
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increasing the stability of Mg2(dobpdc) metal–organic framework in air through solvent removal†
Jenny G.
Vitillo
 * and
Silvia
Bordiga
* and
Silvia
Bordiga
Department of Chemistry, NIS Center and INSTM, University of Turin, via Gioacchino Quarello 15A, I-10135 Torino, Italy. E-mail: jenny.vitillo@unito.it; Fax: +39-011-6707855; Tel: +39-011-6708375
First published on 11th November 2016
Abstract
Mg2(dopbdc) (H4-dobpdc = 4,4′-dihydroxy-(1,1′-biphenyl)-3,3′-dicarboxylic acid) is an important MOF for its astonishing properties in gas separation and storage, in particular for applications related to CO2 separation. Its technological relevance is limited by its reputation for fast decomposition in air. In this article, this reputation is denied by showing that, if fully desolvated, the MOF is stable in air for 24 h (25 °C and 28–35 RH%), a time sufficiently long to allow its processing in a non-inert environment and its use as a CO2 scrubber in a wet post-combustion flow. In contrast, the material solvated with methanol showed a 93% decrease in surface area under the same conditions. Material activation is proposed here as a simple way to increase the stability of Mg2(dobpdc) in air, by the removal of preadsorbed polar molecules that can act as seeds for water condensation. This work allows us to make some important general considerations which can be taken into account in studies on material sensitivity to air. The importance of quantifing the aging time and testing the materials at different relative humidity is stressed. These parameters are important to determine, when possible, the conditions of use and storage, suitable for large scale processing and applications. Among the adopted characterization techniques (XRD, IR spectroscopy, N2 and CO2 volumetry), N2 volumetry at 77 K turned out to be the most suitable to evidence small variations in the structure due to decomposition. In contrast, CO2 uptake at RT and 1 bar was not a reliable quantity to evaluate MOF stability, because of the significant change in the (apparent) MOF affinity toward CO2 upon damaging.
Metal organic frameworks are a class of materials that have been successfully proposed for applications in quite different fields, from catalysis to gas separation1,2 and photovoltaics.3–5 At the basis of the interest in these materials, there is the possibility of tailoring their structure to target applications by appropriately changing their structural building blocks (organic ligands and metal nodes). With respect to applications, a strong obstacle is represented by their well-earned reputation as air and moisture sensitive materials.6–9 The iconic and most famous MOF, MOF-5, decomposes after exposure to air for less than 10 min.8,10 Several MOFs have been reported since then,7,11,12 which have moisture stabilities going from fair (HKUST-1)13 to exceptional (e.g. ZIF-8, MIL-101 and UiO-66).12–14 Particularly sensitive to moisture are MOF structures held on by metal–oxygen bonds, constituting the largest class of MOFs synthetized since then. In this case,15 the degradation in air has a common mechanism, starting with water adsorption followed by the hydrolysis of those bonds, which results in the formation of metal–OH species and the contemporaneous protonation of the linker.15,16 Only in the second step, when the number of broken bonds is larger than the critical value, does the structure collapse. It is therefore evident that any factor delaying water adsorption in MOFs would increase their air stability. Synthesis optimization,14 ligand functionalization17 or metal exchange18 are among the most common strategies adopted to increase their stability. Besides a few exceptions,19 MOFs based on s-block metals are characterized by very poor air stability.6 This is related to the higher ionic character of the metal–ligand bonds, with respect to those based on transition metal cations,9 making them more reactive towards atmospheric water. Nevertheless, the high abundance on Earth of some of these elements makes them more affordable from an economic point of view, and allows widespread application of these MOFs, with respect to those based on transition metal elements.6,9 Moreover, the large polarity of their metal–ligand bonds has been proven to be beneficial to the affinity of MOFs towards adsorbates such as CH420 and CO2.6,21
Among the MOFs proposed for CO2 separation and storage, MOF-74-Mg or Mg2(dobdc) (H4dobdc = 2,5-dihydroxytereftalic acid; also known as IRMOF-74-I-Mg, Mg/DOBDC or CPO-27-Mg) has been reported to possess an exceptional CO2 capacity of 8.0 mol kg−1 at 298 K and 1 bar.22 It is also characterized by a very high CO2/N2 selectivity.23 The honeycomb-like structure of MOF-74-Mg is reported in Fig. S1 (ESI†), where each vertex of the hexagonal channels is occupied by a filar of Mg–O units linked between them by dobdc4− units. After thermal treatment, solvent molecules are removed leaving one coordinative unsaturation on the Mg2+ sites. These open metal sites constitute the main adsorption sites for incoming adsorbates. In particular it has been demonstrated that the high polarity of the Mg-linker bond is on the basis of the larger affinity of MOF-74-Mg for CO2 with respect to MOFs based on other metals.6,21 Unfortunately, this MOF is known to be water unstable, retaining less than 10–20% of its surface area after exposure to water (95% relative humidity, RH%).13,18,24 To overcome this problem, Mg substitution with Ni or Co has been proposed,18,25 although this strategy is detrimental to the CO2 loadings. At this point, it is worth noticing that the stability studies reported in the literature are in general performed at 0 or at >80 RH%, whereas intermediate moisture values are less commonly considered.26–28 Nevertheless, intermediate values of relative humidity are more commonly verified in working environments and then they are more interesting to determine the conditions for an easy handling and processing of the materials. For those concerning MOF-74-Mg, the studies conducted at intermediate RH% are contradictory. Britt et al.27 reported that this MOF would be stable in air for at least 2 days but the temperature and RH% were not reported and no details on sample preparation were given. Moreover, the sample stability was checked only by measuring the CO2 uptake before and after exposure to air. Tan et al.16 showed that water adsorption in MOF-74-Mg was fully reversible for RH < 40% at RT for a very short time (20 min). All the other studies indicate a strong modification of the MOFs also at 9 RH%.26,28
Even more interesting than MOF-74-Mg is the isomorphous Mg2(dopbdc) (H4-dobpdc = 4,4′-dihydroxy-(1,1′-biphenyl)-3,3′-dicarboxylic acid) (see Fig. S1, ESI†). This MOF is identical to MOF-74-Mg, besides having a biphenyl linker instead of a monophenyl one. It possesses almost the same adsorption properties of MOF-74-Mg towards CO2, with open Mg2+ sites also being the main CO2 adsorption sites for Mg2(dopbdc) (see also Section S8.2, ESI†).6 Moreover, when used as a support for N,N′-dimethylethylenediamine (mmen-Mg2(dobpdc)),29 it showed a significant improvement in its performances and in its moisture resistance, representing one of the most promising materials for carbon capture and sequestration (CCS) applications.29–31 Unfortunately, the amine-grafting procedures29,30 are complicated by precautionary measures aimed at avoiding the contact of the support with air, mainly due to the reputation of Mg2(dobpdc) as an air sensitive material. In fact, the MOF quickly assumes a blue coloration when extracted from a methanol solution and left exposed to the air. Moisture stability studies analogous to MOF-74-Mg have not been reported for Mg2(dobpdc). Although a similar decomposition process can be expected for the two MOFs, at least in the first, crucial step, different kinetics cannot be excluded because of the higher hydrophobicity of the dobpdc linker.
In this work, a new strategy to increase the stability of Mg2(dobpdc) in air has been tested, consisting of the complete removal of polar solvent molecules preadsorbed in the pores. It has been previously reported that the kinetics of water adsorption in MOFs can be strongly enhanced by the presence of preadsorbed polar molecules.12 Nevertheless, their removal has never been tested as an easy strategy to increase MOF stability in air. Methanol has been considered as a polar solvent because it is not only a solvent commonly used in MOF synthesis but also in routine solvent exchange procedures. We have tested the air stability of Mg2(dobpdc) by using different aging conditions. The MOF was aged in air for different (i) relative humidities and (ii) in the presence (or absence) of precoordinated solvent molecules in the pores. To evaluate this last point, two samples were preactivated at 180 °C for 15 h in a vacuum, to remove all traces of the solvent (act) before the degradation test. The complete removal of the solvent was tested by means of IR spectroscopy (see Fig. S11a, ESI†). Two additional samples, on the contrary, were just extracted from the mother solution (sol). Two different RH% conditions were adopted: 25–50 RH% (indoor, that is the average RH% range in our laboratory) and >86 RH% (humid, see the ESI†). The aging temperature was in all cases close to room temperature (22–25 °C). A list of all the samples considered is reported in Table 1, with the corresponding aging conditions. After aging, the possible structure loss was evaluated by means of X-ray powder diffraction (XRPD). Moreover, after reactivation, the samples were characterized for their surface area and CO2 capacity. The degradation for the indoor samples was also monitored over time for 20 h using an in situ XRPD study (see Fig. S3–S6, ESI†).
| T (°C) | RH% | Time (h) | S Langmuir [SBET] (m2 g−1) | V pores (cm3 g−1) | CO2@1 bar (mol kg−1) | SA%loss | Color (before/after activation) | |
|---|---|---|---|---|---|---|---|---|
| a V pores evaluated at p/p0 = 0.95. b It turns to dark blue if left for at least one hour at RH < 60%. c The material has also been prepared at 42–49% RH, not showing any significant difference. | ||||||||
| act/sol | 22 | 0 | — | 3895 [2941] | 1.47 | 6.4 | — | White/white |
| act-indoorc | 22–23 | 28–35 | 24 | 3814 [2883] | 1.42 | 6.3 | 2 | White/white |
| act-humid | 22–23 | >86 | 24 | 680 [513] | 0.29 | 4.8 | 83 | Whiteb/green |
| sol-indoor | 22–23 | 28–35 | 24 | 291 [214] | 0.25 | 4.4 | 93 | Dark blue/green |
| sol-humid | 22–23 | >86 | 24 | 532 [398] | 0.24 | 4.1 | 86 | Whiteb/green |
In Fig. 1, the patterns obtained for the starting materials (act, red line; sol, grey line) are reported and compared with those collected after 24 h in air at 29–40 RH% (act-indoor, light violet; sol-indoor, blue) and at >86 RH% (act-humid, violet; sol-humid, dark blue). All the patterns are dominated by the peaks at 4.7° and 8.2°, associated with the (010) and (110) reflections. The changes were quite small for all the samples. Besides sol-humid, which has a pattern essentially identical to the sol one, in the other cases the most important changes in time were observed by the increase of the relative intensity of the peaks at 4.7 and 8.2° with respect to those at 9.4° and 12.5°, associated with the reflections of the planes having index (020) and (210), respectively. This would suggest a decrease in the order along the (010) and (100) directions, due to solvation or to damaging of the material. A phenomenological description of the changes observed in the pattern is reported in Section S5 of the ESI.† A Pawley refinement of the patterns indicate a slight decrease in cell volume of about 5% with respect to the act volume in all the tested conditions after 24 h (see Table S1 and Section S5 (ESI†) for further details) besides sol-humid decreasing of only 1%. This can be imputed (as with all the other changes in the patterns) to a different degree of solvation or to a partial structure loss.32
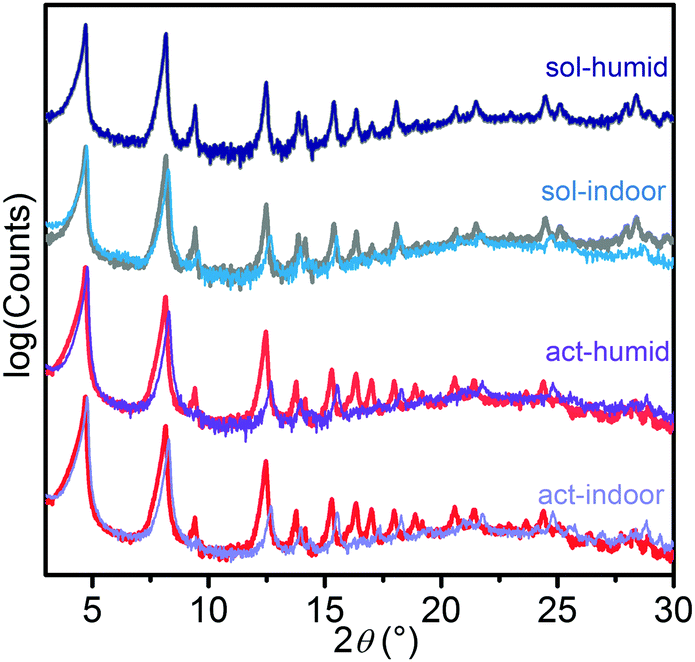 | ||
| Fig. 1 Ex situ XRD patterns of act and sol samples after exposure to air at 22–25 °C and 29–40 RH% (act-indoor, light violet line; sol-indoor, light blue line) or >86 RH% (act-humid, violet line; sol-humid, dark blue line) for 24 h. The patterns of the pristine act (red line) and sol (grey line) samples are also reported. The patterns have been normalised with respect to the peak at 4.7°. | ||
A completely different scenario was described using N2 volumetry. The small differences in the XRD patterns did not portend the >80% decrease in the surface area of all the samples (see Table 1 and Fig. 2), with the only exception of act-indoor. It is interesting to notice that after activation the material was almost unaffected by exposure to the atmosphere for 24 h for RH < 40%, at difference of sol sample (sol indoor). If a higher RH% is considered (humid samples), a strong decrease in porosity was observed (>80%) for both the sol (dark blue line in Fig. 2) and act (violet line) samples. Nevertheless, also in this case the preactivated sample still showed a higher stability (see Table 1). It is surprising that a lower structure loss was observed for sol-humid than for sol-indoor. This can be explained by its lower tendency to evaporate the solvent filling in the pores in the presence of a water saturated atmosphere, allowing slightly less damage of the sample to occur at high RH%. Material damaging was accompanied by a change in color of the material, as reported in Fig. S2 (ESI†) and in Table 1. The difference in the results of XRD and N2 volumetry suggests a lower sensitivity of XRD to evidencing structure degradation. This confirms what was recognized in the MOF literature, that surface area measurements are more effective than diffraction in evaluating the material integrity.13 The capping of the metal sites by solvent can cause a decrease of up to 40% in the surface area (see Section S7, ESI†). XRD inadequacy essentially originates in the disordered structure of the defects and their random distribution in the MOF, making the defects undetectable by means of diffraction and significantly hindering their identification using this technique. The general mechanism on the basis of MOF decomposition has a local character, especially in the first steps, followed only in a second step by structure collapse.15 The decomposition mechanism of the isomorph MOF-74-Mg has been recently studied by Tan et al.,16 showing that this MOF is not an exception from this point of view. In the first step of the process, a water molecule is adsorbed on Mg2+ through its oxygen atom. Water splitting then happens with proton transfer to the alcoholate group of the linker and the formation of a –OH species capping the metal site (Mg–OH).16 Because of the local nature of the process and the disordered location of these defects, the initial structure modifications would not be visible in the XRD patterns, but it will affect N2 volumetry results. XRD inadequacy can be also related to the rigid structure of Mg2(dobpdc) that would require a large number of missing Mg–O bonds before an extensive structure collapse (verified for Mg2(dobpdc) after only 48 h of exposure to air, see Fig. S6, ESI†). It can be hypothesised that in an intermediate step of the process, the missing of some Mg–O bonds would result in a partial fragmentation of the MOF crystals resulting in channel mismatch and/or pore blocking (see Fig. S13, ESI†). Such a process would be hardly detectable using XRD and IR (see Fig. S11b and Sections S8 and S9 in the ESI†), whereas it would significantly affect the surface area of the material and it would explain the observed decrease in the area, which is definitely larger than 40%.
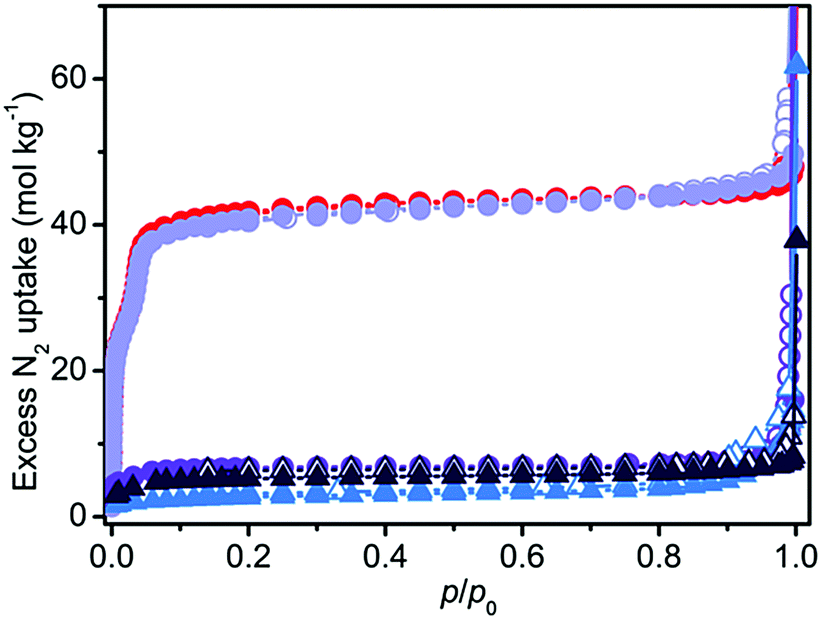 | ||
| Fig. 2 N2 isotherms obtained at 77 K for act (circles, red), act-indoor (circles, light violet), act-humid (circles, violet), sol-indoor (triangles, light blue) and sol-humid (triangles, dark blue). Filled and empty scatters refer to adsorption and desorption points, respectively. | ||
CO2 capacity at 25 °C and 1 bar did not present the trend expected on the basis of the surface area measurements (see Table 1 and Fig. S9, ESI†). The damaged materials present a CO2 capacity significantly larger than expected, retaining more than 60% of the initial uptake, instead of the 10% predicted from the surface area values. This can be appreciated in Fig. 3, where the CO2 capacity is reported as a function of the Langmuir surface area. It is therefore evident that CO2 capacity at a pressure lower than saturation cannot be used as a parameter to correctly evaluate the stability of MOFs. The reason for this discrepancy was investigated by means of IR spectroscopy (see Fig. S12a, ESI†).33 At RT for pressures up to 1 bar the main adsorption sites for CO2 are represented by open Mg2+ in the undamaged Mg2(dobpdc). IR spectroscopy evidenced that they also remain as the main adsorption sites after damaging (see Fig. S12, ESI†). In particular, IR suggests a decrease of only 30% of the Mg2+ population, in agreement with CO2 volumetry. The reactivation of the material, performed before volumetry, would allow the removal of adsorbed water molecules and the condensation of hydroxyls where possible, with a partial restoration of open Mg2+ sites. A discussion on the possible nature of the defects created by damaging in Mg2(dobpdc) is reported in the ESI† (see Sections S8 and S9).33
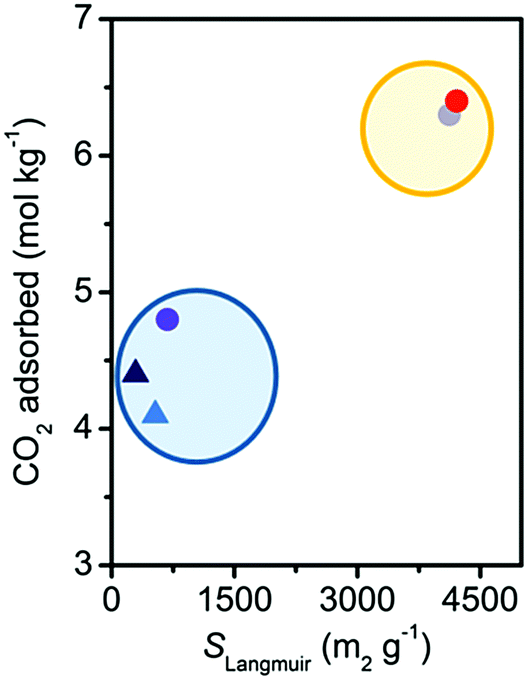 | ||
| Fig. 3 Dependence on SLangmuir of the CO2 uptake at 1 bar and 298 K for the Mg2(dobpdc) samples. Color coding as in Fig. 2. The blue and the yellow ellipses surround the damaged and undamaged samples, respectively. | ||
In conclusion, this study has evidenced that the resistance to atmospheric water of Mg2(dopbdc), a highly moisture sensitive MOF, can be significantly enhanced by careful preactivation. If preactivated, the MOF was stable in air at 25 °C and <50 RH% for up to 1 day, which is a sufficient amount of time to allow for easy processing of the material in a non-inert atmosphere. Moreover, relative humidity values in post-combustion flue gases from a coal-fired plant are in the 5–7% range,1 which is in a RH% range that would not affect the MOF structure (if carefully activated after each cycle). In this sense, the stability of the MOF was also checked by exposing it to 15 mbar of water at 30 °C for 1 h in a microbalance three times, fully reactivating it after each cycle (results not reported). These findings are in agreement with the results reported by Britt et al.27 for MOF-74-Mg. The material fully solvated with methanol showed a surface area loss of 93% under the same conditions. This difference can be explained by the enhanced hydrophilicity of the MOF if residual methanol molecules are present in its pores.12 Solvent molecules would facilitate water condensation in the MOF pores, accelerating the degradation process. Similar mechanisms have been previously described for water adsorption in MOFs.12 Moreover, methanol coordinated to Mg2+ ions is expected to be more easily displaced by water than other solvents as dimethylformamide, not further protecting the MOF against degradation. Differences with previous studies34 can be related to the different operating conditions but also to a lower defectivity of the present material.25,35 The absence of hydroxyls in the synthesized material was in fact verified using IR spectroscopy (Fig. S8, ESI†). An analogous mechanism to that described for precoordinated solvent molecules would work if defects are present in the materials, in general bearing –OH functionalities. These sites would increase the degradation kinetics acting as nucleation centers for water condensation. From this study it is evident that the moisture sensitivity of a MOF has to be retested if the synthesis conditions are modified.14 Similar results are expected also by applying other activation procedures, as for example supercritical CO2 drying.36 Nevertheless, the simplicity of thermal treatment would make this method more advantageous and it has been chosen to test this methodology in the present study.
Acknowledgements
Prof. Jeffrey R. Long and Dr Jeffrey D. Martell are acknowledged for the training on Mg2(dobpdc) synthesis. Cesare Atzori is acknowledged for help in using TOPAS.Notes and references
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- H. Furukawa, Y. B. Go, N. Ko, Y. K. Park, F. J. Uribe-Romo, J. Kim, M. O'Keeffe and O. M. Yaghi, Inorg. Chem., 2011, 50, 9147–9152 CrossRef CAS PubMed.
- W. A. Maza, A. J. Haring, S. R. Ahrenholtz, C. C. Epley, S. Y. Lin and A. J. Morris, Chem. Sci., 2016, 7, 719–727 RSC.
- J. X. Liu, W. C. Zhou, J. X. Liu, I. Howard, G. Kilibarda, S. Schlabach, D. Coupry, M. Addicoat, S. Yoneda, Y. Tsutsui, T. Sakurai, S. Seki, Z. B. Wang, P. Lindemann, E. Redel, T. Heine and C. Woll, Angew. Chem., Int. Ed., 2015, 54, 7441–7445 CrossRef CAS PubMed.
- J. X. Liu, W. C. Zhou, J. X. Liu, Y. Fujimori, T. Higashino, H. Imahori, X. Jiang, J. J. Zhao, T. Sakurai, Y. Hattori, W. Matsuda, S. Seki, S. K. Garlapati, S. Dasgupta, E. Redel, L. C. Sunag and C. Woll, J. Mater. Chem. A, 2016, 4, 12739–12747 CAS.
- J. G. Vitillo, RSC Adv., 2015, 5, 36192–36239 RSC.
- J. G. Vitillo, L. Regli, S. Chavan, G. Ricchiardi, G. Spoto, P. D. C. Dietzel, S. Bordiga and A. Zecchina, J. Am. Chem. Soc., 2008, 130, 8386–8396 CrossRef CAS PubMed.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed.
- D. Banerjee and J. B. Parise, Cryst. Growth Des., 2011, 11, 4704–4720 CAS.
- B. Panella and M. Hirscher, Adv. Mater., 2005, 17, 538–541 CrossRef CAS.
- G. Férey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guegan, Chem. Commun., 2003, 2976–2977 RSC.
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC and references therein.
- P. M. Schoenecker, C. G. Carson, H. Jasuja, C. J. J. Flemming and K. S. Walton, Ind. Eng. Chem. Res., 2012, 51, 6513–6519 CrossRef CAS.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- K. Tan, N. Nijem, Y. Gao, S. Zuluaga, J. Li, T. Thonhauser and Y. J. Chabal, CrystEngComm, 2015, 17, 247–260 RSC.
- K. Tan, S. Zuluaga, Q. Gong, P. Canepa, H. Wang, J. Li, Y. J. Chabal and T. Thonhauser, Chem. Mater., 2014, 26, 6886–6895 CrossRef CAS.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle Iii, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- Y. Jiao, C. R. Morelock, N. C. Burtch, W. P. Mounfield, J. T. Hungerford and K. S. Walton, Ind. Eng. Chem. Res., 2015, 54, 12408–12414 CrossRef CAS.
- Y. Liu, Y.-P. Chen, T.-F. Liu, A. A. Yakovenko, A. M. Raiff and H.-C. Zhou, CrystEngComm, 2013, 15, 9688–9693 RSC.
- P. D. C. Dietzel, V. Besikiotis and R. Blom, J. Mater. Chem., 2009, 19, 7362–7370 RSC.
- L. Valenzano, B. Civalleri, S. Chavan, G. Turnes Palomino, C. Otero Areán and S. Bordiga, J. Phys. Chem. C, 2010, 114, 11185–11191 CAS.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS PubMed.
- T.-H. Bae, M. R. Hudson, J. A. Mason, W. L. Queen, J. J. Dutton, K. Sumida, K. J. Micklash, S. S. Kaye, C. M. Brown and J. R. Long, Energy Environ. Sci., 2013, 6, 128–138 CAS.
- J. Liu, A. I. Benin, A. M. B. Furtado, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2011, 27, 11451–11456 CrossRef CAS PubMed.
- J. Kahr, R. E. Morris and P. A. Wright, CrystEngComm, 2013, 15, 9779–9786 RSC.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637–20640 CrossRef CAS PubMed.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS PubMed.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- W. R. Lee, H. Jo, L.-M. Yang, H. Lee, D. W. Ryu, K. S. Lim, J. H. Song, D. Y. Min, S. S. Han, J. G. Seo, Y. K. Park, D. Moon and C. S. Hong, Chem. Sci., 2015, 6, 3697–3705 RSC.
- B. Vlaisavljevich, S. O. Odoh, S. K. Schnell, A. L. Dzubak, K. Lee, N. Planas, J. B. Neaton, L. Gagliardi and B. Smit, Chem. Sci., 2015, 6, 5177–5185 RSC.
- D. D. Johnson, Phys. Rev. B, 1988, 38, 12807–12813 CrossRef.
- J. G. Vitillo, et al. , J. Phys. Chem. C, 2016 Search PubMed , in preparation.
- E. Mangano, J. Kahr, P. Wright and S. Brandani, Faraday Discuss., 2016, 192, 181–195 RSC.
- R. E. Morris and J. Cejka, Nat. Chem., 2015, 7, 381–388 CrossRef CAS PubMed.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D'Arco, Y. Noel, M. Causa, M. Rerat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287–1317 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Atomistic representations of Mg2(dobdc) and Mg2(dobpdc) structures, pictures of damaged samples, in situ XRPD patterns for act-indoor and sol-indoor, ex situ XRPD patterns for all the samples, FTIR spectra of activated act and act-humid in the presence of H2O and CO2, respectively; CO2 adsorption isotherms at 25 °C for all the samples, and the scheme of a possible degradation mechanism. See DOI: 10.1039/c6qm00220j |
| This journal is © the Partner Organisations 2017 |