
Clinical application of polymeric micelles for the treatment of cancer
Aida
Varela-Moreira
ab,
Yang
Shi
 c,
Marcel H. A. M.
Fens
a,
Twan
Lammers
bcd,
Wim E.
Hennink
b and
Raymond M.
Schiffelers
*a
c,
Marcel H. A. M.
Fens
a,
Twan
Lammers
bcd,
Wim E.
Hennink
b and
Raymond M.
Schiffelers
*a
aDepartment of Clinical Chemistry and Haematology, University Medical Centre Utrecht, Utrecht, 3584 CX, The Netherlands. E-mail: R.Schiffelers@umcutrecht.nl
bDepartment of Pharmaceutics, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Utrecht, 3584 CG, The Netherlands
cDepartment of Nanomedicine and Theranostics, Institute for Experimental Molecular Imaging, RWTH Aachen University Clinic, 52074 Aachen, Germany
dDepartment of Targeted Therapeutics, MIRA Institute for Biomedical Technology and Technical Medicine, University of Twente, Enschede, 7522 NB, The Netherlands
First published on 13th February 2017
Abstract
The in vivo administration of chemotherapeutic drugs is a challenge due to their poor pharmacokinetic (PK) and biodistribution profiles. For this reason, the development of delivery systems capable of targeting these compounds to pathological sites is of great importance. Polymeric micelles (PMs) are good systems for the encapsulation of hydrophobic compounds because their hydrophobic core can accommodate these types of drugs whereas their hydrophilic corona, usually poly(ethylene glycol), enables PMs to circulate for an extended period of time in the bloodstream which allows them to reach tumour tissues by means of the enhanced permeability and retention (EPR) effect. The first generation of PMs was rather unstable and essentially used to solubilize hydrophobic drugs for intravenous (i.v.) administration. More recently, the next-generation of PMs has been developed to achieve high encapsulation and retention of drugs while maintaining prolonged circulation after i.v. administration. These systems are suitable for both passive and active drug targeting. Different approaches have been employed to achieve the abovementioned goals: both non-covalent (hydrophobic and π–π interactions) and chemical (covalent binding of the drug to the polymer backbone and/or crosslinking of the core/shell) strategies have been used to improve the stability in the circulation and to retain the loaded drug in the PM. This will result in the accumulation of the drug at the target site to a greater extent than in healthy tissues and will, in principle, lead to improved therapeutic outcome. Several PM-based formulations are currently being evaluated in clinical trials. In this review, the pre-clinical and clinical outcomes of these PMs are summarized along with the strategies to translate PMs to patients.
 Aida Varela-Moreira | Aida Varela-Moreira received her BSc (2011) from the Faculty of Sciences and MSc (2014) from the Faculty of Medicine, both at Porto University. In 2015 she started her PhD at University Medical Centre Utrecht and at Biopharmacy department at Utrecht University under the supervision of Professor Raymond M. Schiffelers and Professor Wim E. Hennink. Her research focuses on the development of polymeric micelles loaded with hydrophobic compounds for the treatment of cancer. |
 Yang Shi | Yang Shi completed his PhD research in 2014 in Prof. Wim E. Hennink's group (Utrecht University, the Netherlands), focusing on chemical synthesis and in vivo studies of polymeric micelles for tumor-targeted drug delivery and imaging. He started working as an Associate Professor at South China University of Technology in 2015. In 2016, he was appointed as the Group Leader “Polymeric Nanomedicines” at The Institute for Experimental Molecular Imaging (ExMI) at RWTH Aachen University Clinic and at the Helmholtz Institute for Biomedical Engineering. His research focuses on polymeric nanomedicines for cancer theranostics and microbubbles for ultrasound-mediated drug delivery. |
 Marcel H. A. M. Fens | Marcel Fens obtained his PhD in the Department of Biopharmacy of Utrecht University in 2009. He moved to the University Medical Center Utrecht to work as a post-doc. From summer 2010 he was a visiting scientist for 3.5 years at Children's Hospital Oakland Research Institute in California and currently he works at the University Medical Center in Utrecht, focusing on nanomedicine and drug delivery. |
 Twan Lammers | Twan Lammers obtained his DSc degree in Radiation Oncology from Heidelberg University in 2008 and PhD degree in Pharmaceutics from Utrecht University in 2009. In the same year, he started the Nanomedicine and Theranostics group at the Institute for Experimental Molecular Imaging RWTH Aachen University. In 2014, he was promoted to full professor at RWTH Aachen. Since 2012, he has also worked as a part-time assistant professor at the Department of Targeted Therapeutics at the University of Twente. His primary research interests include drug targeting to tumors, image-guided drug delivery and tumor-targeted combination therapies. |
 Wim E. Hennink | Wim Hennink obtained his PhD degree in 1985 at the Twente University of Technology (The Netherlands) with a thesis on biomaterials research. From 1985 to 1992 he had different positions in industry. In 1992, he was appointed as a Professor at the Faculty of Pharmacy of the University of Utrecht. Currently, he is the Head of the Department of Pharmaceutics, a position he took in 1996. Since 1997 he has been an editor of the Journal of Controlled Release. His main research interests are in the field of polymeric drug delivery systems. He has published ≈600 papers and book chapters, and holds 25 patents. |
 Raymond M. Schiffelers | Raymond Schiffelers obtained his PhD degree in 2001 at the Erasmus University Medical Center, Rotterdam (The Netherlands) on liposomal targeting of antimicrobial agents to bacterial infections. After a post-doctoral period (2002–2003) at Intradigm Co (Rockville USA) working on siRNA delivery, he worked at Utrecht University as an assistant and later an associate professor. In 2015, he was appointed as the Professor of Nanomedicine at University Medical Center Utrecht. He has published ≈150 papers and was involved in the discovery and development of OncoCort®, a product currently entering clinical trials. |
1. Introduction
Nanoparticulate systems for the delivery of drugs to tumours have been extensively studied to facilitate drug accumulation at the target site and simultaneously reduce the toxicity related to the systemic delivery of free drugs.1,2 In other words, these strategies aim to improve the therapeutic outcome of anticancer drugs. PMs are attractive systems as they can accommodate a great variety of hydrophobic cytostatic drugs.3,4 PMs are nano-sized colloidal particles with a core–shell structure, which are spontaneously formed by the self-assembly of amphiphilic macromolecules in a block-selective solvent into nanoaggregates above a certain concentration, the critical micelle concentration (CMC). PMs contain several features that endow them with potential for delivering drugs.5–8 Namely, PMs present small and narrow sizes, usually below 100 nm which enables them to accumulate in tumours/inflamed tissues by means of extravasation as a result of the so-called enhanced permeability and retention (EPR) effect.9,10 The EPR effect is based on the fact that tumours, and inflamed tissues in general, comprise abnormal, angiogenic blood vessels with a wide fenestrated endothelium. Therefore, if PMs circulate long enough in the blood stream, they will accumulate in tumour/inflamed tissues to a higher extent than in healthy tissues. Moreover, lymphatic drainage is impaired in these tissues, which also contributes to the retention of the PM at the target site. PMs present a unique core–shell structure with hydrophobic segments of amphiphilic block copolymers forming the inner core of the nanoparticles while the hydrophilic segments are positioned in the aqueous milieu to form the shell. Hydrophobic molecules can be solubilized in the core while the hydrophilic corona stabilizes the PM and provides a prolonged circulation time in the blood. The shell of the PM further provides the possibility of decorating the surface with targeting moieties, exploited for specific targeting to cancer cells.11,12 Finally, the chemical composition and molecular weight of the blocks can be altered in order to obtain PMs with a tailorable size, loading and retention of the drug and drug release profile.The core segregation that occurs in an aqueous solution is the driving force for micelle formation and is accompanied by intermolecular forces, such as hydrophobic interaction, electrostatic interaction, metal complexation and hydrogen bonding. Consequently, a myriad of bioactive molecules can be entrapped in the micellar core by means of a strong interaction between the core-forming block and the drug molecules. Besides hydrophobic drugs, nucleic acids,13,14 antisense oligonucleotides15 and diagnostic agents16,17 can also be entrapped in the core. As mentioned before, PMs are most frequently used for the delivery of hydrophobic molecules that are physically entrapped in their core or chemically attached to micelle forming block copolymers.
In vivo, the administration of free chemotherapeutic drugs presents a low therapeutic value. On the one hand, the rapid extraction of the drug from the systemic circulation reduces the tumour accumulation and retention of the drug. On the other hand, these drugs are i.v. administered in relatively high doses and accumulate in healthy tissues/organs causing severe toxic effects. Also, the large volume of distribution of these drugs leads to their accumulation in healthy tissues/organs. In other words, the poor PK profile of chemotherapeutic drugs after systemic administration leads to their poor therapeutic outcome.18,19
Cremophor® is one of the most frequently used vehicles for the solubilization of hydrophobic drugs.20,21 This excipient is based on a polyoxyethylated castor oil and is clinically used for the solubilization of anaesthetics, photosensitizers, sedatives and anticancer drugs, such as paclitaxel. The drugs are solubilized with Cremophor® to be injected i.v., however the drugs are rapidly transferred to the blood proteins and subsequently cleared from the circulation.22–24 Moreover, the use of Cremophor is associated with several side effects such as dyspnoea, flushing, rash and generalized urticaria.25–27 For example, Taxol is a formulation of paclitaxel with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Cremophor and ethanol.28 Many complications have been reported after the administration of this formulation. The main problem, as mentioned before, is the toxicity associated with the administration of paclitaxel in the presence of the vehicle. Moreover, a large volume of Cremophor is required to deliver a therapeutically effective dose of paclitaxel. Given this, alternative formulations that do not require Cremophor and could be i.v. injected have been explored.
:1 mixture of Cremophor and ethanol.28 Many complications have been reported after the administration of this formulation. The main problem, as mentioned before, is the toxicity associated with the administration of paclitaxel in the presence of the vehicle. Moreover, a large volume of Cremophor is required to deliver a therapeutically effective dose of paclitaxel. Given this, alternative formulations that do not require Cremophor and could be i.v. injected have been explored.
The first generation of PMs aimed at solubilizing hydrophobic compounds. PEG-b-poly(D,L-lactic acid) (PEG-PLA) is an example of a block copolymer used for the solubilization of hydrophobic compounds. Paclitaxel loaded in PEG-PLA (Genexol™-PM) is a micellar formulation able to solubilize paclitaxel in its core, and that decreases the toxicity associated with Cremophor administration.29–32
Although such studies support the pharmaceutical potential of PM-based formulations, there are still some challenges regarding the clinical translation of these nanomedicines. Currently, the main challenges in the PM field are the in vivo stability and drug retention upon i.v. administration, both of which are essential for EPR effect-mediated passive drug delivery. As PMs are dynamic systems, upon i.v. administration and concomitant micellar dilution in the bloodstream, the copolymer concentration will decrease below the CMC. Consequently, there will be a shift in the equilibrium towards the unimer state that leads to (partial) micellar dissolution. The binding of the block copolymers to blood components such as albumin and apolipoproteins can also initiate micelle dissociation and premature drug loss.33 Therefore, to be more than a solubilisation vehicle, PMs should be able to retain the drug for a certain period of time and also present a long circulation time in the blood to guarantee that the PM with the loaded drug will reach the target site at reasonable concentrations to exert its function.
Different strategies can be employed to enhance drug retention in the PM core and to improve its circulation time in the blood. In the first generation of PMs the drug was formulated mainly by physical (hydrophobic) interactions between the core-forming block and the loaded molecules. In recent years, methods have been developed to chemically crosslink either the core or the shell of PMs to enhance their stability in the circulation. Also the chemical coupling of the drug via reversible bonds to the block copolymer has been exploited to retain the drug in the micelles during transport and gradually release the drug at the site of action.34–37
In 2007, the first micellar formulation was approved in South Korea for the treatment of breast and non-small cell lung (NSCL) cancer.38 Genexol™-PM is a formulation based on the block copolymer mPEG-b-poly(D,L-lactic acid) with paclitaxel encapsulated in the core of the micelles. Following the approval of Genexol™-PM, several PMs have entered clinical trials. Poly(ethylene glycol)-block-poly(aspartic acid) (PEG-b-pAsp) loaded with doxorubicin (NK911) or with paclitaxel (NK105) and poly(ethylene glycol)-block-poly(glutamic acid) (PEG-b-pGlu) entrapped with cisplatin (NC-6004), SN-38 (NK012) or oxaliplatin (NC-4016) are some examples of formulations that are under clinical investigation.
In this review, we summarize the PM formulations that are in clinical evaluation for the treatment of cancer.
2. First generation of polymeric micelles
2.1. SP1049C
SP1049C is a micellar formulation composed of doxorubicin physically loaded in a blend of two micellar forming non-ionic Pluronic® block copolymers, namely Pluronic® L61 and L127.39 This formulation is prepared by reconstitution of freeze dried doxorubicin in 0.9% sodium chloride solution with 0.25% (w/v) of L61 and 2% (w/v) of L127. The formulation has an average particle size of 30 nm and the drug loading capacity is 8.2%.In vitro data demonstrated that SP1049C was effective against multidrug resistant cells and presented superior activity compared to free doxorubicin.40In vivo PK shows that the area under the curve (AUC) of the PM is two times higher than that of free doxorubicin, 14.6 μg h ml−1 and 7.1 μg h ml−1, respectively, in mice bearing Lewis lung carcinoma 3LL M-27 cells. SP1049C formulation inhibited tumour growth more than 50% in nine different tumour models, which represents a superior antitumor activity when compared to free doxorubicin.
Antitumor activity revealed that the inhibition of tumour growth was maintained longer when mice were treated with the micelle formulation when compared to the free drug at the same dose. Moreover, the antitumor activity appeared to be dependent on the ratio between hydrophilic/hydrophobic blocks. Higher activity was observed when the copolymer had longer hydrophobic blocks.41
SP1049C antitumor activity was also assessed in mice bearing leukaemia cells intraperitoneally inoculated and found to decrease the tumorigenesis and aggressiveness of tumour cells in vivo.42
This formulation proceeded to phase I clinical trials in patients with advanced solid tumours.43 The starting dose was 5 mg m−2 up to 90 mg m−2 administered every three weeks until a maximum of six cycles. From the total number of patients that enrolled this trial, 11.5% had a partial or complete response after SP1049C treatment and 30.8% of the treated patients had stable disease with a median time for disease progression of 17.5 weeks. The dose limiting toxicity was myelosuppression which was reached at a dose of 90 mg m−2. The maximum tolerated dose was 70 mg m−2 which was the recommended dose for the following clinical trials.
A phase II trial was conducted in patients with advanced adenocarcinoma of the oesophagus or gastroesophageal junction. After SP1049C reconstitution in 0.9% sodium chloride, it was administered at a dose of 75 mg m−2. Each patient received a median number of four cycles, ranging from one to six. The most commonly observed toxicity was neutropenia of grade 3 or 4 in 62% of the patients. The antitumor activity of this formulation resulted in nine patients with partial response, whereas eight patients showed minor response or stable disease and two had progressive disease. The median overall survival and median progression-free survival were 10 and 6.6 months, respectively.44
This formulation recently obtained FDA approval for a pivotal phase III trial in patients with cancer of the gastrointestinal tract.
2.2. NK911
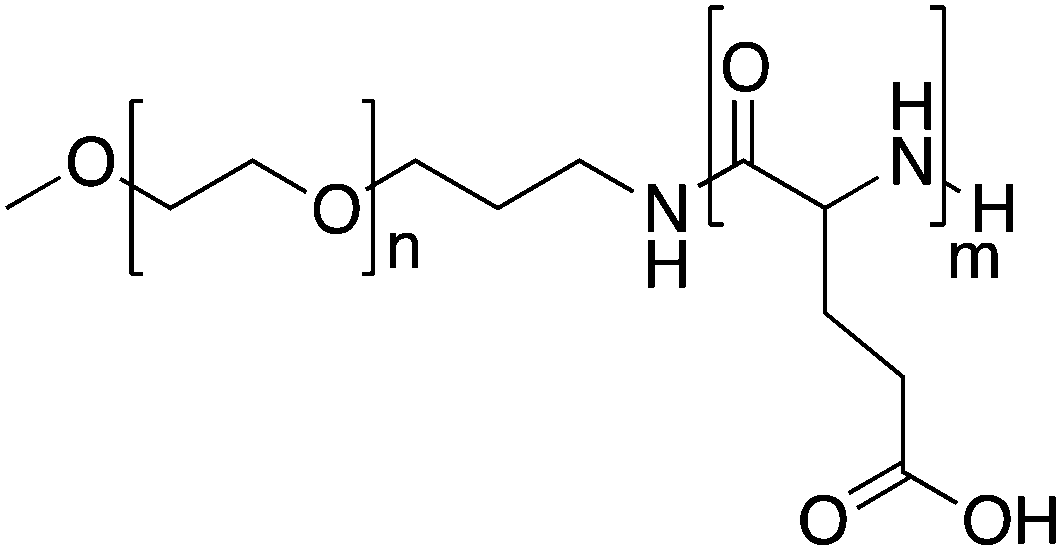
Kataoka and colleagues developed a hybrid drug delivery system based on a covalent-physical loading strategy. A block copolymer of poly(ethylene glycol)-b-poly(α,β-aspartic acid) was synthesised to target solid tumours. To increase the hydrophobicity of the core forming block, doxorubicin was covalently bound to 50% of carboxylic groups of the poly-aspartate block (PEG-b-p(Asp-Dox)), see Table 1. As chemically conjugated doxorubicin did not exert any antitumor activity, likely because it was coupled in the polymer backbone via a hydrolytically stable amide bond, doxorubicin was also physically loaded into the PM. Free doxorubicin molecules interact with covalently bound doxorubicin essentially through π–π interactions. This results in the stabilization of the micellar core and retention of the physically loaded doxorubicin into nano-sized PMs with an average diameter of 40 nm.45 Therefore, both chemically and physically loaded doxorubicin contributed to the low leakage of doxorubicin in the circulation, leading to a reduction of side effects and prolonged antitumor activity due to the sustained release of physically entrapped doxorubicin upon disposition of the PM in the tumour.| Name | Polymer structure | Compound | Indication | Phase | Company |
|---|---|---|---|---|---|
| NK911 |
PEG-b-p(Asp-DOX)
|
Doxorubicin | Pancreatic and colorectal cancer | II | Nippon Kayaku, Japan |
| NK105 |
PEG-b-p(Asp-4-phenyl-1-butanol)*
|
Paclitaxel | Stomach, breast cancer | III | NanoCarrier/Nippon Kayaku, Japan |
| NK012 |
PEG-b-p(Glu-SN-38)
|
SN-38 | Breast, lung, colorectal cancer | II | Nippon Kayaku, Japan |
| NC-6004 |
PEG-b-p(Glu)
|
Cisplatin | Pancreatic, head and neck, lung, bladder and bile duct cancer | III | NanoCarrier, Japan/Orient EuroPharma (co-development) |
| NC-6300 |
PEG-b-p(Asp-hydrazone)
|
Epirubicin | Solid tumors | I | NanoCarrier, Japan/Kowa (co-development) |
| NC-4016 | PEG-b-p(Glu)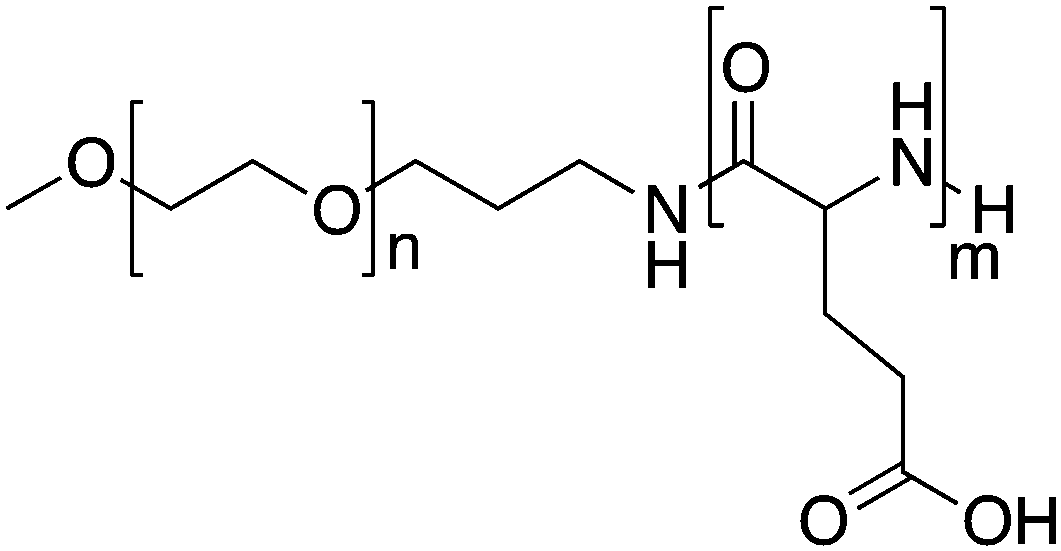 |
DACH-Platinum | Solid tumors and lymphoma | I | NanoCarrier, Japan |
| CriPec® docetaxel |
mPEG5000-b-p(HPMAm-Lacn)
|
Docetaxel | Solid tumors | I | Cristal Therapeutics, The Netherlands |
| SP1049C |
Pluronics®
|
Doxorubicin | Advanced adenocarcinoma of the esophagus or gastroesophageal junction | II | Supratek Pharma Inc. |
In preclinical studies, these PMs displayed a prolonged circulation time in a mouse model with subcutaneously implanted colon 26 tumour cells, with a 29-fold higher AUC in blood when compared to free doxorubicin (120 μg h ml−1versus 4 μg h ml−1, respectively). As a result, these PMs accumulate 3.4 times more in the tumour than the free drug, with tumour AUCs of 1605 μg h g−1versus 474 μg h g−1, respectively. PMs showed good antitumor activity in mouse models of sarcoma, lung, colon and breast cancer.45
In 2001, it was the first micellar formulation to progress into clinical trials. A phase I clinical trial to study PEG-b-(pAsp-Dox) polymeric micelle (NK911) toxicity was performed with twenty three patients that previously received chemotherapeutic treatment. Treatment started with a dosage of 6 mg m−2 doxorubicin and increased up to 67 mg m−2, comprising 6 escalating doses in total.46 NK911 administration was performed every 3 weeks using an infusion pump at a rate of 10 mg min−1. NK911 injection was in general well tolerated and the non-haematological toxicities reported were nausea, vomiting and anorexia. However, neutropenia was the predominant haematological toxicity, with grade 3 and 4 neutropenia at dosing level 5 (50 mg m−2). At dose level 6 of 67 mg m−2, all patients developed neutropenia, which in half of the patients lasted for more than 5 days. Therefore, 67 mg m−2 was considered the maximum tolerated dose due to grade 4 neutropenia being the dose limiting toxicity and 50 mg m−2 was the recommended dose to proceed with phase II clinical trials.
The AUC of NK911 at the recommended dose was 3.2 μg h ml−1 whereas in the case of free doxorubicin at an equivalent dose the AUC was 1.6 μg h ml−1 (Table 2). At the end of this study, eight patients exhibited stable disease for more than 4 weeks. One patient with metastatic pancreatic cancer showed partial response, with a 50% decrease of the liver metastasis.
| Pharmacokinetic parameters | Formulation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NK91146 | NK105 | NK01293 | NC-6004 | SP1049C43 | ||||||
| Free doxorubicin | Micelles | Free paclitaxel51 | Micelles52 | Free SN-38 | Micelles | Free cisplatin109,110 | Micelles75 | Free doxorubicin | Micelles | |
| a Calculated by considering that the body surface area equals 1.8 m2. | ||||||||||
| Dose (mg m−2) | 50 | 50 | 210 | 150 | 28 | 28 | 80–100 | 90 | 50 | 70 |
| t 1/2 (h) | 0.8 ± 1.1 | 2.8 ± 0.3 | 13.3 ± 1.5 | 10.6 ± 1.3 | 209 ± 25 | 137 ± 19 | 0.5 | 129 ± 40 | 0.8 ± 1.1 | 28.95 ± 52.03 |
| AUC (μg h ml−1) | 1.6 ± 1.1 | 3.2 ± 0.4 | 23.2 ± 10.7 | 369.8 ± 35.2 | 2.12 ± 0.83 | 294 ± 62 | 45.89 ± 4.62 | 2836 ± 554 | 1.6 ± 1.1 | 3.42 ± 0.93 |
| Max. concentration in blood (μg ml−1) | n.a. | 3.9 ± 0.7 | 6.74 ± 2.7 | 40.2 ± 5.5 | 0.114 ± 0.031 | 19.1 ± 3.9 | 3.31 ± 0.29 | 60.8 ± 12.5 | n.a. | n.a. |
| Total clearance (ml h−1 m−2) | 33600 ± 13066a |
15633 ± 2566a |
10740 ± 4860 |
408.6 ± 37.3 | n.a. | 98.8 ± 20.6 | 20700 ± 5820 |
33.8 ± 11.1a | 33600 ± 13066a |
27638.9 ± 10712.72a |
| Volume of distribution (ml m−2) | 933333 ± 466666a |
579444 ± 14000a |
58900 ± 24700 |
4527.1 ± 639.5 | n.a. | 2020 ± 530 | 12000 |
6555 ± 3833a | 933333 ± 466666a |
1077.1 ± 759.8a |
This formulation proceeded to Phase II clinical trials but the results are pending.
2.3. NK105
Paclitaxel is one of the most effective anticancer compounds and it has been shown to be efficacious in different types of solid tumours, like breast and ovarian cancers.47–49 Due to its poor water solubility, however, its use in clinical practice is hampered by the adverse effects of the solubilizing agents needed for i.v. injection of this drug. The development of a biocompatible drug delivery system could reduce these adverse effects and potentially increase the therapeutic potential of paclitaxel.NK105 is a polymeric micellar nanoformulation composed of an amphiphilic block copolymer, containing PEG as a hydrophilic block and modified pAsp as a hydrophobic block (Fig. 1). Half of the carboxylic groups of pAsp were modified with 4-phenyl-1-butanol by an esterification reaction to render this block hydrophobic and make micelle formation possible. Paclitaxel was physically loaded into the PM through hydrophobic interactions and the formulation was subsequently freeze dried. Upon reconstitution, PMs had an average diameter of 85 nm and had a paclitaxel loading of 23% (w/w).
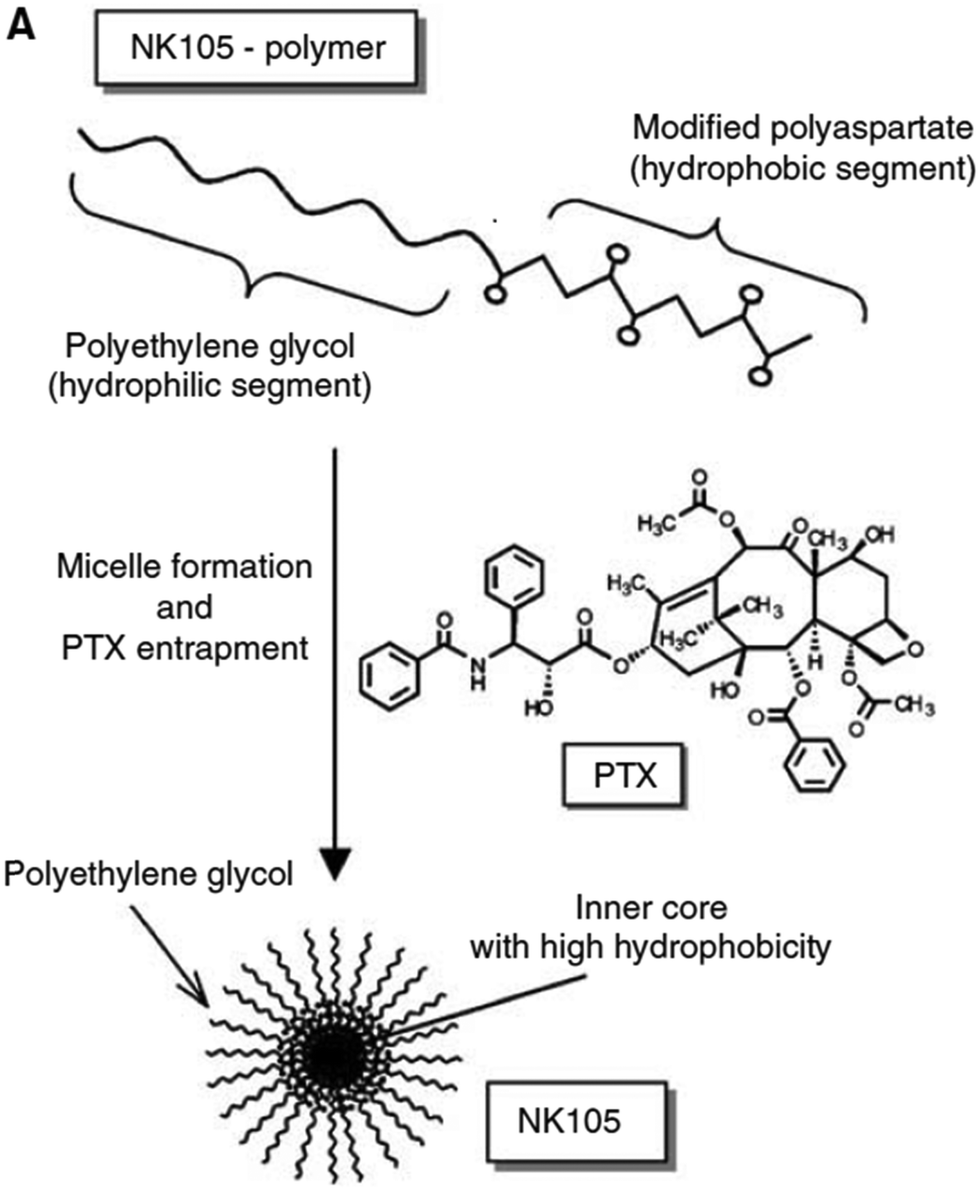 | ||
| Fig. 1 Illustration of NK105 formulation. NK105 micelles are formed by the assembly of the amphiphilic copolymer (composed of polyethylene glycol and polyaspartate modified with 4-phenyl-1-butanol) and Paclitaxel. Paclitaxel is loaded in the hydrophobic core of the micelles via physical interaction, mainly π–π stacking. Image reproduced from ref. 50, with permission of Nature Publishing Group, 2005. | ||
Preclinical data on the PK of NK105 in HT-29 colon tumour-bearing mice showed that paclitaxel was present in the circulation for up to 72 h after injection, whereas free paclitaxel could not be detected 24 h post administration. With the i.v. injection at a dose of 50 mg kg−1 the plasma AUC was 7860.9 and 90.2 μg h ml−1 for NK105 and free paclitaxel, respectively.50 In terms of tumour accumulation, the NK105 formulation achieved a 25-fold higher tumour AUC and 3-fold higher maximum concentration of paclitaxel when compared to free paclitaxel. Moreover, NK105 remained in tumours at least until 72 h after injection.
The therapeutic efficacy of the micellar formulation in mice bearing subcutaneous HT-29 colon tumours showed reduced tumour growth rates for both free paclitaxel and NK105. Still, the antitumor activity of NK105 was superior when compared to that of free drug. The antitumor efficacy at a dose of 25 mg kg−1 of NK105 was equivalent to 100 mg kg−1 of free dosed paclitaxel. Tumour suppression was dose dependent and mice were cured after a single injection of NK105 at a dose of 100 mg kg−1.
Following the promising outcome of preclinical studies, phase I clinical trials were conducted in patients with advanced solid tumours to determine the PK, the maximum tolerated dose and the recommended dose for phase II studies.
NK105 was supplied as a freeze-dried formulation containing 30 mg of paclitaxel. After reconstitution in 5% glucose, the formulation was stable for 24 h at room temperature. NK105 was infused i.v. once every three weeks at a speed of 250 ml h−1, for 1 h. The starting dose was 10 mg m−2 escalating up to 180 mg m−2. A total of nineteen cancer patients who had previously received chemotherapy (not including Taxanes) were recruited. Generally, no major toxicity was noted upon NK105 administration, apart from grade 1 or 2 neuropathy, irrespective of the dosing schedule. No haematological toxicity was observed until dose level 4 (80 mg m−2). Dose-limiting toxicity was reached at level 7 (180 mg m−2) in two out of five patients, who showed grade 4 neutropenia for more than five days. Therefore, 180 mg m−2 was considered the maximum tolerated dose, with neutropenia as the dose-limiting toxicity.
In subsequent phase II trials, a dosage of 150 mg m−2 was selected as the recommended dose. NK105 PK was assessed in patients that received the 150 mg m−2 dose. The AUC of NK105 was 15-fold higher than that of free paclitaxel given at a conventional dose of 210 mg m−2.51 Six patients had stable disease for more than four weeks after the completion of the study. A partial response was observed in a patient with metastatic pancreatic cancer, in whom liver metastasis decreased more than 90%. A patient with stomach cancer experienced a reduction of 40% in peritoneal metastasis.
A phase II trial was performed from 2007 to 2010 to assess the efficacy and safety of the NK105 formulation in patients with advanced gastric cancer who did not respond to first-line chemotherapy consisting of fluoropyrimidine and/or cisplatin. NK105 was administered to fifty seven patients every three weeks at a dose of 150 mg of paclitaxel m−2.52 The median treatment duration was 2.8 months and patients received an average of four cycles of treatment.
The overall response rate was 25% with two patients showing complete response and twelve patients showing partial responses (out of fifty six evaluable patients). Additionally, ∼30% of the patients (seventeen out of fifty six) experienced stable disease for several months. The disease control rate of 55% is encouraging in patients that had previously received chemotherapy. The median time to response was 1.8 months and the duration of the response was 3.7 months. The progression free survival, overall survival and time to treatment failure were 3.0, 14.4 and 2.8 months, respectively.53
The major adverse effects were neurotoxicity of grade 1 or 2 and the most common haematological toxicity was grade 4 neutropenia. The PK profile of NK105 (Table 2) was also assessed at a dose of 150 mg m−2. The AUC was found to be 9-fold higher than that of free paclitaxel at a conventional dose (210 mg m−2).
A multi-national phase III clinical study to confirm NK105 therapeutic efficacy in patients with metastatic or recurrent breast cancer was started in 2012 and is estimated to be completed at the end of 2017 (Study NCT01644890). A recent press release stated that the Phase III clinical trial failed to provide substantial benefits for patients. This was because one of the primary endpoints – progression free survival (PFS) – was not achieved, exemplifying the need for improved PM formulations.
3. Next generation of polymeric micelles
3.1. NC-6300
Progress in the drug delivery field has led to the development of systems capable of “sensing” and responding to stimuli such as pH. Taking this into consideration, pH sensitive PMs were developed by Kataoka and colleagues by the modification of the existing formulation, NK911. NK911 is formed by the self-assembly of the block copolymer with doxorubicin covalently bound and doxorubicin physically loaded, and only the latter presents cytotoxic effects. In NC-6300 doxorubicin is covalently conjugated to the carboxylic acid groups of PEG-b-p(b-Asp) via a hydrolysable linker, a hydrazone bond, to enable drug release upon pH stimuli (pH < 5).This block copolymer was synthesized by means of ring-opening polymerization of β-benzyl-L-aspartate N-carboxyl-anhydride using c-methoxy-ω-amino poly(ethylene glycol) as an initiator.54 The benzyl groups were subsequently removed in order to attach the hydrazone groups. The resulting polymer, PEG-p(Asp-Hyd), had an average of 37 repeating units of aspartate of which 28 were modified with hydrazone. Finally, doxorubicin was conjugated to the polymer backbone via a hydrazone bond. Although the starting poly(ethylene glycol)-poly(β-benzyl-L-aspartate) is fully water-soluble, after derivatization with doxorubicin the PEG-p(Asp-Hyd-dox) block is hydrophobic to make micelle formation possible. PMs were prepared via a dialysis method and had a mean size of 65 nm. Release experiments showed that the PMs were stable at neutral pH and released 30% of their payload at pH 5 within 72 h due to the acid sensitivity of the hydrazine bonds.
The stability of the PM at neutral pH enabled a 4-fold higher dose of doxorubicin when i.v. administered in mice bearing C26 tumour cells, compared to the free drug.55 The PK profile of doxorubicin either administered as a free drug or loaded in the PM at the same dose (10 mg kg−1) showed that the PM circulated 15-fold longer in blood than free doxorubicin (the AUC was 58.86 μg h ml−1 and 858.54 μg h ml−1, respectively). This was paralleled by a 4 times higher tumour accumulation of doxorubicin administered as PMs compared to free doxorubicin. C26-tumour bearing CDF1 mice were treated with free doxorubicin at doses ranging from 5 to 15 mg kg−1 whereas the micellar formulation was administered at a starting dose of 5 mg kg−1 up to 60 mg kg−1.
The antitumor activity of free doxorubicin was demonstrated at a dose of 10 mg kg−1 however mice treated at 15 mg kg−1 died due to toxicity. The PM antitumor effect was observed for doses ≥20 mg kg−1. Complete cure was obtained for 2/6 and 3/6 of the treated mice with a dose of 20 and 40 mg kg−1 of doxorubicin-loaded PMs, respectively.55 Mice treated at 60 mg kg−1 of doxorubicin in the PM showed toxic death. Additionally, at a therapeutic dose for the free doxorubicin (at 10 mg kg−1) or the micellar formulation (at 20 and 40 mg kg−1), the decrease in tumour volume was more pronounced in mice treated with PMs compared to the free drug treatment. In terms of toxicity, the maximum tolerated doses were established at 40 mg kg−1 and 10 mg kg−1, for the micellar and the free doxorubicin, respectively. Taking into account that the therapeutic window for the micellar formulation is broader than that for the free drug and the fact that doxorubicin can be dosed higher when formulated in the PM without showing toxic effects, it presents an advantage towards the application of these PMs.
For further development of this nanoformulation, doxorubicin was substituted by epirubicin which differs from doxorubicin in the spatial orientation of the hydroxyl group at the 4′ carbon of the sugar moiety. Epirubicin presents the same efficacy as doxorubicin, however, the toxicity is lower for epirubicin.56,57 Several clinical trials compared the equimolar administration of doxorubicin and epirubicin in patients with breast cancer and showed that the percentage of patients with cardiotoxicity, nausea, vomiting and neutropenia was higher when treated with doxorubicin than when patients were treated with epirubicin.57
Additionally, the block copolymer was partially substituted with benzyl groups in order to increase the micellar stability, circulation kinetics and ultimately the therapeutic efficacy.58 The block copolymer with epirubicin covalently coupled via a hydrazone bond, known as NC-6300 (Fig. 2), also spontaneously assembles into micellar structures with a diameter of 40–80 nm in an aqueous milieu.
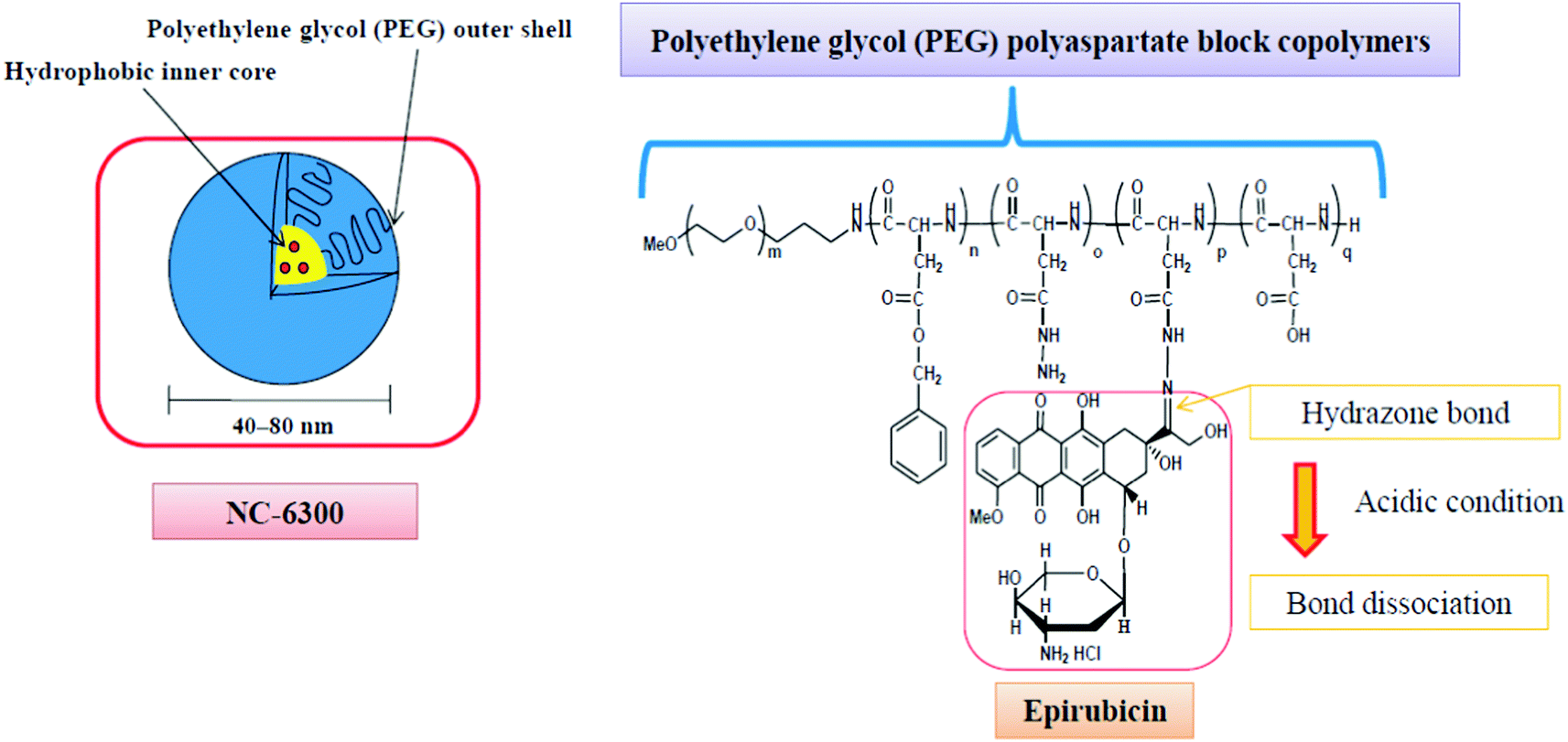 | ||
| Fig. 2 The NC-6300 formulation is spontaneously formed when the poly(ethylene glycol)-b-poly(benzyl-aspartate-hydrazone) copolymer containing an acid-labile linker is disposed in aqueous media; epirubicin is covalently bound to the polymer via a hydrazone bond. The micelles have an average diameter of 40–80 nm and are stable under physiological conditions. Under acidic conditions, the hydrazone bond dissociates and the drug is released.61 | ||
In preclinical experiments, NC-6300 was administered to mice bearing human hepatic Hep3B tumours at doses of 15 or 20 mg kg−1 epirubicin compared to 7 mg kg−1 of the free drug, which is the maximum tolerated dose.59 This formulation was able to decrease the tumour mass by 95% and 99% at doses of 15 and 20 mg kg−1, respectively, whereas free epirubicin decreased the tumour weight by a mere 53%, when compared to untreated control mice.
PK analyses performed in healthy male Wistar rats showed that the half-life of epirubicin when dosed in NC-6300 formulation was substantially prolonged to 4 h, whereas free epirubicin was completely cleared within 0.4 h. The plasma AUC was 2200-fold increased for NC-6300 as compared to free epirubicin. In nude mice bearing MDA-MB-231 breast tumours, free epirubicin was detected up to 48 h after i.v. injection whereas micelle-loaded epirubicin was detected at higher concentrations for up to 168 h after administration. Additionally, 24 h after injection 74% of the total epirubicin found was in its free form in tumour tissue, while in the case of other tissues (spleen, kidneys, liver, lungs, and heart), only around 20–40% was found as the free drug. This indicates that epirubicin is efficiently released in the tumour tissue likely due to the slight acidic pH in tumours.60
Cardiotoxicity is known as the major adverse effect related to the use of epirubicin.61 Mice treated with free epirubicin indeed showed a weakened cardiac function after 9 weeks of treatment. Importantly, mice given the NC-6300 formulation did not show any cardiac malfunction which confirms the ability of PMs to reduce drug accumulation in toxicity–sensitive tissues.
To further improve the selectivity of the targeting approach, NC-6300 was conjugated with an antibody against tissue factor (TF), which is overexpressed in various cancer cells.62In vitro studies demonstrated that the extent of cell binding of the targeted versus the non-targeted (control) formulation to a gastric cancer cell line (44As3) and a pancreatic cancer cell line (BxPC3) was two-fold higher. Moreover, targeted PMs were internalized faster and more efficiently than non-targeted PMs.
In mice bearing tumours with high TF expression, the efficacy of the anti-TF-NC-6300 formulation was better than that of the control formulation (the tumour size at day thirty five was 1200 mm2 and 2000 mm2, respectively). However, the untargeted formulation did not perform better than free epirubicin; free epirubicin presented a higher therapeutic effect than the control mice (untreated). In tumours with low TF expression, the efficacy of both formulations was comparable, which means that the superior efficacy of the targeted formulation in high TF expressing tumours is due to the targeting ligand. The AUC of free epirubicin in the tumour after the administration of the targeted or non-targeted PM was also similar irrespective of the expression level of TF. This shows that the formulations had the same circulation kinetics resulting in the same dose of tumour deposited PMs. The intratumoral distribution of the PM was highly affected by the level of TF expression. Anti-TF-NC-6300PMs reached most of the tumour cells in the malignant tissue and were homogeneously distributed over tumours with high TF expression. In the case of low TF expressing tumours, the targeted PMs were mostly located in the areas surrounding the tumour blood vessels. The distribution of the non-targeted formulation in high or low TF-expressing tumours was not studied.
NC-6300 entered a phase I clinical trial in 2013 to assess tolerability and the recommended dose in patients with advanced solid tumours.38 To date, no results of this study are available.
3.2. NC-6004
Platinum based anticancer drugs are widely used in the clinic as part of the treatment of a variety of tumours, such as melanoma, ovarian cancer and lymphomas.63 Cisplatin is one of the most extensively used drugs of its class. This drug is a platinum chelate complex that targets DNA, forming platinum–DNA adducts that change the structure of the DNA molecule by unwinding and bending it, and ultimately destabilizing DNA.64 Platinum based drugs present major limitations such as systemic toxicities, like nephrotoxicity65 and neurotoxicity66 featured mainly by peripheral neuropathy.Kataoka and colleagues developed the first PM loaded with cisplatin. PMs were formed via polymer–metal complex formation between the carboxylic groups of PEG-b-p(α,β-aspartic acid) and cisplatin in water.67 Cisplatin-loaded PMs had an average diameter of 16 nm at a molar ratio between cisplatin and aspartic acid residues of the polymer of 1:1.68 PMs were stable in distilled water for more than 24 h of incubation. When incubated in an aqueous buffer containing NaCl, the PMs were stable for 10 h, after which the drug was gradually released. This suggests that the core of the PM is not fully packed and the chloride ions can permeate the core and cause the disassembly of the polymer–metal complex.67,69
Cisplatin-loaded PMs were administered i.v. in lung carcinoma bearing mice to assess their PK profile. The AUC for micellar cisplatin was 5.2 and 4.6-fold higher in plasma and tumour, respectively, in comparison to the free drug.70 However, the fast structural decay of the micelles led to the accumulation of the drug in the liver and spleen at high levels which resulted in a similar antitumor efficacy as the free drug.
To improve both the stability of the PM and the drug release profile, a modified carrier system composed of PEG-b-p(L-glutamic acid) (PEG-p(Glu)) was developed.71 Cisplatin and PEG-p(Glu) formed PMs with an average diameter of 30 nm and a drug content of 39% (w/w). These PMs were very stable in water. In physiological saline, the PM size was stable (25 nm) for 50 h and the drug release followed gradually over 150 h. Preclinical data demonstrated that the drug level in plasma was over 50% of the injected dose (I.D.) at 8 h, and at 24 h after injection 13% I.D. was still in the circulation. This is a significant improvement compared to the pAsp PM, for which only 1.5% of the I.D. was recovered in the circulation at 24 h after administration.70 This favourable PK profile was ascribed by the authors to the higher stability of the pGlu based formulation compared to that based on pAsp.
Cisplatin delivered by PEG-pGlu PM accumulated in the tumour 20-fold more than free cisplatin. In C26-tumour bearing mice treated with 4 mg kg−1 of cisplatin, four out of ten mice showed complete tumour regression.71 Similarly, in human gastric cancer (MKN-45)72 and in pancreatic adenocarcinoma (BxPC3)-bearing mice,73 administration of cisplatin in PEG-pGlu PM resulted in improved antitumor activity. Moreover, PMs were able to overcome toxicities related to the administration of free drug, such as nephrotoxicity, decrease in nerve conduction velocity, and degeneration of the sciatic nerve.72 In a guinea pig model, cisplatin-loaded PMs displayed less ear toxicity in comparison to free cisplatin.74
Due to the favourable outcome in preclinical studies, cisplatin loaded in PEG-pGlu PMs entered phase I clinical trials under the name NC-6004. A phase I clinical trial was started in the UK with seventeen patients.75 The administration of NC-6004 commenced at a dose of 10 mg m−2 escalating to 120 mg m−2. A sterile formulation of NC-6004 with an equivalent dose of 2.5 mg ml−1 of cisplatin was i.v. injected once every 3 weeks. The plasma AUC was more than 60-fold higher for NC-6004 than the AUC for the free drug at an equivalent dose. In general, NC-6004 was well tolerated with no occurrence of haematological toxicities. Regarding non-haematological toxicities, the most common were fatigue, nausea, anorexia and vomiting. From six patients treated at 90 mg m−2, one presented grade 3 fatigue as the dose limiting toxicity. In addition, renal toxicity of grade 2 was observed at 90 and 120 mg m−2 even though prophylaxis was employed. Therefore, 120 mg m−2 was concluded to be the maximum tolerated dose and the recommended dose for Phase II studies was 90 mg m−2. At the recommended dose for phase II, 50% of the patients presented stable disease. The median progression-free survival time was forty nine days and fourteen patients (82%) died or presented tumour progression.
A phase I/II study (Study NCT00910741) was conducted in Taiwan and Singapore to determine the maximum tolerated dose and recommended dose of NC-6004 in combination with gemcitabine in patients with pancreatic cancer.76 In total nineteen patients received NC-6004 at doses of 30 up to 120 mg m−2 once every three weeks, and gemcitabine at 1000 mg m−2 twice every 3 weeks. Dose limiting toxicity occurred at 120 mg m−2, and therefore 120 mg m−2 was considered the maximum tolerated dose and 90 mg m−2 the recommended dose of NC-6004 in combination with gemcitabine. From seventeen patients evaluated, one patient had a partial response and ten patients experienced stable disease. No cases of complete response were noted. Overall the disease control percentage in this study was 65%.
A phase III trial was started in 2014 to assess the efficacy of the combined therapy of NC-6004 with gemcitabine versus gemcitabine alone in patients with locally advanced or metastatic pancreatic cancer (Study NCT02043288). The results have not been published yet.
A number of phase I trials are currently running to determine the maximum tolerated dose and recommended dose of NC-6004 in combination with several other drugs. NC-6004 in combination with 5-FU and cetuximab is being tested in patients with recurrent or metastatic squamous cell carcinoma in the head and neck, in Taiwan (Study NCT02817113).
Also in Japan and the U.S.A., NC-6004 safety and tolerability are being tested in patients with head and neck cancer.
A Phase I/II trial of NC-6004 in combination with gemcitabine for the treatment of advanced solid tumours or non-small cell lung, biliary tract, and bladder cancer is being conducted in the U.S.A. (Study NCT02240238).
A phase III clinical trial of the combination of NC-6004 and gemcitabine versus gemcitabine alone in patients with locally advanced metastatic pancreatic cancer started in 2014 (Study NCT02043288).
3.3. NC-4016
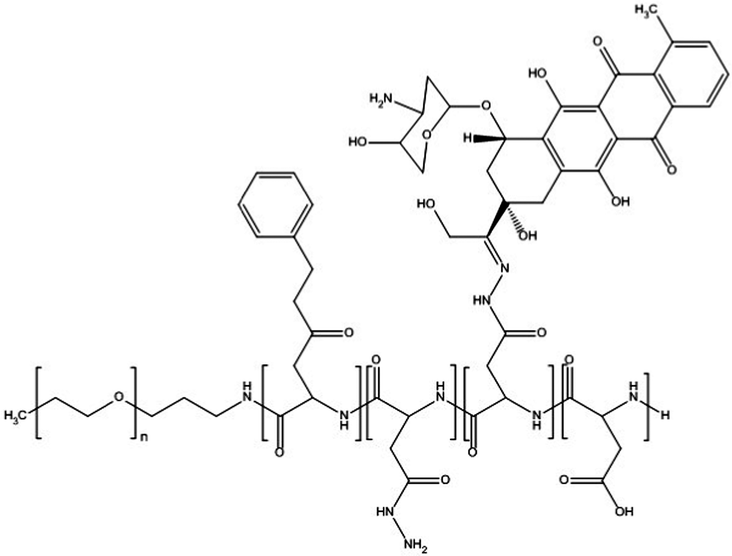
Cisplatin-loaded PMs, as mentioned above, showed good stability and efficient disposition of the drug in the tumour. However, cisplatin administration is associated with acute toxicity and acquired resistance. Therefore, other platinum based drugs, like dichloro(1,2-diaminocyclohexane)platinum(II) (DACHPt), the oxaliplatin parent complex, were developed to enhance drug accumulation in the tumour, and DACHPt is more hydrophobic than cisplatin and has lower toxicity.77DACHPt was loaded in the PEG-poly(γ-benzyl-L-glutamate) block copolymer via polymer–metal complexation between the platinum of DACHPt and the carboxylic groups of the block copolymer (Fig. 4). The PM with an average size of 40 nm showed a platinum loading of 75% (w/w). No drug release occurred in water, whereas in phosphate buffered saline the drug was released in a sustained manner with 60% being released after 96 h of incubation at 37 °C.
 | ||
| Fig. 3 Schematic representation of the formation of core-crosslinked PMs based on mPEG-b-pHEMA-Lacn. The amphiphilic block copolymer assembles into micelles when above the CMC; crosslinking of the micellar core is performed subsequently to prevent destabilization and drug release. The image was reproduced from ref. 98, with permission of Elsevier, Copyright 2007. | ||
 | ||
| Fig. 4 NC-4016 micellar formulation. Micelles are formed by coordination interaction between poly(ethylene glycol)-b-poly(glutamic acid) and DACHPt in an aqueous environment. The image was reproduced from ref. 108, with permission of Nature Publishing Group, 2011. | ||
The good stability of DACHPt-loaded PMs resulted in good circulation kinetics with 80% of the I.D. in the circulation 9 h post administration and 16% of the I.D., 27 h after injection in CDF1 mice bearing C-26 tumours.77 In these mice, DACHPt-loaded PM accumulation in the tumour peaked at 11% of the ID 48 h post injection.78 Compared to free oxaliplatin, this represents a 25-fold increase in tumour accumulation. Antitumor activity of DACHPt-loaded PMs was achieved at doses of 2 up to 6 mg kg−1 whereas free oxaliplatin at doses up to 10 mg kg−1 did not show any reduction in the tumour growth rate. Noticeably, no body weight loss was detected at the therapeutic effective doses of 2 and 4 mg kg−1 of the micellar formulations. Mice treated with 10 mg kg−1 of free oxaliplatin experienced toxic death after the fourth injection. Mice were inoculated intraperitoneally with bioluminescent HeLa (HeLa-Luc) cells and subsequently treated with either free oxaliplatin or DACHPt-loaded PMs. The therapeutic efficacy, determined by bioluminescence, points to a 10- to 50-fold decrease in fluorescence in the group of mice treated with DACHPt-loaded PMs, meaning that these PMs are able to reduce the metastatic character of tumours.78
Antitumor efficacy was demonstrated in tumour models of colon carcinoma,78 orthotopic scirrhous gastric cancer,79 melanoma,80 lung metastasis,80 intraperitoneal metastases,78 and lymph node metastasis.79
The effect of combined treatment with epirubicin and oxaliplatin-loaded micelle formulations (NC-6300 and NC-4016, respectively) was assessed in a gastric cancer xenograft model.81 The antitumor effect was superior when both formulations were administered, as compared to the combination of the free drugs. The combination of epirubicin, oxaliplatin and capecitabine is one of the standard treatments in Europe for gastric cancer. The good outcome of NC-6300 and NC-4016 formulations is encouraging to further study this combination in gastric cancer patients.
Regarding NC-4016, a phase I clinical trial was started in the U.S.A. for the treatment of patients with advanced solid tumours and lymphoma (Study NCT01999491).
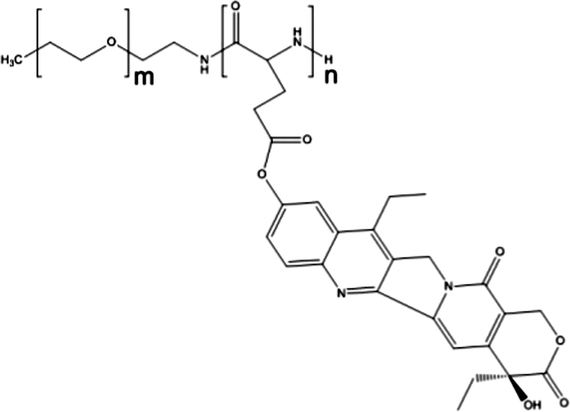
3.4. NK012
NK012 is a polymeric prodrug of SN-38 forming a core–shell structure. SN-38 (7-ethyl-10-hydroxycamptothecin) is covalently conjugated to PEG-b-poly(L-glutamic acid) by esterification of the drug's phenol groups and the carboxylic acid groups of the polymer.82NK012 is a freeze dried formulation containing 20% (w/w) of SN-38 with an average size of 20 nm. The drug release in phosphate buffered saline pH 7.4 is reported to be 57% over 24 h and in 5% glucose (pH 4.6) only 1%, indicating that the release is likely dictated by the pH of the solution.
PK of NK012 in HT-29 tumour-bearing mice showed a much slower plasma clearance of SN-38 when compared to free drug, presenting an AUC that was 200-fold higher, even when the dose of SN-38 was more than two-fold lower for the micellar formulation (30 mg kg−1versus 66.7 mg kg−1, respectively). The prolonged circulation time of NK012 enabled increased drug accumulation in tumour tissue, which in mice bearing HT-29 tumours was 23-fold higher 24 h after administration when compared to the free drug. Moreover, the tumour-to-plasma concentration ratio of the drug was found to be 100 times higher for NK012 than for the free drug, at 168 h after injection. These PK characteristics resulted in a successful pharmacodynamic antitumor activity of the NK012 formulation. A significant decrease in HT-29 tumour volume was observed 60 days after the first injection, at doses of 15 and 30 mg kg−1 for NK012.82
The NK012 formulation was also tested in VEGF-secreting tumours (SBC-3/VEGF) to evaluate the antitumor effect in hypervascularized tumours. In mice bearing slowly vascularising SBC-3/Neo tumours, administration of this drug at 10 and 20 mg kg−1 led to a significant decrease in tumour volume in comparison to the free drug at a higher dose (15 and 30 mg kg−1). For mice with hypervascularized SBC-3/VEGF tumours, the tumours were completely eradicated after 20 days of the first injection and for a total of three injections (10 and 20 mg kg−1). This indicates that high vascularization enhanced the activity of the nanoformulation, likely through increased EPR effects.
This formulation was additionally tested in several human tumour xenograft models, such as glioma,83,84 renal cancer,85 gastric cancer,86 pancreatic cancer87 and small cell lung cancer.88 In all these models superior antitumor activity over the free drug was demonstrated.
NK012 was employed in combination with different conventional drugs to further improve the efficacy of the monotherapy regimen. In combination with 5-fluoruracil (5-FU) improved efficacy in the treatment of colorectal cancer was shown.89 Similarly, with cisplatin the treatment of small cell lung cancer was improved.90
NK012 was also evaluated in a mouse model of multiple myeloma. The combination of this formulation with the clinically approved proteasome inhibitor bortezomib91 showed improved antitumor efficacy when compared with bortezomib alone.92 These pre-clinical studies provided strong support for clinical testing of the NK012 formulation.
Two independent phase I clinical trials, one in Japan93 and the other in the United States (Study NCT00542958),94 were conducted in patients with solid tumours refractory to standard therapy or in patients for whom no alternative therapies were available. NK012, containing 20% (w/w) of SN-38, was diluted in 5% glucose solution and administered as a 30 minute infusion every twenty one days.
In one of the studies,93 the starting dose was 2 mg m−2 equivalent to SN-38 and increased up to 28 mg m−2. The plasma AUC of SN-38 in NK012 was approximately 150-fold higher than that of free SN-38, being 294 μg h ml−1 and 2.12 μg h ml−1 for NK012 and SN-38 at a dose of 28 mg m−2, respectively. The total clearance and the volume of distribution were 150- and 80-fold lower than those of the free drug,93,95 respectively.
Out of twenty three patients that received the formulation two patients presented a partial response and nine patients had stable disease. Moreover, from twelve patients who had colorectal cancer, five presented partial response. The median number of cycles administered was 3.5, ranging from one to twelve cycles. Twenty-two patients received more than two administrations. The common toxicities were grade three/four leukopenia and neutropenia, which were reduced to grade one in four to sixteen days. The dose limiting toxicity for neutropenia was 28 mg m−2. The maximum tolerated dose was not determined due to haematological toxicities. The recommended dose for phase II was 28 mg m−2.
In the other study,94 NK012 was administered at a dose starting from 9 to 37 mg m−2. The NK012 formulation at a dose of 28 mg m−2 and the free drug at a dose of 250 mg m−2 presented an AUC of 287 and 27.86 μg h ml−1, respectively, which accounts for a 10-fold higher AUC of NK012 compared to the free drug and at an almost ten times lower concentration of I.D.
Of a pool of thirty seven patients, six patients had partial response, eighteen presented stable disease and twelve patients showed disease progression; the median number of cycles administered per patient was four; and six or more cycles were administered to sixteen patients. In terms of toxicity, NK012 was generally well tolerated and the dose limiting toxicity was grade three neutropenia. The maximum tolerated dose was 28 mg m−2. Therefore, the recommended dose for phase II was set at 28 mg m−2. These combined data demonstrate that the NK012 formulation is stable upon systemic injection and the PK data support EPR-mediated accumulation at the target site.38
Given the promising outcome in preclinical studies in which the combination of NK012 with 5-FU was examined,89 a phase I trial (Study NCT01238939) of NK012 in combination with 5-FU and leucovorin (or folinic acid, a compound with the same chemical structure as vitamin B9 that also exerts the same vitamin activity) was conducted. Leucovorin is used in combination with chemotherapeutic drugs to either enhance their therapeutic efficacy or decrease their toxic side effects. The number of patients that enrolled in this trial is estimated to be thirty five. The primary outcome measures were set as the number of patients with dose limiting-toxicity, which will determine the maximum tolerated and recommended dose. The outcome has not been reported yet.
Another phase I study of NK012 in combination with carboplatin in patients with advanced solid tumours, following a dose expansion in patients with triple negative metastatic breast cancer (Study NCT01238952), was started at the end of 2010. Thirty-five patients were expected to enroll in this trial that ended in 2014. Also in this case, the results are not yet available.
A phase II trial was performed in the U.S.A. to assess the efficacy of NK012 in patients with relapsed small cell lung cancer (Study NCT00951613). Patients with sensitive relapsed and refractory relapsed lung cancer were enrolled in this study. In patients with sensitive relapsed lung cancer, the overall response rate was 22%, with two patients with complete response; the disease control rate (sum of complete response, partial response and stable disease) was 68%.96 The PMs were well tolerated, with neutropenia as the dominant toxicity observed (44%).
NK012 was also evaluated in a phase II trial, in the U.S.A., in patients with locally advanced non-resectable and metastatic breast cancer (Study NCT00951054). This study was completed in 2015, but no results are available at present.
In Japan, a phase II trial was conducted in fifty eight patients with recurrent or metastatic colorectal cancer who had previously received oxaliplatin-based chemotherapy (Study JapicCTI-090780). The overall response rate was 3.8% and the disease control rate was 56%. The median for progression free survival and overall survival was 99 and 451 days, respectively.97
The phase I trials discussed above show that the NK012 formulation presents a satisfactory safety profile. NK012 was well tolerated for doses up to 28 mg m−2 and the toxicity profile was markedly different from the one exhibited by the free drug. Grade 3 and 4 diarrhea, nausea and vomiting, which are common toxicities of SN-38 systemic administration, were not reported in patients that received the NK012 formulation. Note that the maximum tolerated dose was not determined due to hematological toxicities.
In a phase II trial, NK012 was shown to be efficacious after administration in patients with lung cancer and showed an overall response rate of 22% and two patients had complete response. In this case the toxicity was mild and as expected.
In another phase II study conducted in patients with colorectal cancer, NK012 showed similar efficacy to the free compound. In terms of toxicity, gastrointestinal toxicity and myelosuppression were observed. The results presented are encouraging to pursue the development of this formulation; however, further optimization of the formulation is necessary to decrease toxicity.
3.5. CriPec docetaxel
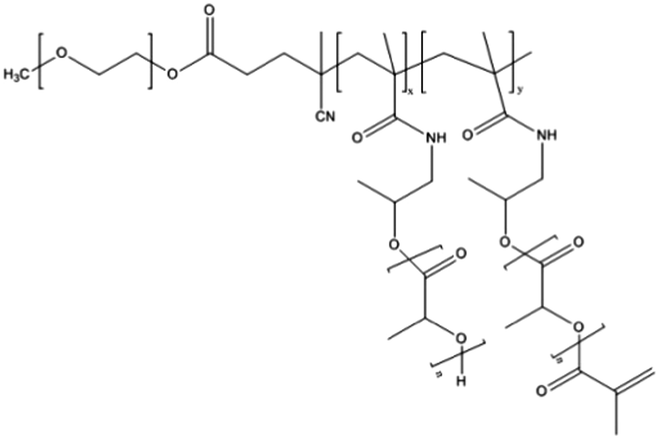
NC-6300 makes use of an acid labile bond to better control drug release after in vivo administration. However, these PMs rely on hydrophobic interactions for stability. Hennink and co-workers proposed chemical crosslinking of the micellar core in order to prevent the disintegration of the PM in the circulation (Fig. 3).98 Partially methacrylated mPEG-b-pHEMAmLacn block copolymers rendered core-crosslinked CCPMs after UV-induced photo-crosslinking. These CCPMs demonstrated superior physicochemical stability when compared to non-crosslinked NCPMs. CCPMs showed a prolonged circulation time with a concomitant increase in tumour accumulation. Half of the I.D. was still in the circulation at 6 h post-injection, which resulted in a high tumour accumulation, as 6% I.D. accumulated in the tumour whereas only 1% I.D. of the non-crosslinked PM was present at the tumour site, at 48 h post injection.98 Even though CCPMs demonstrated an excellent PK and tissue distribution profile, the encapsulated drug was rapidly released from the micellar core. Already at 30 min after administration, 95% of the initially loaded drug was released.99 This study clearly indicates that prolonged blood circulation and target tissue accumulation of PMs do not necessarily represent the same kinetics for the encapsulated drug.To overcome premature drug release, Hennink's group covalently coupled the drug in the micellar core to warrant an efficient delivery of the drug to the target site. For this, mPEG-b-p(HPMAm-Lacn) was co-crosslinked with doxorubicin functionalized with a methacrylamide group (DOX-MA)100 through a pH sensitive linker – hydrazone – that degrades under acidic conditions.101 DOX-MA was loaded into the PM and then copolymerized with the methacrylate groups of the thermosensitive block. In principle, doxorubicin covalently entrapped through the hydrazone linker is preferentially released in the tumour microenvironment or intracellularly in lysosomes where the pH is below 5. The PMs had an average size of 80 nm and 30–40% (w/w) of the added drug was covalently entrapped. Due to the proton catalysed hydrolysis of the hydrazine bond, at pH 7.4 only 5% release was observed whereas 100% of the drug was released within 24 h at pH 5. The therapeutic efficacy of this micellar nanoformulation was assessed in mice bearing B16 melanoma carcinoma and compared to that of the free drug at an equivalent dose. A significant decrease in tumour growth was observed in mice treated with free and micellar doxorubicin compared to control (untreated) mice. Importantly, the mice treated with doxorubicin loaded PMs showed a slower rate of tumour growth when compared to mice treated with free doxorubicin.100 The same formulation was coupled with an anti-EGFR nanobody EGa1 to specifically target and increase the uptake of the PM by cancer cells. An in vitro binding assay was performed with rhodamine labelled PMs showing that after 1 h of incubation with EGFR overexpressing tumour cell lines (A431 and UM-SCC-14C) there were significantly more PMs associated with the cell surface in the case of EGa1-coupled PMs than in the case of untargeted PMs.102 This micellar formulation was subsequently evaluated in mice bearing 14C tumour xenografts and both untargeted and targeted micellar formulations were shown to be more effective than free doxorubicin in inhibiting tumour growth and increasing animal survival.103 Moreover, targeted micelles were able to inhibit tumour growth even in the absence of the drug due to the intrinsic anticancer activity of the nanobody.
The release kinetics of the drug can be tailored through the derivatization of the drug coupled to the polymer backbone with different linkers. For instance, dexamethasone was derivatized with three different methacrylated spacers via ester bonds to create dexamethasone derivatives capable of polymerization. The sulphide, sulfoxide and sulfone linkers presented different hydrolysis rates, enabling control of the release rate of the drug from the PM.104
Recently, docetaxel was methacrylated via an ester bond and chemically loaded into core-crosslinked PMs composed of mPEG-b-p(HPMAm-Lacn).105 PMs were formed with a mean diameter of 66 nm and had a docetaxel encapsulation efficiency of 75% (w/w) at a loading capacity of 12% (w/w). In mice with the MDA-MB-231 breast tumour model, the half-life of docetaxel in CCPMs in the circulation was 16.2 h105 whereas that of free docetaxel was only 0.88 h.106 The tumour accumulation of docetaxel was strongly enhanced in the case of CCPMs. After a single injection of 30 mg kg−1 of docetaxel-loaded CCPM, the docetaxel concentration in the tumours of MDA-MB-231 tumour bearing mice was 20- and 50-fold higher after 2 and 4 days, respectively, when compared to an equivalent dose of the free docetaxel formulation Taxotere (docetaxel in polysorbate 80). The therapeutic efficacy of docetaxel-loaded CCPMs was assessed after a single i.v. injection at a dose of 30 or 60 mg kg−1. At a dose of 30 mg kg−1 the decrease in tumour growth as compared to the saline control was the same for either the docetaxel in CCPMs or Taxotere. At a dose of 60 mg kg−1 CCPMs significantly inhibited the tumour growth whereas the Taxotere formulation did not show any improvement compared to the 30 mg kg−1 concentration. Importantly, a dose of 125 mg kg−1 of docetaxel in the micellar formulation led to the complete eradication of the tumour growth in mice and to the survival of all mice after 62 days.
This formulation, CriPec® docetaxel, was the first in Europe to enter phase I clinical trials started in 2015 (Study NCT02442531).
The same micellar strategy was applied for the formulation of a therapeutic peptide, leuprolide. Leuprolide was covalently coupled to CCPMs through hydrolysable ester bonds.107 Leuprolide entrapped in CCPMs showed a substantially improved (14.4 h) half-life in circulation in comparison to the free peptide, leading to an AUC that was 100-fold higher for leuprolide-CCPMs than for free leuprolide. Importantly, the drug released from the PM was still biologically active.
The conjugation of drug molecules to the polymer backbone by chemical bonds results in a good retention of the drug in the PM during circulation and allows tailoring of drug release kinetics by means of different linkers. Although this approach shows promising results, it also demands extra steps in the formulation of the micelles. Moreover, by crosslinking either the copolymer and/or the drug the principle of micellar formation as a dynamic equilibrium between unimers is compromised.
4. Conclusions
PMs have been extensively studied in the last few decades due to their ability to efficiently and stably load hydrophobic drugs in their core. As many of the anticancer drugs have hydrophobic character, much attention has been paid to the development of PM-based therapies focusing on the treatment of cancer. This resulted in several formulations being investigated in clinical studies and to date one of them has been FDA approved (Genexol™-PM). The first class of PMs aimed to be used as solubilizers of hydrophobic compounds. However, these formulations lack good stability when present in the circulation and, importantly, the loaded drug is rapidly extracted from the PM core which compromised the therapeutic efficacy of these formulations. Different strategies have been successfully employed to achieve improved retention and stability of drug-loaded PMs upon i.v. injection. These strategies include chemical crosslinking of the PM core and/or drug, covalent coupling of the drug to the polymer chains, and hydrophobic interactions between the drug and the hydrophobic block of the copolymer, as π–π interactions. Promising preclinical studies performed with these PMs have translated into several formulations that are currently under clinical investigation for the treatment of different types of cancer with encouraging results. Depending on the pharmacodynamic profile of the incorporated drug, triggered release, e.g. by exogenous light or sound, or endogenous microenvironmental cues, like pH or enzymatic activity, could be envisioned to further improve the therapeutic efficacy. Also, modulation of cell specificity by the introduction of targeting ligands may be an avenue to steer the formulation within the tumour environment, which is especially important for drug molecules that have difficulty in crossing cellular membranes. These developments could render new classes of pathway-specific therapeutic molecules to be attractive candidates for PM encapsulation.References
- T. Lammers, Int. J. Pharm., 2013, 454(1), 527–529 CrossRef CAS PubMed.
- M. Talelli, C. J. F. Rijcken, W. E. Hennink and T. Lammers, Curr. Opin. Solid State Mater. Sci., 2012, 16(6), 302–309 CrossRef CAS.
- N. Nishiyama, Y. Matsumura and K. Kataoka, Cancer Sci., 2016, 107(7), 867–874 CrossRef CAS PubMed.
- D. J. A. Crommelin and J. S. B. de Vlieger, Non-Biological Complex Drugs: The Science and the Regulatory Landscape, Springer International Publishing, 2015 Search PubMed.
- G. S. Kwon, Crit. Rev. Ther. Drug Carrier Syst., 2003, 20(5), 357–403 CrossRef CAS PubMed.
- S. R. Croy and G. S. Kwon, Curr. Pharm. Des., 2006, 12(36), 4669–4684 CrossRef CAS PubMed.
- S. Eetezadi, S. N. Ekdawi and C. Allen, Adv. Drug Delivery Rev., 2015, 91, 7–22 CrossRef CAS PubMed.
- C. Deng, Y. Jiang, R. Cheng, F. Meng and Z. Zhong, Nano Today, 2012, 7(5), 467–480 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65(1–2), 271–284 CrossRef CAS PubMed.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS PubMed.
- T. Lammers, F. Kiessling, W. E. Hennink and G. Storm, J. Controlled Release, 2012, 161(2), 175–187 CrossRef CAS PubMed.
- R. van der Meel, L. J. Vehmeijer, R. J. Kok, G. Storm and E. V. van Gaal, Adv. Drug Delivery Rev., 2013, 65(10), 1284–1298 CrossRef CAS PubMed.
- S. Katayose and K. Kataoka, Bioconjugate Chem., 1997, 8(5), 702–707 CrossRef CAS PubMed.
- S. Katayose and K. Kataoka, J. Pharm. Sci., 1998, 87(2), 160–163 CrossRef CAS PubMed.
- K. Kataoka, H. Togawa, A. Harada, K. Yasugi, T. Matsumoto and S. Katayose, Macromolecules, 1996, 29(26), 8556–8557 CrossRef CAS.
- V. S. Trubetskoy, Adv. Drug Delivery Rev., 1999, 37(1-3), 81–88 CrossRef CAS PubMed.
- V. S. Trubetskoy, M. D. Frank-Kamenetsky, K. R. Whiteman, G. L. Wolf and V. P. Torchilin, Acad. Radiol., 1996, 3(3), 232–238 CrossRef CAS PubMed.
- N. Bertrand and J. C. Leroux, J. Controlled Release, 2012, 161(2), 152–163 CrossRef CAS PubMed.
- T. Lammers, Adv. Drug Delivery Rev., 2010, 62(2), 203–230 CrossRef CAS PubMed.
- A. Sparreboom, S. D. Baker and J. Verweij, J. Clin. Oncol., 2005, 23(31), 7765–7767 CrossRef CAS PubMed.
- H. Gelderblom, J. Verweij, K. Nooter and A. Sparreboom, Eur. J. Cancer, 2001, 37(13), 1590–1598 CrossRef CAS PubMed.
- G. N. Kumar, U. K. Walle, K. N. Bhalla and T. Walle, Res. Commun. Chem. Pathol. Pharmacol., 1993, 80(3), 337–344 CAS.
- S. M. Longnecker, R. C. Donehower, A. E. Cates, T. L. Chen, R. B. Brundrett, L. B. Grochow, D. S. Ettinger and M. Colvin, Cancer Treat. Rep., 1987, 71(1), 53–59 CAS.
- P. H. Wiernik, E. L. Schwartz, J. J. Strauman, J. P. Dutcher, R. B. Lipton and E. Paietta, Cancer Res., 1987, 47(9), 2486–2493 CAS.
- E. K. Rowinsky, E. A. Eisenhauer, V. Chaudhry, S. G. Arbuck and R. C. Donehower, Semin. Oncol., 1993, 20(4 Suppl. 3), 1–15 CAS.
- R. B. Weiss, R. C. Donehower, P. H. Wiernik, T. Ohnuma, R. J. Gralla, D. L. Trump, J. R. Baker Jr., D. A. Van Echo, D. D. Von Hoff and B. Leyland-Jones, J. Clin. Oncol., 1990, 8(7), 1263–1268 CAS.
- J. Szebeni, F. M. Muggia and C. R. Alving, J. Natl. Cancer Inst., 1998, 90(4), 300–306 CrossRef CAS PubMed.
- A. K. Singla, A. Garg and D. Aggarwal, Int. J. Pharm., 2002, 235(1–2), 179–192 CrossRef CAS PubMed.
- H. K. Ahn, M. Jung, S. J. Sym, D. B. Shin, S. M. Kang, S. Y. Kyung, J. W. Park, S. H. Jeong and E. K. Cho, Cancer Chemother. Pharmacol., 2014, 74(2), 277–282 CrossRef CAS PubMed.
- D. W. Kim, S. Y. Kim, H. K. Kim, S. W. Kim, S. W. Shin, J. S. Kim, K. Park, M. Y. Lee and D. S. Heo, Ann. Oncol., 2007, 18(12), 2009–2014 CrossRef PubMed.
- J. L. Lee, J. H. Ahn, S. H. Park, H. Y. Lim, J. H. Kwon, S. Ahn, C. Song, J. H. Hong, C. S. Kim and H. Ahn, Invest. New Drugs, 2012, 30(5), 1984–1990 CrossRef CAS PubMed.
- K. S. Lee, H. C. Chung, S. A. Im, Y. H. Park, C. S. Kim, S. B. Kim, S. Y. Rha, M. Y. Lee and J. Ro, Breast Cancer Res. Treat., 2008, 108(2), 241–250 CrossRef CAS PubMed.
- S. Kim, Y. Shi, J. Y. Kim, K. Park and J.-X. Cheng, Expert Opin. Drug Delivery, 2010, 7(1), 49–62 CrossRef CAS PubMed.
- Y. Shi, T. Lammers, G. Storm and W. E. Hennink, Macromol. Biosci., 2017, 17(1) DOI:10.1002/mabi.201600160.
- M. Talelli, M. Barz, C. J. Rijcken, F. Kiessling, W. E. Hennink and T. Lammers, Nano Today, 2015, 10(1), 93–117 CrossRef CAS PubMed.
- Y. H. Bae and H. Yin, J. Controlled Release, 2008, 131(1), 2–4 CrossRef CAS PubMed.
- W. Zhou, C. Li, Z. Wang, W. Zhang and J. Liu, J. Nanopart. Res., 2016, 18(9), 1–18 Search PubMed.
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed.
- A. Venne, S. Li, R. Mandeville, A. Kabanov and V. Alakhov, Cancer Res., 1996, 56(16), 3626–3629 CAS.
- V. Alakhov, E. Klinski, S. Li, G. Pietrzynski, A. Venne, E. Batrakova, T. Bronitch and A. Kabanov, Colloids Surf., B, 1999, 16(1–4), 113–134 CrossRef CAS.
- E. V. Batrakova, T. Y. Dorodnych, E. Y. Klinskii, E. N. Kliushnenkova, O. B. Shemchukova, O. N. Goncharova, S. A. Arjakov, V. Y. Alakhov and A. V. Kabanov, Br. J. Cancer, 1996, 74(10), 1545–1552 CrossRef CAS PubMed.
- D. Y. Alakhova, Y. Zhao, S. Li and A. V. Kabanov, PLoS One, 2013, 8(8), 1–14 Search PubMed.
- S. Danson, D. Ferry, V. Alakhov, J. Margison, D. Kerr, D. Jowle, M. Brampton, G. Halbert and M. Ranson, Br. J. Cancer, 2004, 90(11), 2085–2091 CAS.
- J. W. Valle, A. Armstrong, C. Newman, V. Alakhov, G. Pietrzynski, J. Brewer, S. Campbell, P. Corrie, E. K. Rowinsky and M. Ranson, Invest. New Drugs, 2011, 29(5), 1029–1037 CrossRef CAS PubMed.
- T. Nakanishi, S. Fukushima, K. Okamoto, M. Suzuki, Y. Matsumura, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, J. Controlled Release, 2001, 74(1-3), 295–302 CrossRef CAS PubMed.
- Y. Matsumura, T. Hamaguchi, T. Ura, K. Muro, Y. Yamada, Y. Shimada, K. Shirao, T. Okusaka, H. Ueno, M. Ikeda and N. Watanabe, Br. J. Cancer, 2004, 91(10), 1775–1781 CrossRef CAS PubMed.
- D. Khayat, E. C. Antoine and D. Coeffic, Cancer Invest., 2000, 18(3), 242–260 CrossRef CAS PubMed.
- E. K. Rowinsky and R. C. Donehower, N. Engl. J. Med., 1995, 332(15), 1004–1014 CrossRef CAS PubMed.
- C. Caldas and W. P. McGuire, 3rd, Semin. Oncol., 1993, 20(4 Suppl. 3), 50–55 CAS.
- T. Hamaguchi, Y. Matsumura, M. Suzuki, K. Shimizu, R. Goda, I. Nakamura, I. Nakatomi, M. Yokoyama, K. Kataoka and T. Kakizoe, Br. J. Cancer, 2005, 92(7), 1240–1246 CrossRef CAS PubMed.
- T. Tamura, Y. Sasaki, Y. Nishiwaki and N. Saijo, Jpn. J. Cancer Res., 1995, 86(12), 1203–1209 CrossRef CAS PubMed.
- T. Hamaguchi, K. Kato, H. Yasui, C. Morizane, M. Ikeda, H. Ueno, K. Muro, Y. Yamada, T. Okusaka, K. Shirao, Y. Shimada, H. Nakahama and Y. Matsumura, Br. J. Cancer, 2007, 97(2), 170–176 CrossRef CAS PubMed.
- K. Kato, K. Chin, T. Yoshikawa, K. Yamaguchi, Y. Tsuji, T. Esaki, K. Sakai, M. Kimura, T. Hamaguchi, Y. Shimada, Y. Matsumura and R. Ikeda, Invest. New Drugs, 2012, 30(4), 1621–1627 CrossRef CAS PubMed.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42(38), 4640–4643 CrossRef CAS PubMed.
- Y. Bae, N. Nishiyama, S. Fukushima, H. Koyama, M. Yasuhiro and K. Kataoka, Bioconjugate Chem., 2005, 16(1), 122–130 CrossRef CAS PubMed.
- V. Bonfante, G. Bonadonna, F. Villani and A. Martini, Recent Results Cancer Res., 1980, 74, 192–199 CAS.
- S. Gluck, Cancer Control, 2002, 9(Suppl. 2), 16–27 Search PubMed.
- I. Bobe, N. Shibata, H. Saito and M. Harada, Block copolymer for drug complex and pharmaceutical composition, 2008 Search PubMed.
- M. Harada, I. Bobe, H. Saito, N. Shibata, R. Tanaka, T. Hayashi and Y. Kato, Cancer Sci., 2011, 102(1), 192–199 CrossRef CAS PubMed.
- Y. Kato, S. Ozawa, C. Miyamoto, Y. Maehata, A. Suzuki, T. Maeda and Y. Baba, Cancer cell Int., 2013, 13(1), 89 CrossRef CAS PubMed.
- A. Takahashi, Y. Yamamoto, M. Yasunaga, Y. Koga, J. Kuroda, M. Takigahira, M. Harada, H. Saito, T. Hayashi, Y. Kato, T. Kinoshita, N. Ohkohchi, I. Hyodo and Y. Matsumura, Cancer Sci., 2013, 104(7), 920–925 CrossRef CAS PubMed.
- Y. Yamamoto, I. Hyodo, Y. Koga, R. Tsumura, R. Sato, T. Obonai, H. Fuchigami, F. Furuya, M. Yasunaga, M. Harada, Y. Kato, A. Ohtsu and Y. Matsumura, Cancer Sci., 2015, 106(5), 627–634 CrossRef CAS PubMed.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99(9), 2451–2466 CrossRef CAS PubMed.
- H. S. Oberoi, N. V. Nukolova, A. V. Kabanov and T. K. Bronich, Adv. Drug Delivery Rev., 2013, 65(13–14), 667–1685 Search PubMed.
- I. Arany and R. L. Safirstein, Semin. Nephrol., 2003, 23(5), 460–464 CrossRef CAS PubMed.
- J. E. Mollman, N. Engl. J. Med., 1990, 322(2), 126–127 CrossRef CAS PubMed.
- M. Yokoyama, T. Okano, Y. Sakurai, S. Suwa and K. Kataoka, J. Controlled Release, 1996, 39(2), 351–356 CrossRef CAS.
- N. Nishiyama and K. Kataoka, J. Controlled Release, 2001, 74(1–3), 83–94 CrossRef CAS PubMed.
- N. Nishiyama, M. Yokoyama, T. Aoyagi, T. Okano, Y. Sakurai and K. Kataoka, Langmuir, 1999, 15(2), 377–383 CrossRef CAS.
- N. Nishiyama, Y. Kato, Y. Sugiyama and K. Kataoka, Pharm. Res., 2001, 18(7), 1035–1041 CrossRef CAS.
- N. Nishiyama, S. Okazaki, H. Cabral, M. Miyamoto, Y. Kato, Y. Sugiyama, K. Nishio, Y. Matsumura and K. Kataoka, Cancer Res., 2003, 63(24), 8977–8983 CAS.
- H. Uchino, Y. Matsumura, T. Negishi, F. Koizumi, T. Hayashi, T. Honda, N. Nishiyama, K. Kataoka, S. Naito and T. Kakizoe, Br. J. Cancer, 2005, 93(6), 678–687 CrossRef CAS PubMed.
- Y. Mochida, H. Cabral, Y. Miura, F. Albertini, S. Fukushima, K. Osada, N. Nishiyama and K. Kataoka, ACS Nano, 2014, 8(7), 6724–6738 CrossRef CAS PubMed.
- M. Baba, Y. Matsumoto, A. Kashio, H. Cabral, N. Nishiyama, K. Kataoka and T. Yamasoba, J. Controlled Release, 2012, 157(1), 112–117 CrossRef CAS PubMed.
- R. Plummer, R. H. Wilson, H. Calvert, A. V. Boddy, M. Griffin, J. Sludden, M. J. Tilby, M. Eatock, D. G. Pearson, C. J. Ottley, Y. Matsumura, K. Kataoka and T. Nishiya, Br. J. Cancer, 2011, 104(4), 593–598 CrossRef CAS PubMed.
- W.-C. Su, L.-T. Chen, C.-P. Li, J.-S. Chen, Y.-L. Lin, S.-P. Choo and Y. Matsumura, ESMO Congress 2012, 2012, abstract 863.
- H. Cabral, N. Nishiyama, S. Okazaki, H. Koyama and K. Kataoka, J. Controlled Release, 2005, 101(1–3), 223–232 CrossRef CAS PubMed.
- H. Cabral, N. Nishiyama and K. Kataoka, J. Controlled Release, 2007, 121(3), 146–155 CrossRef CAS PubMed.
- M. Rafi, H. Cabral, M. R. Kano, P. Mi, C. Iwata, M. Yashiro, K. Hirakawa, K. Miyazono, N. Nishiyama and K. Kataoka, J. Controlled Release, 2012, 159(2), 189–196 CrossRef CAS PubMed.
- S. Deshayes, H. Cabral, T. Ishii, Y. Miura, S. Kobayashi, T. Yamashita, A. Matsumoto, Y. Miyahara, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2013, 135(41), 15501–15507 CrossRef CAS PubMed.
- Y. Yamamoto, I. Hyodo, M. Takigahira, Y. Koga, M. Yasunaga, M. Harada, T. Hayashi, Y. Kato and Y. Matsumura, Int. J. Cancer, 2014, 135(1), 214–223 CrossRef CAS PubMed.
- F. Koizumi, M. Kitagawa, T. Negishi, T. Onda, S. Matsumoto, T. Hamaguchi and Y. Matsumura, Cancer Res., 2006, 66(20), 10048–10056 CrossRef CAS PubMed.
- J. Kuroda, J. Kuratsu, M. Yasunaga, Y. Koga, Y. Saito and Y. Matsumura, Int. J. Cancer, 2009, 124(11), 2505–2511 CrossRef CAS PubMed.
- J. Kuroda, J. Kuratsu, M. Yasunaga, Y. Koga, H. Kenmotsu, T. Sugino and Y. Matsumura, Clin. Cancer Res., 2010, 16(2), 521–529 CrossRef CAS PubMed.
- M. Sumitomo, F. Koizumi, T. Asano, A. Horiguchi, K. Ito, T. Asano, T. Kakizoe, M. Hayakawa and Y. Matsumura, Cancer Res., 2008, 68(6), 1631–1635 CrossRef CAS PubMed.
- T. E. Nakajima, K. Yanagihara, M. Takigahira, M. Yasunaga, K. Kato, T. Hamaguchi, Y. Yamada, Y. Shimada, K. Mihara, T. Ochiya and Y. Matsumura, Cancer Res., 2008, 68(22), 9318–9322 CrossRef CAS PubMed.
- Y. Saito, M. Yasunaga, J. Kuroda, Y. Koga and Y. Matsumura, Cancer Sci., 2008, 99(6), 1258–1264 CrossRef CAS PubMed.
- H. Kenmotsu, M. Yasunaga, K. Goto, T. Nagano, J. Kuroda, Y. Koga, A. Takahashi, Y. Nishiwaki and Y. Matsumura, Cancer, 2010, 116(19), 4597–4604 CrossRef CAS PubMed.
- T. E. Nakajima, M. Yasunaga, Y. Kano, F. Koizumi, K. Kato, T. Hamaguchi, Y. Yamada, K. Shirao, Y. Shimada and Y. Matsumura, Int. J. Cancer, 2008, 122(9), 2148–2153 CrossRef CAS PubMed.
- T. Nagano, M. Yasunaga, K. Goto, H. Kenmotsu, Y. Koga, J. Kuroda, Y. Nishimura, T. Sugino, Y. Nishiwaki and Y. Matsumura, Clin. Cancer Res., 2009, 15(13), 4348–4355 CrossRef CAS PubMed.
- J. Adams, Semin. Oncol., 2001, 28(6), 613–619 CrossRef CAS PubMed.
- O. Miyazaki, K. Sekine, N. Nakajima, E. Ichimura, K. Ebara, D. Nagai, T. Onda, Y. Miyakawa, K. Okamoto and T. Morino, Int. J. Cancer, 2014, 134(1), 218–223 CrossRef PubMed.
- T. Hamaguchi, T. Doi, T. Eguchi-Nakajima, K. Kato, Y. Yamada, Y. Shimada, N. Fuse, A. Ohtsu, S. Matsumoto, M. Takanashi and Y. Matsumura, Clin. Cancer Res., 2010, 16(20), 5058–5066 CrossRef CAS PubMed.
- H. A. Burris, J. R. Infante, F. Anthony Greco, D. S. Thompson, J. H. Barton, J. C. Bendell, Y. Nambu, N. Watanabe and S. F. Jones, Cancer Chemother. Pharmacol., 2016, 77(5), 1079–1086 CrossRef CAS PubMed.
- D. Abigerges, G. G. Chabot, J. P. Armand, P. Herait, A. Gouyette and D. Gandia, J. Clin. Oncol., 1995, 13(1), 210–221 CAS.
- E. Raefsky, D. R. Spigel, J. R. Infante, J. C. Bendell, S. F. Jones, A. J. Lipman; D. Trent, S. Kawamura, F. A. Greco, J. D. Hainsworth and H. A. Burris, Phase II study of NK012 in relapsed small cell lung cancer. In 2011 ASCO Annual Meeting, 2011, vol. 29 Search PubMed.
- A. Tsuji, T. Hamaguchi, K. Yamaguchi, K. Takeda, H. Uetake, T. Esaki, K. Amagai, N. Sugimoto, H. Baba, M. Kimura, Y. Matsumura and Y. Nambu, ASCO Meeting Abstracts, 2015, 33, (Suppl. 15), 3527.
- C. J. Rijcken, C. J. Snel, R. M. Schiffelers, C. F. van Nostrum and W. E. Hennink, Biomaterials, 2007, 28(36), 5581–5593 CrossRef CAS PubMed.
- C. J. F. Rijcken, Tuneable & degradable polymeric micelles for drug delivery: from synthesis to feasibility in vivo, PhD thesis, Utrecht University, 2007 Search PubMed.
- M. Talelli, M. Iman, A. K. Varkouhi, C. J. Rijcken, R. M. Schiffelers, T. Etrych, K. Ulbrich, C. F. van Nostrum, T. Lammers, G. Storm and W. E. Hennink, Biomaterials, 2010, 31(30), 7797–7804 CrossRef CAS PubMed.
- T. Etrych, M. Jelinkova, B. Rihova and K. Ulbrich, J. Controlled Release, 2001, 73(1), 89–102 CrossRef CAS PubMed.
- M. Talelli, C. J. Rijcken, S. Oliveira, R. van der Meel, P. M. van Bergen En Henegouwen, T. Lammers, C. F. van Nostrum, G. Storm and W. E. Hennink, J. Controlled Release, 2011, 151(2), 183–192 CrossRef CAS PubMed.
- M. Talelli, S. Oliveira, C. J. Rijcken, E. H. Pieters, T. Etrych, K. Ulbrich, R. C. van Nostrum, G. Storm, W. E. Hennink and T. Lammers, Biomaterials, 2013, 34(4), 1255–1260 CrossRef CAS PubMed.
- B. J. Crielaard, C. J. Rijcken, L. Quan, S. van der Wal, I. Altintas, M. van der Pot, J. A. Kruijtzer, R. M. Liskamp, R. M. Schiffelers, C. F. van Nostrum, W. E. Hennink, D. Wang, T. Lammers and G. Storm, Angew. Chem., Int. Ed., 2012, 51(29), 7254–7258 CrossRef CAS PubMed.
- Q. Hu, C. J. Rijcken, R. Bansal, W. E. Hennink, G. Storm and J. Prakash, Biomaterials, 2015, 53, 370–378 CrossRef CAS PubMed.
- D. L. Gustafson, M. E. Long, J. A. Zirrolli, M. W. Duncan, S. N. Holden, A. S. Pierson and S. G. Eckhardt, Cancer Chemother. Pharmacol., 2003, 52(2), 159–166 CrossRef CAS PubMed.
- Q. Hu, E. V. van Gaal, P. Brundel, H. Ippel, T. Hackeng, C. J. Rijcken, G. Storm, W. E. Hennink and J. Prakash, J. Controlled Release, 2015, 205, 98–108 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6(12), 815–823 CrossRef CAS PubMed.
- P. A. Reece, I. Stafford, R. L. Abbott, C. Anderson, J. Denham, S. Freeman, R. G. Morris, P. G. Gill and C. L. Olweny, J. Clin. Oncol., 1989, 7(2), 270–275 CAS.
- K. Ikeda, M. Terashima, H. Kawamura, I. Takiyama, K. Koeda, A. Takagane, N. Sato, K. Ishida, T. Iwaya, C. Maesawa, H. Yoshinari and K. Saito, Jpn. J. Clin. Oncol., 1998, 28(3), 168–175 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2017 |