
Surfactants as additives make the structures of organic–inorganic hybrid bromoplumbates diverse†
Guangfeng
Liu‡
a,
Jie
Liu‡
b,
Xutang
Tao
b,
Dong-sheng
Li
*c and
Qichun
Zhang
*ad
aSchool of Materials Science and Engineering, Nanyang Technological University, Singapore. E-mail: qczhang@ntu.edu.sg
bState Key Laboratory of Crystal Materials, Shandong University, China
cCollege of Materials and Chemical Engineering, Hubei Provincial Collaborative Innovation Center for New Energy Microgrid, China. and Three Gorges University, China. E-mail: lidongsheng1@126.com
dDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore
First published on 5th September 2016
Abstract
Although surfactant-facilitated crystallization has been widely used in pharmaceuticals, metal–organic frameworks (MOFs), chalcogenides and colloidal nanocrystals, using surfactants as additives to control the crystal growth of metal halides in the macroscale is unexplored. In this contribution, we used N-methylate 4,4′-bipyridine bromoplumbates as a model system to prove that surfactants as additives could make the structures of organic–inorganic hybrid bromoplumbates diverse. By only changing different surfactants, four new hybrid bromoplumbates including the first novel 3D open framework hybrid bromoplumbate [(C12N2H14)2Pb7Br18] (except perovskites) and a pair of 1D polymorphs were obtained. The related physical properties including optical band gaps, FT-IR and TGA-DSC for the as-obtained compounds have been carefully investigated. In addition, an interesting mother-liquor-facilitated single-crystal-to-single-crystal (SCSC) phase transition is also revealed. Our strategy to employ surfactants to control the crystal growth of metal halides could offer exciting opportunities for discovering novel organic–inorganic hybrid crystals with diverse structures and distinctive properties.
Developing new strategies to grow crystalline materials in the macroscale is of importance in both fundamental sciences and technological applications.1 Normally, crystallization of components from a solution to a crystalline state is strongly affected by the crystal-growing parameters (e.g. evaporation speed, cooling pace, and stirring rate),2 and each of these adjustments could result in new structures. However, the most effective way to obtain diverse structures during crystal growth is to introduce different additives into the crystal-growing system because the subtle changes during the nucleation stage can lead to spectacular changes in structures. Although polymer-induced phase selection has been widely investigated in pharmaceuticals and metal–organic frameworks (MOFs),3 the use of surfactants as additives to approach new phases or particular structures of organic–inorganic hybrid halogenated plumbates is unprecedented. Only recently, we have demonstrated that surfactants can act as reaction media to prepare new crystalline chalcogenides and MOF materials.4 However, in order to deeply understand if surfactants can directly induce the formation of new structures at the initial nucleation stage, we decided to only alter the surfactant additives while keeping other conditions identical (e.g. organic and inorganic components, solvents, reaction temperatures and times, extent of supersaturation) during the crystal-growing process.
The system we have chosen in this study is organic–inorganic hybrid halogenated plumbates because these materials have promising applications in optoelectronics and high-efficiency solar cells.5 To our surprise, no example about the 3D hybrid halogenated plumbates (except perovskites) constructed from purely PbX6 octahedra has been reported before this work. Only one exceptional case is the hybrid iodoplumbate (to our best knowledge)6 and no 3D bromoplumbate is documented in the literature. More importantly, controlling the selection of polymorphs and understanding phase transitions in hybrid halogenated plumbates should be very helpful for researchers to discover new structures with specific chemical and physical properties because different spatial arrangements of PbX6 components could lead to various electrical properties as well as the optical band gaps of these materials.7
In this research, we chose 4,4′-bipyridine (4,4′-bpy), PbBr2 and HI as starting materials, CH3OH as a solvent and different surfactants (PVP, SDS and PEG-400) as additives through in situ N-methylation of 4,4′-bpy under solvothermal conditions to synthesize N-methylated 4,4′-bpy bromoplumbates. In every reaction route, all crystallization parameters except the types of surfactant additives are kept the same (the details are described in the Experimental section and Table S1†). Single-crystal X-ray diffraction (SCXRD) analyses reveal that four types of hybrid bromoplumbate crystals (CCDC numbers for 1–4: 1486414–1486417), namely, C6NH7PbBr3 (1), C6NH7PbBr3 (2), (C12N2H14)2Pb7Br18 (3) and (C12N2H14)2Pb3Br10 (4) have been successfully obtained (Fig. 1 and Table S2†). All of these compounds employ the same N-methylated 4,4′-bpy cations as charge-balance species while inorganic components exhibit variable structural features based on the connecting modes of the octahedral PbBr6 units. These as-obtained crystals demonstrated the feasibility of surfactant-induced heteronucleations in hybrid bromoplumbates. It is noteworthy that compound 3 is the first 3D open-framework hybrid bromoplumbate (except perovskites). Moreover, compounds 1 and 2 are a pair of polymorphs with a SCSC phase transition, which will be discussed later.
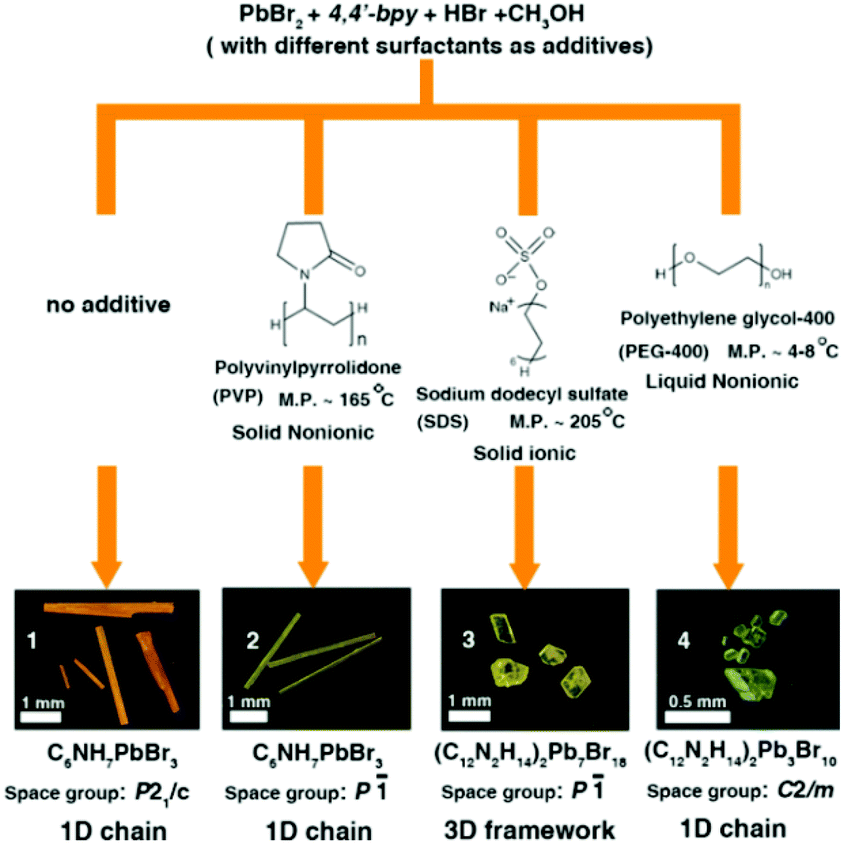 | ||
| Fig. 1 Reaction routes, pictures, chemical formulae, space group and dimensionality of the as-obtained hybrid bromoplumbates 1–4. | ||
Fig. 1 shows the synthetic routes and the images of the as-resulted crystals by this surfactant-induced strategy. Three different surfactants (PVP, SDS and PEG-400) have been chosen as additives based on their melting points (liquid, low or high melting point) and types (ionic or nonionic). At first, we implemented all reactions at 120 °C. Compound 1 was prepared without any additive, which can be considered as the homogeneous nucleation process. However, the processes of surfactant-induced crystallization in 2–4 mainly depended on heterogeneous nucleation. Compounds 1, 2 and 3 can be obtained in relatively high yields (more than 50%) while the yield of 4 is only about 10% (based on the raw material PbBr2, the details described in the Experimental section†). The low yield of compound 4 can be attributed to the fact that the PEG-400 will partly decompose to ethyoxyl groups in a low PH value and high temperature environment. Then, the as-formed ethyoxyl groups can react with 4,4′-bpy molecules, which will further consume an extra amount of 4,4′-bpy raw materials. For comparison, we chose 100 °C and 80 °C as the reaction temperatures for all routes. We found that compounds 1 and 2 can still be obtained with remarkably lower yields (<20%) than those at 120 °C. The yield of compound 4 with a little bit increase (about 15%) may be due to the inhibition of the PEG-400 decomposition at a lower temperature. Unfortunately, we didn't obtain compound 3 through route 3 at 100 °C. It is worth noting that all compounds cannot be obtained at 80 °C. These results further confirm that the surfactant-induced heterogeneous nucleation is the main factor to obtain the diverse crystal structures of these hybrid bromoplumbates and we believe that this method could open up a new pathway for the design of novel hybrid materials.
Compounds 1–4 not only show different colours and morphologies (1, red and prism; 2, gold and prism; 3, yellow and plate; 4, pale yellow and bulk, Fig. 1) but also exhibit diverse architectures of structures. Because all four compounds have the same cations as charge-balance species, different colours are not from the charge-transfer mechanism as previously reported in the CdS system,8 instead, we believe that they should come from the packing mode of PbBr3 units. Fig. 2 shows the packing patterns of compounds 1–4. It can be clearly seen that all the structures employ N-methylated 4,4′-bpy organic species as charge-balance cations. The dihedral angles between the two pyridine rings for compounds 1–4 ranging from 0° to 31.02° (0°, 1; 27.79°, 2; 31.02°, 3; 2.18°, 4, Fig. S1†) indicate that these organic cations are flexible enough to satisfy the spatial arrangement of inorganic clusters. Moreover, N-methylated 4,4′-bpy shows a perfect planarity in the structure of 1 (the dihedral angle = 0°), suggesting that the surfactant-free homogeneous nucleation pathway is smoother than the surfactant-induced heterogeneous nucleation process.
 | ||
| Fig. 2 (a) and (b) View of the 1D anionic [PbBr3]2n−n chains in 1 and 2, respectively, along the a axis. (c) The 16-ring channels along the a axis in the 3D open-framework structure of 3. (d) View of the packing of 4 along the c axis separated by 1D chains [Pb3Br10]4n−n. Color scheme: Pb(II), gold; Br, pink; C, black; N, blue. H atoms are omitted for clarity. | ||
The SCXRD results reveal that compounds 1 and 2 have the same chemical formula as C6NH7PbBr3 but crystallize in different space groups as P21/c and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , respectively, which means 1 and 2 are a pair of polymorphs. Fig. 2a and b show that although 1D anionic [PbBr3]2n−n chains are formed in both phases, the architectures of the 1D chains are totally different. The polyhedral patterns of 1 and 2 exhibit this difference more clearly: the chains in 1 adopt a close packed mode through all edge-sharing PbBr6 octahedra while the chains in 2 show a much looser feature resulting from the coexistence of point-sharing and edge-sharing modes of PbBr6 octahedra (Fig. 3a and b). These factors suggest that 2 should be a metastable phase compared with 1. Moreover, as shown in Fig. 3a and b, the asymmetric unit of 1 consists of only one lead atom, three bromine atoms and a half N-methylation of the 4,4′-bpy molecule, while for 2, there are four crystallographically independent lead atoms, which results in the serious distortion of PbBr6 octahedra. Actually, the inorganic species in 2 obtained from the PVP-induced heterogeneous nucleation are more complicated than those in 1.
, respectively, which means 1 and 2 are a pair of polymorphs. Fig. 2a and b show that although 1D anionic [PbBr3]2n−n chains are formed in both phases, the architectures of the 1D chains are totally different. The polyhedral patterns of 1 and 2 exhibit this difference more clearly: the chains in 1 adopt a close packed mode through all edge-sharing PbBr6 octahedra while the chains in 2 show a much looser feature resulting from the coexistence of point-sharing and edge-sharing modes of PbBr6 octahedra (Fig. 3a and b). These factors suggest that 2 should be a metastable phase compared with 1. Moreover, as shown in Fig. 3a and b, the asymmetric unit of 1 consists of only one lead atom, three bromine atoms and a half N-methylation of the 4,4′-bpy molecule, while for 2, there are four crystallographically independent lead atoms, which results in the serious distortion of PbBr6 octahedra. Actually, the inorganic species in 2 obtained from the PVP-induced heterogeneous nucleation are more complicated than those in 1.
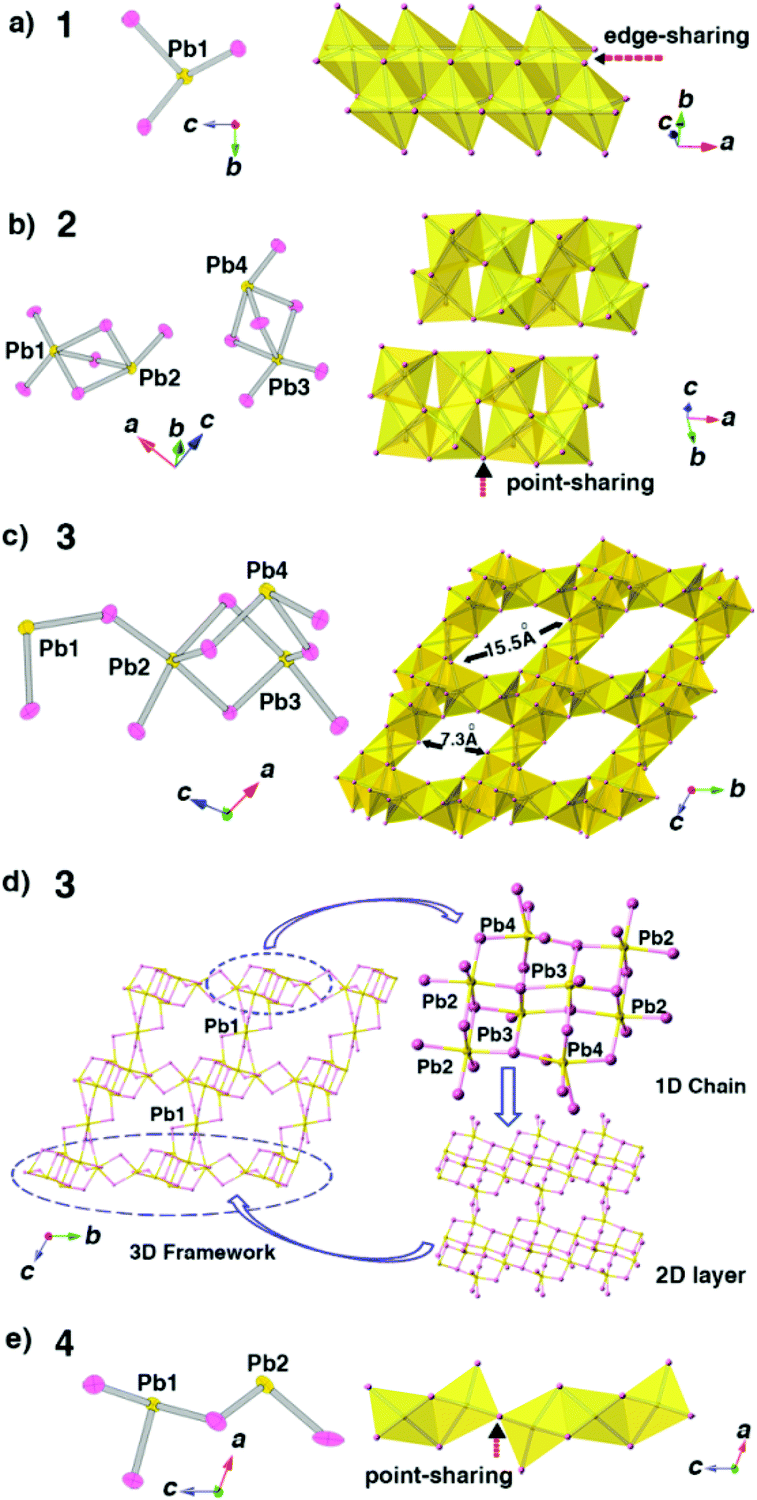 | ||
| Fig. 3 (a), (b), (c) and (e) The asymmetric units and polyhedral stacking patterns of inorganic species in the compounds 1–4. (d) Architectural presentation of the 3D framework structure in 3. Color scheme: Pb(II), gold; Br, pink. | ||
With surfactant SDS as an additive, compound 3 crystallizes in the P space group with three crystallographically independent lead atoms (Pb2, Pb3 and Pb4) and one lead atom (Pb1) occupying the inversion position in the asymmetric unit. Compound 3 features a 3D open-framework structure, in which the inorganic framework walls are constructed by PbBr6 octahedra (Fig. 3c). Fig. 3d exhibits the architectural presentation of this 3D framework structure. The PbBr6 octahedra, centered at Pb2, Pb3 and Pb4 atoms, stack themselves through the edge-sharing and point-sharing modes to form the double incomplete cubane-like chains parallel to the a axis. Then, these chains further connect to each other through Br atoms along the b axis to form 2D layers. Finally, Pb1 atoms link these 2D layers together along the c axis to form a 3D framework. The as-formed 3D framework possesses 1D 16-ring channels with the approximately rhombic shape having dimensions of 7.3 × 15.5 Å2 (Fig. 2c and 3c). It is noteworthy that 3 is the first 3D open-framework bromoplumbate (except perovskites), in which all of the polyhedral units are PbBr6 octahedra.
When PEG-400 is used as the additive, pale yellow crystals of 4 can be obtained. SCXRD analysis reveals that 4 belongs to the space group C2/m with the formula of (C12N2H14)2Pb3Br10. Compound 4 features anionic 1D [Pb3Br10]4−n chains parallel to the c axis (Fig. 2d). There are two Pb atoms and two Br atoms in a mirror plane, one Br atom in a two-fold axis and one crystallographically independent Br atom in the asymmetric unit of 4. The polyhedral stacking pattern exhibits the coexistence of edge-sharing modes and point-sharing modes of PbBr6 octahedra in the structure, showing a non-close-packed feature of these 1D [Pb3Br10]4−n chains (Fig. 3e). Clearly, the distinct structural variability of 1–4 confirms the initial assumption: surfactant additives can directly induce the formation of a new structure or phase at the early nucleation stage in organic–inorganic hybrid systems.
Before measuring the related optical and thermal properties of the as-obtained compounds, the purities of 1–4 have been tested by powder X-ray diffraction. All XRD patterns of 1–4 are matched very well with the corresponding simulated results from the single-crystal diffraction data (Fig. S2†). The solid-state UV-vis diffuse-reflectance spectra of 1–4 were recorded at room temperature and converted to optical absorption data in Fig. 4 according to the Kubelka–Munk function.9 Their optical band gaps (Eg) determined by the extrapolation method are 1.93, 2.08, 2.15 and 2.24 eV, respectively. The clear increase of Eg from 1 to 4 is consistent with their color ranging from red (1) to pale yellow (4). Such changes should mainly contribute from the structural differences of inorganic moieties because all cations in the four samples are the same. FT-IR spectra of 1–4 have been provided in Fig. S3.† The strong or medium peaks ranging from 1175 cm−1 to 1190 cm−1 result from the N–Cmethyl and metric N–Cring stretching vibrations for all compounds.9 The strong peaks between 1627 and 1640 cm−1 can be assigned to the in-plane vibrations of two C–H groups in N-methylate pyridine rings (the C–H groups that are closest to the N-methyl group) for 1–4.10 And the peaks around 3000 cm−1 result from the C–H stretching vibrations in pyridine rings and methyl groups of all compounds.11 These results further confirmed that the N-methylation reactions are successfully fulfilled in all routes. TGA and DSC curves of the polycrystalline samples 1–4 are shown in Fig. S4.† Compounds 1–4 show almost no weight loss up to ∼270 °C and 2–4 show no thermal anomalies below 290 °C, exhibiting their good thermal stability. The small endothermic peak at 225 °C in the DSC curve of 1 may be assigned to a high-temperature phase transition. However, this temperature is beyond the capabilities of the heating device mounted on the single-crystal diffractometer.
 | ||
| Fig. 4 Solid-state optical absorption spectra of 1–4. | ||
Because 1 and 2 are a pair of polymorphs, we also tried to investigate the phase transition between these two phases. Interestingly, yellow phase 2 can be converted into red phase 1 in the presence of a liquid interface (i.e. inside the mother liquor) at room temperature in a few days and the transformation is irreversible. Fig. 5 shows that the intermediate-state crystals have clear boundaries between yellow and red, suggesting that the phase transition has a typical SCSC feature.12 However, no obvious variation of crystal systems can be observed by the variable-temperature unit-cell-data measurements for 2 (ranging from 100 to 450 K) and no thermal anomalies appear before the decomposition of 2 (Fig. S4†), indicating that either the phase transition cannot be induced by changing the environmental temperatures or the transformation might be very slow. As mentioned above, the inorganic chains in 2 arising from the PVP-induced heteronucleation are looser and more unstable than the close-packed inorganic chains in 1. Thus, the mother liquor can supply an environment to accelerate the improvement of the stability of the inorganic 1D chains in 2 and induce this phase transition. Similar mother-liquor-facilitated phase transitions have been reported in HC(NH2)2PbI3 crystals.7a
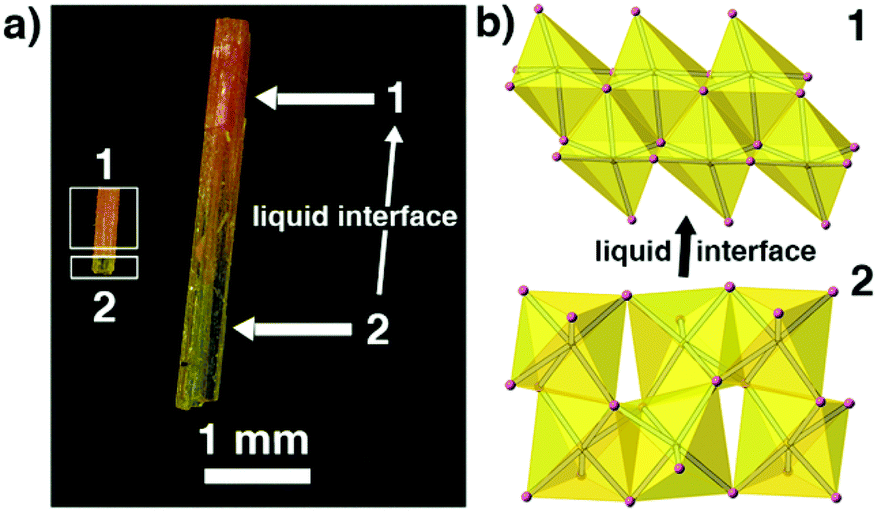 | ||
| Fig. 5 The observed SCSC phase transition between 1 and 2. (a) In the presence of a liquid interface (i.e. inside the mother liquor) and (b) the structure changes when 2 converts into 1 gradually. | ||
Conclusions
In summary, the discovery of new structures or phases using surfactants as additives has been illustrated for organic–inorganic hybrid bromoplumbates. By employing this strategy, four new hybrid bromoplumbates (including the first 3D hybrid bromoplumbate (except perovskites) and a pair of polymorphs of C6NH7PbBr3 with a SCSC phase transition) have been successfully synthesized. More importantly, the substantivity of surfactant-induced heteronucleation in the hybrid system is confirmed because other crystallization parameters are identical. Thus, the formation of a particular structure of organic–inorganic hybrids only depends on the compositions of the surfactants, which is very important to discover new hybrid crystals through altering the functionality and the polarity of surfactants. We believe that this surfactant-induced heteronucleation method could be employed as a promising strategy to prepare new crystalline organic–inorganic hybrids with distinctive physical and chemical properties.Acknowledgements
Q. Z. acknowledges financial support from AcRF Tier 1 (RG133/14 and RG 13/15) from MOE. This work is also financially supported by the NSF of China (no. 21373122 and 51321091).Notes and references
- (a) A. Mersmann, Crystallization Technology Handbook, MarceL Dekker Inc., New York, 2nd edn, 2001 Search PubMed; (b) P. A. Meenan, S. R. Andrson and D. L. Klug, Handbook of industrial Crystallization, Elsevier Science & Technology Books, Amsterdam, 2nd edn, 2002 Search PubMed; (c) W.-W. Xiong, G. Zhang and Q. Zhang, Inorg. Chem. Front., 2014, 1, 292–301 RSC.
- (a) C. Lindenberg, M. Krättli, J. Cornel, M. Mazzotti and J. Brozio, Cryst. Growth Des., 2009, 9, 1124–1136 CrossRef CAS; (b) M. Kempkes, T. Vetter and M. Mazzotti, Chem. Eng. Res. Des., 2010, 88, 447–454 CrossRef CAS; (c) K. Biswas, Q. Zhang, I. Chung, J.-H. Song, J. Androulakis, A. Freeman and M. Kanatzidis, J. Am. Chem. Soc., 2010, 132, 14760–14762 CrossRef CAS PubMed; (d) Q. Zhang, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2009, 48, 10910–10912 CrossRef CAS PubMed; (e) Q. Zhang, G. Armatas and M. G. Kanatzidis, Inorg. Chem., 2009, 48, 8665–8667 CrossRef CAS PubMed; (f) Q. Zhang, I. Chung, J. I. Jang, J. B. Ketterson and M. G. Kanatzidis, Chem. Mater., 2009, 21, 12–14 CrossRef CAS; (g) Y. Liu, P. D. Kanhere, C. L. Wong, Y. Tian, Y. Feng, F. Boey, T. Wu, H. Chen, T. J. White, Z. Chen and Q. Zhang, J. Solid State Chem., 2010, 183, 2644 CrossRef CAS.
- (a) M. Lang, A. L. Grzesiak and A. J. Matzger, J. Am. Chem. Soc., 2002, 124, 14834–14835 CrossRef CAS PubMed; (b) C. P. Price, A. L. Grzesiak and A. J. Matzger, J. Am. Chem. Soc., 2005, 127, 5512–5517 CrossRef CAS PubMed; (c) A. L. Grzesiak, F. J. Uribe, N. W. Ockwig, O. M. Yaghi and A. J. Matzger, Angew. Chem., Int. Ed., 2006, 45, 2553–2556 ( Angew. Chem. , 2006 , 118 , 2615–2618 ) CrossRef CAS PubMed.
- (a) W.-W. Xiong and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 11616–11623 ( Angew. Chem. , 2015 , 127 , 556–560 ) CrossRef CAS PubMed; (b) W.-W. Xiong, E. U. Athresh, Y. T. Ng, J. Ding, T. Wu and Q. Zhang, J. Am. Chem. Soc., 2013, 135, 1256–1259 CrossRef CAS PubMed; (c) J. Gao, K. Ye, L. Yang, W.-W. Xiong, L. Ye, Y. Wang and Q. Zhang, Inorg. Chem., 2014, 53, 691–693 CrossRef CAS PubMed; (d) H.-S. Lu, L. Bai, W.-W. Xiong, P. Li, J. Ding, G. Zhang, T. Wu, Y. Zhang, J. Lee, Y. Yang, B. Geng and Q. Zhang, Inorg. Chem., 2014, 53, 8529–8537 CrossRef CAS PubMed; (e) W.-W. Xiong, J. Miao, K. Ye, Y. Wang, B. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 546–550 ( Angew. Chem. , 2015 , 127 , 556–560 ) CAS; (f) J. Zhao, Y. Wang, W. Dong, Y. Wu, D. Li, B. Liu and Q. Zhang, Chem. Commun., 2015, 51, 9479–9482 RSC; (g) J. Gao, Q. Tay, W.-W. Xiong, P.-Z. Li, Y. Zhao, Z. Chen and Q. Zhang, Chem. – Asian J., 2014, 9, 131–134 CrossRef CAS PubMed; (h) J. Gao, M. He, Z. Y. Lee, W. Cao, W.-W. Xiong, Y. Li, R. Ganguly, T. Wu and Q. Zhang, Dalton Trans., 2013, 42, 11367–11370 RSC.
- (a) A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed; (b) H. Zhou, Q. Chen, G. Li, S. Luo, T.-B. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed; (c) Q. Zhang and X. Liu, Small, 2012, 8, 3711–3713 CrossRef CAS PubMed; (d) F. Hao, C. C. Stoumpos, Z. Liu, R. P. H. Chang and M. G. Kanatzidis, J. Am. Chem. Soc., 2014, 136, 16411–16419 CrossRef CAS PubMed; (e) Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed; (f) P.-Y. Gu, N. Wang, A. Wu, Z. Wang, M. Tian, Z. Fu, X. W. Sun and Q. Zhang, Chem. – Asian J., 2016, 11, 2135–2138 CrossRef CAS PubMed.
- Z.-J. Zhang, S.-C. Xiang, G.-C. Guo, G. Xu, M.-S. Wang, J.-P. Zou, S.-P. Guo and J.-S. Huang, Angew. Chem., Int. Ed., 2008, 47, 4149–4152 ( Angew. Chem. , 2008 , 120 , 4217–4220 ) CrossRef CAS PubMed.
- (a) C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019 CrossRef CAS PubMed; (b) Z. Wang, Y. Zhou, S. Pang, Z. Xiao, J. Zhang, W. Chai, H. Xu, Z. Liu, N. P. Padture and G. Cui, Chem. Mater., 2015, 27, 7149–7155 CrossRef CAS; (c) A. Binek, F. C. Hanusch, P. Docampo and T. Bein, J. Phys. Chem. Lett., 2015, 6, 1249–1253 CrossRef CAS PubMed; (d) G. Liu, J. Liu, Z. Sun, Z. Zhang, L. Chang, J. Wang, X. Tao and Q. Zhang, Inorg. Chem., 2016, 55, 8025–8030 CrossRef CAS PubMed.
- Q. Zhang, T. Wu, X. Bu, T. Tran and P. Feng, Chem. Mater., 2008, 20, 4170–1472 CrossRef CAS.
- W. M. Wendlandt and H. G. Hecht, Reflectance Spectroscopy, Interscience, New York, 1966 Search PubMed.
- K. M. Marzec, A. Jaworska, K. Malek, A. Kaczor and M. Baranska, J. Raman Spectrosc., 2013, 44, 155–165 CrossRef CAS.
- P. Griffiths and J. A. de Hasseth, Fourier Transform Infrared Spectrometry, Wiley-Blackwell, New Jersey, 2nd edn, 2007 Search PubMed.
- (a) H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 Search PubMed; (b) T. Seki, K. Sakurada and H. Ito, Angew. Chem., Int. Ed., 2013, 52, 12828–12832 ( Angew. Chem. , 2013 , 125 , 13062–13066 ) CrossRef CAS PubMed; (c) G. Liu, J. Liu, Y. Liu and X. Tao, J. Am. Chem. Soc., 2014, 136, 590–593 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and tables, Experimental section, characterization of the 1–4 compounds, PXRD, FT-IR, TGA-DSC. CCDC 1486414–1486417. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00292g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |