and finally calcined at 600, 700, 800, 900, or 1000 °C for 8 h. Among the MOF derived electrocatalysts, the one calcined at 900 °C showed the highest onset potential of 0.83 V, compared with 0.95 V for the commercial Pt/C catalyst according to cyclic voltammetry measurements. The high ORR catalytic activity was associated with the large number of active basic N sites and the high surface area, which provided proper channels with easy mass diffusion.
Along a similar line, research by Zhang et al. showed that the introduction of ordered mesopores by using Te nanowires (Te-NWs) as a templating agent dramatically increased the performance of cathode ORR in alkaline media (Fig. 6).224 Te-NWs were added to the ZIF-8 synthesis mixture, and after crystal growth on the NW surface, the composite was pyrolyzed at 1000 °C to vaporize both the Te and the Zn phase. Comparison of the microporous ZIF-8 derived carbon with the Te-NW MOF-templated mesoporous carbon showed large differences in performance. The ORR activity of the templated carbon was much higher and close to that of commercial Pt-catalysts. The hollow carbon nanofibers were subsequently P-doped to create more ORR active sites, and the resulting catalyst showed the best catalytic activity, outperforming the commercial 20 wt% Pt/C.
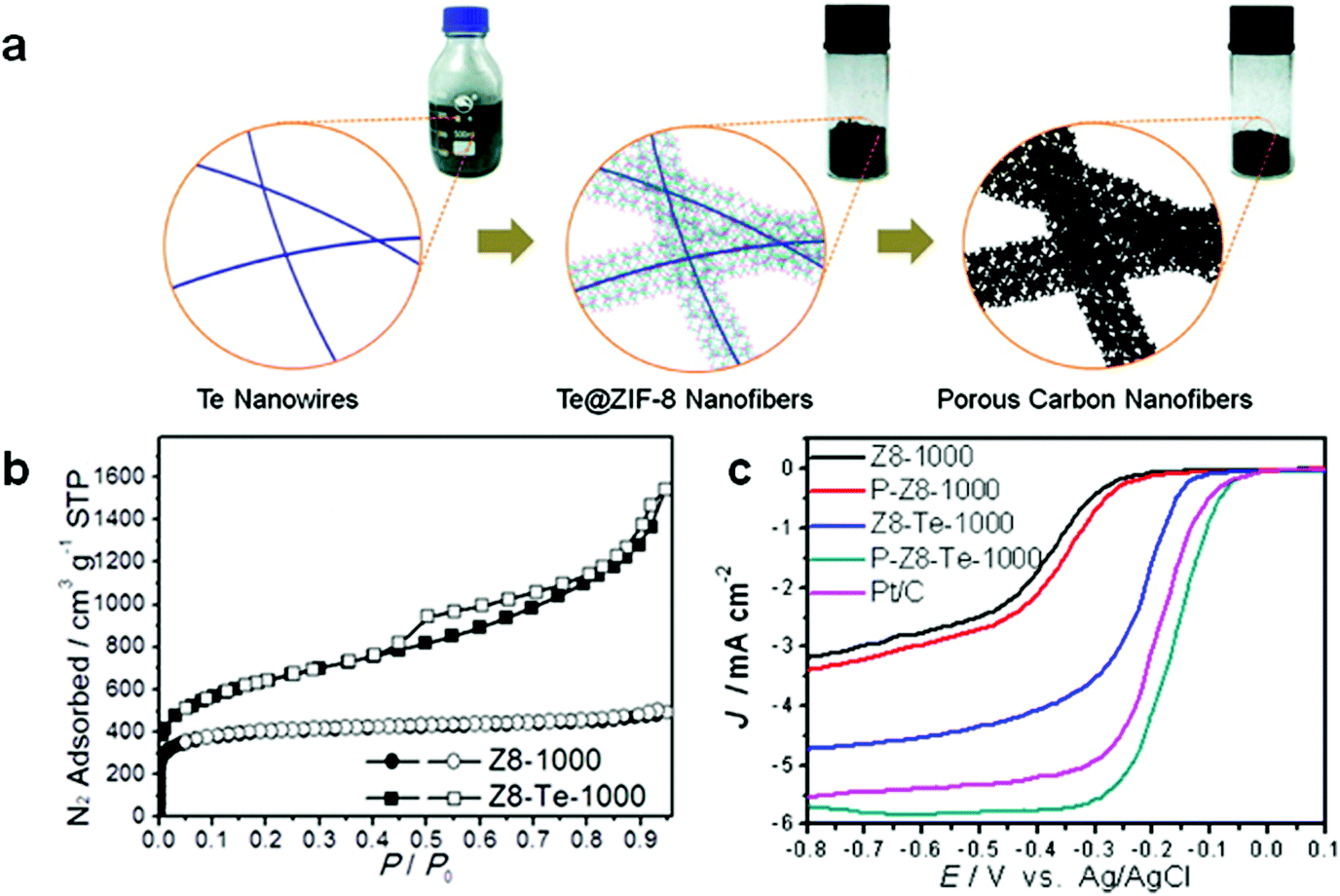 | ||
| Fig. 6 (a) Illustration of the nanowire-directed templated synthesis of ZIF-8 nanofibers and derived porous doped carbon nanofibers; (b) N2 sorption isotherms (@77 K) of Z8-Te-1000 and Z8-1000; (c) linear sweep voltammetry (LSV) curves of all catalysts in O2-saturated 0.1 M KOH with a sweep rate of 10 mV s−1 and electrode rotation speed of 1600 rpm. Reprinted with permission from Zhang et al., 2014.224 Copyright 2016, American Chemical Society. | ||
Apart from their application in electrocatalysis, nanostructured carbons from Zn-MOFs find their application in other fields of catalysis.21,225 For instance, Feng et al. prepared nanoporous carbons by carbonization of MOF-5 in a N2 atmosphere at different temperatures, followed by the introduction of silver and palladium precursors using incipient wetness impregnation.225 Ag3Pd12/NPCs obtained through pyrolysis at 900 °C exhibited high activity and selectivity in dehydrogenation of formic acid in sodium formate under ambient conditions in comparison with reported heterogeneous catalysts for this reaction. The authors ascribed the enhanced catalytic performance of Ag3Pd12/NPCs to its Pd0, Ag0 and AgPd alloy composition as well as to the small particle size and good dispersion of AgPd nanoparticles on the carbon support.
The analysis here is clear. Both the intriguing properties of ZnO for photocatalysis together with the low boiling point of Zn have certainly boosted research on the MOFMS of Zn-MOFs in order to afford nanostructured ZnO and hierarchical carbons for their catalytic application. The simplicity of the technique for the preparation of both catalytic materials makes Zn-MOFMS up-and-coming for its future industrial implementation. In the case of ZnO, the technique implies pyrolysis in an inert atmosphere, calcination in air or a combination of both at temperatures that range from 400–600 °C. Although the technique has been largely applied in the field of photocatalysis, shifting its application to other catalytic processes will require greater effort with regard to both preparation of the materials and catalytic testing. Regarding the preparation of carbon supports from Zn-MOFs by pyrolysis at 900–1000 °C, the MOFMS technique has been mainly applied in electrocatalysis for supporting precious and non-precious metal nanoparticles and also for electrocatalytically active metal-free carbons. However, the number of possibilities seem to be huge in this field which allows metal-free carbons with improved textural properties to be obtained by simple pyrolysis of the parental Zn-MOF.
(m) Zirconium (Zr)-MOFMS
Zirconium occurs in about 30 mineral species, the major ones being zircon and baddeleyite. Zirconium metal is produced commercially by first converting zircon to zirconium chloride, and then reducing the chloride with magnesium. The melting point of Zr is 1855 °C and its boiling point is 4409 °C. The most common oxidation state of zirconium is IV and the reduction potential of Zr4+ is −1.54 V.Zirconia is a very important ceramic material with growing applications in heterogeneous catalysis both as an active material and a support. As a support, ZrO2 has attracted considerable interest in a variety of catalytic systems since it is characterized to be the only one metal oxide that can possess four different chemical properties on the surface: acidic, basic, oxidizing and reducing properties.226 Accordingly, it has been extensively used in catalysis, for example supporting Au, Ag, and Cu catalysts in the water–gas shift reaction, methanol synthesis from CO2 and H2, CO oxidation and in the form of nanostructures, in photocatalytic reactions like CO2 reduction.227 Besides, decomposition of NO has been suggested to occur in Lewis acid sites of zirconia nanoparticles,228 synthesis of aromatic aldehydes by the hydrogenation of carboxylic acids with a ZrO2 catalyst has been already established229 and various authors have reported on the catalytic activity of zirconia for the selective dehydration of secondary alcohols to terminal alkenes as well as for the hydrogenation of CO to isobutene.230–232
The strength of ZrIV–O bonds has been exploited to develop highly stable metal–organic frameworks (MOFs) that are tolerant to extreme conditions, including chemical functionalization and catalytic reactions and they possess high mechanical strength and high thermal and chemical stability (up to ∼500 °C, pH 1–10).233 UiO-66 is composed of [Zr6(μ3-O)4(μ3-OH)4]12+ nodes, each linked to 12 carboxylates of terephthalate ligands to form tetrahedral and octahedral cages; however, there are many defects from missing linkers in this MOF, with the resulting otherwise open metal sites occupied by hydroxo ligands. The node topology of the more highly porous233 NU-1000 has been determined to be [Zr6(μ3-O)4(μ3-OH)4(OH)4(OH2)4]8+ and it is coordinated to eight tetratopic linkers of 1,3,6,8-tetrakis(p-benzoic-acid)pyrene (H4TBAPy) to form triangular and hexagonal pores.234
Very recently Malonzo et al. demonstrated the effectiveness of nanocasting of NU-1000 with silica for the preparation of highly thermally stable MOF derived single site catalytic clusters for high temperature reactions (Fig. 7).25 NU-1000 is an excellent Lewis acid catalyst owing to the high concentration of the oxozirconium clusters in its structure, which are easily accessible through its mesoporous channels. By condensing tetramethylorthosilicate within the NU-1000 pores via vapour phase HCl treatment, they created a silica layer on the inner walls of the MOF which was subsequently calcined in air at 500 °C for 1 h, giving place to the Zr6@SiO2 composite. The silica layer provided anchoring sites for the oxozirconium clusters in NU-1000 after the organic linkers were removed at high temperatures whereas the Lewis acidity was enhanced by dehydration of the clusters during the heat treatment at 500 °C.235 FT-IR spectroscopy of pyridine adsorption showed that it contained appreciable amounts of Brønsted and Lewis acid sites, and both sites remained accessible to pyridine molecules. Besides, the catalytic activity of Zr6@SiO2 was demonstrated through the isomerization of glucose to fructose and compared to that of NU-1000, calcined NU-1000 and control samples of oxozirconium clusters in non-templated silica supports. The authors concluded that the ability to retain catalytic activity, compared to other materials, through site isolation and the templated porosity despite being heated to high temperatures makes nanocasting a useful technique for applying MOF-based catalytic clusters for catalytic processes that may require high temperatures.
 | ||
| Fig. 7 Stabilization of the oxozirconium clusters in NU-1000 by nanocasting with silica. The left image represents the NU-1000 structure with cluster nodes (red) and linkers (black). The white layer in the middle image represents silica. Reprinted with permission from ref. 25. Copyright 2016, American Chemical Society. | ||
Tungstated zirconia WOx/ZrO2 (WZ) is receiving increasing attention as a solid superacid in petrochemistry including alkane's isomerization,236 dehydration of alcohols237 and so on. Generally, the tetragonal phase of zirconia (t-ZrO2) can be generated with the aid of tungstate species after calcination at 600–900 °C forming WOx species and WO3 microcrystals on the surface of zirconia. The existence of hydroxyl groups and missing linker defects make the surface structures of zirconium octahedrons in UiO-66 similar to those in amorphous zirconium hydroxide, the commonly used supports in the synthesis of WZ.238 In this context, Wang et al. addressed the synthesis of tungstate immobilized UiO-66-derived WZ as strong solid acids toward high catalytic performance for acetalization of benzaldehyde.239 Ammonium meta-tungstate was first immobilized in the pores of UiO-66 by the double-solvent technology utilizing water and hexane and the as-prepared WOx@UiO-66 was heated at different temperatures (550, 650, 750 and 850 °C) in air for 6 h. Among all the catalysts, UiO-2-650 (where 2 refers to the adsorbed amount of ammonium meta-tungstate and 650 to the calcination temperature) led to the highest conversion (86.0%) toward benzaldehyde's acetalization in comparison with the 44.9% conversion obtained under the same conditions with the traditional zirconium hydroxide-prepared WZ, besides retaining its original activity for six cycles. The authors concluded that at moderate tungsten contents and calcination temperature, the MOF-derived WZ had various acid sites with different acid strength, and owned the highest amounts of acids due to highly concentrated amorphous polytungsten species. As a consequence, these solid acids of WZ exhibited enhanced catalytic activity compared to the traditional WZ prepared from precipitated zirconium hydroxide.
The development of dual-heteroatom-doped nano-porous carbon electrocatalysts for the ORR is particularly attractive as a feasible solution for the commercialization of fuel-cell technology. Heteroatom-doping approaches with N240 and P241 among others have been employed widely to improve the electrocatalytic performances of target non-precious-metal catalysts. On the other hand, amine-tagged MOFs can be used to graft functional groups.242 In view of these facts, Fu et al. took advantage of post-synthetic modification (PSM) and one-step pyrolysis to produce UiO-66-NH2-derived phosphorus–nitrogen co-doped porous carbon materials (P–N–carbons) from (Zr) UiO-66-NH2 and used them as electrocatalysts in the ORR.243 The as-synthesized UiO-66-NH2 was added to aqueous solutions of the phosphorus source glyphosine, which reacts with the –NH2 groups, to obtain the UiO-66-NH2-based P–N-rich precursors through vigorous stirring for several hours at 50 °C and then the P–N-rich precursor was washed with excess water to remove unreacted glyphosine. Since the UiO-66-NH2 framework possesses high thermal stability up to 500 °C, the P–N-rich precursor was pyrolyzed at a preset temperature (T = 600, 800–900, 950, and 1000 °C) in Ar for 3 h and finally soaked with HF solution to remove Zr and its derivatives thus leading to P–N-rich porous carbons. Among all the prepared samples, P–N–carbon-950 exhibited the best electrocatalytic activity similar to that of commercial 20% Pt/C and also better stability and increased tolerance to the methanol crossover effects. Characterization indicated that both the high electrical conductivity and the N and P content of the prepared P–N–carbon-950 played a key role in its electrocatalytic activity for the ORR, because the two factors significantly affect the electron transportation in electrodes and active sites for oxygen reduction.
In the case of Zr-MOFMS, calcination in air is deemed the optimal technique to produce Zr oxides that can act both as support or active phase, and are especially promising in acid catalysis. Grafting on UiO-66 comprises another possibility to incorporate different functionalities on the MOF and therefore on the resulting MOF derived catalyst.
(n) Bimetallic MOFMS
This last section does not deal with a particular metal but with bimetallic MOF derived catalysts. Bimetallic catalysts may open the door to solids with enhanced selectivity, activity, and stability, since they often show electronic and chemical properties that are distinct from those of their parent metals due to “synergistic” effects between the two metals. Bimetallic catalysts started to gain considerable commercial interest in the 1960s for their use in hydrocarbon reforming and since then have inspired extensive investigations on their possible applications.244Hetero-bimetallic MOFs have gained particular attention as a subclass of MOFs because of their designable framework structures that are modularly built from two transition-metal clusters as nodes and organic ligands as struts.245 Hetero-bimetallic MOFs not only combine the advantages of homogeneously located metal species and tuned concentrations of different metals in the frameworks, but they also expand the metal range to construct various pseudo-mixed-component MOFs bearing “on-demand” functionalities and applications, as the metals are not restricted to the same valence and coordination geometry.246 All this boosts the application of hetero-bimetallic MOFs as precursors to prepare metal/mesoporous carbon composite catalysts for electro-, photo and also conventional heterogenous catalysis.
It is remarkable, however, that the vast majority of bimetallic MOF mediated synthesis reactions reported so far employ Zn as a sacrificial template in bimetallic MOFs in order to gain further control of the porosity of the resulting monometallic material or sometimes bimetallic but after impregnation of an additional metal not in the framework of the parent MOF. You et al. reported a bimetal-MOF (bi-MOF) self-adjusted synthesis of support-free porous Co–N–C nanopolyhedron electrocatalysts by pyrolysis of a Zn/Co bi-MOF without any post-treatments.247 Different mixtures of Co(NO3)2·6H2O and Zn(NO3)2·6H2O were dissolved in methanol and subsequently poured into methanol containing 2-methylimidazole (MeIM). After thorough mixing and incubation at room temperature, the as-obtained precipitates were centrifuged and washed with ethanol several times, resulting in the corresponding ZnxCo1−x(MeIM)2 bi-MOFs. Finally, the corresponding bi-MOF was heated to the desired temperature (800, 900, and 1000 °C) for 3 h under Ar to obtain the self-supported Co–N–C-x–y, where x represents the molar content of Zn ions in the initial bi-MOF precursor and y represents the carbonization temperature. Electrocatalytic studies showed that the carbonization at 900 °C of Zn0.8Co0.2(MeIM)2 resulted in the most active Co–N–C for the ORR with a half-wave potential more positive than that of commercial 20 wt% Pt/C and a kinetic current density 3.1 times that of Pt/C in 0.1 M KOH, and excellent stability and methanol tolerance. It also exhibited ORR activity comparable to and stability much higher than those of Pt/C in acidic and neutral electrolytes. The authors argue that the presence of initial Zn forms a spatial isolation of Co that suppresses its sintering during pyrolysis, and Zn evaporation also promotes the surface area of the resultant catalysts besides its composition, and morphology, and hence the ORR activity of Co–N–C could be tuned by the Zn/Co ratio. Various characterization techniques revealed that the superior activity and strong stability of Co–N–C originated from the intense interaction between Co and N, the high content of ORR active pyridinic and pyrrolic N, and the large specific surface area.
Similarly, Wang et al. reported an analogous and highly efficient Co–N–C catalyst with highly dispersed Co sites derived from a ZnCo bimetallic ZIF and successfully tested it in the ORR.248 In this case the authors prepared ZnxCo nanocrystals (where x is defined as the molar percentage of Co/(Co + Zn) in the starting materials) by a surfactant mediated growth method and the resultant bimetallic ZIF was calcined at higher temperature. Typically, a mixture of surfactants and 2-methylimidazole were dissolved in deionized water under sonication to give a homogeneous emulsion and then the zinc and cobalt mixed metal nitrate solution was poured into the emulsion. The solution was stirred at 60 °C for 4 h to give a turbid suspension and the ZIF–(100 − x)ZnxCo nanocrystals were separated by centrifugation, washed three times with deionized water and two times with ethanol and then dried at 80 °C under vacuum for about 12 h. Finally, the ORR catalysts were prepared by pyrolysis of the ZIF–(100 − x)ZnxCo nanocrystals in Ar at 1000 °C at 5 °C min−1 for 60 min and allowed to cool down to room temperature. In this case, the authors show that the best ORR performance is achieved when 5% Zn is substituted by Co in the MOF precursor. The resulting C–95Zn5Co catalyst outperformed the commercial Pt/C catalyst with 20 wt% Pt loading in terms of half-wave potential. The authors suggested that the homogeneous distribution of Zn and Co in the bimetallic ZIF was critical for achieving high dispersion of Co in the derived catalyst, which was facilitated by both the isomorphous nature of ZIF-67 and ZIF-8 and the surfactant mediated growth method.
For the ORR, a core–shell structure may improve the performance and concurrently increase methanol tolerance and stability of the catalysts65 and for the HER, the core–shell structure is anticipated to not only enhance the corrosion resistance of the metals in acidic electrolytes but also improve the HER activity via the synergistic contributions from both the core and shell.249 No sooner said than done, Lu et al. prepared core–shell nanocomposites based on Au nanoparticle@zinc–iron embedded porous carbons (Au@Zn–Fe–C) derived from bifunctional MOFs as electrocatalysts for the ORR and HER.250 Experimentally, Au nanoparticles were prepared by reducing HAuCl4 at 140 °C with DMF both as a solvent and as a reducing reagent in the presence of poly(vinylpyrrolidone) (PVP) which stabilized the Au nanoparticles.251 Then, a Zn–Fe-MOF shell was grown on the Au nanoparticle surface by using iron(III) acetylacetonate (Fe(acac)3) as the iron source, Zn(NO3)2 as the zinc source, H2bdc as the organic linker, and a DMF–ethanol mixed solution as the solvent. PVP was also added in the above mixture as a stabilizing reagent for the formation of a uniform Zn–Fe-MOF shell. Finally, the Zn–Fe-MOF shell was converted to porous carbons embedded with zinc–iron compounds by calcination in Ar at 600 °C for 2 h at a heating rate of 2 °C min−1, forming core–shell Au@Zn–Fe–C hybrids. As a result, single Au nanoparticles of 50–100 nm diameter were encapsulated within porous carbon shells embedded with Zn–Fe compounds. The resulting Au@Zn–Fe–C hybrids exhibited excellent electrocatalytic performance for both the ORR and HER. The encapsulated Au nanoparticles played an important role in determining the electrocatalytic activity for the ORR and HER by promoting electron transfer to the zinc–iron-embedded porous carbon layer, and the electrocatalytic activity was found to vary with both the loading of the gold nanoparticle cores and the thickness of the metal–carbon shells.
Xu et al. synthesized a series of carbon composite materials containing metal oxides (ZnO and/or TiOx) by pyrolyzing a Zn–Ti heterometallic MOF as the precursor and the photocatalytic activities of these composites were evaluated by the photodegradation of MB in aqueous solutions.252 In a previous work, the same authors developed porous TiOx/C composite materials by pyrolyzing a Ti-containing MIL-125 which exhibited better photocatalytic activities than the benchmark P-25 TiO2 during the photodegradation of MB.253 In order to improve the accessibility to TiOx active sites, the authors addressed the preparation of highly microporous photocatalysts by pyrolyzing a previously reported Zn–Ti heterometallic MOF ZTOF-1.254 The as-prepared ZTOF-1 was held at 80 °C for 45 minutes in Ar and then pyrolyzed under the same Ar atmosphere at different temperatures of 400, 600, 800, and 1000 °C for 5 h affording ZTOF-1-P4, ZTOF-1-P6, ZTOF-1-P8, and ZTOF-1-P10 materials, respectively. ZTOF-1-P10 demonstrated the best catalytic activity, under a UV light source outperforming benchmark P-25 TiO2. ZTOF-1-P10 exhibited a dramatically increased surface area due to the carbothermal reduction of Zn2+ into metallic Zn followed by vaporization as an extra pore-forming mechanism. Because Zn and Ti are adjacent in the heterometallic secondary building units (SBUs) of the MOF precursor, the catalytically active TiOx sites generated during pyrolysis would remain in the pores and channels formed by Zn vaporization and were readily accessible to MB, leading to a huge increase of photocatalytic activity. According to the authors, the excellent photocatalytic activity of ZTOF-1-P10 lies in (i) the substantial amounts of TiO and Ti2O3 (TiOx) which have been reported to have a better photocatalytic activity than pure TiO2 because of the higher density of oxygen vacancies for better trapping of electrons, (ii) the highest Ti content among all the pyrolyzed samples and (iii) the dramatically increased surface area and porosity due to the reduction and evaporation of metallic Zn. In addition, the special prearrangement of Zn and Ti in the SBUs of the ZTOF-1 precursor makes TiOx formed during pyrolysis remain in the pores and channels generated by Zn evaporation thus making TiOx active sites easily accessible by substrates leading to high reaction kinetics.
Yang et al. worked on the preparation of Ni/nanoporous carbon (Ni/NPC) composites by direct pyrolysis of nonporous heterobimetallic zinc–nickel–terephthalate frameworks (Zn1−xNixMOF, x ≈ 0–1) for the 4-nitrophenol reduction reaction.245 Mixing different amounts of Zn(NO3)2·6H2O and Ni(CH3COO)2·4H2O with terephthalic acid (TPA) in DMF afforded nonporous even crystals of Zn1−xNixMOF which were carbonized up to 1223 K in N2 at a rate of 5 K min−1 and maintained for 2 h to yield Nix/NPC metal/mesoporous carbon composites with uniform pore sizes and evenly dispersed metal sites. Both the rate constants and the TOF values suggested higher catalytic efficiency of Ni1.0/NPC and Ni0.1/NPC, which were more active than the previously reported NiCo2 alloy255 and the Ni NPs in a spherical polyelectrolyte brush nanoreactor256 and the values are also comparable to those of soft-templated Ni NCs on mesoporous carbons.257 The Ni1.0/NPC catalyst was facilely separated from the reaction solution by using a magnet and exhibited similar catalytic performance without any visible reduction in the conversion within 20 min even after recycling over six runs. The authors concluded that their work not only introduces a new class of material with the highest possible precursor site density and compatible textural properties, but also provides a distinct platform with abundant model compounds that can be used to study the correlation between precursor composition and catalytic performance.
Cu/ZnO catalysts are widely used in industrial production, environmental protection and biorefineries, therefore finding a new and efficient method for the controlled synthesis of Cu/ZnO hybrids is of great importance. Recently, Zheng et al. have prepared a novel Cu/ZnO catalyst with nanosized ZnO particles dotted on Cu via the calcination and reduction of the Cu(Zn)–HKUST-1 precursor.258 ZnO was dispersed in deionized water and sonicated for 10 min and then mixed with DMF. Subsequently Cu(NO3)2·3H2O dissolved in deionized water and benzene-1,3,5-tricarboxylic acid (H3btc) dissolved in ethanol were consecutively added under vigorously stirring. Then the solid product was immediately filtered, washed with ethanol and dried. The as-synthesized CuxZn3−x–HKUST-1 was slowly heated (2 °C min−1) to 400 °C in air for 4 h yielding CuxZn3–xO3. The calcined CuxZn3−xO3 was reduced in 80 mL min−1 H2 at 350 °C for 1 h before the hydrogenolysis reaction, and these reduced catalysts were identified as Cux/ZnO. It was confirmed that all the catalysts synthesized from HKUST-1 were efficient for the hydrogenolysis of glycerol. Moreover the authors found that the Cu1.1/ZnO catalyst derived from Cu1.1Zn1.9(btc)2·9.4(H2O) was more active and stable than Cu/ZnO prepared via solvent-free grinding and co-precipitation methods and inferred that the interface between Cu and ZnO played a crucial role in its catalytic performance, making ZnO doped Cu more stable than ZnO plate supported Cu particles.
Some other authors made use of bimetallic MOFMS to prepare bimetallic catalysts, retaining and thus taking advantage of both metals in the framework. Kong et al. prepared heterometallic carbide nanoparticles (Co3InC0.75) embedded in nitrogen-enriched carbon by pyrolysis of an indium-MOF with entrapped cobalt dimers for the ORR.259 Co3InC0.75 nanoparticles with a CaTiO3-typecubic structure embedded within carbon (Co3InC0.75@C) were prepared by pyrolysis of CPM-16–Co–In, which is a MOF with the cobalt–indium–btc framework. In CPM-16–Co–In, V-shaped Co2(OH) dimers were grafted onto zeolitic In–btc networks by carboxyl groups, and uniformly arranged in the channels of the framework. By directly heating these crystals at 700 °C for 2 h in Ar the purple rod crystals of CPM-16–Co–In were transformed into Co3InC0.75@C. To enrich the nitrogen content in Co3InC0.75@C, melamine was introduced as the nitrogen source. The pyrolysis of the mixture, prepared by grinding CPM-16–Co–In and melamine with a mass ratio of 1:2, at 700 °C for 2 h in Ar, led to the formation of Co3InC0.75 in the CNx matrix instead of the C matrix. These Co3InC0.75 heterometallic carbide nanoparticles embedded in N-enriched carbon exhibited an impressive ORR activity comparable to the commercial Pt/C (10 wt%) catalyst in alkaline medium and had superior durability and methanol tolerance. The authors proposed that the presence of indium in Co3InC0.75 prevented the decomposition of heterometallic carbides into metallic cobalt and carbon at high temperature and the carbon derived from the btc–indium framework coated on the formed Co3InC0.75 particles prevented them from excessively aggregating leading to high electrocatalytic performance.
Long et al. reported a facile and general approach to fabricate bimetallic and trimetallic transition alloy NPs embedded in an N-doped carbon matrix by a direct pyrolysis method and their performance in hydrogenation reactions.260 With this in mind, the authors firstly developed a series of novel N-donor multimetallic M–M′-MOFs [M–M′(1,4-bdc)2(dabco)]·4DMF·1/2H2O, M/M′ = Co, Ni, Cu. The M–M′-MOFs were prepared by a facile mixed-metal approach. Typically, M(NO3)2·6H2O and M′(NO3)2·6H2O, DABCO (triethylenediamine) and DMF were added to a sealed round-bottom flask. The mixture was treated by ultrasonic dispersion until the solids were dissolved, and then heated in an oil bath for 40 hours at 120 °C after stirring for 8 h. The products were washed with DMF and methanol, and then dried under vacuum at 150 °C for 12 h. The thermolysis was carried out in He with a heating rate of 1 °C min−1. The samples were first heated from room temperature to 200 °C and then to 500 °C and kept at this temperature for 8 h to obtain M–M′@C–N black catalysts. Because of the easy oxidation of metal NPs in air, the samples were reduced at 200 °C under a H2 atmosphere for 2 h prior to being used as catalysts. As a result, the MOF-derived transition-metal alloy NPs were highly dispersed and embedded in the highly ordered N-doped graphene layers with an average size of ca. 20 nm. Moreover, the complete alloying of transition metal elements and the homogeneous distribution of N-doped carbon and metal alloy NPs was confirmed on every nanoparticle as well as and the existence of an obvious synergetic activation of different transition metals and strong coordination interactions between metals of alloy NPs and N atoms. When used in the transfer hydrogenation of nitriles in the absence of basic additives, Co–Ni(3:1)@C–N showed the best catalytic performance being almost 5 times more active than its monometallic counterparts and showed excellent recyclability and extensive applicability.
The general approach in bimetallic MOFMS consists of (i) using Zn as the sacrificial secondary metal for its evaporation at high temperatures under inert conditions, thus improving the porosity of the resulting carbon based material and (ii) preparing bimetallic active sites/promoters as both unsupported and supported in carbon catalytic materials in oxidizing and inert atmospheres, respectively. Furthermore, bimetallic MOFMS allows more than that. During the thermal process that MOFMS basically is, the properties and behaviour of a certain metal will definitely affect the other metal's final state and vice versa, thus providing an extra control parameter to play with during this promising and relatively novel synthetic process.
4. Conclusions and future perspectives
Metal organic frameworks have emerged as exemplary precursors for the manufacture of highly catalytically active materials through the so-called MOF mediated synthesis. From basic to advanced catalytic design, MOFMS embraces a wide range of synthetic techniques. Keeping it simple, the basic definition of MOFMS refers to a single-step heat treatment of the parent MOF. In this process, the properties of the MOF together with the heat treatment conditions (activating atmosphere, temperature, time and heating ramp) dictate the final properties of the MOF derived catalyst, allowing for an unprecedented freedom in design. As highlighted by the examples above, when obtained through MOFMS, metal oxides, carbons, carbides, and nanoparticles in carbon have unique structural properties that result, in many cases, in improved catalytic performance.In an inert atmosphere, when the decomposition temperature of the framework is exceeded, the Gibbs free energy/reduction potential of metal ions predict the metallic species (metal, oxide, and carbide) which are likely to be formed. Once formed, the Tamman temperature of the metallic species needs to be considered in order to control the size of the resulting nanoparticles in the carbon matrix. Finally, according to the boiling point of the metal species, the metal phase will be retained in the material or, in contrast, will evaporate. Although we decided to organize this review according to the MOF metal component, the linker also plays a decisive role, not only defining the final porosity of the carbon matrix and its composition (oxidic, nitridic), but also the oxidation state of the metal species in the catalyst. For instance, oxygenated linkers will favour oxidation of species whereas nitrogen in the linker may result in the formation of highly dispersed M–N species in the final catalyst.
Calcination in air of the pristine MOF is a more straightforward procedure in the sense that it always leads to the formation of the corresponding carbon-free metal oxide. In this case, when the decomposition temperature of the framework is surpassed, the organic linker has been already burnt away. Further increasing the temperature allows the porosity of the material to be tuned. In some cases, a pyrolysis step prior to calcination is applied to enhance the porosity of the resulting material. The boiling point of oxides is usually too high to consider possible evaporation during MOFMS under an oxidizing atmosphere.
In conventional supported catalysts the most stable oxides such as MgO, ZrO2, Al2O3, TiO2, SiO2, MnO, Cr2O3, and ZnO act as supports and/or promoters. In MOFMS, these are achieved by calcination in air and also by pyrolysis in an inert atmosphere up to a given temperature, yielding non-supported and carbon supported metal oxides, respectively. In the latter case, according to their reduction potential and when oxygen available, these ions tend to form their corresponding oxides up to the temperature when carbon starts reducing the metal oxide to form the corresponding metal. However, this usually takes place at a relatively high temperature. These Mg, Zr, Al, Ti or Zn MOF derived catalysts are typically used as metal oxides for photocatalysis and/or as templates for preparing nanostructured carbons. In contrast, metals such as Fe, Co, Ni and Cu are likely to form metallic nanoparticles under an inert atmosphere at a relatively low temperature and these metals are highly effective in catalyzing the graphitization of carbon surrounding them,261 which needs to be taken into consideration for the targeted catalytic application.
The biggest advantage of this new methodology lies in the number of degrees of freedom that it offers. When compared to traditional synthesis methods, the large metal dispersion in the original MOF allows for very high metal dispersion and for exquisite control over the final size of the supported/encapsulated nanoparticles. Moreover, further development and modification of the technique via utilization of different activation atmospheres, introduction of additional components to the MOF by ion exchange or impregnation, intermediate pyrolysis steps and others, has already led to sophisticated catalytic systems that cannot be achieved by any other means. This approach does not only lead to improved catalytic systems but is an excellent tool for the design of model systems that help generate knowledge through understanding.
Probably the aspect that seems to raise more concern regarding the MOFMS approach is the fact that highly engineered, beautiful, very often expensive, materials are simply destroyed and this may look like something unaffordable from an industrial point of view. However, this is not always the case. Some of the aforementioned MOF mediated materials are highly competitive from an economic point of view compared to other more sophisticated synthetic procedures and their counterparts obtained by conventional methods.95 Moreover, keeping in mind a certain level of pragmatism, the main target of catalyst synthesis is the design and preparation of outstanding catalysts and MOFMS is an excellent tool to this end. On the other hand, one should also consider that a large part of the final price of a MOF comes from extensive washing steps needed to purify the as synthesized materials. Obviously, the nature and depth of these steps required for MOFMS are far milder and this should lead to moderate prices of the hard template. Moreover, if the stability of the resulting catalysts is increased by orders of magnitude, the impact in terms of catalyst makeup cost and less process operation interruptions has the potential of bringing additional economic attractiveness. Nevertheless, specific viability studies apply depending on the MOF precursor and synthesis conditions utilized in different cases.
Overall, we are certain that over the next few years this approach will be adopted by more research groups and that new methodologies will be developed. At this stage we safely state that our imagination is the only limit when it comes to the design possibilities in MOF mediated synthesis.
Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 704473.References
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities, Chem. Rev., 2016, 116(18), 10346–10413 CrossRef CAS PubMed.
- F. Akhtar, L. Andersson, S. Ogunwumi, N. Hedin and L. Bergström, Structuring adsorbents and catalysts by processing of porous powders, J. Eur. Ceram. Soc., 2014, 34(7), 1643–1666 CrossRef CAS.
- R. W. J. Scott, O. M. Wilson and R. M. Crooks, Synthesis, Characterization, and Applications of Dendrimer-Encapsulated Nanoparticles, J. Phys. Chem. B, 2005, 109(2), 692–704 CrossRef CAS PubMed.
- A. E. Danks, S. R. Hall and Z. Schnepp, The evolution of ‘sol-gel’ chemistry as a technique for materials synthesis, Mater. Horiz., 2016, 3(2), 91–112 RSC.
- R. W. Johnson, A. Hultqvist and S. F. Bent, A brief review of atomic layer deposition: from fundamentals to applications, Mater. Today, 2014, 17(5), 236–246 CrossRef CAS.
- S. G. Kwon, G. Krylova, P. J. Phillips, R. F. Klie, S. Chattopadhyay and T. Shibata, et al., Heterogeneous nucleation and shape transformation of multicomponent metallic nanostructures, Nat. Mater., 2015, 14(2), 215–223 CrossRef CAS PubMed.
- F. Zaera, Nanostructured materials for applications in heterogeneous catalysis, Chem. Soc. Rev., 2013, 42(7), 2746–2762 RSC.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, Metal Organic Framework Catalysis: Quo vadis?, ACS Catal., 2014, 4(2), 361–378 CrossRef CAS.
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Metal–Organic Frameworks for Energy Applications, Chemistry, 2017, 2(1), 52–80 CrossRef CAS.
- M. G. Goesten, F. Kapteijn and J. Gascon, Fascinating chemistry or frustrating unpredictability: observations in crystal engineering of metal–organic frameworks, CrystEngComm, 2013, 15(45), 9249–9257 RSC.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, A chromium terephthalate-based solid with unusually large pore volumes and surface area, Science, 2005, 309(5743), 2040–2042 CrossRef PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti and S. Bordiga, et al., A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability, J. Am. Chem. Soc., 2008, 130(42), 13850–13851 CrossRef PubMed.
- V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero and G. Palmisano, et al., High thermal and chemical stability in pyrazolate-bridged metal–organic frameworks with exposed metal sites, Chem. Sci., 2011, 2(7), 1311–1319 RSC.
- Z.-W. Wang, M. Chen, C.-S. Liu, X. Wang, H. Zhao and M. Du, A Versatile AlIII-Based Metal–Organic Framework with High Physicochemical Stability, Chem. – Eur. J., 2015, 21(48), 17215–17219 CrossRef CAS PubMed.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, Metal–Organic Framework as a Template for Porous Carbon Synthesis, J. Am. Chem. Soc., 2008, 130(16), 5390–5391 CrossRef CAS PubMed.
- L. Zhang, C. Feng, S. Gao, Z. Wang and C. Wang, Palladium nanoparticle supported on metal–organic framework derived N-decorated nanoporous carbon as an efficient catalyst for the Suzuki coupling reaction, Catal. Commun., 2015, 61, 21–25 CrossRef CAS.
- W. Zhong, H. Liu, C. Bai, S. Liao and Y. Li, Base-Free Oxidation of Alcohols to Esters at Room Temperature and Atmospheric Conditions using Nanoscale Co-Based Catalysts, ACS Catal., 2015, 5(3), 1850–1856 CrossRef CAS.
- K. Shen, L. Chen, J. Long, W. Zhong and Y. Li, MOFs-Templated Co@Pd Core–Shell NPs Embedded in N-Doped Carbon Matrix with Superior Hydrogenation Activities, ACS Catal., 2015, 5(9), 5264–5271 CrossRef CAS.
- I. Mondal and U. Pal, Synthesis of MOF templated Cu/CuO@TiO2 nanocomposites for synergistic hydrogen production, Phys. Chem. Chem. Phys., 2016, 18(6), 4780–4788 RSC.
- R. Li, S. Wu, X. Wan, H. Xu and Y. Xiong, Cu/TiO2 octahedral-shell photocatalysts derived from metal–organic framework@semiconductor hybrid structures, Inorg. Chem. Front., 2016, 3(1), 104–110 RSC.
- Z. Dong, X. Le, Y. Liu, C. Dong and J. Ma, Metal organic framework derived magnetic porous carbon composite supported gold and palladium nanoparticles as highly efficient and recyclable catalysts for reduction of 4-nitrophenol and hydrodechlorination of 4-chlorophenol, J. Mater. Chem. A, 2014, 2(44), 18775–18785 CAS.
- V. P. Santos, T. A. Wezendonk, J. J. D. Jaén, A. I. Dugulan, M. A. Nasalevich and H.-U. Islam, et al., Metal organic framework-mediated synthesis of highly active and stable Fischer-Tropsch catalysts, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- D. Chen, M. Huang, S. He, S. He, L. Ding and Q. Wang, et al., Ru-MOF enwrapped by montmorillonite for catalyzing benzene hydrogenation, Appl. Clay Sci., 2016, 119(Part 1), 109–115 CrossRef CAS.
- X. Cao, B. Zheng, X. Rui, W. Shi, Q. Yan and H. Zhang, Metal Oxide-Coated Three-Dimensional Graphene Prepared by the Use of Metal–Organic Frameworks as Precursors, Angew. Chem., Int. Ed., 2014, 53(5), 1404–1409 CrossRef CAS PubMed.
- C. D. Malonzo, S. M. Shaker, L. Ren, S. D. Prinslow, A. E. Platero-Prats and L. C. Gallington, et al., Thermal Stabilization of Metal–Organic Framework-Derived Single-Site Catalytic Clusters Through Nanocasting, J. Am. Chem. Soc., 2016, 138(8), 2739–2748 CrossRef CAS PubMed.
- L. Zhang, X. Wang, R. Wang and M. Hong, Structural Evolution from Metal–Organic Framework to Hybrids of Nitrogen-Doped Porous Carbon and Carbon Nanotubes for Enhanced Oxygen Reduction Activity, Chem. Mater., 2015, 27(22), 7610–7618 CrossRef CAS.
- S. Deng, M. Kurttepeli, D. J. Cott, S. Bals and C. Detavernier, Porous nanostructured metal oxides synthesized through atomic layer deposition on a carbonaceous template followed by calcination, J. Mater. Chem. A, 2015, 3(6), 2642–2649 CAS.
- M. Enterría and J. L. Figueiredo, Nanostructured mesoporous carbons: Tuning texture and surface chemistry, Carbon, 2016, 108, 79–102 CrossRef.
- C. T. Campbell, S. C. Parker and D. E. Starr, The Effect of Size-Dependent Nanoparticle Energetics on Catalyst Sintering, Science, 2002, 298(5594), 811–814 CrossRef CAS PubMed.
- J.-K. Sun and Q. Xu, Functional materials derived from open framework templates/precursors: synthesis and applications, Energy Environ. Sci., 2014, 7(7), 2071–2100 CAS.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Metal–organic frameworks and their derived nanostructures for electrochemical energy storage and conversion, Energy Environ. Sci., 2015, 8(7), 1837–1866 CAS.
- H. J. T. Ellingham, Reducibility of oxides and sulfides in metallurgical processes, J. Soc. Chem. Ind., London, 1944, 63, 9 Search PubMed.
- R. Das, P. Pachfule, R. Banerjee and P. Poddar, Metal and metal oxide nanoparticle synthesis from metal organic frameworks (MOFs), finding the border of metal and metal oxides, Nanoscale, 2012, 4(2), 591–599 RSC.
- N. Birks, G. H. Meier and F. S. Pettit, Introduction to the High Temperature Oxidation of Metals, Cambridge University Press, Cambridge, 2nd edn, 2006, 2006/003/17 Search PubMed.
- Elements MaBPoC. http://www.chemistry.patent-invent.com/chemistry/melting_boiling_point.html, updated April 2011.
- L. B. Railsback, http://www.gly.uga.edu/railsback/Fundamentals/ElementalAbundanceTableP.pdf, 2006.
- M. Trueba and S. P. Trasatti, γ-Alumina as a Support for Catalysts: A Review of Fundamental Aspects, Eur. J. Inorg. Chem., 2005, 3393–3403 CrossRef CAS.
- T. Ahnfeldt, N. Guillou, D. Gunzelmann, I. Margiolaki, T. Loiseau and G. Férey, et al., [Al4(OH)2(OCH3)4(H2N-bdc)3]·xH2O: A 12-Connected Porous Metal–Organic Framework with an Unprecedented Aluminum-Containing Brick, Angew. Chem., Int. Ed., 2009, 48(28), 5163–5166 CrossRef CAS PubMed.
- E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo and H. Furukawa, et al., Metal Insertion in a Microporous Metal–Organic Framework Lined with 2,2′-Bipyridine, J. Am. Chem. Soc., 2010, 132(41), 14382–14384 CrossRef CAS PubMed.
- F. Afsahi, H. Vinh-Thang, S. Mikhailenko and S. Kaliaguine, Electrocatalyst synthesized from metal organic frameworks, J. Power Sources, 2013, 239, 415–423 CrossRef CAS.
- Q.-L. Zhu, W. Xia, T. Akita, R. Zou and Q. Xu, Metal–Organic Framework-Derived Honeycomb-Like Open Porous Nanostructures as Precious-Metal-Free Catalysts for Highly Efficient Oxygen Electroreduction, Adv. Mater., 2016, 28(30), 6391–6398 CrossRef CAS PubMed.
- S. S. Shendage, A. S. Singh and J. M. Nagarkar, Facile approach to the electrochemical synthesis of palladium-reduced graphene oxide and its application for Suzuki coupling reaction, Tetrahedron Lett., 2014, 55(4), 857–860 CrossRef CAS.
- H.-L. Jiang, B. Liu, Y.-Q. Lan, K. Kuratani, T. Akita and H. Shioyama, et al., From Metal–Organic Framework to Nanoporous Carbon: Toward a Very High Surface Area and Hydrogen Uptake, J. Am. Chem. Soc., 2011, 133(31), 11854–11857 CrossRef CAS PubMed.
- M. Schubert, U. Müller and N. Trukhan, Metal oxides from metal–organic framework materials, Google Patents, US 8501150 B2, 2013 Search PubMed.
- M. Celebi, M. Yurderi, A. Bulut, M. Kaya and M. Zahmakiran, Palladium nanoparticles supported on amine-functionalized SiO2 for the catalytic hexavalent chromium reduction, Appl. Catal., B, 2016, 180, 53–64 CrossRef CAS.
- M. Yadav and Q. Xu, Catalytic chromium reduction using formic acid and metal nanoparticles immobilized in a metal–organic framework, Chem. Commun., 2013, 49(32), 3327–3329 RSC.
- R. Cheng, Z. Liu, L. Zhong, X. He, P. Qiu and M. Terano, et al., Phillips Cr/Silica Catalyst for Ethylene Polymerization, in Polyolefins: 50 years after Ziegler and Natta I: Polyethylene and Polypropylene, ed. W. Kaminsky, Berlin, Heidelberg, Springer Berlin Heidelberg, 2013, pp. 135–202 Search PubMed.
- A. Budnyk, A. Damin, C. Barzan, E. Groppo, C. Lamberti and S. Bordiga, et al., Cr-doped porous silica glass as a model material to describe Phillips catalyst properties, J. Catal., 2013, 308, 319–327 CrossRef CAS.
- D.-W. Lee, M. S. Lee, J. Y. Lee, S. Kim, H.-J. Eom and D. J. Moon, et al., The review of Cr-free Fe-based catalysts for high-temperature water-gas shift reactions, Catal. Today, 2013, 210, 2–9 CrossRef CAS.
- H. Zhao, H. Song, L. Xu and L. Chou, Isobutane dehydrogenation over the mesoporous Cr2O3/Al2O3 catalysts synthesized from a metal–organic framework MIL-101, Appl. Catal., A, 2013, 456, 188–196 CrossRef CAS.
- X. Qiu, X. Wang and Y. Li, Controlled growth of dense and ordered metal–organic framework nanoparticles on graphene oxide, Chem. Commun., 2015, 51(18), 3874–3877 RSC.
- S. Bhattacharjee, C. Chen and W.-S. Ahn, Chromium terephthalate metal–organic framework MIL-101: synthesis, functionalization, and applications for adsorption and catalysis, RSC Adv., 2014, 4(94), 52500–52525 RSC.
- G. Wang, C. Li and H. Shan, Highly Efficient Metal Sulfide Catalysts for Selective Dehydrogenation of Isobutane to Isobutene, ACS Catal., 2014, 4(4), 1139–1143 CrossRef CAS.
- C.-B. Wang, C.-C. Lee, J.-L. Bi, J.-Y. Siang, J.-Y. Liu and C.-T. Yeh, Study on the steam reforming of ethanol over cobalt oxides, Catal. Today, 2009, 146(1–2), 76–81 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Advances in the Development of Novel Cobalt Fischer–Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels, Chem. Rev., 2007, 107(5), 1692–1744 CrossRef CAS PubMed.
- F. Hebrard and P. Kalck, Cobalt-Catalyzed Hydroformylation of Alkenes: Generation and Recycling of the Carbonyl Species, and Catalytic Cycle, Chem. Rev., 2009, 109(9), 4272–4282 CrossRef CAS PubMed.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa and M. O'Keeffe, et al., High-Throughput Synthesis of Zeolitic Imidazolate Frameworks and Application to CO2 Capture, Science, 2008, 319(5865), 939–943 CrossRef CAS PubMed.
- J. Qian, F. Sun and L. Qin, Hydrothermal synthesis of zeolitic imidazolate framework-67 (ZIF-67) nanocrystals, Mater. Lett., 2012, 82, 220–223 CrossRef CAS.
- H. Jing, X. Song, S. Ren, Y. Shi, Y. An and Y. Yang, et al., ZIF-67 Derived Nanostructures of Co/CoO and Co@N-doped Graphitic Carbon as Counter Electrode for Highly Efficient Dye-sensitized Solar Cells, Electrochim. Acta, 2016, 213, 252–259 CrossRef CAS.
- Y.-Q. Tian, Z.-X. Chen, L.-H. Weng, H.-B. Guo, S. Gao and D. Y. Zhao, Two Polymorphs of Cobalt(II) Imidazolate Polymers Synthesized Solvothermally by Using One Organic Template N,N-Dimethylacetamide, Inorg. Chem., 2004, 43(15), 4631–4635 CrossRef CAS PubMed.
- S. Ma, G. A. Goenaga, A. V. Call and D.-J. Liu, Cobalt Imidazolate Framework as Precursor for Oxygen Reduction Reaction Electrocatalysts, Chem. – Eur. J., 2011, 17(7), 2063–2067 CrossRef CAS PubMed.
- F. Jaouen, J. Herranz, M. Lefèvre, J.-P. Dodelet, U. I. Kramm and I. Herrmann, et al., Cross-Laboratory Experimental Study of Non-Noble-Metal Electrocatalysts for the Oxygen Reduction Reaction, ACS Appl. Mater. Interfaces, 2009, 1(8), 1623–1639 CAS.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis, Carbon, 1995, 33(11), 1641–1653 CrossRef CAS.
- W. Xia, J. Zhu, W. Guo, L. An, D. Xia and R. Zou, Well-defined carbon polyhedrons prepared from nano metal–organic frameworks for oxygen reduction, J. Mater. Chem. A, 2014, 2(30), 11606–11613 CAS.
- W. Xia, R. Zou, L. An, D. Xia and S. Guo, A metal–organic framework route to in situ encapsulation of Co@Co3O4@C core@bishell nanoparticles into a highly ordered porous carbon matrix for oxygen reduction, Energy Environ. Sci., 2015, 8(2), 568–576 CAS.
- X. Wang, J. Zhou, H. Fu, W. Li, X. Fan and G. Xin, et al., MOF derived catalysts for electrochemical oxygen reduction, J. Mater. Chem. A, 2014, 2(34), 14064–14070 CAS.
- A. Kong, C. Mao, Q. Lin, X. Wei, X. Bu and P. Feng, From cage-in-cage MOF to N-doped and Co-nanoparticle-embedded carbon for oxygen reduction reaction, Dalton Trans., 2015, 44(15), 6748–6754 RSC.
- S.-T. Zheng, T. Wu, B. Irfanoglu, F. Zuo, P. Feng and X. Bu, Multicomponent Self-Assembly of a Nested Co24@Co48 Metal–Organic Polyhedral Framework, Angew. Chem., Int. Ed., 2011, 50(35), 8034–8037 CrossRef CAS PubMed.
- S. H. Oh, R. Black, E. Pomerantseva, J.-H. Lee and L. F. Nazar, Synthesis of a metallic mesoporous pyrochlore as a catalyst for lithium–O2 batteries, Nat. Chem., 2012, 4(12), 1004–1010 CrossRef CAS PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, Metal–Organic Framework Derived Hybrid Co3O4-Carbon Porous Nanowire Arrays as Reversible Oxygen Evolution Electrodes, J. Am. Chem. Soc., 2014, 136(39), 13925–13931 CrossRef CAS PubMed.
- S. Bao, N. Yan, X. Shi, R. Li and Q. Chen, High and stable catalytic activity of porous Ag/Co3O4 nanocomposites derived from MOFs for CO oxidation, Appl. Catal., A, 2014, 487, 189–194 CrossRef CAS.
- X. Wang, W. Zhong and Y. Li, Nanoscale Co-based catalysts for low-temperature CO oxidation, Catal. Sci. Technol., 2015, 5(2), 1014–1020 CAS.
- X. Xie, Y. Li, Z.-Q. Liu, M. Haruta and W. Shen, Low-temperature oxidation of CO catalysed by Co3O4 nanorods, Nature, 2009, 458(7239), 746–749 CrossRef CAS PubMed.
- I. Stará, V. Nehasil and V. Matolín, The influence of particle size on CO oxidation on Pdalumina model catalyst, Surf. Sci., 1995, 331–333(Part A), 173–177 CrossRef.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, Low-Temperature Oxidation of CO over Gold Supported on TiO2, α-Fe2O3, and Co3O4, J. Catal., 1993, 144(1), 175–192 CrossRef CAS.
- H. Einaga, S. Futamura and T. Ibusuki, Complete Oxidation of Benzene in Gas Phase by Platinized Titania Photocatalysts, Environ. Sci. Technol., 2001, 35(9), 1880–1884 CrossRef CAS PubMed.
- J. Li, W. Li, G. Liu, Y. Deng, J. Yang and Y. Chen, Tricobalt tetraoxide-supported palladium catalyst derived from metal organic frameworks for complete benzene oxidation, Catal. Lett., 2016, 146(7), 1300–1308 CrossRef CAS.
- Y.-X. Zhou, Y.-Z. Chen, L. Cao, J. Lu and H.-L. Jiang, Conversion of a metal–organic framework to N-doped porous carbon incorporating Co and CoO nanoparticles: direct oxidation of alcohols to esters, Chem. Commun., 2015, 51(39), 8292–8295 RSC.
- C. Bai, X. Yao and Y. Li, Easy Access to Amides through Aldehydic C–H Bond Functionalization Catalyzed by Heterogeneous Co-Based Catalysts, ACS Catal., 2015, 5(2), 884–891 CrossRef CAS.
- K.-Y. Andrew Lin, F.-K. Hsu and W.-D. Lee, Magnetic cobalt-graphene nanocomposite derived from self-assembly of MOFs with graphene oxide as an activator for peroxymonosulfate, J. Mater. Chem. A, 2015, 3(18), 9480–9490 CAS.
- C. Xing, Y. Liu, Y. Su, Y. Chen, S. Hao and X. Wu, et al., Structural Evolution of Co-Based Metal Organic Frameworks in Pyrolysis for Synthesis of Core–Shells on Nanosheets: Co@CoOx@Carbon-rGO Composites for Enhanced Hydrogen Generation Activity, ACS Appl. Mater. Interfaces, 2016, 8(24), 15430–15438 CAS.
- S. Enthaler, D. Addis, K. Junge, G. Erre and M. Beller, A General and Environmentally Benign Catalytic Reduction of Nitriles to Primary Amines, Chem. – Eur. J., 2008, 14(31), 9491–9494 CrossRef CAS PubMed.
- D. Pingen, C. Müller and D. Vogt, Direct Amination of Secondary Alcohols Using Ammonia, Angew. Chem., Int. Ed., 2010, 49(44), 8130–8133 CrossRef CAS PubMed.
- J. Long, Y. Zhou and Y. Li, Transfer hydrogenation of unsaturated bonds in the absence of base additives catalyzed by a cobalt-based heterogeneous catalyst, Chem. Commun., 2015, 51(12), 2331–2334 RSC.
- Z.-R. Dong, Y.-Y. Li, J.-S. Chen, B.-Z. Li, Y. Xing and J.-X. Gao, Highly Efficient Iridium Catalyst for Asymmetric Transfer Hydrogenation of Aromatic Ketones under Base-Free Conditions, Org. Lett., 2005, 7(6), 1043–1045 CrossRef CAS PubMed.
- X. Wang and Y. Li, Chemoselective hydrogenation of functionalized nitroarenes using MOF-derived co-based catalysts, J. Mol. Catal. A: Chem., 2016, 420, 56–65 CrossRef CAS.
- X. Ma, Y.-X. Zhou, H. Liu, Y. Li and H.-L. Jiang, A MOF-derived Co-CoO@N-doped porous carbon for efficient tandem catalysis: dehydrogenation of ammonia borane and hydrogenation of nitro compounds, Chem. Commun., 2016, 52(49), 7719–7722 RSC.
- B. Chen, Z. Yang, Y. Zhu and Y. Xia, Zeolitic imidazolate framework materials: recent progress in synthesis and applications, J. Mater. Chem. A, 2014, 2(40), 16811–16831 CAS.
- M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang and R. Silva, et al., Cu and Cu-Based Nanoparticles: Synthesis and Applications in Catalysis, Chem. Rev., 2016, 116(6), 3722–3811 CrossRef CAS PubMed.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n, Science, 1999, 283(5405), 1148–1150 CrossRef CAS PubMed.
- K.-S. Lin, A. K. Adhikari, C.-N. Ku, C.-L. Chiang and H. Kuo, Synthesis and characterization of porous HKUST-1 metal organic frameworks for hydrogen storage, Int. J. Hydrogen Energy, 2012, 37(18), 13865–13871 CrossRef CAS.
- C. G. Carson, K. Hardcastle, J. Schwartz, X. Liu, C. Hoffmann and R. A. Gerhardt, et al., Synthesis and Structure Characterization of Copper Terephthalate Metal–Organic Frameworks, Eur. J. Inorg. Chem., 2009, 2338–2343 CrossRef CAS.
- H. K. Kim, W. S. Yun, M.-B. Kim, J. Y. Kim, Y.-S. Bae and J. Lee, et al., A Chemical Route to Activation of Open Metal Sites in the Copper-Based Metal–Organic Framework Materials HKUST-1 and Cu-MOF-2, J. Am. Chem. Soc., 2015, 137(31), 10009–10015 CrossRef CAS PubMed.
- C. Xu, Y. Zhang, L. Wang, L. Xu, X. Bian and H. Ma, et al., Nanotubular Mesoporous PdCu Bimetallic Electrocatalysts toward Oxygen Reduction Reaction, Chem. Mater., 2009, 21(14), 3110–3116 CrossRef CAS.
- Y. Luo, L. A. Estudillo-Wong, L. Cavillo, G. Granozzi and N. Alonso-Vante, An easy and cheap chemical route using a MOF precursor to prepare Pd–Cu electrocatalyst for efficient energy conversion cathodes, J. Catal., 2016, 338, 135–142 CrossRef CAS.
- J.-B. Raoof, S. R. Hosseini, R. Ojani and S. Mandegarzad, MOF-derived Cu/nanoporous carbon composite and its application for electro-catalysis of hydrogen evolution reaction, Energy, 2015, 90(Part 1), 1075–1081 CrossRef CAS.
- Z. Xiang, D. Cao, X. Shao, W. Wang, J. Zhang and W. Wu, Facile preparation of high-capacity hydrogen storage metal–organic frameworks: A combination of microwave-assisted solvothermal synthesis and supercritical activation, Chem. Eng. Sci., 2010, 65(10), 3140–3146 CrossRef CAS.
- A. Razeghi, A. Khodadadi, H. Ziaei-Azad and Y. Mortazavi, Activity enhancement of Cu-doped ceria by reductive regeneration of CuO–CeO2 catalyst for preferential oxidation of CO in H2-rich streams, Chem. Eng. J., 2010, 164(1), 214–220 CrossRef CAS.
- J. M. Zamaro, N. C. Pérez, E. E. Miró, C. Casado, B. Seoane and C. Téllez, et al., HKUST-1 MOF: A matrix to synthesize CuO and CuO–CeO2 nanoparticle catalysts for CO oxidation, Chem. Eng. J., 2012, 195–196, 180–187 CrossRef CAS.
- S. Qian, C. Wang, W. Liu, Y. Zhu, W. Yao and X. Lu, An enhanced CdS/TiO2 photocatalyst with high stability and activity: Effect of mesoporous substrate and bifunctional linking molecule, J. Mater. Chem., 2011, 21(13), 4945–4952 RSC.
- H. Liu, S. Zhang, Y. Liu, Z. Yang, X. Feng and X. Lu, et al., Well-Dispersed and Size-Controlled Supported Metal Oxide Nanoparticles Derived from MOF Composites and Further Application in Catalysis, Small, 2015, 11(26), 3130–3134 CrossRef CAS PubMed.
- H. Niu, S. Liu, Y. Cai, F. Wu and X. Zhao, MOF derived porous carbon supported Cu/Cu2O composite as high performance non-noble catalyst, Microporous Mesoporous Mater., 2016, 219, 48–53 CrossRef CAS.
- T. Zeng, X. Zhang, S. Wang, Y. Ma, H. Niu and Y. Cai, A double-shelled yolk-like structure as an ideal magnetic support of tiny gold nanoparticles for nitrophenol reduction, J. Mater. Chem. A, 2013, 1(38), 11641–11647 CAS.
- T. Wu, L. Zhang, J. Gao, Y. Liu, C. Gao and J. Yan, Fabrication of graphene oxide decorated with Au–Ag alloy nanoparticles and its superior catalytic performance for the reduction of 4-nitrophenol, J. Mater. Chem. A, 2013, 1(25), 7384–7390 CAS.
- S. Gao, X. Jia, Z. Li and Y. Chen, Hierarchical plasmonic-metal/semiconductor micro/nanostructures: green synthesis and application in catalytic reduction of p-nitrophenol, J. Nanopart. Res., 2012, 14(3), 1–11 CrossRef.
- B. R. Kim, J. S. Oh, J. Kim and C. Y. Lee, Robust Aerobic Alcohol Oxidation Catalyst Derived from Metal–Organic Frameworks, Catal. Lett., 2016, 146(4), 734–743 CrossRef CAS.
- Y. Yang, Z. Du, Y. Huang, F. Lu, F. Wang and J. Gao, et al., Conversion of furfural into cyclopentanone over Ni-Cu bimetallic catalysts, Green Chem., 2013, 15(7), 1932–1940 RSC.
- Y. Wang, S. Sang, W. Zhu, L. Gao and G. Xiao, CuNi@C catalysts with high activity derived from metal–organic frameworks precursor for conversion of furfural to cyclopentanone, Chem. Eng. J., 2016, 299, 104–111 CrossRef CAS.
- M. Munoz, P. Domínguez, Z. M. de Pedro, J. A. Casas and J. J. Rodriguez, Naturally-occurring iron minerals as inexpensive catalysts for CWPO, Appl. Catal., B, 2017, 203, 166–173 CrossRef CAS.
- K. E. deKrafft, C. Wang and W. Lin, Metal–Organic Framework Templated Synthesis of Fe2O3/TiO2 Nanocomposite for Hydrogen Production, Adv. Mater., 2012, 24(15), 2014–2018 CrossRef CAS PubMed.
- S.-w. Lee, J. Drwiega, C.-Y. Wu, D. Mazyck and W. M. Sigmund, Anatase TiO2 Nanoparticle Coating on Barium Ferrite Using Titanium Bis-Ammonium Lactato Dihydroxide and Its Use as a Magnetic Photocatalyst, Chem. Mater., 2004, 16(6), 1160–1164 CrossRef CAS.
- E. Kim and M. Yoon, Facile synthesis of γ-Fe2O3@porous carbon materials using an Fe-based metal–organic framework: structure and porosity study, J. Porous Mater., 2015, 22(6), 1495–1502 CrossRef CAS.
- A. Banerjee, R. Gokhale, S. Bhatnagar, J. Jog, M. Bhardwaj and B. Lefez, et al., MOF derived porous carbon-Fe3O4 nanocomposite as a high performance, recyclable environmental superadsorbent, J. Mater. Chem., 2012, 22(37), 19694–19699 RSC.
- F.-X. Qin, S.-Y. Jia, Y. Liu, X. Han, H.-T. Ren and W.-W. Zhang, et al., Metal–organic framework as a template for synthesis of magnetic CoFe2O4 nanocomposites for phenol degradation, Mater. Lett., 2013, 101, 93–95 CrossRef CAS.
- J. Li, C.-Y. Liu and Y. Liu, Au/graphene hydrogel: synthesis, characterization and its use for catalytic reduction of 4-nitrophenol, J. Mater. Chem., 2012, 22(17), 8426–8430 RSC.
- Y. Li, Y.-X. Zhou, X. Ma and H.-L. Jiang, A metal–organic framework-templated synthesis of [gamma]-Fe2O3 nanoparticles encapsulated in porous carbon for efficient and chemoselective hydrogenation of nitro compounds, Chem. Commun., 2016, 52, 4199–4202 RSC.
- H. J. Lee, W. Cho, E. Lim and M. Oh, One-pot synthesis of magnetic particle-embedded porous carbon composites from metal–organic frameworks and their sorption properties, Chem. Commun., 2014, 50(41), 5476–5479 RSC.
- A. Morozan, M. T. Sougrati, V. Goellner, D. Jones, L. Stievano and F. Jaouen, Effect of Furfuryl Alcohol on Metal Organic Framework-based Fe/N/C Electrocatalysts for Polymer Electrolyte Membrane Fuel Cells, Electrochim. Acta, 2014, 119, 192–205 CrossRef CAS.
- G. Zhang, R. Chenitz, M. Lefèvre, S. Sun and J.-P. Dodelet, Is iron involved in the lack of stability of Fe/N/C electrocatalysts used to reduce oxygen at the cathode of PEM fuel cells?, Nano Energy, 2016, 29, 111–125 CrossRef CAS.
- L. Yang, N. Larouche, R. Chenitz, G. Zhang, M. Lefèvre and J.-P. Dodelet, Activity, Performance, and Durability for the Reduction of Oxygen in PEM Fuel Cells, of Fe/N/C Electrocatalysts Obtained from the Pyrolysis of Metal–Organic-Framework and Iron Porphyrin Precursors, Electrochim. Acta, 2015, 159, 184–197 CrossRef CAS.
- C. Mao, A. Kong, Y. Wang, X. Bu and P. Feng, MIL-100 derived nitrogen-embodied carbon shells embedded with iron nanoparticles, Nanoscale, 2015, 7(24), 10817–10822 RSC.
- F. Afsahi and S. Kaliaguine, Non-precious electrocatalysts synthesized from metal–organic frameworks, J. Mater. Chem. A, 2014, 2(31), 12270–12279 CAS.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian and J. Herranz, et al., Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- J.-S. Li, S.-L. Li, Y.-J. Tang, M. Han, Z.-H. Dai and J.-C. Bao, et al., Nitrogen-doped Fe/Fe3C@graphitic layer/carbon nanotube hybrids derived from MOFs: efficient bifunctional electrocatalysts for ORR and OER, Chem. Commun., 2015, 51(13), 2710–2713 RSC.
- S. Zhao, H. Yin, L. Du, L. He, K. Zhao and L. Chang, et al., Carbonized Nanoscale Metal–Organic Frameworks as High Performance Electrocatalyst for Oxygen Reduction Reaction, ACS Nano, 2014, 8(12), 12660–12668 CrossRef CAS PubMed.
- V. P. Santos, T. A. Wezendonk, J. J. D. Jaén, A. I. Dugulan, M. A. Nasalevich and H.-U. Islam, et al., Metal organic framework-mediated synthesis of highly active and stable Fischer-Tropsch catalysts, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- T. A. Wezendonk, V. P. Santos, M. A. Nasalevich, Q. S. E. Warringa, A. I. Dugulan and A. Chojecki, et al., Elucidating the Nature of Fe Species during Pyrolysis of the Fe-BTC MOF into Highly Active and Stable Fischer–Tropsch Catalysts, ACS Catal., 2016, 3236–3247 CrossRef CAS.
- B. An, K. Cheng, C. Wang, Y. Wang and W. Lin, Pyrolysis of Metal–Organic Frameworks to Fe3O4@Fe5C2 Core–Shell Nanoparticles for Fischer–Tropsch Synthesis, ACS Catal., 2016, 6(6), 3610–3618 CrossRef CAS.
- T. Wezendonk, Q. S. E. Warringa, V. Da Costa Oliveira Santos Silva, A. Chojecki, M. Ruitenbeek and G. Meima, et al., Structural and elemental influence from various MOFs on the performance of Fe@C catalysts for Fischer-Tropsch synthesis, Faraday Discuss., 2017 10.1039/C6FD00198J.
- W. Peng, J. Li, B. Chen, N. Wang, G. Luo and F. Wei, Mesoporous MgO synthesized by a homogeneous-hydrothermal method and its catalytic performance on gas-phase acetone condensation at low temperatures, Catal. Commun., 2016, 74, 39–42 CrossRef CAS.
- A. Mallick, S. Saha, P. Pachfule, S. Roy and R. Banerjee, Selective CO2 and H2 adsorption in a chiral magnesium-based metal organic framework (Mg-MOF) with open metal sites, J. Mater. Chem., 2010, 20(41), 9073–9080 RSC.
- N. Salehifar, Z. Zarghami and M. Ramezani, A facile, novel and low-temperature synthesis of MgO nanorods via thermal decomposition using new starting reagent and its photocatalytic activity evaluation, Mater. Lett., 2016, 167, 226–229 CrossRef CAS.
- K. Mageshwari, S. S. Mali, R. Sathyamoorthy and P. S. Patil, Template-free synthesis of MgO nanoparticles for effective photocatalytic applications, Powder Technol., 2013, 249, 456–462 CrossRef CAS.
- A. Feinle, A. Heugenhauser and N. Hüsing, Impact of surfactants and acids on the sol–gel synthesis of MgO aerogels, J. Supercrit. Fluids, 2015, 106, 133–139 CrossRef CAS.
- A. Hazra Chowdhury, I. Hazra Chowdhury and M. Kanti Naskar, A facile synthesis of grainy rod-like porous MgO, Mater. Lett., 2015, 158, 190–193 CrossRef CAS.
- A. J. Brown, P. S. Francis, J. L. Adcock, K. F. Lim and N. W. Barnett, Manganese(III) and manganese(IV) as chemiluminescence reagents: A review, Anal. Chim. Acta, 2008, 624(2), 175–183 CrossRef CAS PubMed.
- N. D. Wasalathanthri, T. M. SantaMaria, D. A. Kriz, S. L. Dissanayake, C.-H. Kuo and S. Biswas, et al., Mesoporous manganese oxides for NO2 assisted catalytic soot oxidation, Appl. Catal., B, 2017, 201, 543–551 CrossRef CAS.
- H. Einaga, S. Yamamoto, N. Maeda and Y. Teraoka, Structural analysis of manganese oxides supported on SiO2 for benzene oxidation with ozone, Catal. Today, 2015, 242(Part B), 287–293 CrossRef CAS.
- L. Peng, J. Zhang, Z. Xue, B. Han, J. Li and G. Yang, Large-pore mesoporous Mn3O4 crystals derived from metal–organic frameworks, Chem. Commun., 2013, 49(99), 11695–11697 RSC.
- P. Zhang, Y. Zhan, B. Cai, C. Hao, J. Wang and C. Liu, et al., Shape-controlled synthesis of Mn3O4 nanocrystals and their catalysis of the degradation of methylene blue, Nano Res., 2010, 3(4), 235–243 CrossRef CAS.
- N. M. Al-Mansi and N. M. Abdel Monem, Recovery of nickel oxide from spent catalyst, Waste Manage., 2002, 22(1), 85–90 CrossRef CAS PubMed.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Recent advances in homogeneous nickel catalysis, Nature, 2014, 509(7500), 299–309 CrossRef CAS PubMed.
- Z. Zhang, L. Han, R. Chai, Q. Zhang, Y. Li and G. Zhao, et al., Microstructured CeO2-NiO-Al2O3/Ni-foam catalyst for oxidative dehydrogenation of ethane to ethylene, Catal. Commun., 2017, 88, 90–93 CrossRef CAS.
- R. K. Singha, A. Shukla, A. Yadav, L. N. Sivakumar Konathala and R. Bal, Effect of metal-support interaction on activity and stability of Ni-CeO2 catalyst for partial oxidation of methane, Appl. Catal., B, 2017, 202, 473–488 CrossRef CAS.
- X. Li, D. Li, H. Tian, L. Zeng, Z.-J. Zhao and J. Gong, Dry reforming of methane over Ni/La2O3 nanorod catalysts with stabilized Ni nanoparticles, Appl. Catal., B, 2017, 202, 683–694 CrossRef CAS.
- S. Rakass, H. Oudghiri-Hassani, P. Rowntree and N. Abatzoglou, Steam reforming of methane over unsupported nickel catalysts, J. Power Sources, 2006, 158(1), 485–496 CrossRef CAS.
- J. Sehested, S. Dahl, J. Jacobsen and J. R. Rostrup-Nielsen, Methanation of CO over Nickel: Mechanism and Kinetics at High H2/CO Ratios, J. Phys. Chem. B, 2005, 109(6), 2432–2438 CrossRef CAS PubMed.
- G. Pina, C. Louis and M. A. Keane, Nickel particle size effects in catalytic hydrogenation and hydrodechlorination: phenolic transformations over nickel/silica, Phys. Chem. Chem. Phys., 2003, 5(9), 1924–1931 RSC.
- A. Wolf, T. Turek and L. Mleczko, Structured Raney® Nickel Catalysts for Liquid-Phase Hydrogenation, Chem. Eng. Technol., 2016, 39(10), 1933–1938 CrossRef CAS.
- F. Devred, A. H. Gieske, N. Adkins, U. Dahlborg, C. M. Bao and M. Calvo-Dahlborg, et al., Influence of phase composition and particle size of atomised Ni–Al alloy samples on the catalytic performance of Raney®-type nickel catalysts, Appl. Catal., A, 2009, 356(2), 154–161 CrossRef CAS.
- Y. Tamaru, Introductory Guide to Organonickel Chemistry, Modern Organonickel Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 1–40 Search PubMed.
- M. J. Vaidya, S. M. Kulkarni and R. V. Chaudhari, Synthesis of p-Aminophenol by Catalytic Hydrogenation of p-Nitrophenol, Org. Process Res. Dev., 2003, 7(2), 202–208 CrossRef CAS.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Muller and R. Schlogl, et al., Graphitic carbon nitride materials: variation of structure and morphology and their use as metal-free catalysts, J. Mater. Chem., 2008, 18(41), 4893–4908 RSC.
- Z. Dong, C. Dong, Y. Liu, X. Le, Z. Jin and J. Ma, Hydrodechlorination and further hydrogenation of 4-chlorophenol to cyclohexanone in water over Pd nanoparticles modified N-doped mesoporous carbon microspheres, Chem. Eng. J., 2015, 270, 215–222 CrossRef CAS.
- W. Zuo, G. Yu and Z. Dong, A MOF-derived nickel based N-doped mesoporous carbon catalyst with high catalytic activity for the reduction of nitroarenes, RSC Adv., 2016, 6(14), 11749–11753 RSC.
- S. Tang, S. Vongehr and X. Meng, Carbon Spheres with Controllable Silver Nanoparticle Doping, J. Phys. Chem. C, 2010, 114(2), 977–982 CAS.
- Y. Chi, Q. Yuan, Y. Li, J. Tu, L. Zhao and N. Li, et al., Synthesis of Fe3O4@SiO2–Ag magnetic nanocomposite based on small-sized and highly dispersed silver nanoparticles for catalytic reduction of 4-nitrophenol, J. Colloid Interface Sci., 2012, 383(1), 96–102 CrossRef CAS PubMed.
- N. Sahiner, H. Ozay, O. Ozay and N. Aktas, New catalytic route: Hydrogels as templates and reactors for in situ Ni nanoparticle synthesis and usage in the reduction of 2- and 4-nitrophenols, Appl. Catal., A, 2010, 385(1–2), 201–207 CrossRef CAS.
- N. Sahiner, H. Ozay, O. Ozay and N. Aktas, A soft hydrogel reactor for cobalt nanoparticle preparation and use in the reduction of nitrophenols, Appl. Catal., B, 2010, 101(1–2), 137–143 CrossRef CAS.
- H. Topsøe, B. S. Clausen and F. E. Massoth, Hydrotreating Catalysis, in Catal.: Sci. Technol., ed. J. R. Anderson and M. Boudart, Berlin, Heidelberg, Springer Berlin Heidelberg, 1996, pp. 1–269 Search PubMed.
- P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellvag, Hydrogen adsorption in a nickel based coordination polymer with open metal sites in the cylindrical cavities of the desolvated framework, Chem. Commun., 2006, 959–961 RSC.
- C. Larabi, P. K. Nielsen, S. Helveg, C. Thieuleux, F. B. Johansson and M. Brorson, et al., Bulk Hydrodesulfurization Catalyst Obtained by Mo(CO)6 Grafting on the Metal–Organic Framework Ni2(2,5-dihydroxoterephthalate), ACS Catal., 2012, 2(4), 695–700 CrossRef CAS.
- J. A. Rodríguez-Manzo, C. Pham-Huu and F. Banhart, Graphene Growth by a Metal-Catalyzed Solid-State Transformation of Amorphous Carbon, ACS Nano, 2011, 5(2), 1529–1534 CrossRef PubMed.
- K. Takeishi and K.-I. Aika, Study of Raney® ruthenium catalyst for methanol synthesis, J. Catal., 1992, 136(1), 252–257 CrossRef CAS.
- S. Dérien, F. Monnier and P. H. Dixneuf, Ruthenium-Catalyzed C–C Bond Formation, in Ruthenium Catalysts and Fine Chemistry: -/-, ed. C. Bruneau and P. H. Dixneuf, Berlin, Heidelberg, Springer Berlin Heidelberg, 2004, pp. 1–44 Search PubMed.
- S. Miao, Z. Liu, B. Han, J. Huang, Z. Sun and J. Zhang, et al., Ru Nanoparticles Immobilized on Montmorillonite by Ionic Liquids: A Highly Efficient Heterogeneous Catalyst for the Hydrogenation of Benzene, Angew. Chem., Int. Ed., 2006, 45(2), 266–269 CrossRef CAS PubMed.
- Y. Ma, Y. Huang, Y. Cheng, L. Wang and X. Li, Biosynthesized ruthenium nanoparticles supported on carbon nanotubes as efficient catalysts for hydrogenation of benzene to cyclohexane: An eco-friendly and economical bioreduction method, Appl. Catal., A, 2014, 484, 154–160 CrossRef CAS.
- T. Kamegawa, N. Suzuki and H. Yamashita, Design of macroporous TiO2 thin film photocatalysts with enhanced photofunctional properties, Energy Environ. Sci., 2011, 4(4), 1411–1416 CAS.
- K. Khaletskaya, A. Pougin, R. Medishetty, C. Rösler, C. Wiktor and J. Strunk, et al., Fabrication of Gold/Titania Photocatalyst for CO2 Reduction Based on Pyrolytic Conversion of the Metal–Organic Framework NH2-MIL-125(Ti) Loaded with Gold Nanoparticles, Chem. Mater., 2015, 27(21), 7248–7257 CrossRef CAS.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin and C. Sanchez, et al., A New Photoactive Crystalline Highly Porous Titanium(IV) Dicarboxylate, J. Am. Chem. Soc., 2009, 131(31), 10857–10859 CrossRef CAS PubMed.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Photocatalytic Reduction of CO2 on TiO2 and Other Semiconductors, Angew. Chem., Int. Ed., 2013, 52(29), 7372–7408 CrossRef CAS PubMed.
- K. Li, X. An, K. H. Park, M. Khraisheh and J. Tang, A critical review of CO2 photoconversion: Catalysts and reactors, Catal. Today, 2014, 224, 3–12 CrossRef CAS.
- B. Mei, A. Pougin and J. Strunk, Influence of photodeposited gold nanoparticles on the photocatalytic activity of titanate species in the reduction of CO2 to hydrocarbons, J. Catal., 2013, 306, 184–189 CrossRef CAS.
- A. Chica, A. Corma and M. E. Dómine, Catalytic oxidative desulfurization (ODS) of diesel fuel on a continuous fixed-bed reactor, J. Catal., 2006, 242(2), 299–308 CrossRef CAS.
- N. D. McNamara, J. Kim and J. C. Hicks, Controlling the Pyrolysis Conditions of Microporous/Mesoporous MIL-125 To Synthesize Porous, Carbon-Supported Ti Catalysts with Targeted Ti Phases for the Oxidation of Dibenzothiophene, Energy Fuels, 2016, 30(1), 594–602 CrossRef CAS.
- J. Kim, G. T. Neumann, N. D. McNamara and J. C. Hicks, Exceptional control of carbon-supported transition metal nanoparticles using metal–organic frameworks, J. Mater. Chem. A, 2014, 2(34), 14014–14027 CAS.
- S.-N. Kim, J. Kim, H.-Y. Kim, H.-Y. Cho and W.-S. Ahn, Adsorption/catalytic properties of MIL-125 and NH2-MIL-125, Catal. Today, 2013, 204, 85–93 CrossRef CAS.
- D. Rehder, Vanadium. Its Role for Humans, in Interrelations between Essential Metal Ions and Human Diseases, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Dordrecht, Springer Netherlands, 2013, pp. 139–169 Search PubMed.
- E. Hryha, E. Rutqvist and L. Nyborg, Stoichiometric vanadium oxides studied by XPS, Surf. Interface Anal., 2012, 44(8), 1022–1025 CrossRef CAS.
- W. Xing, F. Meng and R. Yu, A new type of vanadium carbide V5C3 and its hardening by tuning Fermi energy, Sci. Rep., 2016, 6, 21794 CrossRef CAS PubMed.
- G. Fang, W. Wu, C. Liu, D. D. Dionysiou, Y. Deng and D. Zhou, Activation of persulfate with vanadium species for PCBs degradation: A mechanistic study, Appl. Catal., B, 2017, 202, 1–11 CrossRef CAS.
- I. E. Wachs, Catalysis science of supported vanadium oxide catalysts, Dalton Trans., 2013, 42(33), 11762–11769 RSC.
- F. Meunier, P. Delporte, B. Heinrich, C. Bouchy, C. Crouzet and C. Pham-Huu, et al., Synthesis and Characterization of High Specific Surface Area Vanadium Carbide; Application to Catalytic Oxidation, J. Catal., 1997, 169(1), 33–44 CrossRef CAS.
- J. Ma, M. Wu, Y. Du, S. Chen, J. Ye and L. Jin, Low temperature synthesis of vanadium carbide (VC), Mater. Lett., 2009, 63(11), 905–907 CrossRef CAS.
- I. J. Kang, N. A. Khan, E. Haque and S. H. Jhung, Chemical and Thermal Stability of Isotypic Metal–Organic Frameworks: Effect of Metal Ions, Chem. – Eur. J., 2011, 17(23), 6437–6442 CrossRef CAS PubMed.
- J. Kim, N. D. McNamara and J. C. Hicks, Catalytic activity and stability of carbon supported V oxides and carbides synthesized via pyrolysis of MIL-47 (V), Appl. Catal., A, 2016, 517, 141–150 CrossRef CAS.
- F. Liu, Y. Yao, H. Zhang, Y. Kang, G. Yin and Z. Huang, et al., Synthesis and characterization of vanadium carbide nanoparticles by thermal refluxing-derived precursors, J. Mater. Sci., 2011, 46(11), 3693–3697 CrossRef CAS.
- X.-F. Wu, Non-Redox-Metal-Catalyzed Redox Reactions: Zinc Catalysts, Chem. – Asian J., 2012, 7(11), 2502–2509 CrossRef CAS PubMed.
- G. Parravano and M. Boudart, Chemisorption and Catalysis on Oxide Semiconductors, in Adv. Catal., ed. W. G. Frankenburg, V. I. Komarewsky and E. K. Rideal, Academic Press, 1955, vol. 7, pp. 47–74 Search PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Design and synthesis of an exceptionally stable and highly porous metal–organic framework, Nature, 1999, 402(6759), 276–279 CrossRef CAS.
- B. Panella, M. Hirscher, H. Pütter and U. Müller, Hydrogen Adsorption in Metal–Organic Frameworks: Cu-MOFs and Zn-MOFs Compared, Adv. Funct. Mater., 2006, 16(4), 520–524 CrossRef CAS.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, Impact of Preparation and Handling on the Hydrogen Storage Properties of Zn4O(1,4-benzenedicarboxylate)3 (MOF-5), J. Am. Chem. Soc., 2007, 129(46), 14176–14177 CrossRef CAS PubMed.
- J. Li, S. Cheng, Q. Zhao, P. Long and J. Dong, Synthesis and hydrogen-storage behavior of metal–organic framework MOF-5, Int. J. Hydrogen Energy, 2009, 34(3), 1377–1382 CrossRef CAS.
- D. Saha, Z. Bao, F. Jia and S. Deng, Adsorption of CO2, CH4, N2O, and N2 on MOF-5, MOF-177, and Zeolite 5A, Environ. Sci. Technol., 2010, 44(5), 1820–1826 CrossRef CAS PubMed.
- Z. Zhao, X. Ma, A. Kasik, Z. Li and Y. S. Lin, Gas Separation Properties of Metal Organic Framework (MOF-5) Membranes, Ind. Eng. Chem. Res., 2013, 52(3), 1102–1108 CrossRef CAS.
- N. T. S. Phan, K. K. A. Le and T. D. Phan, MOF-5 as an efficient heterogeneous catalyst for Friedel–Crafts alkylation reactions, Appl. Catal., A, 2010, 382(2), 246–253 CrossRef CAS.
- W. Zhen, B. Li, G. Lu and J. Ma, Enhancing catalytic activity and stability for CO2 methanation on Ni@MOF-5 via control of active species dispersion, Chem. Commun., 2015, 51(9), 1728–1731 RSC.
- N. A. Rodriguez, R. Parra and M. A. Grela, Structural characterization, optical properties and photocatalytic activity of MOF-5 and its hydrolysis products: implications on their excitation mechanism, RSC Adv., 2015, 5(89), 73112–73118 RSC.
- Y.-R. Lee, M.-S. Jang, H.-Y. Cho, H.-J. Kwon, S. Kim and W.-S. Ahn, ZIF-8: A comparison of synthesis methods, Chem. Eng. J., 2015, 271, 276–280 CrossRef CAS.
- L. H. Wee, N. Janssens, S. P. Sree, C. Wiktor, E. Gobechiya and R. A. Fischer, et al., Local transformation of ZIF-8 powders and coatings into ZnO nanorods for photocatalytic application, Nanoscale, 2014, 6(4), 2056–2060 RSC.
- S. R. Venna and M. A. Carreon, Highly Permeable Zeolite Imidazolate Framework-8 Membranes for CO2/CH4 Separation, J. Am. Chem. Soc., 2010, 132(1), 76–78 CrossRef CAS PubMed.
- G. Lu, O. K. Farha, W. Zhang, F. Huo and J. T. Hupp, Engineering ZIF-8 Thin Films for Hybrid MOF-Based Devices, Adv. Mater., 2012, 24(29), 3970–3974 CrossRef CAS PubMed.
- C. Chizallet, S. Lazare, D. Bazer-Bachi, F. Bonnier, V. Lecocq and E. Soyer, et al., Catalysis of Transesterification by a Nonfunctionalized Metal–Organic Framework: Acido-Basicity at the External Surface of ZIF-8 Probed by FTIR and ab Initio Calculations, J. Am. Chem. Soc., 2010, 132(35), 12365–12377 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Reticular synthesis and the design of new materials, Nature, 2003, 423(6941), 705–714 CrossRef CAS PubMed.
- T. Lee, H. Kim, W. Cho, D.-Y. Han, M. Ridwan and C. W. Yoon, et al., Thermosensitive Structural Changes and Adsorption Properties of Zeolitic Imidazolate Framework-8 (ZIF-8), J. Phys. Chem. C, 2015, 119(15), 8226–8237 CAS.
- Y. Kimitsuka, E. Hosono, S. Ueno, H. Zhou and S. Fujihara, Fabrication of Porous Cubic Architecture of ZnO Using Zn-terephthalate MOFs with Characteristic Microstructures, Inorg. Chem., 2013, 52(24), 14028–14033 CrossRef CAS PubMed.
- R. Mohan, K. Krishnamoorthy and S.-J. Kim, Enhanced photocatalytic activity of Cu-doped ZnO nanorods, Solid State Commun., 2012, 152(5), 375–380 CrossRef CAS.
- D. Lin, H. Wu, R. Zhang and W. Pan, Enhanced Photocatalysis of Electrospun Ag–ZnO Heterostructured Nanofibers, Chem. Mater., 2009, 21(15), 3479–3484 CrossRef CAS.
- S. J. Yang, J. H. Im, T. Kim, K. Lee and C. R. Park, MOF-derived ZnO and ZnO@C composites with high photocatalytic activity and adsorption capacity, J. Hazard. Mater., 2011, 186(1), 376–382 CrossRef CAS PubMed.
- L. Pan, T. Muhammad, L. Ma, Z.-F. Huang, S. Wang and L. Wang, et al., MOF-derived C-doped ZnO prepared via a two-step calcination for efficient photocatalysis, Appl. Catal., B, 2016, 189, 181–191 CrossRef CAS.
- Z.-Y. Yuan and B.-L. Su, Insights into hierarchically meso-macroporous structured materials, J. Mater. Chem., 2006, 16(7), 663–677 RSC.
- A. Fukuoka, J.-I. Kimura, T. Oshio, Y. Sakamoto and M. Ichikawa, Preferential Oxidation of Carbon Monoxide Catalyzed by Platinum Nanoparticles in Mesoporous Silica, J. Am. Chem. Soc., 2007, 129(33), 10120–10125 CrossRef CAS PubMed.
- M. Haruta, Catalysis: Gold rush, Nature, 2005, 437(7062), 1098–1099 CrossRef CAS PubMed.
- O. Pozdnyakova, D. Teschner, A. Wootsch, J. Kröhnert, B. Steinhauer and H. Sauer, et al., Preferential CO oxidation in hydrogen (PROX) on ceria-supported catalysts, part I: Oxidation state and surface species on Pt/CeO2 under reaction conditions, J. Catal., 2006, 237(1), 1–16 CrossRef CAS.
- B. Liu, S. Han, K. Tanaka, H. Shioyama and Q. Xu, Metal–Organic Framework (MOF) as a Precursor for Synthesis of Platinum Supporting Zinc Oxide Nanoparticles, Bull. Chem. Soc. Jpn., 2009, 82(8), 1052–1054 CrossRef CAS.
- D. Banham, F. Feng, K. Pei, S. Ye and V. Birss, Effect of carbon support nanostructure on the oxygen reduction activity of Pt/C catalysts, J. Mater. Chem. A, 2013, 1(8), 2812–2820 CAS.
- I. A. Khan, Y. Qian, A. Badshah, M. A. Nadeem and D. Zhao, Highly Porous Carbon Derived from MOF-5 as a Support of ORR Electrocatalysts for Fuel Cells, ACS Appl. Mater. Interfaces, 2016, 8(27), 17268–17275 CAS.
- W. Chen, J. Zhao, J. Y. Lee and Z. Liu, Microwave heated polyol synthesis of carbon nanotubes supported Pt nanoparticles for methanol electrooxidation, Mater. Chem. Phys., 2005, 91(1), 124–129 CrossRef CAS.
- I. A. Khan, A. Badshah, N. Haider, S. Ullah, D. H. Anjum and M. A. Nadeem, Porous carbon as electrode material in direct ethanol fuel cells (DEFCs) synthesized by the direct carbonization of MOF-5, J. Solid State Electrochem., 2014, 18(6), 1545–1555 CrossRef CAS.
- H. A. Gasteiger and N. M. Marković, Just a Dream—or Future Reality?, Science, 2009, 324(5923), 48–49 CrossRef CAS PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More and C. Yu, et al., Lattice-strain control of the activity in dealloyed core–shell fuel cell catalysts, Nat. Chem., 2010, 2(6), 454–460 CrossRef CAS PubMed.
- X. Liu and L. Dai, Carbon-based metal-free catalysts, Nat. Rev. Mater., 2016, 16064 CrossRef CAS.
- A. Aijaz, N. Fujiwara and Q. Xu, From Metal–Organic Framework to Nitrogen-Decorated Nanoporous Carbons: High CO2 Uptake and Efficient Catalytic Oxygen Reduction, J. Am. Chem. Soc., 2014, 136(19), 6790–6793 CrossRef CAS PubMed.
- W. Zhang, Z.-Y. Wu, H.-L. Jiang and S.-H. Yu, Nanowire-Directed Templating Synthesis of Metal–Organic Framework Nanofibers and Their Derived Porous Doped Carbon Nanofibers for Enhanced Electrocatalysis, J. Am. Chem. Soc., 2014, 136(41), 14385–14388 CrossRef CAS PubMed.
- C. Feng, Y. Wang, S. Gao, N. Shang and C. Wang, Hydrogen generation at ambient conditions: AgPd bimetal supported on metal–organic framework derived porous carbon as an efficient synergistic catalyst, Catal. Commun., 2016, 78, 17–21 CrossRef CAS.
- K. Tanabe, Surface and catalytic properties of ZrO2, Mater. Chem. Phys., 1985, 13(3), 347–364 CrossRef CAS.
- A. R. Puigdollers, F. Illas and G. Pacchioni, Structure and Properties of Zirconia Nanoparticles from Density Functional Theory Calculations, J. Phys. Chem. C, 2016, 120(8), 4392–4402 CAS.
- T. M. Miller and V. H. Grassian, Environmental Catalysis: Adsorption and Decomposition of Nitrous Oxide on Zirconia, J. Am. Chem. Soc., 1995, 117, 10969–10975 CrossRef CAS.
- T. Yokoyama, T. Setoyama, N. Fujita, M. Nakajima, T. Maki and K. Fujii, Novel direct hydrogenation process of aromatic carboxylic acids to the corresponding aldehydes with zirconia catalyst, Appl. Catal., A, 1992, 88(2), 149–161 CrossRef CAS.
- I. Ferino, M. F. Casula, A. Corrias, M. G. Cutrufello, R. Monaci and G. Paschina, 4-Methylpentan-2-ol dehydration over zirconia catalysts prepared by sol–gel, Phys. Chem. Chem. Phys., 2000, 2(8), 1847–1854 RSC.
- Y. Nakano, T. Iizuka, H. Hattori and K. Tanabe, Surface properties of zirconium oxide and its catalytic activity for isomerization of 1-butene, J. Catal., 1979, 57(1), 1–10 CrossRef CAS.
- M.-Y. He and J. G. Ekerdt, Methanol formation on zirconium dioxide, J. Catal., 1984, 90(1), 17–23 CrossRef CAS.
- A. E. Platero-Prats, A. Mavrandonakis, L. C. Gallington, Y. Liu, J. T. Hupp and O. K. Farha, et al., Structural Transitions of the Metal-Oxide Nodes within Metal–Organic Frameworks: On the Local Structures of NU-1000 and UiO-66, J. Am. Chem. Soc., 2016, 138(12), 4178–4185 CrossRef CAS PubMed.
- D. Yang, S. O. Odoh, T. C. Wang, O. K. Farha, J. T. Hupp and C. J. Cramer, et al., Metal–Organic Framework Nodes as Nearly Ideal Supports for Molecular Catalysts: NU-1000- and UiO-66-Supported Iridium Complexes, J. Am. Chem. Soc., 2015, 137(23), 7391–7396 CrossRef CAS PubMed.
- J. E. Mondloch, M. J. Katz, W. C. Isley Iii, P. Ghosh, P. Liao and W. Bury, et al., Destruction of chemical warfare agents using metal–organic frameworks, Nat. Mater., 2015, 14(5), 512–516 CrossRef CAS PubMed.
- W. Zhou, E. I. Ross-Medgaarden, W. V. Knowles, M. S. Wong, I. E. Wachs and C. J. Kiely, Identification of active Zr–WOx clusters on a ZrO2 support for solid acid catalysts, Nat. Chem., 2009, 1(9), 722–728 CrossRef CAS PubMed.
- G. Larsen, E. Lotero, L. M. Petkovic and D. S. Shobe, Alcohol Dehydration Reactions over Tungstated Zirconia Catalysts, J. Catal., 1997, 169(1), 67–75 CrossRef CAS.
- F. Di Gregorio and V. Keller, Activation and isomerization of hydrocarbons over WO3/ZrO2 catalysts: I. Preparation, characterization, and X-ray photoelectron spectroscopy studies, J. Catal., 2004, 225(1), 45–55 CrossRef CAS.
- P. Wang, J. Feng, Y. Zhao, S. Wang and J. Liu, MOF-Derived Tungstated Zirconia as Strong Solid Acids toward High Catalytic Performance for Acetalization, ACS Appl. Mater. Interfaces, 2016, 8(36), 23755–23762 CAS.
- P. Zhang, F. Sun, Z. Xiang, Z. Shen, J. Yun and D. Cao, ZIF-derived in situ nitrogen-doped porous carbons as efficient metal-free electrocatalysts for oxygen reduction reaction, Energy Environ. Sci., 2014, 7(1), 442–450 CAS.
- Z.-W. Liu, F. Peng, H.-J. Wang, H. Yu, W.-X. Zheng and J. Yang, Phosphorus-Doped Graphite Layers with High Electrocatalytic Activity for the O2 Reduction in an Alkaline Medium, Angew. Chem., Int. Ed., 2011, 50(14), 3257–3261 CrossRef PubMed.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Zr-based metal–organic frameworks: design, synthesis, structure, and applications, Chem. Soc. Rev., 2016, 45(8), 2327–2367 RSC.
- Y. Fu, Y. Huang, Z. Xiang, G. Liu and D. Cao, Phosphorous–Nitrogen-Codoped Carbon Materials Derived from Metal–Organic Frameworks as Efficient Electrocatalysts for Oxygen Reduction Reactions, Eur. J. Inorg. Chem., 2016, 2100–2105 CrossRef CAS.
- W. Yu, M. D. Porosoff and J. G. Chen, Review of Pt-Based Bimetallic Catalysis: From Model Surfaces to Supported Catalysts, Chem. Rev., 2012, 112(11), 5780–5817 CrossRef CAS PubMed.
- Y. Yang, Y. Zhang, C. J. Sun, X. Li, W. Zhang and X. Ma, et al., Heterobimetallic Metal–Organic Framework as a Precursor to Prepare a Nickel/Nanoporous Carbon Composite Catalyst for 4-Nitrophenol Reduction, ChemCatChem, 2014, 6(11), 3084–3090 CrossRef CAS.
- A. D. Burrows, Mixed-component metal–organic frameworks (MC-MOFs), enhancing functionality through solid solution formation and surface modifications, CrystEngComm, 2011, 13(11), 3623–3642 RSC.
- B. You, N. Jiang, M. Sheng, W. S. Drisdell, J. Yano and Y. Sun, Bimetal–Organic Framework Self-Adjusted Synthesis of Support-Free Nonprecious Electrocatalysts for Efficient Oxygen Reduction, ACS Catal., 2015, 5(12), 7068–7076 CrossRef CAS.
- X. Wang, X. Fan, H. Lin, H. Fu, T. Wang and J. Zheng, et al., An efficient Co–N–C oxygen reduction catalyst with highly dispersed Co sites derived from a ZnCo bimetallic zeolitic imidazolate framework, RSC Adv., 2016, 6(44), 37965–37973 RSC.
- W. Zhou, J. Zhou, Y. Zhou, J. Lu, K. Zhou and L. Yang, et al., N-Doped Carbon-Wrapped Cobalt Nanoparticles on N-Doped Graphene Nanosheets for High-Efficiency Hydrogen Production, Chem. Mater., 2015, 27(6), 2026–2032 CrossRef CAS.
- J. Lu, W. Zhou, L. Wang, J. Jia, Y. Ke and L. Yang, et al., Core–Shell Nanocomposites Based on Gold Nanoparticle@Zinc–Iron-Embedded Porous Carbons Derived from Metal–Organic Frameworks as Efficient Dual Catalysts for Oxygen Reduction and Hydrogen Evolution Reactions, ACS Catal., 2016, 6(2), 1045–1053 CrossRef CAS.
- L. He, Y. Liu, J. Liu, Y. Xiong, J. Zheng and Y. Liu, et al., Core–Shell Noble-Metal@Metal–Organic-Framework Nanoparticles with Highly Selective Sensing Property, Angew. Chem., Int. Ed., 2013, 52(13), 3741–3745 CrossRef CAS PubMed.
- Q. Xu, Z. Guo, M. Zhang, Z. Hu, Y. Qian and D. Zhao, Highly efficient photocatalysts by pyrolyzing a Zn-Ti heterometallic metal–organic framework, CrystEngComm, 2016, 18(22), 4046–4052 RSC.
- Z. Guo, J. K. Cheng, Z. Hu, M. Zhang, Q. Xu and Z. Kang, et al., Metal–organic frameworks (MOFs) as precursors towards TiOx/C composites for photodegradation of organic dye, RSC Adv., 2014, 4(65), 34221–34225 RSC.
- K. Hong, W. Bak and H. Chun, Unique Coordination-Based Heterometallic Approach for the Stoichiometric Inclusion of High-Valent Metal Ions in a Porous Metal–Organic Framework, Inorg. Chem., 2013, 52(10), 5645–5647 CrossRef CAS PubMed.
- K.-L. Wu, X.-W. Wei, X.-M. Zhou, D.-H. Wu, X.-W. Liu and Y. Ye, et al., NiCo2 Alloys: Controllable Synthesis, Magnetic Properties, and Catalytic Applications in Reduction of 4-Nitrophenol, J. Phys. Chem. C, 2011, 115(33), 16268–16274 CAS.
- Z. Zhu, X. Guo, S. Wu, R. Zhang, J. Wang and L. Li, Preparation of Nickel Nanoparticles in Spherical Polyelectrolyte Brush Nanoreactor and Their Catalytic Activity, Ind. Eng. Chem. Res., 2011, 50(24), 13848–13853 CrossRef CAS.
- Y. Yang, Y. Ren, C. Sun and S. Hao, Facile route fabrication of nickel based mesoporous carbons with high catalytic performance towards 4-nitrophenol reduction, Green Chem., 2014, 16(4), 2273–2280 RSC.
- L. Zheng, X. Li, W. Du, D. Shi, W. Ning and X. Lu, et al., Metal–organic framework derived Cu/ZnO catalysts for continuous hydrogenolysis of glycerol, Appl. Catal., B, 2017, 203, 146–153 CrossRef CAS.
- A. Kong, Q. Lin, C. Mao, X. Bu and P. Feng, Efficient oxygen reduction by nanocomposites of heterometallic carbide and nitrogen-enriched carbon derived from the cobalt-encapsulated indium-MOF, Chem. Commun., 2014, 50(98), 15619–15622 RSC.
- J. Long, K. Shen, L. Chen and Y. Li, Multimetal-MOF-derived transition metal alloy NPs embedded in an N-doped carbon matrix: highly active catalysts for hydrogenation reactions, J. Mater. Chem. A, 2016, 4(26), 10254–10262 CAS.
- W. Weisweiler, N. Subramanian and B. Terwiesch, Catalytic influence of metal melts on the graphitization of monolithic glasslike carbon, Carbon, 1971, 9(6), 755–761 CrossRef CAS.
| This journal is © the Partner Organisations 2017 |