
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Plasmonic substrates comprising gold nanostars efficiently regenerate cofactor molecules†
Ana
Sánchez-Iglesias
a,
Javier
Barroso
b,
Diego M.
Solís
c,
José M.
Taboada
d,
Fernando
Obelleiro
c,
Valeri
Pavlov
b,
Andrey
Chuvilin
 ef and
Marek
Grzelczak
*af
ef and
Marek
Grzelczak
*af
aBionanoplasmonics Laboratory, CIC biomaGUNE, Paseo de Miramón 182, 20009 Donostia-San Sebastián, Spain. E-mail: mgrzelczak@cicbiomagune.es
bBiosensing Laboratory, CIC biomaGUNE, Paseo de Miramón 182, 20009 Donostia-San Sebastián, Spain
cDepartamento de Teoría de la Señal y Comunicaciones, University of Vigo, 36301 Vigo, Spain
dDepartamento de Tecnología de los Computadores y de las Comunicaciones, University of Extremadura, 10003 Cáceres, Spain
eCIC nanoGUNE, Av. De Tolosa 76, 20018 Donostia-San Sebastián, Spain
fIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
First published on 22nd March 2016
Abstract
The light harvesting capacity of plasmonic nanoparticles is a fundamental feature for catalysing chemical reactions close to their surface. The efficiency of the photochemical processes depends not only on the geometrical aspects on a single particle level but also on the complexity of the multiparticle architectures. Although, the effect of the particle geometry is progressively understood in the relevant photochemical processes (water splitting and hydrogen evolution), there are experimental and theoretical needs for understanding the role of the shape in the multiparticle systems in the photocatalytic processes. Here we have shown that macroscopic plasmonic substrates comprising gold nanostars exhibit better efficiencies than nanorods or cubes in the photoregeneration of cofactor molecules. We performed photochemical and photoelectrochemical measurements, supported by theoretical simulations, showing that the unique geometry of nanostars – radially distributed spikes – contributes to stronger light absorption by the plasmonic film containing that type of nanoparticles.
Introduction
The unique properties of surface plasmon resonance ensure the consolidation of noble metal nanoparticles in photochemistry, a field of research and technology commanded currently by semiconductors.1–15 Some of the photochemical processes have been proposed recently in which plasmonic nanoparticles autonomously catalyse chemical reactions, such as water oxidation,16 hydrogen evolution,17 Suzuki coupling,18,19 alcohol oxidation20,21 or reduction of nitroaromatic compounds.22In a conventional photocatalytic system based on semiconductors, the performance in the catalytic process depends on the band gap energy of the material. In plasmon-based photochemistry, the photocatalytic capacity of particles is ruled by the energy of the surface plasmons, which is in turn related to the shape and size of the nanoparticles.23 In general, the isotropic particles with a diameter below 20 nm in the colloidal phase absorb light in the visible spectral range (400–600 nm) while anisotropic particles, of similar volume, absorb/scatter light in the infrared spectral range (700–1200 nm). The optical features of the particles can be further altered in a multiparticle scenario, in which strong plasmon coupling enables intense electric field enhancement, which benefits light–mater interactions. Thus, the spectroscopic sensitivity to geometry on single- and multi-particle levels can be exploited to construct devices that absorb across the full solar spectrum.24,25
According to recent studies, plasmonic nanoparticles can increase the rate of chemical reactions by multiple mechanisms including the photo-thermal effect, electric near-field enhancement, and generation of hot electrons.11,15 In all of these mechanistic pathways, the particle shape plays an important role. Long et al.26 demonstrated that the plasmonic Pd concave nanostructures exhibit superior catalytic performance than cubes or octahedra. They ascribed such behavior to the photothermal effect and local heat at the reaction sites. Trinh et al.19 showed that plasmonic Pd hexagons are better than, again, cubes and octahedra in plasmon-assisted Suzuki coupling, driven by the hot electron pathway. From a broader perspective, it becomes straightforward that the photocatalytic efficiency of plasmonic nanoparticles increases with a decrease in the symmetry and the volume of the nanocrystal.27–29 For example, gold nanorods exhibit better photocatalytic properties than isotropic nanoparticles (spheres, cubes or plates), which is due to the strong enhancement of the field inside the nanocrystal.30 Thus, inspired by recent theoretical and experimental developments, we expect that the branched nanoparticles can bring a significant contribution to the plasmon-assisted photochemistry, in a similar manner to their contribution to the field of surfaced enhanced spectroscopy.31 The presence of multiple and non-evenly distributed spikes in the gold nanostars can improve light harvesting that in turn alters the enhancement of electric near-field and the generation of energetic charge carriers.32
The presence of co-catalyst on the particle surface can additionally improve the rate of the desired reaction. While bare plasmonic particles may exhibit negligible catalytic activity it can be improved in the presence of a surface metal, such as Pt,33 Pd,34 Co,16 or Ni.35 These surface metals facilitate binding events of the molecules that undergo chemical conversion, thereby decreasing the kinetic energy required to run the reaction. The spatial distribution of the co-catalyst on the photocatalyst metal is also an important factor. Weng et al.36 have shown that non-centrosymmetric architecture in the CdSe–Au–Pt hybrid system improves hot electron transfer from Au and Pt subunits, minimising their thermalisation. Thus, finding the most optimal shape and surface properties of the plasmonic nanoparticles is currently a key task in the strongly progressing field of all-metal photochemistry.
The present work focuses on the comparative study of the shape effect of the gold nanoparticles on the photoregeneration of the coenzyme molecules (nicotinamide adenine dinucleotide – NADH) using triethanolamine (TEAOH) as a sacrificial agent. We have recently shown that Pt-decorated gold nanorods can regenerate NADH when irradiated with visible or infrared light.33 Here, we tested cubes, rods and star-like nanoparticles in photochemical and photoelectrochemical experiments, showing that gold nanostars are the most catalytically active. We attribute such behaviour to the greater capacity of light harvesting by nanostars in aggregated state – plasmonic substrates – as compared to rods or cubes. Numerical simulations of the multiparticle system confirmed experimental evidence.
Fabrication of plasmonic substrates containing particles of different shapes
Prior to the synthesis of the particles we assumed that the star-like geometry consists of isotropic cores and one-dimensional spikes pointing outwards. Thus, to perform a comparative photochemical study as precise as possible, we decided to synthesise cube-like nanoparticles with dimensions similar to that of nanostar cores, and nanorods with dimensions similar to that of nanostar spikes. We chose a seed-mediated growth approach to obtain nanoparticles of three desired shapes: cubic,37 rodlike,38 and star-like39 (Fig. 1). For gold nanocubes, the mean value of the edge was equal to 31.9 ± 1.9 nm. For gold nanorods, the average width and length were 45.2 ± 5.1 and 10.9 ± 1.0 nm, respectively (aspect ratio = 4.1). In the case of nanostars, the core diameter was 39.9 ± 5.9 nm. Regarding the spikes, the length and width at the base were 41.8 ± 9.6 and 13.1 ± 2.8 nm, respectively. Detailed geometrical characterization and TEM images of the nanoparticles are provided in the ESI (see Fig. S1 and S2†). The as-prepared particles were stabilised with a cationic surfactant (cethyltrimethylammonium bromide – CTAB) in water. Since the presence of Pt is essential in the photochemical reaction,33 we subjected the as-prepared particles toward the overgrowth with Pt. The reduction of Pt took place in the presence of Ag+ ions that ensured the formation of small domains, partially covering the surface. In the absence of silver, the platinum reduces in the form of a compact shell hindering the interaction of light with gold and thus decreasing the photochemical performance.33,35 | ||
| Fig. 1 Electron microscopy analysis of Pt-coated gold nanoparticles with different shapes: cube (a–c), rod (d–f) and star (g–i). SEM images (a, d, and g) and EDX mapping (b, e, and h) confirming the presence of Pt on the gold surface. (c, f, i) HRTEM images of the nanoparticles showing epitaxial overgrowth of the Pt domains. All particles were covered with 10% mol of Pt. | ||
The analysis by Scanning Electron Microscopy (SEM) confirmed the presence of Pt on Au particles. As shown in Fig. 1a, d and g, the presence of bright spots on the particle surface correspond to Pt domains. The EDX mapping on the individual particle further revealed that Pt domains distribute mostly on the edges of cubes and stars, while in the case of gold nanorods, Pt locates only on the tips. Such a difference in spatial distribution relates to the crystalline structure of the initial gold nanoparticles. Single-crystalline gold nanorods exhibit higher surface anisotropy that allows Pt deposition only on the tips. The lateral part of the rods is blocked by silver-based species.40 Although cubes are also single-crystalline, the surface energy is rather isotropic – equally covered by {100} facets. Thus, the Pt reduction takes places simultaneously on the edges and the flat parts. In the case of nanostars, the Pt reduction takes place on the whole spike surface. Although the star tips are highly anisotropic, the twinned planes (visible on HRTEM images) serve as nucleation points for Pt,41 leading to the homogenous distribution of co-catalyst domains.
The HRTEM analysis revealed that Pt domains are crystalline (Fig. 1c, f and i), thus, confirming the presence of tight heterojunctions between both metals (for more detailed characterisation see ESI, Fig. S3†). Importantly, the size of Pt domains was similar in all samples: 2.0 ± 0.4 nm (nanostars), 2.4 ± 0.5 nm (nanocubes) and 2.8 ± 0.5 nm (nanorods).
The UV-vis-NIR measurements of the colloidal solutions of bare and Pt-coated particles confirmed successful Pt reduction. Regardless of the initial shape of gold particles, the presence of platinum caused the redshift of localised surface plasmon bands (LSPB) that is due to the relative changes in the aspect ratio. For the gold nanocubes, the plasmon band shifted from 557 to 570 nm (Fig. 2a), while in the case of nanorods and nanostars the longitudinal plasmon bands redshifted by 57 and 36 nm, respectively (Fig. 2b and c). To further confirm such optical behavior we performed numerical simulations of the particles with different morphologies using water as a medium. We employed the methodology that implements accelerated calculations based on surface integral equations with the method of moments formulation and the multilevel fast multipole algorithm.42,43 The input parameters of individual particles were based on the TEM and SEM measurements. For nanocubes, Pt was located on the edges, while for nanorods, on the tips (Fig. 2d and e − insets). In the case of nanostars, the Pt shell was placed on the entire surface of spikes (Fig. 2f – inset) (for detailed parameterisation of each particle see Fig. S4†). The simulated spectra correlate well with experimental results, showing the expected redshift and dumping of the plasmon band, confirming the strong interaction between both metals.44,45
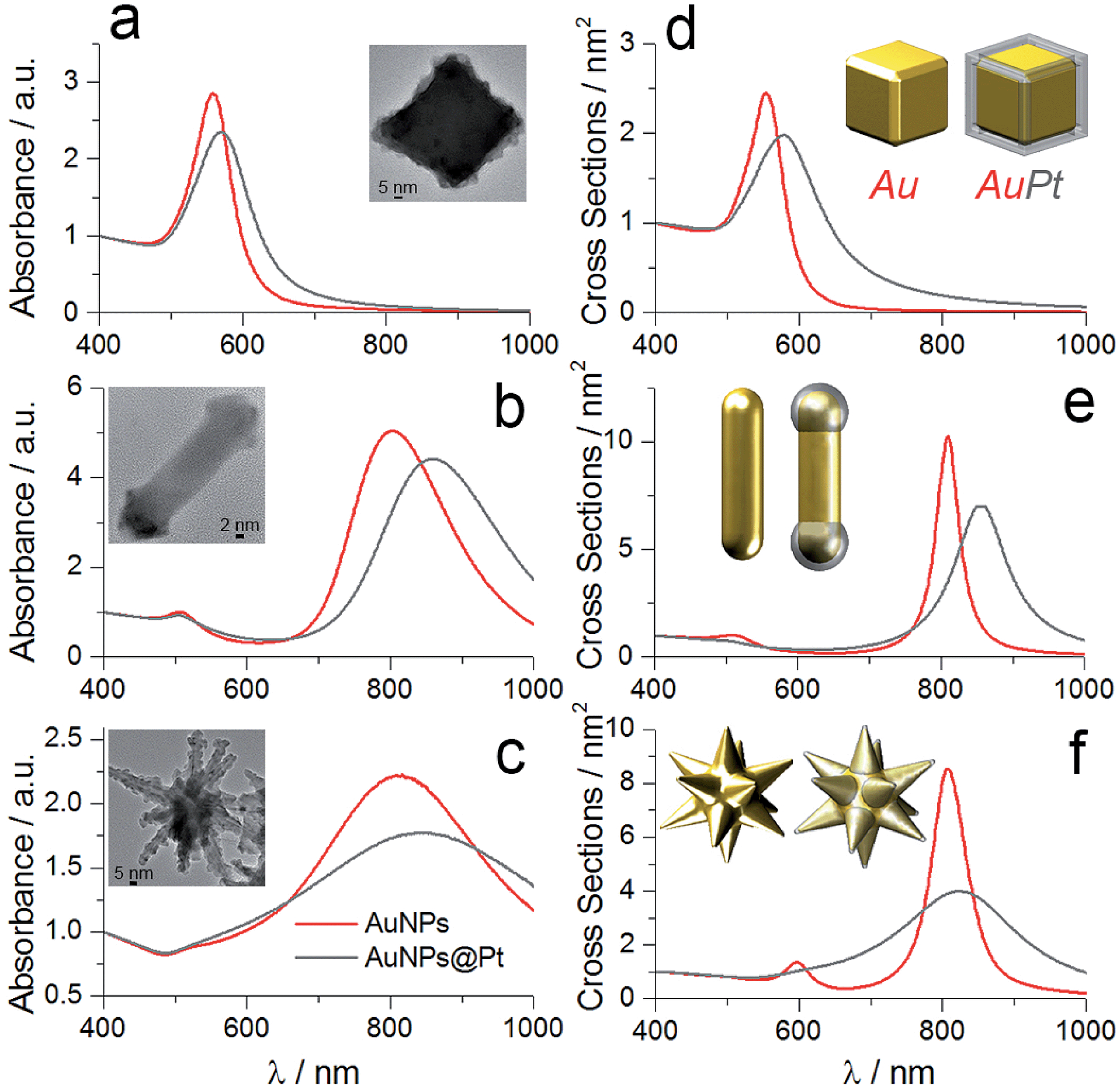 | ||
| Fig. 2 Optical characterisation of the nanoparticles in solution. (a–c) Experimental and (d–f) simulated spectra of the bare gold (red) and Pt-coated (grey) nanoparticles with cubic (a, d), rod-like (b, e) and star-like morphology (c, f). Insets in (d–f) represent three-dimensional images of the particles used in simulations. | ||
Photochemistry and photoelectrochemical characterisation of plasmonic substrates
Prior to photochemical characterisation, the particles were immobilised on the solid substrate (plasmonic substrate).33 Slow evaporation of the solution containing bare gold particles or Pt-coated on a glass substrate led to the formation of opaque films consisting of randomly distributed particles (Fig. S7†). In all cases we maintained a constant amount of metallic gold (5 mg).Plasmonic substrates containing different particle shapes were subjected toward NADH photoregeneration. In a typical experiment, the substrate was immersed in a mixture (1 mL) containing NAD+ (1 mM), phosphate buffer (pH 8) and TEAOH (1 M). The reduction of NAD+ was initiated under light irradiation (400–1200 nm, 150 mW cm2) at 35 °C. The plasmonic substrates could be removed from the solution at any time for spectroscopic validation of the regeneration process (Fig. S5†). Since NADH exhibits a characteristic absorption band at 340 nm (molar absorption coefficient 6220 M−1 cm−1), UV-vis spectroscopy provides a convenient qualitative control of the process. Overall, six substrates were evaluated, three containing bare gold nanoparticles and other three containing Pt-coated nanoparticles. Regardless of the particle shape, the regeneration of NADH was found to be ∼6 times higher for Pt-coated particles as compared to the bare ones. Importantly, the best performing nanoparticles were gold nanostars both without and with Pt on the surface, suggesting that branched geometry is more efficient in the present process (Fig. 3).
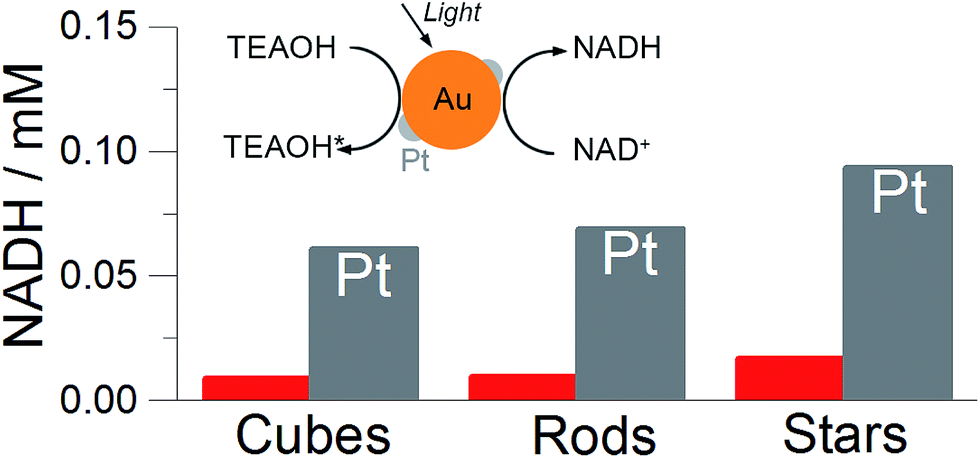 | ||
| Fig. 3 Photochemical regeneration of cofactor molecules (NADH) under visible light irradiation on the plasmonic substrate containing different particles shapes. (inset) Scheme showing the reaction of simultaneous oxidation of TEAOH and reduction of NAD+ to NADH. The final concentration of NADH after a two hour reaction in the presence of plasmonic substrate containing different particle shapes. Red bars represented the concentration of the NADH when bare nanoparticles were used; the grey columns correspond to Pt-coated nanoparticles. In all experiments, the substrates were immersed in the solution containing TEAOH (1 M), NAD+ (1 mM) and phosphate buffer (pH 8) and irradiated with light (400–1200 nm, 150 mW cm−2) at 35 °C for two hours. | ||
Typically, the photoregeneration of NADH involves the transfer of 2e− and H+ from TEAOH to NADH, which is accomplished by simultaneous oxidation and reduction reactions. To ensure that the process takes place on the particle surface, we evaluated the interaction of TEAOH molecules with plasmonic nanoparticles by comparing the position of the maximum of LSPB in water and 0.1 M TEAOH. The LSPB blue shifted 2 nm in the case of cubes and 3 nm for nanorods and stars (Fig. S6†). Such behavior is typical for the particles that are exposed to the electron-rich environment and proves the spontaneous electron injection from the molecules to particles.46 Although the Pt domains are crucial for the oxidation of TEAOH molecules, it is hard to assess to what extent the reduction of NAD+ to NADH takes place on platinum.33 Since the photooxidation of TEAOH is a multistep process involving the formation of many intermediate species,47 we found it appropriate to perform separate photoelectrochemical analysis of the oxidation reaction.
Photoelectrochemical measurements confirmed the trends observed for different particle shapes in photochemical reactions. We performed photoelectrochemical studies of TEAOH oxidation on the electrode coated with nanoparticles. We used a three-electrode system (DropSens) consisting of a carbon working electrode, silver pseudo-reference, and carbon counter electrode. The solutions containing particles were drop-cast on the working electrodes and slowly evaporated under ambient conditions to maximise their homogenous distribution. Although we noticed the inevitable formation of coffee rings on the electrode edges, the surface coverage was similar in all samples containing particles of different shapes (Fig. S7†).
The electrode was covered with a 0.05 mL of the solution containing TEAOH (1 M) and phosphate buffer. To obtain the amperometric i–t curve, the current was stabilised for 6 min at circuit potential (ca. −0.36 V). Then the light was switched on for 10 seconds and an immediate increase in the current was observed (Fig. 4). We noticed that the presence of Pt on the gold surface was crucial to oxidise TEAOH. In the presence of Pt, the photocurrent was approximately ten times higher as compared to the photocurrent obtained from the bare gold nanoparticles regardless of their shape (red line in Fig. 4a, S8 and S9†). Such improvement is due to the low interfacial energy barrier between Pt and Au and suggests the contribution of the photoelectrocatalytic process.36
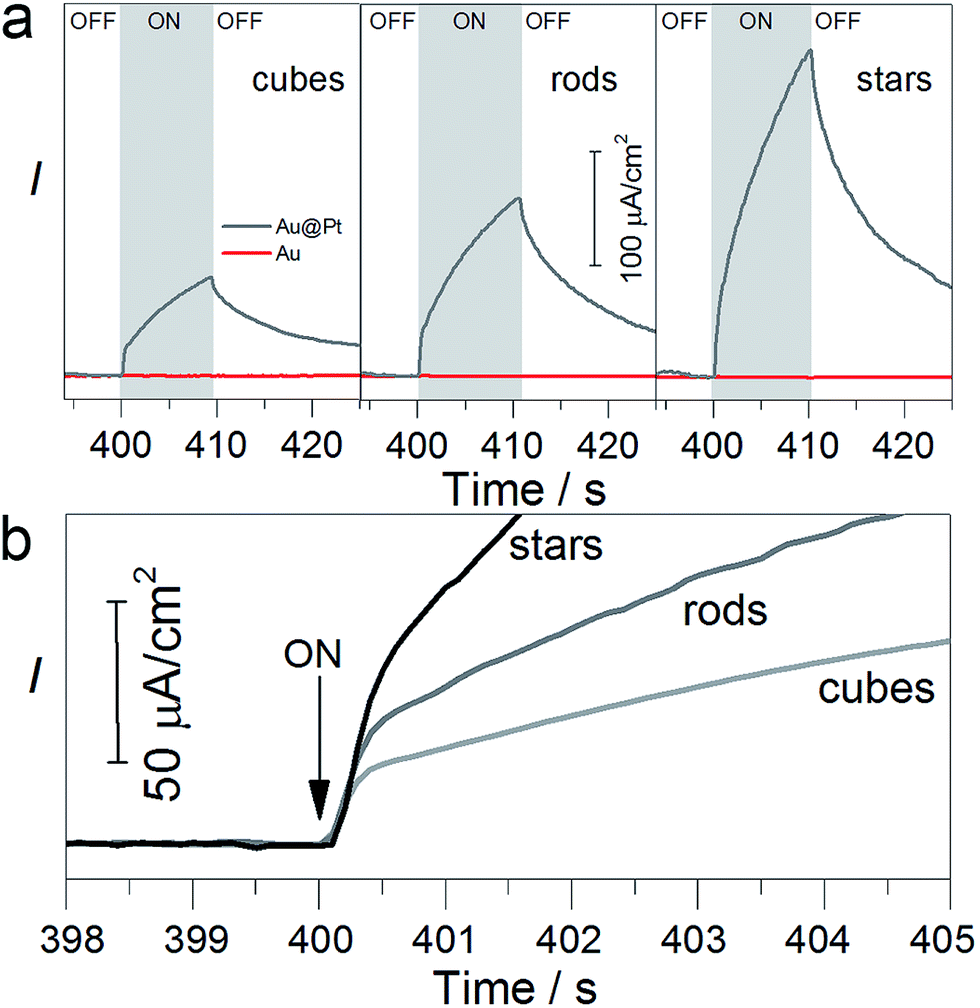 | ||
| Fig. 4 Photoelectrochemical characterisation of TEAOH oxidation on the nanoparticles with different shapes. (a) Amperometric i–t curves for bare cubes, rods and stars (red lines) and platinum-coated (grey lines). Photocurrent exhibits ‘fast’ (0.5 s) and ‘slow’ components (10 s). (b) Fast current component for three samples containing Pt-coated particles of different shapes showing better performance of stars than rods and cubes. In all experiments, the electrodes were immersed in the solution containing TEAOH (1 M), phosphate buffer (pH 8) and irradiated with light (400–1200 nm, 150 mW cm−2) at 0 V at 25 °C. Each electrode was covered by the same amount of nanoparticles in terms of metallic gold. | ||
For all the particles coated with Pt, the photocurrent consisted of two temporal components: a fast component that increased with time constant ∼0.5 s and an additional slow component that constantly increased with a time of ∼10 s (Fig. 4a). The origin of the “fast” components is the contribution of the photoelectrocatalytic process that can involve hot electron generation that immediately flow to the counter electrode.48 The “slow” components are a result of the photothermal effect of the illumination.49 Importantly, the magnitude of photocurrent, for both slow and fast components, depends on the particle shape and increases in the following order: cubes, rods, and stars, which is in good agreement with photochemical reactions (Fig. 3). The photocurrent of the nanostars added 250 μA cm−2 after 10 s of illumination, which is 3.5 and 1.5 times higher than that of nanocubes and nanorods, respectively. A similar trend has been observed for the “fast” component: the photocurrent of the nanostars was 2.5 and 1.5 times higher than that of the nanocubes and nanorods, respectively. SEM inspection confirmed morphological stability of the nanoparticles (Fig. S10†). Overall, the photoelectrochemical measurements correlate with photochemical reactions, showing that the photoelectrocatalytic effect plays an important role and is strictly related to the particle shape.
Since the presence of Pt on the gold particle surface was crucial for achieving the photochemical reaction, we measured the relative amount of this metal for different plasmonic substrates. For estimation of the Pt/Au ratio we used XPS techniques (Fig. S11†), showing that stars and cubes contained a similar amount of surface platinum with respect to gold: Pt/Au = 0.22 (stars) and Pt/Au = 0.20 (cubes). Gold nanorods contained less Pt (Pt/Au = 0.09) that is due to the limited surface area accessible for Pt – tips. Relatively similar values of Pt and Au for different shapes suggest that the morphology of the initial particles is a key parameter that determines the distribution of the particles in the film and defines their interaction with incident light.
Optical properties of plasmonic substrates – experimental and numerical characterisation
Based on the experimental results we assume that better light absorption determines the superior photocatalytic performance of the substrate containing nanostars, as compared to the nanorods or cubes. Visual inspection shows that the substrates containing cubes and rods are shinier than the substrates containing nanostars, which are practically black (see inset in Fig. 5b). Diffuse reflection measurements confirmed such observations, revealing that the substrates containing gold nanorods or cubes reflect more light in the spectral range above 500 nm (Fig. 5b). The difference in light reflection for all shapes is even more pronounced at the maximum of the intensity of the incident light (ca. 700 nm) that we used in all experiments (Fig. 5a), additionally affecting the photochemical performance.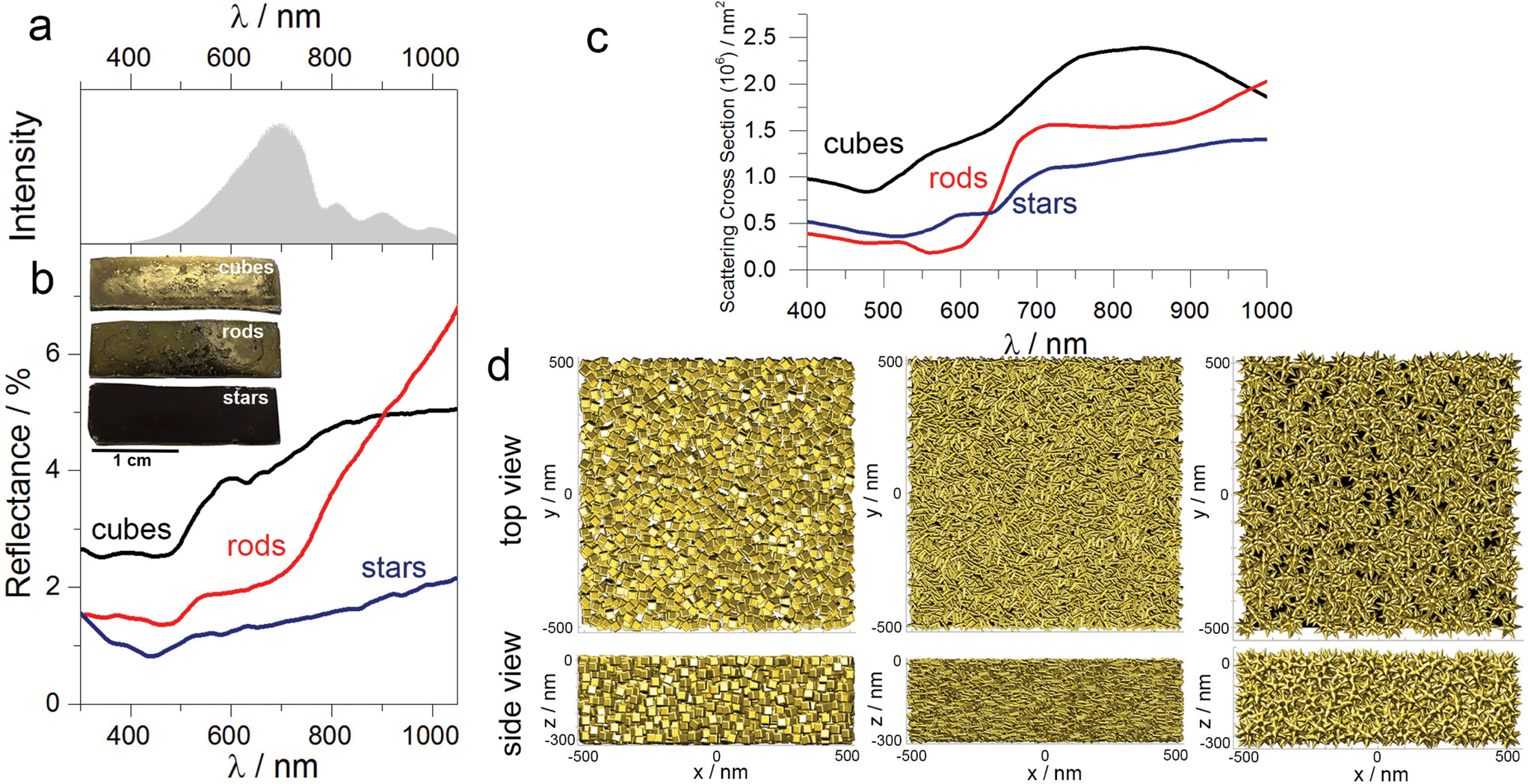 | ||
| Fig. 5 Optical characterisation of the plasmonic substrate. (a) Spectral feature of the lamp used in all experiments. (b) Diffuse reflection spectra of substrates comprising cubes, rods and stars, showing better light harvesting capacity by star-containing plasmonic substrates. (inset) Images of the substrates comprising gold nanoparticles. (c) Simulated scattering cross-section spectra of the substrate comprising three different morphologies. (d) Top view (upper panel) and side view (lower panel) of modelled architectures used for spectra simulations in (c): nanoparticles with different geometries compacted in a 0.3 μm3 box (minimum surface-to-surface separation is 1 nm). | ||
To further correlate our experimental results, we simulated the scattering cross-section spectra of substrates comprising nanoparticles with different shapes. Each model plasmonic substrate contained randomly packed particles: 2194 (cubes), 8071 (rods) and 501 (nanostars), filling a film-like box of 0.03 μm3 (Fig. 5d), with a minimum distance between the particle surface of 1 nm. The calculated scattering spectra displayed typical broadening, suggesting multiple plasmon couplings, which are in good agreement with the experimental reflectance spectra (compare Fig. 5b and c). These results indicate that nanostars in the aggregated mode scatter less amount of light as compared to the nanocubes or nanorods. We hypothesise, then, that such an optical response is related to the intrinsic shape of the individual building blocks. During the drying process, the nanorods or cubes sediment in-parallel to the substrate surface, pointing sidewise toward the incident light, thereby increasing the contribution of the specular reflection. In the case of nanostars, however, radial distribution of spikes increases the diffuse reflection, and, therefore, minimise the amount of light that is bounced back to the medium. Such difference in optical properties in the aggregated state is especially visible in the case of gold nanorods and nanostars. Although the maximum of LSPB for both geometries in solution is very similar (Fig. 2), the reflectance above 800 nm is much larger in the case of nanorods than for stars, confirming more efficient light absorption by the plasmonic substrate containing nanostars.
It is important to stress, that spike-like, or conical shape is more appropriate for photochemical application than nanorods, or cylinders. Although, we cannot compare, at the present stage, the performance of spike only and nanorods, we can support our speculation by the recent work by Weng et al.,36 who showed that nanoparticles that lack the central symmetry exhibit better performance due to the more efficient charge separation. Single spike located on the star core is a non-centrosymmetric object because of its unequal width on the tips. Therefore, we postulate that the unique geometry of the spike tips can render better photochemical conversion on nanostars than other shapes.
Finally, the unique geometry of nanostars – radial spike distribution – hinders close contact between the particles in the plasmonic film, facilitating the formation of the pores that improves the access of the molecules to the particle surface. Thus, the active surface area of the plasmonic substrate containing nanostars can be higher than that of the nanorods and cubes (Fig. S12†).
Conclusions
In conclusion, we studied the shape effect of the platinum coated gold nanoparticles on the photoregeneration of cofactor molecules. In photochemical and photoelectrochemical measurements, we found that substrates comprising branched nanoparticles are better photocatalysts as compared to the rod- or cube-like particles. We explain such behaviour by the geometrical features of gold nanostars that efficiently harvest the light improving photoelectrocatalytic effect. Future studies envisage the study of photochemical performance using nanostars with different spike lengths. This work opens up possibilities for efficient all-metal photochemistry and establish the basement for the design of plasmonic catalytic materials.Experimental
Materials
Gold(III) chloride hydrate (HAuCl4), sodium citrate tribasic dihydrate, hexadecyltrimethylammonium bromide (CTAB), hexadecyltrimethylammonium chloride (CTAC), silver nitrate (AgNO3), hydrochloric acid (HCl), ascorbic acid, sodium borohydride (NaBH4) and potassium(II) tetrachloroplatinate (K2PtCl4) were purchased from Sigma-Aldrich and were used without further purification.Characterisation
For the realistic simulation of the large-scale plasmonic systems considered here, comprising assemblies of more than 8000 nanoparticles, SIE-MoM was expedited via the multilevel fast multipole algorithm-fast Fourier transform (MLFMA-FFT),51,52 and combined with robust Schwarz domain decomposition preconditioning,53 resulting in a high algorithmic efficiency—computational cost of O(N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logN)—along with high-scalability via parallelization using multicore computer clusters.
logN)—along with high-scalability via parallelization using multicore computer clusters.
The simulations were carried out on a workstation with four 16-core Intel Xeon E7-8867v3 45 MB Smart-Cache processors at 2.50 GHz. A high surface mesh density was considered leading to dense matrix systems of 5.7, 7.6 and 4.1 million unknowns for the substrates comprising cubes, rods and stars, respectively. A relative error norm of 10−5 was set to stop the Krylov iterative solver. Considering the above, the computation times per wavelength ranged from 402 s (at 400 nm) to 2450 s (at 1000 nm) for the cube substrate, 382 to 2057 s for the rod substrate, and 466 to 3138 s for the star substrate. The memory usage ranged from 27 to 93 GB for the cube substrate, 71 to 149 GB for the rod substrate, and 23.5 to 67 GB for the star substrate.
Synthetic procedures
Acknowledgements
This work was supported by the Spanish Ministry of Economy and Competitiveness (MAT2013-49375-EXP, BIO2014-59741-R) and BBVA Foundation – “Primera convocatoria de ayudas fundación BBVA a investigadores, innovadores y creadores culturales”. D. M. S., J. M. T, and F. O acknowledge funding from the European Regional Development Fund (ERDF) and the Spanish Ministry of Economy and Competitiveness (Projects MAT2014-58201-C2-1-R, MAT2014-58201-C2-2-R), from the ERDF and the Galician Regional Government under agreement for funding the Atlantic Research Center for Information and Communication Technologies (AtlantTIC), and from the ERDF and the Extremadura Regional Government (Junta de Extremadura) under Project IB13185. We thank Dr Luis Yate for assistance with XPS analysis.Notes and references
- S. Gao, K. Ueno and H. Misawa, Acc. Chem. Res., 2011, 44, 251–260 CrossRef CAS PubMed.
- S. C. Warren and E. Thimsen, Energy Environ. Sci., 2012, 5, 5133–5146 CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- J. Y. Park, L. R. Baker and G. A. Somorjai, Chem. Rev., 2015, 115, 2781–2817 CrossRef CAS PubMed.
- S. Linic, P. Christopher, H. Xin and A. Marimuthu, Acc. Chem. Res., 2013, 46, 1890–1899 CrossRef CAS PubMed.
- J. C. Scaiano and K. Stamplecoskie, J. Phys. Chem. Lett., 2013, 4, 1177–1187 CrossRef CAS PubMed.
- M. Xiao, R. Jiang, F. Wang, C. Fang, J. Wang and J. Yu, J. Mater. Chem. A, 2013, 1, 5790–5805 CAS.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2013, 4, 116–128 CrossRef.
- G. Baffou and R. Quidant, Chem. Soc. Rev., 2014, 43, 3898–3907 RSC.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- J. G. Smith, J. A. Faucheaux and P. K. Jain, Nano Today, 2015, 10, 67–80 CrossRef CAS.
- R. Long, Y. Li, L. Song and Y. Xiong, Small, 2015, 11, 3873–3889 CrossRef CAS PubMed.
- P. Zhang, T. Wang and J. Gong, Adv. Mater., 2015, 27, 5328–5342 CrossRef CAS PubMed.
- S. Mubeen, J. Lee, N. Singh, S. Krämer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247–251 CrossRef CAS PubMed.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 136, 6870–6873 CrossRef CAS PubMed.
- F. Wang, C. Li, H. Chen, R. Jiang, L.-D. Sun, Q. Li, J. Wang, J. C. Yu and C.-H. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef CAS PubMed.
- T. T. Trinh, R. Sato, M. Sakamoto, Y. Fujiyoshi, M. Haruta, H. Kurata and T. Teranishi, Nanoscale, 2015, 7, 12435–12444 RSC.
- X. Huang, Y. Li, Y. Chen, H. Zhou, X. Duan and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 6063–6067 CrossRef CAS PubMed.
- G. L. Hallett-Tapley, M. J. Silvero, C. J. Bueno-Alejo, M. González-Béjar, C. D. McTiernan, M. Grenier, J. C. Netto-Ferreira and J. C. Scaiano, J. Phys. Chem. C, 2013, 117, 12279–12288 CAS.
- W. Xie and S. Schlücker, Nat. Commun., 2015, 6, 7570 CrossRef PubMed.
- L. M. Liz-Marzán, Langmuir, 2006, 22, 32–41 CrossRef PubMed.
- M. Grzelczak and L. M. Liz-Marzán, Langmuir, 2013, 29, 4652–4663 CrossRef CAS PubMed.
- M. Moskovits, Nat. Nanotechnol., 2015, 10, 6–8 CrossRef CAS PubMed.
- R. Long, Z. Rao, K. Mao, Y. Li, C. Zhang, Q. Liu, C. Wang, Z.-Y. Li, X. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2015, 54, 2425–2430 CrossRef CAS PubMed.
- A. O. Govorov, H. Zhang and Y. K. Gun'ko, J. Phys. Chem. C, 2013, 117, 16616–16631 CAS.
- A. O. Govorov, H. Zhang, H. V. Demir and Y. K. Gun'ko, Nano Today, 2014, 9, 85–101 CrossRef CAS.
- A. O. Govorov and H. Zhang, J. Phys. Chem. C, 2015, 119, 6181–6194 CAS.
- C. S. Kumarasinghe, M. Premaratne, Q. Bao and G. P. Agrawal, Sci. Rep., 2015, 5, 12140 CrossRef CAS PubMed.
- A. Guerrero-Martínez, S. Barbosa, I. Pastoriza-Santos and L. M. Liz-Marzán, Curr. Opin. Colloid Interface Sci., 2011, 16, 118–127 CrossRef.
- S. Kumar, et al. , Nanotechnology, 2008, 19, 015606 CrossRef PubMed.
- A. Sánchez-Iglesias, A. Chuvilin and M. Grzelczak, Chem. Commun., 2015, 51, 5330–5333 RSC.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 137, 948–957 CrossRef PubMed.
- C. Kim, Y. Kwon and H. Lee, Chem. Commun., 2015, 51, 12316–12319 RSC.
- L. Weng, H. Zhang, A. O. Govorov and M. Ouyang, Nat. Commun., 2014, 5, 4792 CrossRef CAS PubMed.
- J. Zhang, M. R. Langille, M. L. Personick, K. Zhang, S. Li and C. A. Mirkin, J. Am. Chem. Soc., 2010, 132, 14012–14014 CrossRef CAS PubMed.
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2005, 109, 22192–22200 CrossRef CAS PubMed.
- H. Yuan, C. G. Khoury, H. Hwang, C. M. Wilson, G. A. Grant and T. Vo-Dinh, Nanotechnology, 2012, 23, 075102 CrossRef PubMed.
- M. Grzelczak, J. Pérez-Juste, B. Rodriguez-Gonzalez and L. Liz-Marzán, J. Mater. Chem., 2006, 16, 3946–3951 RSC.
- M. R. Langille, J. Zhang, M. L. Personick, S. Li and C. A. Mirkin, Science, 2012, 337, 954–957 CrossRef CAS PubMed.
- D. M. Solís, J. M. Taboada, F. Obelleiro, L. M. Liz-Marzán and F. J. García de Abajo, ACS Nano, 2014, 8, 7559–7570 CrossRef PubMed.
- C. Hamon, S. M. Novikov, L. Scarabelli, D. M. Solís, T. Altantzis, S. Bals, J. M. Taboada, F. Obelleiro and L. M. Liz-Marzán, ACS Photonics, 2015, 2, 1482–1488 CrossRef CAS.
- M. Grzelczak, J. Pérez-Juste, F. J. García de Abajo and L. M. Liz-Marzán, J. Phys. Chem. C, 2007, 111, 6183–6188 CAS.
- H.-J. Jang, S. Hong, S. Ham, K. L. Shuford and S. Park, Nanoscale, 2014, 6, 7339–7345 RSC.
- C. Novo, A. M. Funston and P. Mulvaney, Nat. Nanotechnol., 2008, 3, 598–602 CrossRef CAS PubMed.
- S. Horikoshi, N. Watanabe, M. Mukae, H. Hidaka and N. Serpone, New J. Chem., 2001, 25, 999–1005 RSC.
- J. Lee, S. Mubeen, X. Ji, G. D. Stucky and M. Moskovits, Nano Lett., 2012, 12, 5014–5019 CrossRef CAS PubMed.
- H. Yang, L.-Q. He, Y.-W. Hu, X. Lu, G.-R. Li, B. Liu, B. Ren, Y. Tong and P.-P. Fang, Angew. Chem., Int. Ed., 2015, 127, 11624–11628 CrossRef.
- D. M. Solís, J. M. Taboada and F. O. Basteiro, IEEE Trans. Antennas Propag., 2015, 63, 2141–2152 CrossRef.
- J. M. Taboada, M. G. Araújo, F. O. Basteiro, J. L. Rodriguez and L. Landesa, Proc. IEEE, 2013, 101, 350–363 CrossRef.
- J. Song, C.-C. Lu and W. C. Chew, IEEE Trans. Antennas Propag., 1997, 45, 1488–1493 CrossRef.
- D. M. Solís, M. G. Araújo, L. Landesa, S. García, J. M. Taboada and F. Obelleiro, IEEE Photonics J., 2015, 7, 1–9 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images, size analysis of the nanoparticles, control experiments for photoelectrochemical measurements, and SEM images. See DOI: 10.1039/c6ta01770c |
| This journal is © The Royal Society of Chemistry 2016 |