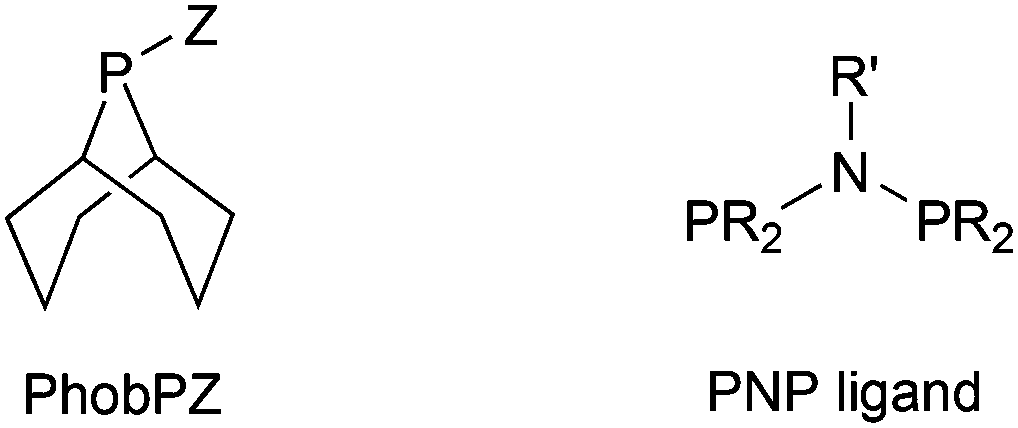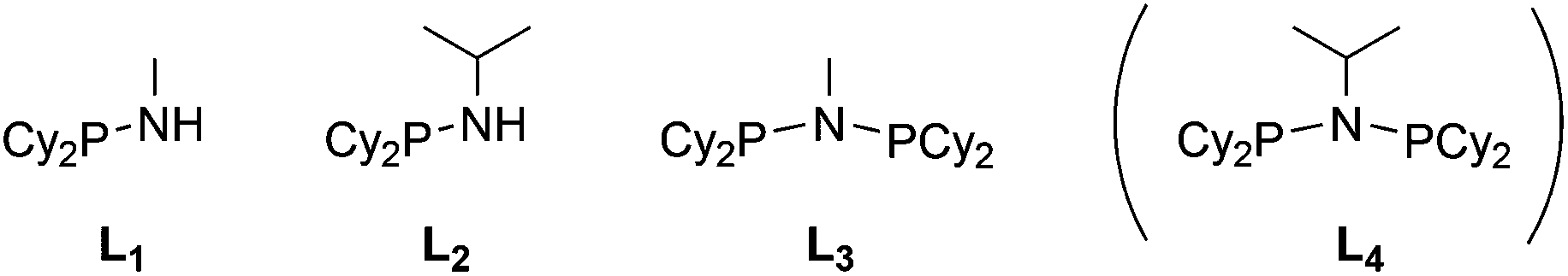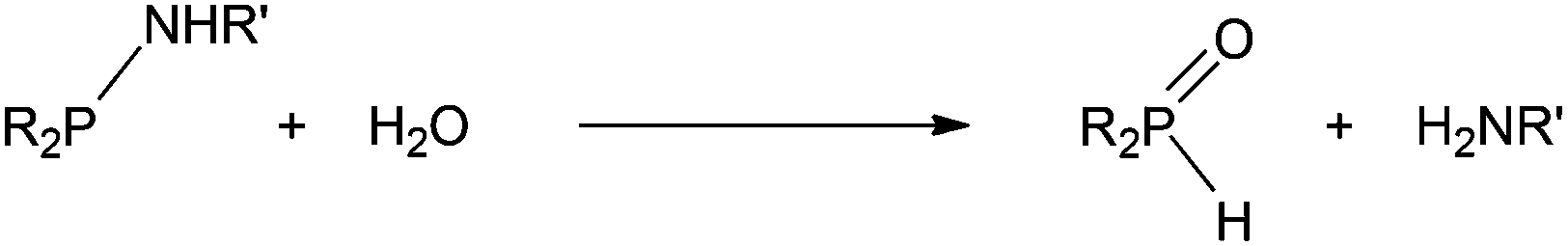DOI:
10.1039/C5DT04394H
(Paper)
Dalton Trans., 2016,
45, 2294-2307
Aminophobanes: hydrolytic stability, tautomerism and application in Cr-catalysed ethene oligomerisation†
Received
6th November 2015
, Accepted 11th January 2016
First published on 11th January 2016
Introduction
Significant differences between the donor properties of phosphacycles and their acyclic analogues are to be expected because of the effects that ring constraints can have on the frontier orbital energies and the steric properties of the P-donor.1 The molecular manifestations of these ring effects include stability (thermodynamic and kinetic) and structural rigidity which can be desirable qualities when considering the design of ligands. As a result, the coordination chemistry of phosphacycles and their applications in catalysis have attracted much academic and industrial attention.2
Phobanes (PhobPZ, in Chart 1) are examples of rigid phosphacycles which have found important applications in homogeneous catalysis3,4 most notably in Co-catalysed hydroformylation.5 We are interested in heterophobanes (PhobPZ where Z = a non-hydrocarbyl group) as ligands and particularly the effect that the phobyl group has on the reactivity of the P–Z bond. For example, fluorophobane (PhobPF) was shown to be a rare example of a fluorophosphine that is thermodynamically more stable to disproportionation and kinetically more stable to hydrolysis than acyclic fluorophosphine analogues; moreover PhobPF shows promise as a ligand for hydroformylation and hydrocyanation catalysis.6
 |
| | Chart 1 | |
Aminodiphosphines R2PNR′PR2 (known as PNP ligands, Chart 1) are excellent ligands for Cr-catalysed ethene tri/tetramerisation. As illustrated in Table 1, the characteristics of the R and R′ groups in R2PNR′PR2 have a decisive effect on the chemoselectivity, productivity and therefore the potential industrial utility of the oligomerisation catalyst.7,8 Increased steric bulk serves to lower the ratio of 1-octene to 1-hexene obtained, whilst changing from aryl to alkyl substituents on phosphorus dramatically reduces activity and increases polymer formation. The data in Table 1 highlight the impact of process conditions such as solvent, temperature and pressure upon the catalysis.
Table 1 Catalytic data for Cr–PNP based tri/tetramerisationa
| Ligand |
P/bar |
T/°C |
TON/kg per g Cr |
TOF/kg per g Cr per h |
1-C8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-C6 :1-C6 |
C6 /wt%(% 1-C6) |
C8 /wt%(% 1-C8) |
C10+/wt% |
PE/wt % |
|
Catalysis data taken from ref. 7 and 8. All wt% values are of total product slate. C6 and C8 refers to the entire C6 and C8 fractions and 1-C6 and 1-C8 refer to the proportion of the linear α-olefin within that fraction.
Catalysis conditions: 33 μmol CrCl3(THF)3; 2 eq. ligand; 300 eq. MMAO-3A; 100 mL toluene.
Catalysis conditions: 33 μmol Cr(acac)3; 2 eq. ligand; 300 eq. MMAO-3A; 100 mL toluene.
Catalysis conditions: 2.5 μmol Cr(acac)3; 1.2 eq. ligand; 300 eq. MMAO-3A; 100 mL methylcyclohexane.
|
| Ph2PNMePPh2b |
30 |
65 |
27 |
54 |
5.7 |
24.8 (39.4) |
59.0 (94.1) |
n/r |
1.4 |
| Ph2PNiPrPPh2b |
30 |
65 |
12 |
24 |
2.1 |
32.7 (86.5) |
60.6 (99.2) |
n/r |
0.3 |
| Ph2PNiPrPPh2c |
45 |
45 |
272 |
544 |
5.7 |
16.9 (70.3) |
68.3 (98.8) |
n/r |
1.1 |
| Et2PNMePEt2c |
45 |
45 |
4 |
8 |
4.1 |
16.8 (64.6) |
45.2 (97.4) |
n/r |
13.6 |
| Ph2PNMePPh2d |
45 |
60 |
482 |
964 |
9.5 |
16.4 (33.4) |
54.0 (95.9) |
24.8 |
4.9 |
| Ph2PNiPrPPh2d |
45 |
60 |
552 |
1104 |
5.4 |
16.8 (75.5) |
69.5 (99.0) |
12.6 |
0.9 |
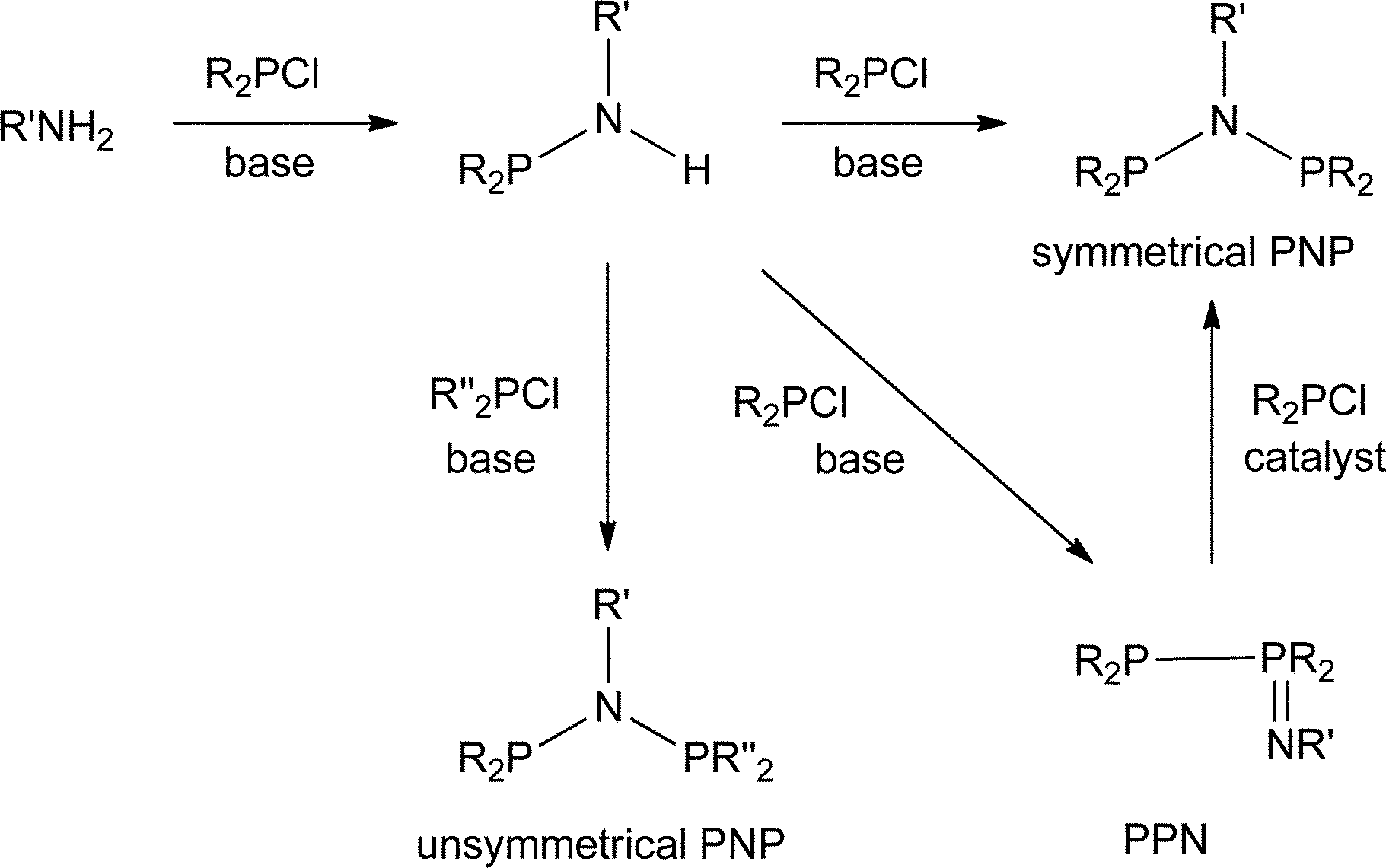
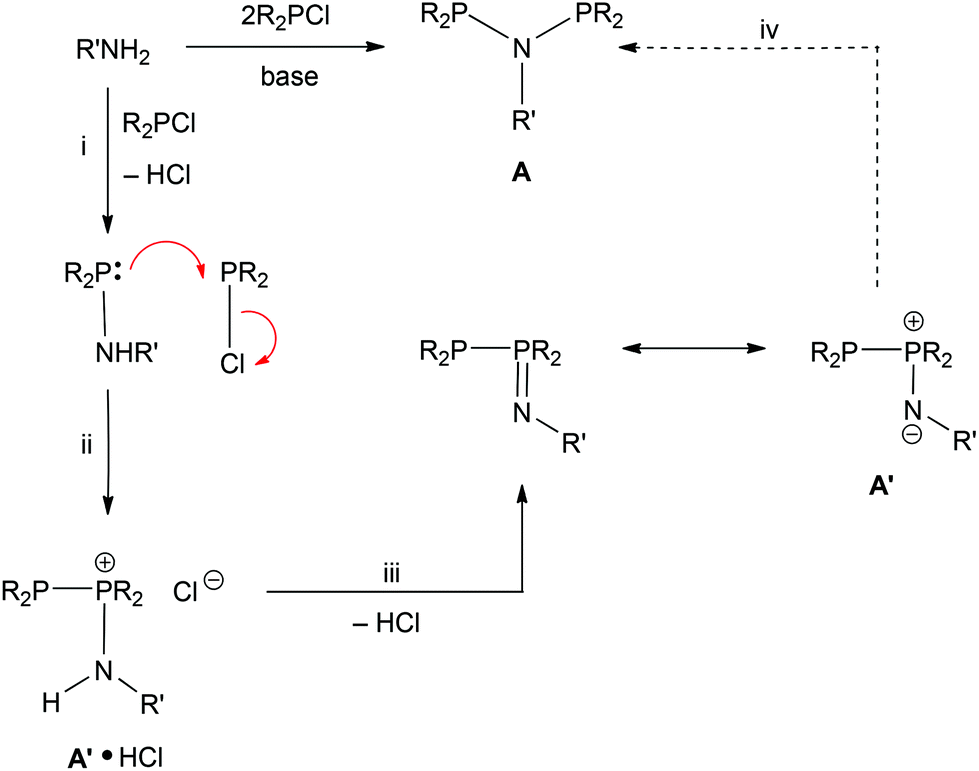
The industrial interest in PNP ligands9 makes it important to have reliable methods for their preparation. As summarised in Scheme 1, the most general route to PNP ligands is the reaction of a primary amine with a chlorophosphine in the presence of a base.10 The monophos R2PNHR′ species are presumed intermediates and when R or R′ is bulky, they are readily isolated and are potential intermediates to unsymmetrical PNP ligands.11 When the substituents in either of the reactants R2PCl or R′NH2 are bulky, a complication is the formation of the phosphinimine PPN compounds (Scheme 1); Maumela et al.10 have shown that when R = Ph and R′ = tBu, the PPN product is the kinetic product whose isomerisation to the thermodynamic PNP product is catalysed by Ph2PCl.
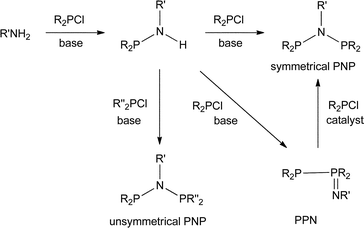 |
| | Scheme 1 | |
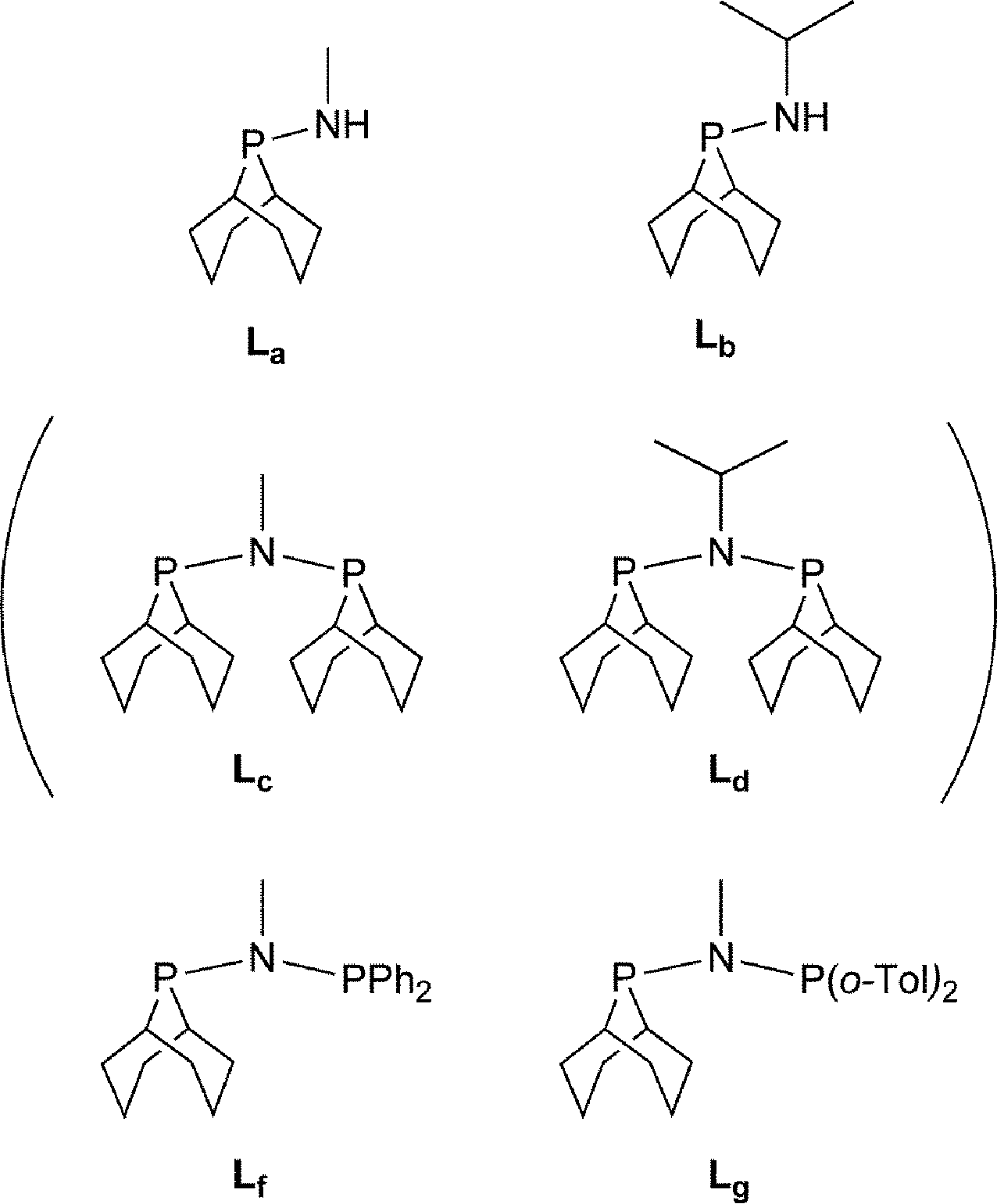
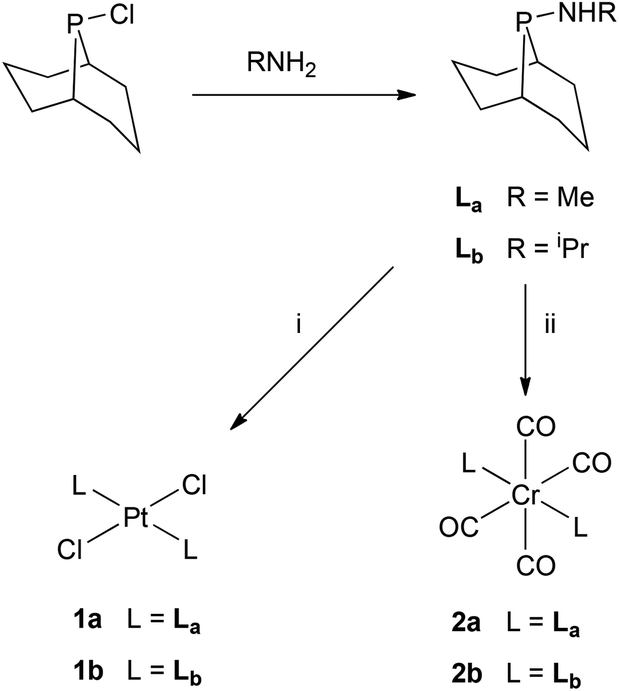
We were interested in investigating PNP ligands such as La–Ld where a phobyl group has been incorporated (Chart 2). It is shown here that the monophosphines La and Lb are readily prepared but their conversions to Lc and Ld has not been achieved. However the mixed diphosphines Lf and Lg are accessible and are shown to be ligands for Cr-catalysed ethene tri/tetramerisation.
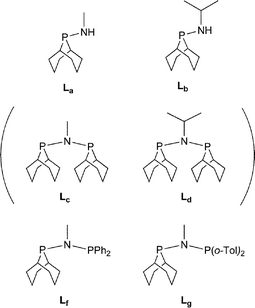 |
| | Chart 2 Aminophobanes targets; Lc and Ld (in parentheses) have not been observed. | |
Results and discussion
Stereoelectronically, a Cy2P group can be viewed as an acyclic analogue of a PhobP group since ostensibly, they are similarly bulky dialkylphosphino groups. However, we have shown previously that the rigidity of the PhobP moiety leads to a larger steric profile than expected12 and the approximately 90° C–P–C bridgehead angle in PhobP has the effect of lowering the HOMO and LUMO energies.13 Ligands L1–L4 (Chart 3) were targeted in the belief that a comparison of their chemistry with the phobane analogues La–Ld (Chart 2) would provide insight into the effect of the bicycle.
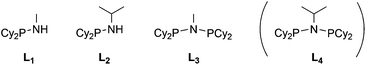 |
| | Chart 3 Aminodicyclohexylphosphines targets; L4 (in parentheses) has not been observed. | |
Monodentate aminophobanes
The monophobanes La and Lb were readily prepared by aminolysis of PhobPCl. The relative lability of La and Lb to hydrolysis (eqn (1)) was gauged by treatment of La, Lb, L1 and L2 with aqueous solutions under the same conditions and monitoring the formation of R2P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)H by 31P NMR spectroscopy. All four aminophosphines eventually underwent complete hydrolysis but at different rates. Comparison of the extents of hydrolysis after 16 h (Table 2) shows that the NHiPr group provides more protection from hydrolysis than the less bulky NHMe. Moreover, the bicyclic compounds PhobPNHR are significantly kinetically stabilised to hydrolysis with respect to the acyclic Cy2PNHR analogues. The resistance to hydrolysis of PhobPNHR is consonant with the phobyl moiety behaving as a bulky group.12
O)H by 31P NMR spectroscopy. All four aminophosphines eventually underwent complete hydrolysis but at different rates. Comparison of the extents of hydrolysis after 16 h (Table 2) shows that the NHiPr group provides more protection from hydrolysis than the less bulky NHMe. Moreover, the bicyclic compounds PhobPNHR are significantly kinetically stabilised to hydrolysis with respect to the acyclic Cy2PNHR analogues. The resistance to hydrolysis of PhobPNHR is consonant with the phobyl moiety behaving as a bulky group.12| |  | (1) |
Table 2 Comparison of some properties of aminophobanes and acyclic analogues
| |
% Hydrolysis at 16 ha |
ν
COb/cm−1 |
|
For the hydrolysis experiments, the aminophosphines (1.4 mmol) were dissolved in a 0.55 M solution of water in MeOH (25 mL) and stirred. The reactions were monitored by periodically taking aliquots of the solution and measuring the 31P NMR spectrum.
The IR spectrum in the 2050–1850 cm−1 region was measured in CH2Cl2 for the trans-[RhCl(CO)(L)2] complexes generated in situ by combining [Rh2Cl2(CO)4] with 4 equiv. of L.
L1 was 50% hydrolysed after 0.5 h.
|
| PhobPNHMe (La) |
5 |
1957 |
| Cy2PNHMe (L1) |
100c |
1955 |
| PhobPNHiPr (Lb) |
1 |
1954 |
| Cy2PNHiPr (L2) |
64 |
1951 |
The donor properties of La and Lb can be compared quantitatively with L1 and L2 from the νCO values for their trans-[RhCl(CO)(L)2] complexes.14 The rhodium complexes were made in situ (see Scheme 2) and the recorded νCO values (Table 2) are consistent with La and Lb being slightly poorer σ-donors/better π-acceptors than their acyclic analogues L1 and L2, as expected.13
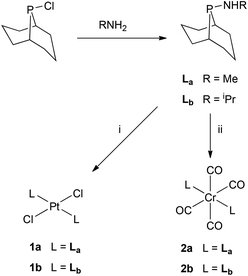 |
| | Scheme 2 Reagents: (i) [PtCl2(NCtBu)2] in CH2Cl2; (ii) [Cr(CO)4(nbd)] (nbd = norbornadiene) in CH2Cl2. | |
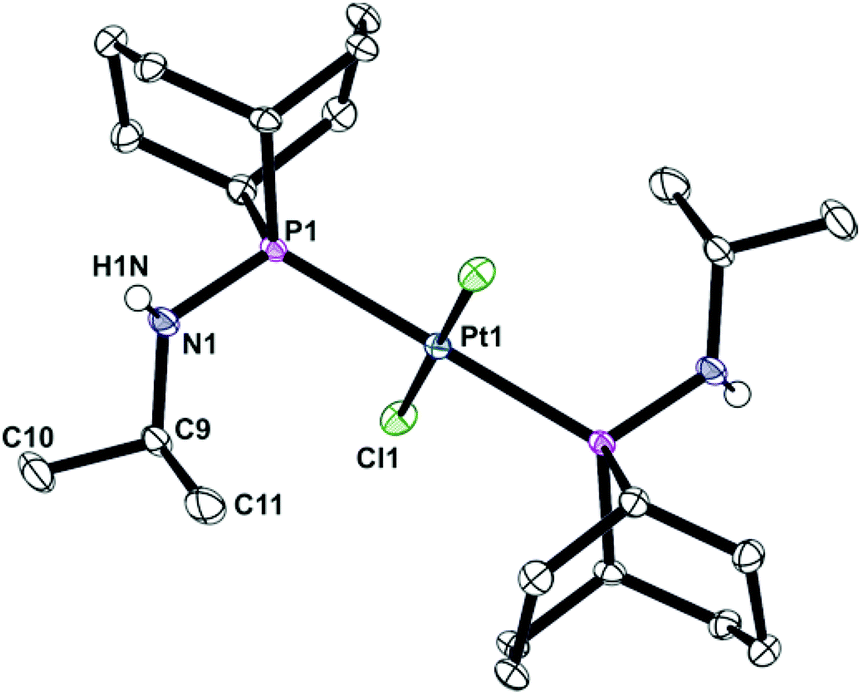
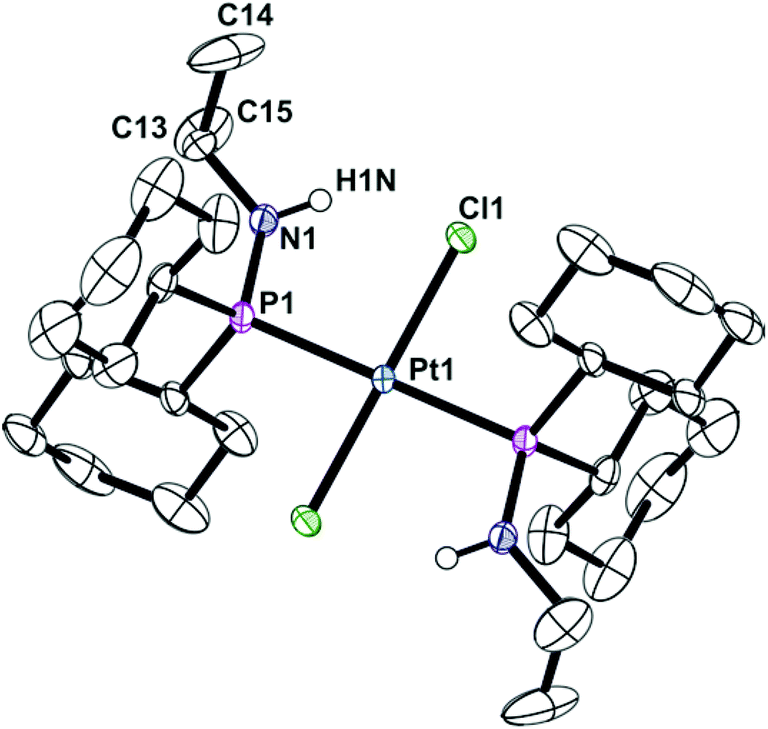
Ligands La and Lb form trans-dichloroplatinum(II) complexes 1a and 1b, and trans-tetracarbonylchromium(0) complexes 2a and 2b (Scheme 2). The crystal structures of 1b and 2b have been determined and are shown in Fig. 1 and 2. In addition, the crystal structure of trans-[PtCl2(L2)2] (3), an acyclic analogue of 1b has been determined (Fig. 3).
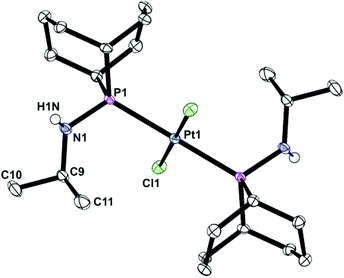 |
| | Fig. 1 Crystal structure of trans-[PtCl2(PhobPNHiPr)2] (1b). All hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Pt(1)–P(1) 2.3241(10), Pt(1)–Cl(1) 2.3102(8), P(1)–N(1) 1.6594(16), Pt(1)–P(1)–N(1) 109.40(6). | |
 |
| | Fig. 2 Crystal structure of trans-[Cr(CO)4(PhobPNHiPr)2] (2b). All hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Cr(1)–P(1) 2.3434(3), Cr(1)–P(2) 2.3499(3), P(1)–N(1) 1.6910(9), P(2)–N(2) 1.6934(10), 114.06(4), Cr(1)–P(1)–N(1) 114.20(2), Cr(1)–P(2)–N(2). | |
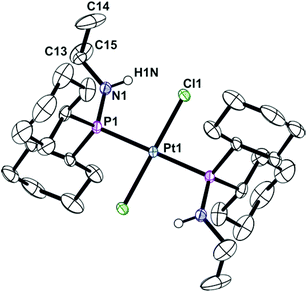 |
| | Fig. 3 Crystal structure of trans-[PtCl2(Cy2PNHiPr)2] (3). All hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Pt(1)–P(1) 2.3178(7), Pt(1)–Cl(1) 2.3193(7), P(1)–N(1) 1.681(3), Pt(1)–P(1)–N(1) 113.91(10). | |
In aminophobane complex 1b and its acyclic analogue 3, the Pt metal centre is square planar. The Pt sits on a crystallographic inversion centre and the asymmetric unit consists of half of the complete molecule, consequently the N–P–P–N torsion angles are 180° in both cases, i.e. the anti conformer is adopted, as in other trans-[PtCl2(PhobPZ)2] complexes.6,12,15 The cone angle of Lb in 1b is 111.8° and of L2 in 3 is larger at 115.8°. In the structure of 2b, the asymmetric unit contains one complete molecule. The cone angle of Lb in 2b is 109.2° which is smaller than in 1b, the compression probably reflecting the greater crowding in the octahedral complex. The N–P–P–N torsion angle in 2b is 108.3(1)° indicating the amino groups are gauche to each other, a conformation not previously observed in PhobPZ complexes.
Bidentate aminophobanes
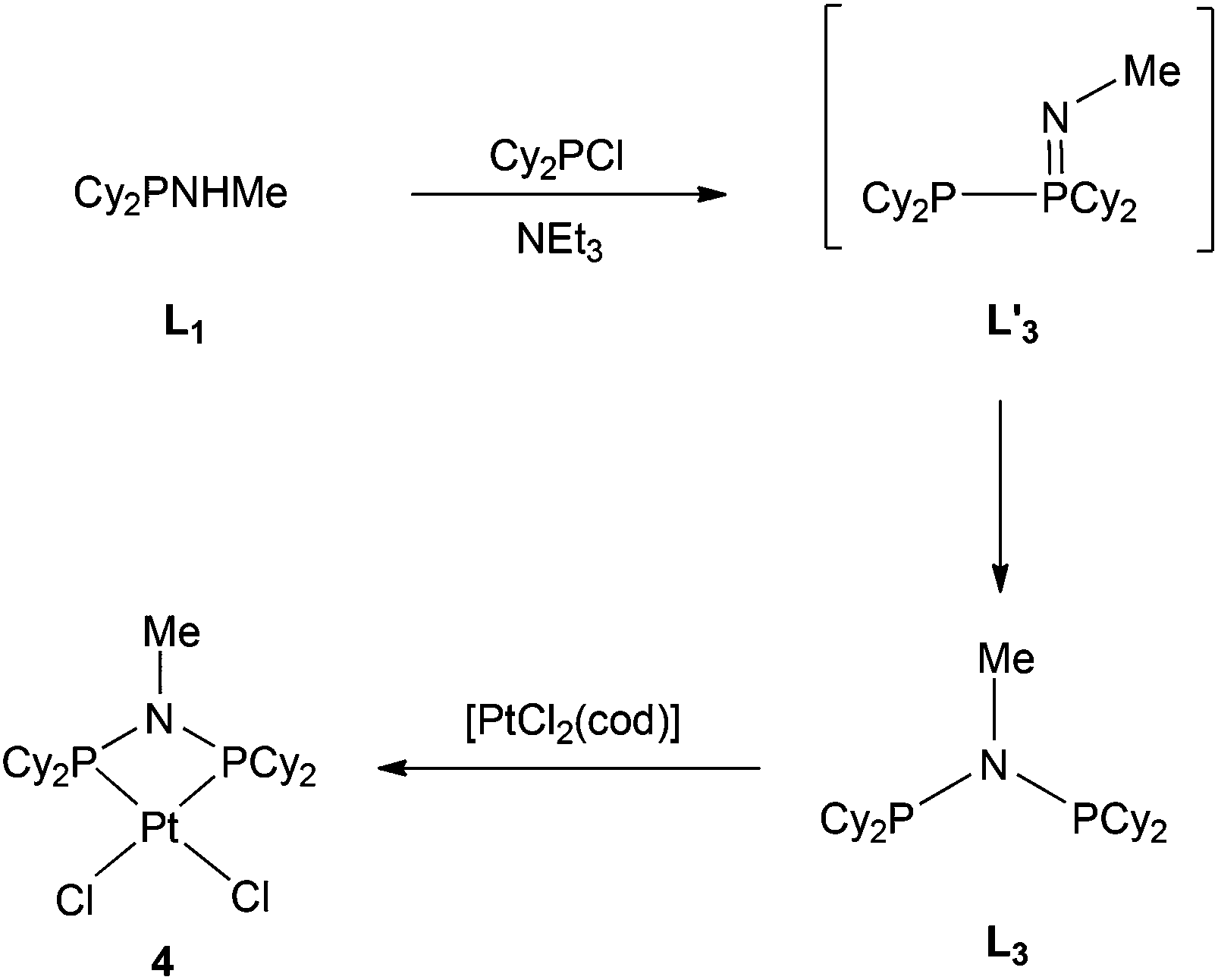
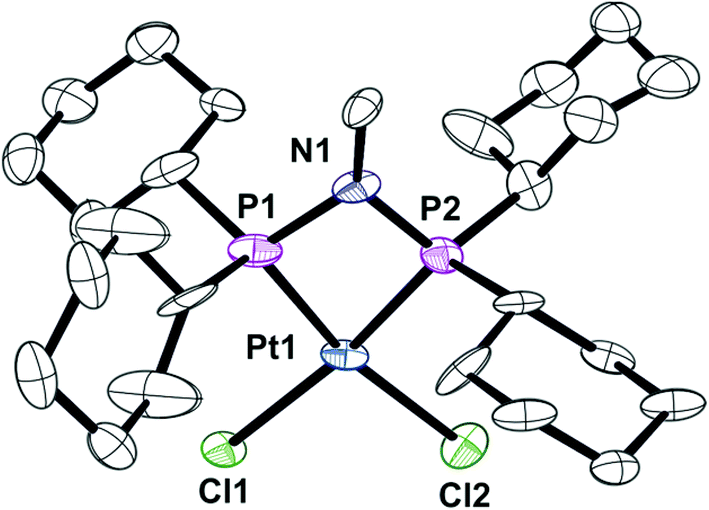
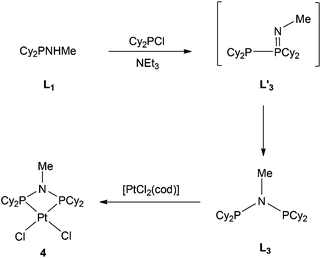
The previously reported16 diphosphinoamine L3 is readily prepared from MeNH2 and Cy2PCl in the presence of Et3N (Scheme 3). The intermediate in this reaction is presumably Cy2PNHMe (L1) and indeed treatment of the isolated L1 with Cy2PCl in the presence of Et3N in CH2Cl2 gave L3 quantitatively according to 31P NMR spectroscopy. The spectrum of the reaction mixture also revealed a transient PPN species (as evidenced by a large JPP of 280 Hz) to which the tautomeric structure L′3 is assigned. The PPN species L′3 smoothly converted over 30 min to PNP ligand L3 whose structure was confirmed by its conversion to the chelate complex 4 (Scheme 3), the crystal structure of which has been determined (Fig. 4).
 |
| | Scheme 3 | |
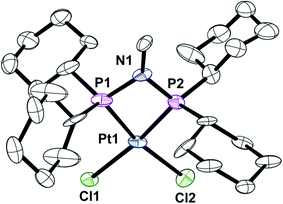 |
| | Fig. 4 Crystal structure of complex [PtCl2(L3)] (4). Only one of the three unique molecules is shown and all hydrogen atoms and six molecules of CHCl3 have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pt(1)–P(2) 2.206(6), Pt(1)–P(1) 2.257(6), Pt(1)–Cl(1) 2.363(5), Pt(1)–Cl(2) 2.376(7), P(1)–N(1) 1.745(17), P(2)–N(1) 1.668(18), P(2)–Pt(1)–P(1) 71.1(3), N(1)–P(1)–Pt(1) 92.8(6), N(1)–P(2)–Pt(1) 96.9(9), P(2)–N(1)–P(1) 99.0(8). | |
The asymmetric unit consists of three independent molecules of 4 along with six chloroform molecules. Although the PtP2Cl2 fragment is approximately planar (rms deviation ∼0.03 Å), the Pt has a distorted square planar geometry due to the constraints of the 4-membered PNP chelate. The three independent Pt–P–N–P rings are approximately planar with rms deviations of ∼0.03 Å.
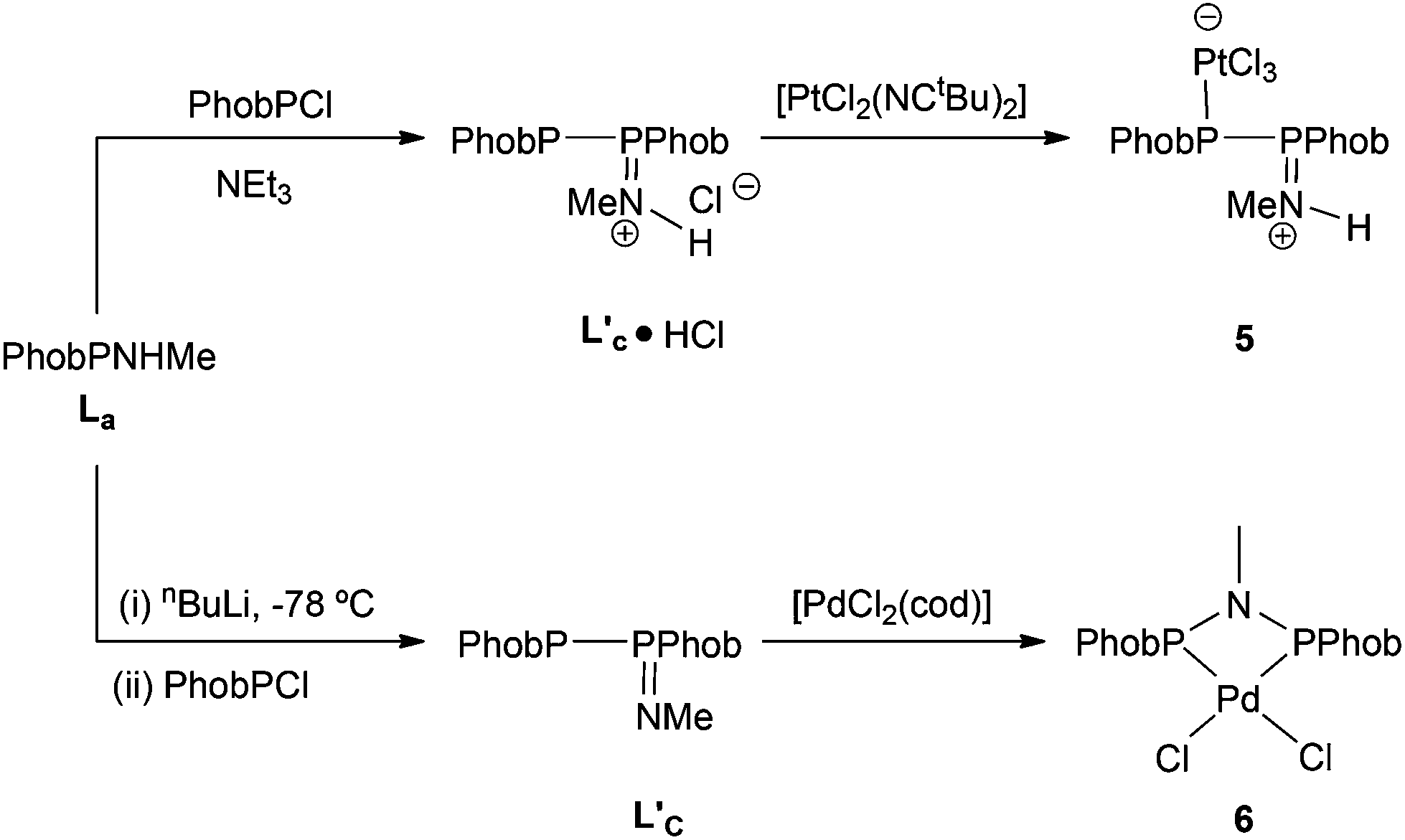
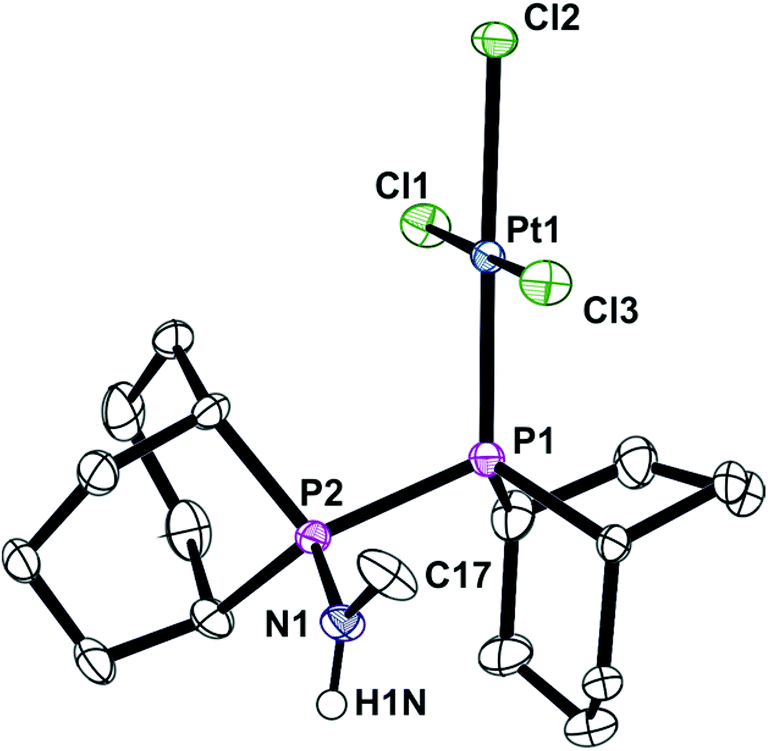
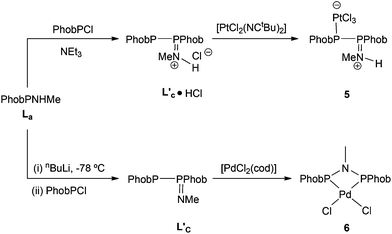
In contrast to the ready reaction of L1 with Cy2PCl to give PNP ligand L3 (Scheme 3), the reaction of PhobPNHMe (La) with PhobPCl in the presence of NEt3 or N-methylpyrrolidine did not give the expected diphosphinoamine Lc. Instead, a PPN species (JPP = 407 Hz) was the exclusive product; this was initially assigned structure L′c but its 1H NMR spectrum (which showed a multiplet at 7.01 ppm integrating for 1H) and mass spectrum (M+ at [L′c + 1]) led to its assignment as the HCl adduct L′c·HCl (Scheme 4). This was supported by its reaction with [PtCl2(NCtBu)2] which yielded crystals of the insoluble, zwitterionic complex [PtCl3(L′c·H)] (5) whose X-ray crystal structure is shown in Fig. 5. The conditions under which L′c·HCl was formed (Scheme 4) indicate that the iminophosphine L′c is more basic than either NEt3 or N-methylpyrrolidine.
 |
| | Scheme 4 | |
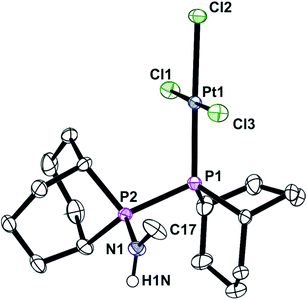 |
| | Fig. 5 Crystal structure of [PtCl3(L′c·H)] (5). All hydrogen (except H1N) atoms, a CH2Cl2 molecule (50% occupied) and one C2H2Cl4 molecule have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pt(1)–P(1) 2.2194(13), Pt(1)–Cl(1) 2.3085(12), Pt(1)–Cl(2) 2.3683(12), Pt(1)–Cl(3) 2.2988(12), P(1)–P(2) 2.2797(19), P(2)–N(1) 1.629(4), N(1)–C(17) 1.468(7), N(1)–H(1N) 0.8800, P(1)–Pt(1)–Cl(3) 88.86(5), P(1)–Pt(1)–Cl(1) 92.80(5), Cl(3)–Pt(1)–Cl(1)177.36(5), P(1)–Pt(1)–Cl(2) 177.89(5), Cl(3)–Pt(1)–Cl(2)89.39(5), Cl(1)–Pt(1)–Cl(2) 88.99(5), Pt(1)–P(1)–P(2) 108.26(6), N(1)–P(2)–P(1) 111.95(19). | |
The crystal structure of 5 has a square planar Pt with an rms deviation of the atoms from the square plane of ∼0.03 Å. The PPN ligand is rotated away from the PtCl3 plane with torsion angles Cl1–Pt1–P1–P2 of −102.4(1)° and Cl3–Pt1–P1–P2 of 75.6(1)°.
Treatment of PhobPNMeH with nBuLi at −78 °C followed by PhobPCl gave a PPN species with a JPP = 327 Hz (significantly smaller than the JPP of 407 Hz for L′c·HCl) that is assigned to the neutral Lc′ which has been isolated. No reaction occurred upon addition of PhobPCl to L′c in CH2Cl2, conditions that might have been expected to tautomerise L′c to Lc.10
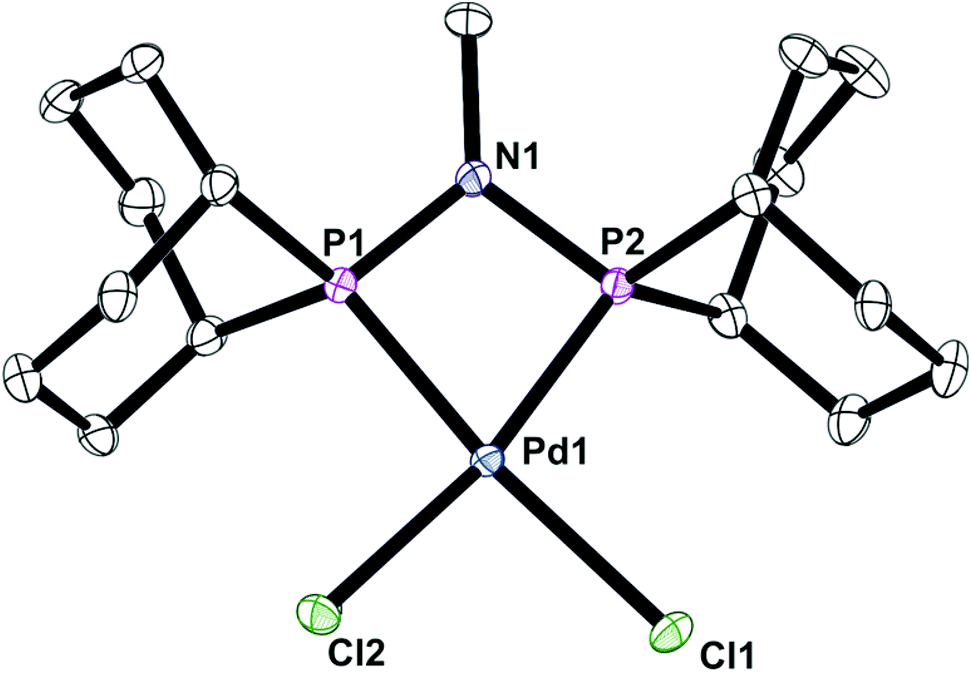
It has previously been shown that some neutral PPN compounds rearrange when they react with [MCl2(cod)] (M = Pd or Pt)17 or [NiBr2(dme)]18 to give PNP chelate complexes. Reaction of [PdCl2(cod)] with L′c gave the chelate [PdCl2(Lc)] (6) whose crystal structure has been determined and is shown in Fig. 6.
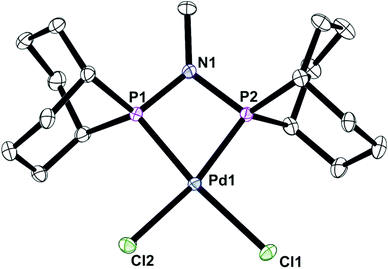 |
| | Fig. 6 Crystal structure of complex [PdCl2(Lc)] (6). All hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pd(1)–P(1) 2.2311(4), Pd(1)–P(2) 2.2353(4), Pd(1)–Cl(2) 2.3735(4), Pd(1)–Cl(1) 2.3748(4), P(1)–N(1) 1.6949(14), P(2)–N(1) 1.7016(14), P(1)–Pd(1)–P(2) 70.851(15). | |
The asymmetric unit contains one molecule of 6, with the PdP2Cl2 fragment being essentially planar (rms ∼ 0.07 Å) although the overall geometry is a distorted square planar due to the constraints of the 4-membered PNP chelate. As seen in the structure of analogue 4, the Pd1–P1–N1–P2 ring is also essentially planar with an rms deviation for the atoms of 0.01 Å.
From the homodiphos products obtained in the reactions of La and L1 with R2PCl (see Schemes 3 and 4), it appears that the PhobP group differs from Cy2P and Ar2P groups in promoting PPN over PNP formation; this raised the question of what would happen when the syntheses of the heterodiphos PNP ligands PhobPNMePR2 where R = Cy (Le), Ph (Lf) or o-Tol (Lg) were attempted?
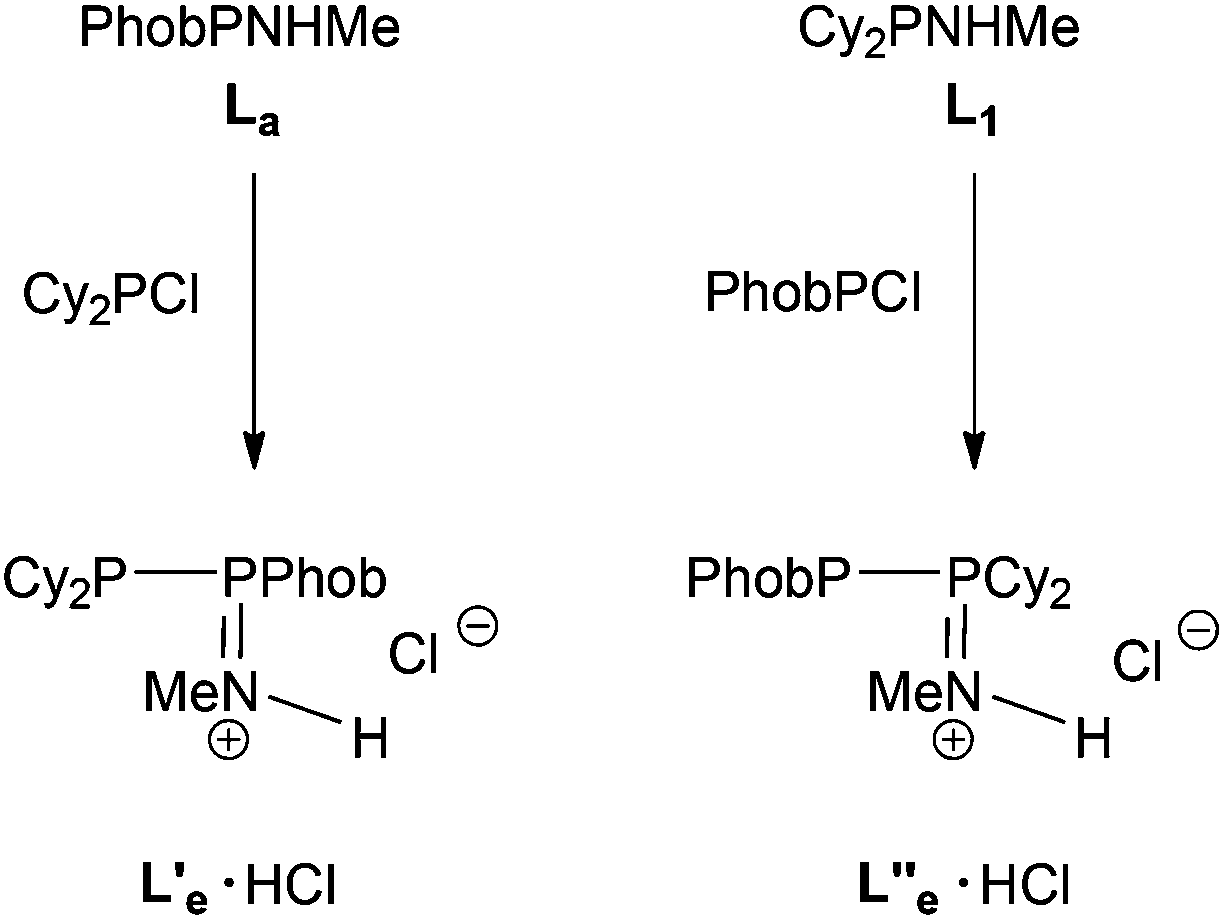
The reaction between PhobPNHMe and Cy2PCl was followed by 31P NMR spectroscopy and it was unambiguously shown that a PPN product was formed which, on the basis of its JPP of 358 Hz, was tentatively assigned to the protonated species L′e·HCl (Scheme 5); addition of Et3N led to multiple P-containing species but there was no evidence for the formation of the neutral PPN (L′e) or PNP (Le) species. The reaction between PhobPCl and Cy2PNHMe was also monitored and in this case, 31P NMR spectroscopy revealed that a PPN product was formed (JPP = 403 Hz) which was assigned to the cationic species L′′e·HCl (Scheme 5), an isomer of L′e·HCl. It therefore appears that the PPN-promoting effect of the PhobP group dominates over the PNP-preference of the Cy2P group.
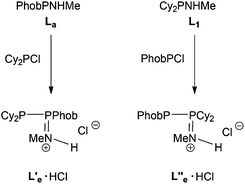 |
| | Scheme 5 | |
The unsymmetrical PNP ligands Lf and Lg (JPP = 80 Hz in both) featuring PhobP groups were successfully prepared upon treatment of PhobPNHMe with Ar2PCl (Ar = Ph or o-Tol) in the presence of Et3N (eqn (2)). It therefore appears that the PPN formation promoted by the PhobP group is superseded by the PNP preference of the Ar2P groups.
| | 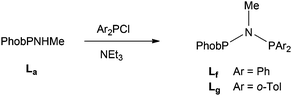 | (2) |
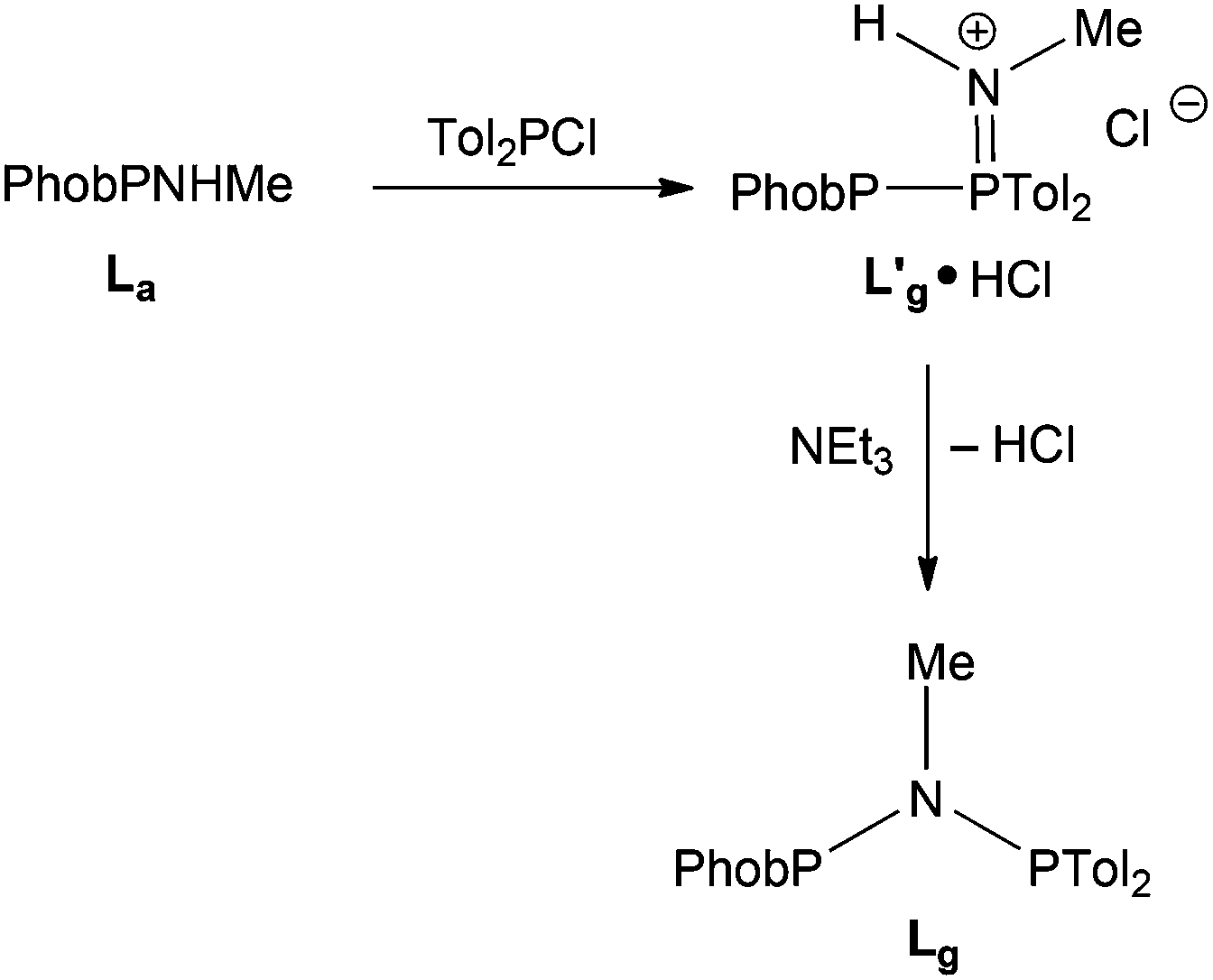
The reaction of PhobPNHMe with Tol2PCl was monitored by 31P NMR spectroscopy. A PPN species (JPP = 331 Hz), tentatively assigned to L′g·HCl (Scheme 6) was formed rapidly which, upon treatment with NEt3, was transformed to Lg (JPP = 80 Hz).
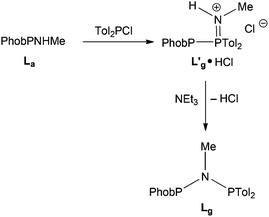 |
| | Scheme 6 | |
Treatment of the bulky R2PNHiPr (Lb or L2) with R2PCl (R2P = Cy2P or PhobP) under the conditions that converted R2PNHMe to the corresponding L3 (Scheme 3) or L′c·HCl/L′c (Scheme 4) gave, according to in situ31P NMR spectroscopy, mixtures of unidentified products as well as the reactants.
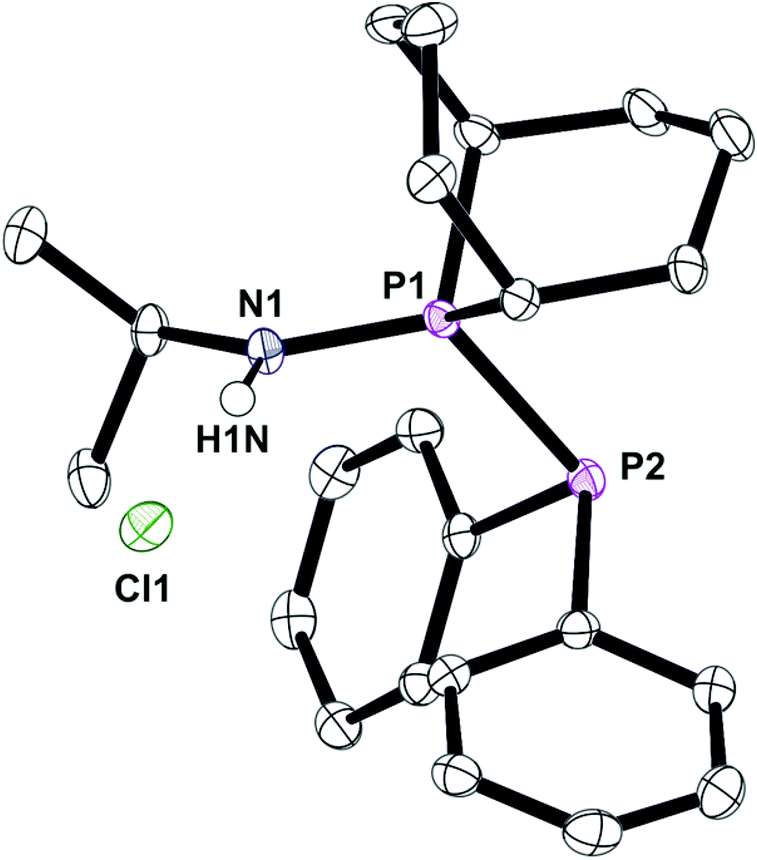
Under the conditions that smoothly led to the mixed PNP ligands Lf and Lg (eqn (2)), Lb reacted with Ar2PCl to give PPN species whose structures were assigned to the protonated L′h·HCl and L′i·HCl (eqn (3)) on the basis of the large JPP values of 338 and 359 Hz respectively. Crystals of L′h·HCl were obtained and the crystal structure shown in Fig. 7 confirms the PPN assignment. The N⋯Cl distance of 3.101(1) Å indicates the presence of hydrogen-bonding between the N–H and Cl.
| | 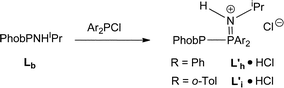 | (3) |
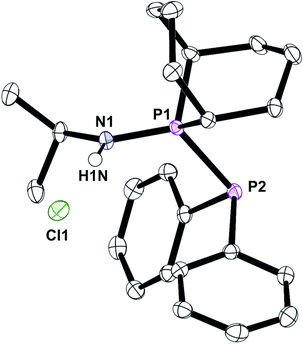 |
| | Fig. 7 Crystal structure of complex L′h·HCl. All hydrogen atoms, except on the N1, have been omitted for clarity. Selected bond lengths (Å) and angles (°); P(1)–N(1) 1.6241(0), P(1)–P(2) 2.2403(4), N(1)–H(1N) 0.863(16), N(1)–P(1)–P(2) 119.93(4). | |
PPN versus PNP preferences
The N- and P-substituents determine whether PNP (A) or PPN (A′) species are formed in the reaction of amines with chlorophosphines (Scheme 7). In some cases, it has been shown10,19 that the PPN can be converted to the PNP tautomer using a R2PCl catalyst and we have observed PPN species as transients en route to the PNP products (e.g. Cy2PNMePCy2 see Scheme 3) showing that the PNP is the thermodynamic product. In other cases (e.g. Cy2PN{SO2Ar}PCy2) the neutral PPN tautomer appears to be the thermodynamic product.17,18,20 An additional element observed in this work is the formation of a protonated A′·HCl product that is resistant to deprotonation by amines.
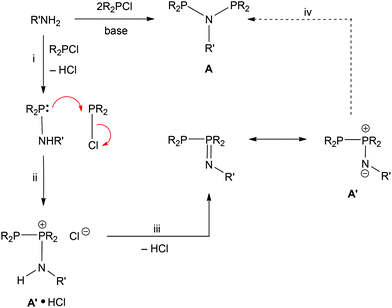 |
| | Scheme 7 Proposed pathway for the conversion of an amine to a PNP ligand. A dotted line is used for step iv since this step is not observed with the aminophobanes. | |
A pathway from chlorophosphine and primary amine to PNP/PPN products that encompasses these empirical observations is shown in Scheme 7. Nucleophilic attack by amine on chlorophosphine with loss of HCl would give the intermediate aminophosphine (step i). Reaction of a second chlorophosphine at the P site of the aminophosphine would give the salt A′·HCl (step ii) which can eliminate HCl to give the neutral A′ (step iii) and finally rearrangement to give PNP (step iv).
The formation of a PPN species when PhobPCl reacts with PhobPNHMe or PhobPNMeLi instead of PhobPNMePPhob contrasts with the smooth formation of Cy2PNMePCy2via a PPN intermediate; furthermore, PhobP(NMe)PPhob does not isomerise to the PNP tautomer in the presence of PhobPCl. At present, it is not known whether these observations are due to PhobP(NMe)PPhob being the thermodynamically preferred tautomer or slow kinetics of interconversion and therefore further investigation of this system is warranted.
Oligomerisation catalysis
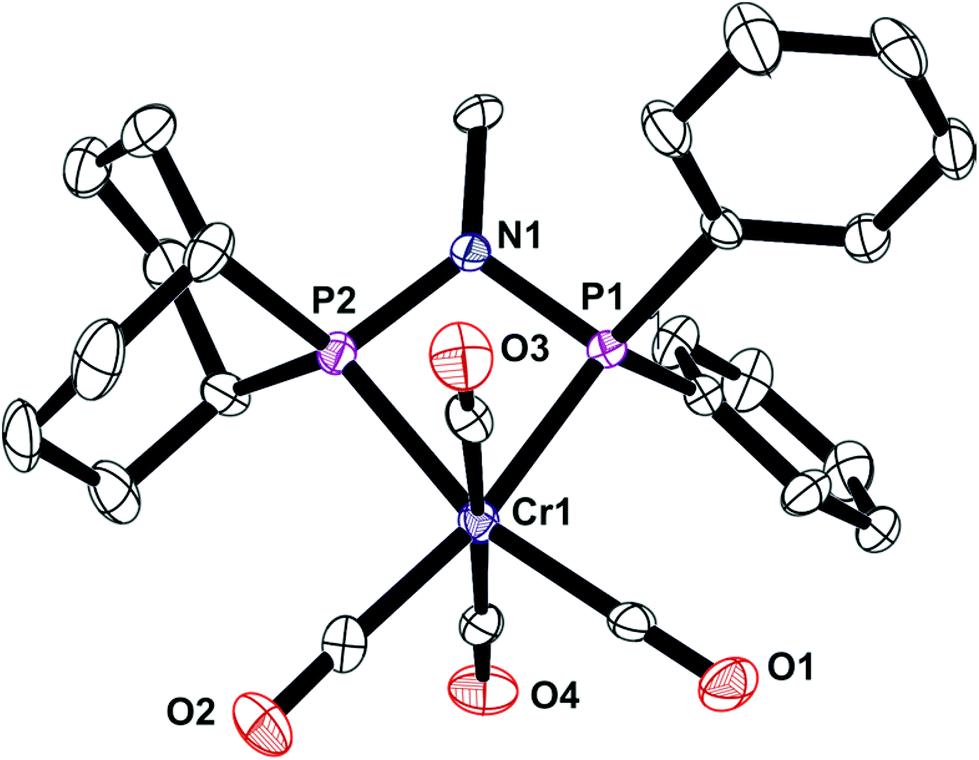
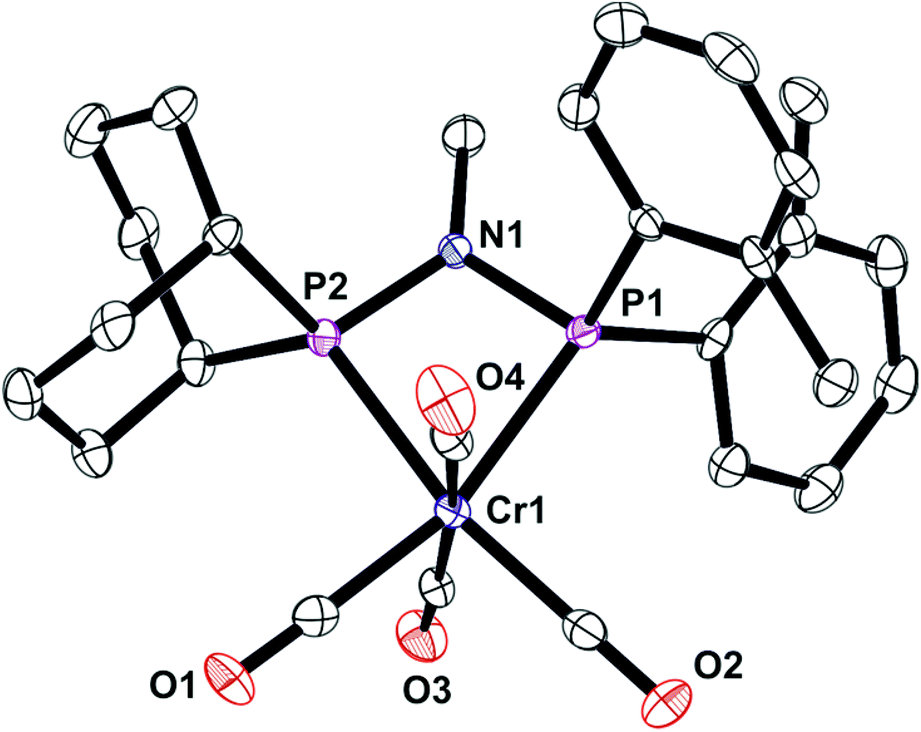
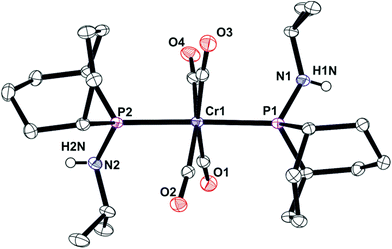
The unsymmetrical PNP ligands Lf and Lg have been screened for Cr-catalysed ethene oligomerisation (see below) and it was therefore appropriate to explore their Cr coordination chemistry. The reaction of [Cr(CO)4(nbd)] with Lf or Lg gave the corresponding Cr(0) complexes 7 and 8 (eqn (4)) which have been fully characterised and their crystal structures have been determined (Fig. 8 and 9).| | 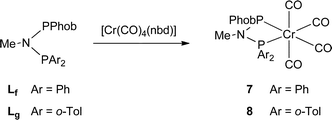 | (4) |
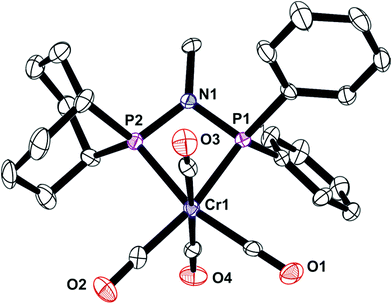 |
| | Fig. 8 Crystal structure of complex [Cr(CO)4(Lf)] (7). All hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°); Cr(1)–P(3) 2.3163 (16), Cr(1)–P(2) 2.3474(16), P(2)–N(1) 1.714(5), P(3)–N(1) 1.701(5), P(3)–Cr(1)–P(2) 68.29(5), P(3)–N(1)–P(2) 100.1(2), N(1)–P(3)–Cr(1) 96.48(17), N(1)–P(2)–Cr(1) 94.99(16), C(17)–P(2)–C(16) 96.0(3), C(7)–P(3)–C(1) 103.5(2). | |
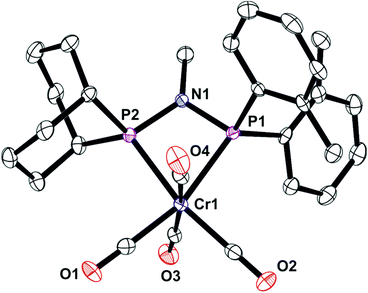 |
| | Fig. 9 Crystal structure of [Cr(CO)4(Lg)] (8). All hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°); Cr(1)–P(1) 2.3477(6), Cr(1)–P(2) 2.3669(6), P(1)–N(1) 1.7146(17), P(2)–N(1) 1.7035(17), P(1)–Cr(1)–P(2) 68.04(4), P(2)–N(1)–P(1) 101.01(9), N(1)–P(1)–Cr(1) 94.67(6), N(1)–P(2)–Cr(1) 94.29(6), C(16)–P(1)–C(13) 95.66(10), C(7)–P(2)–C(4) 105.17(9). | |
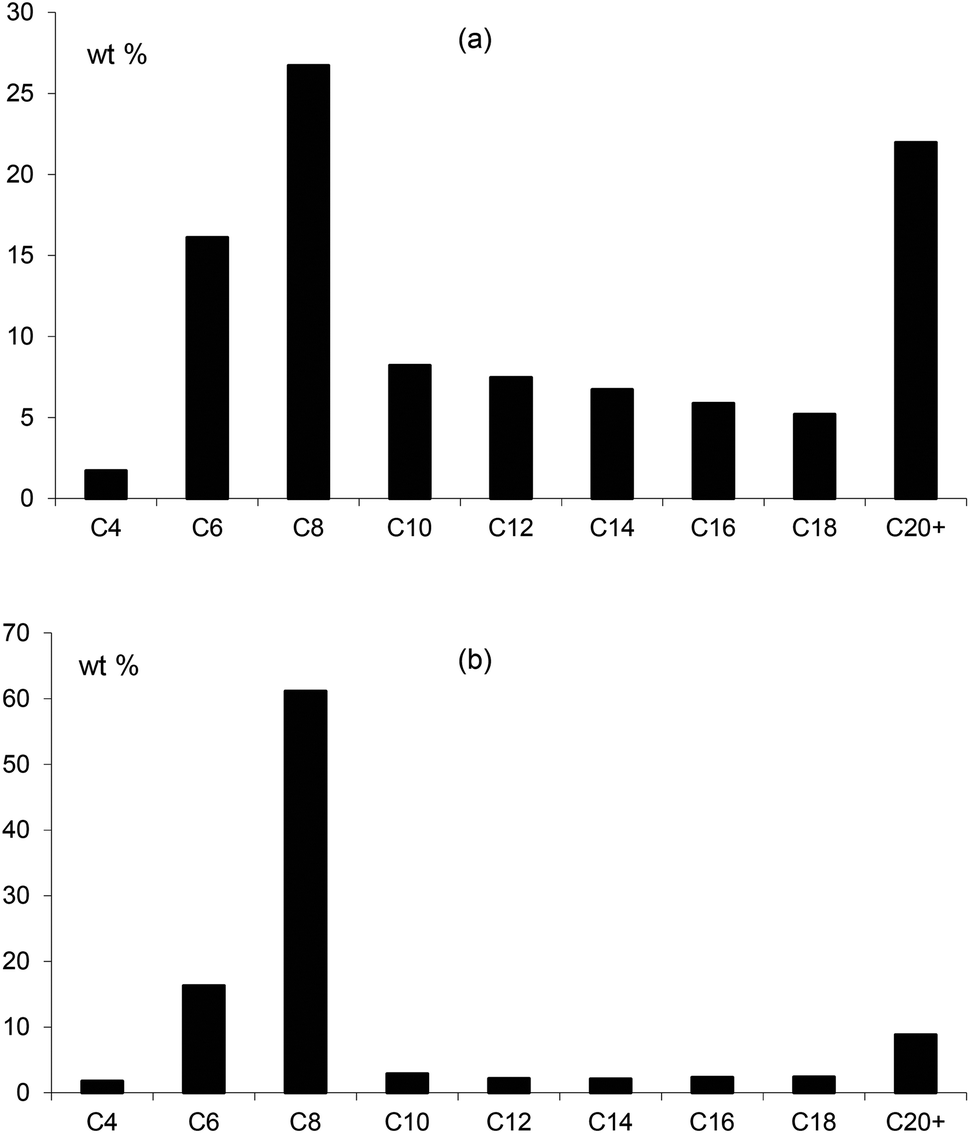
In combination with chromium, the ligands Lf and Lg gave moderate activities towards ethylene oligomerisation but the formation of polymer was high, as can be seen from Table 3. Within the liquid fraction, it is clear that a degree of selective oligomerisation to 1-hexene and 1-octene did occur (particularly for Lg) but concurrently with Schulz-Flory selectivity (Fig. 10). The 1-octene to 1-hexene ratios obtained for both ligands is high.
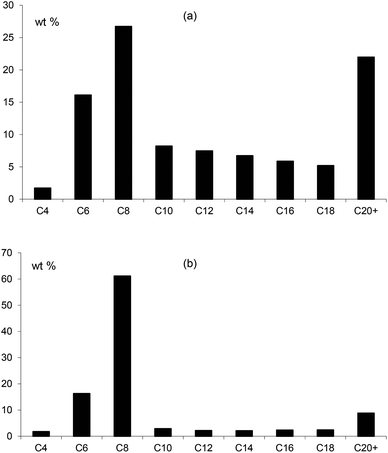 |
| | Fig. 10 The distribution of products in the liquid fraction formed in the ethene oligomerisation catalysed by Cr catalysts of (a) Lf and (b) Lg showing the preferential formation of C6 and C8 alkenes. | |
Table 3 Ethene oligomerisation resultsa
| L |
Rxn time/min |
TON/kg per g Cr |
TOF/kg per g Cr per h |
Liquid product/wt% |
Solid product (PE)/wt% |
C4/wt% |
C6/wt%(% 1-C6) |
C8/wt%(% 1-C8) |
1-C8:1-C6 |
C10–14 /wt% |
C15+/wt% |
|
Conditions: Cr(acac)3 (2.5 μmol), 1.2 eq. L, 960 eq. MMAO-3A (800 eq. added to pre-activation, 160 eq. added to autoclave vessel), PhCl solvent (70 mL), 53 bar ethylene, 60 °C. The wt% values for the carbon number fractions refer to the liquid fraction.
|
|
Lf
|
24.8 |
393 |
951 |
89.5 |
10.5 |
1.7 |
16.1 (20.7) |
26.7 (93.0) |
7.5 |
22.4 |
33.0 |
|
Lg
|
16.7 |
31 |
110 |
56.6 |
43.4 |
1.8 |
16.3 (70.2) |
61.1 (99.4) |
5.3 |
7.2 |
13.6 |
Conclusions
The monodentate aminophobanes PhobPNHR (R = Me or iPr) have been readily prepared and are more resistant to hydrolysis than their Cy2PNHR analogues consistent with the PhobP group having a greater effective steric bulk than Cy2P. Attempts to make the free ligand PhobPNMePPhob have been thwarted by formation of PPN species which resist tautomerisation although a rearrangement takes place in the presence of [PdCl2(cod)] to give the desired PNP–Pd chelate. The readily prepared mixed diphos ligands PhobPNMePAr2 (Ar = Ph or o-Tol) in combination with Cr, catalysed the oligomerisation of ethylene with a partial selectivity to tri/tetramerisation, the remainder of the selectivity appearing to be Schulz-Flory in nature; the activities were moderate, but the polymer formation was high.
Experimental
Unless otherwise stated, all reactions were carried out under a dry nitrogen atmosphere using standard Schlenk-line techniques. Dry N2-saturated solvents were collected from a Grubbs system21 in flame and vacuum-dried glassware. MeOH was dried over 3 Å molecular sieves, pentane was dried over 4 Å molecular sieves and both were deoxygenated by N2 saturation. The starting materials PhobPCl,13 [Cr(CO)4(η4-norbornadiene)],22 [PtCl2(NCtBu)2],23 [PdCl2(cod)],24 were prepared by literature methods. All other reagents were used as received from Aldrich, Strem or Lancaster. The aminophosphines were stored under nitrogen at room temperature. NMR spectra were recorded on a Jeol Delta 270, Jeol Eclipse 300, Jeol Eclipse 400, Varian 400 or Lambda 300. Infrared spectroscopy was carried out on a Perkin Elmer 1600 Series FTIR. Mass spectra were recorded on a MD800 by the Mass Spectrometry Service, University of Bristol. Elemental analyses were carried out by the Microanalytical Laboratory of the School of Chemistry, University of Bristol.
Preparation of PhobPNHMe (La)
PhobPCl (2.00 g, 11.3 mmol) was dissolved in a 2 M THF solution of MeNH2 (22.9 mL, 45.3 mmol). The resulting suspension was stirred at room temperature for 16 h. The solvent was removed under reduced pressure to give a white residue which was extracted with diethyl ether (35 mL). The ethereal solution was filtered to remove the methylammonium chloride and then the solvent was evaporated to dryness to give a white solid (1.42 g, 75%). 31P{1H} NMR (121 MHz; CDCl3) δ 26.8. 1H NMR (270 MHz; CDCl3) δ 1.48–2.20 (14H, m, phobane), 2.67 (3H, d, JHP = 15.0 Hz, CH3N). 13C{1H} NMR (67 MHz; CDCl3) δ 21.5 (d, JCP = 2.0 Hz, CH2), 22.9 (d, JCP = 4.6 Hz, CH2), 23.7 (d, JCP = 3.1 Hz, CH2), 28.1 (d, JCP = 10.9 Hz, CH3N), 31.2 (d, JCP = 14.0 Hz, CH2), 33.0 (d, JCP = 26.9 Hz, CH). Elemental analysis: Found (Calc. for C9H18NP) C, 63.5 (63.1); N, 8.1 (8.1); H, 10.4 (10.6) %. MS (ESI: m/z 171 (M+).
Preparation of PhobPNHiPr (Lb)
PhobPCl (0.88 g, 5.0 mmol) was dissolved in a solution of iPrNH2 (1.54 mL, 20.1 mmol) in THF (9.0 mL). The resulting suspension was stirred at room temperature for 16 h and the filtered to remove the isopropylammonium chloride. The solvent was removed under reduced pressure to give an oily residue which was dissolved in toluene (10 mL) to precipitate any remaining isopropylammonium chloride. This toluene solution was filtered to remove the methylammonium chloride and then the solvent was evaporated to dryness to give a colourless oil (0.57 g, 63%). 31P{1H} NMR (121 MHz; CDCl3) δ 15.5. 1H NMR (270 MHz; CDCl3) δ 1.47–2.15 (14H, m, phobane), 1.14 (6H, d, JHP = 6.2 Hz, CH3N), 3.1–3.3 (1H, m, CH). 13C{1H} NMR (75 MHz; CDCl3) δ 21.6 (s, CH2), 22.9 (s, CH2), 24.0 (d, JCP = 3.1 Hz, CH3), 26.7 (d, JCP = 6.9 Hz, CH2), 29.5 (d, JCP = 9.2 Hz, CH), 31.4 (d, JCP = 13.8 Hz, CH2), 48.6 (d, JCP = 24.3 Hz, CH). Elemental analysis: Found (Calc. for C11H22NP) C, 66.7 (66.3); N, 6.9 (7.0); H, 11.3 (11.1) %. HRMS (EI): Found (Calc. for C11H22NP 199.1496 (199.1490).
Preparation of [PhobP(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) NHMe)PPhob]Cl (L′c·HCl)
NHMe)PPhob]Cl (L′c·HCl)
A solution of PhobPCl (0.530 g, 3.03 mmol) in CH2Cl2 (2.0 mL) was added in portions to a solution of La (0.510 g, 3.00 mmol)) and N-methylpyrrolidine (2.40 g, 28.2 mmol) in CH2Cl2 (2 mL). The reaction mixture was stirred for 3 h and then the solvent was removed under reduced pressure. The resulting white solid was recrystallised from hot MeCN to afford white crystals (0.43 g, 45%). 31P {1H} NMR (121 MHz; CDCl3) δ 46.5 (d, JPP = 407 Hz), −24.2 (d, JPP = 407 Hz). 1H NMR (400 MHz; CDCl3) δ 1.60–2.54 (28H, m, phobane), 2.66 (3H, dd, JHP = 12.3 Hz, JHP = 5.68 Hz, CH3N), 7.01 (1H, m, HCl). 13C {1H} NMR (100 MHz; CDCl3) δ 20.4 (d, JCP = 6.9 Hz), 20.7 (d, JCP = 6.9 Hz), 20.8 (d, JCP = 6.9 Hz), 21.2 (d, JCP = 1.5 Hz), 21.9 (d, JCP = 6.9 Hz), 24.9 (d, JCP = 6.1 Hz), 25.1 (d, JCP = 6.1 Hz), 26.2 (d, JCP = 6.1 Hz), 26.9 (d, JCP = 6.1 Hz), 27.3 (d, JCP = 6.1 Hz), 27.5 (dd, JCP = 9.2 Hz, JCP = 1.5 Hz) 27.7 (d, JCP = 3.8 Hz), 28.9 (t, JCP = 3.84 Hz), 32.7 (d, JCP = 14.6 Hz), 32.9 (d, JCP = 15.3 Hz). Elemental analysis: Found (Calc. for C17H32ClNP2) C, 59.1 (58.7); N, 3.8 (4.0); H, 9.4 (9.3) %. HRMS (EI): Found (Calc. for C17H32NP2) 312.2004 (312.2011).
Preparation of PhobP(NMe)PPhob (L′c)
To the solution of La (0.45 g, 2.6 mmol) in THF (3.0 mL), a 1.6 M solution of nBuLi (4.5 mL, 7.20 mmol) in hexane was added at −78 °C over 5 min. The reaction mixture was stirred at −78 °C for 40 min. PhobPCl (0.45 g, 2.6 mmol) in THF (2 mL) was added in portions to the cooled reaction mixture. The mixture was allowed to warm to room temperature and stirred for 2 h. The solvent was then removed under reduced pressure and the residue was triturated with diethyl ether (10 mL). The solid was filtered off and dried. satisfactory elemental analysis was not obtained and the product was used without further purification (0.52 g, 65%) 31P{1H} NMR (121 MHz, C6D6) δ 30.7 (d, JPP = 320 Hz), −32.3 (d, JPP = 320 Hz). 1H NMR (300 MHz, C6D6) δ 1.25–2.52 (14H, m, phobane), 2.62 (3H, dd, JHP = 14.5 Hz, JHP = 5.7 Hz, CH3N).
Preparation of PhobPNMePPh2 (Lf)
Aminophobane La (0.280 g, 1.64 mmol) and NEt3 (0.220 g, 2.17 mmol) were dissolved in MeCN (1 mL). To this stirred mixture, Ph2PCl (0.380 g, 2.12 mmol) was added dropwise over 5 min. The reaction mixture was stirred for 2 h to give a white solid, which was filtered off and recrystallised from hot MeCN to afford white needles (0.44 g, 69%). 31P {1H} NMR (121 MHz; CD2Cl2) NMR δ 57.6 (d, JPP = 80 Hz), 51.1 (d, JPP = 80 Hz). 1H NMR (270 MHz; CD2Cl2) δ 1.21–2.24 (14H, m, phobane), 2.32 (3H, dd, JHP = 4.3 Hz, JHP = 8.5 Hz CH3N), 7.05–7.22, 7.55–7.69 (10H, 2 m, ArH). 13C {1H} NMR (67 MHz; CD2Cl2) δ 21.8 (d, JCP = 1.5 Hz, CH2), 23.2 (d, JCP = 2.6 Hz, CH2), 28.7 (dd, JCP = 20.7 Hz, JCP = 11.9 Hz, CH3N), 30.2 (d, JCP = 17.1 Hz, CH2), 33.0 (dd, JCP = 16.6 Hz, JCP = 7.2 Hz, CH), 24.1 (t, JCP = 3.1 Hz, CH2), 128.2 (d, JCP = 5.7 Hz, CH), 128.4 (s, CH) 132.2 (d, JCP = 19.2 Hz, CH), 138.4 (dd, JCP = 16.6 Hz, JCP = 1.0 Hz, C). Elemental analysis: Found (Calc. for C21H27NP2) C, 71.8 (70.9); N, 4.5 (4.0); H, 7.8 (7.6) %. HRMS (EI): Found (Calc. for C21H27NP2) 355.1613 (355.1619).
Preparation of PhobPNMeP(o-Tol)2 (Lg)
Aminophobane La (0.200 g, 1.16 mmol) and NEt3 (0.240 g, 2.32 mmol) were dissolved in MeCN (1 mL). To this stirred mixture, (o-Tol)2PCl (0.580 g, 1.52 mmol) was added dropwise over 5 min. The reaction mixture was stirred for 16 h to give the white solid product, which was filtered off and recrystallized from hot MeCN to afford white needles (0.17 g, 38%). 31P {1H} NMR (121 MHz; CD2Cl2) δ 60.2, (d, JPP = 80 Hz), 36.8 (d, JPP = 80 Hz). 1H NMR (300 MHz; CD2Cl2) δ 1.44–2.59 (14H, m, phobane), 2.34 (3H, dd, JHP = 3.9 Hz, JHP = 8.9 Hz, CH3N), 2.35 (6H, s, CH3), 7.07–7.42 (8H, m, HAr). 13C {1H} (100 MHz; CD2Cl2) 21.0 (d, JCP = 5.3 Hz, CH3), 21.2 (d, JCP = 5.3 Hz, CH3), 21.7 (d, JCP = 1.5 Hz, CH2), 23.1 (d, JCP = 3.0 Hz, CH2), 24.3 (t, JCP = 2.8 Hz, CH2), 28.7 (dd, JCP = 20.7 Hz, JCP = 11.5 Hz, CH3N), 30.5 (dd, JCP = 16.9 Hz, JCP = 1.1 Hz CH2), 33.9 (dd, JCP = 16.1 Hz, JCP = 6.1 Hz, CH), 128.5 (s, CH) 125.5 (s, CH), 130.3 (d, JCP = 3.8 Hz, CH), 131.6 (d, JCP = 2.3 Hz, CH), 136.7 (dd, JCP = 17.6 Hz, JCP = 1.5 Hz, C), 141.7 (d, JCP = 26.9 Hz, C). Elemental analysis: Found (Calc. for C23H31NP2) C, 72.2 (72.0); N, 3.8 (3.6); H, 8.1 (8.1) %. HRMS (EI): Found (Calc. for C23H31NP2) 383.1931 (383.1932).
Preparation of [PhobP(NHiPr)PPh2]Cl (L′h·HCl)
Ph2PCl (0.90 g, 5.0 mmol) was added dropwise to a solution of Lb (0.83 g, 4.2 mmol) and N-methylpyrrolidine (0.39 g, 4.2 mmol) in CH2Cl2 (5 mL). The reaction mixture was stirred for 1 h and the solvent was then removed under reduced pressure. The resulting white solid was recrystallised from hot MeCN to yield white crystals (0.71 g, 44%) suitable for X-ray crystallography. 31P{1H} NMR (121 MHz; CDCl3) δ 50.0 (d, JPP = 338 Hz), 39.1 (d, JPP = 338 Hz). 1H NMR (400 MHz; CDCl3) δ 0.94 (6H, d, JHH = 6.2 Hz, CH3), 1.35 (1H, d, JHH = 6.4 Hz, CH), 1.74–2.94 (14H, m, phobane), 7.46–7.54, 7.64–7.77 (10H, 2 m, HAr). 13C{1H} NMR (100 MHz; CD2Cl2) δ 20.5 (d, JCP = 5.4 Hz), 20.9 (d, JCP = 4.6 Hz), 25.1 (d, JCP = 4.6 Hz), 26.6 (d, JCP = 5.3 Hz), 27.7 (d, JCP = 2.3 Hz), 28.1 (s), 46.6 (s), 128.5 (d, JCP = 8.5 Hz), 130.2 (d, JCP = 7.6 Hz), 131.4 (s), 134.9 (d, JCP = 2.3 Hz). HRMS (EI): Found (Calc. for C23H32NP2) 384.2004 (384.1999).
Preparation of [PhobP(NHiPr)P(o-Tol)2]Cl (L′i·HCl)
A solution of (o-Tol)2PCl (0.620 g, 2.51 mmol) in MeCN (5.0 mL) was added in portions to PhobPNH(iPr) (Lb) (0.530 g, 2.67 mmol) and N-methylpyrrolidine (3.20 g, 37.5 mmol) in MeCN (5 mL). The reaction mixture was stirred for 1 h and then the solvent was removed under reduced pressure. The remaining white solid was recrystallised from hot MeCN to yield white crystals (0.71 g, 62%). 31P {1H} NMR (161 MHz; CDCl3) δ 49.3 (d, JPP = 359 Hz), −39.8 (d, JPP = 359 Hz). 1H (400 MHz; CDCl3) δ 0.89 (6H, d, JHH = 6.36 Hz, CH3iPr), 1.65–2.40 (14H, m, phobane), 2.53 (6H, s, CH3), 7.20–7.40 (8H, m, HAr). 13C{1H} NMR (100 MHz; CDCl3) δ 19.5 (dd, JCP = 14.1 Hz, JCP = 5.4 Hz), 21.1 (d, JCP = 24.1 Hz), 23.8 (d, JCP = 4.6 Hz), 25.8 (d, JCP = 6.2 Hz), 26.8 (d, JCP = 10.9 Hz), 28.5 (d, JCP = 3.8 Hz), 28.9 (d, JCP = 4.6 Hz), 45.6 (d, JCP = 4.6 Hz), 123.8 (dd, JCP = 14.0 Hz, JCP = 3.8 Hz), 126.7 (s), 130.4 (d, JCP = 7.8 Hz), 135.9 (d, JCP = 7.0 Hz), 142.3 (dd, JCP = 31.9 Hz, JCP = 7.0 Hz). Elemental analysis: Found (Calc. for C25H36ClNP2) C, 67.0 (67.0); N, 3.5 (3.1); H, 8.5 (8.1)%.
Preparation of Cy2PNMePCy2 (L3)
Cy2PCl (2.79 g, 11.9 mmol) was dissolved in CH2Cl2 (6.0 mL). To this, a 2 M THF solution of MeNH2 (3.0 mL, 6.0 mmol) and NEt3 (1.30 g, 12.8 mmol) were added dropwise over 5 min. The resulting suspension was stirred for 16 h. The solvent was then removed under reduced pressure and the residue was redissolved in toluene (30 mL). The [Et3NH]Cl salt was filtered off to give a clear solution. The solvent was removed under reduced pressure to give the crude product, which was recrystallized from hot acetonitrile (0.77 g, 32%). 31P{1H} NMR (121 MHz; CDCl3) δ 86.8. 1H NMR (300 MHz; CDCl3) δ 1.10–1.60 (44H, m, CH and CH2), 2.64 (s, CH2), 27.2 (s, CH2), 29.5 (t, JCP = 9.2 Hz, CH3N). 13C{1H} NMR (300 MHz; CDCl3) δ 26.7 (d, JCP 36.9 Hz, CH3), 26.8 (s, CH2), 24.9 (s, CH2), 32.2 (t, JCP 8.6 Hz, CH), 45.7 (s, CH3). Elemental analysis: Found (Calc. for C25H47NP2) C, 70.4 (70.9); N, 2.8 (3.3); H, 11.4 (11.2) %. HRMS (CI): Found (Calc. for [C25H47NP2H]) 424.3262 (424.3262).
Preparation of trans-[PtCl2(PhobPNHMe)2] (1a)
PhobPNHMe (0.069 g, 0.45 mmol) was dissolved in CH2Cl2 (2.0 mL). [PtCl2(NCtBu)2] (0.086 g, 0.23 mmol) was added and the resulting yellow solution was stirred for 2 h. The solvent was reduced to ca. 1 mL and then hexane (4 mL) was added to give the yellow precipitated product (0.080 g, 57%). 31P{1H} NMR (121 MHz; CDCl3) δ 46.3 (JPtP = 2638 Hz). 1H NMR (300 MHz; CDCl3) δ 1.52–2.16 (28H, m, phobane), 2.98 (6H, t, JHP = 6.6 Hz, CH3N). 13C{1H} NMR (75 MHz; CDCl3) δ 21.3 (s), 22.1 (s), 24.2 (t, JCP = 6.6 Hz), 25.8 (s), 29.2 (s), 30.8 (s). Elemental analysis: Found (Calc. for C18H36Cl2N2P2Pt) C, 35.4 (35.5); H, 5.5 (6.0); N, 4.6 (5.0) %. HRMS (EI): Found (Calc. for [C18H36Cl2N2P2Pt]H) 608.1451 (608.1438).
Preparation of trans-[PtCl2(PhobPNHiPr)2] (1b)
PhobPNHiPr (0.18 g, 0.90 mmol) was dissolved in CH2Cl2 (2.0 mL). [PtCl2(NCtBu)2] (0.20 g, 0.46 mmol) was added and the resulting yellow solution was stirred for 2 h. The solvent was reduced to ca. 1 mL and then hexane (4 mL) was added to give the yellow precipitated product (0.26 g, 85%). 31P{1H} NMR (121 MHz; CDCl3) δ 40.4 (JPtP = 2646 Hz). 1H NMR (300 MHz; CDCl3) δ 1.56–2.15, 2.55–2.76 (28H, m, phobane), 1.31 (12H, d, JHH = 6.6 Hz, CH3iPr), 4.14–4.26 (2H, m, CH). 13C{1H} NMR (75 MHz; CDCl3) δ 21.4 (t, JCP = 2.3 Hz), 22.0 (t, JCP = 2.3 Hz), 25.4 (t, JCP = 24.3 Hz). Elemental analysis: Found (Calc. for C22H44Cl2N2P2Pt) C, 39.7 (39.7); N, 3.9 (4.2); H, 6.6 (6.6)%. MS (ESI): m/z 665.21 (M + 2H)+.
Preparation of trans-[Cr(CO)4(PhobPNHMe)2] (2a)
PhobPNHMe (0.048 g, 0.29 mmol) was dissolved in CH2Cl2 (4.0 mL). [Cr(CO)4(η4-norbornadiene)] (0.048 g, 0.15 mmol) was added and the resulting yellow solution was stirred for 24 h. The solvent was removed under reduced pressure and the resulting yellow solid product was washed with hexane (0.023 g, 26%). 31P{1H} NMR (121 MHz; CDCl3) δ 102.5. 1H NMR (300 MHz; CDCl3) δ 1.53–2.57 (28H, m, phobane), 2.70 (6H, d, JHH = 8.4 Hz, CH3N). 13C{1H} NMR (75 MHz; CDCl3) δ 21.5 (d, JCP = 52.2 Hz, CH3), 26.4 (s, CH2), 28.3 (s, CH2), 29.9 (t, JCP = 8.6 Hz, CH). HRMS (ES): Found (Calc. for C22H36CrN2O2P2) 506.1552 (506.1555). IR (CH2Cl2): v(CO) 1869, 1858 cm−1.
Preparation of trans-[Cr(CO)4(PhobPNHiPr)2] (2b)
PhobPNHiPr (0.078 g, 0.39 mmol) was dissolved in CH2Cl2 (4.0 mL). [Cr(CO)4(η4-norbornadiene)] (0.050 g, 0.19 mmol) was added and the resulting yellow solution was stirred for 24 h. The solvent was removed under reduced pressure and the resulting yellow solid product was washed with hexane (0.042 g, 36%). 31P{1H} NMR (121 MHz; CDCl3) δ 94.9. 1H NMR (300 MHz; CDCl3) d 1.24 (12H, d, JHH = 6.4 Hz, CH3iPr), 1.49–2.65 (12H, m, phobane). 13C{1H} NMR (75 MHz; CDCl3) δ 21.3 (d, JCP = 36.9 Hz, CH3), 26.8 (s, CH2), 24.9 (s, CH2), 32.2 (t, JCP = 8.6 Hz, CH), 45.7 (s, CH3). IR (CH2Cl2): v(CO) 1863 cm−1.
Preparation of [PtCl2(Cy2PNMePCy2)] (4)
To a solution of L3 (0.032 g, 0.070 mmol) in CH2Cl2 (2 mL), [PtCl2(NCtBu)2] (0.034 g, 0.070 mmol) in CH2Cl2 (2 mL) was added and the mixture was stirred for 2 h. The solvent was then reduced to ca. 2 mL, hexane was added (4 mL) and a yellow solid was obtained (0.023 g, 47%). 31P{1H} NMR (121 MHz; CDCl3) δ 37.1 (JPtP = 3211 Hz). 1H NMR (300 MHz; CDCl3) δ 1.20–1.97 (44H, m, CH and CH2), 2.72 (3H, t, JHP = 9.4 Hz, CH3N). 13C {1H} NMR (75 MHz; CDCl3) δ 25.7 (s), 26.9 (m) 28.7 (s), 27.9 (s), 38.0 (t, JCP = 15.2 Hz). Elemental analysis: Found (Calc. for C25H47Cl2NP2Pt) C, 43.3 (43.5); N, 2.3 (2.0); H, 6.8 (6.9) %. HRMS (EI): Found (Calc. for C25H47ClNP2Pt) 653.2529 (653.2520).
Preparation of [PtCl3(PhobP(NHMe)PPhob)] (5)
A mixture of L′c·HCl (0.025 g, 0.070 mmol) and [PtCl2(NCtBu)2] (0.034 g, 0.070 mmol) were dissolved in CH2Cl2 (5 mL) and stirred for 2 h to give a yellow solution. Warming this solution to 40 °C led to the slow formation of yellow crystals of the product 5 suitable for X-ray crystallography. Satisfactory elemental analysis was not obtained and the crystals were insoluble in common organic solvents which precluded further characterisation by NMR spectroscopy.
Preparation of [PdCl2(PhobPNMePPhob)] (6)
To a suspension of L′c (0.031 g, 0.099 mmol) in toluene (3 mL), [PdCl2(cod)] (0.031 g, 0.12 mmol) was added. The suspension was stirred at 50 °C for 5 min. The clear reaction mixture was then cooled to room temperature and the resulting yellow precipitate was filtered off and washed with hexane (0.010 g, 20%). Crystals suitable for X-ray crystallography were grown from CDCl3 although satisfactory elemental analysis was not obtained. 31P{1H} NMR (121 MHz; CDCl3) δ 37.0. 1H NMR (300 MHz; CDCl3) δ 1.65–2.40 (28H, m, phobane), 2.58 (d, JHP = 8.3 Hz). HRMS (EI): Found (Calc. for [C17H31NP2ClPd]+) 452.0672 (452.0649).
Preparation of [Cr(CO)4(PhobPNMePPh2)] (7)
To a solution of Lf (0.030 g, 0.070 mmol) in CH2Cl2 (2 mL), [Cr(CO)4(η4-norbornadiene)] (0.018 g, 0.070 mmol) in CH2Cl2 (2 mL) was added. The resulting yellow solution was stirred for 24 h. The solvent was then removed under reduced pressure to yield a yellow solid, which was washed with hexane (5 mL) (0.014 g, 27%). 31P{1H} NMR (121 MHz, CDCl3) δ 105.7 (d, JPP = 42 Hz), 108.1 (d, JPP = 42 Hz). 1H NMR (300 MHz, CDCl3) δ 1.50–2.65 (14H, m, phobane), 2.97 (3H, t, JHP = 6.9 Hz, CH3N), 7.33–7.59 (10H, m, HAr). 13C {1H}NMR (125 MHz; CD2Cl2) δ 20.3 (d, JCP = 7.8 Hz), 21.2 (d, JCP = 6.3 Hz), 23.4 (d, JCP = 4.4 Hz), 23.6 (d, JCP = 8.3 Hz), 29.6 (m), 34.7 (m), 126.1 (d, JCP = 10.7 Hz), 126.7 (d, JCP = 12.2 Hz) 132.7 (m). HRMS (EI): Found (Calc. for C25H27CrNO4P2) 519.0812 (519.0820). IR (in CH2Cl2): ν(CO) 2003, 1910, 1885, 1873 cm−1.
Preparation of [Cr(CO)4(PhobPNMeP(o-Tol)2)] (8)
To a solution of Lg (0.030 g, 0.060 mmol) in CH2Cl2 (2 mL), [Cr(CO)4(η4-norbornadiene)] (0.017 g, 0.060 mmol) in CH2Cl2 (2 mL) was added. The resulting yellow solution was stirred for 24 h. The solvent was then removed under reduced pressure to yield a yellow solid, which was washed with hexane (5 mL) (0.021 g, 30%). 31P{1H} NMR (121 MHz, CDCl3) δ 104.1 (d, JPP = 27 Hz), 104.6 (d, JPP = 27 Hz). 1H NMR (300 MHz, CDCl3) δ 1.24 (6H, s, CH3), 1.52–2.65 (14H, m, phobane), 2.97 (3H, dd, JHP = 7.4 Hz, JHP = 8.1 Hz, CH3N), 7.11–7.56 (8H, m, HAr). HRMS (EI): Found (Calc. for C27H31CrNO4P2) 547.1130 (547.1133). IR (in CH2Cl2): ν(CO) 2003, 1908, 1885, 1869 cm−1.
Oligomerisation catalysis
A rigorously cleaned autoclave was heated (130 °C) under vacuum for 60 min, then cooled to reaction temperature and back-filled with Ar (1 bar). Solvent was then added via syringe. The autoclave was pressurised with ethylene to 10 bar and vented. On a Schlenk line, a pre-activated catalyst solution was prepared by stirring the Cr source, ligand and modified methylaluminoxane (MMAO) together for 60 s, then transferred to the autoclave via syringe. The autoclave was pressurised and the pressure kept constant throughout the reaction by the continuous addition of ethylene, which was monitored via flow-meter. Once ethylene uptake had ceased or the autoclave was filled, the gas supply was closed and the reactor cooled to 5 °C. The reactor was carefully vented. The reactor contents were treated with 1000 μL of nonane (GC internal standard) and 10% HCl (aq). A sample of the organic phase was taken for GC-FID analysis. Any solid formed was collected, washed repeatedly with EtOH, then acetone and dried overnight and weighed.
GC-FID analysis was performed on an Agilent Technologies 6890N GC system equipped with PONA (50 m × 0.20 mm × 0.50 μm) and MDN-12 (60 m × 0.25 mm × 0.25 μm) columns. Catalysis was performed in a stainless steel 300 mL volume AE autoclave with Viton-ETP seals, equipped with a customised gas-entraining mechanical stirrer, internal cooling coil (tap water) and fluidised jacket (connected to an external thermostatic bath). Ethylene was passed through moisture and oxygen scrubbing columns prior to use and the flow measured using a Siemens Sitrans F C Massflo system (Mass 6000-Mass 2100) and the data logged.
Crystal structure determinations
X-ray diffraction experiments for 1b, 3, 4, 5, 6 and Lh·HCl were carried out at 100 K and for 2b at 173 K on a Bruker APEX II diffractometer using Mo-Kα radiation (λ = 0.71073 Å). 7 was collected at 120 K on a Bruker Nonius FR591 Rotating Anode using Mo-Kα radiation (λ = 0.71073 Å)25 and 8 was collected on EH1 of Station I19 of Diamond Light Source (λ = 0.71073 Å) at 120 K.26 Data collections were performed using a CCD area detector from a single crystal mounted on a glass fibre. Intensities were integrated using SAINT with a multi-scan absorption correction preformed using SADABS.27 All structures were solved using SHELXS and refined against all Fo2 using SHELXL and OLEX2.28 All non-hydrogen atoms were refined anisotropically and hydrogen atoms were located geometrically and refined using a riding model. The structure of 4 was refined as a racemic twin and restraints were applied to the thermal displacement parameters to maintain sensible values. Crystal structure and refinement data are given in Table 4. The structures are shown in Fig. 1–8 with thermal ellipsoids drawn at the 50% probability level.
Table 4 Crystal data and structure refinement for all structures in the paper
| Identification code |
1b_LR302 |
2b_jj142 |
3_lr305 |
4_jj228 |
5_jj200 |
| Empirical formula |
C22H44Cl2N2P2Pt |
C26H44CrN2O4P2 |
C30H60Cl2N2P2Pt |
C27H49NP2Cl8Pt |
C19.5H37Cl6NP2Pt |
| Formula weight |
664.52 |
562.57 |
776.72 |
928.30 |
755.23 |
| Temperature/K |
99.99 |
173(2) |
100.0 |
99.99 |
100.0 |
| Crystal system |
Monoclinic |
Triclinic |
Triclinic |
Orthorhombic |
Hexagonal |
| Space group |
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
Pna21 |
P65 |
|
a/Å |
9.9057(7) |
9.5739(2) |
9.448(4) |
21.9631(6) |
16.7268(3) |
|
b/Å |
7.2983(5) |
12.2783(2) |
9.493(5) |
27.7581(8) |
16.7268(3) |
|
c/Å |
17.7434(13) |
13.2759(2) |
10.788(5) |
17.7763(5) |
17.0407(4) |
|
α/° |
90 |
69.2670(10) |
109.190(10) |
90 |
90.00 |
|
β/° |
92.139(2) |
89.1370(10) |
107.174(9) |
90 |
90.00 |
|
γ/° |
90 |
71.601 (10) |
93.167(15) |
90 |
120.00 |
| Volume/Å3 |
1281.86(16) |
1376.68(4) |
860.5(7) |
10837.4(5) |
4128.99(14) |
|
Z
|
2 |
2 |
1 |
12 |
6 |
|
ρ
calc/g cm−3 |
1.722 |
1.357 |
1.499 |
1.707 |
1.822 |
|
μ/mm−1 |
5.819 |
0.566 |
4.346 |
4.585 |
5.806 |
|
F(000) |
664.0 |
600.0 |
396.0 |
5544.0 |
2226.0 |
| Crystal size/mm3 |
0.6 × 0.25 × 0.1 |
0.16 × 0.15 × 0.08 |
0.4 × 0.25 × 0.2 |
0.1 × 0.21 × 0.27 |
0.3 × 0.1 × 0.1 |
| Radiation |
MoKα (λ = 0.71073) |
MoKα (λ = 0.71073) |
MoKα (λ = 0.71073) |
MoKα (λ = 0.71073) |
Mo Kα (λ = 0.71074) |
| 2Θ range for data collection/° |
4.594 to 66.432 |
6.328 to 61.144 |
4.238 to 66.332 |
2.72 to 55.062 |
3.7 to 68.3 |
| Index ranges |
−15 ≤ h ≤ 15, −11 ≤ k ≤ 10, −27 ≤ l ≤ 26 |
−13 ≤ h ≤ 13, −17 ≤ k ≤ 17, −18 ≤ l ≤ 18 |
−14 ≤ h ≤ 14, −14 ≤ k ≤ 14, −16 ≤ l ≤ 16 |
−28 ≤ h ≤ 28, −36 ≤ k ≤ 36, −23 ≤ l ≤ 23 |
−26 ≤ h ≤ 26, −26 ≤ k ≤ 26, −17 ≤ l ≤ 26 |
| Reflections collected |
46419 |
56825 |
36290 |
300715 |
137345 |
| Independent reflections |
4710 [Rint = 0.0365, Rsigma = 0.0203] |
8447 [Rint = 0.0291, Rsigma = 0.0188] |
6208 [Rint = 0.0186, Rsigma = 0.0126] |
24919 [Rint = 0.0572, Rsigma = 0.0279] |
9590 [Rint = 0.0716, Rsigma = 0.0407] |
| Data/restraints/parameters |
4710/0/139 |
8447/0/328 |
6208/0/175 |
24919/955/1060 |
9590/1/290 |
| Goodness-of-fit on F2 |
1.078 |
1.055 |
1.075 |
1.085 |
1.155 |
| Final R indexes [I ≥ = 2σ (I)] |
R
1 = 0.0304, wR2 = 0.0717 |
R
1 = 0.0282, wR2 = 0.0745 |
R
1 = 0.0167, wR2 = 0.0426 |
R
1 = 0.0561, wR2 = 0.1363 |
R
1 = 0.0278, wR2 = 0.0668 |
| Final R indexes [all data] |
R
1 = 0.0357, wR2 = 0.0745 |
R
1 = 0.0332, wR2 = 0.0777 |
R
1 = 0.0167, wR2 = 0.0426 |
R
1 = 0.0846, wR2 = 0.1581 |
R
1 = 0.0367, wR2 = 0.0909 |
| Largest diff. peak/hole/e Å−3 |
4.08/−1.91 |
0.45/−0.55 |
1.34/−1.00 |
4.17/−2.14 |
1.38/−1.76 |
| Flack parameter |
— |
— |
— |
— |
−0.021(6) |
| Identification code |
6_jj380 |
Lh·HCl _jj353 |
7_jj68 |
8_jj82 |
| Empirical formula |
C17H31Cl2NP2Pd |
C23H32ClNP2 |
C25H27CrNO4P2 |
C27H31CrNO4P2 |
| Formula weight |
488.67 |
419.89 |
519.41 |
547.47 |
| Temperature/K |
100(2) |
100(2) |
120(2) |
120.0 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Orthorhombic |
| Space group |
P21/c |
P21/c |
P21/c |
P212121 |
|
a/Å |
11.9640(3) |
16.8271(8) |
11.5207(3) |
10.05490(10) |
|
b/Å |
12.5989(4) |
8.5130(4) |
13.1975(5) |
10.8831(2) |
|
c/Å |
13.8207(4) |
15.6725(8) |
16.7912(6) |
23.6863(4) |
|
α/° |
90.00 |
90.00 |
90 |
90 |
|
β/° |
112.2130(10) |
100.569(2) |
106.502(2) |
90 |
|
γ/° |
90.00 |
90.00 |
90 |
90 |
| Volume/Å3 |
1928.63(10) |
2206.98(19) |
2447.85(14) |
2591.96(7) |
|
Z
|
4 |
4 |
4 |
4 |
|
ρ
calc/g cm−3 |
1.683 |
1.264 |
1.409 |
1.403 |
|
μ/mm−1 |
1.404 |
0.327 |
0.630 |
0.599 |
|
F(000) |
1000.0 |
896.0 |
1080.0 |
1144.0 |
| Crystal size/mm3 |
0.26 × 0.11 × 0.08 |
0.35 × 0.28 × 0.28 |
0.1 × 0.06 × 0.02 |
0.24 × 0.2 × 0.15 |
| Radiation |
MoKα (λ = 0.71073) |
MoKα (λ = 0.71073) |
MoKα (λ = 0.71073) |
MoKα (λ = 0.71073) |
| 2Θ range for data collection/° |
3.68 to 55.08 |
2.46 to 55.08 |
5.902 to 55.066 |
6.376 to 55.032 |
| Index ranges |
−13 ≤ h ≤ 15, −12 ≤ k ≤ 16, −17 ≤ l ≤ 17 |
−21 ≤ h ≤ 21, −8 ≤ k ≤ 11, −19 ≤ l ≤ 20 |
−13 ≤ h ≤ 14, −16 ≤ k ≤ 17, −21 ≤ l ≤ 21 |
−12 ≤ h ≤ 13, −14 ≤ k ≤ 14, −30 ≤ l ≤ 30 |
| Reflections collected |
19685 |
19574 |
25383 |
30546 |
| Independent reflections |
4437 [Rint = 0.0231, Rsigma = 0.0191] |
5066 [Rint = 0.0194, Rsigma = 0.0169] |
5589 [Rint = 0.1046, Rsigma = 0.1035] |
5929 [Rint = 0.0548, Rsigma = 0.0494] |
| Data/restraints/parameters |
4437/0/220 |
5066/1/249 |
5589/0/299 |
5929/0/319 |
| Goodness-of-fit on F2 |
1.040 |
1.034 |
1.082 |
1.041 |
| Final R indexes [I ≥ = 2σ (I)] |
R
1 = 0.0178, wR2 = 0.0446 |
R
1 = 0.0277, wR2 = 0.0707 |
R
1 = 0.0979, wR2 = 0.1670 |
R
1 = 0.0328, wR2 = 0.0712 |
| Final R indexes [all data] |
R
1 = 0.0198, wR2 = 0.0458 |
R
1 = 0.0300, wR2 = 0.0724 |
R
1 = 0.1546, wR2 = 0.1940 |
R
1 = 0.0404, wR2 = 0.0742 |
| Largest diff. peak/hole/e Å−3 |
0.90/−0.30 |
0.43/−0.23 |
0.55/−0.58 |
0.27/−0.36 |
| Flack parameter |
— |
— |
— |
0.020(10) |
Acknowledgements
We thank Sasol for a Ph.D. studentship (JJ). We thank the Bristol Chemical Synthesis Centre for Doctoral Training, funded by EPSRC (EP/G036764/1), and the University of Bristol, for a Ph.D. studentship (LR).
References
-
(a) M. F. Haddow, A. J. Middleton, A. G. Orpen, P. G. Pringle and R. Papp, Dalton Trans., 2009, 202–209 RSC and references therein;
(b) J. A. Gillespie, D. L. Dodds and P. C. J. Kamer, Dalton Trans., 2010, 39, 2751–2764 RSC;
(c)
F. Matthey, in Phosphorus-Carbon Heterocyclic Chemistry, Elsevier, 2001 Search PubMed.
- L. Kollár and G. Keglevich, Chem. Rev., 2010, 110, 4257–4302 CrossRef PubMed.
- D. L. Dodds, J. Floure, M. Garland, M. F. Haddow, T. R. Leonard, C. L. McMullin, A. G. Orpen and P. G. Pringle, Dalton Trans., 2011, 40, 7137–7146 RSC and references therein.
- For recent examples see:
(a) C. Schotten, D. Plaza, S. Manzini, S. P. Nolan, S. V. Ley, D. L. Browne and A. Lapkin, ACS Sustainable Chem. Eng., 2015, 3, 1453–1459 CrossRef CAS PubMed;
(b) D. Schweitzer and K. D. Snell, Org. Process Res. Dev., 2015, 19, 715–720 CrossRef CAS;
(c) S. Manzini, A. Poater, D. J. Nelson, L. Cavallo, A. M. Z. Slawin and S. P. Nolan, Angew. Chem., Int. Ed., 2014, 53, 8995–8999 CrossRef CAS PubMed;
(d) S. Manzini, D. J. Nelson, T. Lebl, A. Poater, L. Cavallo, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2014, 50, 2205–2207 RSC;
(e) J. A. Bailey, M. F. Haddow and P. G. Pringle, Chem. Commun., 2014, 50, 1432–1434 RSC;
(f) S. Raoufmoghaddam, E. Drent and E. Bouwman, Adv. Synth. Catal., 2013, 355, 717–733 CrossRef CAS;
(g) M. Czapiewski, O. Kreye, H. Mutlu and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2013, 115, 76–85 CrossRef;
(h) A. Behr, S. Krema and A. Kämper, RSC Adv., 2012, 2, 12775–12781 RSC;
(i) M. Yoshida, T. Nemoto, Z. Zhao, Y. Ishige and Y. Hamada, Tetrahedron: Asymmetry, 2012, 23, 859–866 CrossRef CAS;
(j) D. M. Ohlmann, N. Tschauder, J.-P. Stockis, K. Gooβen, M. Dierker and L. Gooβen, J. Am. Chem. Soc., 2012, 134, 13716–13729 CrossRef CAS PubMed.
-
(a)
J. P. Mulders, Neth. Patent, 660409 to Shell, 1966 Search PubMed;
(b)
J. L. V. Winkle and R. F. Mason, U. S. Patent, 3400163 to Shell, 1968 Search PubMed;
(c)
J. L. V. Winkle, R. C. Morris and R. F. Mason, Ger. Patent, 1909620 to Shell, 1969 Search PubMed;
(d) P. N. Bungu and S. Otto, Dalton Trans., 2007, 2876 RSC;
(e) J. M. Birbeck, A. Haynes, H. Adams, L. Damoesense and S. Otto, ACS Catal., 2012, 2, 2512–2523 CrossRef CAS;
M. de Boer-Wildschut, M. Charernsuk, C. A. Krom and P. G. Pringle, World Patent, WO2012072594, 2012 Search PubMed.
- N. Fey, M. Garland, J. P. Hopewell, C. L. McMullin, S. Mastroianni, A. G. Orpen and P. G. Pringle, Angew. Chem., Int. Ed., 2012, 51, 118–122 CrossRef CAS PubMed.
- K. Blann, A. Bollmann, H. de Bod, J. T. Dixon, E. Killian, P. Nongodlwana, M. C. Maumela, H. Maumela, A. E. McConnell, D. H. Morgan, M. J. Overett, M. Prétorius, S. Kuhlmann and P. Wasserscheid, J. Catal., 2007, 249, 244–249 CrossRef CAS.
- A. Bollmann, K. Blann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. S. McGuinness, D. H. Morgan, A. Neveling, S. Otto, M. Overett, A. M. Z. Slawin, P. Wasserscheid and S. Kuhlmann, J. Am. Chem. Soc., 2004, 126, 14712–14713 CrossRef CAS PubMed.
-
(a) D. F. Wass, Dalton Trans., 2007, 816–819 RSC;
(b) P. W. N. M. van Leeuwen, N. D. Clement and M. J.-L. Tschan, Coord. Chem. Rev., 2011, 255, 1499–1517 CrossRef CAS;
(c) D. S. McGuinness, Chem. Rev., 2011, 111, 2321 CrossRef CAS PubMed;
(d) T. Agapie, Coord. Chem. Rev., 2011, 255, 861 CrossRef CAS;
(e) G. P. Belov, Pet. Chem., 2012, 52, 139–154 CrossRef CAS;
(f) G. J. P. Britovsek, R. Malinowski, D. S. McGuiness, J. D. Nobbs, A. K. Tomov, A. W. Wadsley and C. T. Young, ACS Catal., 2015, 5, 6922–6925 CrossRef CAS.
-
(a) M. C. Maumela, K. Blann, H. de Bod, J. T. Dixon, W. F. Gabrielli and D. B. G. Williams, Synthesis, 2007, 3863–3867 CrossRef CAS and references therein;
(b) K. G. Gaw, M. B. Smith, J. B. Wright, A. M. Z. Slawin, S. J. Coles, M. B. Hursthouse and G. J. Tizzard, J. Organomet. Chem., 2012, 699, 39–47 CrossRef CAS and references therein.
- L. E. Bowen, M. Charernsuk, T. W. Hey, C. L. McMullin, A. G. Orpen and D. F. Wass, Dalton Trans., 2010, 39, 560–567 RSC.
- J. M. Lister, M. Carreira, M. F. Haddow, A. Hamilton, C. L. McMullin, A. G. Orpen, P. G. Pringle and T. E. Stennett, Organometallics, 2014, 33, 702–714 CrossRef CAS.
- M. Carreira, M. Charernsuk, M. Eberhard, N. Fey, R. van Ginkel, A. Hamilton, W. P. Mul, A. G. Orpen, H. Phetmung and P. G. Pringle, J. Am. Chem. Soc., 2009, 131, 3078–3092 CrossRef CAS PubMed.
-
(a) A. Roodt, S. Otto and G. Steyl, Coord. Chem. Rev., 2003, 245, 121–137 CrossRef CAS;
(b) M. L. Clarke, D. J. Cole-Hamilton, A. M. Z. Slawin and J. D. Woollins, Chem. Commun., 2000, 2065–2066 RSC.
- M. R. Eberhard, E. Carrington-Smith, E. E. Drent, P. S. Marsh, A. G. Orpen, H. Phetmung and P. G. Pringle, Adv. Synth. Catal., 2005, 347, 1345–1348 CrossRef CAS.
- I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779–2786 CrossRef CAS.
- Z. Fei, R. Scopelliti and P. J. Dyson, Eur. J. Inorg. Chem., 2004, 530–534 CrossRef CAS.
- P. Boulens, M. Lutz, E. Jeanneau, H. Olivier-Bourbigou, J. N. H. Reek and P.-A. R. Breuil, Eur. J. Inorg. Chem., 2014, 3754–3762 CrossRef CAS.
- V. L. Foss, Y. A. Veits, T. E. Chernykh and I. F. Lutsenko, Zh. Obshch. Khim., 1984, 54, 2670–2684 CAS.
-
(a) Z. Fei, R. Scopelliti and P. J. Dyson, Dalton Trans., 2003, 2272–2779 Search PubMed;
(b) Z. Fei, N. Biricik, D. Zhao, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2004, 43, 2228–2230 CrossRef CAS PubMed.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- M. A. Bennett, L. Pratt and G. Wilkinson, J. Chem. Soc., 1961, 2037 RSC.
- D. Fraccarollo, R. Bertani, M. Mozzon, U. Belluco and R. A. Michelin, Inorg. Chim. Acta, 1992, 201, 15–22 CrossRef CAS.
- D. Drew and J. R. Doyle, Inorg. Synth., 1991, 28, 346–349 CrossRef.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- H. Nowell, S. A. Barnett, K. E. Christensen, S. J. Teat and D. R. Allan, J. Synchrotron Radiat., 2012, 19, 435–441 CrossRef CAS PubMed.
-
Bruker-Nonius, SAINT version 7.32A, 2006, Bruker-AXS, Madison, Wisconsin, USA Search PubMed;
G. M. Sheldrick, SADABS V2008/1, University of Göttingen, Germany Search PubMed.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed;
(b) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a