
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of quinones for high power density biofuel cells†
Ross D.
Milton
,
David P.
Hickey
,
Sofiene
Abdellaoui
,
Koun
Lim
,
Fei
Wu
,
Boxuan
Tan
and
Shelley D.
Minteer
*
Departments of Chemistry and Materials Science and Engineering, University of Utah, 315 S 1400 E Room 2020, Salt Lake City, UT 84112, USA. E-mail: minteer@chem.utah.edu
First published on 8th June 2015
Abstract
Enzymatic fuel cells (EFCs) are devices that can produce electrical energy by enzymatic oxidation of energy-dense fuels (such as glucose). When considering bioanode construction for EFCs, it is desirable to use a system with a low onset potential and high catalytic current density. While these two properties are typically mutually exclusive, merging these two properties will significantly enhance EFC performance. We present the rational design and preparation of an alternative naphthoquinone-based redox polymer hydrogel that is able to facilitate enzymatic glucose oxidation at low oxidation potentials while simultaneously producing high catalytic current densities. When coupled with an enzymatic biocathode, the resulting glucose/O2 EFC possessed an open-circuit potential of 0.864 ± 0.006 V, with an associated maximum current density of 5.4 ± 0.5 mA cm−2. Moreover, the EFC delivered its maximum power density (2.3 ± 0.2 mW cm−2) at a high operational potential of 0.55 V.
Introduction
Enzymatic fuel cells (EFCs) are devices that employ enzymes as bioelectrocatalysts to produce electrical energy from energy-dense fuels (i.e. sugars).1–6 Typically, strategies to improve EFC performance seek to improve upon the open circuit potential (OCP) or maximum catalytic current density (Jmax) of devices,7–9 since these two variables are usually mutually exclusive, whereby increased potential difference between the redox cofactor of an enzyme and the employed electron mediator typically results in an increased Jmax.Since the OCP of a biofuel cell is increased by expanding the potential difference between the bioanode and biocathode of a device,10 it is theoretically favorable to employ enzymes with wider potential windows. Nicotinamide adenine dinucleotide-dependent (NAD) enzymes are commonly used due to their relatively low redox potential, although the oxidation of reduced NADH often requires sacrificial overpotentials and the cofactor is not bound to the enzyme. To this end flavin adenine dinucleotide-dependent enzymes (FAD-) such as glucose oxidase (GOx) are commonly employed at the bioanode since they offer relatively low cofactor redox potentials and the cofactor is tightly bound.
In contrast, laccases11 or bilirubin oxidases12 (BOx) are typically utilized at the biocathode to reduce O2 to H2O, since they exhibit high catalytic onset potentials compared to traditional O2 reduction catalysts based on precious metals13 with the additional ability to undergo O2 reduction in mild conditions (neutral pH, temperature, etc.).14,15
In early research, NAD-dependent glucose dehydrogenase (NAD-GDH) was used within an amperometric biosensor configuration in which reduced NADH was oxidized by phenothiazine-modified redox polymers.16 Additionally, it was also previously used within a high-power EFC that was capable of producing approximately 1.45 mW cm−2 at 0.3 V from glucose and O2, using non-immobilized electron mediators that required an additional enzyme (diaphorase).17 High performance has also been reported for a fructose/O2 EFC containing a FAD-dependent dehydrogenase, whereby direct electron transfer (DET) at the bioanode and at a BOx-based biocathode enabled the production of 4.9 mA cm−2 and 0.87 mW cm−2 at an operational voltage of 0.3 V.18 Ferrocene-based redox hydrogel polymers have also been demonstrated as alternative immobilized MET systems which are capable of achieving high Jmax values for bioelectrocatalytic glucose oxidation by GOx, where performances as high as approximately 15 mA cm−2 have been reported on porous carbon felt electrodes.19 Presently, the highest GOx EFCs produce approximately 1.5 mW cm−2.20
The modification of polymers with phenothiazine-type electron mediators has been demonstrated as a strategy to mediate electrons from the FAD-dependent dehydrogenase domain of cellobiose dehydrogenase (CDH, E.C.: 1.1.99.18), at relatively low potentials.21,22 These polymers were able to result in the formation of an EFC with large OCPs, close to 0.7 V, although low power densities of less than 10 μW cm−2 were reported.
FAD-dependent glucose dehydrogenase (GDH) is emerging as an alternative glucose-oxidizing enzyme.23,24 GDH was recently demonstrated to be capable of producing very large Jmax values for glucose oxidation at a porous carbon electrode, although the Jmax was observed at approximately +0.7 V (vs. Ag/AgCl) at a scan rate of 10 mV s−1 and in the presence of 500 mM glucose.25 Nevertheless, such high Jmax values highlight GDH's superior suitability within glucose EFCs.
Although phenothiazines and organometallic complexes are popular mediators, various quinones (cyclic dione-containing species whose reduction and reorganization yields equal alcohol functionalities) are becoming more prevalent within bioelectrochemical applications. Non-covalently immobilized naphthoquinone-based (NQ) derivatives have been demonstrated as an alternative redox mediator to reported osmium- and ferrocene-based redox polymer hydrogels, capable of contributing towards power densities as high as 0.8 ± 0.2 and 1.54 mW cm−2.20,24 Additionally, a ruthenium complex-based redox mediator which utilized a quinone-type ligand (9,10-phenthraquinone) has also been demonstrated as an electron mediator for NAD-GDH.26 Benzoquinone-derived polymers have also been demonstrated in photobioelectrochemical applications.27,28 Benzoquinone was self-polymerized (without an additional covalent polymer support and spatial linker) and covalently attached (via an amine functionality) to poly(lysine) to yield effective mediator matrices, although the formal redox potentials associated with these polymers are more positive (between 0 and +370 mV vs. Ag/AgCl) than the formal redox potentials associated with NQ species with reported current densities of up to 120 μA cm−2. The presence of a “spacer” between a polymer and a redox species has been shown to be of importance.29 Additionally, the results presented below from our research highlight the significance of the functionality of quinone species, where amine-linkers result in ineffective electron mediators of FAD-dependent GDH.
A recently-reported viologen redox hydrogel demonstrated how the functionalization of hydrogel polymers with electron mediators tailored to have specifically-designed chemical properties can yield favorable immobilization matrices,30 suggesting a specifically-tailored NQ and covalent immobilization strategy could be favorable.
The covalent immobilization of NQ is not trivial, however; as reported below, NQ is able to quickly react with primary and secondary amines under very mild conditions, which yields the product ineffective as an electron mediator for FAD-dependent GDH. Therefore, we report the synthesis of a rationally designed NQ derivative that was subsequently successfully covalently immobilized at an EFC bioanode, by the formation of a NQ-modified linear polyethyleneimine (NQ-LPEI) redox hydrogel and by direct attachment to GDH. The resulting NQ derivative was capable of facilitating efficient MET between GDH at a simple carbon electrode, while enabling a glucose/O2 EFC with a high OCP (0.864 ± 0.006 V) to deliver a Jmax of 5.4 ± 0.5 mA cm−2, with an associated maximum power density of 2.3 ± 0.2 mW cm−2 at 0.55 V.
Results and discussion
Naphthoquinone derivative design
Compared to previously established technologies designed on osmium- and ferrocene-based redox polymer mediators,19,31 the use of NQs as low potential organic electron mediators for glucose-oxidizing enzymes is increasing.17,20,24,32,33 The redox potential of quinones can be sufficiently tailored to suit the environment, which increases its applicability within EFCs.34 However, the covalent immobilization of a NQ derivative in an enzymatic FAD-dependent glucose-oxidizing application has not yet been demonstrated. Initial investigation into the ability for NQ to mediate electrons from GDH determined that two methoxy-NQ derivatives were able to undergo MET at low potentials. Thus, two NQ derivatives with epoxide functionalities were rationally designed and synthesized (Fig. 1a) to allow for the covalent modification of a polymer and of GDH directly without any additional reagents, since NQ easily undergoes undesirable side reactions under very mild conditions. LPEI and GDH were successfully modified with 1 and 2 to yield NQ-modified LPEI (NQ-4-LPEI and NQ-2-LPEI) and NQ-labeled GDH (NQ-4-GDH and NQ-2-GDH). The epoxy functionalities of 1,2-NQ-epoxy (1) and 1,4-NQ-epoxy (2) allow for multiple-site enzyme modification, whereby epoxides are able to react with all surface nucleophilic amino acid residues (lysine, histidine, cysteine, tyrosine, etc.).35 Due to the ability for NQ to undergo self-exchange36 (the ability for electron transport across proximal NQ moieties), it was predicted that NQ-polymer modification (Fig. 1b) and NQ-enzyme labeling (Fig. 1c) could provide effective platforms for mediated glucose oxidation by GDH. Initially, the ability for NQ to undergo MET with GDH was evaluated with soluble NQ salts, followed by methoxy-NQ derivatives (Fig. 2a).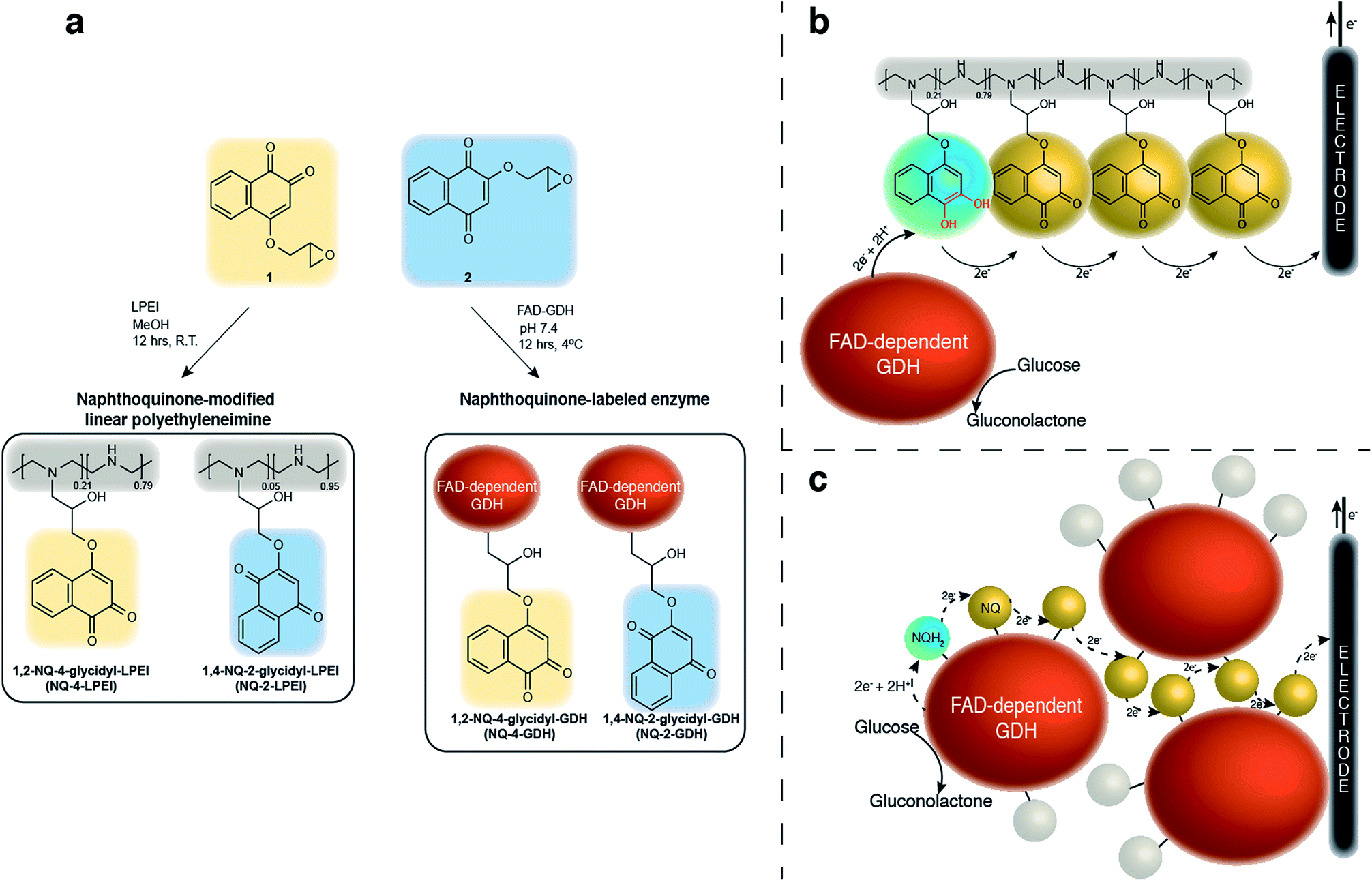 | ||
| Fig. 1 The rational design of naphthoquinone (NQ) derivatives allows for desirable covalent immobilization by two methods. Mediated bioelectrocatalytic glucose oxidation can subsequently occur by flavin adenine dinucleotide-dependent glucose dehydrogenase (GDH). (a) NQ derivatives were designed and synthesized to have epoxide functionalities, allowing their covalent attachment to a linear polyethyleneimine backbone (forming a NQ redox hydrogel, NQ-LPEI) and direct labeling of GDH. (b) NQs are able to undergo self-exchange whereby electrons and protons diffuse along a functionalized polymer backbone to an electrode surface. (c) Epoxide functionalities permit multipoint covalent NQ attachment to GDH. | ||
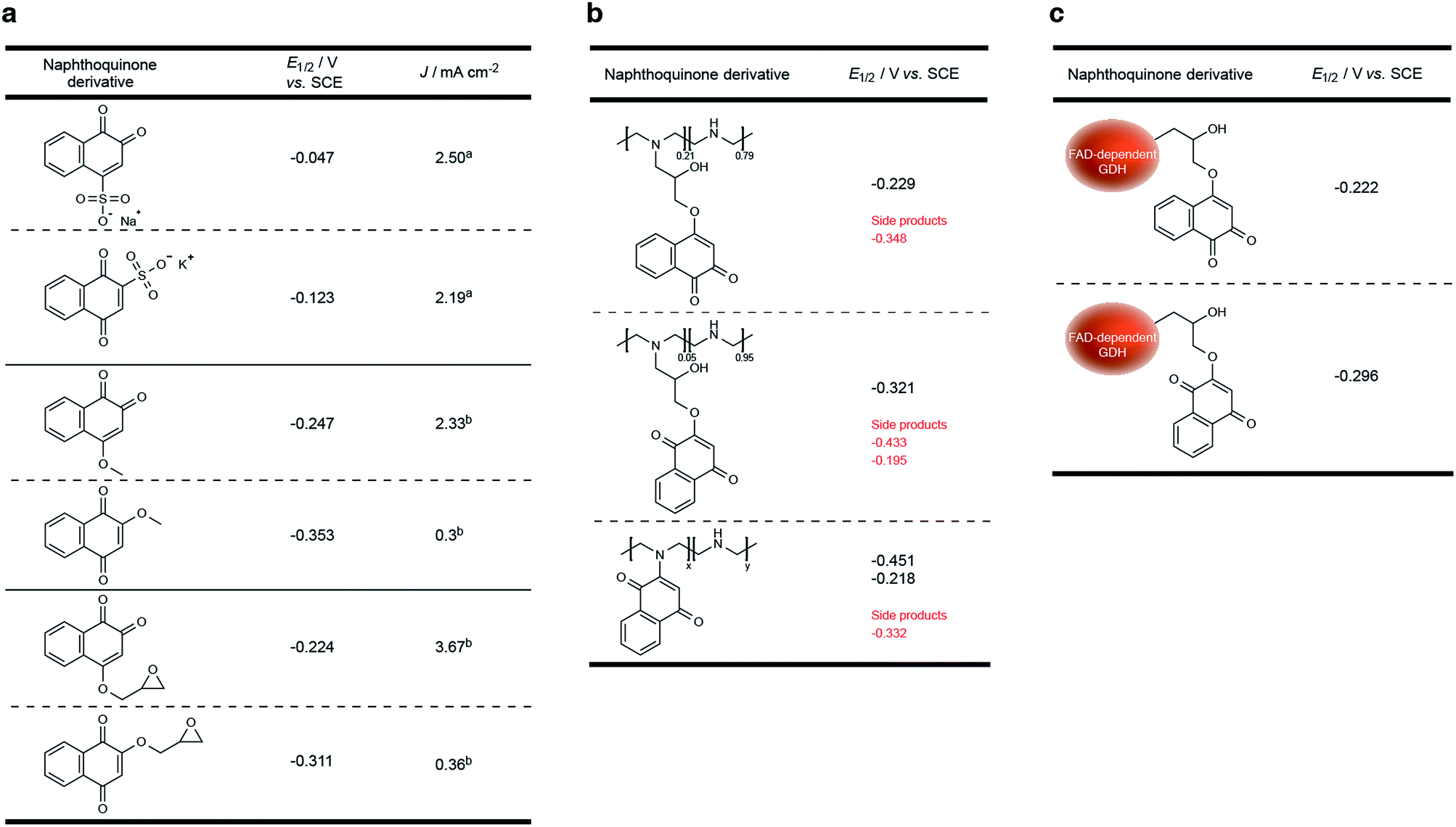 | ||
| Fig. 2 Direct substitution significantly alters the redox potential of NQ. (a) Three NQ derivative couples were comparatively evaluated for their ability to mediate electrons between GDH and an electrode (following enzymatic glucose oxidation). Each NQ couple was evaluated as its 2- or 4-functionalized derivative. Soluble NQsa were evaluated as diffusive moieties at concentrations of 2 mM, where GDH was entrapped on a glassy carbon electrode. Partially soluble NQsb were evaluated by co-entrapment at a glassy carbon electrode surface. (b) Redox potential of two desired NQ-LPEI redox hydrogels (NQ-4-LPEI and NQ-2-LPEI) and one undesired by-product. (c) Redox potential of two NQ derivatives covalently attached to GDH. All NQ derivatives were evaluated at pH 7.4 (citrate/phosphate buffer, 0.2 M). Glassy carbon electrodes were modified with carbon nanotubes, prior to enzyme entrapment. J values were determined by background subtraction (blank solution) at a potential of 100 mV more positive than the oxidative peak potential (cyclic voltammetry performed at 10 mV s−1). | ||
The redox potential of NQ derivatives also varies depending on the position of introduced chemical functionality, as well as the position of the ketone groups (1,2-NQ or 1,4-NQ). Additionally, 1,4-NQ derivatives were found to possess lower redox potentials, in all cases investigated. Interestingly, 1,2-NQ derivatives were found to have larger catalytic current densities (Jmax), which could be a structural reorganization effect, an enzymatic affinity effect, or simply a result of increased overpotential between the potential of the enzyme redox cofactor (FAD) and the NQ derivative. Following covalent immobilization of the 1,2-NQ-epoxy(1) and 1,4-NQ-epoxy(4) onto their respective LPEI polymers, NQ-4-LPEI and NQ-2-LPEI, small shifts in their redox potentials of −5 mV and −10 mV were observed (Fig. 2b). The modification of GDH with 1 and 2 resulted in small respective potential shifts of +2 mV and +15 mV (Fig. 2c).
Mediated glucose bioelectrocatalysis
As expected, LPEI and GDH modification with the 1,2-NQ-epoxy (1) derivatives (NQ-4-LPEI and NQ-4-GDH, Fig. 3a and b) yielded greater Jmax values than their 1,4-NQ-epoxy (2) derivative counterparts (NQ-2-LPEI and NQ-2-GDH, ESI Fig. 1a and b†) at the expense of slightly increased onset potentials of mediated bioelectrocatalysis. Nevertheless, the NQ-4-LPEI redox hydrogel was able to facilitate mediated bioelectrocatalytic glucose oxidation by GDH from an approximate onset potential of −0.25 V (vs. SCE at pH 6.5) while reaching 1.95 mA cm−2 at −0.09 V. The NQ-labeled GDH (NQ-4-GDH) was able to mediate enzymatic glucose oxidation from an onset potential of approximately −0.23 V (vs. SCE at pH 6.5) while reaching 1.94 mA cm−2 at 0 V, although approximately 5.8× more GDH was employed per electrode for the NQ-4-GDH bioelectrode to reach similar Jmax values. Along with the ability to produce similar Jmax values using significantly lower enzyme quantities per electrode, the NQ-4-LPEI method can be characterized with more ease. Thus, the NQ-4-LPEI method was chosen to be scaled up onto larger planar carbon electrodes (Toray paper), which yielded Jmax values of 3.3 mA cm−2 at 0 V vs. SCE (Fig. 3c) for mediated glucose oxidation by GDH. This ability to generate high current at low potentials is desirable, since it contributes to increased EFC OCP values and results in larger power densities.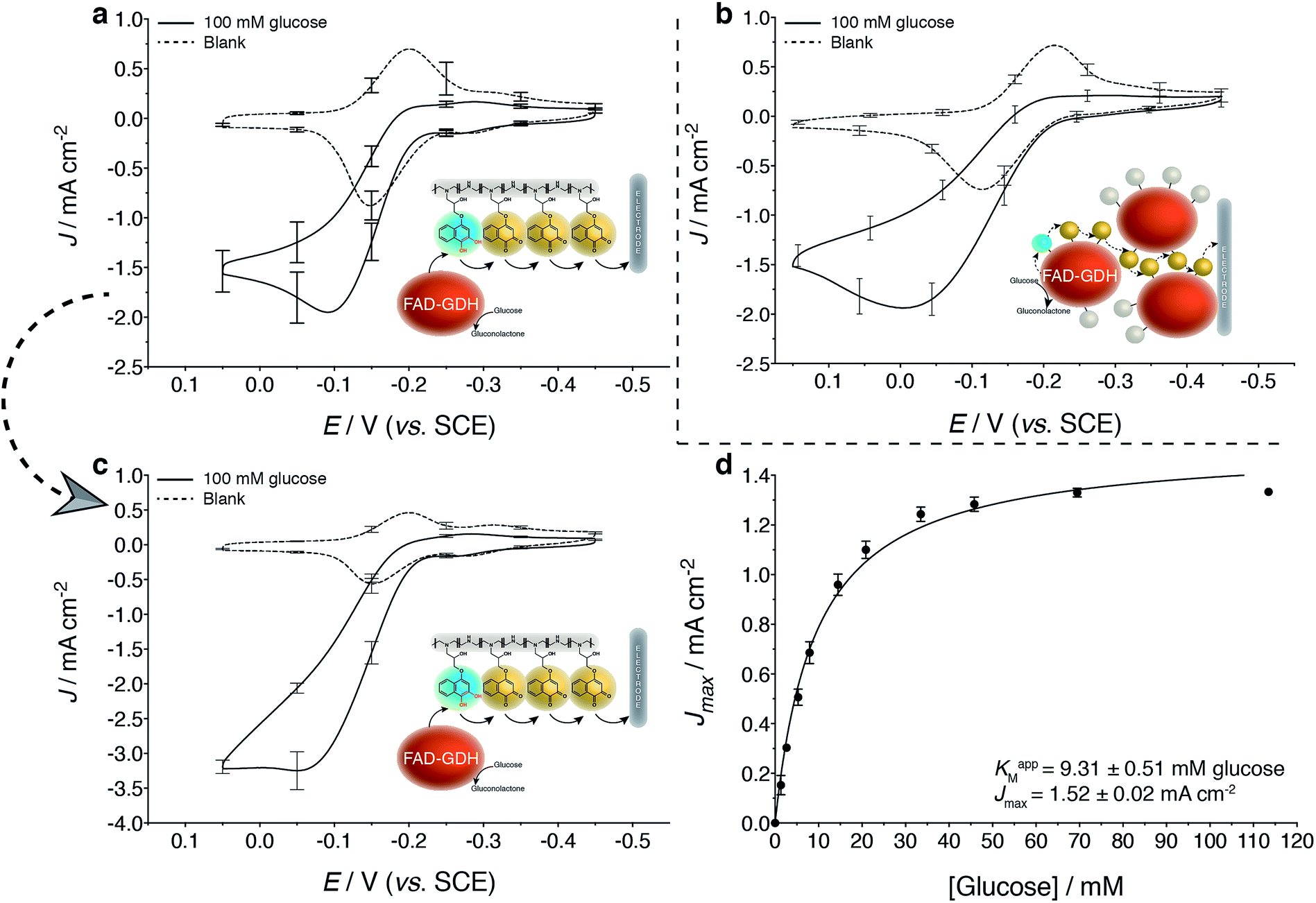 | ||
| Fig. 3 Electrochemical evaluation of carbon electrodes modified with a NQ redox hydrogel (NQ-4-LPEI) and NQ-labeled GDH (NQ-4-GDH). (a) Cyclic voltammogram of NQ-4-LPEI GDH bioelectrodes in the absence and presence of glucose. (b) Cyclic voltammogram of NQ-4-GDH bioelectrodes in the absence and presence of glucose. NQ-4-GDH was covalently immobilized with hydrophobically modified linear polyethyleneimine (C8LPEI). (c) NQ-4-LPEI GDH redox hydrogels were scaled to simple carbon paper electrodes (Toray) to demonstrate performance on an applicable and disposable electrode. (d) Apparent Michaelis–Menten kinetics of the resulting NQ-4-LPEI GDH bioelectrode (from c). All electrodes were evaluated at pH 6.5 (citrate/phosphate buffer, 0.2 M). Cyclic voltammograms were performed at 10 mV s−1 and amperometric i–t experiments were performed at 0 V (vs. SCE). Error bars report one standard deviation (n = 3). Electrodes were modified with carbon nanotubes prior to functionalization. | ||
Evaluation of the Michaelis–Menten kinetics (Fig. 3d) of the NQ-4-LPEI bioelectrodes by non-linear regression at this scaled electrode reported an apparent Michaelis–Menten constant (KM) of 9.3 mM, with an associated Jmax of 1.5 mA cm−2 measured in pH 6.5 buffer at 0 V (vs. SCE). Apparent KM values were also calculated for the NQ-4-LPEI and NQ-4-GDH bioelectrodes on glassy carbon electrodes (ESI Fig. 2a and b†). The apparent KM obtained for the NQ-4-LPEI GDH bioelectrodes were almost a factor of 10 lower than those of the NQ-4-GDH bioelectrodes, with improved Jmax values. The apparent KM obtained in Fig. 3d compares favorably to the value reported by the supplier (50 mM), as well as to two previous studies at ferrocene- and dimethylferrocene-modified redox hydrogel electrodes (33 mM at pH 5.5 and 22 mM at pH 7.4, respectively).37,38 Control bioelectrodes for the NQ-4-GDH technique were prepared with NQ-labeled bovine serum albumin (NQ-4-BSA) and with GDH (unlabeled), which demonstrated the necessity for the combination of GDH and NQ to undergo bioelectrocatalytic oxidation of glucose (ESI Fig. 2c†).
In contrast, although bioelectrodes prepared with the NQ-2-LPEI hydrogel outperformed bioelectrodes prepared with NQ-2-GDH, only negligible mediated bioelectrocatalytic currents were observed when compared to their NQ-4-LPEI and NQ-4-GDH counterparts. Thus, bioelectrodes and biofuel cells were evaluated with the NQ-4-LPEI and NQ-4-GDH derivatives, since they offered improved performance over NQ-2-LPEI and NQ-2-GDH.
Characterization of NQ immobilization methods
To create the NQ-4-LPEI polymer, a 25% substitution ratio of the LPEI polymer with 1 was targeted. Washing of the polymer to remove unreacted 1 suggested a final substitution of approximately 22%. 1H NMR analysis of the NQ-4-LPEI polymer calculated a substitution of approximately 21% (ESI Fig. 12 and 13†).29 Additionally, an average molecular mass of 91.3 g mol−1 was calculated per average repeating unit (previously determined from substitution approximation). Potentiostatic evaluation of the NQ-4-LPEI bioelectrodes at varying scan rates demonstrated a linear relationship between current and scan rate, which was used to calculate a total NQ quantity of 14.4 nmol cm−2 by the Laviron equation (ESI Fig. 3a†).39In order to estimate an apparent electron diffusion coefficient, it was necessary to determine the approximate thickness of the NQ-4-LPEI redox polymer. To this end, equivalent bioelectrodes were prepared on indium–tin oxide (ITO) electrodes with the inclusion of fluorescein (as a fluorescent probe) and the approximate film thickness was determined by confocal laser-scanning microscopy (ESI†). Bioelectrodes were rinsed with DI water and pretreated by cyclic voltammetry (at a scan rate of 50 mV s−1) in citrate/phosphate buffer (pH 6.5), to induce any film swelling due to hydration. An estimated film thickness of 210 μm was determined, which yielded an apparent electron diffusion coefficient of 1.26 × 10−11 cm2 s−1, with the use of the Randles–Sevčik equation for a planar glassy carbon electrode, which exhibited a dependence of current on the square-root of the scan rate in the absence of multi-walled carbon nanotubes (MWCNTs) that were used previously.40 To enable these calculations, the volume of the NQ-4-LPEI redox polymer matrix was calculated by assuming a cylindrical volume, and by assuming all NQ-4-LPEI is immobilized and not removed during rinsing of the electrodes prior to testing.
As shown in Fig. 3, a smaller set of redox peaks are identified that have an E1/2 of −0.295 V (vs. SCE), which shift to −0.348 V (vs. SCE at pH 7.4) and do not facilitate MET by GDH. These peaks are attributed to ability of the NQ-derivatives to react directly with the secondary amines of LPEI, forming a NQ with a tertiary amine in place of the introduced epoxide functionality. This is rationalized by the use of water-soluble 1,2-naphthoquinone-4-sulfonate and its ability to quantify amines under mild conditions,41,42 and was demonstrated by adding 2-methoxy-1,4-naphthoquinone to a stirred solution of LPEI (Fig. 2b) or N-ethylmethylamine (in the absence of additional base). 1H and 13C NMR and ESI-MS were used to characterize the red product formed between 2-methoxy-1,4-naphthoquinone and N-ethylmethylamine, which confirmed direct amine substitution of the NQ in place of the methoxy functionality. Nevertheless, analysis by the Laviron equation determined only approximately 22% of the total NQ loading per electrode to be due to this side reaction (ESI Fig. 3a†). The LPEI polymer formed from the side reaction with 2-methoxy-1,4-naphthoquinone (Fig. 2b) was used to create a GDH bioelectrode (ESI Fig. 3b†). Mediated bioelectrocatalysis was not observed, although NQ redox peaks were observed (along with trace peaks associated with residual 2-methoxy-1,4-naphthoquinone). It is hypothesized that the redox couple observed at −0.218 V (vs. SCE) is due to electrochemistry of the tertiary amine at the NQ.
Analysis of the NQ-4-GDH bioelectrodes at differing scan rates demonstrated only trace amounts of this amine side-product; however, a low naphthoquinone loading of 3.1 nmol cm−2 was calculated (ESI Fig. 3c†). Unreacted 1, denatured protein, and dissociated FAD were removed from the product by centrifugal spin-column size exclusion. Electrochemistry of the filtrate demonstrated the presence of 1 (or hydrolyzed 1 product), confirming the ability for this technique to efficiently remove NQ that had not covalently attached to GDH.
The NQ-4-LPEI and NQ-4-GDH bioelectrodes were evaluated between pH 5.5–7.4 (ESI Fig. 4a–f†). Both bioelectrode architectures were able to oxidize glucose by mediated bioelectrocatalysis, with pH 6.5 appearing optimal for the NQ-4-LPEI bioelectrodes and pH 7.4 appearing optimal for the NQ-4-GDH bioelectrodes. Between pH 5.5–7.4, the E1/2 was related to pH by a shift in potential of −58.4 mV (NQ-4-LPEI) and −68.5 mV (NQ-4-GDH) per pH unit. A shift of −59.2 mV was expected per pH unit, since the redox potential of the NQ derivatives below the pKa of reduced NQ (NQH2) is coupled to two protons.43 For later evaluation within a complete glucose/O2 EFC, pH 6.5 was chosen as a compromise for efficient biocathodic O2 reduction by bilirubin oxidase (BOx).
FAD-dependent GDH versus GOx
Bioelectrodes were prepared with GOx from Aspergillus niger, to evaluate its ability (when compared to GDH) to undergo mediate bioelectrocatalytic glucose oxidation at the NQ-4-LPEI redox polymer. Interestingly, the addition of glucose to GOx-based bioelectrodes did not result in an oxidative catalytic wave (Fig. 4a). Further, GOx did not exhibit activity towards a glucose assay performed with 1,2-naphthoquinone-4-sulfonate (Fig. 4b). Enzyme activity is observed at the GOx-based bioelectrodes, however, in the form of the depletion of dissolved O2 at the electrode surface. These results further demonstrate the unique applicability of FAD-dependent GDH within glucose EFC technologies as well as glucose biosensor applications.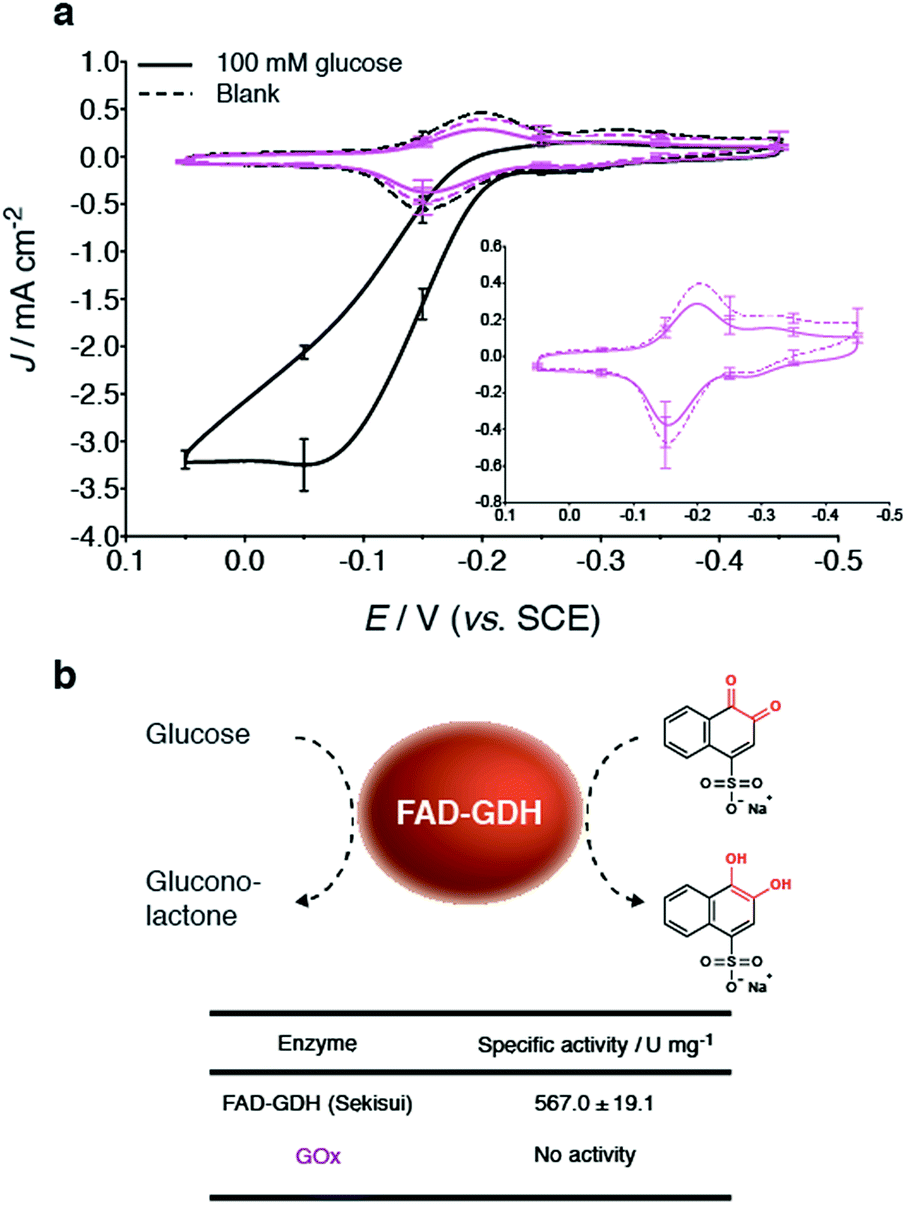 | ||
| Fig. 4 (a) Cyclic voltammograms of NQ-4-LPEI bioelectrodes prepared with GOx (Aspergillus niger) compared to their GDH counterpart electrodes. The inset presents increased zoom of the NQ-4-LPEI GOx bioelectrode. (b) Specific enzymatic activities of GDH and GOx to glucose, using 1,2-naphthoquinone-4-sulfonate as the redox indicator. Absorbance changes were measured at 500 nm. BSA controls showed no activity. All electrochemical experiments were performed at pH 6.5 (citrate/phosphate buffer, 0.2 M). Specific activities were determined at pH 6.5 (citrate/phosphate buffer, 0.05 M). Error bars represent one standard deviation (n = 3). Cyclic voltammograms were performed at 10 mV s−1. Electrodes were prepared on carbon paper electrodes (Toray) that were pretreated with carbon nanotubes. | ||
Additionally, the specific activity of the three different GDHs obtained from different commercial sources were determined bioelectrochemically as well as by using 1,2-naphthoquinone-4-sulfonate as a water-soluble indicator while having direct applicability to the MET system presented in this research (ESI Fig. 5†). Of the three commercial sources evaluated, GDH obtained from Sekisui Diagnostics (UK) demonstrated the largest Jmax values, under the given experimental conditions.
High power density enzymatic fuel cell operating on glucose
Finally, the applicability of the NQ immobilization strategies presented within this research were evaluated within glucose/O2 EFCs (Fig. 5a). Direct electron transfer-type (DET) O2 reducing BOx-based biocathodes were prepared from a previously reported procedure, whereby MWCNTs are modified with anthracene moieties to enable orientation of BOx towards the electrode surface to undergo DET via the type-1 copper center (Fig. 5a and b).11,44–46 BOx bioelectrodes prepared on carbon felt were capable of producing Jmax values of 2 mA cm−2 for O2 reduction in quiescent solutions (stationary and not purged with O2). EFCs were characterized by galvanostatically drawing current from the device while measuring the potential difference of the EFC. Increasing current was drawn from the device at small increments, until the potential difference between the bioanode and biocathode reached 0 V.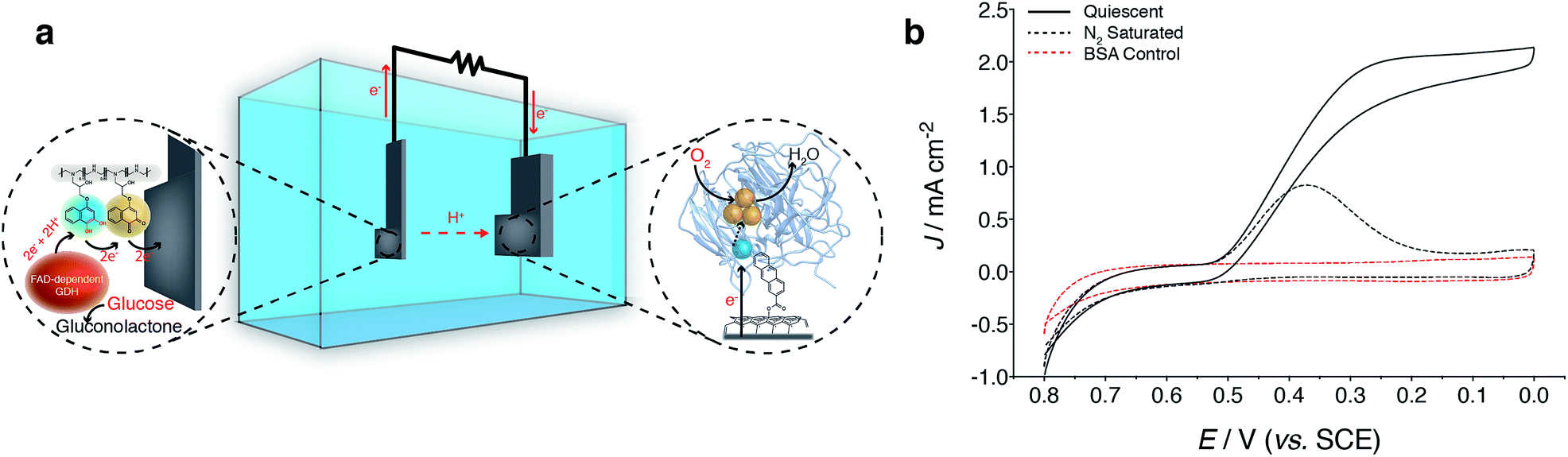 | ||
| Fig. 5 NQ-4-LPEI GDH bioelectrodes were operated as bioanodes in EFCs and coupled with O2-reducing direct electron transfer-type (DET) biocathodes employing bilirubin oxidase (BOx, Myrothecium sp.). (a) EFC configuration for a glucose/O2 EFC, where the NQ-4-LPEI hydrogel mediated bioelectrocatalytic glucose oxidation by GDH at the bioanode. Carbon nanotubes modified with anthracene moieties were used to orientate BOx and permit direct bioelectrocatalytic O2 reduction. EFCs were operated in buffer containing 100 mM glucose at pH 6.5 (citrate/phosphate, 0.2 M) that was not gas purged. In all cases, the bioanodes were limiting, whereby the absolute currents from the bioanodes (uncorrected to surface area) were smaller than the absolute currents obtained at the biocathodes. (b) Cyclic voltammograms of BOx biocathodes in quiescent solutions demonstrate direct bioelectrocatalytic O2 reduction with high Jmax values. Blank experiments were performed in buffer that was purged with N2 and control experiments were performed in quiescent buffer using bovine serum albumin (BSA). Cyclic voltammograms were performed at 1 mV s−1 and under hydrostatic conditions. Biocathodes were prepared on carbon felt with a geometric facial area of 1 cm2. | ||
EFCs prepared with NQ-4-LPEI bioanodes exhibited large OCPs of 0.821 ± 0.008 V and large Jmax values of 2.4 ± 0.1 mA cm−2. This resulted in power densities of 1.1 ± 0.1 mW cm−2 under stirred conditions, although the EFCs were not purged with any additional air or oxygen and the bioanode was limiting (Fig. 6). Small charging contributions were expected from the electrochemical procedure used to characterize the EFCs. These were determined to be only 0.20 ± 0.02 mA cm−2 and 0.02 ± 0.01 mW cm−2 in the absence of glucose.
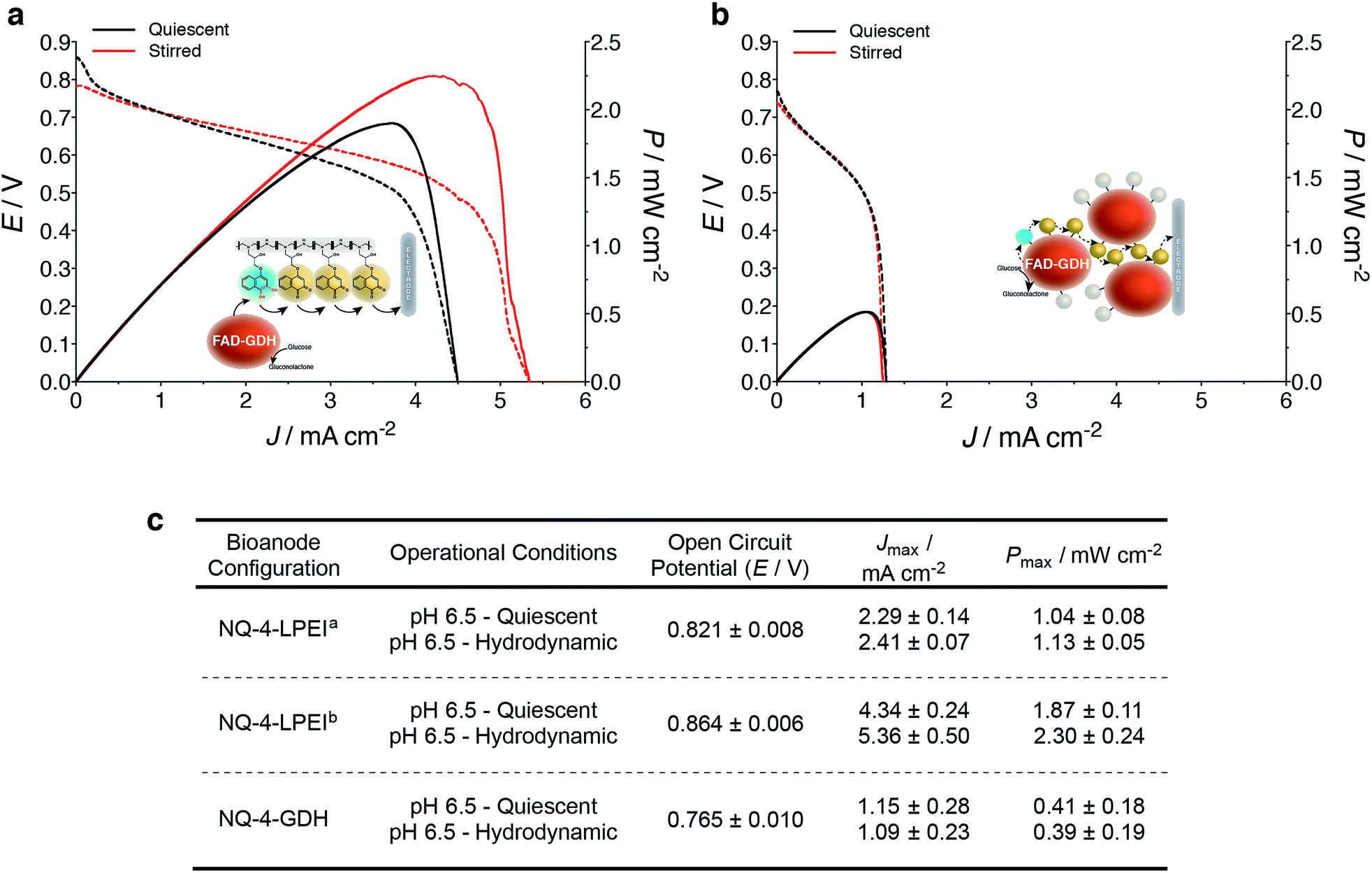 | ||
| Fig. 6 Evaluation of glucose/O2 EFCs employing the NQ-4-LPEI GDH and NQ-4-GDH bioanodic configurations. (a) EFCs employing NQ-4-LPEI GDH bioelectrodes as the bioanodes were evaluated in quiescent and stirred buffer containing 100 mM glucose (pH 6.5, citrate/phosphate, 0.2 M). Bioanodes were prepared on carbon paper electrodes (Toray) modified with carbon nanotubes and biocathodes were prepared on carbon felt coated with anthracene-modified carbon nanotubes. The bioanodes were the limiting components. (b) EFCs employing NQ-4-GDH bioelectrodes as the bioanodes were evaluated in quiescent and stirred buffer containing 100 mM glucose (pH 6.5, citrate/phosphate, 0.2 M). Bioanodes were prepared on glassy carbon electrodes modified with carbon nanotubes and biocathodes were prepared on carbon paper (Toray) coated with anthracene-modified carbon nanotubes. The bioanodes were the limiting components. (c) Comparison of alternative EFC configurations where NQ-4-LPEI GDH bioanodes were one-sideda and two-sidedb. EFC performance measured by galvanostatically drawing increasing current from the EFC, until short circuit. Power densities were calculated from the current and associated potential of the EFC. | ||
EFCs were then prepared with bioanodes with increased biocatalyst loadings, whereby both sides of the electrode support were coated with the NQ-4-LPEI/GDH matrix. The OCP of the EFC increased to 0.821 ± 0.008 V and larger Jmax values of 5.4 ± 0.5 mA cm−2 (Fig. 6a and c) were obtained. This resulted in the EFC being able to produce power densities of 2.3 ± 0.2 mW cm−2 at a high operational voltage of 0.55 V.
EFCs were also prepared with the NQ-4-GDH bioanodes, which yielded an OCP of 0.765 ± 0.010 V (Fig. 6b and c). The device was capable of producing Jmax values of 1.2 ± 0.3 mA cm−2 and an associated power density of 0.4 ± 0.2 mW cm−2 under quiescent conditions, where stirring did not statistically improve performance.
In light of reports that demonstrate the importance of carbon nanotubes in bioelectrode construction,47–50 it is hypothesized that the incorporation of MWCNTs at both the bioanode and biocathode (albeit with different functionalities and roles) is vital to obtaining high current and associated power densities.
To evaluate the stability of the devices, low current was continually drawn (10 μA cm−2) from an EFC. This demonstrates the device's ability to continually power devices that require low current feeds, as well as the ability to stack devices in parallel to increase current densities over larger operational windows. The device was continually operated at room temperature for 24 hours, where an initial potential difference of 0.86 V dropped to only 0.71 V (ESI Fig. 6†).
Conclusions
Organic redox species are emerging as alternative mediators in EFCs, whose redox potentials can be tailored according to the intended enzyme system and application. We report the covalent immobilization of a rationally designed naphthoquinone mediator (Fig. 1a–c) alongside an FAD-dependent GDH that is able to produce high current (5.4 ± 0.5 mA cm−2) and power (2.3 ± 0.2 mW cm−2) densities from glucose, at high operational voltages (0.55 V). Furthermore, these exceptionally large values are obtained from the use of only one anodic enzyme performing only a single 2e− oxidation. Thus, this redox hydrogel could be applied to additional enzymes to further enhance performance.The covalent immobilization of NQ was demonstrated by two methods. First, NQ was immobilized onto LPEI to yield a NQ-functionalized redox hydrogel polymer (NQ-4-LPEI or NQ-2-LPEI). Such hydrogel polymers are desirable due to their unique ability to swell in aqueous environments, which can enhance substrate/product diffusion to/from electrodes. Second, GDH was directly labeled with NQ and the resulting enzyme was able to mediate bioelectrocatalytic glucose oxidation.
Suitable functionalization and subsequent covalent attachment of NQ onto polymer supports is not trivial. Relatively mild reaction conditions are all that are required to enable NQ to undergo undesired side reactions with primary and secondary amines, which are then rendered inactive in respect to MET with FAD-dependent GDH. The epoxy-functionalized NQ derivatives presented within this research article (1 and 2) are covalently attached to the polymer backbone (LPEI) without the need for any additional reagents, which minimized undesired side reactions (only 22% inefficiency is reported). Although the absence of MET with amine-functionalized NQs could simply be a function of potential, 2-methoxy-1,4-naphthoquinone was found to have a comparable redox potential to that of the amino-NQ side product, which suggests enzyme active site rejection or electronic reorganizational properties of these amino-NQ side products may be responsible for inactivity (Fig. 2a and b).
Interestingly, GOx did not exhibit any activity to any of the NQ derivatives and resulting NQ-polymers (Fig. 4a). Additionally, the NQ-4-LPEI hydrogel demonstrated variable performances when combined with GDH from three different commercial suppliers, which were determined to have specific activities to 1,2-naphthoquinone-4-sulfonate that were not too dissimilar.
The stability of the NQ-4-LPEI based EFCs was also evaluated over time, by continually drawing a low current density and measuring the potential difference over time. The EFC continued to deliver 10 μA cm−2 over an extended time period of 24 hours, where the potential difference of the EFC comfortably continued to operate at high potential differences of over 0.7 V. The initial potential difference of 0.86 V dropped only to 0.71 V after 24 hours of continuous operation (ESI Fig. 6†).
Acknowledgements
We thank National Science Foundation for funding and Dr Jim Muller for performing mass spectrometry.References
- D. Leech, P. Kavanagh and W. Schuhmann, Electrochim. Acta, 2012, 84, 223–234 CrossRef CAS PubMed.
- S. C. Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867–4886 CrossRef CAS.
- A. Heller, Phys. Chem. Chem. Phys., 2004, 6, 209–216 RSC.
- M. Falk, C. W. Narváez Villarrubia, S. Babanova, P. Atanassov and S. Shleev, ChemPhysChem, 2013, 14, 2045–2058 CrossRef CAS PubMed.
- M. Holzinger, A. Le Goff and S. Cosnier, Front. Chem., 2014, 2, 63 Search PubMed.
- M. Opallo and R. Bilewicz, Adv. Phys. Chem., 2011, 947637 Search PubMed.
- C. F. Blanford, R. S. Heath and F. A. Armstrong, Chem. Commun., 2007, 1710–1712, 10.1039/b703114a.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366–4413 CrossRef CAS PubMed.
- E. Katz and I. Willner, J. Am. Chem. Soc., 2003, 125, 6803–6813 CrossRef CAS PubMed.
- N. Mano, F. Mao, W. Shin, T. Chen and A. Heller, Chem. Commun., 2003, 518–519, 10.1039/b211796g.
- C. F. Blanford, C. E. Foster, R. S. Heath and F. A. Armstrong, Faraday Discuss., 2008, 140, 319–335 RSC.
- J. A. Cracknell, T. P. McNamara, E. D. Lowe and C. F. Blanford, Dalton Trans., 2011, 40, 6668–6675 RSC.
- V. Soukharev, N. Mano and A. Heller, J. Am. Chem. Soc., 2004, 126, 8368–8369 CrossRef CAS PubMed.
- N. Mano and L. Edembe, Biosens. Bioelectron., 2013, 50, 478–485 CrossRef CAS PubMed.
- N. Mano, Appl. Microbiol. Biotechnol., 2012, 96, 301–307 CrossRef CAS PubMed.
- B. Persson, H. L. Lan, L. Gorton, Y. Okamoto, P. D. Hale, L. I. Boguslavsky and T. Skotheim, Biosensors'92 Proceedings, 1992, pp. 111–118 Search PubMed.
- H. Sakai, T. Nakagawa, Y. Tokita, T. Hatazawa, T. Ikeda, S. Tsujimura and K. Kano, Energy Environ. Sci., 2009, 2, 133–138 CAS.
- K. Murata, K. Kajiya, N. Nakamura and H. Ohno, Energy Environ. Sci., 2009, 2, 1280–1285 CAS.
- D. P. Hickey, A. J. Halmes, D. W. Schmidtke and D. T. Glatzhofer, Electrochim. Acta, 2014, 149, 252–257 CrossRef CAS PubMed.
- B. Reuillard, A. Le Goff, C. Agnes, M. Holzinger, A. Zebda, C. Gondran, K. Elouarzaki and S. Cosnier, Phys. Chem. Chem. Phys., 2013, 15, 4892–4896 RSC.
- S. Pöller, M. Shao, C. Sygmund, R. Ludwig and W. Schuhmann, Electrochim. Acta, 2013, 110, 152–158 CrossRef PubMed.
- F. Conzuelo, J. Vivekananthan, S. Pöller, J. M. Pingarrón and W. Schuhmann, ChemElectroChem, 2014, 1, 1854–1858 CrossRef CAS PubMed.
- M. N. Zafar, N. Beden, D. Leech, C. Sygmund, R. Ludwig and L. Gorton, Anal. Bioanal. Chem., 2012, 402, 2069–2077 CrossRef CAS PubMed.
- D. Fapyane, Y. Lee, C. Y. Lim, J.-H. Ahn, S.-W. Kim and I. S. Chang, ChemElectroChem, 2014, 1, 1844–1848 CrossRef CAS PubMed.
- S. Tsujimura, K. Murata and W. Akatsuka, J. Am. Chem. Soc., 2014, 136, 14432–14437 CrossRef CAS PubMed.
- B. Reuillard, A. Le Goff and S. Cosnier, Anal. Chem., 2014, 86, 4409–4415 CrossRef CAS PubMed.
- O. Yehezkeli, R. Tel-Vered, J. Wasserman, A. Trifonov, D. Michaeli, R. Nechushtai and I. Willner, Nat. Commun., 2012, 3, 742 CrossRef PubMed.
- O. Yehezkeli, R. Tel-Vered, D. Michaeli, R. Nechushtai and I. Willner, Small, 2013, 9, 2970–2978 CrossRef CAS PubMed.
- S. A. Merchant, M. T. Meredith, T. O. Tran, D. B. Brunski, M. B. Johnson, D. T. Glatzhofer and D. W. Schmidtke, J. Phys. Chem. C, 2010, 114, 11627–11634 CAS.
- N. Plumeré, O. Rüdiger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef PubMed.
- N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2003, 125, 6588–6594 CrossRef CAS PubMed.
- F. Giroud, R. D. Milton, B.-X. Tan and S. D. Minteer, ACS Catal., 2015, 5, 1240–1244 CrossRef CAS.
- Z. Zhu, T. Kin Tam, F. Sun, C. You and Y. H. P. Zhang, Nat. Commun., 2014, 5, 3026 Search PubMed.
- E. Yu. Katz, V. V. Borovkov and R. P. Evstigneeva, J. Electroanal. Chem., 1992, 326, 197–212 CrossRef.
- C. Mateo, V. Grazú, B. C. Pessela, T. Montes, J. M. Palomo, R. Torres, F. López-Gallego, R. Fernández-Lafuente and J. M. Guisán, Biochem. Soc. Trans., 2007, 35, 1593–1601 CrossRef CAS PubMed.
- G. Grampp and W. Jaenicke, J. Electroanal. Chem. Interfacial Electrochem., 1987, 229, 297–303 CrossRef CAS.
- R. D. Milton, K. Lim, D. P. Hickey and S. D. Minteer, Bioelectrochemistry, 2015 DOI:10.1016/j.bioelechem.2015.04.005.
- R. D. Milton, F. Giroud, A. E. Thumser, S. D. Minteer and R. C. T. Slade, Phys. Chem. Chem. Phys., 2013, 15, 19371–19379 RSC.
- E. Laviron and L. Roullier, J. Electroanal. Chem., 1980, 115, 65–74 CrossRef CAS.
- F. Mao, N. Mano and A. Heller, J. Am. Chem. Soc., 2003, 125, 4951–4957 CrossRef CAS PubMed.
- O. Folin and W. T. A. O. H. Wu, J. Biol. Chem., 1922, 51, 377–391 CAS.
- J. H. Ahn, S. Y. Cho, J. D. Ha, S. Y. Chu, S. H. Jung, Y. S. Jung, J. Y. Baek, I. K. Choi, E. Y. Shin, S. K. Kang, S. S. Kim, H. G. Cheon, S.-D. Yang and J.-K. Choi, Bioorg. Med. Chem. Lett., 2002, 12, 1941–1946 CrossRef CAS.
- H. Nivinskas, S. Staškevičienė, J. Šarlauskas, R. L. Koder, A.-F. Miller and N. Čėnas, Arch. Biochem. Biophys., 2002, 403, 249–258 CrossRef CAS.
- M. T. Meredith, M. Minson, D. Hickey, K. Artyushkova, D. T. Glatzhofer and S. D. Minteer, ACS Catal., 2011, 1, 1683–1690 CrossRef CAS.
- F. Giroud and S. D. Minteer, Electrochem. Commun., 2013, 34, 157–160 CrossRef CAS PubMed.
- R. D. Milton, F. Giroud, A. E. Thumser, S. D. Minteer and R. C. T. Slade, Chem. Commun., 2014, 50, 94–96 RSC.
- Y. Yan, W. Zheng, L. Su and L. Mao, Adv. Mater., 2006, 18, 2639–2643 CrossRef CAS PubMed.
- K. Gong, X. Zhu, R. Zhao, S. Xiong, L. Mao and C. Chen, Anal. Chem., 2005, 77, 8158–8165 CrossRef CAS PubMed.
- Y. Yan, W. Zheng, M. Zhang, L. Wang, L. Su and L. Mao, Langmuir, 2005, 21, 6560–6566 CrossRef CAS PubMed.
- I. Osadebe and D. Leech, ChemElectroChem, 2014, 1, 1988–1993 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Fig. 1–18, supplementary Tables 1 and 2, 1H NMR and 13C NMR spectral data, HRMS (ESI), full experimental procedures, and enzymatic activity assays. See DOI: 10.1039/c5sc01538c |
| This journal is © The Royal Society of Chemistry 2015 |