DOI:
10.1039/C5QI00233H
(Research Article)
Inorg. Chem. Front., 2016,
3, 142-149
Heterotrimetallic sandwich complexes supported by sulfonamido ligands†
Received
31st October 2015
, Accepted 25th November 2015
First published on 8th December 2015
Abstract
CoII complexes bearing sulfonamido ligands derived from tris(2-aminoethyl)amine (H6tren) assemble into complex architectures in the presence of Group II ions through interactions between the Group II ion and the sulfonyl oxygens. Novel ligands where the sulfonyl groups bear tert-butyl substituents afford well-defined isostructural heterotrimetallic sandwich complexes where two anionic [LCoII]− fragments are bridged by a Group II metal ion (Mg2+, Ca2+, Sr2+, or Ba2+). Solution studies on these novel sandwich complexes by cyclic voltammetry and electronic absorption spectroscopy indicate that these structures are not dramatically altered in solution, opening the door to the exploration of reactivity at CoII that is modified by Group II ions.
 Christopher C. Scarborough | Chris Scarborough was born in Orange County, California. He obtained his B.S. in Chemistry in 2003 from the University of California – Irvine and conducted undergraduate research under Gregory Weiss. His graduate studies at the University of Wisconsin under the supervision of Shannon Stahl focused on the development of axially chiral 7-membered N-heterocyclic carbene ligands and their application to palladium catalysis. After completion of his PhD in 2008, Chris was an Alexander von Humboldt postdoctoral researcher at the Max Planck Institute for Bioinorganic Chemistry under Karl Wieghardt, where he examined the electronic structure of electron-rich metal–bipyridine complexes. In 2011, Chris began his independent career at Emory University where he has developed a research program exploring novel methods of activating O2 and H2O2 for oxidation catalysis. |
Introduction
The properties and reactivity of transition metals can be dramatically modified by the presence of second-sphere redox-inactive metals. This is most notably seen in the oxygen-evolving complex (OEC) of photosystem II,1 where second-sphere calcium or strontium2 is required for the formation of O2 from water at the manganese cluster. Synthetic models of the OEC3 have corroborated the importance of the redox-inert ion by demonstrating a dependence of the redox potentials4 and reactivity5 of manganese clusters on the identity of the redox-inert ion. The effects of second-sphere redox-inert metals have been demonstrated in other systems as well, for example by affecting the properties and reactivity of metal–oxo6,7 and metal–peroxo8 species, by altering the rate of O2 activation by MnII and FeII,9,10 by controlling the accessibility and reactivity of copper-nitrene species,11 by changing the reactivity of a CuIII–OH species,12 and by modulating the water-oxidation reactivity of cobalt and manganese oxides.13 Clearly, redox-inert metals can have a profound effect on the properties and reactivity of nearby transition metals, and new methods to access well-defined complexes with second-sphere redox-inert metals are expected to enable the discovery of altered and novel transition-metal reactivity. While heterometallic systems that involve either a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of redox-active and redox-inert metal ions6,8–12 or the interaction of a redox-inert metal ion with a cluster of redox-active metals3–5,7 are common, we have been targeting systems with a 2:1 ratio of redox-active and redox-inert metal ions that could be well suited for splitting symmetric small molecules (e.g. O2, N2, or RNNR). Recently, Borovik demonstrated that redox-inert ions may be brought into close proximity to transition- and main-group metals coordinated by sulfonamido ligands, which bind the redox-inert metal through the electron-rich sulfonyl oxygens.9,10,14–18 Accordingly, we have pursued ligands that facilitate the assembly of heterotrimetallic coordination compounds where two anionic redox-active metal moieties coordinated by sulfonamido ligands are brought together through interactions with a Group II ion (e.g. Mg2+, Ca2+, Sr2+, or Ba2+) to form sandwich compounds. In particular, we have been targeting systems where the redox-active metal ion maintains an open coordination site, as the availability of this site would form the focus of reactivity studies. In this contribution, we describe the assembly of heterotrimetallic (CoII2M) sandwich complexes (M = Mg2+, Ca2+, Sr2+, or Ba2+) supported by a new sulfonamido ligand that feature open axial coordination sites on each CoII ion.
:1 ratio of redox-active and redox-inert metal ions6,8–12 or the interaction of a redox-inert metal ion with a cluster of redox-active metals3–5,7 are common, we have been targeting systems with a 2:1 ratio of redox-active and redox-inert metal ions that could be well suited for splitting symmetric small molecules (e.g. O2, N2, or RNNR). Recently, Borovik demonstrated that redox-inert ions may be brought into close proximity to transition- and main-group metals coordinated by sulfonamido ligands, which bind the redox-inert metal through the electron-rich sulfonyl oxygens.9,10,14–18 Accordingly, we have pursued ligands that facilitate the assembly of heterotrimetallic coordination compounds where two anionic redox-active metal moieties coordinated by sulfonamido ligands are brought together through interactions with a Group II ion (e.g. Mg2+, Ca2+, Sr2+, or Ba2+) to form sandwich compounds. In particular, we have been targeting systems where the redox-active metal ion maintains an open coordination site, as the availability of this site would form the focus of reactivity studies. In this contribution, we describe the assembly of heterotrimetallic (CoII2M) sandwich complexes (M = Mg2+, Ca2+, Sr2+, or Ba2+) supported by a new sulfonamido ligand that feature open axial coordination sites on each CoII ion.
Results and discussion
We reasoned that heterotrimetallic sandwich compounds would be accessible from ligands of the type [(RSO2NCH2CH2)3N]3− ([(RSO2)3tren]3−), which have been demonstrated by Borovik to furnish heterobimetallic complexes where the sulfonyl oxygens of [((RSO2)3tren)MII]− coordinate redox-active16 or redox-inert9,10,14,17 metal ions in the second sphere of MII.18 Our first efforts employed Ts3H3tren (R = p-tolyl), a readily accessible ligand that has been used in the synthesis of heterobimetallic species.10 [Ts3tren]3− is smaller than the R = mesityl derivative employed in the synthesis of heterobimetallic cobalt complexes,14 and we reasoned that this diminished size might be important for assembly of heterotrimetallic systems.
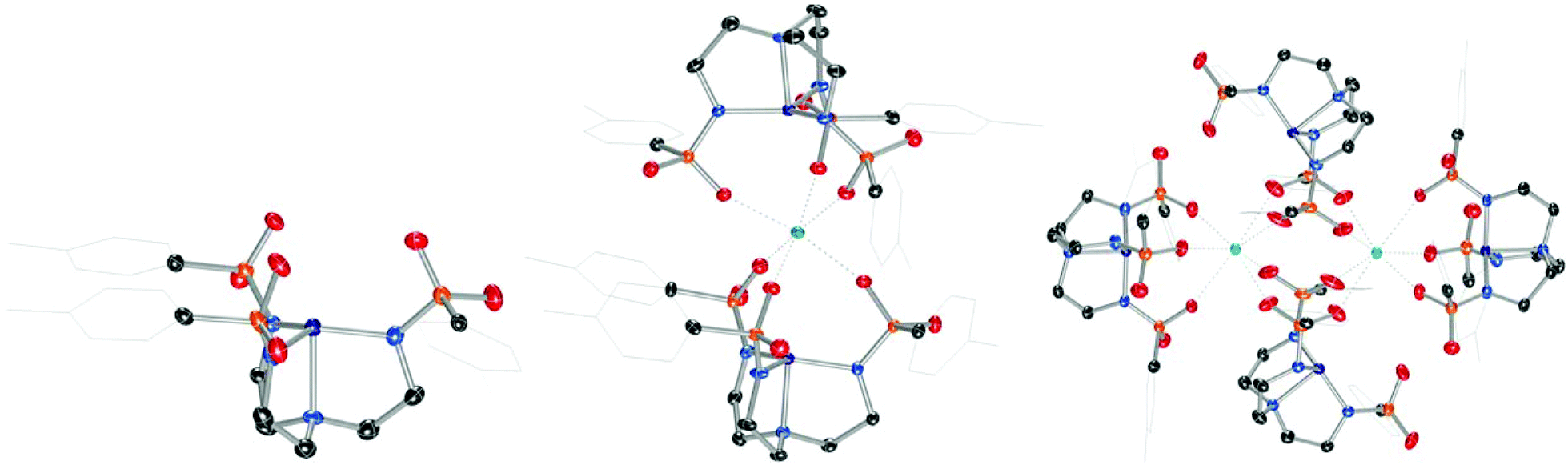
Combining Ts3H3tren with CoIICl2 and NaH in DMF, followed by addition of [nBu4N][Br] and aqueous workup provided [nBu4N][(Ts3tren)CoII(OH2)] ([nBu4N][1-OH2]) in 77% crystalline yield (Scheme 1). Combining [nBu4N][1-OH2] with 3 Å molecular sieves generated the four-coordinate species [nBu4N][1], which formed crystals of X-ray quality from CH2Cl2/Et2O (Fig. 1).‡ Heating Ca(OTf)2 and [nBu4N][1] in methanol/glyme to 85 °C provided a novel sandwich compound that crystallized upon cooling (Fig. 1), wherein two [1]− units are bonded to Ca2+ through one sulfonyl oxygen of each ligand arm to afford the sandwich compound Ca12, a species that is octahedral at Ca2+ and trigonal monopyramidal at the two CoII ions. Attempts to prepare structurally related species using Mg(OTf)2 were unsuccessful. A related procedure employing Ba(OTf)2 in place of Ca(OTf)2 afforded crystalline [Ba12]2 (Fig. 1), which features two seven-coordinate Ba2+ ions bridging four [1]− units. We were also unsuccessful in accessing Sr12, but we were able to crystalize a partially hydrated form of this species (see ESI†).
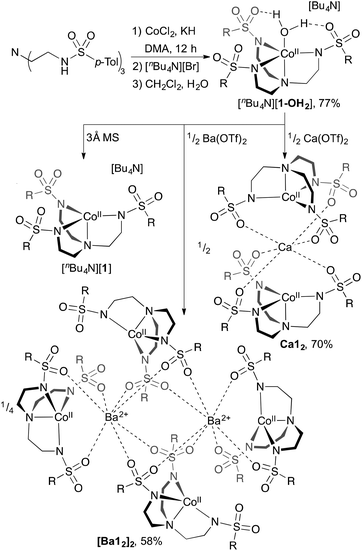 |
| | Scheme 1 Synthesis of [nBu4N][1-OH2], [nBu4N][1], Ca12, and [Ba22]2. Yields are of crystalline product. R = p-tolyl. | |
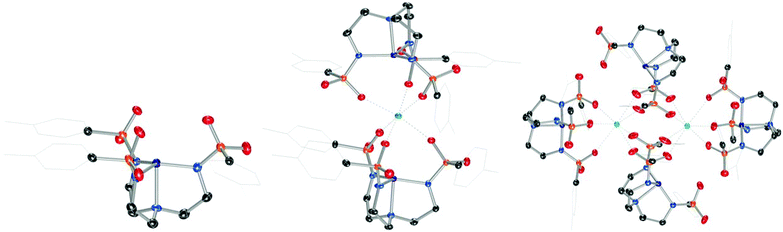 |
| | Fig. 1 Solid-state molecular structures of the anion in [nBu4N][1] (left), Ca12 (middle), and [Ba12]2 (right). | |
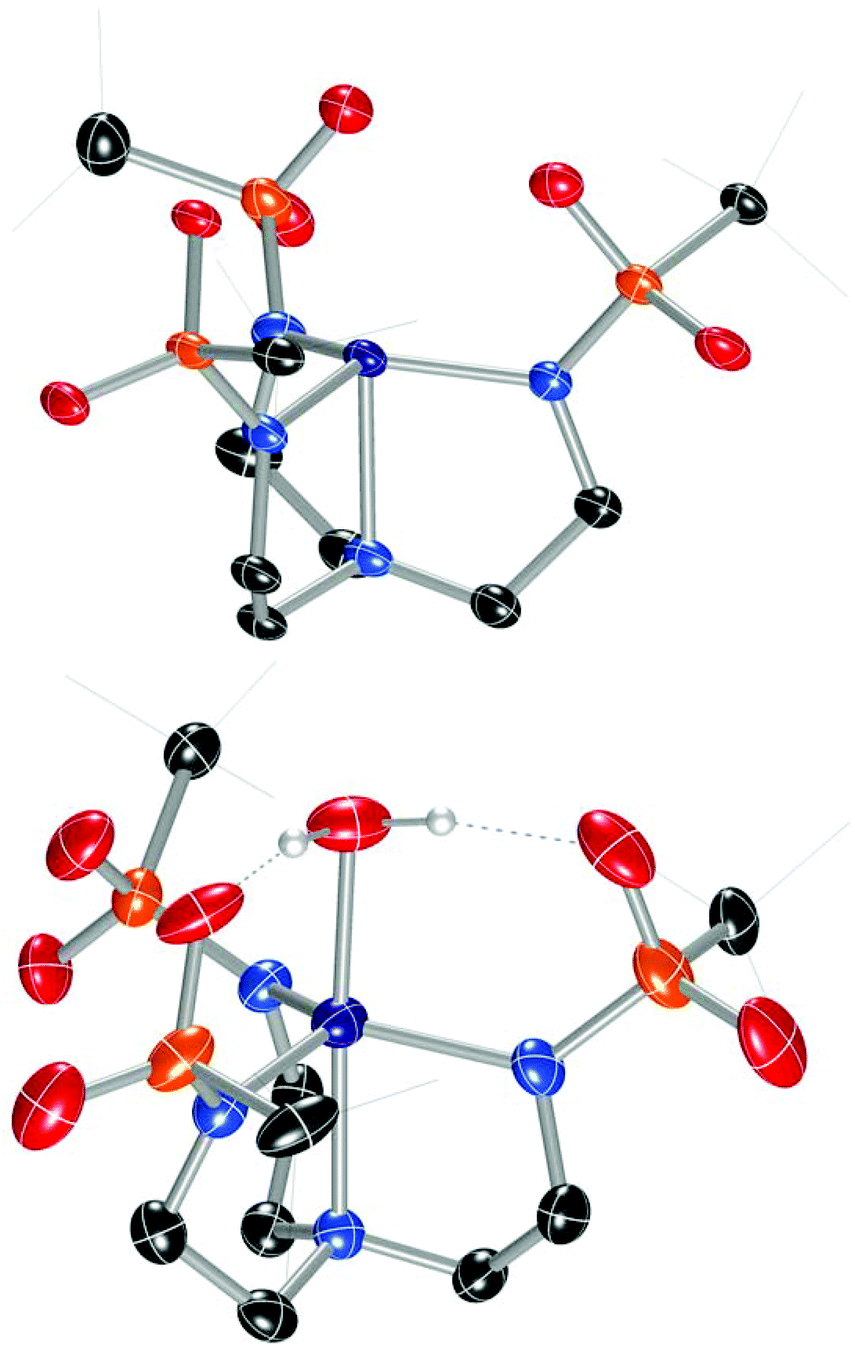
In order to facilitate a direct comparison between sandwich complexes with different Group II ions, we reasoned that a ligand with more electron-rich sulfonyl oxygens would be beneficial for accessing compounds where the Group II metals all feature the same coordination number. This led us to explore alkylsulfonyl groups that would provide more electron-rich oxygen donors than the arylsulfonyl ligands in Ca12. This reasoning is supported by the higher acidity of PhSO2NH2 than CH3SO2NH2 (pKa = 16.1 vs. 17.5, respectively).19 Ms3H3tren20 was not targeted owing to its water solubility; instead, we targeted (tBuSO2NHCH2CH2)3N (Bus3H3tren, Bus = busyl). Bus3H3tren had not been reported, and accessing this ligand in an analogous manner to Ts3H3tren is not feasible because tBuSO2Cl is not an effective sulfonyl transfer agent (tBuSO2Cl decomposes into isobutylene, SO2, and HCl on addition to amines).21 However, busyl amides are accessible from amine sulfinylation with tBuSOCl22 followed by oxygen-atom transfer to sulphur with m-CPBA.23 We accessed Bus3H3tren from commercially available tBuSStBu over four steps in 21% overall yield (Scheme 2). This ligand was metallated analogously to Ts3H3tren, providing crystalline [nBu4N][(Bus3tren)CoII(OH2)] ([nBu4N][2-OH2]) in 47% yield (Scheme 2 and Fig. 2). As with [nBu4N][1-OH2], [nBu4N][2-OH2] could be readily dehydrated with 3 Å molecular sieves to afford the crystalline four-coordinate complex [nBu4N][2] (Fig. 2).
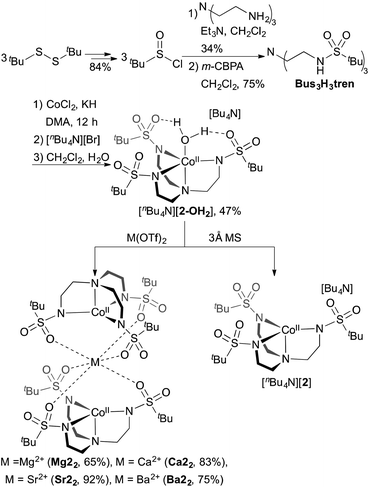 |
| | Scheme 2 Synthesis of [nBu4N][2-OH2], [nBu4N][2], Mg22, Ca22, Sr22, and Ba22. Yields are of crystalline products. | |
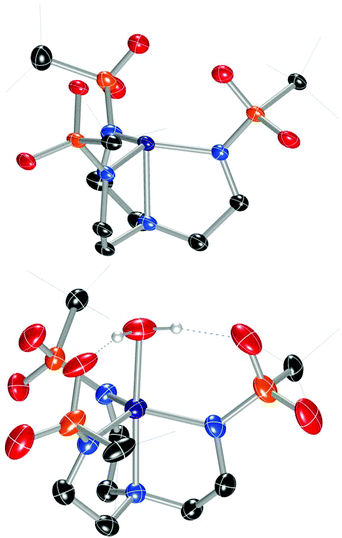 |
| | Fig. 2 Solid-state molecular structures of the anions in [nBu4N][2] (top) and [nBu4N][2-OH2] (bottom). | |
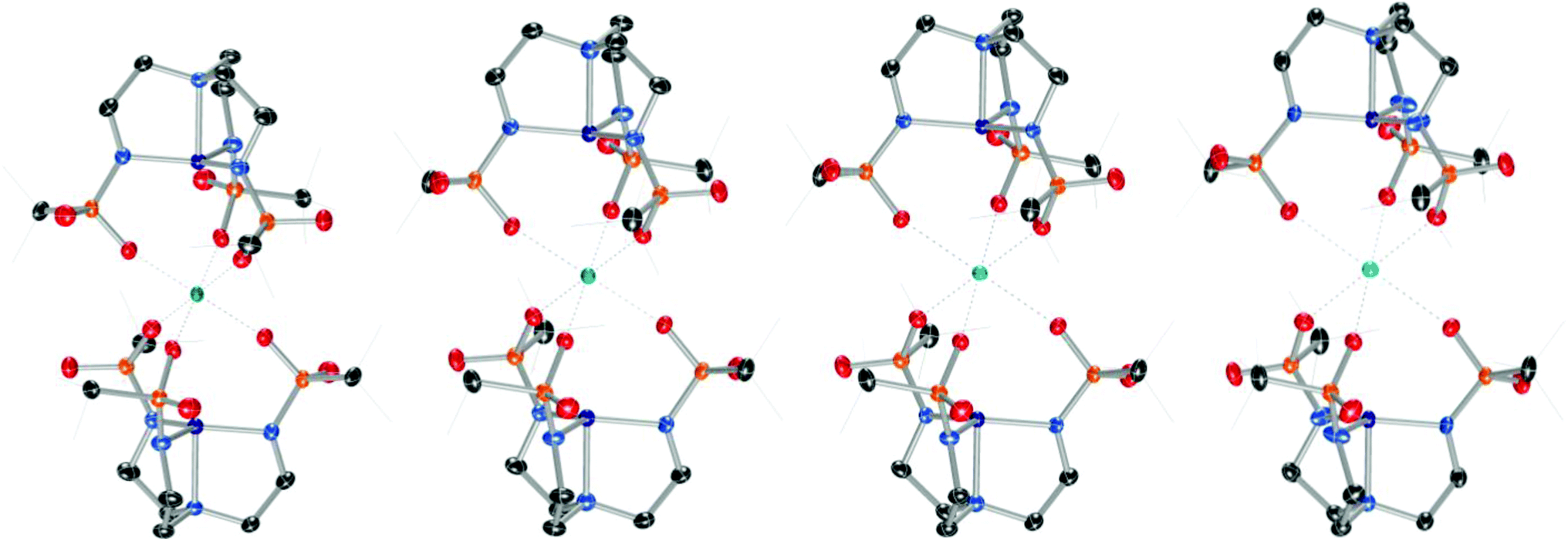
Addition of M(OTf)2 to [nBu4N][2] with heat provided crystalline samples of M22, where M = Mg, Ca, Sr, and Ba (Scheme 2 and Fig. 3). In contrast to compounds containing [1]−, the Group II ion in each of these species is octahedral, and each member of the series is isostructural: the redox-inert metal ion is bound symmetrically by one sulfonyl oxygen of each busyl group. The ability to access this isostructural series that was inaccessible with [1]− may at least partially result from the increased electron density of the sulfonyl oxygens of busyl vs. tosyl groups. Table 1 highlights relevant crystallographic distances. The Co–Nax distance is sensitive to the coordination number at cobalt, where this bond in species containing [2]− elongates by ∼0.1 Å upon coordination of H2O. Differences between the Co–Nax distances for all four-coordinate species containing [2]− are within 3σ. The Co–Neq bond lengths are sensitive to the coordination number of cobalt, but to a lesser extent than the Co–Nax distance. No clear trends in Co–N bond lengths are observed for the M22 series; however, the Co⋯M distance increases monotonically with increasing size of the Group II ion.
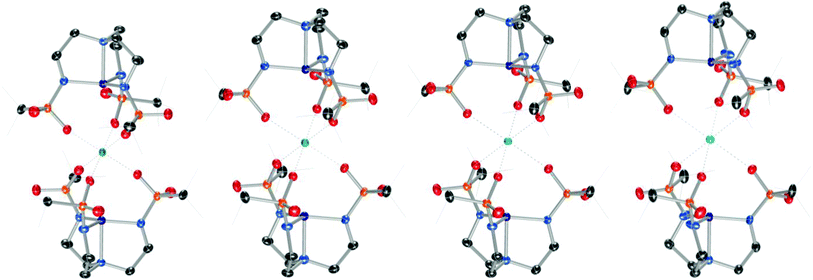 |
| | Fig. 3 From left to right, solid-state molecular structures of Mg22, Ca22, Sr22, and Ba22. | |
Table 1 Crystallographic distances for compounds containing [2]−
| Compound |
Co–Naxa |
Co–Neqb |
Co–OH2 |
Co⋯M |
|
Nax is the central nitrogen of the H6tren-derived ligand.
Neq depicts the three nitrogens in the trigonal plane. The presence of one value indicates that these bonds are equivalent by symmetry.
|
| [nBu4N][2-OH2] |
2.246(3) Å |
2.038(3) Å, 2.014(3) Å, 2.030(3) Å |
2.134(3) Å |
— |
| [nBu4N][2] |
2.109(6) Å |
1.933(5) Å, 1.937(6) Å, 1.991(6) Å |
— |
— |
|
Mg22
|
2.1069(10) Å |
1.9440(7) Å |
— |
3.4101(9) Å |
|
Ca22
|
2.0951(19) Å |
1.9344(11) Å |
— |
3.5485(11) Å |
|
Sr22
|
2.087(3) Å |
1.965(6) Å |
— |
3.6411(10) Å |
|
Ba22
|
2.087(5) Å |
1.942(3) Å |
— |
3.7490(7) Å |
We next explored the CoII/CoIII redox couple of [nBu4N][1-OH2], [nBu4N][2-OH2], [nBu4N][1], and [nBu4N][2] in CH2Cl2 using cyclic voltammetry to probe the effect of coordination number and ligand identity on redox potential as a benchmark for probing the members of the M22 series. Borovik recently demonstrated that the FeII/FeIII redox couple in [Me4N][((ArSO2))3tren)FeII(OH2)] is sensitive to the electronic properties of the aryl group,15 so we wanted to probe the extent to which the CoII/CoIII couple is sensitive to the nature of the sulfonyl substituent (p-tolyl vs. tert-butyl). The electrochemical data are summarized in Table 2. Both species containing [2]− are more easily oxidized than the corresponding [1]− species, where the onset of oxidation was shifted by −30 and −60 mV for the respective hydrated and anhydrous [2]− species compared to the [1]− species in CH2Cl2, consistent with busyl amides being more electron rich than tosyl amides. Furthermore, the onset of oxidation for the four-coordinate species [nBu4N][1] and [nBu4N][2] were shifted by +270 and +240 mV from the corresponding hydrated species, revealing a strong dependence of the CoII/CoIII couple on coordination number.§ These data clearly demonstrate the stronger donor strength of [Bus3tren]3− compared to [Ts3tren]3−, and also provide a benchmark for electrochemical studies of the members of the M22 series.
Table 2 Electrochemical dataa
| Compound |
Solvent |
E
anodic (mV) |
E
cathodic (mV) |
E
1/2 (mV) |
|
Scan rate = 100 mV s−1 with [nBu4N][PF6] as the supporting electrolyte. All values vs. Fc/Fc+. See Fig. 4 for electrochemical traces.
Features of aqua complexes consistently appear as minor impurities, likely arising from adventitious H2O in the experimental setup.
The return oxidation wave of [nBu4][1] is not discernible.
Electrochemically irreversible, but cycling current does not result in changes to the voltammogram (chemically reversible).
Multiple oxidation events.
Position of final (lowest-potential) reduction wave.
|
| [nBu4][1-OH2] |
CH2Cl2 (TFE) |
+125 (+465) |
+30 (+380) |
+78 (+422) |
| [nBu4][1]b |
CH2Cl2 |
+395 |
—c |
Irr.d |
| [nBu4][2-OH2] |
CH2Cl2 (TFE) |
+95 (+450) |
–40 (+335) |
+28 (+392) |
| [nBu4][2]b |
CH2Cl2 |
+335 |
+230 |
+282 |
|
Ca22
|
TFE |
Multiplee |
+405f |
Irr.d |
|
Sr22
|
TFE |
Multiplee |
+385f |
Irr.d |
|
Ba22
|
TFE |
Multiplee |
+370f |
Irr.d |
We next interrogated how the CoII/CoIII redox couple was affected by the presence and identity of a second-sphere Group II ion by examining the electrochemical properties of the members of the M22 series (M = Ca, Sr, and Ba).¶ These species are not soluble in CH2Cl2, but they are soluble in 2,2,2-trifluoroethanol (TFE). We therefore examined the electrochemical behaviour of [nBu4N][1-OH2] and [nBu4N][2-OH2] in TFE to benchmark the M22 data. We were unable to measure the electrochemical properties of [nBu4N][1] and [nBu4N][2] in TFE because UV-vis indicated that these complexes were always 5-coordinate in TFE (see below) and electrochemical studies of these compounds in TFE provided data that matched the hydrated species, indicating either that these species bind residual water in the TFE despite distillation from MgSO4 or that TFE occupies the fifth coordination site on CoII in these compounds. Nevertheless, all M22 species explored maintained a four-coordinate geometry at cobalt by absorption spectroscopy (see below), suggesting that these species remain intact in solution to the extent that they prevent coordination of H2O or TFE to the CoII ions. As seen in Table 2, solvent has a strong effect on the position of the CoII/CoIII redox couple in [nBu4N][2-OH2] (+28 mV in CH2Cl2 and +390 mV in TFE), likely reflecting hydrogen bonding between TFE and the sulfonyl oxygens that pulls electron density away from the anionic nitrogen donors. The cyclic voltammograms of the members of the M22 series are distinct from the voltammogram of [nBu4N][2-OH2] even in the presence of a large excess of [nBu4N]+, suggesting that these M22 species are not ionized in TFE. The voltammograms of M22 species are complicated by the presence of multiple oxidation events, which is consistent with the M22 species remaining intact in solution. However, the final return reduction events of Ca22, Sr22, and Ba22 (Table 2) approach the position of the return reduction event of [nBu4N][2-OH2] in TFE, where Ba22 is the closest and Ca22 is the furthest of this series. This suggests that (1) the identity of the metal ion affects the potential of the return reduction event indicating that these redox-inert metal ions remain in close contact to the [2]− ions throughout the electrochemical event, and (2) electrochemical oxidation results in occupation of the fifth coordination site on the electrophilic four-coordinate CoIII centers either by water or TFE. Electrochemical cycling experiments indicate that the overall processes are all chemically reversible, so the complexity of the oxidation events likely reflects reversible and oxidation-state-dependent occupation of the fifth coordination site on cobalt, although mixed valency and partial disruption of M⋯OS interactions may also play a role. From these data, we see that the interactions between the M2+ ion and the [2]− units are still present in solution, although it is unclear whether these interactions are the same as in the solid-state structures. We next turned to electronic absorption spectroscopy to probe the extent to which the CoII ions in the M22 series remain four-coordinate in solution.
[nBu4N][2] and [nBu4N][2-OH2] are clearly distinguishable by their electronic absorption profiles in CH2Cl2 (Fig. 5), most notably by the presence of a transition of the anhydrous species at 414 nm that is absent in the hydrated species, providing a benchmark for assessing the coordination number of the cobalt ions in the members of the M22 series in solution. As described above, Ca22, Sr22, and Ba22 are all soluble in TFE but are insoluble in CH2Cl2. Consistent with our electrochemical studies, dissolution of [nBu4N][2] in TFE furnishes an absorption spectrum that is indistinguishable from the absorption spectrum of [nBu4N][2-OH2] in TFE, suggesting that [nBu4N][2] is binding either residual water from the TFE or TFE itself in the fifth coordination site of CoII. However, the positions of the peaks of [nBu4N][2-OH2] are nearly solvent independent (no solvatochromism), giving us confidence in our ability to distinguish between the spectra of four- and five-coordinate CoII species in both CH2Cl2 and TFE. All three soluble members of the M22 series give nearly identical electronic absorption spectra in TFE, and these spectra clearly align with the spectrum of [nBu4][2] in CH2Cl2, including the presence of a transition around 400 nm, indicating that four-coordinate CoII centers are present in each sandwich complex even though no efforts were made to dry the TFE and the samples were prepared open to air. Given our inability to obtain spectra of [nBu4N][2] in TFE, the presence of four-coordinate CoII ions in the members of the M22 series indicates that [2]− binds preferentially to the Group II ions examined instead of water, and that these interactions between [2]− and M2+ prevent H2O or TFE coordination at CoII. Furthermore, the positions of the d–d bands are largely unaffected by the identity of the countercation ([nBu4N]+, Ca2+, Sr2+, or Ba2+), even though the electrochemical properties of [2]− are affected by the identity of the countercation (Fig. 4 and Table 2). Taken together, the electrochemical and electronic absorption data reveal that M22 species are not ionized in solution, but retain four coordinate CoII ions and interactions between the Group II ions and [2]− even in wet TFE.
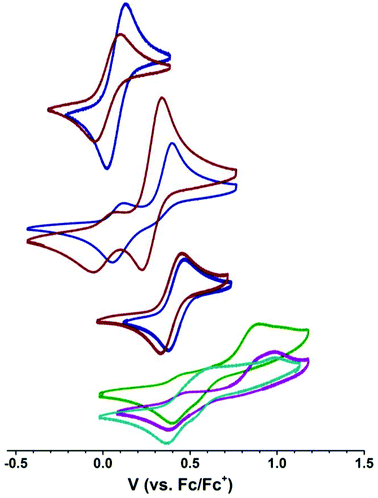 |
| | Fig. 4 Cyclic voltammograms of [nBu4N][1-OH2] ( ) and [nBu4N][2-OH2] ( ) and [nBu4N][2-OH2] ( ) in CH2Cl2 (top); [nBu4N][1] ( ) in CH2Cl2 (top); [nBu4N][1] ( ) and [nBu4N][2] ( ) and [nBu4N][2] ( ) in CH2Cl2 (second from top, smaller features are from hydrated forms derived from adventitious water in the experimental setup); [nBu4N][1-OH2] ( ) in CH2Cl2 (second from top, smaller features are from hydrated forms derived from adventitious water in the experimental setup); [nBu4N][1-OH2] ( ) and [nBu4N][2-OH2] ( ) and [nBu4N][2-OH2] ( ) in TFE (second from bottom); and Ca22 ( ) in TFE (second from bottom); and Ca22 ( ), Sr22 ( ), Sr22 ( ), and Ba22 ( ), and Ba22 ( ) in TFE. Scan rate = 100 mV s−1. ) in TFE. Scan rate = 100 mV s−1. | |
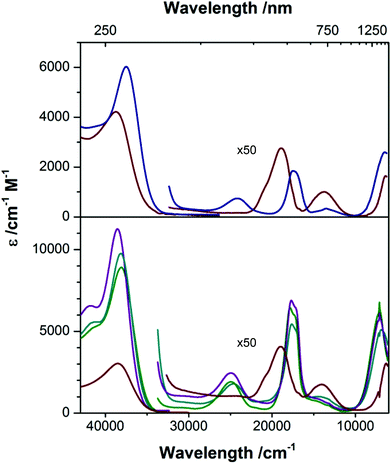 |
| | Fig. 5 Electronic absorption spectra of [nBu4N][2] ( ) and [nBu4N][2-OH2] ( ) and [nBu4N][2-OH2] ( ) in CH2Cl2 (top), and [nBu4N][2-OH2] ( ) in CH2Cl2 (top), and [nBu4N][2-OH2] ( ) Ca22 ( ) Ca22 ( ), Sr22 ( ), Sr22 ( ), and Ba22 ( ), and Ba22 ( ) in TFE (bottom). ) in TFE (bottom). | |
In an effort to quantify the extent to which the M22 and Ca12 species remain intact in solutions containing H2O, we explored the hydration reactivity of the four-coordinate CoII ions in samples of M22 and Ca12 in dried TFE by titrating H2O and measuring the change in absorption profile consistent with conversion from four- to five-coordinate CoII (see ESI† for details). Fig. 6 shows the decay of four-coordinate CoII upon titatration with H2O, and we have verified that the product in each case corresponds to five-coordinate CoII derived from hydration of the CoII ion. As shown in Fig. 6, there is a strong dependence of the extent of hydration of M22 and Ca12 on the identity of both the redox-inert metal ion and the ligand (aryl- vs. alkylsulfonamide). Both Ca22 and Sr22 contain essentially all four-coordinate CoII ions in dried TFE, whereas both Ba22 and Ca12 contain both four- and five-coordinate CoII ions in dried TFE. The extent of hydration in the M22 series is strongly dependent on the nature of M, where hydration of Ca22 is most difficult (half of the CoII ions are five-coordinate when 209 equivalents of H2O are added), followed by hydration of Sr22 (half of the CoII ions are five-coordinate when 116 equivalents of H2O are added), and Ba22 is already a mixture of four- and five-coordinate CoII species in the absence of added H2O, consistent with Ca2+ having the strongest interaction with the sulfonyl oxygens and Ba2+ having the weakest. While the extent of hydration is dependent on the identity of the redox-inert metal ion in the M22 series, the starkest difference in hydration is derived from changing the ligand from an arylsulfonamide ligand in Ca12 ([Ts3tren]3−) to an alkylsulfonamide ligand in Ca22 ([Bus3tren]3−), where hydration of Ca12 occurs much more readily than hydration of Ca22. This is consistent with the solid-state behaviour of members of the M12 and M22 series, where isostructural members of the M22 series were accessible for M = Mg2+, Ca2+, Sr2+ and Ba2+, whereas with [1]− anions, the only accessible sandwich compound was Ca12. The relative ease of hydration of Ca12 and Ca22 is also a clear indication that the alkylsulfonyl oxygens are more electron rich than arylsulfonyl oxygens, and accordingly bind Ca2+ more tightly. Taken together, the solid-state, solution-phase, and redox behaviour of CoII ions in complexes of [1]− and [2]− reveal that [Bus3tren]3− is a significantly improved ligand for interactions with second-sphere redox-inert metal ions than [Ts3tren]3−.
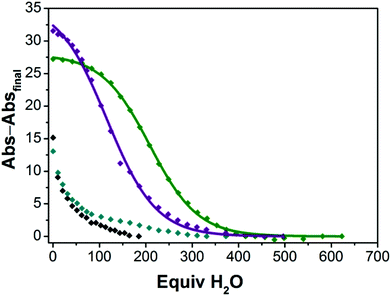 |
| | Fig. 6 Decay of four-coordinate CoII ions in Ca22 ( ), Sr22 ( ), Sr22 ( ), Ba22 ( ), Ba22 ( ) and Ca12 (◆) into five-coordinate CoII in TFE with added water. Abs–Absfinal represents the absorption of Ca22 at 401 nm, Sr22 at 401 nm, Ba22 at 406 nm, and Ca12 at 406 nm as a function of added equivalents of H2O, with the final absorption value for each species subtracted from each point. The curves for Ca22 and Sr22 are fits to the data using sigmoidal logistic functions (see ESI† for details). ) and Ca12 (◆) into five-coordinate CoII in TFE with added water. Abs–Absfinal represents the absorption of Ca22 at 401 nm, Sr22 at 401 nm, Ba22 at 406 nm, and Ca12 at 406 nm as a function of added equivalents of H2O, with the final absorption value for each species subtracted from each point. The curves for Ca22 and Sr22 are fits to the data using sigmoidal logistic functions (see ESI† for details). | |
Conclusions
We have detailed the synthesis of novel heterotrimetallic sandwich complexes involving two CoII ions bridged by redox-inert metal ions (Mg2+, Ca2+, Sr2+, and Ba2+). These M22 assemblies, which are made readily accessible by the new ligand [Bus3tren]3−, are isostructural across the entire series. Furthermore, electrochemical and electronic absorption studies indicate that Ca22, Sr22, and Ba22 retain interactions between the Group II ions and [2]− in solution. The solid-state, solution-phase, and redox behaviour of members of the M22 series compared to related complexes derived from [1]− demonstrates the improved properties of alkylsulfonamide ([Bus3tren]3−) vs. arylsulfonamides ([Ts3tren]3−) ligands for formation of discrete heterotrimetallic sandwich complexes. These results highlight the rational design of complex heterotrimetallic clusters with significant solution integrity, and open the door to the exploration of the effect of Group II ions on substrate reactivity at the open coordination site of CoII in these systems. Our studies into the reactivity of these sandwich species will be published in due course.
Acknowledgements
The authors thank the National Science Foundation (CHE-1455211), the American Chemical Society Petroleum Research Fund (53254-DNI3) and Emory University for financial support of this research.
Notes and references
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- N. Cox, L. Rapatskiy, J.-H. Su, D. A. Pantazis, M. Sugiura, L. Kulik, P. Dorlet, A. W. Rutherford, F. Neese, A. Boussac, W. Lubitz and J. Messinger, J. Am. Chem. Soc., 2011, 133, 3635–3648 CrossRef CAS PubMed; Y. Pushkar, J. Yano, K. Sauer, A. Boussac and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1879–1884 CrossRef PubMed.
- S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Chem. Rev., 2004, 104, 3981–4026 CrossRef CAS PubMed.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed; E. Y. Tsui and T. Agapie, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10084–10088 CrossRef PubMed; E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef PubMed.
- S. Nayak, H. P. Nayek, S. Dehnen, A. K. Powell and J. Reedijk, Dalton Trans., 2011, 40, 2699–2702 RSC; J. S. Kanady, J. L. Mendoza-Cortes, E. Y. Tsui, R. J. Nielsen, W. A. Goddard and T. Agapie, J. Am. Chem. Soc., 2013, 135, 1073–1082 CrossRef CAS PubMed; J. S. Kanady, P.-H. Lin, K. M. Carsch, R. J. Nielsen, M. K. Takase, W. A. Goddard and T. Agapie, J. Am. Chem. Soc., 2014, 136, 14373–14376 CrossRef PubMed.
- C. G. Miller, S. W. Gordon-Wylie, C. P. Horwitz, S. A. Strazisar, D. K. Peraino, G. R. Clark, S. T. Weintraub and T. J. Collins, J. Am. Chem. Soc., 1998, 120, 11540–11541 CrossRef CAS; S. Fukuzumi, Y. Morimoto, H. Kotani, P. Naumov, Y.-M. Lee and W. Nam, Nat. Chem., 2010, 2, 756–759 CrossRef PubMed; Y. Morimoto, H. Kotani, J. Park, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 403–405 CrossRef PubMed; J. Park, Y. Morimoto, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 5236–5239 CrossRef PubMed; J. Park, Y. Morimoto, Y.-M. Lee, Y. You, W. Nam and S. Fukuzumi, Inorg. Chem., 2011, 50, 11612–11622 CrossRef PubMed; F. F. Pfaff, S. Kundu, M. Risch, S. Pandian, F. Heims, I. Pryjomska-Ray, P. Haack, R. Metzinger, E. Bill, H. Dau, P. Comba and K. Ray, Angew. Chem., Int. Ed., 2011, 50, 1711–1715 CrossRef PubMed; P. Leeladee, R. A. Baglia, K. A. Prokop, R. Latifi, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2012, 134, 10397–10400 CrossRef PubMed; J. Chen, Y.-M. Lee, K. M. Davis, X. Wu, M. S. Seo, K.-B. Cho, H. Yoon, Y. J. Park, S. Fukuzumi, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2013, 135, 6388–6391 CrossRef PubMed; H. Yoon, Y.-M. Lee, X. Wu, K.-B. Cho, R. Sarangi, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 9186–9194 CrossRef PubMed; S. Hong, F. F. Pfaff, E. Kwon, Y. Wang, M.-S. Seo, E. Bill, K. Ray and W. Nam, Angew. Chem., Int. Ed., 2014, 53, 10403–10407 CrossRef PubMed.
- D. E. Herbert, D. Lionetti, J. Rittle and T. Agapie, J. Am. Chem. Soc., 2013, 135, 19075–19078 CrossRef CAS PubMed.
- S. Yao, Y. Xiong, M. Vogt, H. Grützmacher, C. Herwig, C. Limberg and M. Driess, Angew. Chem., Int. Ed., 2009, 48, 8107–8110 CrossRef CAS PubMed; Y.-M. Lee, S. Bang, Y. M. Kim, J. Cho, S. Hong, T. Nomura, T. Ogura, O. Troeppner, I. Ivanovic-Burmazovic, R. Sarangi, S. Fukuzumi and W. Nam, Chem. Sci., 2013, 4, 3917–3923 RSC; F. Li, K. M. Van Heuvelen, K. K. Meier, E. Münck and L. Que, J. Am. Chem. Soc., 2013, 135, 10198–10201 CrossRef PubMed; S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Nat. Chem., 2014, 6, 934–940 CrossRef PubMed; Y.-M. Lee, S. Bang, H. Yoon, S. H. Bae, S. Hong, K.-B. Cho, R. Sarangi, S. Fukuzumi and W. Nam, Chem. – Eur. J., 2015, 21, 10676–10680 CrossRef PubMed.
- Y. J. Park, J. W. Ziller and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 9258–9261 CrossRef CAS PubMed; Y. J. Park, S. A. Cook, N. S. Sickerman, Y. Sano, J. W. Ziller and A. S. Borovik, Chem. Sci., 2013, 4, 717–726 RSC.
- S. A. Cook, J. W. Ziller and A. S. Borovik, Inorg. Chem., 2014, 53, 11029–11035 CrossRef CAS PubMed.
- S. Kundu, E. Miceli, E. Farquhar, F. F. Pfaff, U. Kuhlmann, P. Hildebrandt, B. Braun, C. Greco and K. Ray, J. Am. Chem. Soc., 2012, 134, 14710–14713 CrossRef CAS PubMed; S.-L. Abram, I. Monte-Pérez, F. F. Pfaff, E. R. Farquhar and K. Ray, Chem. Commun., 2014, 50, 9852–9854 RSC; I. Monte-Pérez, S. Kundu and K. Ray, Z. Anorg. Allg. Chem., 2015, 641, 78–82 CrossRef.
- M. R. Halvagar and W. B. Tolman, Inorg. Chem., 2013, 52, 8306–8308 CrossRef CAS PubMed.
- M. Risch, K. Klingan, F. Ringleb, P. Chernev, I. Zaharieva, A. Fischer and H. Dau, ChemSusChem, 2012, 5, 542–549 CrossRef CAS PubMed; M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Chem. Sci., 2012, 3, 2330–2339 RSC.
- D. C. Lacy, Y. J. Park, J. W. Ziller, J. Yano and A. S. Borovik, J. Am. Chem. Soc., 2012, 134, 17526–17535 CrossRef CAS PubMed.
- N. Lau, J. W. Ziller and A. S. Borovik, Polyhedron, 2015, 85, 777–782 CrossRef CAS PubMed.
- Y. Sano, A. C. Weitz, J. W. Ziller, M. P. Hendrich and A. S. Borovik, Inorg. Chem., 2013, 52, 10229–10231 CrossRef CAS PubMed.
- N. S. Sickerman, R. M. Henry, J. W. Ziller and A. S. Borovik, Polyhedron, 2013, 58, 65–70 CrossRef CAS PubMed.
- S. A. Cook and A. S. Borovik, Acc. Chem. Rev., 2015, 48, 2407–2414 CrossRef CAS PubMed.
- F. G. Bordwell and D. Algrim, J. Org. Chem., 1976, 41, 2507–2508 CrossRef CAS; F. G. Bordwell, H. E. Fried, D. L. Hughes, T. Y. Lynch, A. V. Satish and Y. E. Whang, J. Org. Chem., 1990, 55, 3330–3336 CrossRef.
- A. D. Schwarz, K. R. Herbert, C. Paniagua and P. Mountford, Organometallics, 2010, 29, 4171–4188 CrossRef CAS.
- R. T. v. Aller, R. B. Scott Jr. and E. L. Brockelbank, J. Org. Chem., 1966, 31, 2357–2365 CrossRef CAS; F. Asinger, P. Laue, B. Fell and G. Gubelt, Chem. Ber., 1967, 100, 1696–1700 CrossRef; J. F. King, J. Y. L. Lam and V. Dave, J. Org. Chem., 1995, 60, 2831–2834 CrossRef.
- A. V. Gontcharov, H. Liu and K. B. Sharpless, Org. Lett., 1999, 1, 783–786 CrossRef CAS PubMed; T. Netscher and H. Prinzbach, Synthesis, 1987, 683–688 CrossRef.
- P. Sun, S. M. Weinreb and M. Shang, J. Org. Chem., 1997, 62, 8604–8608 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data and full crystallographic information. CCDC 1434432–1434441. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00233h |
| ‡ Crystallographic data deposited to the Cambridge Structural Database: [nBu4][1] (1434440), Ca12 (1434437), Sr12(OH2) (1434441), [Ba12]2 (1434432), [nBu4][2-OH2] (1434433), [nBu4][2] (1434434), Mg22 (1434438), Ca22 (1434436), Sr22 (1434439) and Ba22 (1434435). |
| § Onset of oxidation is used here instead of E1/2 because some events are electrochemically irreversible. Each of these electrochemical events may be cycled without changes to the voltammogram, demonstrating chemical reversibility. |
| ¶ Mg22 is not sufficiently soluble for study by CV or electronic absorption spectroscopy. |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 



 ) and [nBu4N][2-OH2] (
) and [nBu4N][2-OH2] ( ) in CH2Cl2 (top); [nBu4N][1] (
) in CH2Cl2 (top); [nBu4N][1] ( ) and [nBu4N][2] (
) and [nBu4N][2] ( ) in CH2Cl2 (second from top, smaller features are from hydrated forms derived from adventitious water in the experimental setup); [nBu4N][1-OH2] (
) in CH2Cl2 (second from top, smaller features are from hydrated forms derived from adventitious water in the experimental setup); [nBu4N][1-OH2] ( ) and [nBu4N][2-OH2] (
) and [nBu4N][2-OH2] ( ) in TFE (second from bottom); and Ca22 (
) in TFE (second from bottom); and Ca22 ( ), Sr22 (
), Sr22 ( ), and Ba22 (
), and Ba22 ( ) in TFE. Scan rate = 100 mV s−1.
) in TFE. Scan rate = 100 mV s−1.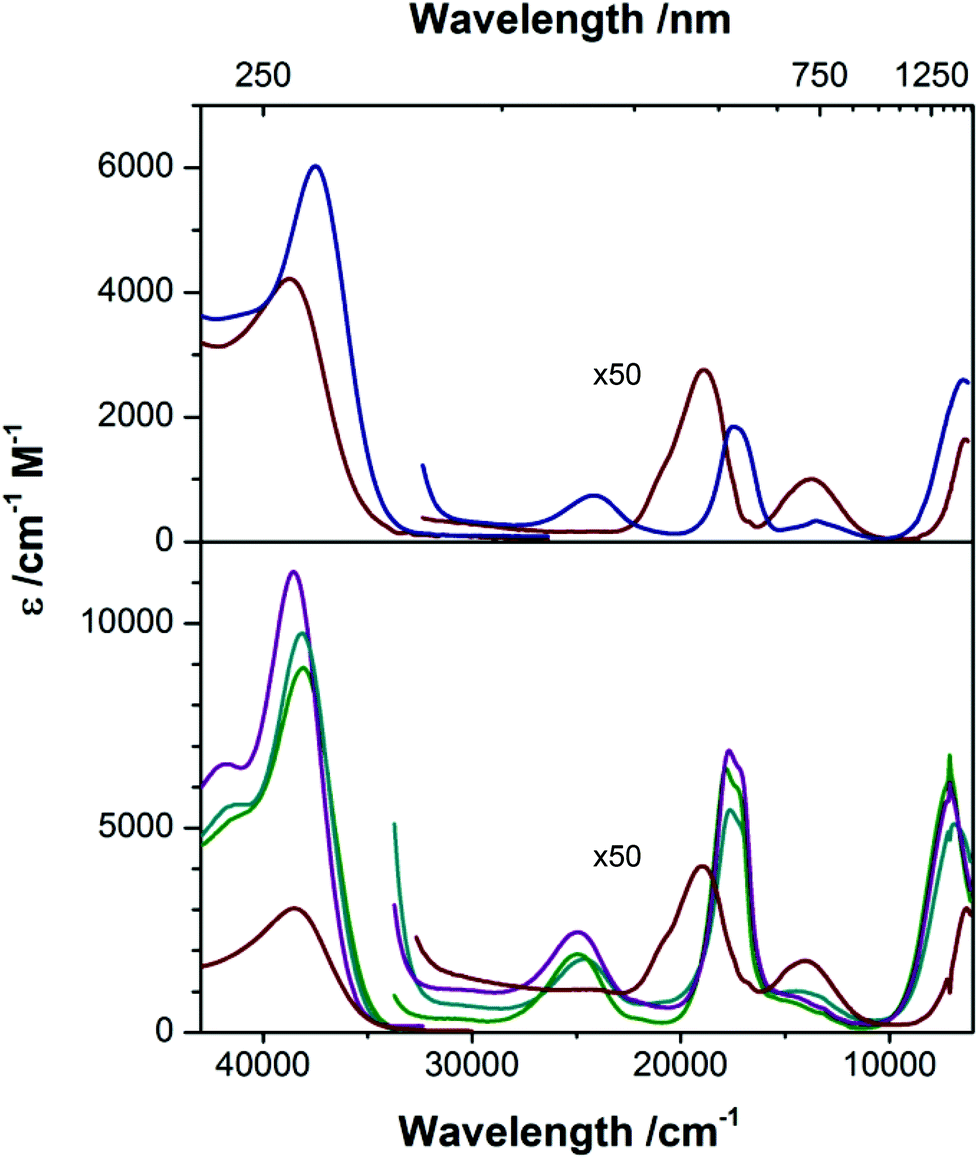
 ) and [nBu4N][2-OH2] (
) and [nBu4N][2-OH2] ( ) in CH2Cl2 (top), and [nBu4N][2-OH2] (
) in CH2Cl2 (top), and [nBu4N][2-OH2] ( ) Ca22 (
) Ca22 ( ), Sr22 (
), Sr22 ( ), and Ba22 (
), and Ba22 ( ) in TFE (bottom).
) in TFE (bottom).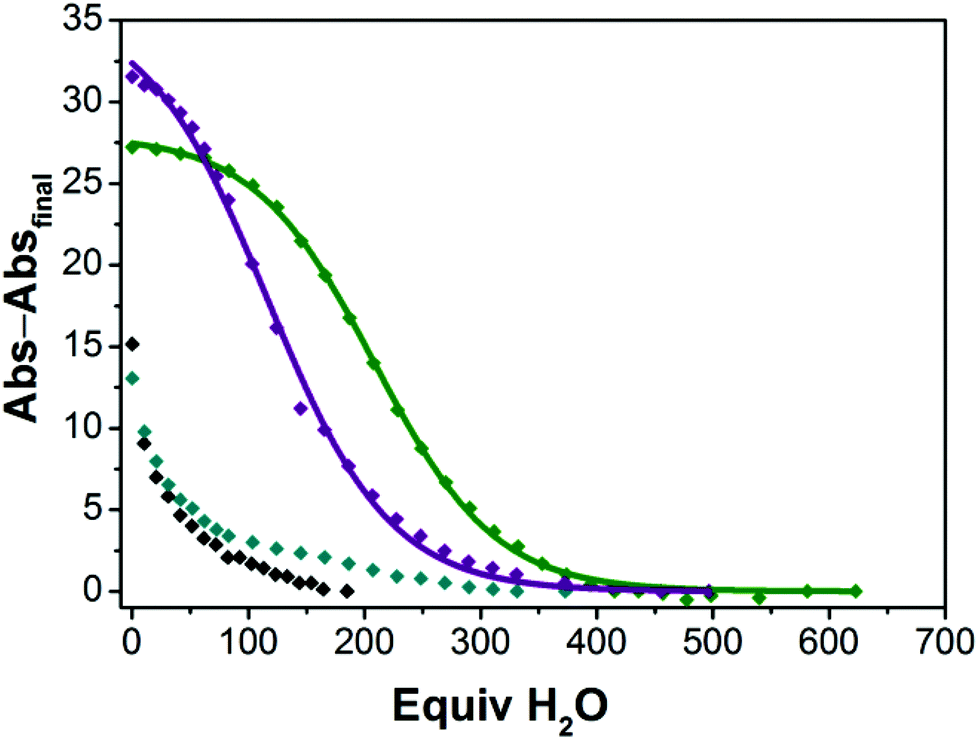
 ), Sr22 (
), Sr22 ( ), Ba22 (
), Ba22 ( ) and Ca12 (◆) into five-coordinate CoII in TFE with added water. Abs–Absfinal represents the absorption of Ca22 at 401 nm, Sr22 at 401 nm, Ba22 at 406 nm, and Ca12 at 406 nm as a function of added equivalents of H2O, with the final absorption value for each species subtracted from each point. The curves for Ca22 and Sr22 are fits to the data using sigmoidal logistic functions (see ESI† for details).
) and Ca12 (◆) into five-coordinate CoII in TFE with added water. Abs–Absfinal represents the absorption of Ca22 at 401 nm, Sr22 at 401 nm, Ba22 at 406 nm, and Ca12 at 406 nm as a function of added equivalents of H2O, with the final absorption value for each species subtracted from each point. The curves for Ca22 and Sr22 are fits to the data using sigmoidal logistic functions (see ESI† for details).