
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 3-stannyl and 3-silyl propargyl phosphanes and the formation of a phosphinoallene†
Amy J.
Saunders
and
Ian R.
Crossley
*
Department of Chemistry, University of Sussex, Brighton, UK. E-mail: i.crossley@sussex.ac.uk; Fax: +44 (0)1273 876687; Tel: +44 (0)1273 877302
First published on 4th November 2015
Abstract
The group 14 chloropropargyls R3EC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2Cl (R3E = nBu3Sn, Ph3Sn, Me2PhSi, iPr3Si, nPr3Si, nBu3Si), obtained by a modified literature procedure, react with LiPPh2 to afford the novel propargyl phosphanes Ph2PCH2CCER3 in high yield, as viscous oils; (Me3Si)2PCH2CCSiPhMe2 is similarly obtained from LiP(SiMe3)2. In contrast, the reaction of PhCCCH2MgCl with ClP(NEt2)2 fails to produce a comparable propargyl phosphane, but generates preferentially (>70%) the novel phosphinoallene (Et2N)2PC(Ph)
CCH2Cl (R3E = nBu3Sn, Ph3Sn, Me2PhSi, iPr3Si, nPr3Si, nBu3Si), obtained by a modified literature procedure, react with LiPPh2 to afford the novel propargyl phosphanes Ph2PCH2CCER3 in high yield, as viscous oils; (Me3Si)2PCH2CCSiPhMe2 is similarly obtained from LiP(SiMe3)2. In contrast, the reaction of PhCCCH2MgCl with ClP(NEt2)2 fails to produce a comparable propargyl phosphane, but generates preferentially (>70%) the novel phosphinoallene (Et2N)2PC(Ph)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2, which is characterised spectroscopically, and through its reaction with HCl. The coordination chemistry of representative phosphanes is explored with respect to platinum and palladium for the first time.
CCH2, which is characterised spectroscopically, and through its reaction with HCl. The coordination chemistry of representative phosphanes is explored with respect to platinum and palladium for the first time.
Introduction
Tertiary phosphanes are both ubiquitous and innumerable, being the subject of exhaustive efforts to control steric and electronic profiles through substituent modification, driven by their utility as ligands. The opportunities to impose steric bulk and/or asymmetry within the metal coordination sphere offer particular impetus, typically directed toward symmetric R3P and chiral PRR′R′′ derivatives respectively. Equally important are derivatives of the type R2PR′ (R = aryl, alkyl) that occupy the intermediate ground, allowing for subtle variation of sterics and electronics (variation of R′), while also imposing some level of asymmetry about the metal. Moreover, the ready availability of R2PX (X = halide, H) renders a convenient scaffold with which to investigate more elaborate and functional substituents (R′).Despite prolific levels of activity in phosphane synthesis,1 particularly systems of the type R2PR′, surprising oversights remain, a case in point being the dearth of systems bearing a propargylic substituent (viz. CH2CCR′). Indeed, while alkynyl phosphanes are common,2 their propargyl counterparts are limited to R2PCH2CCR′ (R = Ph, R′ = H,3 Me,4 Ph;5 R = Cy, iPr, R′ = H, SiMe3;6), typically isolated as stabilised BH3 adducts, R2PCH{OSiMe3}CCPh (R = Ph, Et),7 {(Me3Si)2N}RPCH2CCSiMe3 (R = Ph,8 Et,8 Cl9), the diphosphane Ph2PCH2CCCH2PPh2,10 and the bis-propargylphosphanes RP{CH2CCR′}2 (R = Np, R′ = H, SiMe3;11 R = CH2CMe2Et, R′ = H;11 R = Ph, R′ = nBu;12 R = Mes, R′ = SiMe313), which are typically putative intermediates in the synthesis of macrocycles. The primary propargyl phosphane H2PCH2CCH has also been reported.14
This lack of activity is surprising given continued interest in developing polyfunctional phosphorus-containing molecules, driven by their utility as ligands, optoelectronically active π-conjugates15 and, topically, frustrated Lewis pairs (FLPs).16 In these contexts, propargylphosphanes should constitute ideal ‘building-block’ substrates, and allow for incorporation of further functionality (e.g. by cycloaddition, hydroboration, hydrophosphination) akin to their more extensively utilised alkynyl, alkenyl and allyl counterparts. Moreover, they embody intrinsic potential to act as σ/π-chelating ligands. Indeed, among very limited coordination chemistry reported to date, the μ-(σ-P,π-CC) bridging mode has been described for [Cp2Rh2(CO)(μ–η1:η1-CF3C2CF3){PPh2CH2CCMe}CO2(CO)6], obtained by reaction of [Co2(CO)8] with the dirhodium complex [Cp2Rh2(CO)(μ–η1:η1-CF3C2CF3){PPh2CH2CCMe}].4
The remaining complexes described to date involve mono-dentate coordination of the phosphane, typically to metals of the mid transition series, with saturated coordination spheres. Thus, [M(CO)5(PR2CCH)] (M = Mo, R = Ph,17 DBP;18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ‡ Cr,19 R = Ph, SiMe3), [Mo(CO)3(PH2CH2CCH)3], [Mo(CO)4(PH2CCH)20] and [CpRMn(CO)2(PPh3−n(CH2CCH)n] (CpR = Cp, n = 1, 2; CpR = CpMe, n = 1).19 have been obtained directly from the respective phosphanes and suitable metal salts, as has the bimetallic complex [{HCCCH2Ph2P}Ru(CO)3(μ-PPh2)Co(CO)3].21 In contrast, [Co(NO)(CO)(PPh2CH2CCH)2]22 and the ruthenium phthalocyaninato (Pc2−) complex [Ru(Pc)(PPh2CH2CCCH3)2]23 are obtained from the respective diphenylphosphane complexes, via in situ deprotonation (BuLi) and quenching with the appropriate propargylic bromide; [CpMo{κ2-O,P-P(O)Mes*(CH2CCH)}] is similarly prepared, but without need for base.24 Finally, [W(CO)5{PPh(OMe)C(H)Me(CCSiiPr3)}] was obtained upon methanolysis of the putative phosphaalkene [W(CO)5{P(Ph)CMe(CCSiiPr3)}].25
‡ Cr,19 R = Ph, SiMe3), [Mo(CO)3(PH2CH2CCH)3], [Mo(CO)4(PH2CCH)20] and [CpRMn(CO)2(PPh3−n(CH2CCH)n] (CpR = Cp, n = 1, 2; CpR = CpMe, n = 1).19 have been obtained directly from the respective phosphanes and suitable metal salts, as has the bimetallic complex [{HCCCH2Ph2P}Ru(CO)3(μ-PPh2)Co(CO)3].21 In contrast, [Co(NO)(CO)(PPh2CH2CCH)2]22 and the ruthenium phthalocyaninato (Pc2−) complex [Ru(Pc)(PPh2CH2CCCH3)2]23 are obtained from the respective diphenylphosphane complexes, via in situ deprotonation (BuLi) and quenching with the appropriate propargylic bromide; [CpMo{κ2-O,P-P(O)Mes*(CH2CCH)}] is similarly prepared, but without need for base.24 Finally, [W(CO)5{PPh(OMe)C(H)Me(CCSiiPr3)}] was obtained upon methanolysis of the putative phosphaalkene [W(CO)5{P(Ph)CMe(CCSiiPr3)}].25
Notably, no complexes of the group 10 metals have been described, though the formally related diphosphane-bridged complexes [LnM{μ–η1:η1-PPh2CH2CCCH2PPh2}]2 (LnM = Cl2Pt, (OC)2Ni) have been reported,26 alongside examples with other metals (LnM = AuCl, CpCoI2, CpFe(CO)2+, CpFeBr(CO), CpMn(NO)(CO), CpMo(CO)2(COCH3),26 Mo(CO)427). The intriguing tetrameric complex [{η2-C,C-Mo(CO)4(η2-P,P-PPh2CH2CCCH2PPh3)}3Mo(CO)], has also been described.27
We have recently been interested in the synthesis and study of reactive and functional phosphanes28 and organometallic phosphacarbons,29 with the goal of developing novel ambiphilic systems28c and molecular conductive and/or optoelectronically active molecules.29b In continuing these works, we have had cause to access propargyl phosphanes of the type R2P(CH2CCER′3) (E = Si, Sn) as intermediates, seeking to exploit their capacity for desilylative/destannylative functionalisation. In view of the limited range of propargyl phosphanes reported previously, we thus undertook to prepare a putative series of such materials; viz. Ph2P(CH2CCER3) (E = Si, Sn), which we describe herein, along with attempts to obtain ‘(Et2N)2P’ derivatives, leading to the generation of a novel, and very rare, phosphinoallene. We also outline the coordination chemistry of representative propargylphosphanes toward Pd and Pt, reporting the first such complexes from group 10, and the first to involve coordinately unsaturated metal centres.
Results and discussion
Phosphane synthesis
The silyl and stannyl chloropropargyl precursors R3ECCCH2Cl were prepared following a modified literature procedure (Scheme 1),30via the low-temperature (−78 °C) lithiation of HCCCH2Cl, quenched with R3SnCl (1 and 2) or R3SiCl (3–7). The silanes were amenable to purification by reduced-pressure distillation, apart from the solid 7 (R = Ph), which was sublimed. However, both silanes and stannanes are typically obtained in adequate purity for further reaction (>95%) upon extraction with pentane. In each case, compound identity was apparent from the 1H NMR spectra, which exhibit resonances associated with the group 14 fragment, integrating consistently against that of the propargyl methylene moiety (δH 3.5–3.7), which is shifted by ca. 0.3 ppm to lower frequency compared with propargyl chloride. Moreover, correlations are observed between the methylenic resonances and respective group 14 centre in each case (1H–X HMBC; X = 119Sn, 29Si); for the stannanes the 4JSnH coupling (∼10 Hz) is also large enough to resolve tin satellites. The 13C{1H}-NMR data are similarly consistent, while bulk purity was confirmed from microanalytical data. It is noted that 1,312,30,31332 and 433 have been previously obtained via alternate methodology.
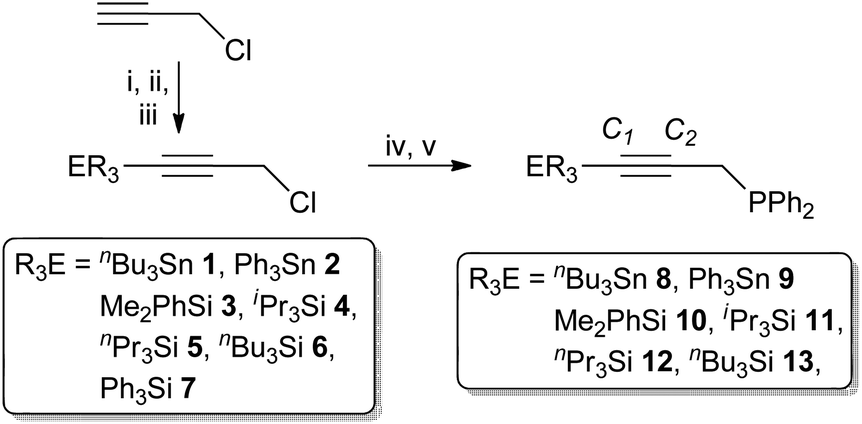 | ||
| Scheme 1 Reagents and conditions: (i) nBuLi, −78 °C, Et2O, 30 min; (ii) R3ECl, −78 °C, 30 min; (iii) warm to ambient, stir 18 h; (iv) LiPPh2, −78 °C, Et2O, 30 min; (v) r.t. 18 h. | ||
Ethereal solutions of 1 to 6 were added (−78 °C) to LiPPh2 in ether (formed by in situ lithiation of HPPh2 with nBuLi) and the mixtures stirred overnight to afford the propargyl phosphanes Ph2P(CH2CCER3) (8–13, Scheme 1). Extraction with pentane afforded the phosphanes as viscous oils, the silyl derivatives 10–13 requiring no further purification. In contrast, stannanes formed in admixture with nBu4Sn (1:4 of 8) or nBuPh3Sn (1:1 with 9), presumably due to metathesis of 1 and 2 with residual nBuLi, as is common among Sn(IV) organyls.34 Both 8 and 9 are unstable toward distillation and were thus only characterised spectroscopically, though for 8, further data were obtained by coordination to platinum (vide infra), which proceeds cleanly. In contrast, 9 forms in a complex, inseparable mixture that includes unidentified by-products; it has not been studied further.
Compounds 8 to 13 are identified from characteristic spectroscopic data (Table 1), the alkynic moieties exhibiting marginal change from those of the parent propargyls. Retention of the group 14 fragments is universally apparent (1H–X HMBC), with 8 and 9 also allowing for resolution of 119Sn satellites (4JSnP ∼ 14 Hz) in the 31P{1H} spectra. The 119Sn spectra of 8 and 9 indicate the presence of nBu4Sn (δSn −12.0)35 and nBuPh3Sn (δSn −98.3)36 by-products respectively.
| δ P | δ H (CH2)[JPH]b | δ C (C1) | δ C (C2) | δ E (E) | |
|---|---|---|---|---|---|
| a As C6D6 solutions. b Couplings in Hz. | |||||
| 8 | −13.4 | 2.87 [1.6] | 85.0 | 107.0 | −68.4 [119Sn] |
| 9 | −13.2 | 2.84 [3.0] | 82.8 | 109.0 | −168.4 [119Sn] |
| 10 | −13.5 | 2.76 [2.9] | 84.7 | 105.0 | −22.9 [29Si] |
| 11 | −13.5 | 2.75 [2.3] | 83.3 | 105.0 | −3.03 [29Si] |
| 12 | −13.6 | 2.76 [2.5] | 85.4 | 103.0 | −14.5 [29Si] |
| 13 | −13.5 | 2.76 [2.3] | 85.5 | 104.0 | −12.9 [29Si] |
| 14 | −158.9 | 2.43 [1.3] | 83.3 | 109.3 | −22.8; 3.7 [29Si] |
Attempts to vary the nature of the phosphanyl substituents met with limited success. Dicyclohexyl analogues failed to form, regenerating HPCy2 as the only phosphorus-containing product, which presumably reflects the greater basicity and steric bulk of ‘PCy2’ (cf. ‘PPh2’), favouring proton-abstraction from the chloropropargyls over SN2 substitution. In contrast, reactions with LiP(SiMe3)2 did afford species consistent with the desired propargylphosphanes, though in admixture with several significant contaminants, which defied separation or characterisation. Nonetheless, Me2PhSiCCCH2P(SiMe3)2 (14) was obtained as the primary product (92% by 31P{1H}-NMR) in admixture with P(SiMe3)3 (4%) and a mono-silylphosphane (δP −84.4; 4%), which presumably result from disproportionation; indeed, the bulk composition is consistent with that of 14.
Given these difficulties, the generation of propargyl Grignard reagents from 1 to 7 was considered as an alternative approach; however, these reactions proved unreliable, presumably reflecting diminished reactivity of the halide in comparison to organo-propargyl derivatives. Indeed, though less favoured than their bromide analogues, propargyl chlorides have been shown to form Grignard reagents,37 and we encountered no difficulty is generating ‘PhCCCH2MgCl’ under comparable conditions. However, our efforts to quench this reagent with (Et2N)2PCl led to an unexpected outcome.
Formation of a phosphino-allene
The addition of freshly prepared ‘PhCCCH2MgCl’ to a cooled (−78 °C) THF solution of (Et2N)2PCl affords, after work-up, a deep red oil comprising one predominant phosphorus-containing product (15; 75%). The spectroscopic features of 15 confirm the presence of a ‘(Et2N)2P’ moiety (δP 90.9; cf. PhP(NEt2)2 97.9,38 H2CC(H)–P(NEt2)2 89.939), the 1H NMR resonances integrating consistently against those for single equivalences of aromatic and methylenic fragments. However, the methylenic moiety is significantly deshielded (δH 4.72. δC 75.0) relative to both PhCCCH2Cl (δH 4.39, δC 31.2) and propargyl phosphanes, and exhibits appreciably greater magnitude coupling to phosphorus (|JPH| = 7.1 Hz) than 8–14. The unsaturated carbon centres are also heavily deshielded (δc 137.4 (JPC 19.0 Hz) C1; 209.9 (JPC 11.3 Hz) C2), the latter in particular being more characteristic of an allenic,40 rather than alkynic centre; indeed, these data are in good agreement with those for the limited range of phosphinoallenes§ (Table 2) described previously.14,41 We thus confidently formulate 15 as (Et2N)2PC(Ph)CCH2 (Scheme 2).
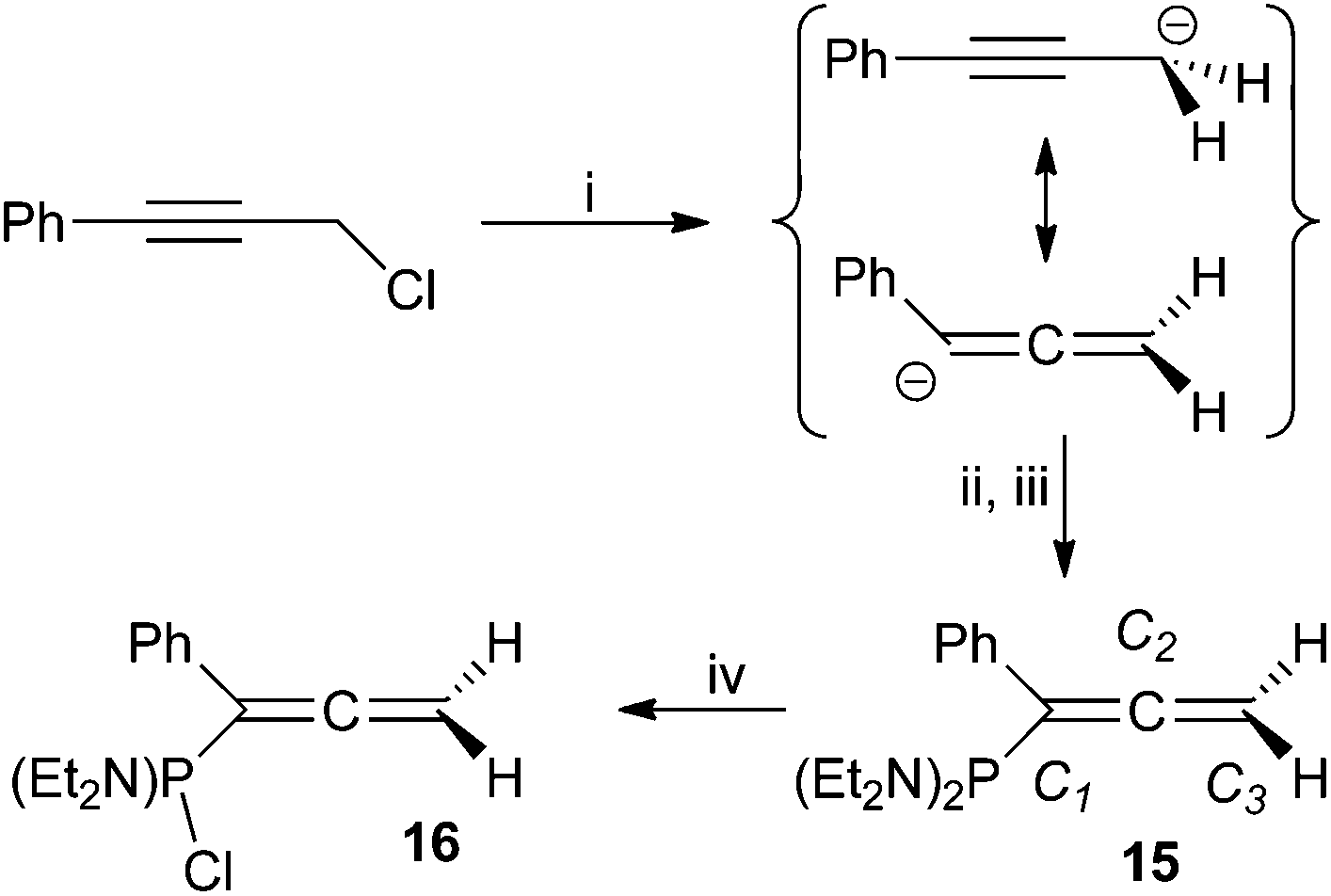 | ||
| Scheme 2 Reagents and conditions: (i) Et2O, HgCl2 (5 mol%), Mg, △, 4 h; (ii) (Et2N)2PCl, −78 °C, 30 min; (iii) r.t. 18 h; (iv) 2 equiv. HCl/Et2O. | ||
|
δ
H (CH2) |
δ
C (CH2) |
δ
C (C) |
|
|---|---|---|---|
| a Chemical shifts in ppm. b Data sourced from ref. 14 and 41. | |||
| Mes(H)PC(Me)CCH2 |
4.40 | 71.12 | 208.0 |
| Mes(Me)PC(Me)CCH2 |
4.64 | 73.26 | 204.9 |
| Mes(Me3Si)PC(Me)CCH2 |
4.55 | 72.35 | 206.4 |
| Mes (Cl)PC(Me)CCH2 |
4.57 | 74.65 | 205.5 |
| Ph2PC(H)CCH2 |
— | 71.7 | 213.2 |
| Ph2PC(Me)CCH2 |
— | 70.7 | 210.3 |
| Ph2PC(H)CC(Me)2 |
— | — | 209.6 |
The reaction of propargyl Grignard reagents with R2PCl has been noted to afford mixtures that include allenyl-phosphanes, their proportion being dependent on the nature of ‘R’.42 However, this is to our knowledge the first example of an allenylphosphane being obtained as the major product (>70%) in such a reaction, with minimal levels (<2%) of the propargyl tautomer. While we have not further studied this reaction, the noted outcome might reasonably be considered to reflect either enhanced stability of the α-phenyl-allenyl carbanion over its propargylic counterpart (localisation at an sp2, rather than sp3 centre) or be the result of conjugate addition, favoured by the relatively ‘soft’ ClP(NEt2)2 electrophile, as compared, for instance, with the notionally ‘harder’ PCl3, with which we encountered significantly greater complexity, yielding a largely intractable mixture.
In order to confirm or dismiss the presence of Cl2PC(Ph)CCH2 within this mixture, we sought to prepare an authentic sample, treating 15 with HCl (2 equiv.). This effected quantitative conversion to (Et2N)(Cl)PC(Ph)CCH2 (16), as evidenced by the 1H NMR spectrum, which indicates loss of one diethylamino moiety (Et2N vs. Ph resonances) and emergence of diasterotopicity for the methylenic ‘CH2’. The phosphorus resonance of 16 is appreciably deshielded from that of 15, consistent with replacement of NEt2 by Cl (δP 122; cf. Ph(Cl)PNEt2 142.143). Upon further treatment with HCl there is superficial evidence for removal of the remaining diethylamino moiety, viz. loss of its 1H NMR resonances, and of diasterotopicity of the ‘CH2’ protons (δH 4.63, d, JPH 3 Hz). However, the 31P shift (δP 58.7, t, JPH 3 Hz) seems inconsistent with a species of the type RPCl2; moreover, several other, unidentified, species are apparent in both the 1H and 31P-NMR spectra, precluding confident assignment of the bulk product.
Coordination chemistry of propargylphosphanes
As previously noted (vide supra) the coordination chemistry of propargylphosphanes is significantly underdeveloped and focused exclusively on coordinately saturated, mid-transition metals. We thus sought to prepare representative complexes featuring the unsaturated group 10 metals Pd and Pt.The propargylphosphanes 8, 11 and 12 react with [PtCl2]n, as a suspension in CH2Cl2, to afford exclusively the complexes cis-[Pt{PPh2(CH2CCER3)}2Cl2] (ER3 = nBu3Sn 17, iPr3Si 18, nPr3Si 19, Scheme 3) in excess of 75% isolated yield. For the silanes, palladium analogues (ER3 = iPr3Si 20, nPr3Si 21) are similarly obtained from [PdCl2]n, forming exclusively as the trans isomers.
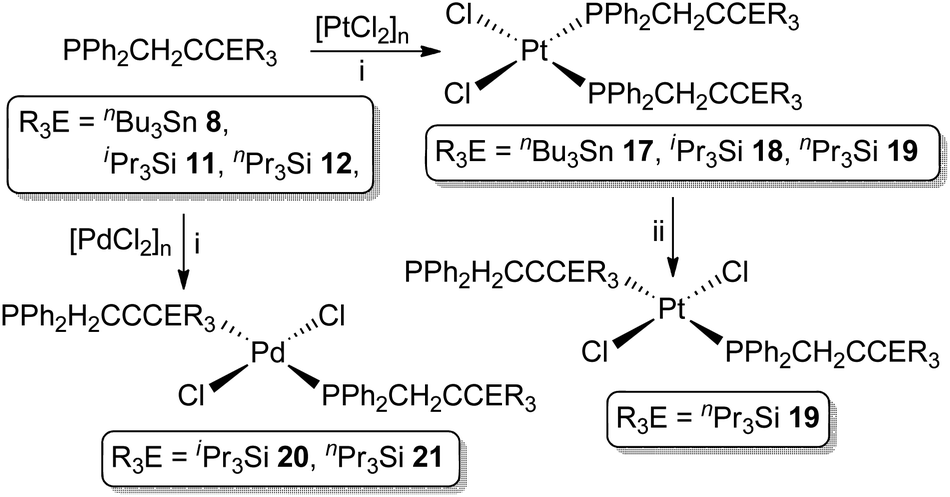 | ||
| Scheme 3 Reagents and conditions: (i) CH2Cl2, 12 h; (ii) C6D6, hν, 30 min. | ||
Complexes 17–21 have, thus far, not yielded X-ray quality single crystals, in common with most of the limited range of precedent examples. Nonetheless, their identities are unequivocally established from characteristic spectroscopic data, which verify the structural integrity of the ligands and coordination of the phosphorus centres (ΔδP ∼ +20). For the platinum complexes 17–19, |1JPtP| values of ca. 3600 Hz are wholly consistent with assignment of a cis geometry, while the palladium complexes exhibit virtual coupling in the 1H and 13C{1H}-NMR resonances associated with the CH2P moiety, consistent with a trans ligand arrangement. Notably, despite coordinative unsaturation of the metals, there is no evidence for either intra or intermolecular association of the pendant alkynyl moieties, the spectroscopic features of these units being little perturbed from the free ligands.
All of the complexes appear robust, both in solution and the solid state, universally resisting attempts to thermally induce cis/trans isomerisation. However, the UV irradiation (broad spectrum) of the platinum complex cis-19 over a period of 30 minutes did result in partial isomerisation, affording a mixture of cis-19 (42%) and trans-19 (58%). The identity of trans-19 was established on the basis of (i) reduced magnitude Pt–P coupling (|1JPtP| = 2601 Hz), consistent with trans-[Pt(PR3)2X2], and (ii) manifestation of virtual coupling for the CH2P centres, as in the palladium systems. However, attempts to effect complete conversion to trans-19 through extended irradiation proved unsuccessful, no further perturbation of the isomeric distribution being achieved.
Conclusions
We have described the synthesis and characterisation of a series of novel propargylphosphanes that feature tin and silicon termini on the alkyne moiety. Attempts to increase the range of phosphanyl termini used via the reaction of R2PCl with propargyl Grignard reagents proved unsuccessful, but allowed for the generation of the novel allenylphosphine (Et2N)2PC(Ph)CCH2, the first time a species of this type has been obtained as the primary product (>70%) of such a reaction.
Representative phosphanes have been shown to form complexes [M(PPh2CH2CCER3)2Cl2] with palladium and platinum, adopting exclusively trans (Pd) or cis (Pt) geometries respectively, though the latter can be partially isomerised under UV irradiation. These are the first examples of propargyl phosphane complexes incorporating group 10, or indeed any unsaturated, metals and are among a very limited number (<25) of coordination compounds known for any such ligands.
Experimental
General methods
All manipulations were performed under strict anaerobic conditions using standard Schlenk line and glovebox (MBraun) techniques, working under an atmosphere of dry argon or dinitrogen respectively. Solvents were distilled from appropriate drying agents and stored over either molecular sieves (4 Å for DCM and THF) or potassium mirrors. Propargyl chloride, group 14 triorganohalides and HPPh2 were obtained from Sigma-Aldrich, purified by appropriate methods and degassed (freeze–thaw) before use. nBuLi (2.5 M in hexanes) was obtained from Sigma-Aldrich and titrated to establish concentration. Precious metal salts (PtCl2, PdCl2) were obtained from STREM and used as supplied. HP(SiMe3)2 was prepared by literature procedure.44 Deuterated solvents were supplied by Goss Scientific and purified by refluxing with potassium (hydrocarbon) or CaH2 (chlorinated) for 3 days prior to use, being vacuum transferred and stored under inert atmosphere. Unless otherwise stated, NMR spectra were recorded on a Varian VNMRS 400 (1H, 399.50 MHz; 13C, 100.46 MHz; 31P, 161.71 MHz; 29Si, 79.37 MHz; 119Sn, 148.97 MHz; 195Pt, 85.53 MHz) or VNMRS 500 (1H 499.91 MHz; 13C, 125.72 MHz) spectrometer. All spectra were referenced to Me4Si, 85% H3PO4, Me4Sn or K2PtCl6 as appropriate. Carbon-13 NMR data were assigned with recourse to the 2D (HSQC, HMBC) spectra; detailed connectivity and 29Si chemical shifts were assessed using 1H−X HMBC spectra (X = 29Si; 119Sn; 31P). Elemental analyses were obtained by Mr S. Boyer of the London Metropolitan University Elemental Analysis Service.Synthesis
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CCH2Cl (1).
In a modification of literature procedure, a solution of propargyl chloride (2.24 g, 3.0 × 10−2 mol) in THF (ca. 20 cm3) was cooled to −78 °C before the drop-wise addition of nBuLi (2.5 M, 6.0 cm3, 1.5 × 10−2 mol). The mixture was stirred for 30 min., after which time nBu3SnCl (4.40 cm3, 1.5 × 10−2 mol) as solution in THF (ca. 10 cm3) was added drop-wise, resulting in formation of a yellow solution. The mixture was held at −78 °C for a further 30 min. with continued stirring before being allowed to warm to ambient temperature overnight. Solvent and excess HCCCH2Cl were removed under reduced pressure and the product extracted with pentane, stripped of volatiles and dried in vacuo as yellow oil. Yield: 5.09 g, 94%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.91 (t, 3JHH 7.3 Hz, 9 H, CH3), 0.97 (t, 3JHH 6 Hz, JSnH 54 Hz, 6H, CH2Sn), 1.34 (m, 6H, CH3CH2), 1.61 (m, 6H, CH2CH2Sn), 3.70 (s, JSnH 9.6 Hz, 2H, CH2Cl). 13C{1H}-NMR: δC 11.3 (s,
CCH2Cl (1).
In a modification of literature procedure, a solution of propargyl chloride (2.24 g, 3.0 × 10−2 mol) in THF (ca. 20 cm3) was cooled to −78 °C before the drop-wise addition of nBuLi (2.5 M, 6.0 cm3, 1.5 × 10−2 mol). The mixture was stirred for 30 min., after which time nBu3SnCl (4.40 cm3, 1.5 × 10−2 mol) as solution in THF (ca. 10 cm3) was added drop-wise, resulting in formation of a yellow solution. The mixture was held at −78 °C for a further 30 min. with continued stirring before being allowed to warm to ambient temperature overnight. Solvent and excess HCCCH2Cl were removed under reduced pressure and the product extracted with pentane, stripped of volatiles and dried in vacuo as yellow oil. Yield: 5.09 g, 94%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.91 (t, 3JHH 7.3 Hz, 9 H, CH3), 0.97 (t, 3JHH 6 Hz, JSnH 54 Hz, 6H, CH2Sn), 1.34 (m, 6H, CH3CH2), 1.61 (m, 6H, CH2CH2Sn), 3.70 (s, JSnH 9.6 Hz, 2H, CH2Cl). 13C{1H}-NMR: δC 11.3 (s, ![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H2Sn, 1J117SnC 365 Hz, 1J119SnC 382 Hz), 13.9 (s, H3), 27.3 (s, H2CH2Sn, 2J117SnC 58 Hz, 2J119SnC 60 Hz), 29.3 (s, CH3H2, 3JSnC 24 Hz), 31.2 (s, JSnC 8 Hz, CH2Cl), 91.1 (s, CCH2Cl), 105 (s, CCH2Cl). 119Sn{1H}-NMR: δSn −65.1. Anal. Found: C, 49.44; H, 7.86. Calcd for C15H29ClSn: C, 49.56; H, 8.04.
CCH2Cl (2).
As for 1, using propargyl chloride (2.03 g, 2.7 × 10−2 mol), nBuLi (2.5 M, 5.4 cm3, 1.3 × 10−2 mol) and Ph3SnCl (5.25 g, 1.3 × 10−2 mol). Isolated as yellow oil. Yield: 3.96 g, 72%. NMR (C6D6, 30 °C): 1H-NMR: δH 3.67 (s, JSnH 10.5 Hz, 2H, CH2Cl), 7.10–7.20 (m, 9H, m/p-C6H5), 7.60–7.65 (m, JSnH 55 Hz, 6H, o-C6H5). 13C{1H}-NMR: δC 30.8 (s, JSnC 10 Hz, CH2Cl), 88.5 (s, CCH2Cl), 106.8 (s, CCH2Cl), 128.8 (s, p-6H5), 129.5 (s, m-6H5), 130.1 (s, i-6H5), 136.7 (s, o-6H5). 119Sn{1H}-NMR: δSn −169.5. Anal. Found: C, 59.63; H, 4.12. Calcd for C20H17ClSn: C, 59.55; H, 4.05.
CCH2Cl (3).
As for 1, using propargyl chloride (3.73 g, 5.0 × 10−2 mol), nBuLi (2.5 M, 10.0 cm3, 2.5 × 10−2 mol) and Me2PhSiCl (4.26 g, 2.5 × 10−2 mol). The crude product was distilled at 66 °C, 8.1 × 10−1 mbar to afford colourless oil. Yield: 4.98 g, 96%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.32 (s, JSiH 8 Hz, 6 H, SiCH3), 3.21 (s, 2H, CH2Cl), 7.14–7.18 (m, 3H, m/p-C6H5), 7.55–7.59 (m, 2H, o-C6H5). 13C{1H}-NMR: δC −1.2 (s, SiH3, 1JSiC 58 Hz,), 30.5 (s, CH2Cl), 90.1 (s, CCH2Cl), 102.0 (s, CCH2Cl). 29Si{1H}-NMR: δSi −21.6. Anal. Found: C, 63.18; H, 6.14. Calcd for C11H13ClSi: C, 63.29; H, 6.28.
CCH2Cl (4).
As for 1, using propargyl chloride (6.24 g, 8.4 × 10−2 mol), nBuLi (2.5 M, 16.8 cm3, 4.2 × 10−2 mol) and iPr3SiCl (8.06 g, 4.2 × 10−2 mol). The crude product was distilled at 52 °C, 3.0 × 10−1 mbar to afford colourless oil. Yield: 5.76 g, 60%. NMR (C6D6, 30 °C): 1H-NMR: δH 1.03 (m,3 H, SiCH), 1.11 (d, 3JHH 6.5 Hz, 18H, CH3), 3.53 (s, 2H, CH2Cl). 13C{1H}-NMR: δC 11.5 (s, SiH, 1JSiC 57 Hz), 18.8 (s, H3), 30.6 (s, CH2Cl), 88.4 (s, CCH2Cl), 102.7 (s, CCH2Cl). 29Si{1H}-NMR: δSi −1.68. Anal. Found: C, 62.38; H, 9.85. Calcd for C12H23ClSi: C, 62.43; H, 10.04.
CCH2Cl (5).
As for 1, using propargyl chloride (1.62 g, 2.2 × 10−2 mol), nBuLi (2.5 M, 4.35 cm3, 1.1 × 10−2 mol) and nPr3SiCl (2.09 g, 1.1 × 10−2 mol). Obtained as orange oil. Yield: 2.33 g, 93%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.60 (m, 6 H, SiCH2), 0.99 (t, 3JHH 7.2 Hz, 9H, CH3), 1.47 (m, 6H, CH3CH2), 3.55 (s, 2H, CH2Cl). 13C{1H}-NMR: δC 16.2 (s, H2Si, 1JSiC 56 Hz,), 17.9 (s, H3), 18.4 (s, H2CH2Si, 2JSiC 6 Hz), 30.7 (s, CH2Cl), 90.2 (s, CCH2Cl), 101.8 (s, CCH2Cl). 29Si{1H}-NMR: δSi −13.0. Anal. Found: C, 62.87; H, 9.79. Calcd for C12H23ClSi: C, 62.43; H, 10.04.
CCH2Cl (6).
As for 1, using propargyl chloride (1.92 g, 2.5 × 10−2 mol), nBuLi (2.5 M, 5.2 cm3, 1.3 × 10−2 mol) and nBu3SiCl (3.02 g, 1.29 × 10−2 mol). Obtained as orange oil. Yield: 3.08 g, 88%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.67 (m, 6H, SiCH2), 0.92 (t, 3JHH 7.3 Hz, 9H, CH3), 1.38 (m, 6H, CH2CH2Si), 1.46 (m, 6H, CH3CH2CH2), 3.56 (s, 2H, CH2Cl). 13C{1H}-NMR: δC 13.3 (s, H2Si, 1JSiC 57 Hz), 14.0 (s, H3), 26.5 (s, CH3H2CH2), 26.8 (s, H2CH2Si, JSiC 6 Hz), 30.7 (s, CH2Cl), 90.3 (s, CCH2Cl), 101.8 (s, CCH2Cl). 29Sn{1H}-NMR: δSi −11.3. Anal. Found: C, 66.39; H, 10.02. Calcd for C15H29ClSi: C, 66.01; H, 10.71.
CCH2Cl (7).
As for 1, using propargyl chloride (1.00 g, 1.03 × 10−2 mol), nBuLi (2.5 M, 2.7 cm3, 6.7 × 10−3 mol) and nBu3SiCl (3.83 g, 1.3 × 10−3 mol). The crude product was sublimed under reduced pressure (23.0 × 10−3 mbar) to afford a colourless solid. Yield: 3.04 g, 89%. NMR (C6D6, 30 °C): 1H-NMR: δH 3.49 (s, 2H, CH2Cl), 7.14–7.16 (m, 9H, m/p-C6H5), 7.73–7.78 (m, 6H, o-C6H5). 13C{1H}-NMR: δC 30.4 (s, CH2Cl), 87.6 (s, CCH2Cl), 104.9 (s, CCH2Cl), 128.4 (s, p-6H5), 130.4 (s, m-6H5), 133.4 (s, i-6H5), 136.0 (s, o-6H5). 29Sn{1H}-NMR: δSi −28.8. Anal. Found: C, 75.68; H, 5.11. Calcd for C21H17ClSi: C, 75.77; H, 5.15.
CCH2PPh2 (8).
To an ethereal solution (ca. 20 cm3) of HPPh2 (0.375 g, 2.02 × 10−3 mol) held at −78 °C was added drop-wise nBuLi (2.5 M, 0.808 cm3, 2.02 × 10−3 mol); the mixture was stirred for 30 min. A solution of 1 (0.733 g, 2.02 × 10−3 mol) in ether (ca. 10 cm3) was then added drop-wise and the mixture maintained at −78 °C while stirring for 30 min. The mixture was allowed to warm to ambient temperature while stirring overnight. Volatiles were removed under reduced pressure and the product extracted with pentane; the solvent was removed and the product dried in vacuo to afford yellow oil. Yield: 0.800 g (4:1 8:SnBu4). NMR (C6D6, 30 °C): 1H-NMR: δH 0.93 (m, CH3), 1.36 (m, 12H, 2 × CH2), 1.58 (m, 6H, CH2), 2.87 (d, JPH 1.6 Hz, J117SnC 8.7 Hz, J119SnC 12.4 Hz, 2H, CH2P), 7.02–7.13 (m, 6H, m/p-C6H5), 7.43–7.51 (m, 4H, o-C6H5). 13C{1H}-NMR: δC 11.3 (s, H2Sn, 1J117SnC 366 Hz, 1J119SnC 383 Hz), 13.9 (s, H3), 20.4 (d, 1JPC 18 Hz,
H2Sn, 1J117SnC 365 Hz, 1J119SnC 382 Hz), 13.9 (s, H3), 27.3 (s, H2CH2Sn, 2J117SnC 58 Hz, 2J119SnC 60 Hz), 29.3 (s, CH3H2, 3JSnC 24 Hz), 31.2 (s, JSnC 8 Hz, CH2Cl), 91.1 (s, CCH2Cl), 105 (s, CCH2Cl). 119Sn{1H}-NMR: δSn −65.1. Anal. Found: C, 49.44; H, 7.86. Calcd for C15H29ClSn: C, 49.56; H, 8.04.
CCH2Cl (2).
As for 1, using propargyl chloride (2.03 g, 2.7 × 10−2 mol), nBuLi (2.5 M, 5.4 cm3, 1.3 × 10−2 mol) and Ph3SnCl (5.25 g, 1.3 × 10−2 mol). Isolated as yellow oil. Yield: 3.96 g, 72%. NMR (C6D6, 30 °C): 1H-NMR: δH 3.67 (s, JSnH 10.5 Hz, 2H, CH2Cl), 7.10–7.20 (m, 9H, m/p-C6H5), 7.60–7.65 (m, JSnH 55 Hz, 6H, o-C6H5). 13C{1H}-NMR: δC 30.8 (s, JSnC 10 Hz, CH2Cl), 88.5 (s, CCH2Cl), 106.8 (s, CCH2Cl), 128.8 (s, p-6H5), 129.5 (s, m-6H5), 130.1 (s, i-6H5), 136.7 (s, o-6H5). 119Sn{1H}-NMR: δSn −169.5. Anal. Found: C, 59.63; H, 4.12. Calcd for C20H17ClSn: C, 59.55; H, 4.05.
CCH2Cl (3).
As for 1, using propargyl chloride (3.73 g, 5.0 × 10−2 mol), nBuLi (2.5 M, 10.0 cm3, 2.5 × 10−2 mol) and Me2PhSiCl (4.26 g, 2.5 × 10−2 mol). The crude product was distilled at 66 °C, 8.1 × 10−1 mbar to afford colourless oil. Yield: 4.98 g, 96%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.32 (s, JSiH 8 Hz, 6 H, SiCH3), 3.21 (s, 2H, CH2Cl), 7.14–7.18 (m, 3H, m/p-C6H5), 7.55–7.59 (m, 2H, o-C6H5). 13C{1H}-NMR: δC −1.2 (s, SiH3, 1JSiC 58 Hz,), 30.5 (s, CH2Cl), 90.1 (s, CCH2Cl), 102.0 (s, CCH2Cl). 29Si{1H}-NMR: δSi −21.6. Anal. Found: C, 63.18; H, 6.14. Calcd for C11H13ClSi: C, 63.29; H, 6.28.
CCH2Cl (4).
As for 1, using propargyl chloride (6.24 g, 8.4 × 10−2 mol), nBuLi (2.5 M, 16.8 cm3, 4.2 × 10−2 mol) and iPr3SiCl (8.06 g, 4.2 × 10−2 mol). The crude product was distilled at 52 °C, 3.0 × 10−1 mbar to afford colourless oil. Yield: 5.76 g, 60%. NMR (C6D6, 30 °C): 1H-NMR: δH 1.03 (m,3 H, SiCH), 1.11 (d, 3JHH 6.5 Hz, 18H, CH3), 3.53 (s, 2H, CH2Cl). 13C{1H}-NMR: δC 11.5 (s, SiH, 1JSiC 57 Hz), 18.8 (s, H3), 30.6 (s, CH2Cl), 88.4 (s, CCH2Cl), 102.7 (s, CCH2Cl). 29Si{1H}-NMR: δSi −1.68. Anal. Found: C, 62.38; H, 9.85. Calcd for C12H23ClSi: C, 62.43; H, 10.04.
CCH2Cl (5).
As for 1, using propargyl chloride (1.62 g, 2.2 × 10−2 mol), nBuLi (2.5 M, 4.35 cm3, 1.1 × 10−2 mol) and nPr3SiCl (2.09 g, 1.1 × 10−2 mol). Obtained as orange oil. Yield: 2.33 g, 93%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.60 (m, 6 H, SiCH2), 0.99 (t, 3JHH 7.2 Hz, 9H, CH3), 1.47 (m, 6H, CH3CH2), 3.55 (s, 2H, CH2Cl). 13C{1H}-NMR: δC 16.2 (s, H2Si, 1JSiC 56 Hz,), 17.9 (s, H3), 18.4 (s, H2CH2Si, 2JSiC 6 Hz), 30.7 (s, CH2Cl), 90.2 (s, CCH2Cl), 101.8 (s, CCH2Cl). 29Si{1H}-NMR: δSi −13.0. Anal. Found: C, 62.87; H, 9.79. Calcd for C12H23ClSi: C, 62.43; H, 10.04.
CCH2Cl (6).
As for 1, using propargyl chloride (1.92 g, 2.5 × 10−2 mol), nBuLi (2.5 M, 5.2 cm3, 1.3 × 10−2 mol) and nBu3SiCl (3.02 g, 1.29 × 10−2 mol). Obtained as orange oil. Yield: 3.08 g, 88%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.67 (m, 6H, SiCH2), 0.92 (t, 3JHH 7.3 Hz, 9H, CH3), 1.38 (m, 6H, CH2CH2Si), 1.46 (m, 6H, CH3CH2CH2), 3.56 (s, 2H, CH2Cl). 13C{1H}-NMR: δC 13.3 (s, H2Si, 1JSiC 57 Hz), 14.0 (s, H3), 26.5 (s, CH3H2CH2), 26.8 (s, H2CH2Si, JSiC 6 Hz), 30.7 (s, CH2Cl), 90.3 (s, CCH2Cl), 101.8 (s, CCH2Cl). 29Sn{1H}-NMR: δSi −11.3. Anal. Found: C, 66.39; H, 10.02. Calcd for C15H29ClSi: C, 66.01; H, 10.71.
CCH2Cl (7).
As for 1, using propargyl chloride (1.00 g, 1.03 × 10−2 mol), nBuLi (2.5 M, 2.7 cm3, 6.7 × 10−3 mol) and nBu3SiCl (3.83 g, 1.3 × 10−3 mol). The crude product was sublimed under reduced pressure (23.0 × 10−3 mbar) to afford a colourless solid. Yield: 3.04 g, 89%. NMR (C6D6, 30 °C): 1H-NMR: δH 3.49 (s, 2H, CH2Cl), 7.14–7.16 (m, 9H, m/p-C6H5), 7.73–7.78 (m, 6H, o-C6H5). 13C{1H}-NMR: δC 30.4 (s, CH2Cl), 87.6 (s, CCH2Cl), 104.9 (s, CCH2Cl), 128.4 (s, p-6H5), 130.4 (s, m-6H5), 133.4 (s, i-6H5), 136.0 (s, o-6H5). 29Sn{1H}-NMR: δSi −28.8. Anal. Found: C, 75.68; H, 5.11. Calcd for C21H17ClSi: C, 75.77; H, 5.15.
CCH2PPh2 (8).
To an ethereal solution (ca. 20 cm3) of HPPh2 (0.375 g, 2.02 × 10−3 mol) held at −78 °C was added drop-wise nBuLi (2.5 M, 0.808 cm3, 2.02 × 10−3 mol); the mixture was stirred for 30 min. A solution of 1 (0.733 g, 2.02 × 10−3 mol) in ether (ca. 10 cm3) was then added drop-wise and the mixture maintained at −78 °C while stirring for 30 min. The mixture was allowed to warm to ambient temperature while stirring overnight. Volatiles were removed under reduced pressure and the product extracted with pentane; the solvent was removed and the product dried in vacuo to afford yellow oil. Yield: 0.800 g (4:1 8:SnBu4). NMR (C6D6, 30 °C): 1H-NMR: δH 0.93 (m, CH3), 1.36 (m, 12H, 2 × CH2), 1.58 (m, 6H, CH2), 2.87 (d, JPH 1.6 Hz, J117SnC 8.7 Hz, J119SnC 12.4 Hz, 2H, CH2P), 7.02–7.13 (m, 6H, m/p-C6H5), 7.43–7.51 (m, 4H, o-C6H5). 13C{1H}-NMR: δC 11.3 (s, H2Sn, 1J117SnC 366 Hz, 1J119SnC 383 Hz), 13.9 (s, H3), 20.4 (d, 1JPC 18 Hz, ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2PPh2), 27.4 (s, H2CH2Sn, JSnC 58 Hz) 85.0 (d, JPC 6 Hz, CCH2PPh2), 106.8 (d, JPC 5 Hz, CCH2PPh2), 128.6 (d, JPC 6 Hz, m-C6H5), 128.9 (s, p-C6H5), 133.2 (d, JPC 19 Hz, o-C6H5), 138.8 (d, JPC 17 Hz, i-C6H5). 31P{1H}-NMR: δP −13.4 (s, JSnP 14.5 Hz). 119Sn{1H}-NMR: δSn −68.5 (d, JSnP 14.5 Hz, 4Sn), −12.0 (s, 1Sn, Bu4Sn).
CCH2PPh2 (9).
As for 8, using HPPh2 (0.309 g, 1.66 × 10−3 mol), nBuLi (2.1 M, 0.80 cm3, 1.66 × 10−3 mol) and 2 (0.876 g, 1.66 × 10−3 mol). Isolated as yellow oil. NMR (C6D6, 30 °C): 1H-NMR: δH 2.84 (d, JPH 3.0 Hz, JSnH 14.8 Hz, 2H, CH2P), 6.89–7.20 (m, Ar, m/p-C6H5), 7.37–7.70 (m, Ar, 6H, o-C6H5). 13C{1H}-NMR: δC 20.2 (d, JPC 21 Hz, JSnC 11.9 Hz, CH2P), 82.8 (d, JPC 6 Hz, JSnC 3.4 Hz, CCH2P), 109.3 (d, JPC 3.4 Hz, CCH2P). 31P{1H}-NMR: δP −13.2 (s). 119Sn{1H}-NMR: δSn −168.4 (JSnP 14.8 Hz, 1Sn), −98.3 (s, BuSnPh3, 1Sn).
CCH2PPh2 (10).
As for 8, using HPPh2 (0.780 g, 4.24 × 10−3 mol), nBuLi (2.5 M, 1.7 cm3, 4.24 × 10−3 mol) and 3 (0.884 g, 4.24 × 10−3 mol). Isolated as brown oil. Yield: 1.19 g, 78%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.30 (s, 6 H, SiCH3), 2.75 (d, JPH 2.9 Hz, 2H, CH2P), 7.01–7.09 (m, 6H, m/p-P(C6H5)2), 7.17–7.22 (m, 4H, o-P(C6H5)2), 7.39–7.46 (m, 3H, m/p-SiC6H5), 7.52–7.58 (m, 2H, o-SiC6H5). 13C{1H}-NMR: δC −0.6 (s, SiH3), 19.9 (d, JPC 21 Hz, H2P), 85.7 (d, JPC 5 Hz, CCH2P), 104.9 (d, JPC 4 Hz, CCH2P), 128.7 (d, JPC 6.5 Hz, m-C6H5), 129.0 (s, p-C6H5), 129.5 (s, p-C6H5), 133.2 (d, JPC 19.5 Hz, o-C6H5), 134.2 (s, o-C6H5), 137.7 (s, i-C6H5), 138.1 (d, JPC 16 Hz, i-C6H5). 31P{1H}-NMR: δP −13.5 (s). 29Si{1H}-NMR: δSi −22.9. Anal. Found: C, 76.89; H, 6.34. Calcd for C23H23PSi: C, 77.06; H, 6.47.
CCH2PPh2 (11).
As for 8, using HPPh2 (0.780 g, 4.24 × 10−3 mol), nBuLi (2.5 M, 1.7 cm3, 4.24 × 10−3 mol) and 4 (0.976 g, 4.24 × 10−3 mol). Isolated as orange oil. Yield: 1.45 g, 90%. NMR (C6D6, 30 °C): 1H-NMR: δH 1.01 (m, 3H, SiCH), 1.09 (d, 3JHH 6.8 Hz, 18H, CH3), 2.75 (d, JPH 2.3 Hz, 2H, CH2P), 7.02–7.12 (m, 6H, m/p-P(C6H5)2), 7.39–7.47 (m, 4H, o-P(C6H5)2). 13C{1H}-NMR: δC 11.7 (s, SiH, 1JSiC 56 Hz), 18.9 (s, JSiC 16 Hz, H3), 19.9 (d, JPC 19.5 Hz, H2P), 83.3 (d, JPC 5 Hz, CCH2P), 104.7 (d, JPC 4 Hz, CCH2P), 128.7 (d, JPC 6.5 Hz, m-C6H5), 129.0 (s, p-C6H5), 133.1 (d, JPC 19 Hz, o-C6H5), 138.3 (d, JPC 16 Hz, i-C6H5). 31P{1H}-NMR: δP −13.5 (s, JSiP 20 Hz). 29Si{1H}-NMR: δSi −3.03. Anal. Found: C, 75.77; H, 8.64. Calcd for C24H33PSi: C, 75.74; H, 8.74.
CCH2PPh2 (12).
As for 8, using HPPh2 (0.650 g, 3.49 × 10−3 mol), nBuLi (2.5 M, 1.4 cm3, 3.49 × 10−3 mol) and 5 (0.805 g, 3.49 × 10−3 mol). Isolated as brown oil. Yield: 1.00 g, 80%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.58 (m, 6H, SiCH2), 0.99 (t, 3JHH 7.0 Hz, 9H, CH3), 1.42 (m, 6H, CH2CH2Si), 2.75 (d, JPH 2.5 Hz, 2H, CH2P), 7.04–7.12 (m, 6H, m/p-P(C6H5)2), 7.40–7.46 (m, 4H, o-P(C6H5)2). 13C{1H}-NMR: δC 16.7 (s, H2Si, 1JSiC 56 Hz,), 18.0 (s, H3), 18.5 (s, H2CH2Si, 2JSiC 8 Hz), 19.9 (d, JPC 20 Hz, H2P), 85.4 (d, JPC 5.4 Hz, CCH2P), 104.7 (d, JPC 4 Hz, CCH2P), 128.5 (d, JPC 6.6 Hz, m-C6H5), 129.0 (s, p-C6H5), 133.2 (d, JPC 19 Hz, o-C6H5), 138.3 (d, JPC 16.5 Hz, i-C6H5). 31P{1H}-NMR: δP −13.6 (s, JSiP 19.7 Hz). 29Si{1H}-NMR: δSi −14.8. Anal. Found: C, 75.77; H, 8.59. Calcd for C24H33PSi: C, 75.74; H, 8.74.
CCH2PPh2 (13).
As for 8, using HPPh2 (0.650 g, 4.24 × 10−3 mol), nBuLi (2.5 M, 1.15 cm3, 2.87 × 10−3 mol) and 6 (0.784 g, 1.87 × 10−3 mol). Isolated as brown oil. Yield: 0.95 g, 79%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.6.3 (m, 6H, SiCH2), 0.93 (t, 3JHH 7.2 Hz, 9H, CH3), 1.37 (m, 6H, CH2CH2Si), 1.41 (m, 6H, CH3CH2CH2), 2.76 (d, JPH 2.3 Hz, 2H, CH2P), 7.04–7.13 (m, 6H, m/p-P(C6H5)2), 7.41–7.46 (m, 4H, o-P(C6H5)2). 13C{1H}-NMR: δC 13.7 (s, H2Si), 14.1 (s, H3), 19.9 (d, JPC 20 Hz, H2P), 26.7 (s, CH3H2CH2), 26.9 (s, H2CH2Si), 85.5 (d, JPC 4.8 Hz, CCH2P), 104.0 (d, JPC 4.2 Hz, CCH2P), 128.6 (d, JPC 6.4 Hz, m-C6H5), 129.0 (s, p-C6H5), 133.2 (d, JPC 19 Hz, o-C6H5), 138.3 (d, JPC 15.5 Hz, i-C6H5). 31P{1H}-NMR: δP −13.5 (s, JSiP 18.0 Hz). 29Si{1H}-NMR: δSi −12.9. Anal. Found: C, 76.78; H, 9.32. Calcd for C27H39PSi: C, 76.73; H, 9.30.
CCH2P(SiMe3)2 (14).
In a manner similar to that described for 8, using HP(SiMe3)2 (1.04 g, 5.84 × 10−3 mol), nBuLi (2.5 M, 2.3 cm3, 5.75 × 10−3 mol) and 3 (1.25 g, 6.00 × 10−3 mol). Isolated as orange oil. Yield: 1.84 g, 90%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.25 (d, JPH 4.8 Hz, 18H, 2 × Si(CH3)3), 0.44 (s, 2 × SiCH3), 2.43 (d, JPH 1.3 Hz, 2H, CH2P), 7.20–7.25 (m, 3H, m/p-SiC6H5), 7.70–7.74 (m, 2H, o-SiC6H5). 13C{1H}-NMR: δC −0.6 (s, SiH3), 1.1 (d JPC 12.5 Hz, P{SiH)3}2), 5.5 (d, JPC 23 Hz, H2P), 83.3 (d, JPC 4 Hz, CCH2P), 109.3 (br., CCH2P), 128.2 (s, m-C6H5), 129.6 (s, p-C6H5), 134.2 (s, o-C6H5), 137.7 (s, i-C6H5). 31P{1H}-NMR: δP −84.4 (s, 5%), −158.9 (s, 14, 93%), −252.0 (s, 2%). 29Si{1H}-NMR: δSi −23.0 (SiMe2Ph), 3.42 (P(SiMe3)2). Anal. Found: C, 58.29; H, 8.86. Calcd for C17H31PSi3: C, 58.23; H, 8.91.
H2PPh2), 27.4 (s, H2CH2Sn, JSnC 58 Hz) 85.0 (d, JPC 6 Hz, CCH2PPh2), 106.8 (d, JPC 5 Hz, CCH2PPh2), 128.6 (d, JPC 6 Hz, m-C6H5), 128.9 (s, p-C6H5), 133.2 (d, JPC 19 Hz, o-C6H5), 138.8 (d, JPC 17 Hz, i-C6H5). 31P{1H}-NMR: δP −13.4 (s, JSnP 14.5 Hz). 119Sn{1H}-NMR: δSn −68.5 (d, JSnP 14.5 Hz, 4Sn), −12.0 (s, 1Sn, Bu4Sn).
CCH2PPh2 (9).
As for 8, using HPPh2 (0.309 g, 1.66 × 10−3 mol), nBuLi (2.1 M, 0.80 cm3, 1.66 × 10−3 mol) and 2 (0.876 g, 1.66 × 10−3 mol). Isolated as yellow oil. NMR (C6D6, 30 °C): 1H-NMR: δH 2.84 (d, JPH 3.0 Hz, JSnH 14.8 Hz, 2H, CH2P), 6.89–7.20 (m, Ar, m/p-C6H5), 7.37–7.70 (m, Ar, 6H, o-C6H5). 13C{1H}-NMR: δC 20.2 (d, JPC 21 Hz, JSnC 11.9 Hz, CH2P), 82.8 (d, JPC 6 Hz, JSnC 3.4 Hz, CCH2P), 109.3 (d, JPC 3.4 Hz, CCH2P). 31P{1H}-NMR: δP −13.2 (s). 119Sn{1H}-NMR: δSn −168.4 (JSnP 14.8 Hz, 1Sn), −98.3 (s, BuSnPh3, 1Sn).
CCH2PPh2 (10).
As for 8, using HPPh2 (0.780 g, 4.24 × 10−3 mol), nBuLi (2.5 M, 1.7 cm3, 4.24 × 10−3 mol) and 3 (0.884 g, 4.24 × 10−3 mol). Isolated as brown oil. Yield: 1.19 g, 78%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.30 (s, 6 H, SiCH3), 2.75 (d, JPH 2.9 Hz, 2H, CH2P), 7.01–7.09 (m, 6H, m/p-P(C6H5)2), 7.17–7.22 (m, 4H, o-P(C6H5)2), 7.39–7.46 (m, 3H, m/p-SiC6H5), 7.52–7.58 (m, 2H, o-SiC6H5). 13C{1H}-NMR: δC −0.6 (s, SiH3), 19.9 (d, JPC 21 Hz, H2P), 85.7 (d, JPC 5 Hz, CCH2P), 104.9 (d, JPC 4 Hz, CCH2P), 128.7 (d, JPC 6.5 Hz, m-C6H5), 129.0 (s, p-C6H5), 129.5 (s, p-C6H5), 133.2 (d, JPC 19.5 Hz, o-C6H5), 134.2 (s, o-C6H5), 137.7 (s, i-C6H5), 138.1 (d, JPC 16 Hz, i-C6H5). 31P{1H}-NMR: δP −13.5 (s). 29Si{1H}-NMR: δSi −22.9. Anal. Found: C, 76.89; H, 6.34. Calcd for C23H23PSi: C, 77.06; H, 6.47.
CCH2PPh2 (11).
As for 8, using HPPh2 (0.780 g, 4.24 × 10−3 mol), nBuLi (2.5 M, 1.7 cm3, 4.24 × 10−3 mol) and 4 (0.976 g, 4.24 × 10−3 mol). Isolated as orange oil. Yield: 1.45 g, 90%. NMR (C6D6, 30 °C): 1H-NMR: δH 1.01 (m, 3H, SiCH), 1.09 (d, 3JHH 6.8 Hz, 18H, CH3), 2.75 (d, JPH 2.3 Hz, 2H, CH2P), 7.02–7.12 (m, 6H, m/p-P(C6H5)2), 7.39–7.47 (m, 4H, o-P(C6H5)2). 13C{1H}-NMR: δC 11.7 (s, SiH, 1JSiC 56 Hz), 18.9 (s, JSiC 16 Hz, H3), 19.9 (d, JPC 19.5 Hz, H2P), 83.3 (d, JPC 5 Hz, CCH2P), 104.7 (d, JPC 4 Hz, CCH2P), 128.7 (d, JPC 6.5 Hz, m-C6H5), 129.0 (s, p-C6H5), 133.1 (d, JPC 19 Hz, o-C6H5), 138.3 (d, JPC 16 Hz, i-C6H5). 31P{1H}-NMR: δP −13.5 (s, JSiP 20 Hz). 29Si{1H}-NMR: δSi −3.03. Anal. Found: C, 75.77; H, 8.64. Calcd for C24H33PSi: C, 75.74; H, 8.74.
CCH2PPh2 (12).
As for 8, using HPPh2 (0.650 g, 3.49 × 10−3 mol), nBuLi (2.5 M, 1.4 cm3, 3.49 × 10−3 mol) and 5 (0.805 g, 3.49 × 10−3 mol). Isolated as brown oil. Yield: 1.00 g, 80%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.58 (m, 6H, SiCH2), 0.99 (t, 3JHH 7.0 Hz, 9H, CH3), 1.42 (m, 6H, CH2CH2Si), 2.75 (d, JPH 2.5 Hz, 2H, CH2P), 7.04–7.12 (m, 6H, m/p-P(C6H5)2), 7.40–7.46 (m, 4H, o-P(C6H5)2). 13C{1H}-NMR: δC 16.7 (s, H2Si, 1JSiC 56 Hz,), 18.0 (s, H3), 18.5 (s, H2CH2Si, 2JSiC 8 Hz), 19.9 (d, JPC 20 Hz, H2P), 85.4 (d, JPC 5.4 Hz, CCH2P), 104.7 (d, JPC 4 Hz, CCH2P), 128.5 (d, JPC 6.6 Hz, m-C6H5), 129.0 (s, p-C6H5), 133.2 (d, JPC 19 Hz, o-C6H5), 138.3 (d, JPC 16.5 Hz, i-C6H5). 31P{1H}-NMR: δP −13.6 (s, JSiP 19.7 Hz). 29Si{1H}-NMR: δSi −14.8. Anal. Found: C, 75.77; H, 8.59. Calcd for C24H33PSi: C, 75.74; H, 8.74.
CCH2PPh2 (13).
As for 8, using HPPh2 (0.650 g, 4.24 × 10−3 mol), nBuLi (2.5 M, 1.15 cm3, 2.87 × 10−3 mol) and 6 (0.784 g, 1.87 × 10−3 mol). Isolated as brown oil. Yield: 0.95 g, 79%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.6.3 (m, 6H, SiCH2), 0.93 (t, 3JHH 7.2 Hz, 9H, CH3), 1.37 (m, 6H, CH2CH2Si), 1.41 (m, 6H, CH3CH2CH2), 2.76 (d, JPH 2.3 Hz, 2H, CH2P), 7.04–7.13 (m, 6H, m/p-P(C6H5)2), 7.41–7.46 (m, 4H, o-P(C6H5)2). 13C{1H}-NMR: δC 13.7 (s, H2Si), 14.1 (s, H3), 19.9 (d, JPC 20 Hz, H2P), 26.7 (s, CH3H2CH2), 26.9 (s, H2CH2Si), 85.5 (d, JPC 4.8 Hz, CCH2P), 104.0 (d, JPC 4.2 Hz, CCH2P), 128.6 (d, JPC 6.4 Hz, m-C6H5), 129.0 (s, p-C6H5), 133.2 (d, JPC 19 Hz, o-C6H5), 138.3 (d, JPC 15.5 Hz, i-C6H5). 31P{1H}-NMR: δP −13.5 (s, JSiP 18.0 Hz). 29Si{1H}-NMR: δSi −12.9. Anal. Found: C, 76.78; H, 9.32. Calcd for C27H39PSi: C, 76.73; H, 9.30.
CCH2P(SiMe3)2 (14).
In a manner similar to that described for 8, using HP(SiMe3)2 (1.04 g, 5.84 × 10−3 mol), nBuLi (2.5 M, 2.3 cm3, 5.75 × 10−3 mol) and 3 (1.25 g, 6.00 × 10−3 mol). Isolated as orange oil. Yield: 1.84 g, 90%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.25 (d, JPH 4.8 Hz, 18H, 2 × Si(CH3)3), 0.44 (s, 2 × SiCH3), 2.43 (d, JPH 1.3 Hz, 2H, CH2P), 7.20–7.25 (m, 3H, m/p-SiC6H5), 7.70–7.74 (m, 2H, o-SiC6H5). 13C{1H}-NMR: δC −0.6 (s, SiH3), 1.1 (d JPC 12.5 Hz, P{SiH)3}2), 5.5 (d, JPC 23 Hz, H2P), 83.3 (d, JPC 4 Hz, CCH2P), 109.3 (br., CCH2P), 128.2 (s, m-C6H5), 129.6 (s, p-C6H5), 134.2 (s, o-C6H5), 137.7 (s, i-C6H5). 31P{1H}-NMR: δP −84.4 (s, 5%), −158.9 (s, 14, 93%), −252.0 (s, 2%). 29Si{1H}-NMR: δSi −23.0 (SiMe2Ph), 3.42 (P(SiMe3)2). Anal. Found: C, 58.29; H, 8.86. Calcd for C17H31PSi3: C, 58.23; H, 8.91.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CCH2 (15).
To a THF suspension (ca. 30 cm3) of excess, pre-activated magnesium turnings containing HgCl2 (0.100 g, 3.68 × 10−4 mol) as initiator, was added drop-wise PhCCCH2Cl (1.00 g, 6.65 × 10−3 mol) as solution in THF (ca. 10 cm3); upon complete addition the mixture was brought to reflux for 4 h. After allowing to cool to ambient temperature, the mixture was filtered (via cannula) directly into a pre-cooled (−78 °C) THF solution of (Et2N)2PCl (1.39 cm3, 6.65 × 10−3 mol). The resulting red solution was stirred for 30 minutes at this temperature, before allowing it to attain ambient temperature and stir overnight. The resulting orange solution was stripped of volatiles under reduced pressure then extracted with pentane; this fraction was taken to dryness and dried in vacuo to afford the product as dark red oil. Yield: 1.46 g, 76%. 15 (74%): NMR (C6D6, 30 °C): 1H-NMR: δH 0.89 (t, 3JHH 7.0 Hz, 12H, CH3), 3.05 (q, 3JHH 7.0 Hz, 8H, CH2), 4.69 (d, JPH 7.0 Hz, 2H, CH2), 7.11–7.15 (m, 3H, m/p-C6H5), 7.63–7.59 (m, 2H, o-C6H5). 13C{1H}-NMR: δC 14.8 (d, 3JPC 3.2 Hz, H3), 43.4 (d, 3JPC 17.4 Hz, NH2), 75.0 (s, H2), 105.9 (d, JPC 13.5 Hz, i-C6H5), 137.4 (d, JPC 19 Hz, Ph{P(NEt2)2}C), 127.8 (s, o-C6H5), 127.9 (overlapped m-/p-C6H5), 209.9 (d, JPC 11.4 Hz, ). 31P{1H}-NMR: δP 91.0 (s, br, 74%). Propargyl tautomer (5%): NMR (C6D6, 30 °C): 1H-NMR: δH 1.02 (t, 3JHH 7.2 Hz, 12H, CH3), 2.71 (d, JPH 5.8 Hz, 2H, CH2P), 2.87 (m, 8H, NCH2). 13C{1H}-NMR: δC 14.0 (d, 3JPC 5 Hz, H3), 19.8 (m, H2P), 42.8 (d, 3JPC 17 Hz, NH2), 81.5 (s, CCH2P), 87.6 (s, CCH2P). 31P{1H}-NMR: δP 83.2 (s, br, 5%).
CCH2 (16).
To an ethereal solution of 15 held at −78 °C was added drop-wise two equivalent of HCl (1 M in ether). The mixture was held at −78 °C while stirring for 20 min, before being allowed to warm to ambient temperature and stir overnight. The resulting suspension was filtered and stripped of volatiles under reduced pressure, the resulting orange oil was dried in vacuo. NMR (C6D6, 30 °C): 1H-NMR: δH 0.81 (t, 3JHH 6.9 Hz, 6H, CH3), 2.94 (q, 3JHH 7.4 Hz, 4H, CH2), 4.89 (dd, 2JHH 13.0 Hz, JPH 5.7 Hz, 1H, CH2), 4.93 (dd, 2JHH 13.0 Hz, JPH 5.7 Hz, 1H, CH2), 6.94–7.02 (m, 1H, p-C6H5), 7.11 (7, JHH 7.8 Hz, 2H, m-C6H5), 7.50 (d, JHH 7.8 Hz, 2H, o-C6H5). 13C{1H}-NMR: δC 13.9 (d, 3JPC 6.2 Hz, H3), 43.9 (d, 3JPC 13 Hz, NH2), 77.6 (s, H2), 105.3 (d, JPC 40 Hz, Ph{PCl(NEt2)}C), 135.4 (d, JPC 24 Hz, i-C6H5), 127.6 (d, JPC 1.5 Hz, o-C6H5), 127.98 (s, p-C6H5) 128.9 (s, m-C6H5), 210.6 (d, JPC 8.4 Hz, ). 31P{1H}-NMR: δP 122.0 (s, br, 77%).
CCH2 (15).
To a THF suspension (ca. 30 cm3) of excess, pre-activated magnesium turnings containing HgCl2 (0.100 g, 3.68 × 10−4 mol) as initiator, was added drop-wise PhCCCH2Cl (1.00 g, 6.65 × 10−3 mol) as solution in THF (ca. 10 cm3); upon complete addition the mixture was brought to reflux for 4 h. After allowing to cool to ambient temperature, the mixture was filtered (via cannula) directly into a pre-cooled (−78 °C) THF solution of (Et2N)2PCl (1.39 cm3, 6.65 × 10−3 mol). The resulting red solution was stirred for 30 minutes at this temperature, before allowing it to attain ambient temperature and stir overnight. The resulting orange solution was stripped of volatiles under reduced pressure then extracted with pentane; this fraction was taken to dryness and dried in vacuo to afford the product as dark red oil. Yield: 1.46 g, 76%. 15 (74%): NMR (C6D6, 30 °C): 1H-NMR: δH 0.89 (t, 3JHH 7.0 Hz, 12H, CH3), 3.05 (q, 3JHH 7.0 Hz, 8H, CH2), 4.69 (d, JPH 7.0 Hz, 2H, CH2), 7.11–7.15 (m, 3H, m/p-C6H5), 7.63–7.59 (m, 2H, o-C6H5). 13C{1H}-NMR: δC 14.8 (d, 3JPC 3.2 Hz, H3), 43.4 (d, 3JPC 17.4 Hz, NH2), 75.0 (s, H2), 105.9 (d, JPC 13.5 Hz, i-C6H5), 137.4 (d, JPC 19 Hz, Ph{P(NEt2)2}C), 127.8 (s, o-C6H5), 127.9 (overlapped m-/p-C6H5), 209.9 (d, JPC 11.4 Hz, ). 31P{1H}-NMR: δP 91.0 (s, br, 74%). Propargyl tautomer (5%): NMR (C6D6, 30 °C): 1H-NMR: δH 1.02 (t, 3JHH 7.2 Hz, 12H, CH3), 2.71 (d, JPH 5.8 Hz, 2H, CH2P), 2.87 (m, 8H, NCH2). 13C{1H}-NMR: δC 14.0 (d, 3JPC 5 Hz, H3), 19.8 (m, H2P), 42.8 (d, 3JPC 17 Hz, NH2), 81.5 (s, CCH2P), 87.6 (s, CCH2P). 31P{1H}-NMR: δP 83.2 (s, br, 5%).
CCH2 (16).
To an ethereal solution of 15 held at −78 °C was added drop-wise two equivalent of HCl (1 M in ether). The mixture was held at −78 °C while stirring for 20 min, before being allowed to warm to ambient temperature and stir overnight. The resulting suspension was filtered and stripped of volatiles under reduced pressure, the resulting orange oil was dried in vacuo. NMR (C6D6, 30 °C): 1H-NMR: δH 0.81 (t, 3JHH 6.9 Hz, 6H, CH3), 2.94 (q, 3JHH 7.4 Hz, 4H, CH2), 4.89 (dd, 2JHH 13.0 Hz, JPH 5.7 Hz, 1H, CH2), 4.93 (dd, 2JHH 13.0 Hz, JPH 5.7 Hz, 1H, CH2), 6.94–7.02 (m, 1H, p-C6H5), 7.11 (7, JHH 7.8 Hz, 2H, m-C6H5), 7.50 (d, JHH 7.8 Hz, 2H, o-C6H5). 13C{1H}-NMR: δC 13.9 (d, 3JPC 6.2 Hz, H3), 43.9 (d, 3JPC 13 Hz, NH2), 77.6 (s, H2), 105.3 (d, JPC 40 Hz, Ph{PCl(NEt2)}C), 135.4 (d, JPC 24 Hz, i-C6H5), 127.6 (d, JPC 1.5 Hz, o-C6H5), 127.98 (s, p-C6H5) 128.9 (s, m-C6H5), 210.6 (d, JPC 8.4 Hz, ). 31P{1H}-NMR: δP 122.0 (s, br, 77%).
Platinum and palladium complexes
In a typical procedure, to a suspension of the [MCl2]n (M = Pt, Pd) in DCM was added a cooled DCM solution of the respective ligand (8, 11 or 12). The mixture was stirred overnight then stripped of volatiles under reduced pressure to afford the complexes as yellow solids, which were recrystallised from DCM/ether.CSnBu3)2Cl2] (17).
Yield: 78%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.81 (m, 12H, SnCH2), 0.88 (m, 18H, CH3), 1.27 (m, 12H, CH2), 1.44 (m, 12H, CH2), 3.78 (m, JPH ∼ 5 Hz, 4H, CH2P), 6.90–7.01 (m, 12H, m/p-C6H5), 7.63–7.77 (m, 8H, o-C6H5). 13C{1H}-NMR: δC 11.1 (s, H2Sn, 1J117SnC 365 Hz, 1J119SnC 381 Hz), 13.9 (s, H3), 23.8 (d, 1JPC 27 Hz, H2PPh2), 27.4 (s, H2CH2Sn, J117SnC 58.8, J119SnC 60.7 Hz), 29.2 (s, JSnC 10 Hz, CH3H2), 88.7 (m, CCH2PPh2), 104.0 (m, CCH2PPh2), 127.9 (br, m-P(C6H5)2), 129.a (br, i-P(C6H5)2), 131.1 (s, p-P(C6H5)2), 134.4 (m, o-P(C6H5)2). 31P{1H}-NMR: δP 6.0 (s, JPtP 3618 Hz). 119Sn{1H}-NMR: δSn −68.2 (m). 195Pt{1H}-NMR: δPt −4407 (t, JPtP 3618 Hz). Anal. Found: C, 50.23; H, 5.95. Calcd for C54H78Cl2P2PtSn2 Si: C, 50.18; H, 6.08.
CSiPri3)2Cl2] (18).
Yield: 86%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.84 (sept., 3JHH 7.1 Hz, 6H, SiCH), 0.93 (d, 3JHH 7.1 Hz, 36H, CH3), 3.87 (d, JPC 10.8 Hz, 4H, CH2P), 6.82–6.88 (m, 8H, m-P(C6H5)2), 6.91–6.95 (m, 4H, p-P(C6H5)2), 7.51–7.57 (m, 8H, o-P(C6H5)2). 13C{1H}-NMR: δC 11.6 (s, SiH), 18.8 (s, H3), 23.9 (d, JPC 40 Hz, H2P), 85.8 (m, CCH2P), 101.9 (m, CCH2P), 127.9 (m, m-C6H5), 131.1 (s, p-C6H5), 134.2 (m, o-C6H5), 134.6 (m, i-C6H5). 31P{1H}-NMR: δP 5.83 (s, JPtP 3614 Hz). 29Si{1H}-NMR: δSi −2.98. 195Pt{1H}-NMR: δPt −4399 (t, JPtP 3614 Hz). Anal. Found: C, 56.03; H, 6.39. Calcd for C48H66Cl2P2PtSi2: C, 56.13; H, 6.48.
CSiPrn3)2Cl2] (19).
Yield: 78%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.41 (m, 12H, SiCH2), 0.93 (t, 3JHH 7.2 Hz, 18H, CH3), 1.23 (m, 12H, CH2CH2Si), 3.81 (d, JPH 10 Hz, 4H, CH2P), 6.86–6.93 (m, 8H, m-P(C6H5)2), 6.94–7.00 (m, 4H, o-P(C6H5)2), 7.54–7.62 (m, 8H, o-P(C6H5)2). 13C{1H}-NMR: δC 16.3 (s, H2Si, 1JSiC 55 Hz,), 17.8 (s, H3), 18.5 (s, H2CH2Si, 2JSiC 7.4 Hz), 23.9 (d, JPC 46 Hz, H2P), 88.0 (m, CCH2Cl), 101.4 (m, CCH2P), 128.2 (m, m-C6H5), 129.0 (s, p-C6H5), 131.1 (s, o-C6H5), 134.3 (m, i-C6H5). 31P{1H}-NMR: δP 5.95 (s, JPtP 3614 Hz). 29Si{1H}-NMR: δSi −13.9. 195Pt{1H}-NMR: δPt −4403 (t, JPtP 3614 Hz). Anal. Found: C, 56.13; H, 6.45. Calcd for C48H66Cl2P2PtSi2: C, 56.13; H, 6.48.
CSiPri3)2Cl2] (20).
Yield: 88%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.90 (m, 6H, SiCH), 0.97 (d, 3JHH 6.7 Hz, 36H, CH3), 3.74 (t, JPH 3.9 Hz, 2H, CH2P), 7.05–7.11 (m, 12H, m/p-P(C6H5)2), 7.92–7.98 (m, 8H, o-P(C6H5)2). 13C{1H}-NMR: δC 11.6 (s, SiH), 18.8 (s, H3), 18.9 (t, JPC 13.6 Hz, H2P), 85.8 (d, JPC 2.9 Hz, CCH2Cl), 101.3 (d, JPC 5.6 Hz, CCH2P), 128.0 (m, m-C6H5), 129.3 (t, JPC 24 Hz, i-C6H5), 130.9 (s, p-C6H5), 134.6 (t, JPC 6 Hz, o-C6H5). 31P{1H}-NMR: δP 16.0 (s). 29Si{1H}-NMR: δSi −2.75. Anal. Found: C, 61.07; H, 6.94. Calcd for C48H66Cl2P2PdSi2: C, 61.43; H, 7.09.
CSiPrn3)2Cl2] (21).
Yield: 89%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.45 (m, 12H, SiCH2), 0.91 (t, 3JHH 7.0 Hz, 18H, CH3), 1.25 (m, 12H, CH2CH2Si), 3.75 (t, JPH 4 Hz, 4H, CH2P), 7.03–7.12 (m, 12H, m/p-P(C6H5)2), 7.89–7.98 (m, 8H, o-P(C6H5)2). 13C{1H}-NMR: δC 16.4 (s, H2Si, 1JSiC 57 Hz,), 17.8 (s, H3), 18.5 (s, H2CH2Si, 2JSiC 6 Hz), 18.8 (t, JPC 13.5 Hz, H2P), 97.9 (d, JPC 2.8 Hz, CCH2Cl), 100.8 (d, JPC 4.9 Hz, CCH2P), 128.2 (m, m-C6H5), 129.2 (t, JPC 24 Hz, i-C6H5), 130.9 (s, p-C6H5), 134.7 (t, JPC 5.5 Hz, o-C6H5). 31P{1H}-NMR: δP 15.9 (s, JSiP 23 Hz). 29Si{1H}-NMR: δSi −13.8. Anal. Found: C, 61.08; H, 7.00. Calcd for C48H66Cl2P2PdSi2: C, 61.43; H, 7.09.
CSiPrn3)2Cl2] (19).
In a borosilicate NMR tube was placed cis-19 as solution in C6D6. The sample was irradiated for 20 min. with a 500 mW full spectrum mercury lamp, resulting in precipitation of an orange solid, which redissolved upon agitation. Yield of trans-19 (by 1H NMR): 58%. NMR (C6D6, 30 °C): 1H-NMR: δH 0.46 (m, 12H, SiCH2), 0.92 (t, 3JHH 7.3 Hz, 18H, CH3), 1.25 (m, 12H, CH2CH2Si), 3.77 (t, JPH 4.3 Hz, 4H, CH2P), 7.03–7.13 (m, 12H, m/p-P(C6H5)2), 7.95–8.01 (m, 8H, o-P(C6H5)2). 13C{1H}-NMR: δC 16.4 (s, H2Si), 17.8 (s, H3), 18.5 (s, H2CH2Si), 23.8 (t, JPC 24 Hz, H2P), 88.0 (m, CCH2Cl), 101.4 (t, JPC 6.3 Hz, CCH2P), 128.2 (m, m-C6H5), 128.8 (s, p-C6H5), 130.9 (s, o-C6H5), 134.7 (t, JPC 6.0 Hz, i-C6H5). 31P{1H}-NMR: δP 11.5 (s, JPtP 2601 Hz). 29Si{1H}-NMR: δSi −13.2. 195Pt{1H}-NMR: δPt −3993 (t, JPtP 2601 Hz).
Acknowledgements
We thank the Royal Society and Leverhulme Trust (F/00 230/AL, studentship to A.J.S.) for financial support. I.R.C. gratefully acknowledges the award of a Royal Society University Research Fellowship.Notes and references
- For recent reviews see: D. W. Allen, Organophosphorus Chem., 2015, 44, 1–55 CAS; D. W. Allen, Organophosphorus Chem., 2014, 43, 1–51 Search PubMed and preceding volumes in the series; D. H. Valentine Jr. and J. H. Hillhouse, Synthesis, 2003, 2437–2460 Search PubMed; D. H. Valentine Jr. and J. H. Hillhouse, Synthesis, 2003, 317–334 Search PubMed.
- For a recent review see: E. Bernoud, R. Veillard, C. Alayrac and A.-C. Gaumont, Molecules, 2012, 17, 14573–14587 CrossRef CAS PubMed.
- R. J. Detz, S. A. Heras, R. de Gelder, P. W. N. M. van Leeuwen, H. Hiemstra, J. N. H. Reek and J. H. van Maarseveen, Org. Lett., 2006, 8, 3227–3230 CrossRef CAS PubMed.
- T. D. Simone, R. S. Dickson, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 1995, 240, 323–333 CrossRef.
- N. Maigrot, M. Melaimi, L. Ricard and P. Le Floch, Heteroatom. Chem., 2003, 14, 326–333 CrossRef CAS.
- M. Gandelman, E. M. Schuster and G. Nisnevich, US PatentUS8318960B2, 2012 Search PubMed.
- M. Reisser, A. Maier and G. Maas, Synlett, 2002, 1459–1462 CAS.
- T. J. Taylor, A. P. Soto, K. Huynh, A. J. Lough, A. C. Swain, N. C. Norman, C. A. Russell and I. Manners, Macromolecules, 2010, 43, 7446–7452 CrossRef CAS.
- T. W. Mackewitz and M. Regitz, Liebigs Ann., 1996, 327 CAS.
- R. B. King and A. Efraty, Inorg. Chim. Acta, 1970, 4, 123–128 CrossRef CAS.
- T. W. Mackewitz, D. Ulrich, U. Bergstrasser, S. Leininger and M. Regitz, Liebigs Ann., 1997, 1827–1839 CrossRef CAS.
- R. Popp, R. Gleiter and F. Rominger, Tetrahedron Lett., 2000, 41, 4075–4078 CrossRef CAS.
- M. Mirza-Aghayan, R. Boukherroub, G. Oba, G. Manuel and M. Koenig, J. Organomet. Chem., 1998, 564, 61–70 CrossRef CAS.
- R. H. Shay, B. N. Diel, D. M. Schubert and A. D. Norman, Inorg. Chem., 1988, 27, 2378–2382 CrossRef CAS.
- T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS PubMed; T. Baumgartner and R. Reau, Chem. Rev., 2006, 106, 4681–4727 CrossRef PubMed and references therein.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed; D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef PubMed and references therein.
- K. Maitra, V. J. Catalano and J. H. Nelson, J. Organomet. Chem., 1997, 529, 409–422 CrossRef CAS.
- K. Maitra, W. L. Wilson, M. M. Jemin, C. Yeung, W. S. Rader, K. D. Redwine, D. P. Striplin, V. J. Catalano, J. H. Nelson, S. Song and E. C. Alyea, Synth. React. Inorg. Met.-Org. Chem., 1996, 26, 967–996 CrossRef CAS; W. L. Wilson, N. W. Alcock, E. C. Alyea, S. Song and J. H. Nelson, Bull. Soc. Chim. Fr., 1993, 130, 673–682 Search PubMed.
- H. Lang, U. Lay, M. Leise and L. Zsolnai, Z. Naturforsch., B: Chem. Sci., 1993, 48, 27–36 CrossRef CAS.
- B. N. Diel, P. F. Brandt, R. C. Haltiwanger, M. L. J. Hackney and A. D. Norman, Inorg. Chem., 1989, 28, 2811–2816 CrossRef CAS.
- R. Regragui and P. H. Dixneuf, New J. Chem., 1988, 12, 547–550 CAS.
- J. T. Lin, S. Y. Wang, Y. C. Chou, M. L. Gong, Y.-M. Shiow, H.-M. Gau and Y. S. Wen, J. Organomet. Chem., 1996, 508, 183–193 CrossRef CAS.
- J.-S. Huang, G.-A. Yu, J. Xie, K.-M. Wong, N. Zhu and C.-M. Che, Inorg. Chem., 2008, 47, 9166–9181 CrossRef CAS PubMed.
- M. Alonso, M. A. Alvarez, E. Garcia, D. Garcia-Vivó and M. A. Ruiz, Inorg. Chem., 2010, 49, 8962–8976 CrossRef CAS PubMed.
- A. I. Arkhypchuk, A. Orthaber, V. A. Mihali, A. Ehlers, K. Lammertsma and S. Ott, Chem. Eur. J., 2013, 19, 13692–13704 CrossRef CAS PubMed.
- R. B. King and A. Efraty, Inorg. Chim. Acta, 1970, 4, 123–128 CrossRef CAS.
- G. Hogarth and J. Y. Pang, J. Organomet. Chem., 1996, 515, 193–203 CrossRef CAS.
- (a) A. J. Saunders, I. R. Crossley, M. P. Coles and S. M. Roe, Chem. Commun., 2012, 48, 5766–5768 RSC; (b) C. E. Averre, M. P. Coles, I. R. Crossley and I. J. Day, Dalton Trans., 2012, 41, 278–284 RSC; (c) V. K. Greenacre, M. B. Ansell, S. M. Roe and I. R. Crossley, Eur. J. Inorg. Chem., 2014, 5053–5062 CrossRef CAS.
- (a) V. K. Greenacre, N. Trathen and I. R. Crossley, Organometallics, 2015, 34, 2533–2542 CrossRef CAS; (b) N. Trathen, M. C. Leech, I. R. Crossley, V. K. Greenacre and S. M. Roe, Dalton Trans., 2014, 43, 9004–9007 RSC; (c) N. Trathen, V. K. Greenacre, I. R. Crossley and S. M. Roe, Organometallics, 2013, 32, 2501–2504 CrossRef CAS.
- K. Ruitenberg, H. Westmijze, H. Klejn and P. Vermeer, J. Organomet. Chem., 1984, 277, 227–234 CrossRef CAS.
- K. Kiyokawa, N. Tachikake, M. Yasuda and A. Baba, Angew. Chem., Int. Ed., 2011, 50, 10393–10396 CrossRef CAS PubMed.
- S. K. Woo, L. M. Geary and M. J. Kirsche, Angew. Chem., Int. Ed., 2012, 51, 7830–7834 CrossRef CAS PubMed.
- O. Tsutsumi and K. Nishiguchi, J. Am. Chem. Soc., 1998, 120, 1938–1939 CrossRef CAS; R. L. Phillips, I.-B. Kim, L. M. Tolbert and U. H. F. Bunz, J. Am. Chem. Soc., 2008, 130, 6952–6954 CrossRef PubMed.
- See for example: H. Gilman and S. Rosenberg, J. Org. Chem., 1959, 24, 2063–2064 CrossRef CAS; D. Seyferth and M. A. Weiner, J. Am. Chem. Soc., 1962, 84, 361–364 CrossRef; D. Seyferth and T. Wada, Inorg. Chem., 1962, 1, 78–83 CrossRef; D. Seyferth, L. G. Vaughan and R. Suzuki, J. Organomet. Chem., 1964, 1, 437–448 CrossRef; D. Seyferth and G. B. Womack, Organometallics, 1986, 5, 2360–2370 CrossRef; J. J. Eisch and J. E. Galle, J. Organomet. Chem., 1988, 341, 293–313 CrossRef; A. K. Brisdon, I. R. Crossley, R. G. Pritchard and J. E. Warren, Inorg. Chem., 2002, 41, 4748–4755 CrossRef PubMed.
- J.-P. Quintard and G. Dumartin, J. Organomet. Chem., 1984, 266, 123–138 CrossRef CAS.
- P. R. Deacon, N. Devylder, M. S. Hill, M. F. Mahon, K. C. Molloy and G. J. Price, J. Organomet. Chem., 2003, 687, 46–56 CrossRef CAS.
- G. Pattenden and D. Whybrow, Tetrahedron Lett., 1979, 20, 1885–1888 CrossRef CAS; K. V. Baker, J. M. Brown, N. Hughes, A. J. Skarnulis and A. Sexton, J. Org. Chem., 1991, 56, 698–703 CrossRef; G. Courtois, M. Harama and L. Miginiac, J. Organomet. Chem., 1980, 198, 1–14 CrossRef.
- K. S. Dunne, S. E. Lee and V. Gouveneur, J. Organomet. Chem., 2006, 691, 5246–5259 CrossRef CAS.
- R. B. King and P. M. Sundaram, J. Org. Chem., 1984, 49, 1784–1789 CrossRef CAS.
- See for example: R. L. Danheiser, Y. M. Choi, M. Menichincheri and E. J. Stoner, J. Org. Chem., 1993, 58, 322–327 CrossRef CAS.
- B. Németh, B. Khater, T. Veszprémi and J.-C. Guillemin, Dalton Trans., 2009, 3526–3535 RSC; J.-C. Guillemin, P. Savignac and J.-M. Denis, Inorg. Chem., 1991, 30, 2170–2173 CrossRef CAS; F. Nief and F. Mathey, Tetrahedron, 1991, 47, 6673–6680 CrossRef; W. Hewertson, I. C. Taylor and S. Trippett, J. Chem. Soc. C, 1970, 1835–1839 RSC.
- M. P. Simonnin and C. Charrier, Org. Magn. Reson., 1969, 1, 27–49 CrossRef.
- N. Allefeld, M. Grasse, N. Ignat'ev and B. Hoge, Chem. Eur. J., 2014, 20, 8615–8620 CrossRef CAS PubMed.
- H. Burfer and U. Groetze, J. Organomet. Chem., 1968, 12, 451 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Representative NMR spectra for compounds 8, 15 and 16, in lieu of bulk elemental analysis data. See DOI: 10.1039/c5dt03558a |
| ‡ DBP = dibenzophosphole. |
| § We note that allenylphosphonates have been more heavily studied; indeed, several of the limited allenylphosphines reported previously have been obtained through reduction of the respective phosphonates. |
| This journal is © The Royal Society of Chemistry 2016 |