
Recent developments in the catalytic hydrogenation of CO2 to formic acid/formate using heterogeneous catalysts
Gunniya Hariyanandam
Gunasekar†
a,
Kwangho
Park†
a,
Kwang-Deog
Jung
b and
Sungho
Yoon
*a
aDepartment of Bio&Nano Chemistry, Kookmin University, 861-1 Jeongneung-dong, Seongbuk-gu, Republic of Korea. E-mail: yoona@kookmin.ac.kr
bClean Energy Research Centre, Korea Institute of Science and Technology, P.O. Box 131, Cheongryang, Seoul 136-791, Republic of Korea
First published on 8th March 2016
Abstract
The conversion of CO2 into value added chemicals is one of the fascinating strategies to mitigate the level of CO2 in the atmosphere. Specifically, the hydrogenation of CO2 into formic acid/formate has received a great deal of attention since the product is a valuable basic chemical as well as a promising energy storage material. However due to the kinetic and thermodynamic limitations of this conversion, developing an efficient catalytic system has become desirable. Therefore various approaches have been implemented for the development of both homogeneous and heterogeneous catalysts. In this context, recent advances in the hydrogenation of CO2 to formic acid/formate using heterogeneous catalysts as well as theoretical investigations are presented.
 Gunniya Hariyanandam Gunasekar | Gunniya Hariyanandam Gunasekar received his Master's degree in Chemistry from Madurai Kamaraj University, India in the year 2009. He has worked as a research executive at the Orchid Chemicals and Pharmaceuticals R&D Centre during 2009–2013. Currently, he is pursuing his Ph.D. degree in the group of Prof. Sungho Yoon at Kookmin University, South Korea. His research interests focus on the transformations of CO2 as well as the development of multifunctional COF materials. |
 Kwangho Park | Kwangho Park obtained his B.S. degree in Chemistry at the Department of Bio and Nano Chemistry of Kookmin University in 2014. Currently, he is a Master's degree student in Kookmin University, working in the Nano Inorganic Lab under Prof. Sungho Yoon. His research interests include CO2 capture, CO2 hydrogenation and the development of covalent triazine framework materials. |
 Kwang-Deog Jung | Dr Kwang-Deog Jung received his Bachelor's degree and Ph.D. in Chemical Engineering from Yonsei University in 1985 and Korea Advanced Institute of Science and Technology in 1996, respectively. He has worked on C1 chemistry and processes at Korea Institute of Science and Technology since 1997. His research interests focus on the synthesis of heterogeneous catalysts and the catalytic processes for hydrogenation of CO and CO2, hydrogen production, carbonylation of CO and electrochemical CO2 reduction. |
 Sungho Yoon | Prof. Sungho Yoon received his B.S. and Ph.D. degrees from Korea University in 1998 and Massachusetts Institute of Technology in 2004, respectively, and worked as a post-doctoral fellow at the University of California, Berkeley during 2004–2005 and as a researcher at LG Chem. Research Park during 2005–2007. He has worked on C1 chemistry in Kookmin University since 2007. His research interests focus on the synthesis of covalent organic frameworks and catalysts for hydrogenation and carbonylation. |
1. Introduction
In recent years, climate change and increasing global temperatures due to the anthropogenic emission of CO2 into the atmosphere have caused increasing concern. The concentration of CO2 in the atmosphere has reached unprecedented levels (∼400 ppm), mainly due to the utilization of carbon-rich fossil fuels, such as coal, oil and natural gas.1,2 Thus, reducing the emission of CO2, utilizing the abundant CO2 in the atmosphere and recycling CO2 have attracted much attention.3–6 Recently, “Lima call for climate action” aimed at achieving a near-zero anthropogenic emission of CO2 by the end of the century, highlighting the concern over atmospheric CO2 levels.7,8 Consequently, research into renewable fuels, CO2 storage and CO2 utilization has gained increased attention from the scientific and technological community.9–12CO2 has emerged as an attractive C1 feedstock for producing various chemicals owing to the fact that it is safe, abundant, nontoxic, economical and renewable. Such CO2-based products are not only profitable, but also alternatives to those derived from petroleum resources, and could therefore lessen the petroleum dependence in society.13–17 However, very few industrial processes, largely limited to those involving urea, salicylic acid and carbonate synthesis, utilize CO2 as a raw material.18
One important chemical out of diverse chemical products derived based on CO2 as a C1 feedstock is formic acid, which is a valuable basic chemical used as a preservative, an antibacterial agent, an insecticide and also as a de-icing agent in various industries.19,20 In addition, it plays a major role in synthetic chemistry as an acid, a reductant and a precursor for syntheses. Moreover, it is being considered as a hydrogen storage material in the energy industries. Even though formic acid has a relatively low H2 content (4.4 wt%), it is easy to store and transport.21 Recently, formic acid has emerged as a promising fuel source in direct liquid fuel cell systems due to its excellent oxidation kinetics, high cell potential and fewer fuel crossover problems.22 Thus, the potentials of formic acid as an energy carrier as well as a chemical building block make the hydrogenation of CO2 to formic acid/formates attractive to the scientific and technological community to a greater extent.
Currently, 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 T of formic acid is produced per year in the industries using toxic carbon monoxide and methanol.23 According to Bardow and coworkers, this process could emit ca. 3076 kg of CO2 for the production of 1 T of formic acid, whereas only 100 kg of CO2 could be emitted if the same is produced by the CO2 hydrogenation process.24 Hence, CO2-based production has enormous potential to reduce the environmental impact when compared to the conventional CO-based method.
000 T of formic acid is produced per year in the industries using toxic carbon monoxide and methanol.23 According to Bardow and coworkers, this process could emit ca. 3076 kg of CO2 for the production of 1 T of formic acid, whereas only 100 kg of CO2 could be emitted if the same is produced by the CO2 hydrogenation process.24 Hence, CO2-based production has enormous potential to reduce the environmental impact when compared to the conventional CO-based method.
The hydrogenation of CO2 to formic acid involves the conversion of gaseous substances into liquid products and hence the reaction is entropically disfavored. Thus, the reaction is endergonic in the gas phase (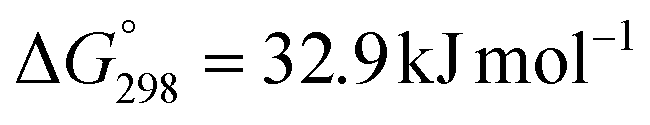 ) and is exergonic in the aqueous (solvation) phase (
) and is exergonic in the aqueous (solvation) phase (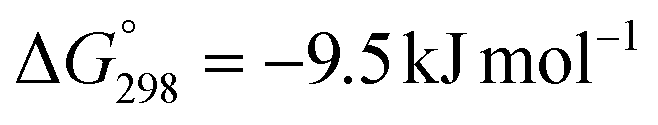 ). In addition, the equilibrium can shift towards the product side using certain bases, such as ammonia (NH3) and triethylamine (NEt3) (eqn (1)–(3)).25 Moreover, the chemical equilibrium of CO2/bicarbonate (HCO3−)/carbonate (CO32−) is influenced by many parameters, such as temperature, CO2 pressure, kind of base, and solution pH. Thus, the actual form of the substrate in this reaction is ambiguous. Henceforth, the term ‘hydrogenation of CO2’ being used in this review and elsewhere may actually involve HCO3−/CO32− as the real substrate.
). In addition, the equilibrium can shift towards the product side using certain bases, such as ammonia (NH3) and triethylamine (NEt3) (eqn (1)–(3)).25 Moreover, the chemical equilibrium of CO2/bicarbonate (HCO3−)/carbonate (CO32−) is influenced by many parameters, such as temperature, CO2 pressure, kind of base, and solution pH. Thus, the actual form of the substrate in this reaction is ambiguous. Henceforth, the term ‘hydrogenation of CO2’ being used in this review and elsewhere may actually involve HCO3−/CO32− as the real substrate.
Since the discovery of phosphine-based Ru complexes by Inoue et al.,26 excellent progress has been achieved in the development of homogeneous catalysts.27–32 In particular, extensive studies on homogeneous Ir, Ru, and Rh complexes have been reported, and recently, half-sandwich Ir derivatives and Ru/Ir-pincer complexes have shown tremendous catalytic activities, with a maximum turnover number (TON) of 3500000 and a maximum turnover frequency (TOF) of 1100000 h−1.33–40
Despite the fact that the homogeneous catalysts exhibit excellent catalytic efficiencies for the hydrogenation of CO2 to formate, industries are reluctant to use them for large-scale production, owing to the difficulty of catalyst separation from the final reaction mixture.41 Importantly, these homogeneous catalysts also promote the decomposition of the generated formate back into CO2 and H2 during the product separation step(s).23,42,43 Because of such limitations, diverse heterogeneous catalysts which have a strong merit in separation have been reported.
There were many publications on the hydrogenation of CO2 to formic acid/formate using homogeneous catalysts,44–47 but only a few studies were reported recently on the hydrogenation of CO2 to formic acid/formate by heterogeneous catalysts. Therefore, the progress achieved in the development of heterogeneous catalysts for this conversion is presented here. In order to explore the prospective design of highly active and industrially viable heterogeneous catalysts, we have segmented this review based on the types of catalysts and the catalyst supports employed. The organization of this review is as follows: it has been categorized into three major sections excluding Introduction and Conclusion. After Introduction, the second section provides the application of heterogeneous metal catalysts. This section is subdivided into two parts based on the type of metal catalyst employed: section 2.1 explores the performances of bulk metal catalysts; section 2.2 presents the catalytic efficiencies of supported metal catalysts. The third section addresses the application of heterogenized molecular catalysts. This section is further classified into two parts based on the kind of heterogenization process followed; section 3.1 deals with the efficiencies of metal complexes heterogenized by grafting methods; section 3.2 explores the performances of metal complexes heterogenized by incorporating into the support's pore wall. Since covalent organic framework (COF) based supports have only been utilized in the literature, we titled this section “Heterogenized COFs catalyst”. Section 4 briefly explores the theoretical investigations on this topic and in the final section, we briefly summarize the status and perspectives of this field.
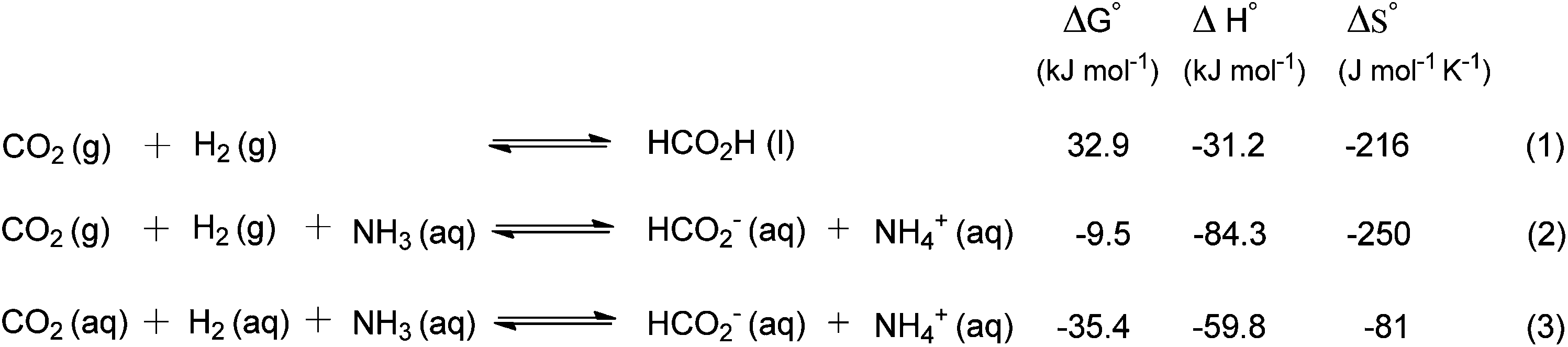
2. Heterogeneous metal catalysts
2.1 Bulk metal catalysts
Research on the hydrogenation of CO2 to formate was started in 1935 by Adkins et al. using RANEY® nickel (Ra-Ni) as a catalyst and they reported 57% yield at 80–150 °C under high pressure (20–40 MPa) in the ethanol (EtOH)/phenol solvent system (Table 1, entry 1).48 Even though the reaction was not catalytic (TON < 1), it initiated a new field of research, which is the conversion of relatively cheap and abundant CO2 into value added chemicals. Later, Stalder et al. employed Pd black as a catalyst for the conversion (entry 2) and observed catalytic conversion (TON 2.1) in 1 M aqueous NaHCO3 solution after long reaction times (53 h).49 Takahashi et al. used a mixture of Fe and Ni powders as a catalyst under hydrothermal conditions (300 °C) using water (H2O) as a hydrogen source; however, the yield of formate was very low (TON 0.022).50 Later, Fachinetti et al. broadened their search to Group 8–11 metal blacks.51 Initially they screened metals that promote the reverse reaction, that is, the decomposition of formate salts to CO2 and H2, as they necessarily promote the hydrogenation of CO2 to formate. Transition metals (Group 8–11), such as Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Ir, Pt and Au, were screened for the decomposition of formic acid–NEt3 (HCOOH/NEt3) adducts to CO2 and H2 at 40 °C, and only Au metal blacks showed gas evolution, indicating that only Au metal had the potential to promote the hydrogenation of CO2 to formate. Since then, Au black was examined as a catalyst for the hydrogenation of CO2 to formate at 40 °C in NEt3 solution, and the results showed that Au black fairly converted CO2 to formate. However, deactivation of the catalyst was observed during the reaction, which was due to the aggregation of catalytic metal particles during the reaction, leading to a decrease of the number of active sites on the surface of the catalyst. This result clearly explains the low catalytic efficiencies of bulk metals and also the inevitability of a support to stabilize metal blacks.| Entry | Catalyst | Temperature (°C) | p(H2)/p(CO2) (MPa) | Solvent | Additive/base | Time (h) | TON/final formate concentration (M) | TOF (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Ra-Ni | 100 | 4–14/6 | EtOH/phenol | 2,2,6,6-Tetra-Me-4-OH piperidine | 6 | —/0.31 | <1 | 48 |
| 2 | Pd black | 25 | 0.1/0 | H2O | NaHCO3 | 53 | 2.1/0.19 | <1 | 49 |
| 3 | AUROlite® | 40 | 9/9 | NEt3 | NEt3 | 52 | 855 | 16.4 | 51 |
| 4 | Au NP/Al2O3 | 70 | 20/20 | EtOH | NEt3 | 20 | 215/0.20 | 11 | 54 |
| 5 | Au NP/TiO2 | 70 | 20/20 | EtOH | NEt3 | 20 | 111/0.09 | 5.5 | 54 |
| 6 | Au NP/ZnO | 70 | 20/20 | EtOH | NEt3 | 20 | 2/<0.01 | <1 | 54 |
| 7 | Au NP/CeO2 | 70 | 20/20 | EtOH | NEt3 | 20 | 8/<0.01 | <1 | 54 |
| 8 | Au NP/MgAl-HT | 70 | 20/20 | EtOH | NEt3 | 20 | 91/0.01 | 4.5 | 54 |
| 9 | Au NP/MgCr-HT | 70 | 20/20 | EtOH | NEt3 | 20 | 52/<0.01 | 2.6 | 54 |
| 10 | Au NP/CuCr2O4 | 70 | 20/20 | EtOH | NEt3 | 20 | 6/<0.01 | <1 | 54 |
| 11 | Pd/BaSO4 | 25 | 0.1/0 | H2O | NaHCO3 | 50 | 19/0.09 | <1 | 49 |
| 12 | Pd/γ-Al2O3 | 25 | 0.1/0 | H2O | NaHCO3 | 53 | 50/0.23 | <1 | 49 |
| 13 | Pd/C | 25 | 0.17/0 | H2O | NaHCO3 | 46 | 115/0.54 | 2.5 | 49 |
| 14 | Pd/AC | 20 | 2.75/0 | H2O | NaHCO3 | 1 | 527/0.28 | 527 | 55 |
| 15 | Pd/AC | 20 | 2.75/0 | H2O | KHCO3 | 1 | 567/0.30 | 567 | 55 |
| 16 | Pd/AC | 20 | 2.75/0 | H2O | NH4HCO3 | 1 | 782/0.42 | 782 | 55 |
| 17 | Pd/AC | 20 | 2.75/0 | H2O | Na2CO3 | 1 | <1/<0.01 | <1 | 55 |
| 18 | Pd/AC | 20 | 2.75/0 | H2O | K2CO3 | 1 | <1/<0.01 | <1 | 55 |
| 19 | Pd/AC | 20 | 2.75/0 | H2O | (NH4)2CO3 | 1 | 278/0.15 | 278 | 55 |
| 20 | Pd/AC | 20 | 2.75/0 | H2O | NH4HCO3 | 15 | 1769/0.95 | 118 | 55 |
| 21 | Pd/AC | 20 | 5.52/0 | H2O | NH4HCO3 | 2 | 1672/0.90 | 836 | 55 |
| 22 | Pd/Al2O3 | 20 | 2.75/0 | H2O | NH4HCO3 | 1 | 278/0.08 | 278 | 55 |
| 23 | Pd/CaCo3 | 20 | 2.75/0 | H2O | NH4HCO3 | 1 | 20/<0.01 | 20 | 55 |
| 24 | Pd/BaSO4 | 20 | 2.75/0 | H2O | NH4HCO3 | 1 | 212/0.02 | 212 | 55 |
| 25 | Pd/AC | 20 | 2.75/0 | EtOH/H2O (7/3) | NH2CO2NH4 | 8 | 845/0.91 | 105 | 56 |
| 26 | Pd/r-GO (1 wt%) | 100 | 4/0 | H2O | KHCO3 | 32 | 7088/4.53 | 221 | 57 |
| 27 | Pd/r-GO (2 wt%) | 100 | 4/0 | H2O | KHCO3 | 10 | 2117/4.06 | 211 | 57 |
| 28 | Pd/r-GO (5 wt%) | 100 | 4/0 | H2O | KHCO3 | 10 | 1658/3.18 | 165 | 57 |
| 29 | PdNi/CNT-GR | 40 | 25/25 | H2O | Nil | 15 | 6.4/0.02 | <1 | 58 |
| 30 | Ru/MgO | 80 | 5/8.5 | EtOH | NEt3 | 1 | 0 | — | 59 |
| 31 | Ru/AC | 80 | 5/8.5 | EtOH | NEt3 | 1 | 10/0.05 | 10 | 59 |
| 32 | Ru/γ-Al2O3 | 80 | 5/8.5 | EtOH | NEt3 | 1 | 91/0.455 | 91 | 59 |
| 33 | Ru/γ-Al2O3(n)) | 80 | 5/8.5 | EtOH | NEt3 | 1 | 731/0.68 | 731 | 60 |
| 34 | 3 | 80 | 5.4/9.3 | EtOH | PPh3/NEt3 | 1 | 1022/0.40 | 1022 | 66 |
| 35 | 4 | 80 | 4/12 | EtOH | PPh3/NEt3 | 1 | 656/0.52 | 656 | 63 |
| 36 | 5 | 80 | 5/8 | EtOH | PPh3/NEt3 | 1 | 151/0.36 | 151 | 65 |
| 37 | 6 | 80 | 5.4/9.3 | EtOH | PPh3/NEt3 | 1 | 723/0.28 | 723 | 66 |
| 38 | 7 | 80 | 5.4/9.3 | EtOH | PPh3/NEt3 | 1 | 537/0.21 | 537 | 66 |
| 39 | 8 | 80 | 4/12 | EtOH | PPh3/NEt3 | 1 | 1384/1.10 | 1384 | 63 |
| 40 | 9 | 80 | 4/12 | EtOH | PPh3/NEt3 | 1 | 868/0.69 | 868 | 63 |
| 41 | 10 | 80 | 5/8 | EtOH | PPh3/NEt3 | 1 | 75/0.18 | 75 | 65 |
| 42 | 11 | 80 | 5/8 | EtOH | PPh3/NEt3 | 1 | 143/0.34 | 143 | 65 |
| 43 | 5 | 80 | 5/8 | EtOH | dppe/NEt3 | 1 | 191/0.45 | 191 | 65 |
| 44 | 5 | 80 | 5/8 | EtOH | AsPh3/NEt3 | 1 | 29/0.06 | 29 | 65 |
| 45 | 3 | 80 | 5.4/9.3 | EtOH | NPh3/NEt3 | 1 | 179/0.07 | 179 | 66 |
| 46 | 3 | 80 | 5.4/9.3 | EtOH | AsPh3/NEt3 | 1 | 171/0.06 | 171 | 66 |
| 47 | 12 | 60 | 2/2 | H2O | NEt3 | 2 | 1300/0.13 | 620 | 70 |
| 48 | 13 | 60 | 2/2 | H2O | NEt3 | 2 | 110/0.01 | 55 | 70 |
| 49 | 14 | 60 | 2/2 | H2O | NEt3 | 2 | 400/0.04 | 200 | 70 |
| 50 | 15 | 60 | 2/2 | H2O | NEt3 | 1 | 0 | 0 | 70 |
| 51 | 16 | 60 | 2/2 | H2O | NEt3 | 2 | 70/<0.01 | 35 | 70 |
| 52 | 12 | 60 | 2/2 | H2O | NEt3 | 1 | 880/0.08 | 880 | 70 |
| 53 | 12 | 90 | 2/2 | H2O | NEt3 | 1 | 1100/0.11 | 1100 | 70 |
| 54 | 12 | 120 | 2/2 | H2O | NEt3 | 2 | 2300/0.23 | 1200 | 70 |
| 55 | 12 | 60 | 2/2 | H2O | NEt3 | 20 | 2700/0.27 | 140 | 70 |
| 56 | 17 | 120 | 2/2 | H2O | NEt3 | 1 | 248/0.04 | 248 | 71 |
| 57 | 18 | 120 | 2/2 | H2O | NEt3 | 1 | 38/<0.01 | 38 | 71 |
| 58 | 19 | 120 | 2/2 | H2O | NEt3 | 1 | 132/0.02 | 132 | 71 |
| 59 | 20 | 40 | 6/6 | NEt3 | NEt3 | 24 | 25 | 1 | 79 |
| 60 | 20 | 60 | 6/6 | NEt3 | PPh3 | 24 | 2254 | 94 | 79 |
| 61 | 21 | 150 | 2/2 | D2O | NEt3 | 24 | 81/0.28 | 3.3 | 80 |
| 62 | 21 | 150 | 2.7/1.3 | D2O | NEt3 | 24 | 106/0.37 | 4.4 | 80 |
| 63 | 21 | 150 | 3/1 | D2O | NEt3 | 24 | 95/0.34 | 3.9 | 80 |
| 64 | 21 | 150 | 3.5/0.5 | D2O | NEt3 | 24 | 77/0.27 | 3.2 | 80 |
| 65 | 23 | 80 | 2/2 | H2O | NEt3 | 2 | 500/0.06 | 250 | 81 |
| 66 | 23 | 120 | 2/2 | H2O | NEt3 | 2 | 3320/0.40 | 1660 | 81 |
| 67 | 23 | 160 | 2/2 | H2O | NEt3 | 2 | 2700/0.33 | 1350 | 81 |
| 68 | 23 | 200 | 2/2 | H2O | NEt3 | 2 | 1320/0.16 | 660 | 81 |
| 69 | 23 | 120 | 4/4 | H2O | NEt3 | 2 | 5000/0.61 | 2500 | 81 |
| 70 | 23 | 120 | 4/4 | H2O | NEt3 | 0.25 | 1300/0.15 | 5300 | 81 |
| 71 | 25 | 120 | 4/4 | H2O | NEt3 | 0.5 | 750/0.01 | 1500 | 82 |
| 72 | 25 | 120 | 4/4 | H2O | NEt3 | 10 | 6400/0.14 | 640 | 82 |
2.2 Supported metal catalysts
Since heterogeneous catalytic reactions are surface reactions, they typically follow the following steps: (1) the adsorption of reactants onto the surface of the catalyst, (2) the surface reaction of adsorbed reactants with the catalyst, and (3) desorption of products from the surface of the catalyst.52 Since the number of metal atoms on the surface of a bulk metal is extremely small compared to the number of metal atoms in the interior, a large number of active metal atoms are not exposed to the reactants, which makes bulk metals economically inefficient to use in catalytic processes. Moreover, as these bulk metals often aggregate under reaction conditions, the number of active metal atoms on the surface of the catalyst is reduced further. To overcome these problems, catalytically active metals are often dispersed on solid supports.Since the Au blacks aggregated under reaction conditions, Fachinetti et al. employed titanium dioxide (TiO2) supported Au particles (1 wt% Au) for this conversion.51 The supported Au particles (AUROlite®) showed superior catalytic conversion to unsupported Au particles, and attained a TON of 855 at 40 °C under a total pressure of 18 MPa in NEt3 solution for 3 days (entry 3). Importantly, 1.3 kg of the HCOOH/NEt3 adduct with an acid to amine ratio of 1.7 was obtained after 37 days of continuous production. This result showed that, unlike bulk Au black, the supported Au particles were very stable during the reaction conditions. However, owing to the Au-catalyzed reverse water gas shift reaction, the accumulation of CO (63 mmol) was observed, which consequently slowed the reaction.53
Recently Pidko and co-workers employed both supported and unsupported Au nanoparticles [Au NP(s)] as a catalyst for this reaction.54 They also observed that the catalytic efficiency of supported Au NPs was superior to that of unsupported Au NPs. They employed a series of supports, namely, Al2O3, TiO2, ZnO, CeO2, MgAl-HT (hydrotalcite), MgCr-HT and CuCr2O4, to stabilize the Au NPs and studied their catalytic efficiencies (entries 4–10). Of the various supports employed, the Au NP supported on Al2O3 (Au NP/Al2O3) have shown better catalytic performance (TON 215) (entry 4). Notably, Au NP/Al2O3 exhibited a two-fold higher catalytic efficiency than the titania-supported Au NP (Au NP/TiO2) (entries 4 and 5). This superior performance of Au NP/Al2O3 is believed to be the synergetic effect of Au NP and Al2O3 supports. The temperature dependent TOF values and the kinetic modeling results revealed that the Au NP/Al2O3 has a near-zero apparent activation energy (1.2 kcal mol−1). Based on the spectroscopic data and previous studies, they proposed a plausible mechanism by which the Au–H species, formed from the heterolytic dissociation of H2, react with the surface bicarbonate species and form Au-formate intermediates. This surface formate species would migrate to the more stable alumina surface and subsequently formates are released from the catalytic cycle.
Previously, Stalder et al. studied the catalytic efficiency of a series of supported Pd species for the conversion of NaHCO3 to formate under ca. 0.1 MPa H2 pressure.49 Three types of supports, namely BaSO4, γ-Al2O3 and carbon, were employed, and all the supported Pd catalysts (Pd/BaSO4, Pd/γ-Al2O3 and Pd/C) showed better activity than unsupported Pd black under similar reaction conditions, again revealing the importance of catalyst supports (entries 2 and 11–13). Among the supported catalysts, Pd/C showed the maximum TON (115) at 25 °C (entries 11–13). Additionally, Ni, Ru, Rh and Pt metals supported on γ-Al2O3 also catalyzed the reaction, but to a lesser extent than Pd.
Recently, Su et al. employed a porous carbon material (activated carbon) as a support (Pd/AC) and systematically studied the conversion of various bicarbonate salts to formates under relatively high H2 pressures (0.69–5.52 MPa).55 The Pd/AC showed a higher TOF (527 h−1) than the Pd/C (TOF 2.5 h−1), indicating the benefits of porous catalyst supports for the reaction (entries 13 and 14). In addition, various carbonates (Na2CO3, K2CO3 and (NH4)2CO3) and bicarbonates (NaHCO3, KHCO3 and NH4HCO3) were screened and it was found that the hydrogenation of carbonate salts was much more difficult than that of bicarbonate salts (entries 14–19). This is because the protonation of carbonate ions was the rate limiting step at this temperature. Of the bicarbonate salts screened, NH4HCO3 gave higher yield (42.4%, TON 782) than NaHCO3 (28.6%, TON 527) and KHCO3 (30.8%, TON 567) (entries 14–16). The high yield of formate from NH4HCO3 salt was thought to be caused by the higher equilibrium concentration of HCO3− ions (0.92 M) over CO32− ions than that of KHCO3 (0.89 M) or NaHCO3 salts (0.61 M).
However, the yield of formate was decreased at higher temperatures because Pd/AC promoted the decomposition of ammonium formate back into CO2, H2 and NH3 to a large extent at higher temperatures. Therefore, the maximum activity was obtained at high H2 pressure (5.52 MPa) and a long reaction time (entries 20 and 21). In addition, various catalyst supports (BaSO4, CaCO3 and γ-Al2O3) were also screened and it was found that Pd/AC exhibited better catalytic performance than the others (entries 16, 22–24). This was attributed to the hydrophobic nature of activated carbon, which results in the accumulation of H2 on the surface of the support. In addition to the above salts (NaHCO3, KHCO3 and NH4HCO3), ammonium carbamate salt (NH2CO2NH4) was also studied.56 It was shown that the activities of these salts were dependent on the solvents employed. For example, in pure H2O solution, NaHCO3 (0.5 M) showed the maximum yield of 23% but in pure EtOH solution, NH2CO2NH4 (0.5 M) exhibited the maximum yield of 40%. The yield of NH2CO2NH4 was improved to 44% in an EtOH–H2O mixture (7:3), and this was expected due to the formation of ethyl carbamate salt in the EtOH–H2O mixture. The Pd/AC showed the maximum TON of 845 using NH2CO2NH4 salt at 20 °C under a H2 pressure of 2.75 MPa (entry 25). Furthermore, the catalytic efficiency of Pd/AC was maintained over repeated recycling.
Yong Cao and co-workers evaluated the catalytic performance of Pd nanoparticles supported on reduced graphitic oxide nanosheets (Pd/r-GO).57 They obtained the highest TON (7088) using 1 wt% Pd/r-GO after a long reaction period (32 h) at 100 °C under a H2 pressure of 4 MPa (entry 26). However, the catalytic efficiency gradually decreased with increasing the Pd loading (entries 26 and 28), and this was attributed to the larger lattice strain of Pd nanoparticles in 1 wt% Pd/r-GO than that in 2 and 5 wt% Pd/r-GO.
Very recently, Nguyen et al. studied the catalytic performance of PdNi alloy supported on the Carbon Nanotube–Graphene (PdNi/CNT–GR) composite for the direct synthesis of formic acid by CO2 hydrogenation (without any base).58 To prevent the stacking of GR and the bundling of CNTs, which usually occur by themselves, the CNT–GR composite has been utilized as a support to expose the entire surface area of CNTs and GR for the catalysis. Interestingly, the bimetallic PdNi/CNT–GR (Pd3Ni7/CNT–GR; Pd-30%, Ni-70%) produced a substantial amount of formic acid (1.92 mmol; TON 6.4) at 40 °C under a total pressure of 5 MPa (entry 29). However, the generation of a small amount of acetic acid was also observed. By comparing with the efficiency of mono-metallic Ni supported on CNT–GR (Ni/CNT–GR) and Pd supported on CNT–GR (Pd/CNT–GR), it was inferred that the synergic effect of Pd–Ni was actually responsible for its catalytic activity.
Hao et al. hypothesized that the hydroxyl groups on the surface of a support may enhance the adsorption of CO2 and the catalytic efficiency of a catalyst.59 To test the hypothesis, ruthenium hydroxide was supported on three different supports: (1) MgO which does not have hydroxyl groups (Ru/MgO); (2) activated carbon which has a limited number of hydroxyl groups on the surface (Ru/AC); and (3) γ-alumina, which has abundant hydroxyl groups on the surface (Ru/γ-Al2O3). As the authors hypothesized, Ru/MgO did not produce formate and Ru/AC showed little activity (TON 10), and Ru/γ-Al2O3 showed higher catalytic activity (TON 91) (entries 30–32). Unlike Pd/AC and Pd/Al2O3, Ru/γ-Al2O3 exhibited higher catalytic efficiency than Ru/AC. This might be due to the synergic effect of the metal and the support. Moreover, they observed that the presence of RuO2 on the surface of the catalyst (formed during catalyst preparation) lowers the efficiency of the catalyst. Later, Liu et al. employed γ-Al2O3 nanorods (γ-Al2O3(n)) to support the ruthenium hydroxide species (Ru/γ-Al2O3(n)) owing to the high surface area, abundant hydroxyl groups, and its increased interaction with ruthenium species.60,61 As expected, the catalytic efficiency of Ru/γ-Al2O3(n) (TON 731) was higher than that of Ru/γ-Al2O3 under similar reaction conditions (entries 32 and 33).
3. Heterogenized molecular catalysts
Heterogeneous metal catalysts demonstrated the possibility of separating catalysts from the reaction mixtures and the continuous use for repeated runs. However, the catalytic efficiencies of these catalysts were poor; for example, the maximum TOF obtained using these metal-based heterogeneous catalysts was 836 h−1 (entry 21).55 Since these catalysts are mainly made from precious metals (Ru, Pd, Au, etc.), achieving a high catalytic efficiency is essential for their economical use. Conversely, homogeneous metal complexes have shown tremendous catalytic efficiencies (TONs up to 3500000,38 TOFs up to 1100000 h−1),40 but are difficult to separate from the reaction mixtures. Therefore, much research on the development of heterogeneous catalysts with high catalytic efficiencies has been undertaken. In this context, strategies involving the heterogenization of homogeneous metal complexes have been developed. These strategies seek to develop materials that combine the activity and selectivity of homogeneous metal catalysts with the advantages of heterogeneous catalysts, i.e., recyclability and easy seclusion from reaction mixtures. Such heterogenization processes, also called ‘immobilization’, have been performed to produce heterogenized catalysts for CO2 hydrogenation to formate.
3.1 Grafted molecular catalysts
Krocher et al. prepared silica tethered Ru complexes (1 and 2) through the covalent bond between the ligand and the support for the synthesis of dimethylformamide from CO2 (Chart 1).62 Later, Zheng and co-workers immobilized the Ru complexes on the surface of supports through the coordination bond between the donor ligand of the support and the metal ion (Chart 1, 3–11).63–67 They extensively studied the effect of various supports (silica, MCM-41 and polystyrene (PS)) and surface-bound donor ligands (amine (–NR2), nitrile (–CN) and thiol (–SH)) for the hydrogenation of CO2 to formate. To study the effect of supports on reactivity, both inorganic and organic supports have been considered. For the inorganic supports, only silica based supports (silica and MCM-41) were employed, which might be due to the ease of functionalization through their surface OH-groups and their rigid structure. Among the inorganic supports, the MCM-41 supported catalyst (3) showed higher activity than the silica supported catalyst (4) (entries 34 and 35). The high surface area (852 m2 g−1) and uniform pores (3.5 nm) of MCM-41 were responsible for this result, indicating that porous catalysts together with a large surface area would be the possible candidates for a better catalytic efficiency. Regarding the organic supports, only PS was investigated. The catalytic efficiency of the PS supported catalyst (5) was found to be very low (TOF 151 h−1) under similar reaction conditions (entries 34 and 36). Considering the amine (3), nitrile (6) and thiol (7) functionalities on the surface of the support, the amine functionalized catalyst (3) exhibited better activity than 6 and 7 (entries 34, 37 and 38). This might be due to the stronger electron donating ability of amine groups to metal ions than those of nitrile and thiol groups. Of the primary (4), secondary (8) and tertiary amine (9) functionalized complexes, the secondary amine functionalized complex (8) showed the highest TOF (1384 h−1) (entries 35, 39 and 40). This again reveals that the strong electron donating ligands increase the efficiency of the catalyst as the secondary amine is a better electron donor than the primary and tertiary amines. A similar trend was obtained while considering catalysts 5, 10 and 11 (entries 35, 41 and 42). Conversely, recycling experiments of 3, 6 and 7 showed that catalysts 3 and 6 deactivated more significantly than catalyst 7. This may be due to the better back donating ability of the thiol group than those of the nitrile and amine functional groups; thus, the bond between metal and thiol donor ligands of the support remains stronger.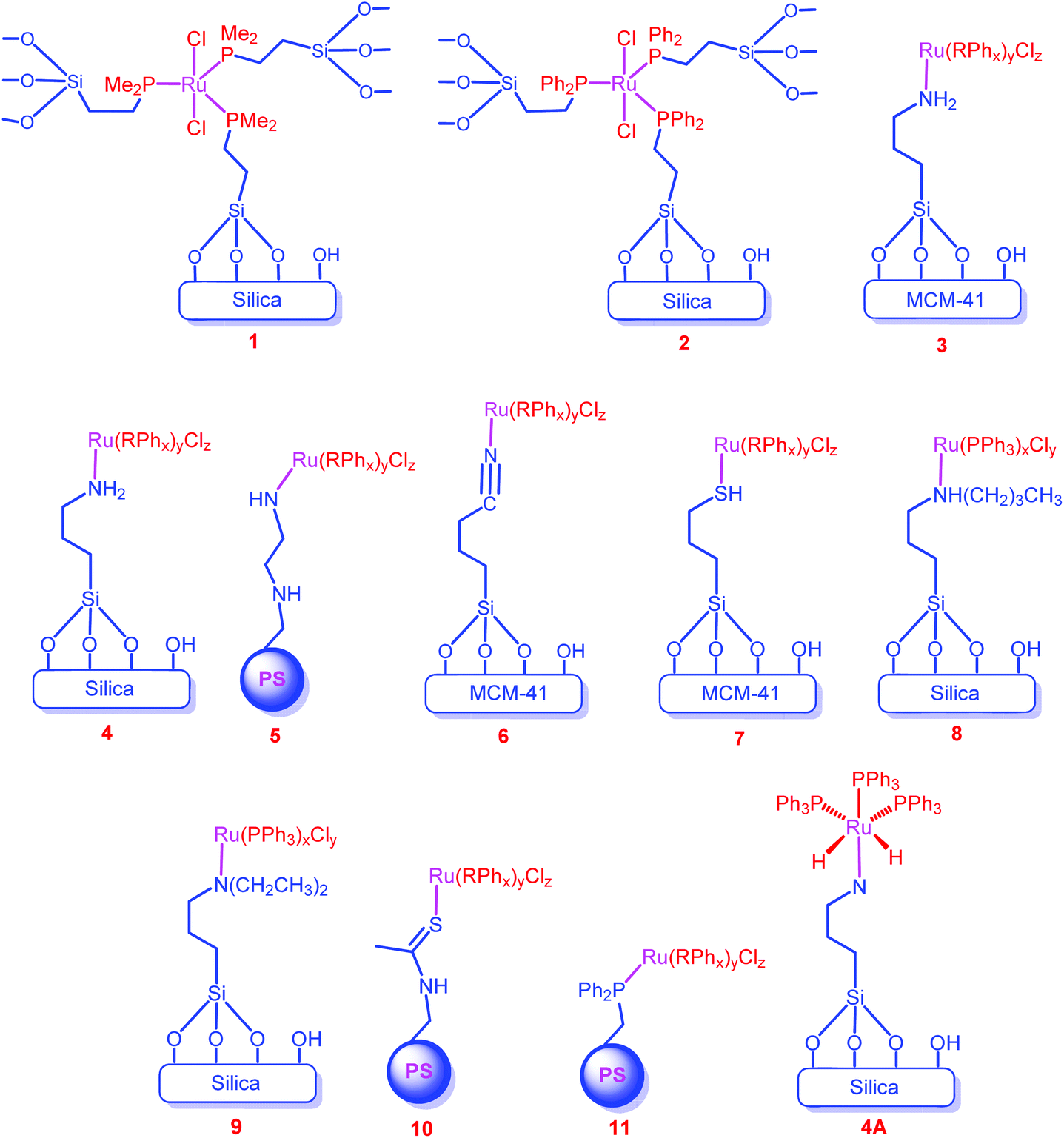 | ||
| Chart 1 Representative structures of catalysts 1–11. | ||
Furthermore, these Ru complexes produced the formate only in the presence of external ligands/additives (PPh3, AsPh3, NPh3 and Ph2P(CH2)2PPh2 (dppe)), suggesting that the in situ formed active catalyst contained an external ligand as one of the ligands (Chart 1, showing the expected active catalyst (4A) for catalyst 4).64 They have also studied the outcome of the reaction with various additives and observed that the bidentate ligand, dppe, exhibited a higher catalytic efficiency than the monodentate ligands (entries 34, 36, and 43–46). This was attributed to the smaller bite angle of the dppe ligand (313.082°, 313.418°) than that of the PPh3 ligand (314.76°), which consequently reduces the steric hindrance of the dppe ligand with the Ru ion. Therefore, the coordination of dppe with metal ions was more favorable than PPh3, which led to the higher activity of dppe containing catalysts.67
In 2008, Zhang et al. employed the ionic liquid (IL) as a reusable base to isolate formic acid from formate.68 In light of Zheng and co-workers’ results,63–67 they have employed the silica supported Ru complex [“Si”-(CH2)3NH(CSCH3)-{RuCl3(PPh3)}] as a catalyst for this purpose. The catalyst in combination with amine functionalized IL promoted the CO2 hydrogenation with a maximum TOF of 103 h−1. Almost 1:1 molar ratio of formic acid to IL (used) was observed, indicating the complete consumption of the added IL. They also prepared diamine-functionalized IL to improve the efficiency of CO2 hydrogenation.69 The maximum TOF of 920 h−1 was obtained at a relatively high temperature (80 °C) and pressure (18 MPa total pressure). As expected, the molar ratio of formic acid to IL reached up to 2:1. Notably, the free formic acid was separated from the IL with the aid of N2 flow at 130 °C and the catalyst and IL were reused for several runs. These unique features of this method offer the opportunity to apply the reaction in commercial processes.
Hicks and co-workers further functionalized the amine groups of the mesoporous silica into imine groups by a Schiff base reaction with o-(diphenylphosphino)benzaldehyde, and immobilized Ir complexes through an imine–phosphine coordination bond (12).70 For comparison, monodentate phosphine complexes (13 and 14), amine precatalyst 15 and the homogeneous counterpart (16) were synthesized (Chart 2). X-ray Photoelectron Spectroscopy (XPS) measurements of 12 and 16 showed that the environments of Ir in 12 (61.6 eV) and 16 (61.8 eV) are similar. Among the catalysts screened (12, 13, 14, 15 and 16), only phosphine containing catalysts (12, 13, 14 and 16) showed activity in CO2 hydrogenation to formate (entries 47–50). Catalyst 12 exhibited a higher TON (1300) than 13 (110) and 14 (400) (entries 47–49). Moreover, 12 showed almost 20 times higher efficiency than the unsupported catalyst (16), revealing the increased stability and activity upon heterogenization of homogeneous complexes (entries 47 and 51). The catalytic efficiency was increased with temperature, and the highest TOF of 1200 h−1 was attained at 120 °C (entries 52–54). Time-dependent formate production studies showed that formate production increased with time, and the catalyst exhibited the maximum TON (2700) and afforded a maximum concentration of formate (0.270 M) at 60 °C after 20 h (entries 47, 52 and 55). Catalyst 12 was recycled at minimum time intervals (0.5 h) under mild conditions and obtained the average TON of 70.
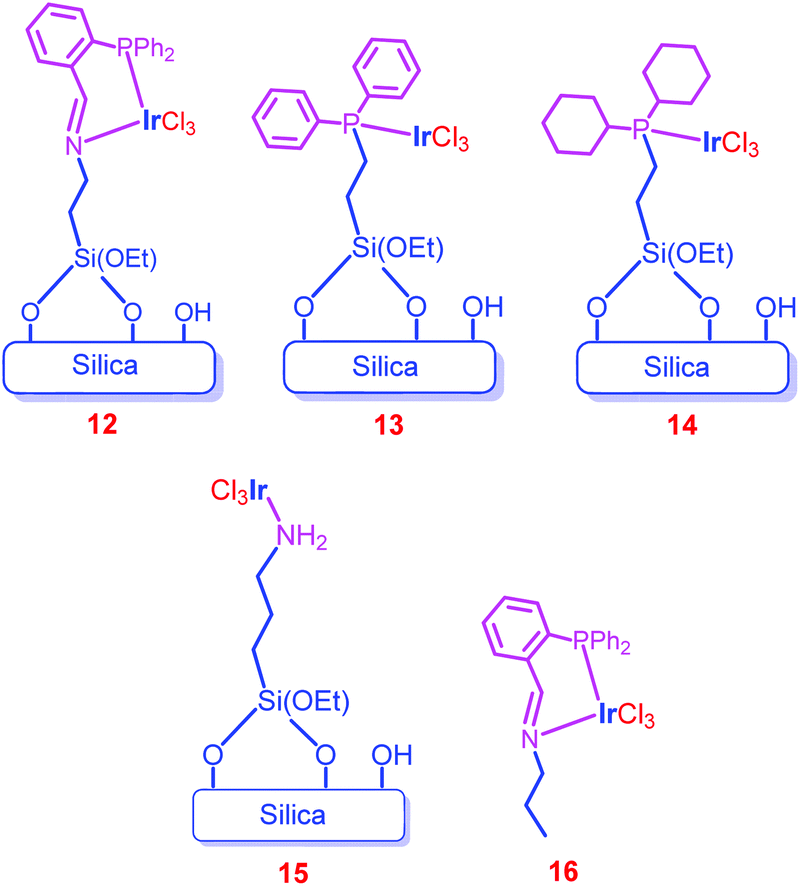 | ||
| Chart 2 Representative structures of catalysts 12–16. | ||
Hicks and co-workers also employed polyethyleneimine (PEI), an aliphatic amine-based organic polymer containing primary, secondary and tertiary amine groups, as a support to immobilize the Ir complexes.71 It was expected that this amine-based support would be multifunctional, acting as a CO2 capturing material, a formate stabilizer and a catalyst support. Hence, catalyst 17 was prepared by tethering complex 16 on this PEI (Chart 3). They compared the activity of 17 with an imine containing catalyst (18) and a phosphine containing catalyst (19) and found that 17 exhibited better activity than 18 and 19 (entries 56–58). However, it is noteworthy that the efficiency of 17 is poor when compared to 12 (entries 54 and 56), indicating the weakness of PEI supports. Additionally, it was found that varying the amount of Ir loading [Ir-25% (PEI-PN/Ir-25), Ir-65% (PEI-PN/Ir-65) and Ir-95% (PEI-PN/Ir-95)] on the PEI backbone affected the efficiency of the catalyst; PEI-PN/Ir-65 exhibited better efficiency (TOF 310 h−1) than PEI-PN/Ir-25 (TOF 94 h−1) and PEI-PN/Ir-95 (TOF 122 h−1). XPS and TEM measurements revealed the existence of agglomerated Ir nanoparticles on the surface of PEI-PN/Ir-25 and PEI-PN/Ir-95, whereas no such agglomerated Ir nanoparticles were observed in PEI-PN/Ir-65. Consequently, they suggested that the Ir nanoparticles are catalytically inactive in the hydrogenation of CO2 to formate. In addition, the efficiency of catalysts was also affected by the PEI molecular weight (PEI-MW). Moreover, recycling experiments demonstrated that the efficiency of catalysts decreases continuously, especially for low PEI-MW catalysts, owing to the increased solubility and formation of inactive Ir species. Furthermore, the catalysts exhibited poor activity in the absence of an external base, suggesting that the amines present in the backbone of PEI no longer significantly stabilize the formate under these conditions.
 | ||
| Chart 3 Representative structures of catalysts 17–19. | ||
3.2 Heterogenized COF catalysts
Since the Ir/Ru complexes heterogenized on conventional silica, PS and aliphatic polymer supports demonstrated low catalytic activity and stability,63–71 further research on new strategies/supports to immobilize the homogeneous complexes is inevitable. Thus, porous organic polymers have been used as catalyst supports owing to their high surface area, well-defined porosity, ease of use and the diverse number of synthetic routes.72 Specifically, crystalline microporous organic polymers with ordered porous structures – so-called ‘covalent organic frameworks’ (COFs) – have drawn particular attention due to their low density, high thermal stability, large surface area, tunable pore size and structure, and versatile covalent combination of building blocks.73 These polymeric materials have been used as gas storage materials, catalyst supports and photonic band gap materials.74–78 Consequently, COFs like Troger's base-derived microporous organic polymers (TB-MOPs),79 graphitic carbon nitrides (g-C3N4),80 triazine-based covalent organic frameworks (CTFs)81 and heptazine-based covalent organic frameworks (HBFs)82 have been employed as catalyst supports for CO2 hydrogenation to formate.Liu and co-workers prepared a TB-MOP supported Ru(III) catalyst (20) through the coordination bond between the N atoms of the TB-MOP and the Ru(III) ions (Chart 4).79 The microporous structure of 20 was confirmed by Brunauer–Emmett–Teller (BET) measurements. Catalyst 20 exhibited a substantial catalytic efficiency (TON 2254) in the presence of PPh3 ligands at 40 °C under a total pressure of 12 MPa (entry 60). However, in the absence of PPh3 ligands the TON was decreased to 25 (entries 59 and 60), suggesting that the active catalyst was formed in situ and that the structure of the active catalyst could be similar to that proposed by Zheng and co-workers.64
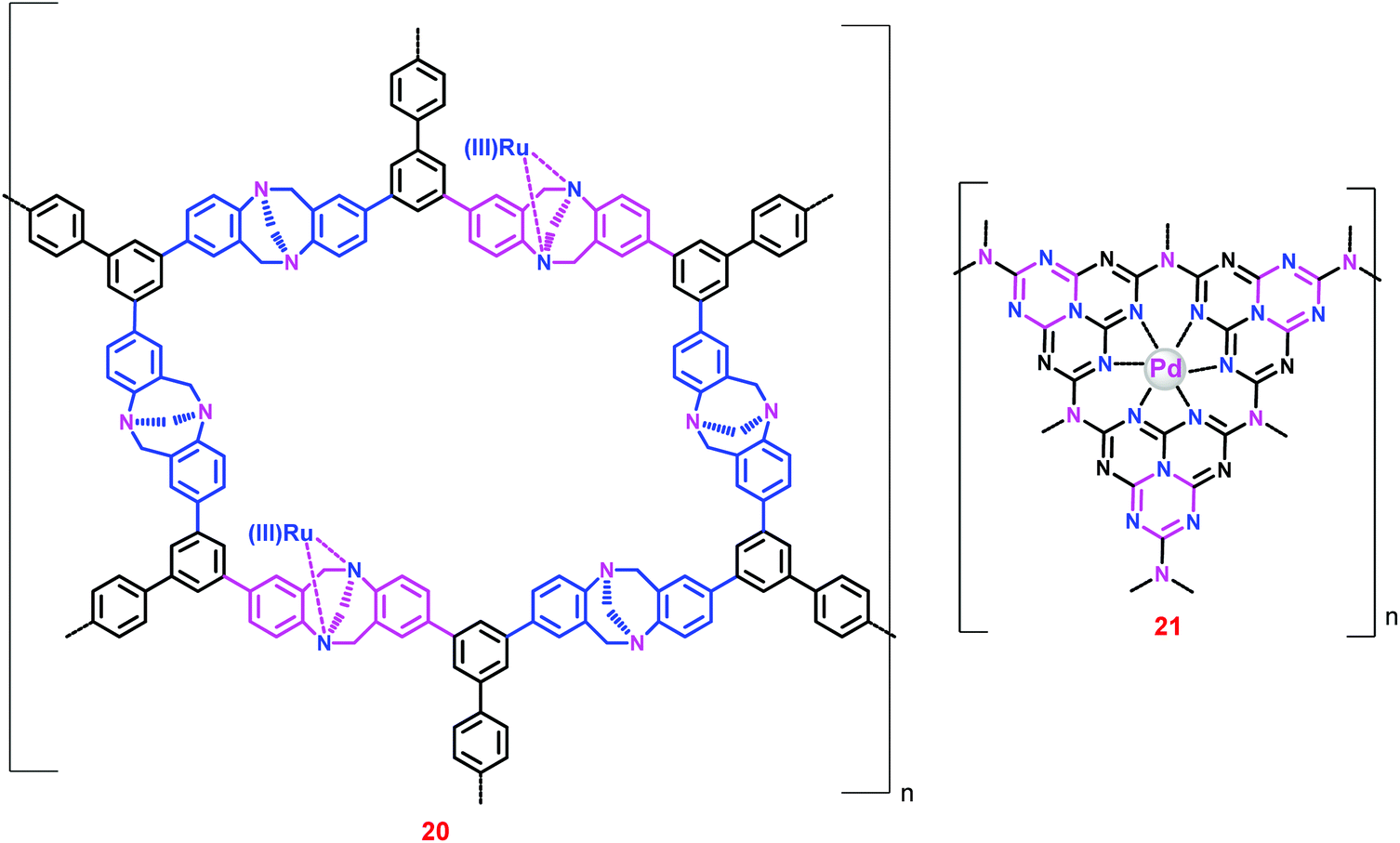 | ||
| Chart 4 Structural representation of catalysts 20 and 21. | ||
Later, Lee et al. demonstrated the catalytic performance of Pd nanoparticles supported on mesoporous g-C3N4 (21) (Chart 4).80 The TON for 21 in a 20% NEt3–deuterium oxide (D2O) solution was found to be 81 at 150 °C under p(H2)/p(CO2) = 1 (entry 61). Interestingly, the relative ratio of gas pressures [p(H2)/p(CO2)] affected the outcome of the reaction; TON was increased to 106 at p(H2)/p(CO2) = 2 (entries 61 and 62). However, TON was reduced on increasing the ratio further (entries 62 and 64).
Over the past ten years, homogeneous [IrCp*(N–N)X]Y complexes (N–N represents bipyridine (bpy), phenanthroline and pyrimidine derivatives, Cp*-1,2,3,4,5-pentamethyl cyclopentadiene, X-Cl/H2O, Y-Cl/SO42−) have shown tremendous catalytic activities and selectivities.83–85 The highest TON (222000) and the maximum TOF (53800 h−1) were obtained with these catalysts by Himeda et al.35,83 Since these catalysts are proton-responsive and pH-switchable, their catalytic activity and H2O solubility can be tuned by the pH of the solution. Thus, at the end of the CO2 hydrogenation (a decrease in the pH of the solution led by the reaction equilibrium), the catalyst can be precipitated because of its low solubility in a weakly acidic solution (pH 5.5).86 However, this unique property is only observed for the Ir-phenanthroline derivative. Recently, Yoon and co-workers developed a new strategy to heterogenize the homogeneous [IrCp*(bpy)Cl]Cl complex (22) (Chart 5).81 They employed a bpy incorporated CTF (bpy-CTF) as a support owing to its high thermal stability, large pore volumes and high surface areas. The bpy-CTF has the potential to form complexes with metal precursors (Chart 5). The authors hypothesized that the complex resulting from the reaction of [IrCp*Cl2]2 and bpy-CTF would have a similar coordination environment to that of complex 22. As they hypothesized, the heterogenized complex, bpy-CTF-[IrCp*Cl]Cl (23), was synthesized and thoroughly characterized to prove its exact coordination environment as that of complex 22. Scanning electron microscopy (SEM) measurements of 23 illustrated the uniform distribution of Ir and Cl atoms throughout the complex, suggesting the uniform metalation of Ir ions onto bpy moieties. In addition, energy dispersive X-ray spectroscopy (EDS) as well as XPS measurements revealed that the atomic ratio of Ir and Cl was close to 1:2. Moreover, XPS measurements of both 22 and 23 showed that they have exactly the same EBE value for Ir 4f7/2 (62.1 eV), reiterating the similar coordination environment of Ir in both the catalysts. Inductively coupled plasma mass spectrometry (ICP-MS) analysis of 23 showed that it has a relatively high Ir content (4.7 wt%) in the framework, indicating that every sixth CTF ring unit contains one {IrCp*} unit.
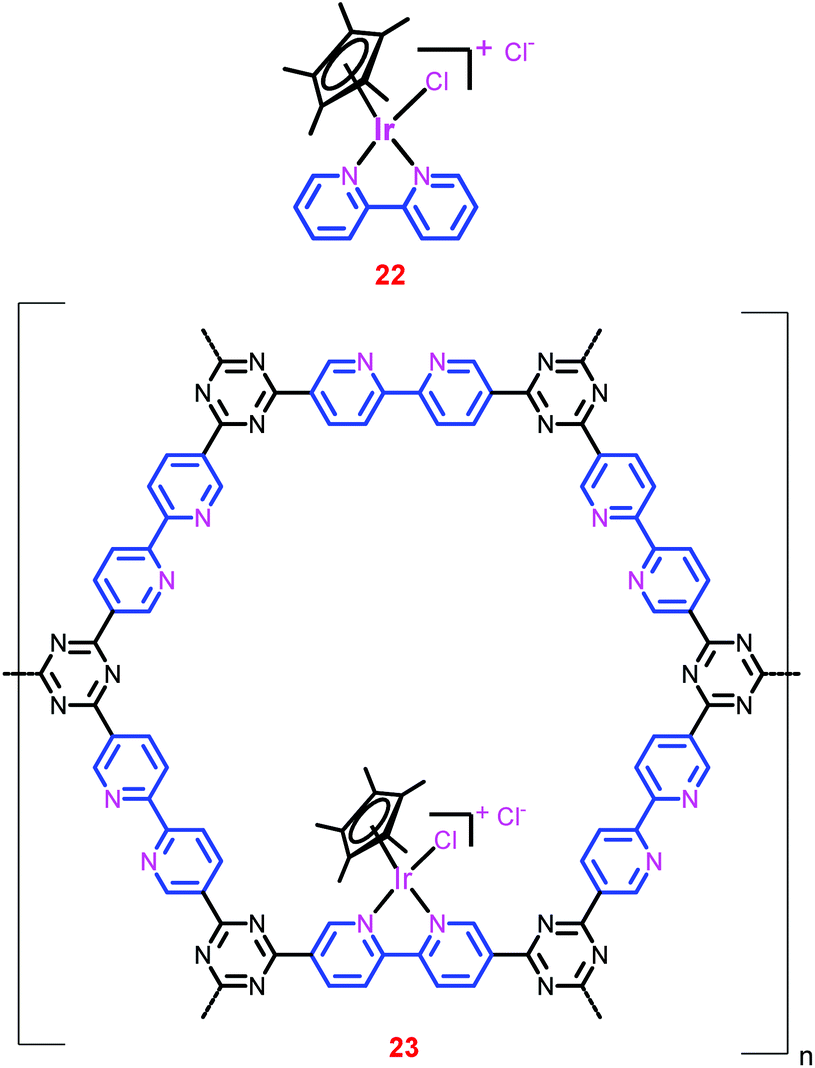 | ||
| Chart 5 Structural representation of catalysts 22 and 23. | ||
The temperature-dependent catalytic performance of 23 showed that the activity increased with temperature up to 120 °C, and thereafter decreased as the temperature increased further (entries 65–68). This is attributed to the exothermic nature of the reaction. However, the catalytic activity increased with pressure and attained a maximum TON of 5000 at 120 °C under a total pressure of 8 MPa (entry 69). The time-dependent formate generation studies revealed that the reaction attained equilibrium (0.6–0.7 M formate concentration) within a relatively short reaction time (2 h). In addition, under relatively mild conditions, catalyst 23 showed the highest ever initial TOF of 5300 h−1 for the conversion of CO2 to formate using heterogeneous catalysts (entry 70). Moreover, catalyst 23 was recycled over five runs without a significant loss of activity and on each recycling, ca. 92% catalytic activity was maintained.
Vey recently, Yoon and co-workers have also heterogenized the [IrCp*(N–N)X]Y complex using g-C3N4 (24) and HBF (25) supports (Chart 6).82 The uniform metalation as well as the ratio of Ir to Cl (1:2) were confirmed by SEM and EDS measurements, respectively. ICP-MS analysis of 25 showed a very low Ir content (0.86 wt%) in the framework. Catalyst 25 exhibited an initial TOF of 1500 h−1 and a TON of 6400 at 120 °C under a total pressure of 8 MPa (entries 71 and 72). The catalyst was recycled without a significant loss of activity and stability, and on each recycling, ca. 90% catalytic activity was maintained with an average TON of 4000.
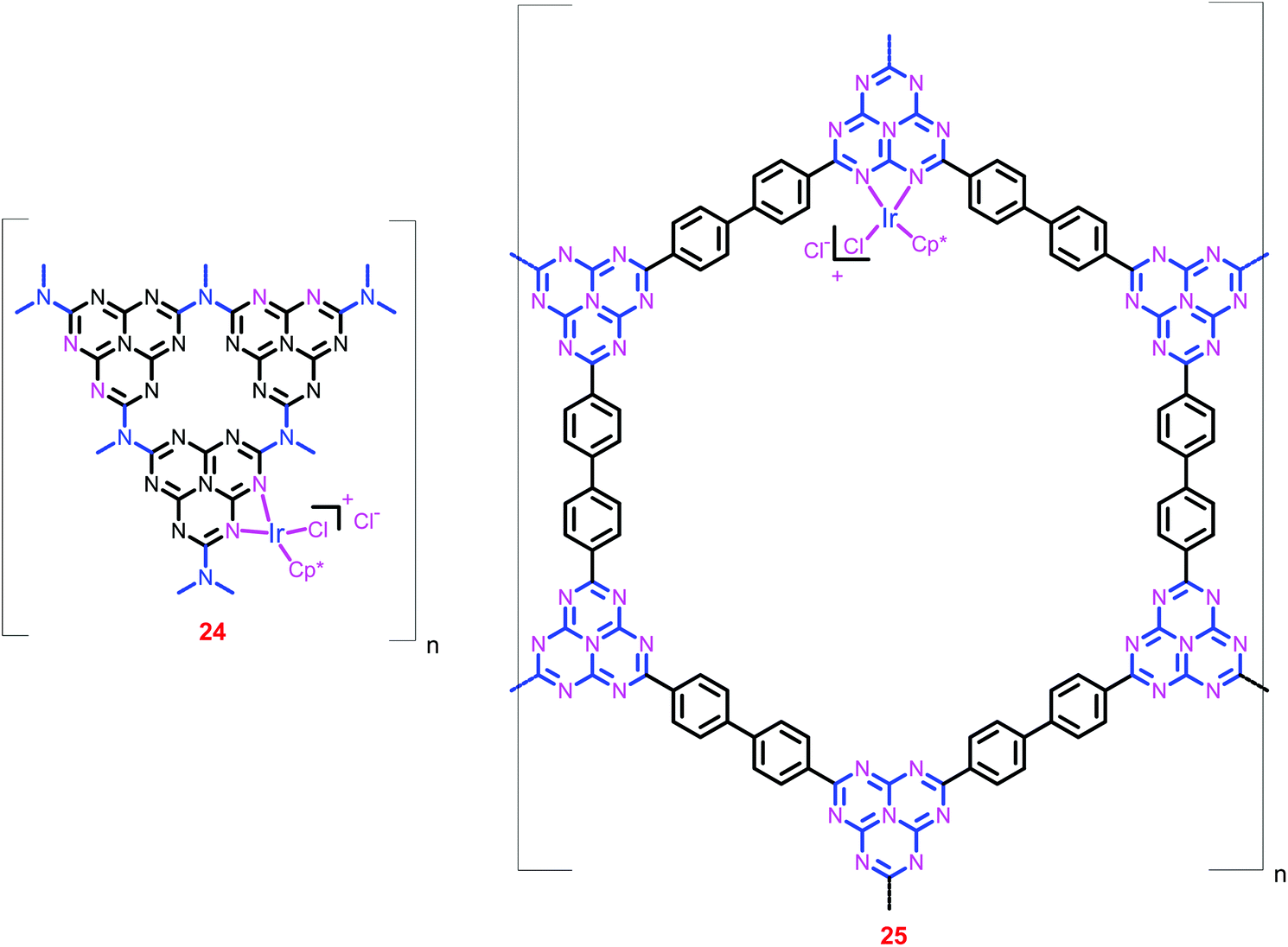 | ||
| Chart 6 Structural representation of catalysts 24 and 25. | ||
4. Theoretical investigations
Even though research on the development of efficient catalysts is in progress, understanding the complete reaction mechanism through experimental research is rather difficult and time consuming. This is due to the fact that finding the substrate–catalyst interactions, energies of the molecules involved, transition states and intermediate structures, and activation barriers for the process is quite difficult.87 To overcome these limitations, theoretical calculations have become powerful tools to investigate the reaction pathway.88 Moreover, the key structure–activity relationship for the rational design of suitable and efficient catalysts could be gained through this study.The synthesis of formic acid and methanol from CO2 and H2 involves formate as one of the intermediates. Hence, many reports have discussed the reaction pathway for the formation of formic acid, the free formate anion and catalyst bound formate from CO2 and H2.89–91 However, the calculations involving only formic acid formation using heterogeneous catalysts are presented here.92–97
Peng et al. theoretically explored CO2 hydrogenation to formic acid on Ni(111) surfaces.92 They investigated the reaction mechanism via two routes, namely the formate intermediate route (HCOO**) in which CO2 can be hydrogenated at its carbon atom and the carboxyl intermediate route (COOH*) in which CO2 can be hydrogenated at its oxygen atom, using Ni(111) surfaces. Their calculations showed that for the formation of HCOO**, a lower activation energy (14.3 kcal mol−1) is required than that for the COOH* (19.1 kcal mol−1). This result clearly suggests that the HCOO** route is more favorable than the COOH* route. Previously, Vesselli et al. concluded that the COOH* route preferably produces CO and H2O instead of formic acid by the Ni(110) surface.93 Peng et al. also studied the second hydrogenation step, that is, the formation of formic acid from HCOO**, and found that it has a very high activation barrier (14.3 kcal mol−1), suggesting that this might be the rate-determining step for formic acid production.92 Overall, the reaction is highly endothermic (12.9 kcal mol−1). Moreover, they also extensively studied ways to reduce such a high activation barrier and concluded that if the subsurface H [absorbed in the subsurface of Ni(111)] was involved in the second hydrogenation step, the activation barrier could be lowered and one can make the overall reaction exothermic (−16.3 kcal mol−1). Similar results were obtained for Ni(110) surfaces.94
Limtrakul and co-workers theoretically investigated the reaction mechanism of hydrogenation of CO2 to formic acid catalyzed by a Cu-alkoxide-functionalized metal organic framework (Cu-MOF-5).95 Two different reaction pathways are proposed, namely concerted and stepwise mechanisms. In both pathways, reactants (CO2 and H2) are initially adsorbed on the Cu-MOF-5 catalyst and form the coadsorption complex. For the concerted mechanism, the reaction is proposed to take place in a single step with the active site of the catalyst not assisting in the H2 splitting. In the transition state of this pathway, the adsorbed CO2 is simultaneously hydrogenated at the C and O atoms to form formic acid. The activation barrier for this pathway is calculated to be 67.2 kcal mol−1. For the stepwise mechanism, in which a part of the active site assists in the H2 splitting, the reaction occurs via two steps involving a formate intermediate. The first step, which is the formation of a formate intermediate, is found to be the rate-determining step with an activation energy of 24.2 kcal mol−1. The more facile second step, in which the proton is transferred to the formate intermediate, has a smaller activation barrier (18.3 kcal mol−1). Because of the smaller activation barriers associated with this pathway (24.2 vs. 67.2 kcal mol−1), it therefore seems to be favored over the concerted one. Furthermore, the catalytic effect of Cu-MOF-5 is also compared with the gas-phase uncatalyzed reaction in which the reaction takes place in one step with a barrier of 73.0 kcal mol−1. Therefore, this investigation clearly reveals the two important design strategies for obtaining efficient catalysts for this reaction: (1) the catalyst would adsorb the reactants and (2) it would assist in the H2 splitting.
Since the frustrated Lewis pairs (FLPs) are capable of activating CO2 and heterolytically dissociating H2, Ye et al. theoretically demonstrated the reaction mechanism of hydrogenation of CO2 to formic acid catalyzed by a Lewis pair-functionalized metal organic framework (UiO-66-P-BF2).96 They investigated the reaction by two different approaches. In the first approach, H2 is activated initially by dissociative adsorption, and then reacted with physisorbed CO2 to produce formic acid. In this pathway, the addition of hydridic and protic hydrogens (chemisorbed H atoms) to C and O of physisorbed CO2, respectively, occurs in a concerted fashion with the activation barrier of 10.8 kcal mol−1. In the second approach, CO2 is activated initially by chemisorption, and then reacted with physisorbed H2. However, in this approach, the undesired formyl and hydroxyl moieties were produced with a high activation barrier rather than the desired formic acid product. This is because the CO2 bound too strongly with the FLP of UiO-66-P-BF2 through chemisorption. Hence, for the UiO-66-P-BF2 catalyst to work in practice, one would have to expose the material first to H2 and then to CO2 to avoid the competing reaction and potential poisoning of the catalyst. This requirement would prohibit its practical use. Therefore, the designed catalyst should weakly bind to CO2 through FLP (P-BF2) and at the same time it would provide a binding site that selectively dissociates H2.
Limtrakul and co-workers also explored the catalytic reaction pathway of Cu embedded in the surface of graphene (Cu/dG) for the CO2 hydrogenation to formic acid.97 Initially, they studied the properties of Cu/dG through their calculations and found that the catalyst Cu/dG binds more strongly to H2 (adsorption energy = −6.12 kcal mol−1) than CO2 (adsorption energy = −5.1 kcal mol−1). In addition, Cu/dG is more selective for the adsorption of the H2 molecule than the CO2 molecule. They investigated the course of the reaction via two paths. In the first path, the reaction takes place in two steps. In the initial step, the first hydrogenation of CO2 takes place, without activating the H2 molecule, through the highest activation barrier (34.6 kcal mol−1) and produces the unstable H–Cu–COOH intermediate. In the later step, the second hydrogenation takes place through the H-transfer from the Cu atom to the carbon of the –COOH moiety via a three-membered ring with an activation barrier of 4.0 kcal mol−1. Finally, formic acid is released from the Cu/dG catalyst with an activation energy of 20.1 kcal mol−1. For the second route, the H2 molecule is activated by dissociating H2 molecules to Cu–H and C–H (from graphene carbon) moieties in the first step with the highest energy barrier of 19.7 kcal mol−1. In the second step, the insertion of CO2 into the Cu–H species to form the formate intermediate (HCOO–Cu/H-dG) is much more favorable (−14.6 kcal mol−1) and requires an activation energy of 13.6 kcal mol−1. Finally, the protonation of the formate intermediate preferably takes place with the second H2 molecule (11.6 kcal mol−1). Therefore this study infers that the H2 activated route would be more facile than the inactivated H2 route.
5. Summary and conclusions
Diverse forms of heterogeneous catalysts for the conversion of CO2 into formate have evolved to meet the requirements for industrial application. The following features have emerged as important for achieving this challenging goal. The heterogeneous catalyst should be sufficiently active to generate enough formate within a relatively short period of reaction time. The catalyst's support framework should be sufficiently robust to prevent deleterious decomposition of catalytic active sites under harsh reaction conditions (pH = 13). The catalysts should be easily separated from the reaction mixture at the end of the reaction and recycled. Most of these requirements are fulfilled by heterogenization of homogeneous Ir complexes by incorporating them onto the pore of the covalent organic frameworks.Although the field has gone through a rapid progression phase, more studies must be carried out to achieve two remaining important goals in the development of catalysts for the conversion of CO2 into formate. The first goal is to obtain a system having extreme stability over repeated recycles. The other goal is to generate the catalyst with cheap transition metal ion(s).
Acknowledgements
We acknowledge the financial support through grants from the Korea CCS R&D Center, funded by the Ministry of Education, Science and Technology of the Korean Government, and from the Global Scholarship Program for Foreign Graduate Students at Kookmin University in Korea.References
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347–3357 RSC.
- D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 CAS.
- http://www.nature.com/news/lima-talks-map-out-path-to-climate-treaty-1.16557 (accessed on 2016.01.06).
- https://unfccc.int/files/meetings/lima_dec_2014/application/pdf/auv_cop20_lima_call_for_climate_action.pdf (accessed on 2016.01.06).
- G. Ferey, C. Serre, T. Devic, G. Maurin, H. Jobic, P. L. Llewellyn, G. De Weireld, A. Vimont, M. Daturi and J. S. Chang, Chem. Soc. Rev., 2011, 40, 550–562 RSC.
- D. Sivanesan, Y. Choi, J. Lee, M. H. Youn, K. T. Park, A. N. Grace, H.-J. Kim and S. K. Jeong, ChemSusChem, 2015, 8, 3977–3982 CrossRef CAS PubMed.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 CAS.
- P. Sudakar, D. Sivanesan and S. Yoon, Macromol. Rapid Commun. DOI:10.1002/marc.201500681.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- M. Peters, B. Kohler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Muller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- A. Baiker, Appl. Organomet. Chem., 2000, 14, 751–762 CrossRef CAS.
- W. Reutemann and H. Kieczka, Formic acid, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2011 Search PubMed.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 CAS.
- H.-L. Jiang, S. K. Singh, J.-M. Yan, X.-B. Zhang and Q. Xu, ChemSusChem, 2010, 3, 541–549 CrossRef CAS PubMed.
- S. Uhm, H. J. Lee and J. Lee, Phys. Chem. Chem. Phys., 2009, 11, 9326–9336 RSC.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CAS.
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- Y. Inoue, H. Izumida, Y. Sasaki and H. Hashimoto, Chem. Lett., 1976, 863–864 CrossRef CAS.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344–355 CrossRef CAS.
- F. Joo, G. Laurenczy, L. Nadasdi and J. Elek, Chem. Commun., 1999, 971–972 RSC.
- P. G. Jessop, F. Joo and C. C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS.
- C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef CAS PubMed.
- M. S. Jeletic, M. T. Mock, A. M. Appel and J. C. Linehan, J. Am. Chem. Soc., 2013, 135, 11533–11536 CrossRef CAS PubMed.
- Y. M. Badiei, W.-H. Wang, J. F. Hull, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2013, 52, 12576–12586 CrossRef CAS PubMed.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, J. Photochem. Photobiol., A, 2006, 182, 306–309 CrossRef CAS.
- Y. Himeda, Eur. J. Inorg. Chem., 2007, 3927–3941 CrossRef CAS.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- E. Fujita, J. T. Muckerman and Y. Himeda, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1031–1038 CrossRef CAS PubMed.
- S. Sanz, M. Benitez and E. Peris, Organometallics, 2010, 29, 275–277 CrossRef CAS.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- G. A. Filonenko, E. J. M. Hensen and E. A. Pidko, Catal. Sci. Technol., 2014, 4, 3474–3485 CAS.
- G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef CAS.
- D. Preti, S. Squarcialupi and G. Fachinetti, Angew. Chem., Int. Ed., 2010, 49, 2581–2584 CrossRef CAS PubMed.
- C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef CAS PubMed.
- T. Schaub and R. A. Paciello, Angew. Chem., Int. Ed., 2011, 50, 7278–7282 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- S. Saeidi, N. A. S. Amin and M. R. Rahimpour, J. CO2 Util., 2014, 5, 66–81 CrossRef CAS.
- A. Behr and K. Nowakowski, Adv. Inorg. Chem., 2014, 66, 223–258 CrossRef CAS.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- M. W. Farlow and H. Adkins, J. Am. Chem. Soc., 1935, 57, 2222–2223 CrossRef CAS.
- C. J. Stalder, S. Chao, D. P. Summers and M. S. Wrighton, J. Am. Chem. Soc., 1983, 105, 6318–6320 CrossRef CAS.
- H. Takahashi, L. H. Liu, Y. Yashiro, K. Ioku, G. Bignall, N. Yamasaki and T. Kori, J. Mater. Sci., 2006, 41, 1585–1589 CrossRef CAS.
- D. Preti, C. Resta, S. Squarcialupi and G. Fachinetti, Angew. Chem., Int. Ed., 2011, 50, 12551–12554 CrossRef CAS PubMed.
- http://authors.library.caltech.edu/25070/6/FundChemReaxEngCh5.pdf (accessed on 2016.01.06).
- D. Preti, S. Squarcialupi and G. Fachinetti, ChemCatChem, 2012, 4, 469–471 CrossRef CAS.
- G. A. Filonenko, W. L. Vrijburg, E. J. M. Hensen and E. A. Pidko, J. Catal. DOI:10.1016/j.jcat.2015.10.002.
- J. Su, L. Yang, M. Lu and H. Lin, ChemSusChem, 2015, 8, 813–816 CrossRef CAS PubMed.
- J. Su, M. Lu and H. Lin, Green Chem., 2015, 17, 2769–2773 RSC.
- Q.-Y. Bi, J.-D. Lin, Y.-M. Liu, X.-L. Du, J.-Q. Wang, H.-Y. He and Y. Cao, Angew. Chem., Int. Ed., 2014, 53, 13583–13587 CrossRef CAS PubMed.
- L. T. M. Nguyen, H. Park, M. Banu, J. Y. Kim, D. H. Youn, G. Magesh, W. Y. Kim and J. S. Lee, RSC Adv., 2015, 5, 105560–105566 RSC.
- C. Y. Hao, S. P. Wang, M. S. Li, L. Q. Kang and X. Ma, Catal. Today, 2011, 160, 184–190 CrossRef CAS.
- N. Liu, R. Du and W. Li, Adv. Mater. Res., 2013, 821–822, 1330–1335 CrossRef.
- N. Liu, J. Lei, M. Li and P. Wang, Adv. Mater. Res., 2014, 881–883, 283–286 CrossRef.
- O. Krocher, R. A. Koppel, M. Froba and A. Baiker, J. Catal., 1998, 178, 284–298 CrossRef CAS.
- Y. Zhang, J. Fei, Y. Yu and X. Zheng, Catal. Commun., 2004, 5, 643–646 CrossRef CAS.
- Y. M. Yu, Y. P. Zhang, J. H. Fei and X. M. Zheng, Chin. J. Chem., 2005, 23, 977–982 CrossRef CAS.
- Y. M. Yu, J. H. Fei, Y. P. Zhang and X. M. Zheng, Chin. Chem. Lett., 2006, 17, 1097–1100 CAS.
- Y. M. Yu, J. H. Fei, Y. P. Zhang and X. M. Zheng, Chin. J. Chem., 2006, 24, 840–844 CrossRef CAS.
- Y. P. Zhang, J. H. Fei, Y. M. Yu and X. M. Zheng, Chin. Chem. Lett., 2006, 17, 261–264 CAS.
- Z. Zhang, Y. Xie, W. Li, S. Hu, J. Song, T. Jiang and B. Han, Angew. Chem., Int. Ed., 2008, 47, 1127–1129 CrossRef CAS PubMed.
- Z. Zhang, S. Hu, J. Song, W. Li, G. Yang and B. Han, ChemSusChem, 2009, 2, 234–238 CrossRef CAS PubMed.
- Z. Xu, N. D. McNamara, G. T. Neumann, W. F. Schneider and J. C. Hicks, ChemCatChem, 2013, 5, 1769–1771 CrossRef CAS.
- N. D. McNamara and J. C. Hicks, ChemSusChem, 2014, 7, 1114–1124 CrossRef CAS PubMed.
- X. Feng, X. S. Ding and D. L. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- A. Nagai, Z. Q. Guo, X. Feng, S. B. Jin, X. Chen, X. S. Ding and D. L. Jiang, Nat. Commun., 2011, 2, 536 CrossRef PubMed.
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. H. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed.
- J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793–1799 CrossRef CAS PubMed.
- X. Zhu, C. Tian, S. M. Mahurin, S. H. Chai, C. Wang, S. Brown, G. M. Veith, H. Luo, H. Liu and S. Dai, J. Am. Chem. Soc., 2012, 134, 10478–10484 CrossRef CAS PubMed.
- L. Hao, J. Ning, B. Luo, B. Wang, Y. Zhang, Z. Tang, J. Yang, A. Thomas and L. Zhi, J. Am. Chem. Soc., 2015, 137, 219–225 CrossRef CAS PubMed.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- Z.-Z. Yang, H. Zhang, B. Yu, Y. Zhao, G. Ji and Z. Liu, Chem. Commun., 2015, 51, 1271–1274 RSC.
- J. H. Lee, J. Ryu, J. Y. Kim, S. W. Nam, J. H. Han, T. H. Lim, S. Gautam, K. H. Chae and C. W. Yoon, J. Mater. Chem. A, 2014, 2, 9490–9495 CAS.
- K. Park, G. H. Gunasekar, N. Prakash, K. D. Jung and S. Yoon, ChemSusChem, 2015, 8, 3410–3413 CrossRef CAS PubMed.
- G. Gunniya Hariyanandam, D. Hyun, P. Natarajan, K. D. Jung and S. Yoon, Catal. Today, 2016, 265, 52–55 CrossRef CAS.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara, H. Arakawa and K. Kasuga, Organometallics, 2004, 23, 1480–1483 CrossRef CAS.
- W.-H. Wang, J. F. Hull, J. T. Muckerman, E. Fujita and Y. Himeda, Energy Environ. Sci., 2012, 5, 7923–7926 CAS.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Organometallics, 2007, 26, 702–712 CrossRef CAS.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, J. Am. Chem. Soc., 2005, 127, 13118–13119 CrossRef CAS PubMed.
- J. K. Norskov, T. Bligaard and J. Kleis, Science, 2009, 324, 1655–1656 CrossRef CAS PubMed.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS PubMed.
- R. Zhang, B. Wang, H. Liu and L. Ling, J. Phys. Chem. C, 2011, 115, 19811–19818 CAS.
- D. Cheng, F. R. Negreiros, E. Apra and A. Fortunelli, ChemSusChem, 2013, 6, 944–965 CrossRef CAS PubMed.
- Y. Li, S. H. Chan and Q. Sun, Nanoscale, 2015, 7, 8663–8683 RSC.
- G. Peng, S. J. Sibener, G. C. Schatz, S. T. Ceyer and M. Mavrikakis, J. Phys. Chem. C, 2012, 116, 3001–3006 CAS.
- E. Vesselli, M. Rizzi, L. De Rogatis, X. Ding, A. Baraldi, G. Comelli, L. Savio, L. Vattuone, M. Rocca, P. Fornasiero, A. Baldereschi and M. Peressi, J. Phys. Chem. Lett., 2010, 1, 402–406 CrossRef CAS.
- G. Peng, S. J. Sibener, G. C. Schatz and M. Mavrikakis, Surf. Sci., 2012, 606, 1050–1055 CrossRef CAS.
- T. Maihom, S. Wannakao, B. Boekfa and J. Limtrakul, J. Phys. Chem. C, 2013, 117, 17650–17658 CAS.
- J. Ye and J. K. Johnson, ACS Catal., 2015, 5, 2921–2928 CrossRef CAS.
- J. Sirijaraensre and J. Limtrakul, Appl. Surf. Sci., 2016, 364, 241–248 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |