
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A structural investigation of ionic liquid mixtures†
Richard P.
Matthews‡
a,
Ignacio J.
Villar-Garcia‡
ab,
Cameron C.
Weber‡
a,
Jeraime
Griffith
a,
Fiona
Cameron
a,
Jason P.
Hallett
ac,
Patricia A.
Hunt
a and
Tom
Welton
*a
aDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: t.welton@imperial.ac.uk
bDepartment of Materials, Imperial College London, London, SW7 2AZ, UK
cDepartment of Chemical Engineering, Imperial College London, London, SW7 2AZ, UK
First published on 26th February 2016
Abstract
The structures of mixtures of ionic liquids (ILs) featuring a common 1-butyl-3-methylimidazolium ([C4C1im]+) cation but different anions have been investigated both experimentally and computationally. 1H and 13C NMR of the ILs and their mixtures has been performed both on the undiluted liquids and those diluted by CD2Cl2. These experiments have been complemented by quantum chemical density functional theory calculations and molecular dynamics simulations. These techniques have identified the formation of preferential interactions between H2 of the imidazolium cation and the most strongly hydrogen bond (H-bond) accepting anion. In addition, a preference for the more weakly H-bond accepting anion to interact above the imidazolium ring through anion–π+ interactions has been identified. The modelling of these data has identified that the magnitude of these preferences are small, of the order of only a few kJ mol−1, for all IL mixtures. No clustering of the anions around a specific cation could be observed, indicating that these interactions arise from the reorientation of the cation within a randomly assigned network of anions. π+–π+ stacking of the imidazolium cations was also studied and found to be promoted by ILs with a strong H-bond accepting anion. Stacking interactions are easily disrupted by the introduction of small proportions (<50 mol%) of a weakly coordinating anion due to their propensity to form anion–π+ interactions. These results suggest that the formation of IL mixtures with different anions leads to subtle structural changes of much lower energy than the Coulombic ordering of ions, accounting for why most IL mixtures exhibit ideal, or nearly ideal, behaviour.
Introduction
Ionic liquids (ILs) are low melting salts that have found application in areas as diverse as organic and inorganic synthesis,1,2 catalysis,3 the preparation of materials,4–6 energy applications,7 separations8 and tribology.9 One of the ideas central to many of these applications has been the ability to modify IL properties through appropriate ion selection, behaviour that has led to these liquids being referred to as ‘designer solvents’.10 An extension of this concept is the fine tuning of IL properties through the use of mixtures.11 To accomplish this in a rational fashion, the impact of mixing ILs on their structure and the effect this has on their physical and chemical properties needs to be fully understood.The structures of ILs are governed by an array of different intermolecular interactions, including Coulombic forces, dispersion interactions, H-bonding and π-interactions.12 While a substantial amount of work has been conducted examining the structure of simple ILs, the impact of mixing on solution structure has received considerably less attention.12 Nonetheless, a major finding consistent across a range of IL mixtures is that the structure of IL mixtures is dominated by the random distribution of ions driven by Coulombic interactions.13–17 Within the ionic environment, more subtle structural perturbations arise as a result of the other (weaker) intermolecular forces present within these complex fluids.
A number of studies have looked at some of the effects of H-bonding interactions on IL mixtures. Brüssel et al. examined [C2C1im]Clx[SCN]1−x mixtures using ab initio molecular dynamics (MD) simulations and NMR experiments.18,19 They found that the [SCN]− anion in the mixture is displaced from interacting with H2 on the imidazolium cation (see Fig. 1 for the labelling of imidazolium cation atoms) by the stronger coordination of the Cl− anion. This illustrates that the cation can preferentially H-bond with one particular anion over another. Similar H-bonding effects have been observed by Rebelo and coworkers for mixtures of [NH4][SCN] with ILs [C4C1im][EtSO3], [C4C1im][EtSO4] and [C4C1im][OAc]. Utilising NMR spectroscopy it was found that the presence of the [NH4]+ cation reduced the strength of H-bonding interaction between the imidazolium ring hydrogens and the stronger H-bond accepting anion, which was inferred to arise from competitive H-bonding interactions between the [NH4]+ cation and this anion.20,21 MD simulations indicated that the preferential H-bonds between these ions did not affect the random distribution of ions in the network. Wang et al. and D'Anna et al. observed preferential H-bonding interactions between imidazolium cations and specific anions that give rise to non-linear NMR behaviour, particularly for H2, in [C2C1im][OAc]x[NTf2]1−x ([NTf2] = bis(trifluoromethanesulfonyl)imide) and 1-benzyl-3-butylimidazolium ([BzC4im][BF4]x[NTf2]1−x) mixtures respectively.22,23
 | ||
| Fig. 1 The [C4C1im]+ cation with atomic sites labelled as they will be referred to within this article. | ||
Other forms of spectroscopy have also observed preferential H-bonding behaviour. Optical Kerr effect (OKE) spectroscopy was used to study [C5C1im]Brx[NTf2]1−x and [C5C1im][PF6]x[TFA]1−x (TFA = trifluoroacetate).24,25 [C5C1im][PF6]x[TFA]1−x was found to exhibit non-additivity in the OKE spectra which was interpreted as random ion association without preferential interactions, however, [C5C1im]Brx[NTf2]1−x yielded additive behaviour. This was inferred as evidence for the formation of a nanostructured random block network, consistent with the development of a H-bonding network based on the more effective H-bond accepting Br− anion. Far-infrared spectroscopy illustrated the formation of preferential H-bonds between the NH group on the protic triethylammonium cation and [OAc]− anions relative to [OTf]− anions for [C2C2C2N][OAc]x[OTf]1−x mixtures.26 Furthermore, the preferential association of the Cl− anion with H2 has been predicted computationally for [C4C1im]Clx[PF6]1−x with no strong preference for either anion determined for [C4C1im][BF4]x[PF6]1−x due to the similar size and H-bond accepting capacity of these anions.13 Collectively these results suggest that preferential H-bonding interactions do occur within IL mixtures. In imidazolium ILs the interaction of the stronger H-bond accepting anion with H2 appears to be favourable although the orientation of the weaker H-bond accepting anion and an understanding of the magnitude of this preference has not been thoroughly established.
A less studied feature of imidazolium ILs is their ability to engage in π–π stacking interactions.27 The term π–π stacking is taken here to include the π+–π+ interactions between cationic aromatic imidazolium rings within an IL. For these interactions, the positive charge acts to both reduce dispersive interactions through the contraction of the π electron cloud, and to introduce a repulsive cation–cation Coulombic contribution. In this context π+–π+ interactions are only observed in the presence of additional molecules (solvent or counter anions) which facilitate and stabilise the interaction.27 Moreover the term “stacking” suggests rings which are positioned directly one above the other, however this is not the lowest energy configuration and possible stacking conformers (depending on the relative orientation of the two imidazolium rings) include stacked-parallel, stacked-rotated, stacked-antiparallel, stacked-displaced and T-shaped conformers.27
Evidence for π+–π+ interactions between imidazolium rings has been observed in the solid state for ILs including [C2C1im][NO3], [C2C1im][NO2], [C2C1im]2[SO4]·H2O, [C1C1im][OTf] and [C1C1im][NTf2].28–31 Despite evidence for π+–π+ stacking in the solid state, [C1C1im][NTf2] was not found to possess these interactions in the liquid state.28,31 Moreover, π+–π+ motifs have been assigned based on NOE data for [C2C1im]Cl in dichloromethane and in neat [C4C1im][BF4] and [C4C1C12im][BF4].32,33 π+–π+ stacking interactions have been examined computationally for imidazolium salts and found to occur in ILs including [C1C1im]Cl and [C1C1im][NO3], but not in ILs with more weakly interacting anions.27,34 This phenomenon was attributed to the stabilisation of the cationic π+–π+ interactions through both Coulombic and H-bonding interactions between the IL cation and anion.35 Further examples of π+–π+ interactions have been determined computationally. These include the observation of parallel and anti-parallel orientated cation rings for [C2C1im]Br36 and further relationships between anion size/type and π+–π+ interactions have been shown,18,37 with a general finding being that π+–π+ interactions are favoured by smaller anions as larger anions induce a greater distance between imidazolium rings, preventing their interaction.
The effect of forming an IL mixture on π+–π+ stacking has not been widely examined, although MD simulations performed by Brüssel et al. discovered that the [SCN]− anion in [C2C1im]Clx[SCN]1−x is capable of disrupting these interactions as the [SCN]− anion preferentially occupies a location above the plane of the imidazolium ring.18 The preference of larger more diffuse anions to locate above the imidazolium ring and engage in anion–π+ interactions may be competitive with π+–π+ stacking, thus limiting the range of ILs that exhibit π+–π+ phenomena, particularly in mixtures.
Several physicochemical studies have demonstrated that mixtures of ILs often show close to ideal mixing behaviours.38–42 A mixture is considered ideal if the chemical potential of its components can be directly related to the chemical potential of the pure component and its mole fraction in the mixture.11 A consequence of this definition is that for ideal mixtures the enthalpy of mixing is zero and properties thermodynamically related to the chemical potential (such as molar volume) scale with the mole fraction of the component. There is no direct relationship between the excess enthalpy or excess molar volume in terms of sign or magnitude although non-zero values of either component are indicative of a non-ideal mixture.
We have recently reported the physical properties of an extensive range of IL mixtures.43 Most of the IL mixtures studied possessed negligibly small excess molar volumes, indicating that they display close to ideal thermodynamic behaviour. Non-ideality was observed for mixtures with a substantial difference in the H-bond accepting abilities of the anions, however, the magnitude of non-ideality was small with excess molar volumes below 0.5% of the total molar volume being observed.
Nevertheless, not all mixtures of ILs will be ideal or nearly ideal and exceptions to the ideality of IL mixtures have been observed. Mixtures based on bulky phosphonium cations, such as trihexyltetradecylphosphonium chloride ([C6C6C6C14P]Cl), which contain large alkyl groups that effectively shield the central phosphorous, exhibit non-ideal behaviour at concentrations where both ILs are miscible.44,45 In such IL mixtures it is anticipated that the steric bulk of the alkyl groups interferes with the ability of the anion to associate closely with the cationic core, resulting in an increased association of anions with the smaller cation, which has a more accessible charge centre. ILs composed of one ion possessing large non-polar groups (typically the cation) have been discovered to be immiscible with ILs containing smaller highly interacting ions, moreover large excess enthalpies of mixing have been calculated for these systems.46
Despite the prevalence of specific interactions such as preferential H-bonding between different ions, close to ideal mixing of ILs has been shown to be remarkably commonplace, warranting a systematic study of the structures of IL mixtures. It is clear from previous studies that perturbations to H-bonding interactions can arise when anions of different H-bond accepting capacity are employed in mixtures. The effect of the anion on phenomena such as the π+–π+ stacking of imidazolium rings within mixtures has also not been studied experimentally to date.
In order to address these issues, six different IL mixtures based on a common [C4C1im]+ cation were studied experimentally using NMR techniques and theoretically through the use of MD simulations. IL mixtures were chosen to give a variety of anion sizes and H-bond accepting differences; [C4C1im][Me2PO4]x[NTf2]1−x, [C4C1im][OAc]x[NTf2]1−x, [C4C1im][OTf]x[NTf2]1−x, [C4C1im]Clx[NTf2]1−x, [C4C1im]Clx[SCN]1−x and [C4C1im]Clx[OTf]1−x. Furthermore, our previous physicochemical investigations found that [C4C1im][Me2PO4]x[NTf2]1−x formed non-ideal mixtures whereas [C4C1im][OTf]x[NTf2]1−x mixtures were very close to ideal, with [C4C1im]Clx[NTf2]1−x being intermediate between these two extremes. Thus, these mixtures were chosen to enable a direct comparison between mixtures of similar ILs that exhibit an increase in non-ideality.43 [C4C1im]Clx[SCN]1−x was included to build upon the computational work of Brüssel et al.18
Results and discussion
NMR of undiluted ionic liquids and their mixtures
The 1H and 13C NMR spectra of the undiluted ILs and their mixtures were obtained to gain an understanding of any preferential interactions, such as H-bonding, between specific cation sites and the anions, as well as the magnitude of these associations. While the absolute chemical shifts obtained for each chemical environment do not provide direct information on the structure of the mixture, the examination of different chemical environments for each mole fraction of a component within the mixture allows for the presence of specific interactions to be inferred (vide infra). For the NMR spectra, a capillary filled with DMSO-d6 was used as a lock and chemical shift reference. As [C4C1im]Cl is solid at room temperature, these investigations were performed at 80 °C to enable all compositions to be explored.Fig. 2 depicts the H2, H4 and H5 signals for [C4C1im][Me2PO4]x[NTf2]1−x, [C4C1im]Clx[SCN]1−x and [C4C1im][OTf]x[NTf2]1−x. All graphs are depicted with the concentration of the most H-bond accepting anion in the mixture on the x-axis to enable a more facile comparison between mixtures. 1H and 13C NMR chemical shifts are provided in ESI,† Tables S28–S50 and Fig. S24–S42, and details of the fitted model are provided in the ESI,† (Section D) and below.
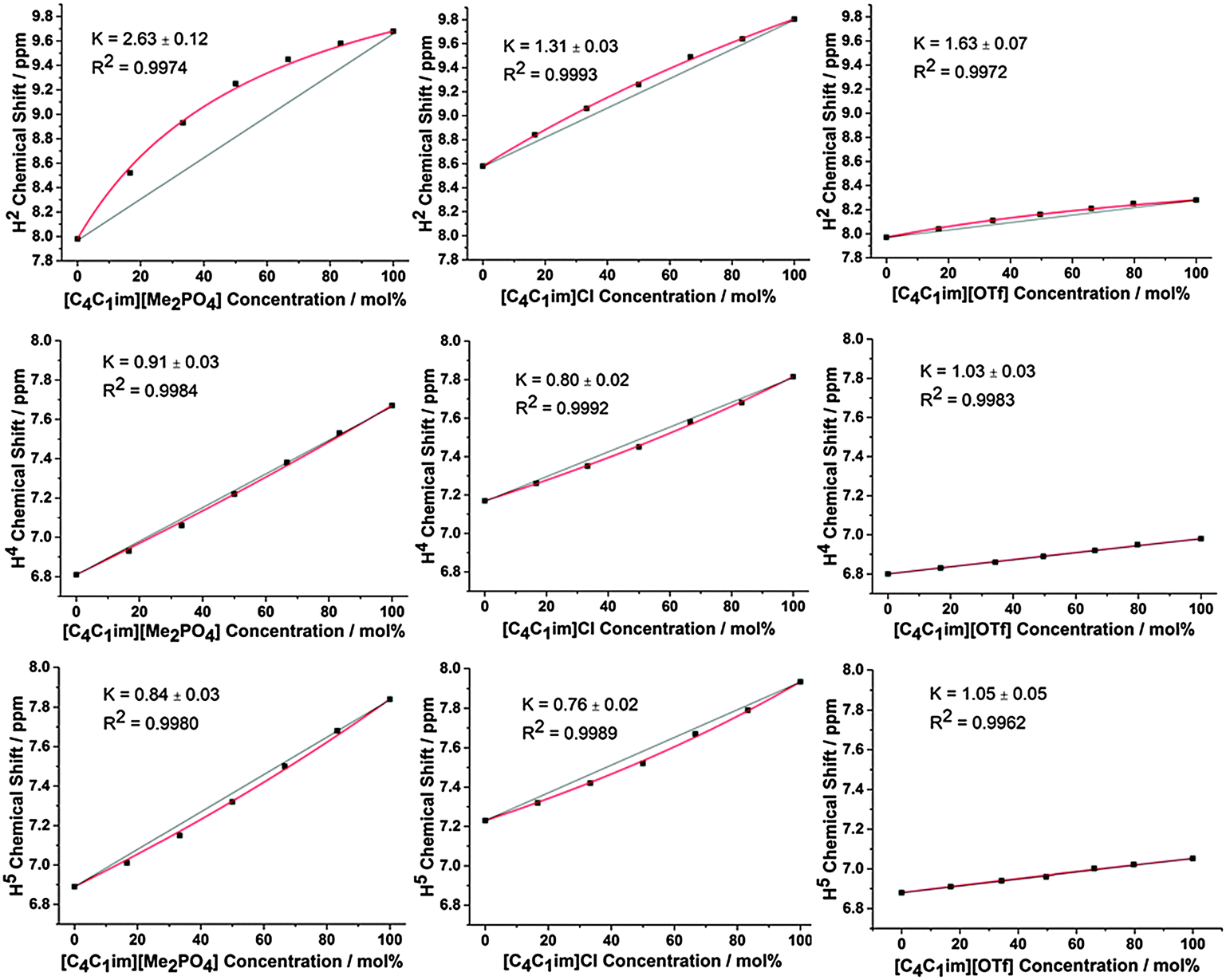 | ||
| Fig. 2 Neat 1H chemical shifts obtained for H2 (top), H4 (middle) and H5 (bottom) of the [C4C1im][Me2PO4]x[NTf2]1−x mixtures (left), [C4C1im]Clx[SCN]1−x mixtures (middle) and [C4C1im][OTf]x[NTf2]1−x mixtures (right). Red lines represent the fitted model with given fitting parameter (see below), straight black lines are drawn between the chemical shifts of the simple ILs as a guide for the eye. | ||
From Fig. 2 and ESI,† Fig. S24–S29 it is apparent that the chemical shifts for H2, H4 and H5 follow a similar trend for all of the IL mixtures. All of the chemical shifts for these resonances lie within the range defined by the simple IL components of the mixture. Increasing the strength of H-bonding interactions has been associated with a downfield shift of the H-bond donating resonance in the 1H NMR, i.e. an increased chemical shift. Hence, displacement of the chemical shift above the black line joining the chemical shift of both simple ILs is indicative of preferential H-bonding between the relevant hydrogen and the stronger H-bond acceptor, i.e. the 1H environment interacts to a greater extent with the stronger H-bond accepting anion over the NMR timescale than would be expected from a random association of anions with that environment. These results indicate that H2 preferentially interacts with the stronger H-bond accepting anion, while H4 and H5 either exhibit no preference for either anion or a slight preference for the more weakly H-bonding anion.
The impact of H-bonding on the 13C NMR signals is more complex as there is an interplay between electron density changes due to the formation of the H-bond, and variations in the energy of the molecular orbitals. Despite experimental and theoretical results for ILs indicating that electron density increases on the carbon atom upon the formation of a stronger C–H⋯A− H-bond, the 13C signals of C2/4/5 move downfield with increasing H-bond accepting ability of the anion.47,48 This phenomenon has been attributed to the reduction in the natural bond order of the C–H bond dominating electron density effects. Correspondingly, the influence of stronger H-bonding on the chemical shift of 13C NMR signals would be expected to follow a similar trend to that of the 1H NMR, i.e. a downfield (higher ppm) shift. The 13C NMR results for C2 (Fig. 3 and Fig. S31–S36, Tables S35–S40, ESI†) are largely consistent with those obtained for H2 for all of the mixtures.
 | ||
| Fig. 3 Neat 13C chemical shifts obtained for C2 (top), C4 (middle) and C5 (bottom) of the [C4C1im][Me2PO4]x[NTf2]1−x mixtures (left), [C4C1im]Clx[SCN]1−x mixtures (middle) and [C4C1im][OTf]x[NTf2]1−x mixtures (right). Red lines represent the fitted model with given fitting parameter (see below), straight black lines are drawn between the chemical shifts of the simple ILs as a guide for the eye. | ||
The 13C NMR results for C4 and C5, however, show a much greater affinity for the more weakly H-bond accepting anion than their H4 and H5 counterparts, i.e. a larger negative deviation from linearity, with the exception of [C4C1im][OTf]x[NTf2]1−x where the chemical shift change between the simple ILs was too small to determine a distinct trend. This suggests that the more weakly H-bond accepting anion has a greater preference for interactions with C4/C5 than H4/H5, consistent with the anion predominantly occupying a position above the imidazolium ring rather than an in-plane interaction with the back of the ring.
A quantum chemical study of ion-pair dimers (IP-dimers) which included [C1C1im]2[OTf]2 found that this IP-dimer only formed stable conformers with an alternating (diagonal anion) configuration, Fig. 4.34 In these conformers the anion is positioned above the ring with one O-atom sitting over the C4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C5 bond at the rear of the ring. This conformation is consistent with the 13C NMR data which suggests preferential interaction of weak H-bonding anions with the C4/5 atoms. This can be rationalised by weakly H-bonding anions being typically more diffuse and better able to form dispersive interactions with the π-cloud of the imidazolium ring than strongly coordinating anions. In these calculated structures, there are no H-bonds with the rear of the ring, but there can be supporting H-bond interactions with the alkyl chain. The calculated structures do not feature a more strongly H-bond accepting anion which would interact preferentially with H2 and could promote the interaction of the weakly coordinating anions with H4/5 as is observed from the NMR data.
C5 bond at the rear of the ring. This conformation is consistent with the 13C NMR data which suggests preferential interaction of weak H-bonding anions with the C4/5 atoms. This can be rationalised by weakly H-bonding anions being typically more diffuse and better able to form dispersive interactions with the π-cloud of the imidazolium ring than strongly coordinating anions. In these calculated structures, there are no H-bonds with the rear of the ring, but there can be supporting H-bond interactions with the alkyl chain. The calculated structures do not feature a more strongly H-bond accepting anion which would interact preferentially with H2 and could promote the interaction of the weakly coordinating anions with H4/5 as is observed from the NMR data.
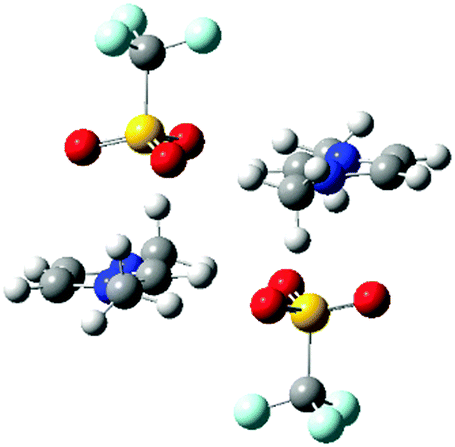 | ||
| Fig. 4 Stable IP-dimer of [C1C1im]2[OTf]2 showing a strong anion–π+ interaction, the effect on C4/5 will be much stronger than the effect of the anion on H4/5. | ||
It is worth noting the contrast between the curves in NMR chemical shift obtained here (for H2, H4 and H5) compared to the linear behaviour reported by Brüssel et al. for [C4C1im]Clx[SCN]1−x mixtures.18 This discrepancy can be rationalised by consideration of the different temperatures used for the experiments which precluded, in their case, the examination of mixtures with mole fractions of Cl− anions greater than 0.5. The very subtle variation from linearity observed in Fig. 2 would not be evident over the reduced composition range, leading to the initial finding of a linear relationship. Nonetheless, in both cases it is evident that within these mixtures any preferential interactions of either anion with a specific position on the cation ring are small in magnitude.
Another important comparison is between the non-linear NMR results obtained here and the linear dependence of the C2–H2 IR stretch obtained for similar mixtures containing the [C4C1im]+ cation.49 One of the major differences between these two techniques is the time scale, with IR providing measurements over a much shorter timescale than NMR. This suggests that perhaps the total number of H-bonds formed with each anion varies consistently with the anion composition of the mixture (leading to the linear dependence in IR) but the lifetime of H-bonds with the better H-bond accepting anion are longer (leading to a non-linear NMR dependence which represents a time weighted average of these interactions). Differences in H-bond dynamics have been investigated computationally for simple ILs previously,50,51 but are beyond the scope of the current study.
To enable trends between mixtures to be more readily compared, a simple model is proposed, based on an equilibrium between the IL anions (A1 and A2) and an interacting atom on the cation (Ci, eqn (1) and (2)). This model is an adaptation of that introduced by Bosch and Rosés as well as Skwierczynski and Connors to analyse the preferential solvation of solvatochromic dyes in mixed solvent systems.52,53 While this model has been used previously to analyse solvatochromic dyes and rate constants in binary IL mixtures,54,55 to the best of our knowledge this is the first time that it has been applied to the modelling of NMR data.
| CiA1 + A2 ⇌ CiA2 + A1 | (1) |
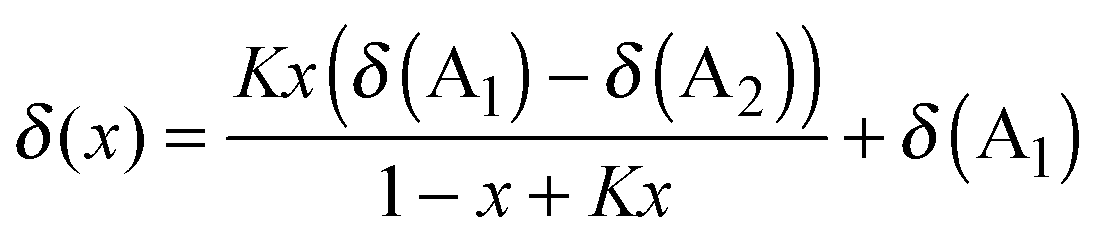 | (2) |
From eqn (1) and (2), A1 and A2 were assigned such that A2 was always the more strongly H-bond accepting anion, as determined by its Kamlet–Taft β value, so that more favourable interactions of this anion with a chemical environment on the cation would result in K > 1 and vice versa. It is important to note that this model implicitly assumes that the chemical shift can be described as an equilibrium between the local structures of the two simple ILs at a given cation environment, regardless of what those structures are, and does not necessarily imply the formation and exchange of well-defined ion pairs. Consequently the model is not able to account for conformations not present at the cation environment in the simple ILs (e.g. a bifurcated H-bond between different anions). However, despite these assumptions and the simplicity of this model, the majority of our data is well modelled by this approach. Full details of the underlying assumptions and limitations of the model are detailed in ESI,† (Section D).
Fig. 2 and 3 detail the equilibrium constants obtained using the model for the H2/4/5 atoms and C2/4/5 atoms of [C4C1im][Me2PO4]x[NTf2]1−x, [C4C1im]Clx[SCN]1−x and [C4C1im][OTf]x[NTf2]1−x. The K values obtained represent a relative preference rather than absolute information. A large K value is not necessarily indicative of stronger H-bonding at that site, only that there is a greater preference for that cation environment to be dominated by interactions with the most strongly H-bond accepting anion. In general, it was found that the strong H-bond accepting anion interacts preferentially with H2 and C2 (K > 1.0) and that the more weakly coordinating H-bonding anion likely forms anion–π+ interactions with C4/5 (K ≪ 1.0) and weak preferential interactions with H4/5 (K < 1.0).
To assess trends in preferential interactions across the range of IL mixtures examined, the free energy changes derived from the fitted values of K (using ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnK) have been plotted against the difference in H-bond accepting strength of the anions, as determined by the difference in Kamlet–Taft β values (Fig. 5). The Kamlet–Taft Δβ values used for the mixtures are given in ESI,† Table S42. The free energies derived from this model should be treated with some caution given the simplicity of the underlying model. Nonetheless, the excellent fits to the chemical shift data obtained from multiple independent experiments suggests that it should be a reasonable approximation of the true free energy difference.
lnK) have been plotted against the difference in H-bond accepting strength of the anions, as determined by the difference in Kamlet–Taft β values (Fig. 5). The Kamlet–Taft Δβ values used for the mixtures are given in ESI,† Table S42. The free energies derived from this model should be treated with some caution given the simplicity of the underlying model. Nonetheless, the excellent fits to the chemical shift data obtained from multiple independent experiments suggests that it should be a reasonable approximation of the true free energy difference.
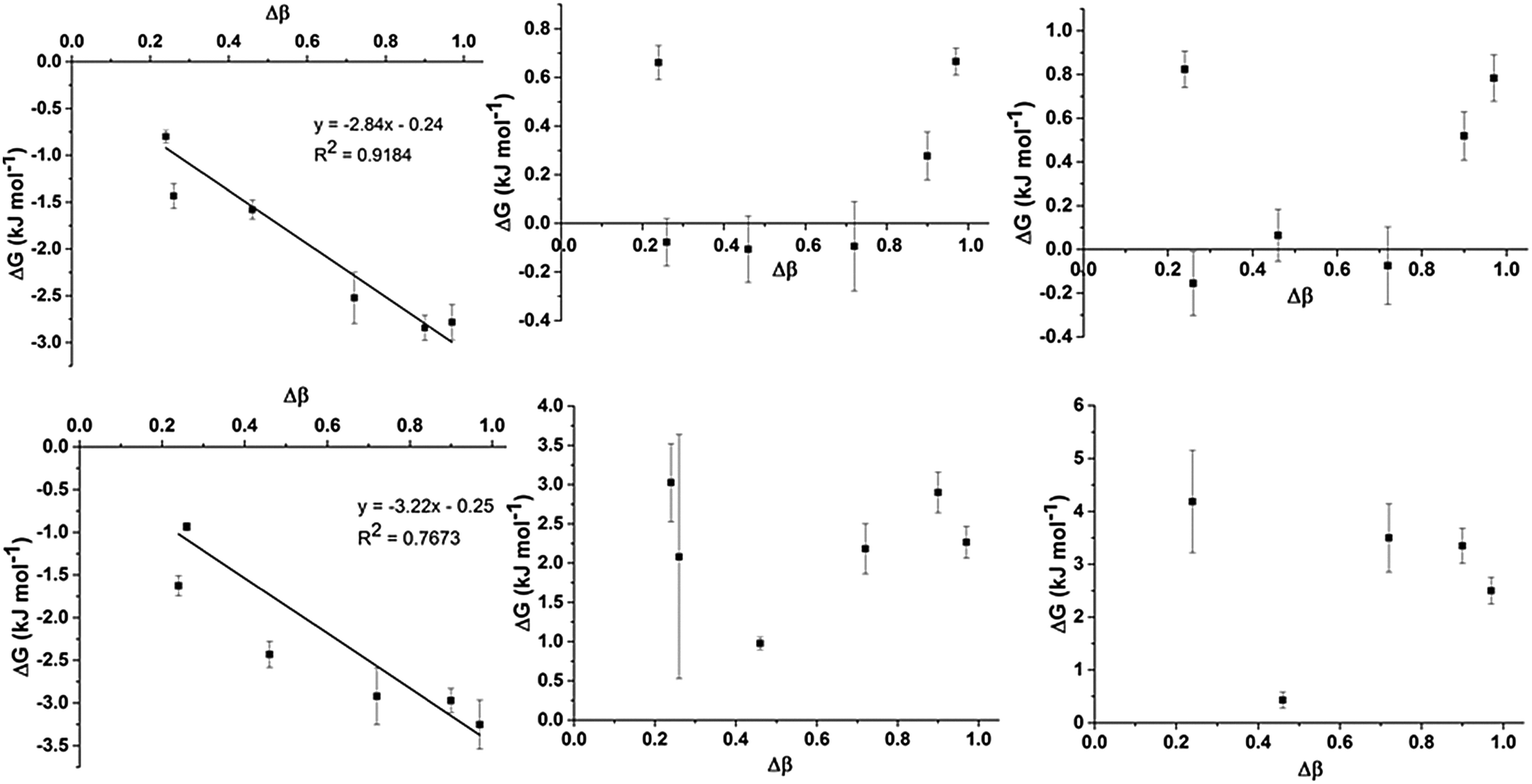 | ||
| Fig. 5 Relationship between ΔG obtained from the preferential interaction model and Δβ of the IL mixture for H2 (top left), H4 (top centre), H5 (top right), C2 (bottom left), C4 (bottom centre) and C5 (bottom right). Where an approximately linear relationship was observed, a linear fit was made and the fitted parameters reported. Reported errors are fitting errors propagated through the free energy calculation. | ||
From Fig. 5, some interesting trends emerge. The preferential interaction of H2 and C2 with the more strongly H-bonding anion increases as the difference in the H-bond acceptor capacity of the anion increases. This result is not surprising given the well-known ability of H2 to be the most effective H-bond donor on the imidazolium cation.33,48 However, it is more remarkable that this preference appears to be very small in magnitude (∼3 kJ mol−1) even for strongly interacting anions such as [Me2PO4]− and [OAc]−. The computed interaction energies for ion pairs of [C1C1im]Cl and [C1C1im][Me2PO4] with H4 (side conformer) and H2 (front conformer) only differ by ≈10 kJ mol−1 in a CH2Cl2 CPCM and an IL SMD model which will be discussed in greater detail in the following section. These energy differences are close to that of thermal energy (RT) at 80 °C suggesting that H2/4/5 will all be approximately equally populated over time, allowing the other anion within the mixture easy access. It is mutually supportive that both experimental and computational methods independently arrive at similar (very small) energy differences.
There is no generalised trend for preferential interactions with H4/5 and C4/5. There is a limited trend for the weaker anion to increasingly associate with H4/5 for Δβ above an apparent threshold of Δβ > 0.72. However, ΔG for these preferential interactions is less than 1 kJ mol−1. A notable outlier is the [SCN]− anion in the [C4C1im]Clx[SCN]1−x mixtures (Δβ = 0.24) which appears to preferentially interact with H4 and H5 despite both [SCN]− and Cl− exhibiting similar H-bond accepting ability. The [OTf]− and [NTf2]− anions have an analogously small Δβ (0.26) but [NTf2]− does not exhibit a stronger preference over [OTf]− for H4/5. The contrast between the pairs Clx[SCN]1−x and [OTf]x[NTf2]1−x may be due to the greater absolute H-bond accepting ability of [SCN]− and Cl−, hence [SCN]− is more likely to favour the formation of H-bonding interactions than [NTf2]−.
C4 and C5 always favour interacting with the weaker H-bond accepting anion (all positive ΔG), although this preference is very small for [C4C1im]Clx[OTf]1−x (ΔG < 1 kJ mol−1). [C4C1im]Clx[OTf]1−x is the only mixture in which [OTf]− is the weakest H-bond acceptor (the others feature [SCN]− or [NTf2]−) and therefore this result may indicate that the positioning of [OTf]− at C4 and C5 may be determined by factors other than simply H-bonding ability. This result could be rationalised by considering that [OTf]− is smaller and less diffuse than [NTf2]−, leading to a reduced propensity of this anion to form anion–π+ interactions compared to [NTf2]− in the Clx[NTf2]1−x mixtures. Compared to the Clx[SCN]1−x mixtures, [OTf]− is a weaker H-bond acceptor and the polarisable sulfur atom is less accessible than that in [SCN]− leading to weaker H-bonding interactions with H4/5 (incorporated in C4/5 changes) as well as weaker dispersive interactions with the π system leading to the comparatively reduced C4/5 preference for [OTf]− in Clx[OTf]1−x mixtures. MD simulations (discussed fully below) show that both the Cl− and [OTf]− interact almost equally with C4 and C5 (ESI,† Fig. S4), consistent with the small preference observed experimentally.
To isolate H-bonding effects from geometric considerations, the 1-butyl-2,3-dimethylimidazolium ([C4C1C12im]+) cation was employed in mixtures with Cl− and [NTf2]− anions. Methylating the C2 position removes H2, the most H-bond acidic site on the imidazolium cation, rendering H4 and H5 the most H-bond acidic positions, albeit with a lower propensity for H-bond donation than H2 in the unsubstituted imidazolium ring. Results for the 2-methyl, ring protons and the corresponding 13C signals are depicted in Fig. 6, with all NMR signals reported in ESI,† Fig. S30, S37, Tables S34 and S41.
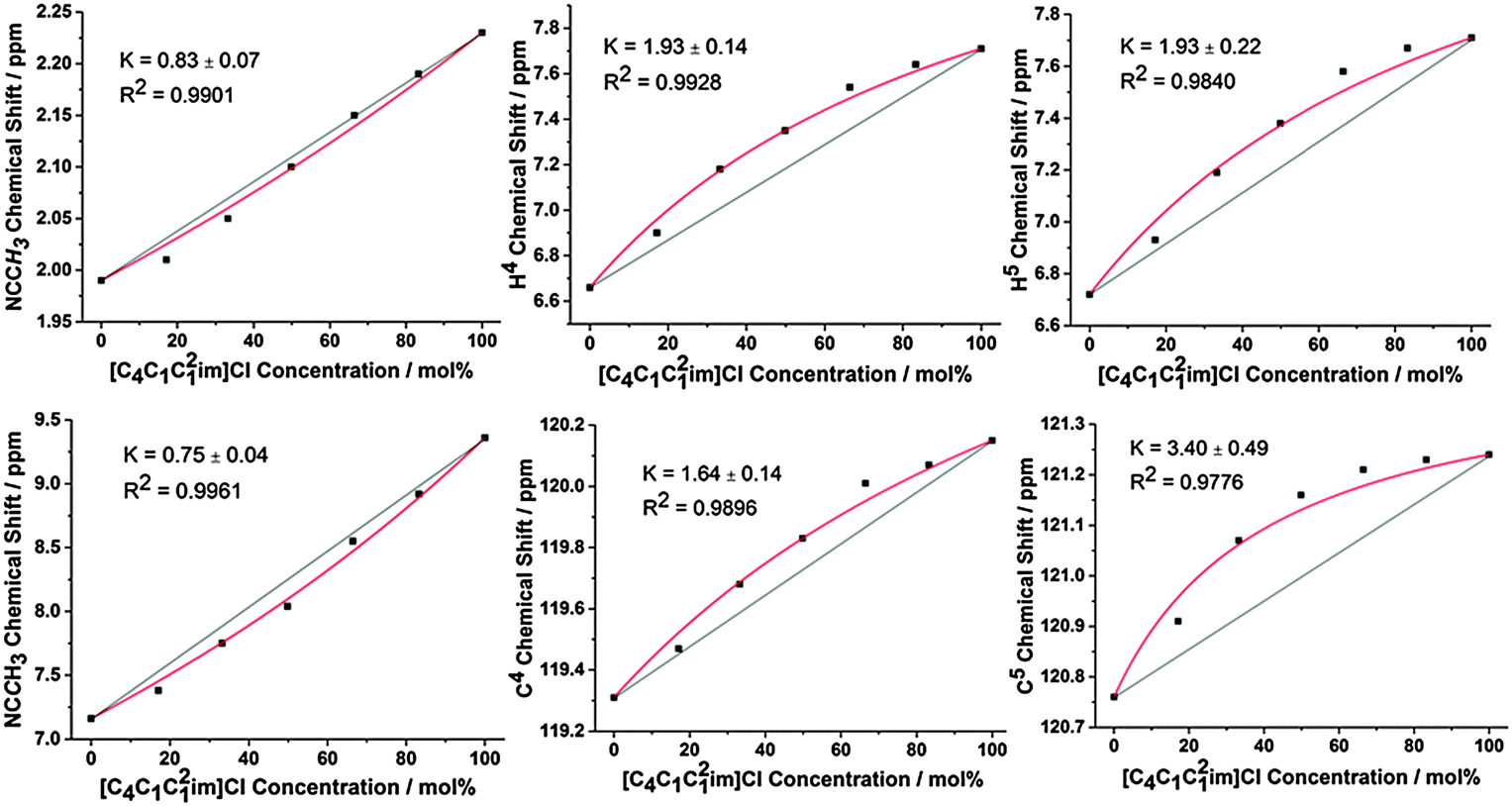 | ||
| Fig. 6 Neat 1H (top) and 13C (bottom) NMR chemical shifts for the 2-methyl (denoted NCCH3), C4/H4 and C5/H5 of the [C4C1C12im]Clx[NTf2]1−x mixtures. Red lines represent the fitted model, straight black lines are drawn between the chemical shifts of the simple ILs as a guide for the eye. | ||
The behaviour for this set of mixtures is different to that of the mixtures based on the [C4C1im]+ cation. Notably, the Cl− anion now preferentially interacts with H4 and H5 at the back of the ring which are now the strongest available H-bond donor sites. Moreover, there is a slight preference for the [NTf2]− anion to interact with the 2-methyl position at the front. The change in preferential interaction is directly in line with the H-bond strength of these positions and illustrates that the H-bond donor ability of the ring positions determines the extent of the preferential association with specific anions in the mixture.
Again, these preferential interactions are very small, with K values for the H4/H5 positions less than 2 (ΔG < 2 kJ mol−1). These values are also smaller than the analogous K and ΔG found for the H2 position of the [C4C1im]Clx[NTf2]1−x mixtures, consistent with the higher H-bond donating ability of H2. The 13C NMR is subtly different from the 1H NMR results with an increased preference of the Cl− anion for C5 compared to C4 whereas identical results were obtained for H4/5. As the C5 position is likely to be more sterically hindered compared to C4 due to the adjacent butyl chain this may simply represent a decrease in the interaction of the much larger [NTf2]− at this position rather than increased Cl− association. The 2-methyl position exhibits a slight preference for [NTf2]−, in line with the 1H NMR, however the C2 resonance has the most favourable interaction with the [NTf2]− anion (K = 0.34 compared to K = 0.75 for 2-methyl), indicative of anion–π+ interactions with the opposite side of the imidazolium ring to the strongest H-bond donating groups.
Results so far have suggested that there is a slight preference for the association of anions with specific locations around the cation due to H-bonding. Stronger H-bond acceptors have a preference for interactions with H2 in the front of the ring, and weaker H-bond acceptors have a preference for an anion–π+ interaction (above the ring) and/or H-bonding with H4 and H5. However, we have not yet probed the mechanism for movement of ions to these locations. Two possibilities are that anions can diffuse into the preferred location or, alternatively, a cation surrounded by a selection of anions can rotate so as to preferentially orientate towards a specific anion.
To probe this more thoroughly, [C4C1im]0.50[C4C1pyrr]0.50Clx[NTf2]1−x and [C4C1im]x[C4C1pyrr]1−xClx[NTf2]1−x mixtures have been examined to determine whether preferential anion clustering around the imidazolium or pyrrolidinium ion could be observed. The [C4C1pyrr]+ cation was chosen because it is a weaker H-bond donor than [C4C1im]+, thus it is anticipated that strongly H-bonding anions such as Cl− would favour interacting with [C4C1im]+ over [C4C1pyrr]+ if ion translation was able to occur. Conversely, if preferential association is simply due to the rotation of the imidazolium cation within an established ionic matrix, the chemical shifts observed should be independent of the cation composition. The results for H2, H4 and H5 and the corresponding 13C signals for the [C4C1im]+ cation are depicted in Fig. 7. The other signals, including those for the [C4C1pyrr]+ cation, can be found in ESI,† Fig. S38, S39, S41, S42 and Tables S43–S50. No reliable data could be obtained for x > 0.50 for the [C4C1im]0.50[C4C1pyrr]0.50Clx[NTf2]1−x mixtures due to their high melting points which prevented a neat NMR spectrum being obtained at 80 °C.
 | ||
| Fig. 7 Comparison of chemical shifts observed for [C4C1im]Clx[NTf2]1−x (black squares), [C4C1im]0.50[C4C1pyrr]0.50Clx[NTf2]1−x (red circles) and [C4C1im]x[C4C1pyrr]1−xClx[NTf2]1−x (blue triangles) mixtures as a function of the chloride ratio. The 1H NMR for H2, H4 and H5 are depicted on the top and 13C NMR for C2, C4 and C5 on the bottom. Black lines are drawn between the chemical shifts of the simple [C4C1im]Cl and [C4C1im][NTf2] ILs as a guide to the eye. | ||
From Fig. 7 it is apparent that the chemical shift observed is dependent only on the anion composition and not the relative proportion of cations present. The chemical shifts of H2/4/5 or C2/4/5 of [C4C1im]+ do not change when [C4C1pyrr]+ is present in the mixture, thus there is no local increase in the concentration of the strongly H-bonding Cl− anions around the better H-bond donor [C4C1im]+. Thus, there is no evidence for the preferential association or “clustering” of specific anions around the [C4C1im]+ cation when the [C4C1pyrr]+ cation is present. A similar outcome is observed from the NMR spectra of [C4C1pyrr]+, with all 1H and 13C NMR signals being dependent only on the anion composition (ESI,† Fig. S41 and S42). This result is important, it reinforces the hypothesis that there is an essentially random distribution of (mutually repulsive) anions and that preferential interactions arise from subtle perturbations to the orientation of the cations within the ionic framework, i.e. there is no net displacement of particular ions.15
However, preferential interactions of the imidazolium ring atoms with specific anions are identified, thus within a given solvation shell the cation is rotating so as to interact through H2 with the best H-bond accepting anion, depicted schematically in Fig. 8. Blue cations represent the stronger H-bond donor and red anions the stronger H-bond acceptors. Within the random distribution of cations and anions, some “blue” cations find a “red” anion within their solvation shell and rotate so as to spend a larger proportion of time forming an H-bond with that anion. Simple rotation of the cation within a diffuse solvation shell is in accordance with the low energy (<3.5 kJ mol−1) determined earlier using the equilibrium model. Fig. 8 illustrates a simplified snapshot and it is likely that the ‘preference’ observed by NMR relates more to H-bond dynamics than the absolute number of H-bonding interactions. Hence, sites with H-bonds between the best H-bond donating/accepting pair are longer lived than those between weaker pairs.
 | ||
| Fig. 8 Schematic ‘snapshot’ illustrating the preferential orientation of H-bonding within a randomly ordered ionic network for a reciprocal binary mixture [C1]x[C2]1−x[A1]x[A2]1−x where blue cations represent those with good H-bond donating groups and red anions are the strongest H-bond acceptors. | ||
NMR of the simple ionic liquids in CD2Cl2
In the previous section, it was found that preferential interactions within the IL environment are relatively weak and relate to the rotation of cations within a randomly organised network of anions. As these effects are of relatively low energy and since ILs are well-known to possess a plethora of intermolecular forces, the influence of the formation of a mixture on other weak intermolecular interactions was studied. In particular, the presence of π+–π+ stacking and the role of the previously discussed anion–π+ interactions are of interest.In order to assess these weak interactions, it is necessary to eliminate contributions from stronger effects such as ion-pairing and H-bonding. Based on NMR and conductivity data, Avent et al. have proposed that imidazolium halide (Cl−, Br− or I−) ILs in non-polar solvents, such as chloroform or dichloromethane, consist of associated ions, i.e. ion-pairs or other larger neutral ion clusters, rather than discrete solvated ions at all concentrations meaning that H-bonding and ion-pairing effects are not observed in NMR spectra with changing concentration.33 However, as the concentration of the IL is increased the effects of clustering and aggregation of the ion-pairs can be observed.
In an NMR spectrum, where π+–π+ stacking occurs, the magnetic cone above and below the aromatic cation rings, induced by the delocalised π electrons, opposes the applied magnetic field, shielding nuclei within the cone and reducing the chemical shift. Nuclei outside of the cone are deshielded and have an increased chemical shift. Previously for IL solutions of [C2C1im]X (X = Cl, Br and I), an upfield shift of the H2 signals with increasing IL concentration (in CD2Cl2) has been associated with the H2 proton lying within the shielding cone of a neighbouring imidazolium ring. The H4/H5 signals typically shift downfield indicating they are outside the shielding cone. Taken together these results have been interpreted as the formation of rotated π stacked structures.33 It is worth emphasising that the NMR spectrum represents a weighted average over all conformations present within the liquid phase, so these results imply that rotated π stacked structures are preferentially formed or are longer lived than other conformations within the liquid.
Theoretical calculations on IL IP-dimers of imidazolium salts have identified two key motifs; a stacked cation conformer where the anions are located on the periphery but are positioned vertically (in the “middle”) between the cations (Fig. 9a), and an alternating anion above or below the cation conformer where the anions are located on the “diagonal” and the cation rings are displaced relative to each other (Fig. 9b).27,34 In the π+–π+ stacked IP-dimer structure, anions are positioned in front of H2 and slightly out of plane of one imidazolium ring while the anions also lie slightly out of plane and interact via an H-bond with the rear H4/5 of the other imidazolium ring. Calculations suggest that this arrangement stabilises an otherwise repulsive π+–π+ interaction and is facilitated by the strength of the H-bonds between the cation and anion.34 Substitution of strong H-bonding anions (e.g. Cl−, [NO3]−) with weakly coordinating anions (e.g. [OTf]− or [BF4]−) leads to a shift in the more favoured conformer from the π+–π+ stacked to alternating cation–anion conformer. A preference for the alternating structure has also been recently established for [C4C1im]2[NTf2]2 IP-dimers as well as the [C1C1im]2[OTf]2 dimers introduced in the previous section.56 In the alternating configuration anions form anion–π+ (dispersive) interactions, thus precluding stacking of the imidazolium π systems.34 In contrast to the stacked conformer, the anions in the alternating conformer exhibit essentially no interaction with H4/5 at the rear of the ring.
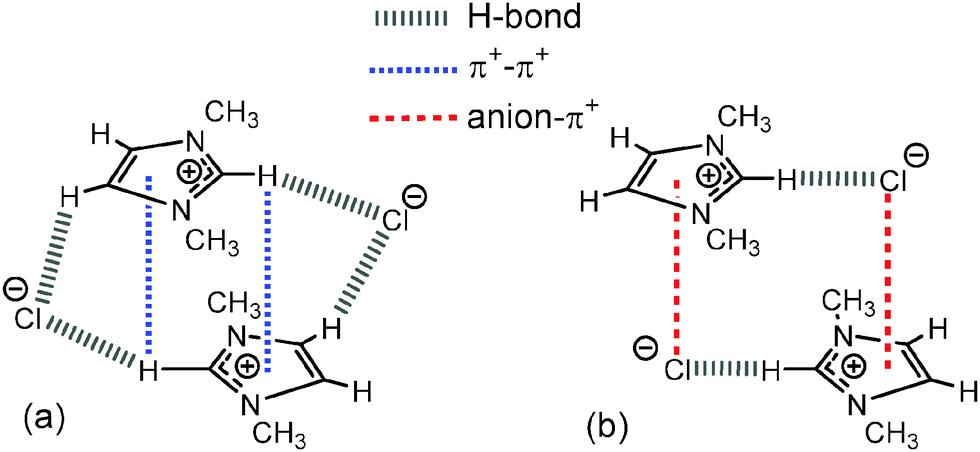 | ||
| Fig. 9 (a) Stacked “middle” structure of [C1C1im]Cl showing C2–H2 positioned in the magnetic cone of the aromatic ring and (b) alternating “diagonal” structure of [C1C1im]Cl showing ion-pairs with a dominant front C2–H2 H-bond and weak anion–π+ interaction.34 Major H-bonding, anion–π+ and π+–π+ interactions are depicted by dashed lines. | ||
Hence, we have investigated the 1H and 13C NMR of ILs and IL mixtures in CD2Cl2 to give insight into the interplay between π+–π+ stacking and the anion–π+ interactions identified as occurring in the undiluted IL mixtures. The NMR spectra for the simple ILs [C4C1im]Cl, [C4C1im][OTf], [C4C1im][SCN], [C4C1im][NTf2] and [C4C1im][Me2PO4] are detailed in Fig. 10. Spectra range from those in highly dilute solutions to those with a concentration approaching 2.5 mol L−1, which is close to that of a pure IL (cf. simple [C4C1im][NTf2] has a concentration of 3.4 mol L−1 at 25 °C). The relative chemical shift between the ions depends primarily on the H-bonding ability of the anion, for example dilute [C4C1im][X] have chemical shifts at 11.13 ppm, 10.89 ppm, 9.55 ppm, 9.18 ppm and 8.68 ppm for X = Cl, Me2PO4, SCN, OTf and NTf2 respectively. No significant change in chemical shift for the most dilute H4/5 resonances are observed between ILs, consistent with an ion-pair H-bonding via H2. The y-axis of Fig. 10 shows a uniform span of 1.4 ppm for H2 and 1 ppm for H4/5, allowing a comparison to be made between anions for the magnitude of the total change in chemical shift as the concentration of the IL is increased.
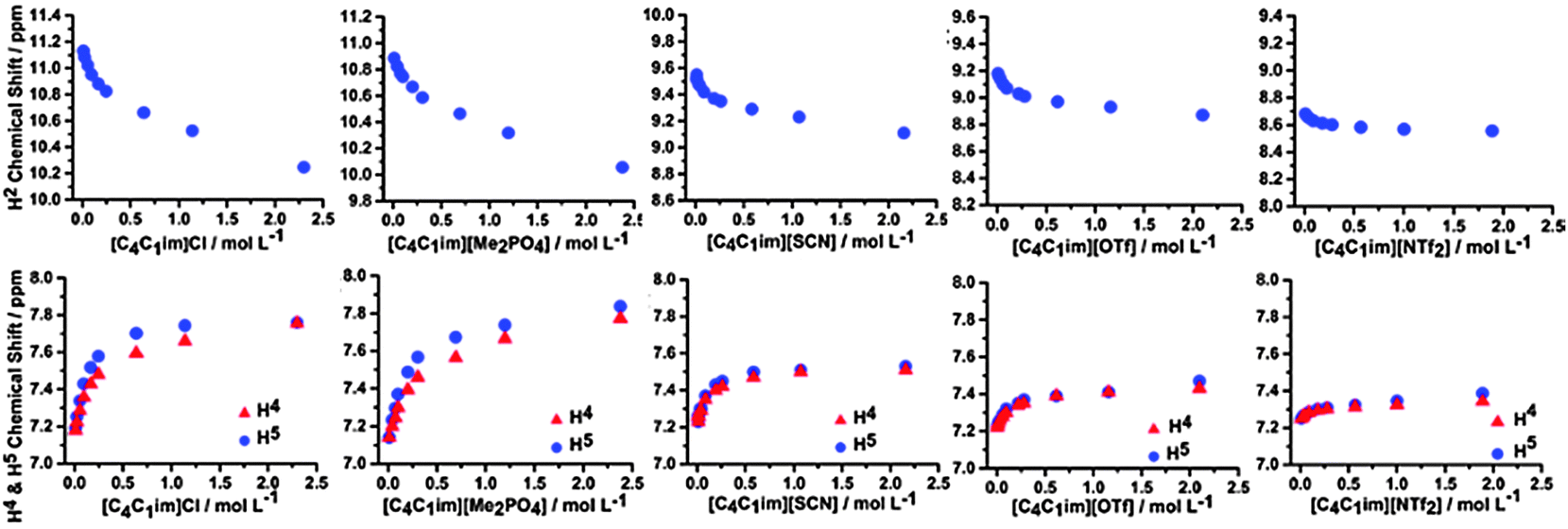 | ||
| Fig. 10 1H chemical shifts of the H2, H4 and H5 signals obtained in CD2Cl2 for various concentrations of the simple ILs. | ||
The ILs studied here show a similar behaviour to that obtained for [C2C1im]X (X = Cl, Br and I) in CD2Cl2.33 The most pronounced changes in the 1H chemical shifts of H2 and H4/5 are observed for [C4C1im]Cl and [C4C1im][Me2PO4], [C4C1im][OTf] and [C4C1im][SCN] show intermediate behaviour, and only a very weak effect is found for [C4C1im][NTf2]. The observed upfield shift of H2 with increasing concentration is indicative of π+–π+ stacking whereby H2 lies above the shielding cone of an adjacent imidazolium ring with the corresponding downfield shift of H4/5 due to positioning of these atoms outside of the shielding cone. However, if the conformers in Fig. 9 are considered it is evident that the transition from a “diagonal” to a “middle” structure is accompanied by an out of plane motion of the anion, which would reduce the H2 H-bond strength (upfield chemical shift), coupled with an increase in H4/5 H-bond strength (downfield chemical shift).
The magnitude of the H2 and H4/5 chemical shift change for [C4C1im][Me2PO4] and [C4C1im]Cl on increasing concentration is approximately 1 ppm which is of a similar order to the difference between the concentrated samples of the ILs containing a weakly ([OTf]−) as opposed to strongly ([Me2PO4]−) coordinating anion. The H-bond perturbation due to the changing anion is likely to be much greater than that due to a change in concentration (the effects of which should be minimal). Thus, the trend for changing chemical shift with concentration can be inferred to arise from the close approach of the imidazolium rings and not subtle H-bond effects.
The 13C NMR results for C2 (ESI,† Fig. S19) follow a similar trend to those obtained for the 1H chemical shifts of H2, exhibiting a substantial upfield shift for [C4C1im]Cl and [C4C1im][Me2PO4] with increasing concentration, while [C4C1im][SCN] and [C4C1im][OTf] exhibit an intermediate shift (notably the shift for [C4C1im][SCN] is greater than for [C4C1im][OTf]), and [C4C1im][NTf2] shows only a very small upfield shift. In contrast to H2 and C2, the 13C NMR shifts of C4/5 vary considerably compared to H4/5 with both downfield and upfield shifts observed with increasing concentration for most 13C signals.
The rationalisation of the C4/5 results is that at low concentrations the imidazolium rings are offset enough for the C4/5 regions to lie (on average) outside of the shielding cone, generating the initial downfield shifts with increasing concentration. Further aggregation appears to bring the C4 and C5 towards the shielding cone, particularly for [C4C1im]Cl and [C4C1im][SCN], as an upfield shift is observed at larger IL concentrations.
Despite significant movement first upfield and then downfield as the concentration is increased, the net change in chemical shift of C4/5 between the most dilute and the most concentrated samples in [C4C1im]Cl and [C4C1im][SCN] is very small. The similarity of the initial and final chemical shifts indicate that C4 and C5 are in line with the shielding cone of the adjacent imidazolium ring. In contrast, [C4C1im][Me2PO4], [C4C1im][OTf] and [C4C1im][NTf2] undergo a net downfield shift indicating that the C4/5 atoms lie outside the shielding cone of the adjacent imidazolium ring. These results suggest that larger anions interfere with the ability of the imidazolium rings to overlap, forcing the rings to lie slightly further offset. This is consistent with the MD findings which will be discussed in detail later.
The magnitude of the cumulative chemical shift changes for [C4C1im][Me2PO4] is much greater than for the other two ILs with large anions indicating that the stronger H-bond accepting anion is more able to stabilise the π+–π+ interaction and enable closer approach of the imidazolium rings despite its size. Unlike the other simple ILs, [C4C1im][Me2PO4] also shows a difference in the chemical shift trends between C4 and C5. C4 shifts only downfield upon increasing the IL concentration while C5 shifts downfield initially and then slightly upfield. Such an effect could arise from the formation of a rotated stacked structure where each parallel alternating imidazolium ring was rotated by less than 180°, positioning C5 nearer the edge of the shielding cone than C4.
As observed for the other signals, the Cl− and [Me2PO4]− favour π+–π+ interactions, [SCN]− and [OTf]− exhibit intermediate behaviour and no significant evidence of π+–π+ interactions can be determined for [NTf2]−. The lack of π+–π+ stacking within [C4C1im][NTf2] could be due either to poor Coulombic stabilisation of the π+–π+ interaction by the diffuse [NTf2]− anion or the formation of more favourable anion–π+ interactions caused by its weak H-bond accepting ability, both increasing the displacement between imidazolium rings (Fig. 11).
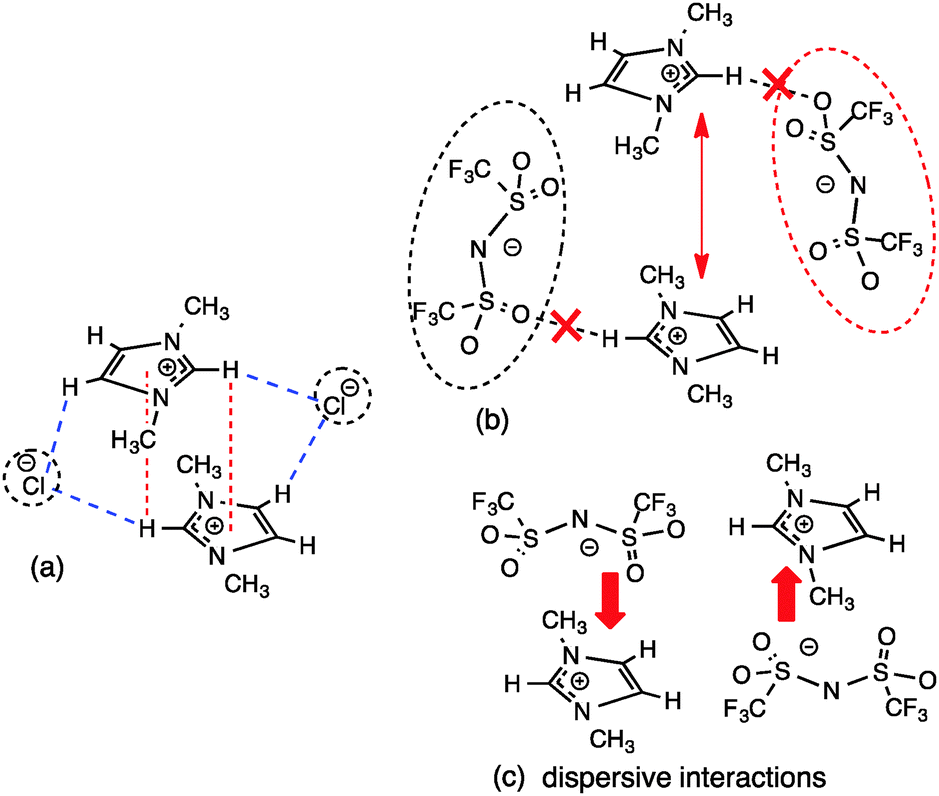 | ||
| Fig. 11 (a) π+–π+ stacking with Cl− (b) no stacking interactions due to larger more diffuse [NTf2]− not stabilising the Coulombic repulsion of the imidazolium rings, forcing them further apart, and (c) the weaker H-bonding ability and stronger anion–π+ interaction leading to [NTf2]− preferring a top position and thus displacing the π+–π+ stacked cation. | ||
In order to verify the nature of the π+–π+ stacking interactions for ILs based on the [Me2PO4]− and Cl− anions, selected low energy π+–π+ stacked (anions in the middle M_FS_SF_A and M_FS_SF_R) and alternating (anions on the diagonal, D_FT_TF_A) structures of [C1C1im]2[Me2PO4]2 and [C1C1im]2Cl2 IP-dimers have been computed at the B3LYP-D3BJ/6-311+G(d,p) level, Fig. 12. The nomenclature for these structures has been described in a previous publication and is reproduced in the ESI,† (Section B).27 The effect of a solvent environment has previously been shown to enhance π+–π+ stacking.57 Thus, conformer energies and NMR parameters were recomputed employing (a) a CH2Cl2 CPCM continuum model to mimic the solvent environment of a dilute solution and (b) an IL SMD model to simulate the solvent environment of a concentrated solution, further computational details can be found in the ESI,† (Section B), energies are reported in ESI,† Table S1.
 | ||
| Fig. 12 Representative [C1C1im]2Cl2 and [C1C1im]2[Me2PO4]2 IP-dimer structures at the B3LYP-D3BJ/6-311+G(d,p) level. ZPE and BSSE corrected conformer energies are set relative to the respective lowest energy structure. | ||
13C NMR shifts are evaluated relative to the respective ion-pair chemical shifts of the front conformer. The computational results for the dilute (CH2Cl2) vs. concentrated (IL) should not be over-interpreted given the current level of theory employed and the limited number of structures examined. The experimental spectrum will be a weighted average of conformers, the stability of which changes as the solvent environment is altered. The NMR calculations presented here can be expected to provide qualitative rather than quantitative information.
Stable π+–π+ stacking structures have been obtained (in the gas phase) for [C1C1im]2[Me2PO4]2, but the energies are ≈14 kJ mol−1 higher than for the alternating structure (ESI,† Table S1). However, in CH2Cl2 the π+–π+ stacking IP-dimers are stabilised relative to the alternating IP-dimer, and under the greater dielectric screening of the concentrated IL the π+–π+ stacking and alternating structures are of comparable energy. For [C1C1im]2Cl2 the π+–π+ stacking and alternating structures are of similar energy in the gas-phase. However, the alternating conformer is increased in energy in CH2Cl2 and becomes unstable in the concentrated IL. Overall, we find that screening of the positive charge in the higher dielectric favours π+–π+ stacking. Thus, as the concentration of the IL increases π+–π+ stacking will be favoured.
For [C1C1im]2Cl2 and [C1C1im]2[Me2PO4]2 the computed 13C NMR chemical shift for C2 and C5 in the π+–π+ stacked conformers are upfield relative to the front ion-pair conformation, consistent with the upfield shift observed in the NMR spectra of the concentrated ILs, although somewhat larger than the small net effects observed experimentally. The computed 13C NMR chemical shift for C4 in the π+–π+ stacked conformers moves downfield, consistent with the downfield shift observed with increasing concentration for C4 of [C4C1im][Me2PO4] and the small net change for [C4C1im]Cl. The alternating structure shows essentially no change. These results support the hypothesis that π+–π+ stacking is causing the shifts in the 13C spectra of C2 and C4/5.
Visual inspection of a plan view of the M_FS_SF_R conformer (Fig. 12), shows that for [C1C1im]2Cl2 the cation rings are essentially overlapped while for [C1C1im]2[Me2PO4]2 the rings are slightly displaced. This is consistent with the trends observed in the 13C NMR C4/5 spectra with increasing concentration (in CD2Cl2). C4/5 of [C4C1im]Cl exhibit similar behaviour because both C4 and C5 overlap the adjacent imidazolium ring. However, C4/5 of [C4C1im][Me2PO4] exhibit disparate behaviour because the rings are displaced slightly resulting in reduced overlap leading to C5 aligning within the aromatic magnetic cone, but C4 remaining outside. The computed 13C NMR chemical shifts of C4 move more substantially further downfield in [C1C1im]2[Me2PO4]2 compared to [C1C1im]2Cl2, in qualitative agreement with the experimental observations.
Two geometric parameters are of interest for the stacked structures; the overlap and the vertical distance between the cation rings. The shortest distance between any two atoms on the rings is closer for [C1C1im]2Cl2 by 0.016 Å and 0.074 Å for M_FS_SF_A and M_FS_SF_R respectively (details in ESI,† (Section B)). Examination of the optimised structures shows that the Cl− anion must be positioned in the “middle” between the rings to enable H-bonding with both rings, this results in a significant loss of linearity in the H-bond (C4/5–H–Cl angles ≈117–132°). In contrast, the oxygen atoms of the [Me2PO4]− anion easily form linear H-bonds due to the size of the anion (C4/5–H–O angles ≈140–164°). However, the H-bonding is strengthened at the cost of forcing the rings further apart to accommodate the distance between the O atoms of [Me2PO4]−, as can be seen in the structure M_FS_SF_A of Fig. 12. Thus both overlap and vertical distance facilitate π+–π+ stacking for [C1C1im]2Cl2 relative to [C1C1im]2[Me2PO4]2. The subtle differences between these ILs are not apparent from the NMR results where the magnitude of π+–π+ stacking was found to be similar for both ILs, nonetheless the stability of these π+–π+ stacked structures is in accordance with the experimental NMR data.
A QTAIM analysis has been performed on gas-phase dimers D_FT_TF_A and M_FS_SF_A for both [C1C1im]2Cl2 and [C1C1im]2[Me2PO4]2. ρBCP(r) has been evaluated for the ring C–H H-bonding interactions (ESI,† Table S2 and Fig. S1). ρBCP(r) reports the electron density at the bond critical point (BCP). The presence of a BCP is used to identify the existence of a bond and the magnitude of ρ(r) at the BCP is a measure of the strength of a bond. The larger ρBCP(r) for H2 H-bonding within the [C1C1im]2[Me2PO4]2 IP-dimers (compared to [C1C1im]2Cl2) is indicative of stronger H-bonding. The existence of ρBCP(r) for H4/5 H-bonding within the IP-dimers indicates an increase in H-bonding relative to the front and top ion-pairs which exhibit no H-bonding at H4/5.
Within [C1C1im]2Cl2 the aromatic rings overlap better, leading to both C4 and C5 being affected by the shielding cone of the adjacent imidazolium ring, whereas within [C1C1im]2[Me2PO4]2 the larger anion forces the aromatic rings to be forced slightly further apart and reduces the overlap, consistent with the different experimental observations for the C4/5 resonances in the 13C NMR. The combined experimental and computational results imply that the electrostatic stabilisation of the π+–π+ interaction provided by stronger H-bonding with the [Me2PO4]− anion is able to offset the steric effects induced by the larger anion which prevents the overlap of the imidazolium rings. This accounts for the similar magnitude of π+–π+ stacking behaviour observed experimentally between [C4C1im]Cl and [C4C1im][Me2PO4] despite the latter anion being significantly bulkier.
Overall, we find that the strong H-bonding ability and shape of an anion plays a distinct role in stabilising π+–π+ stacked interactions, consistent with the NMR results. There is experimental and computational evidence that strongly H-bond accepting anions such as [Me2PO4]− and Cl− promote the closer approach of the imidazolium cations. Similar conclusions regarding the prevalence of π+–π+ stacking for Cl− relative to [SCN]− based ILs have been obtained through MD simulations.18 [SCN]− is smaller in size but a weaker H-bond acceptor than [Me2PO4]−, hence the intermediate behaviour exhibited by [SCN]− indicates the extent to which H-bonding is required to stabilise π+–π+ interactions. The [NTf2]− anion is both the most bulky and the weakest H-bond acceptor of the anions, hence it is not able to stabilise π+–π+ interactions electrostatically and if positioned between the imidazolium rings will increase their separation. Consequently, no evidence for π+–π+ stacking interactions is observed in this IL.
NMR of the ionic liquid mixtures in CD2Cl2
Similar concentration dependent NMR studies were conducted for mixtures of ILs within CD2Cl2. These mixtures include [C4C1im][Me2PO4]x[NTf2]1−x, [C4C1im]Clx[NTf2]1−x and [C4C1im][OTf]x[NTf2]1−x, as well as [C4C1im]Cl0.50[OTf]0.50. The 1H and 13C NMR data for [C4C1im][Me2PO4]x[NTf2]1−x and [C4C1im][OTf]x[NTf2]1−x are depicted in Fig. 13 and ESI,† Fig. S20 and S21 respectively, the 1H NMR and 13C NMR data for the other mixtures can be found in ESI,† Tables S7–S27 and Fig. S17–S23.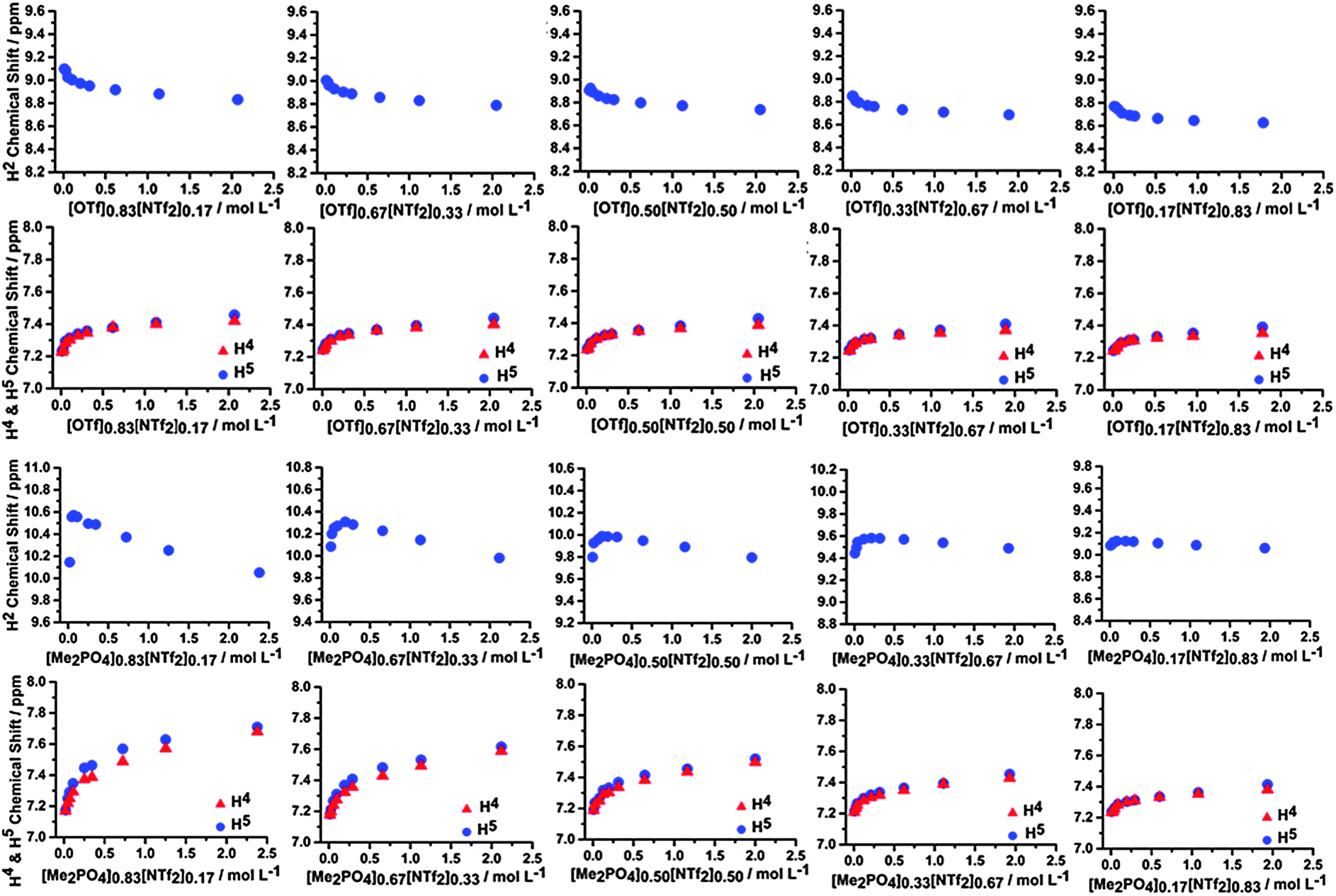 | ||
| Fig. 13 1H NMR chemical shifts of H2, H4 and H5 of the [C4C1im][OTf]x[NTf2]1−x mixtures (top) and [C4C1im][Me2PO4]x[NTf2]1−x mixtures (bottom) at various concentrations in CD2Cl2. | ||
From Fig. 13 it is evident that the H2, H4 and H5 signals in the [C4C1im][OTf]x[NTf2]1−x mixtures do not vary significantly with composition, and exhibit only a small change in curvature and chemical shift across the range of compositions examined. The magnitude of the chemical shifts and shapes of the curves are as expected based on an appropriately weighted average of the simple [C4C1im][OTf] and [C4C1im][NTf2] ILs. The 13C NMR results (ESI,† Fig. S20 and S21) are similar with the only noticeable change being the slow disappearance of the upfield shift feature with increasing concentration for the C4 and C5 spectra as the proportion of [NTf2]− increases. The magnitude of the chemical shift changes are small in all cases indicating that the π+–π+ interactions are not significant and that the distance between imidazolium cations does not change dramatically compared to [C4C1im][OTf] and [C4C1im][NTf2].
In contrast, the [C4C1im][Me2PO4]x[NTf2]1−x mixtures are not simply additive and the H2 signal exhibits substantially different behaviour compared to either of the simple ILs. In these mixtures, the H2 signal initially shifts downfield then moves upfield at higher concentrations whereas it monotonically shifts upfield in the simple ILs. This is not observed in any of the other 1H NMR spectra. The magnitude of the initial downfield shift decreases with a decreasing proportion of [C4C1im][Me2PO4] within the mixture, while the concentration of the mixture in CD2Cl2 at which the maximum downfield shift is observed increases. The C2 chemical shift of the mixture does not move downfield, instead only an upfield shift is observed. C4 and C5 follow the pattern of an initial downfield shift, before either flattening out or moving slightly upfield with increasing IL concentration. The shape of the chemical shift change, particularly for the H4/H5 and C4/C5 positions, varies more rapidly with increasing [NTf2]− up to [C4C1im][Me2PO4]0.50[NTf2]0.50 where further increasing the [NTf2]− anion concentration in the mixture leads to curve shapes consistent with simple [C4C1im][NTf2]. The upfield shift of C2 indicates there is π+–π+ stacking, however the initial downfield shift of H2 indicates that either H2 is outside of the magnetic cone (which is unlikely) or that a corresponding and more pronounced increase in H-bonding is occurring which dominates leading to an overall downfield shift. [Me2PO4]− containing ILs have much greater β-values than those based on [NTf2]−, indicating the improved H-bond accepting capacity of the [Me2PO4]−.58 A range of quantum chemical descriptors have also found H-bonding interactions with [Me2PO4]− to be much stronger than with [NTf2]−.59 A possible rationalisation for the data is that as isolated ion pairs and ion clusters coalesce, the more strongly interacting [Me2PO4]− anion displaces [NTf2]− anions from around H2 leading to a downfield chemical shift at H2 which is characteristic of strong H-bonding.60,61 Once the H2 sites have become saturated by [Me2PO4]− the aggregation of the IL proceeds in the conventional fashion with ring current effects leading to an upfield chemical shift.
The comparatively rapid changes in the shape of the curves with increasing [NTf2]− proportion in the mixture indicate that the [NTf2]− anion significantly disrupts the close interaction of the imidazolium rings. This disruption likely arises from favourable anion–π+ interactions, such as those found for the [NTf2]− anions in the undiluted NMR results, which then cause an increase in imidazolium ring displacement. The loss of a [Me2PO4]− anion may also reduce the stability of π+–π+ stacked structures (via the loss of Coulombic stabilisation and supportive H-bonding interactions), resulting in the translation of the imidazolium rings facilitated by a stabilising interaction with [NTf2]− (ESI,† Fig. S2).
The [C4C1im]Clx[NTf2]1−x mixtures (ESI,† Fig. S17 and S22) follow a similar, but less pronounced, trend to those observed for the [C4C1im][Me2PO4]x[NTf2]1−x mixtures. Mixtures with [NTf2]− proportions above [C4C1im]Cl0.67[NTf2]0.33 all exhibit changes in chemical shift on increasing the concentration similar to those found for [C4C1im][NTf2], reiterating the disruptive nature of the bulky [NTf2]− anion on mixtures, and particularly those with more tightly coordinating anions. In a similar fashion, [C4C1im]Cl0.50[OTf]0.50 (ESI,† Fig. S18 and S23) displays behaviour reminiscent of [C4C1im][OTf] in terms of the reduced magnitude of chemical shift. This signifies that the [OTf]− anion is capable of disrupting the interaction of imidazolium rings in a similar manner to [NTf2]−. The disruption of these interactions occurs despite: the closer approach of imidazolium cations in simple [C4C1im][OTf], the smaller size and better H-bond accepting capacity of the [OTf]− anion, and the reduced propensity of the [OTf]− anion to engage in anion–π+ interactions as determined by the undiluted NMR results. The behaviour of the [C4C1im]Cl0.50[OTf]0.50 mixture illustrates that π+–π+ stacking interactions in mixtures are readily disrupted by the presence of weakly coordinating anions, even those that are smaller and better H-bond acceptors than [NTf2]−.
In summary, the quantum chemical calculations of selected IP-dimers and the CD2Cl2 experiments indicate that the strongly coordinating anions (Cl− and [Me2PO4]−) lead to close interactions of the imidazolium cations, suggestive of π+–π+ stacking interactions. When present in mixtures with weakly coordinating anions, such as [OTf]− or [NTf2]−, the magnitude of ring current effects decrease dramatically even at relatively low (<50 mol%) proportions of the weakly coordinating anion. This illustrates that these anions disrupt the close stacking arrangement. Combined with the previous results for the undiluted ILs, the implication is that the interaction of the more strongly H-bond accepting anion with H2 leads to the displacement of the weakly coordinating anion above and behind the ring, facilitating the greater displacement of imidazolium rings through anion–π+ type interactions. When two weakly coordinating anions are present then additive behaviour is observed for the mixtures and no dramatic changes in imidazolium cation spacing are observed.
Molecular dynamics simulation of ionic liquid mixtures
To elucidate the preferential interactions indicated by the NMR studies, MD simulations have been carried out for the mixtures exhibiting the most significant deviations from ideality. Thus the simple ILs and IL mixtures [C4C1im][Me2PO4]x[NTf2]1−x and [C4C1im]Clx[OTf]1−x have been simulated. Simulation details are provided in the ESI,† (Section B).The spatial distribution functions (SDF) for [C4C1im][Me2PO4]x[NTf2]1−x (Fig. 14) provide a graphical representation of the relative position of anions around a cation. The shaded surfaces in Fig. 14 represent where the density of anions is at least 2.5 times greater than the background density. SDF for [C4C1im]Clx[OTf]1−x are provided in the ESI,† Fig. S4.
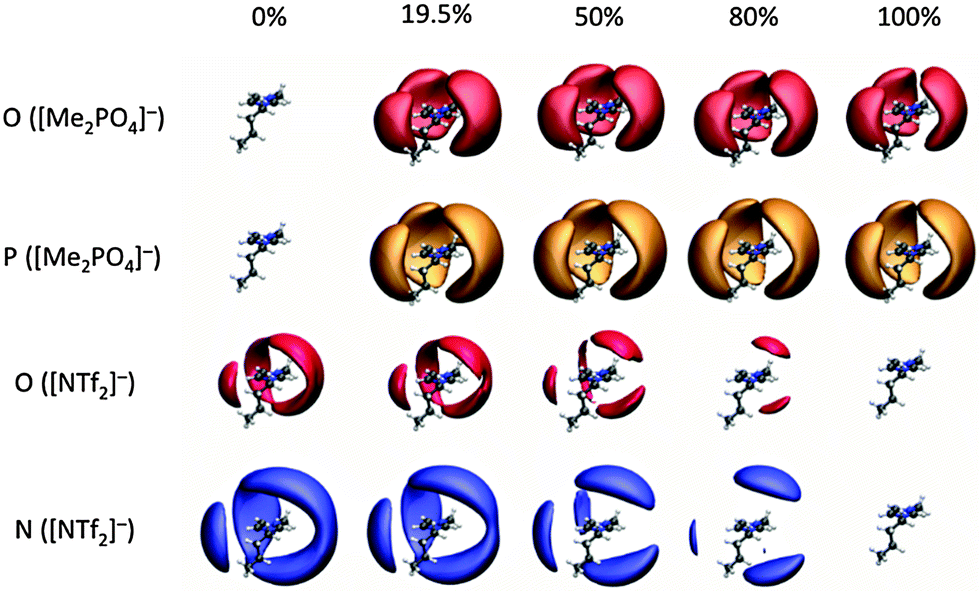 | ||
| Fig. 14 Spatial density maps of selected atoms on the anions around the cation for several [Me2PO4]− concentrations in the [C4C1im][Me2PO4]x[NTf2]1−x mixtures. Isosurfaces: free O([Me2PO4]−) in red, P([Me2PO4]−) in orange, O([NTf2]−) in matt red and N([NTf2]−) in blue. Isosurfaces are shown at 2.5 times the normalised atomic density with the more strongly H-bonding anion to the front, and to have the weaker H-bonding anion positioned above or below the ring. | ||
It is clear that in the mixture both anions interact with the ring H2, H4 and H5 atoms. The strongly H-bond accepting anions can interact “in-plane” or in the “middle” between two rings, leading to the extension of the SDF surfaces above and below the plane around the periphery of an imidazolium cation. The more weakly H-bond accepting anions tend to occupy positions directly above and below the centre of the aromatic ring. This is more easily visualised in the related SDF of [C4C1im]Cl0.50[OTf]0.50 shown for increasing isovalue of an anion around the cation (ESI,† Fig. S5). The SDF for the N of [NTf2]− (blue in Fig. 14) also shows a clear preference for [NTf2]− to locate above or below the cation ring, particularly for low concentrations of [NTf2]− in [Me2PO4]−.
Fig. 14 shows that [Me2PO4]− is closely associated with the cation even in very low proportions. In contrast, the smaller surfaces for [NTf2]− in Fig. 14 and [OTf]− in ESI,† Fig. S5 demonstrate the much weaker interaction of these anions with the cation. These data are consistent with the NMR data observed for [C4C1im][Me2PO4]x[NTf2]1−x mixtures, where the addition of [NTf2]− led to stronger apparent preferential interactions with C4/5 rather than H4/5, indicating an anion–π+ interaction is dominating, rather than in-plane H-bonding at the rear of the ring. These data are also consistent with the NMR data observed for the [C4C1im]x[C4C1pyrr]1−xClx[NTf2]1−x mixtures. The first solvation shell of any cation is a random mixture of the anions present, this can be seen in snap shots from a single trajectory frame, as shown in ESI,† Fig. S6 and S7. Within a solvation shell the cation will orient so as to interact given the similarity in shape and size of [Me2PO4]− and [OTf]− it is possible to question why [C4C1im][Me2PO4]x[NTf2]1−x shows larger deviations from ideality than [C4C1im][OTf]x[NTf2]1−x. Some insight can be obtained from the charge parameters for the MD potentials; [Me2PO4]− is significantly more polarised than [OTf]−. The charge on the H-bond accepting O atoms in [Me2PO4]− is −0.92 e, while those in [OTf]− are −0.63 e and those of [NTf2]− carry even less charge (−0.53 e). Thus while the overall charge on the ion is −1 e, the polarisability of the central atom (P (+1.62) or S (+1.02)) can lead to a significantly larger charge present on the periphery of the anion.
Atom-atom pair radial distribution functions (RDFs) plot the distance between specific pairs of atoms over a simulation, and provide more detail than the SDFs. RDFs between the free O's (not OMe) of both [Me2PO4]− and [NTf2]− with ring atoms (H2, H4 and H5) for the various [C4C1im][Me2PO4]x[NTf2]1−x mixtures are presented in ESI,† Fig. S8 and those for C2, C4 and C5 are presented in ESI,† Fig. S9. Qualitatively similar results are found for the [C4C1im]Clx[OTf]1−x mixtures (ESI,† Fig. S10 and S11). The variation of cation–anion RDFs within IL mixtures as a function of the proportion of a constituent is not unique to these systems.13,18,19
In general, each RDF exhibits a first maximum at either 2–3 Å (Hn–X) or 3–4 Å (Cn–X) n = 2, 4, 5. The first peak corresponds to direct interaction of the anion with the respective hydrogen/carbon atom. The second peak mainly represents anions coordinated to the other ring atomic (H/C) sites. The position of the first maximum for each of the cation–anion RDFs is found to be independent of anion composition, indicating that the relative cation–anion distances of the simple ILs are retained on mixing. Thus the presence of a second anion does not weaken or strengthen the interactions to an extent large enough to influence the distance between the first anion and cation.
The higher g(r) maxima for [Me2PO4]− compared to [NTf2]− indicate that the interaction of [Me2PO4]− with [C4C1im]+ is much stronger than that of [NTf2]−. As the concentration changes, a smaller maximum in the RDF indicates a reduced probability of finding the anion in that specific location. For example, in the [C4C1im][Me2PO4]x[NTf2]1−x mixtures (ESI,† Fig. S8), the intensity of the first peak maximum of the distance between H2 and the O of [Me2PO4]− systematically decreases from ≈9 to ≈5 with an increasing proportion of [Me2PO4]−. Thus with increasing [Me2PO4]− concentration there is less interaction between the anion and all of the cation ring C–H groups. A decrease in H2–O ([NTf2]−) from ≈2.5 to ≈1.5 is also observed with increasing [Me2PO4]− concentration. Thus, in contrast to [Me2PO4]−, at high [NTf2]− concentrations the [NTf2]− anion is more likely to be interacting with the cation.
These data can be rationalised; for dilute [Me2PO4]− in [C4C1im][NTf2] there are strong directional H-bonds between the cation and [Me2PO4]−, however as the concentration of [Me2PO4]− increases the cation becomes surrounded by [Me2PO4]− anions, reducing the preference for each site on [C4C1im]+ to interact with a specific [Me2PO4]− anion. In contrast, for small amounts of [NTf2]− in [C4C1im][Me2PO4], the [Me2PO4]− dominates H-bonding interactions with the cation, and the weaker H-bonding [NTf2]− is excluded from direct association, thus there is a reduced proportion of [NTf2]− directly interacting with specific sites on the imidazolium ring. As the proportion of [NTf2]− increases, the number of [Me2PO4]− anions H-bonding with, thereby restricting access to, the cation is reduced and the [NTf2]− can undertake more structured H-bonding. Hence [NTf2]− structuring around the imidazolium ring is more pronounced with a high proportion of [C4C1im][NTf2] while [Me2PO4]− structuring around the imidazolium ring is less pronounced as the ratio of [C4C1im][Me2PO4] is increased.
Changes in the RDFs are less pronounced for H5 than H2, and are reduced even further for H4. This result is in-line with the differences observed in the 13C NMR of C4 and C5 for the mixtures in CD2Cl2. The differences between H4 and H5 are more pronounced for [Me2PO4]− than [NTf2]−. The stronger relative structuring around H5 may be due to additional H-bonding or dispersive interactions of the anion with butyl chain groups facilitating anion association or the role of alkyl chain aggregation promoted by the more coordinating anion.
To gain further insight into the local cation–anion structure, angular distribution functions (ADFs) have been computed for each mixture relative to the cation ring protons. The data and insights obtained support the rationalisations presented here, and are discussed more fully in the ESI,† (Section B). The normalised first-shell coordination number of anions surrounding H2 has also been computed in a similar manner to Payal et al. for these mixtures (ESI,† Fig. S15 and S16).13 The trends in these values accord with the previous discussion with slightly increased coordination of Cl− and [Me2PO4]− with H2 relative to [OTf]− and [NTf2]− respectively. Further details and discussion are provided in the ESI,† (Section B).
The potential for π+–π+ stacking (or a change in the overall IL structure) has been investigated by computing RDFs between the centre-of-geometry (COG) of cation rings (i.e. excluding the alkyl chains) for both the [C4C1im]Clx[OTf]1−x and [C4C1im][Me2PO4]x[NTf2]1−x (Fig. 15). There are three distinct features, a pre-peak ≈4 Å, where π+–π+ stacking is observed, a primary maximum at ≈7 Å representing the first “solvation” shell of cations, and a second broad peak due to the second solvation shell of cations. The pre-peak has been assigned to π+–π+ stacking as imidazolium rings positioned closer than in the purely charge ordered structures would require an attractive interaction (most likely π+–π+ stacking) for stabilisation. This assignment is consistent with that proposed by others.13,18 As the proportion of Cl− or [Me2PO4]− increases there is an increase in π+–π+ stacking, consistent with QM calculations,56 and the NMR data reported here. The effect is significantly larger for [Me2PO4]x[NTf2]1−x compared to Clx[OTf]1−x. As the proportion of [NTf2]− increases there is a substantial increase in the cation–cation distances (not just those involved in π+–π+ stacking), for example, the first solvation shell peak shifts from ≈7 Å to broad flat topped peak ≈8–10 Å. This result is consistent with the NMR data which indicate that the [NTf2]− rapidly disrupts π+–π+ stacking (<50 mol%). In addition, the increase in the first shell cation–cation distances, not observed within the [C4C1im]Clx[OTf]1−x mixtures, is consistent with a move from π+–π+ stacked structures to the alternating (diagonal anion) structural motifs found for the QC determined IP-dimers, Fig. 12 and as motifs within the MD simulations (ESI,† Fig. S14). In these structures, the positioning of the [NTf2]− anion above and below the imidazolium ring would substantially increase the cation–cation distance.
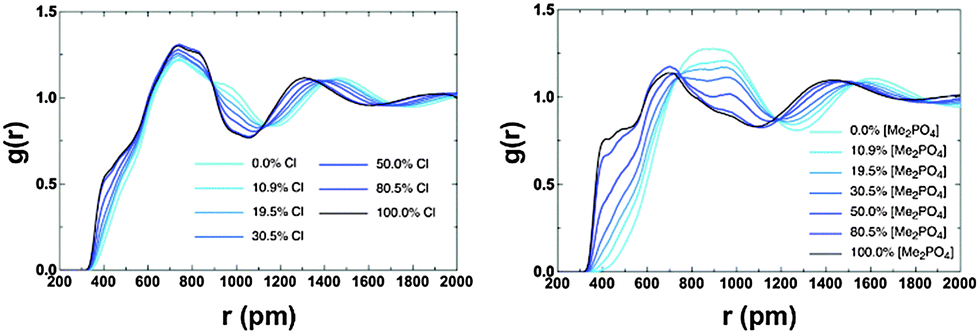 | ||
| Fig. 15 Calculated RDFs between the centre of geometries of the cation rings for (left) [C4C1im]Clx[OTf]1−x and (right) [C4C1im][Me2PO4]x[NTf2]1−x. | ||
Conclusions
Preferential H-bonding between H2 and the most strongly H-bond accepting anion has been identified in a range of IL mixtures through the analysis of NMR spectra and MD simulations of the undiluted ILs. This effect was more pronounced when the difference in H-bond accepting ability between the anions in the mixture was greater. These preferential H-bonds resulted in the more weakly H-bond accepting anion engaging in anion–π+ interactions above the rear of the imidazolium ring and in some mixtures also participating in weak preferential H-bonding with H4/5. The magnitude of these preferential interactions was small and it was found that the proportion of anions around a given cation was random with no evidence of anion clustering around favourable H-bonding sites. There is no evidence for the formation of new bonds with the imidazolium ring atoms within the mixture that cannot be found in the individual IL components of the mixture.These results give rise to a visualisation of the structures of mixtures of the ILs studied in which there is a random distribution of anions and cations through the structure, as previously suggested.15 Each cation is surrounded, on average, by a cage of anions (and vice versa) within which it is rotating. However, this rotation is not entirely random and the most strongly hydrogen bond donating protons will associate for a greater time with the most strongly hydrogen bond accepting anions than with the other anions in forming the cage.
The role of IL anions in π+–π+ stacking was also investigated by using NMR spectroscopy on IL solutions at various concentrations in CD2Cl2, quantum chemical calculations and MD simulations. It was established that strong H-bond accepting anions are required to provide the Coulombic stabilisation necessary for π+–π+ interactions to occur and that these interactions were enhanced in the presence of a higher dielectric. Larger more diffuse anions are both less able to provide the necessary level of Coulombic stabilisation and their propensity to form anion–π+ interactions pushes the rings apart. The disruption of π+–π+ stacking by less coordinating anions occurs readily and at relatively low proportions (<50 mol%) of the weakly coordinating anion in mixtures. This was observed both for the bulky [NTf2]− anion and the smaller [OTf]− and is consistent with previous computational findings for Clx[SCN]1−x mixtures. This disruption is promoted by the formation of anion–π+ interactions by these anions.
As one of the motivating factors of this study, it is worth considering how these structures are linked with the physical properties of such mixtures. In particular, it was recently reported that [C4C1im][Me2PO4]x[NTf2]1−x mixtures are non-ideal whereas [C4C1im][OTf]x[NTf2]1−x mixtures gave ideal behaviour.43 The results obtained here indicate that the non-ideality of the former mixtures may be due to the larger than anticipated ion separation at low proportions of [NTf2]− due to the preferential positioning of [NTf2]− above or below the imidazolium ring. This would account for the positive excess volumes observed. The cation–cation distances in [C4C1im][OTf]x[NTf2]1−x mixtures vary much less dramatically due to the greater initial distance between imidazolium rings for [C4C1im][OTf] arising from the poorly stabilised π+–π+ interaction. Additionally, there is a weaker preferential association of each anion with specific positions on the cation which results in less reorganisation of the cation conformation on varying the composition of the mixture. This accords with the ideality of the measured physical properties.
The magnitude of non-ideality identified for most IL mixtures to date has generally been small despite the complexity of IL structure.38–43 The fact that such effects are small can be accounted for by the structural considerations highlighted here. Mixtures of anions with quite substantial differences in H-bond accepting capacity only exhibit a slight preferential association with specific cation locations. These preferential associations are likely to be the main driving force for non-ideality in these solutions and the free energy changes estimated are approximately 2 orders of magnitude smaller than the average binding energies determined for similar ions. The main contributor to the structure of IL mixtures are the Coulombic interactions which are isotropic and are unlikely to change dramatically depending on anion choice (assuming the charges are the same). This was illustrated by the lack of ion clustering observed in the NMR experiments and MD simulations. Secondary effects such as H-bonding are much smaller in magnitude, particularly in the context of an IL mixture where it is not the absolute H-bond strength but the difference in H-bond strength between different ion combinations which governs the molecular orientation. Correspondingly, IL mixtures that consist of equivalently charged anions would be expected, compared to conventional organic molecules, to lead to mixtures that behave in a more ideal fashion. This also accounts for the IL mixtures that exhibit the greatest non-ideality arising from ions where ideal inter-ion distances are affected by steric factors, i.e. a disruption of the ionic network.
The dispersive interactions that form a component of π+–π+ stacking and anion–π+ interactions are small but also competitive in magnitude to weak H-bonding interactions. This gives rise to a diversity of subtle local structural effects in these mixtures which could be of interest for various applications but do not greatly influence the bulk liquid properties outside of those that could be predicted by modelling the liquid as an ideal solution. There are likely to be some further exceptions to these considerations as discussed in the introduction, for example, ILs that possess a large degree of asymmetry, long alkyl chains leading to large dispersive interactions, fluorous phases or ILs that are functionalised resulting in the formation of very strong H-bonds. In these cases the liquids would behave less as mixtures of ions. Further investigation into these types of IL mixtures are currently underway within our laboratory.
Acknowledgements
The authors acknowledge an ERC Advanced Investigator Grant held by TW and PAH for funding.Notes and references
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- C. C. Weber, A. F. Masters and T. Maschmeyer, Green Chem., 2013, 15, 2655–2679 RSC.
- T. Torimoto, T. Tsuda, K.-i. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196–1221 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliot, J. H. Davis, Jr., M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 CAS.
- X. Han and D. W. Armstrong, Acc. Chem. Res., 2007, 40, 1079–1086 CrossRef CAS PubMed.
- I. Minami, Molecules, 2009, 14, 2286–2305 CrossRef CAS PubMed.
- M. Freemantle, Chem. Eng. News, 1998, 76, 32–37 Search PubMed.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780–7802 RSC.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- R. S. Payal and S. Balasubramanian, Phys. Chem. Chem. Phys., 2013, 15, 21077–21083 RSC.
- J.-M. Andanson, M. J. Beier and A. Baiker, J. Phys. Chem. Lett., 2011, 2, 2959–2964 CrossRef CAS.
- M. Y. Lui, L. Crowhurst, J. P. Hallett, P. A. Hunt, H. Neidermeyer and T. Welton, Chem. Sci., 2011, 2, 1491–1496 RSC.
- A. P. Abbott, G. Frisch, H. Garrett and J. Hartley, Chem. Commun., 2011, 47, 11876–11878 RSC.
- O. Hollóczki, F. Malberg, T. Welton and B. Kirchner, Phys. Chem. Chem. Phys., 2014, 16, 16880–16890 RSC.
- M. Brüssel, M. Brehm, A. S. Pensado, F. Malberg, M. Ramzan, A. Stark and B. Kirchner, Phys. Chem. Chem. Phys., 2012, 14, 13204–13215 RSC.
- M. Brüssel, M. Brehm, T. Voigt and B. Kirchner, Phys. Chem. Chem. Phys., 2011, 13, 13617–13620 RSC.
- A. B. Pereiro, J. M. M. Araújo, F. S. Oliveira, C. E. S. Bernardes, J. M. S. S. Esperança, J. N. Canongia Lopes, I. M. Marrucho and L. P. N. Rebelo, Chem. Commun., 2012, 48, 3656–3658 RSC.
- F. S. Oliveira, A. B. Pereiro, J. M. M. Araújo, C. E. S. Bernardes, J. N. Canongia Lopes, S. Todorovic, G. Feio, P. L. Almeida, L. P. N. Rebelo and I. M. Marrucho, Phys. Chem. Chem. Phys., 2013, 15, 18138–18147 RSC.
- F. D'Anna, S. Marullo, P. Vitale and R. Noto, ChemPhysChem, 2012, 13, 1877–1884 CrossRef PubMed.
- H. Wang, S. P. Kelley, J. W. Brantley III, G. Chatel, J. Shamshina, J. F. B. Pereira, V. Debbeti, A. S. Myerson and R. D. Rogers, ChemPhysChem, 2015, 16, 993–1002 CrossRef CAS PubMed.
- D. Xiao, J. R. Rajian, L. G. Hines, S. Li, R. A. Bartsch and E. L. Quitevis, J. Phys. Chem. B, 2008, 112, 13316–13325 CrossRef CAS PubMed.
- D. Xiao, J. R. Rajian, S. Li, R. A. Bartsch and E. L. Quitevis, J. Phys. Chem. B, 2006, 110, 16174–16178 CrossRef CAS PubMed.
- K. Fumino, A.-M. Bonsa, B. Golub, D. Paschek and R. Ludwig, ChemPhysChem, 2015, 16, 299–304 CrossRef CAS PubMed.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2014, 16, 3238–3253 RSC.
- J. D. Holbrey, W. M. Reichert and R. D. Rogers, Dalton Trans., 2004, 2267–2271 RSC.
- J. S. Wilkes and M. J. Zaworotko, Supramol. Chem., 1993, 1, 191–193 CrossRef CAS.
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965–967 RSC.
- M. Deetlefs, C. Hardacre, M. Nieuwenhuyzen, A. A. H. Padua, O. Sheppard and A. K. Soper, J. Phys. Chem. B, 2006, 110, 12055–12061 CrossRef CAS PubMed.
- A. Mele, G. Romanò, M. Giannone, E. Ragg, G. Fronza, G. Raos and V. Marcon, Angew. Chem., Int. Ed., 2006, 45, 1123–1126 CrossRef CAS PubMed.
- A. G. Avent, P. A. Chaloner, M. P. Day, K. R. Seddon and T. Welton, J. Chem. Soc., Dalton Trans., 1994, 3405–3413 RSC.
- R. P. Matthews, C. Ashworth, T. Welton and P. A. Hunt, J. Phys.: Condens. Matter, 2014, 26, 284112 CrossRef PubMed.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2015, 17, 14437–14453 RSC.
- B. Aoun, A. Goldbach, S. Kohara, J.-F. Wax, M. A. González and M.-L. Saboungi, J. Phys. Chem. B, 2010, 114, 12623–12628 CrossRef CAS PubMed.
- J. de Andrade, E. S. Böes and H. Stassen, J. Phys. Chem. B, 2008, 112, 8966–8974 CrossRef CAS PubMed.
- A. Podgoršek, A. S. Pensado, C. C. Santini, M. F. Costa Gomes and A. A. H. Pádua, J. Phys. Chem. C, 2013, 117, 3537–3547 Search PubMed.
- D. Song and J. Chen, J. Chem. Thermodyn., 2014, 77, 137–143 CrossRef CAS.
- J. N. Canongia Lopes, T. C. Cordeiro, J. M. S. S. Esperança, H. J. R. Guedes, S. Huq, L. P. N. Rebelo and K. R. Seddon, J. Phys. Chem. B, 2005, 109, 3519–3525 CrossRef PubMed.
- A. Stoppa, R. Buchner and G. Hefter, J. Mol. Liq., 2010, 153, 46–51 CrossRef CAS.
- P. Navia, J. Troncoso and L. Romani, J. Chem. Eng. Data, 2007, 52, 1369–1374 CrossRef CAS.
- M. T. Clough, C. R. Crick, J. Gräsvik, P. A. Hunt, H. Niedermeyer, T. Welton and O. P. Whitaker, Chem. Sci., 2015, 6, 1101–1114 RSC.
- A. Arce, M. J. Earle, S. P. Katdare, H. Rodríguez and K. R. Seddon, Chem. Commun., 2006, 2548–2550 RSC.
- G. Annat, M. Forsyth and D. R. MacFarlane, J. Phys. Chem. B, 2012, 116, 8251–8258 CrossRef CAS PubMed.
- S. Omar, J. Lemus, E. Ruiz, V. R. Ferro, J. Ortega and J. Palomar, J. Phys. Chem. B, 2014, 118, 2442–2450 CrossRef CAS PubMed.
- T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, R. Wölfel, P. S. Schulz, P. Wasserscheid, H. Weber, J. Thar, B. Kirchner, F. Maier and H.-P. Steinrück, Chem. – Eur. J., 2010, 16, 9018–9033 CrossRef CAS PubMed.
- P. A. Hunt, C. R. Ashworth and R. P. Matthews, Chem. Soc. Rev., 2015, 44, 1257–1288 RSC.
- S. Cha and D. Kim, Phys. Chem. Chem. Phys., 2015, 17, 29786–29792 RSC.
- W. Zhao, F. Leroy, B. Heggen, S. Zahn, B. Kirchner, S. Balasubramanian and F. Müller-Plathe, J. Am. Chem. Soc., 2009, 131, 15825–15833 CrossRef CAS PubMed.
- I. Skarmoutsos, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2014, 16, 3675–3685 RSC.
- R. D. Skwierczynski and K. A. Connors, J. Chem. Soc., Perkin Trans. 2, 1994, 467–472 RSC.
- E. Bosch and M. Rosés, J. Chem. Soc., Faraday Trans., 1992, 88, 3541–3546 RSC.
- C. C. Weber, A. F. Masters and T. Maschmeyer, Org. Biomol. Chem., 2013, 11, 2534–2542 CAS.
- C. C. Weber, A. F. Masters and T. Maschmeyer, J. Phys. Chem. B, 2012, 116, 1858–1864 CrossRef CAS PubMed.
- B. A. Marekha, O. N. Kalugin and A. Idrissi, Phys. Chem. Chem. Phys., 2015, 17, 16846–16857 RSC.
- E. Buisine, K. de Villiers, T. J. Egan and C. Biot, J. Am. Chem. Soc., 2006, 128, 12122–12128 CrossRef CAS PubMed.
- M. A. Ab Rani, A. Brant, L. Crowhurst, A. Dolan, M. Y. Lui, N. H. Hassan, J. P. Hallett, P. A. Hunt, H. Niedermeyer, J. M. Perez-Arlandis, M. Schrems, T. Welton and R. Wilding, Phys. Chem. Chem. Phys., 2011, 13, 16831–16840 RSC.
- H. Niedermeyer, C. Ashworth, A. Brandt, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2013, 15, 11566–11578 RSC.
- R. Lungwitz and S. Spange, New J. Chem., 2008, 32, 392–394 RSC.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp00156d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2016 |