
Hydrosilylation reaction of olefins: recent advances and perspectives
Y. Nakajima and
S. Shimada*
National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: s-shimada@aist.go.jp
First published on 12th February 2015
Abstract
This review covers the recent advances in hydrosilylation chemistry mainly published in the last decade. Hydrosilylation of olefins is an important reaction for the production of various organosilicon compounds such as industrially important silicone products. Although the utility of platinum catalysts, Speier's and Karstedt's catalysts, has been widely recognized in this field for decades, development of more efficient, selective, and cheaper catalyst systems are still desired for more economical production of organosilicon materials having superior properties. In these contexts, much progress has been made in recent years. In the platinum catalysis systems, continuous progress has been made to further improve selectivity and activity. Several non-precious metal catalysts, such as Fe and Ni catalysts, with good efficiency and selectivity have been developed. Furthermore, unique chemo- and regioselectivity have been achieved not only by precious metal catalysts but also by non-precious metal catalysts. The utility of non-transition metal catalysts including early main group metals, Lewis acidic alane, borane and phosphonium salts as well as N-heterocyclic carbenes has also been disclosed.
1. Introduction
The hydrosilylation reaction, which enables the addition of silicon hydrides across C–C multiple bonds, is an efficient method for the formation of organosilicon compounds and represents one of the most important reactions in silicon chemistry. The process is widely applied in industry to produce silane coupling agents and silicone polymers such as oils, rubbers and resins.1 The hydrosilylation reactions can also produce various organosilicon reagents, which are used in fine chemical synthesis for stereospecific oxidation, cross-coupling reactions, etc.2–4The first hydrosilylation reaction was reported in 1947 by Sommer in which trichlorosilane and 1-octene reacted in the presence of peroxide in a free-radical mechanism although the reaction selectivity was quite low.5 In the late 1950s, hexachloroplatinic acid [H2PtCl6]·H2O was revealed to be a very effective homogeneous transition metal catalyst (Speier's catalyst), in which system the selectivity was improved to a large extent.6 An important breakthrough has been made in early 70's. Thus, in 1973, Karstedt developed the platinum(0) complex containing vinyl-siloxane ligands (Karstedt's catalyst, Fig. 1),7 which exhibits extremely improved activity and selectivity as well as high solubility in polysiloxane compositions. As a result, the finding lead to the wider application of this process in the silicone chemistry, for example, the manufacture of diverse commodities including lubricating oils, pressure-sensitive adhesives, liquid injection moulding products and paper release coatings, etc.
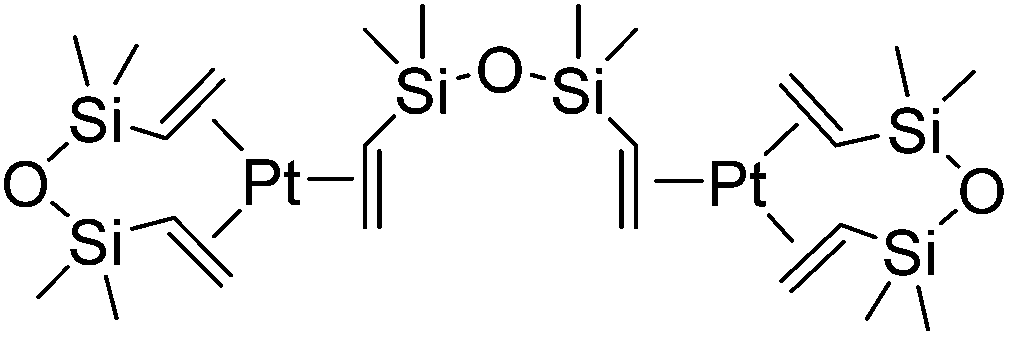 | ||
| Fig. 1 Karstedt's catalyst. | ||
In spite of the high utility in industry, however, it is known that the platinum catalysts still suffer from a number of drawbacks. For example, platinum-catalysed hydrosilylation of olefins is often accompanied by side reactions, such as dehydrogenative silylation, hydrogenation of olefins, isomerization of olefins, olefin oligomerization and redistribution of hydrosilanes (Scheme 1).
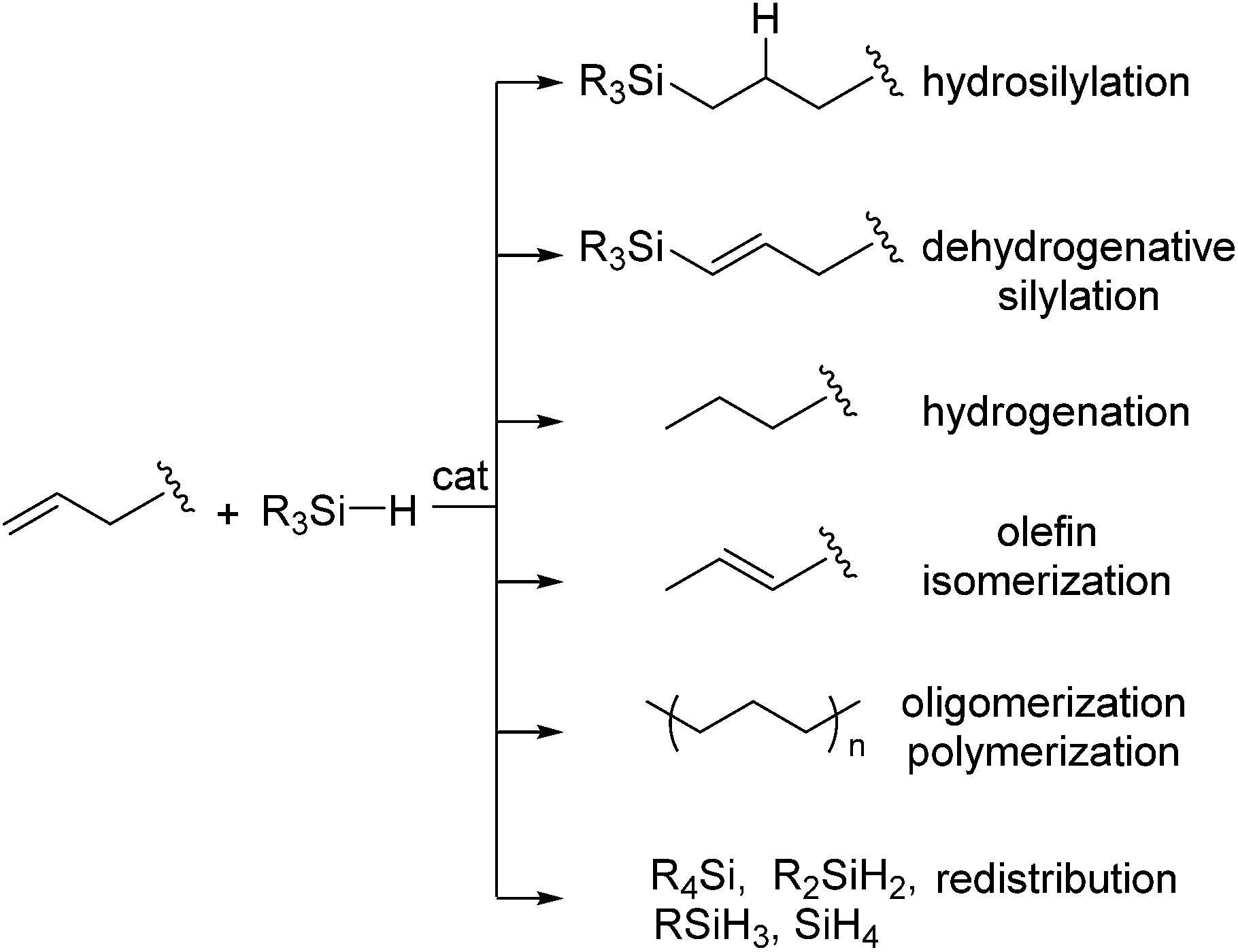 | ||
| Scheme 1 Reactions of olefin with hydrosilane in the presence of a catalyst. | ||
These side reactions lead to significant yield losses of the desired product. Furthermore, additional processes to remove the undesirable impurities are necessary, which would cause the deleterious effects on the quality or properties of the final silicone materials. Therefore, development of a well-designed catalytic system with higher selectivity and activity is still an intensive research topic to attain more economical production of silicone materials with superior quality and/or properties.
So far, numerous transition metal-based catalysts have been developed mainly using groups 8, 9, and 10 metals, and many reviews on these studies have been already reported especially in the last 20 years.8,9 Most recently, Troegel and Stohrer reviewed the advances of hydrosilylation processes covering the period from 2000 to 2010, in particular from industrial point of view.10 Active studies have been continued until now in this field using various transition metal catalysts including earth-abundant and environmental benign metals, which recently attract strong interest due to the high cost, environmental concerns, and uncertainty of the long-term supply of platinum. In addition, new types of catalysts are recently intensively investigated including not only transition metal catalysts but also other metals or non-metal catalyst systems. Thus, the present review mainly deals with important relevant contributions appearing in the last 10 years.
2. Precious metal catalysts
2.1. Platinum catalysts
The platinum catalysts have been utilized in industry for more than half a century due to the high catalytic activity and selectivity, and the high stability towards heat, oxygen and moisture. One of the most industrially important platinum catalysts, Karstedt's catalyst, has been established as the benchmark for other hydrosilylation catalysts in industrial hydrosilylation processes. Therefore, it seems that the chemistry of platinum catalysts has already reached a high level of sophistication. On the other hand, the platinum catalysts are known to possess some disadvantages as mentioned in the introduction section. One of them is the formation of platinum black during the hydrosilylation reaction, which is responsible for some of the side products and leads to a contamination of the hydrosilylated product. Also the platinum catalyst used for the curing of silicone products cannot be recovered, which leads to significant consumption of platinum metal in silicone industry (5.6 t in 2007,11 ∼3% of annual worldwide platinum production). Therefore, the development of a new Pt-catalyst system, which attains remarkable efficiency and selectivity, still draws lots of attentions.Recently, the efficiency of Pt(0) complexes supported by the N-heterocyclic carbene (NHC) as a robust σ-donor ligand was reported (Fig. 2).12 The catalytic activities of 1a–c were examined by performing hydrosilylation of 1-octene with a commercial monomer silane, bis(trimethylsiloxy)methylsilane (MD′M) (eqn (1)). Although these catalysts are slightly less active than Karstedt's catalyst, good reaction rates were obtained even at a catalyst loading of 30 ppm or less. Furthermore, no colloidal platinum species was formed and the amount of undesired by-products was greatly reduced with 1a–c. The reactivity and selectivity of these catalysts is dependent on the steric and electronic properties of the carbene substituents. An optimum was reached with cyclohexyl substituted 1b, leading to the selective formation of primary alkylsilanes with less than 1% yield of side-products, while Karstedt's catalyst provides the alkylsilane in only 78% yield with isomerized or reduced products in ca. 20%. These Pt–NHC catalysts is tolerant toward various reactive functionalities, such as free alcohols, protected alcohols, silyl ethers, ketones, and esters. It is to be noted that 1b catalysed hydrosilylation of vinylcyclohexeneoxide, which is sensitive to side reactions such as ring-opening and polymerization reactions, gave the corresponding hydrosilylated product in excellent yield (eqn (2)). The reaction is of significant importance relevant to the preparation of an polyepoxy silicone oil from vinylcyclohexeneoxide and Me3SiO(Me2SiO)80(MeHSiO)7SiMe3. Based on detailed kinetic studies, a plausible catalytic cycle, which is initiated by the dissociation of divinyltetramethyldisiloxane, was proposed (Scheme 2).13
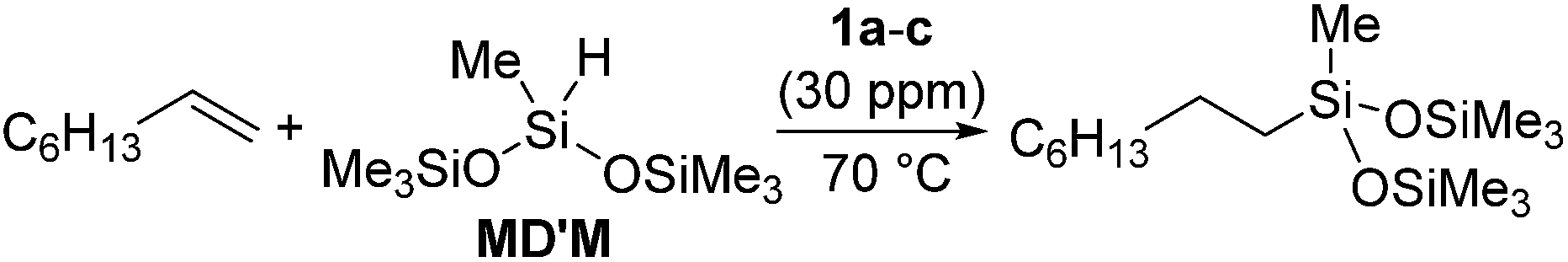 | (1) |
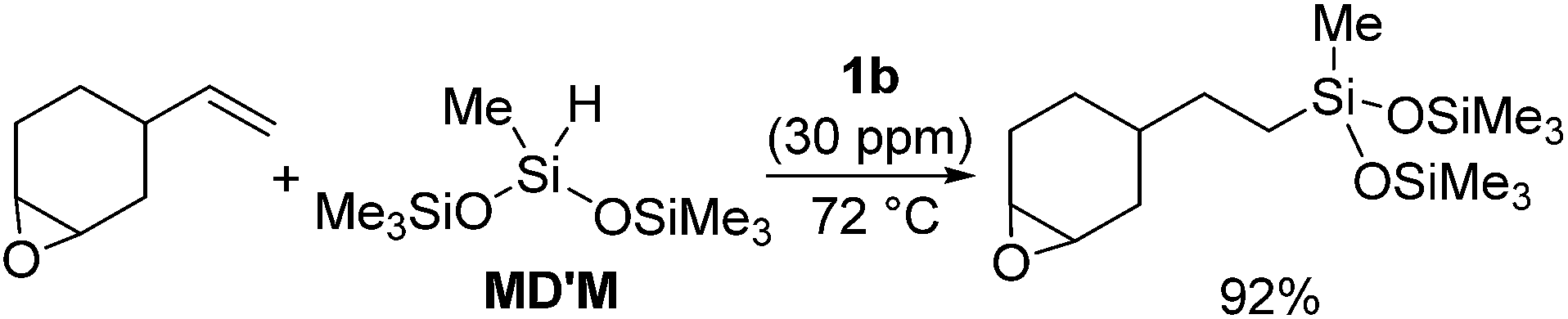 | (2) |
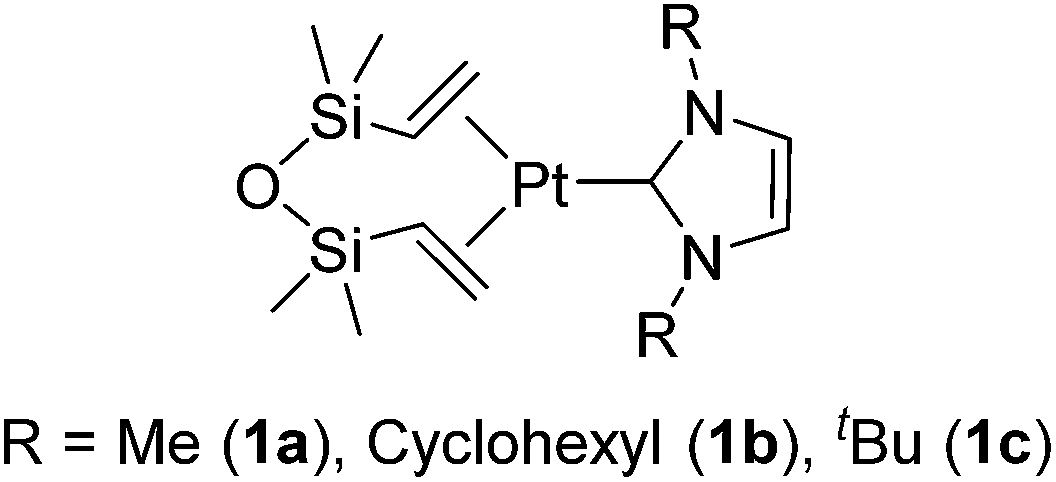 | ||
| Fig. 2 Structures of 1a–c. | ||
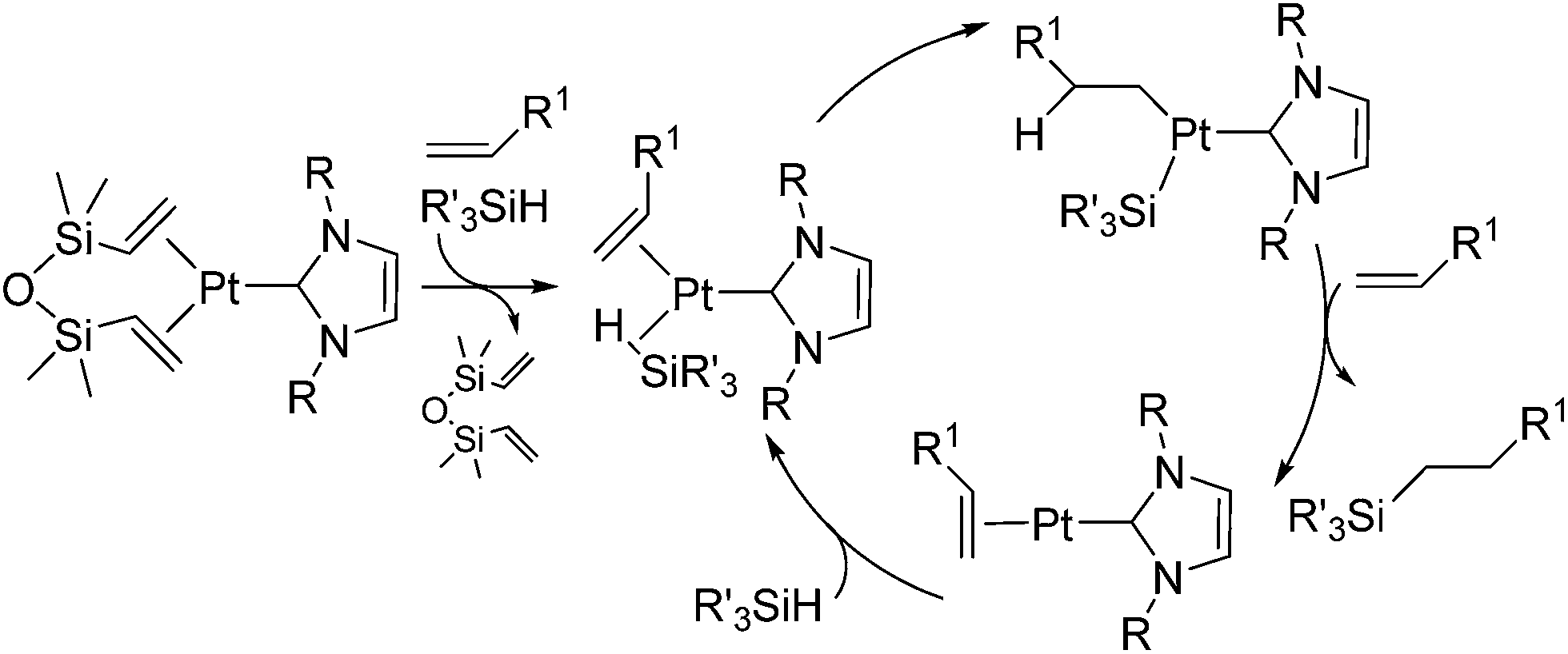 | ||
| Scheme 2 Proposed hydrosilylation mechanism catalysed by 1. | ||
Related to the above examples, many platinum complexes with a carbene ligand have been examined and revealed to be active towards catalytic hydrosilylation of styrenes.14 Cavell and co-workers reported Pt(0) complexes 2a–c coordinated with sterically demanding and stronger σ-donating 6-membered NHC ligands (Fig. 3).15
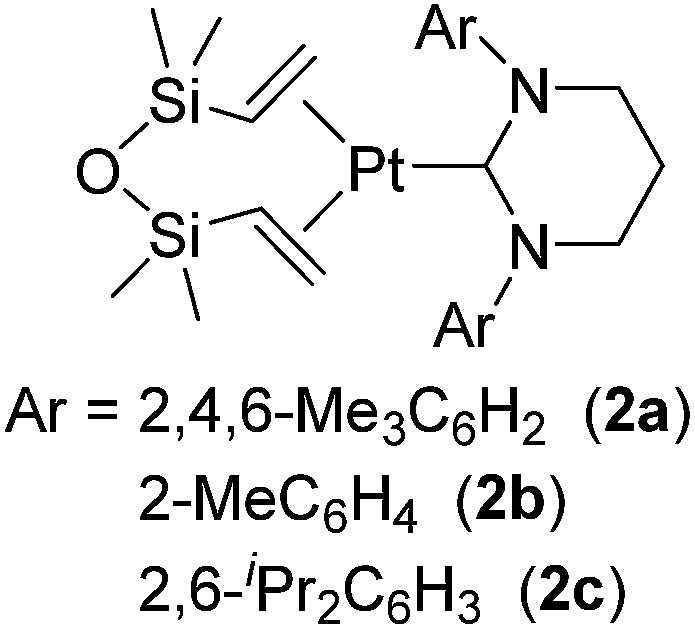 | ||
| Fig. 3 Structures of 2a–c. | ||
Although these complexes catalysed olefin isomerization in the presence of Et3SiH to give internal olefins, 1-octene was successfully hydrosilylated with MD′M at 72 °C in the presence of 0.005 mol% catalyst, resulting in the selective formation of the corresponding β-hydrosilylated product. The catalytic activity was revealed to be dependent on the steric hindrance of the complexes; i.e. the least sterically hindered 2b is the most active.
Pt(0) complexes supported by a bidentate-carbene ligand, which are expected to exhibit higher stability under harsh reaction conditions and do not form platinum metal species, were also developed (eqn (3)).16 The catalysts are active towards hydrosilylation of styrene with MD′M above 50 °C. Among the complexes investigated, it has been revealed that the Pt(II) dichloride coordinated with 4-methoxyphenyl-substituted bidentate-carbene ligand 3 (eqn (3)) turned out to be the most active. Thus, hydrosilylation of styrene with MD′M proceeded at 140 °C in the presence of 0.5 mol% of 3 to give two hydrosilylated product isomers in total yield of 69% (A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B = 22:78) in 30 s (eqn (3)). The high activity of 3 is comparable to the activity of the Karstedt's catalyst.
:B = 22:78) in 30 s (eqn (3)). The high activity of 3 is comparable to the activity of the Karstedt's catalyst.
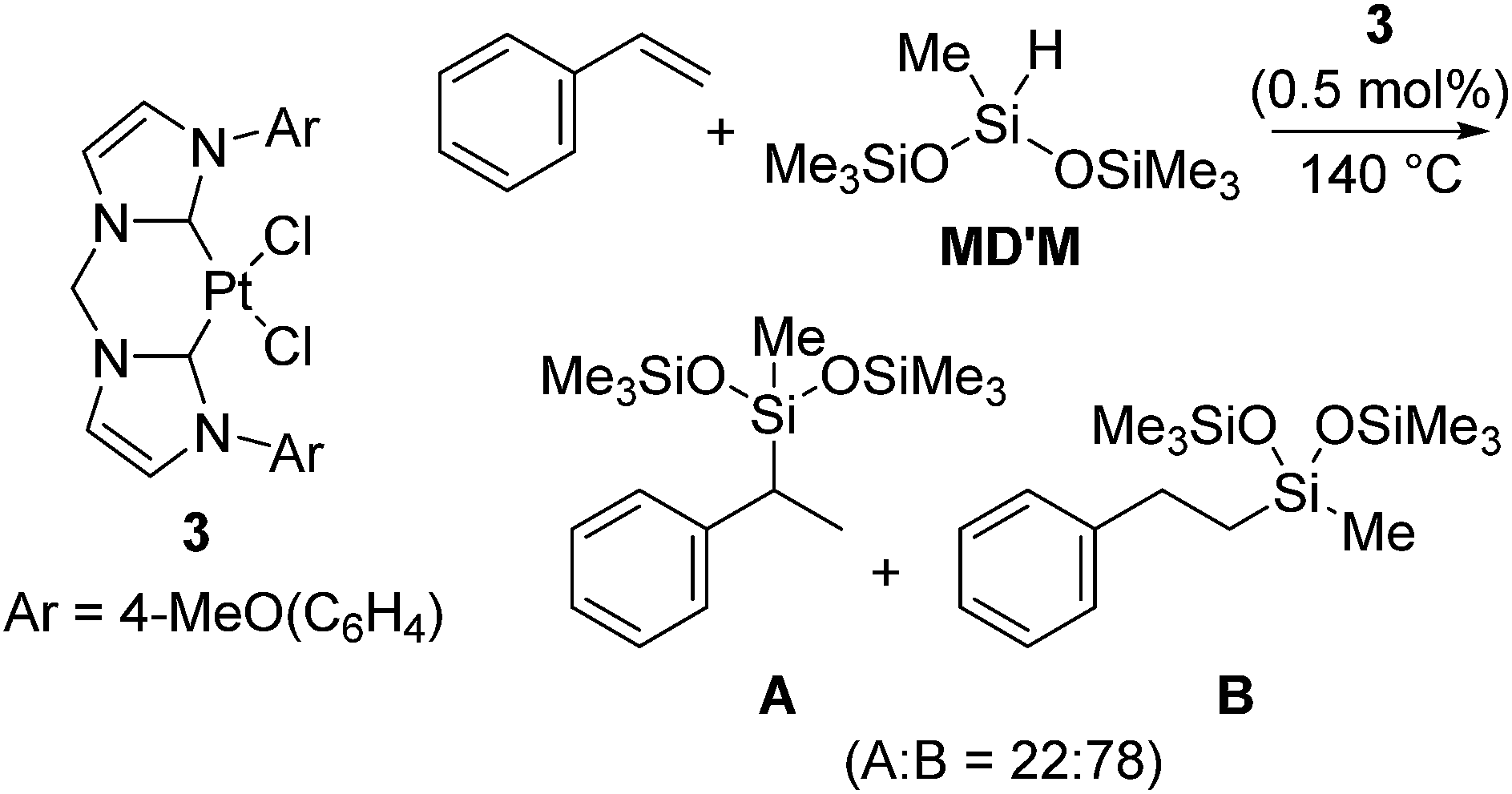 | (3) |
New trinuclear Pt(0) catalysts 4a–d with η2-alkyne ligands have recently been developed (eqn (4)).17 These complexes have a unique bent trinuclear chain skeleton, in which the Pt atoms are supported only by weak η2-alkyne ligands. It has been revealed that the trinuclear chain structures are maintained even in solution. These complexes (10−4 mol% loading) exhibit comparative or even higher catalytic activity towards hydrosilylation of 1-hexene or vinylmethylbis(trimethylsiloxy)silane with MD′M at 50 °C compared to Karstedt's catalyst.
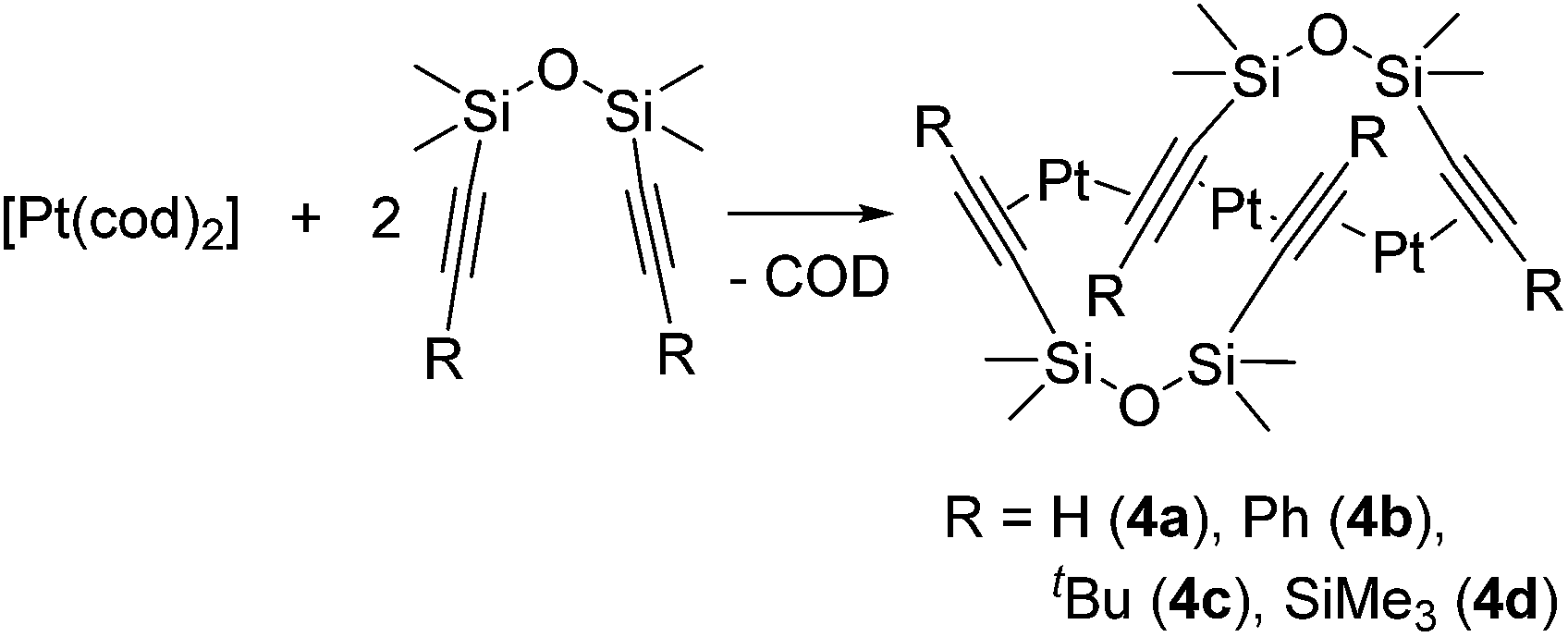 | (4) |
As a part of a longstanding endeavour to improve platinum catalyst systems, several more attempts have recently been performed. One interesting example is the development of platinum catalysts possessing an anti-sulfur-poisoning character. It has been widely recognized that sulfur compounds strongly coordinate to Pt(0) and therefore suppress the catalytic activity. On the other hand, Kung and co-workers reported that the addition of excess diethylsulfide to Karstedt's catalyst prevents formation of platinum colloids, resulting in enhancement of the catalyst activity.18 In addition to the above finding, Peng et al. recently revealed that trans-[Pt(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)2(PPh3)2] (5a), trans-[Pt{CC(CH3)2OH}2(PPh3)2] (5b), and trans-[Pt{CC(C6H10)(OH)}2(PPh3)2] (5c) exhibit higher hydrosilylation catalytic activity at 0.05 mol% catalyst loading in the presence of small amount of benzothiazole (0.02–0.5 mol%).19
CPh)2(PPh3)2] (5a), trans-[Pt{CC(CH3)2OH}2(PPh3)2] (5b), and trans-[Pt{CC(C6H10)(OH)}2(PPh3)2] (5c) exhibit higher hydrosilylation catalytic activity at 0.05 mol% catalyst loading in the presence of small amount of benzothiazole (0.02–0.5 mol%).19
2.2. Precious metal catalysts other than platinum
Apart from platinum complexes, rhodium complexes ([RhCl(PPh3)3] etc.) have been widely applied as effective hydrosilylation catalysts. Continuous attempts using rhodium complexes have been performed, such as the use of various phosphine and phosphonium ligands20 and strongly donating N-heterocyclic carbene (NHC) as the alternative of phosphine ligands21 and super critical CO2 (ref. 22) or ionic liquid as a solvent.23The iridium catalyst system [IrCl(cod)]2 (6) with 4 equiv. cod exhibits interesting chemo-selectivity; selective hydrosilylation of olefins in the presence of alkynes was achieved, in spite that alkynes are normally more reactive compared to olefins.24 Thus, hydrosilylation of allyl compounds with alkynylhydrosilanes were successfully attained at ambient temperature (eqn (5)). It is to be noted that the system demonstrates good chemo- and regioselectivity, allowing efficient and easy access to highly functionalized alkynylsilane tethers. The iridium complex 6 was also used for the selective hydrosilylation of allyl acetate with octakis(dimethylsiloxy)octasilsesquioxane [(HSiMe2O)8(SiO1.5)8].25
The utility of rhenium complex [ReBr2(NO)(CH3CN)3] (7) has been reported by Berke and co-workers.26 Although 7 catalysed dehydrogenative silylation of 4-methylstyrene with PhMe2SiH affording vinyl silanes in good to excellent yields, moderately regioselective hydrosilylation of nitrile substituted olefins has been attained in the presence of catalytic amount (1.5 mol%) of 7 (eqn (6)).
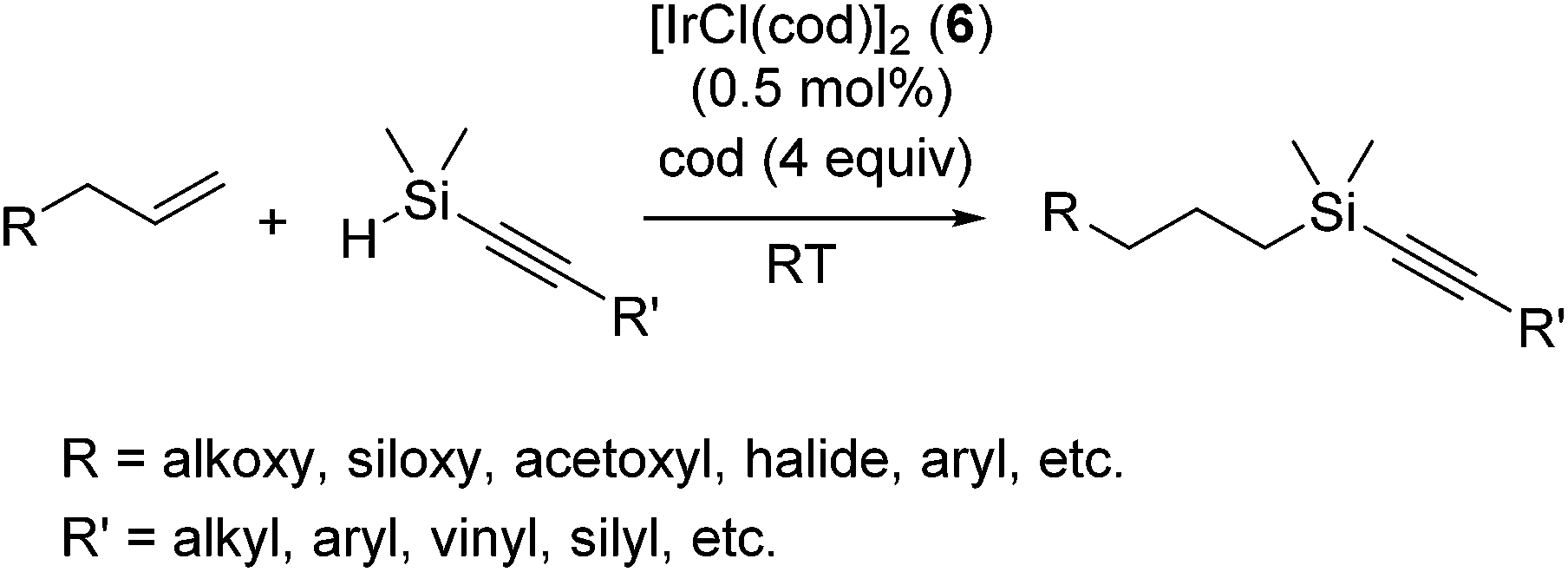 | (5) |
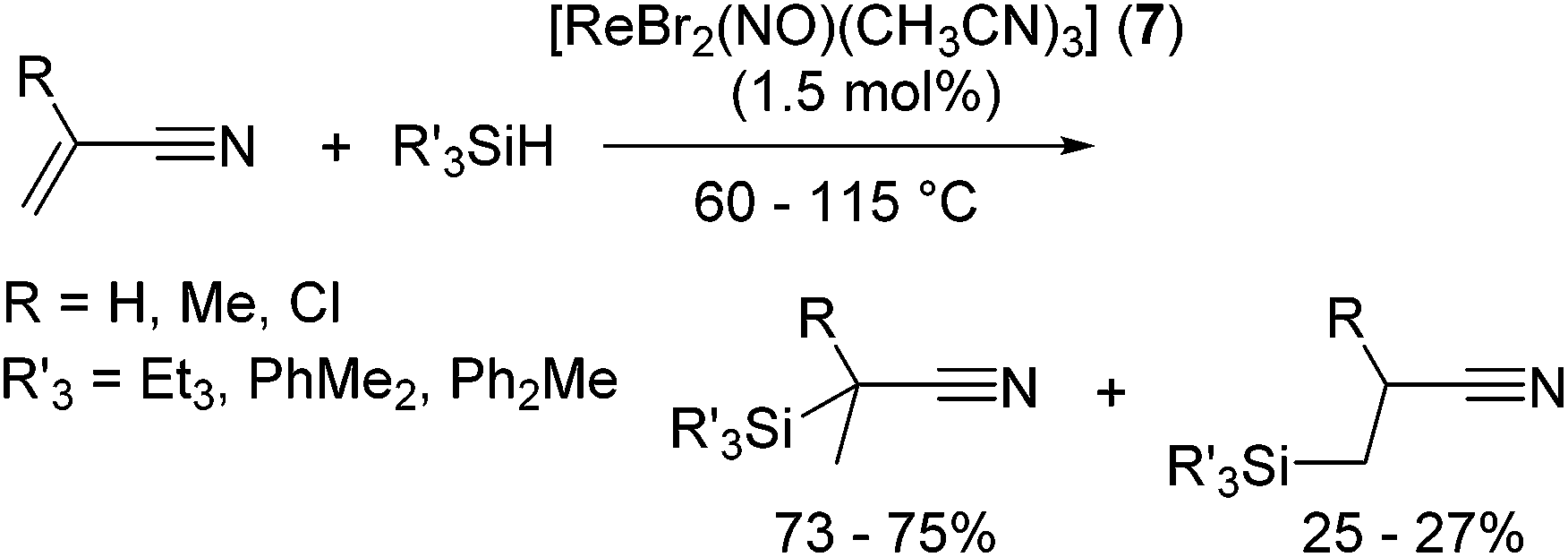 | (6) |
It has been reported that ligand substitution reaction of molybdenum carbonyls with silanes can easily takes place upon photo irradiation to form η2-silane complexes.27 These results have led to the finding that photochemical activation of molybdenum(0) complexes [Mo(CO)6] (8) and [Mo(CO)4(η4-nbd)] (9) (nbd = norbornadiene) can initiate the hydrosilylation of dienes.28 For examples, norbornadiene underwent hydrosilylation with tertiary and secondary hydrosilanes to initially give 5-silyl-2-norbornene, which is successively transformed to polynorbornene via ROMP (ring opening metathesis polymerization), while double hydrosilylation proceeded when Et2SiH2 was utilized as a substrate (Scheme 3). It was suggested by control experiments that the η2-norbornene ligand attached to the Mo atom could easily rearrange to give a metal carbene species, which was active towards ROMP.
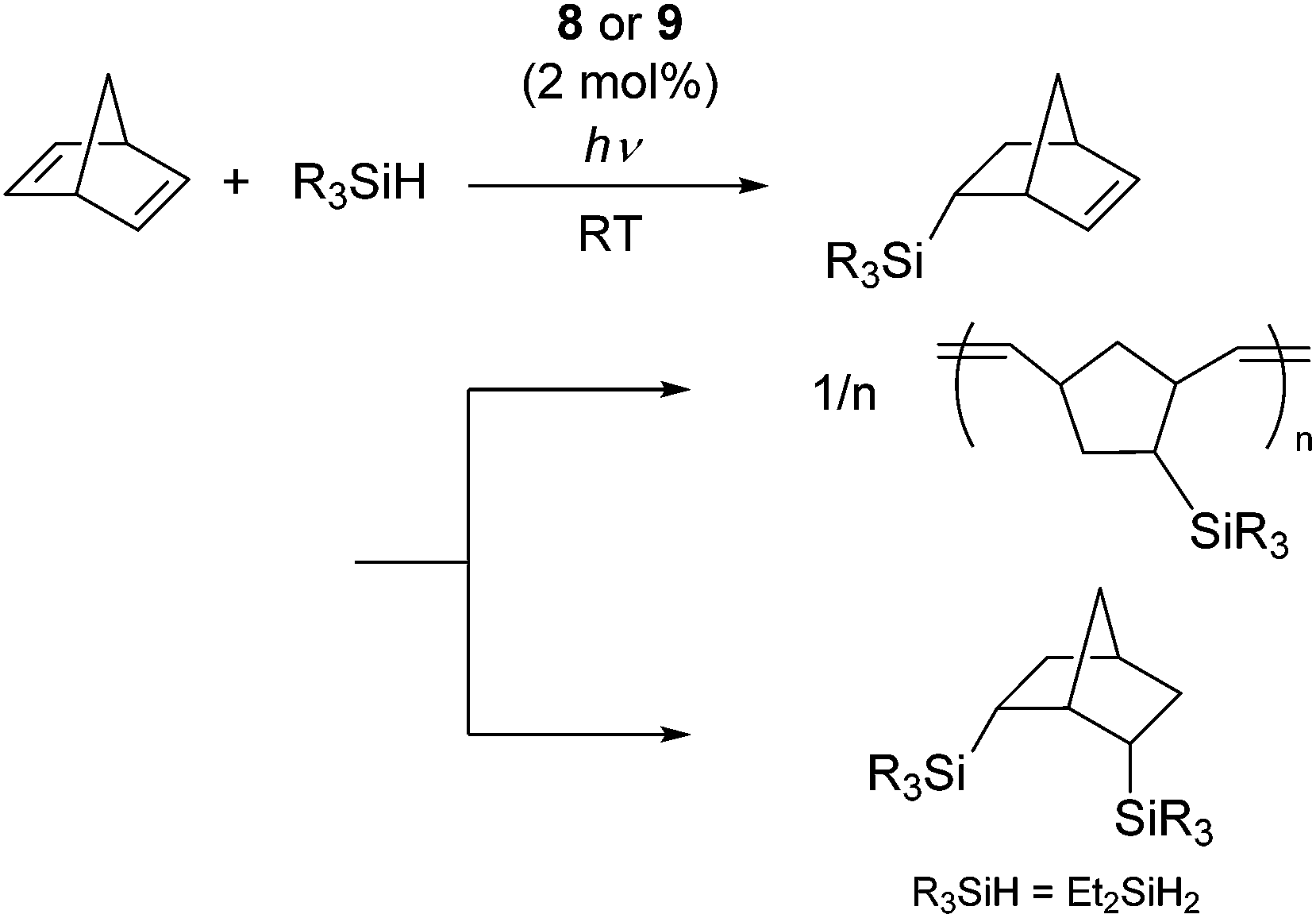 | ||
| Scheme 3 Transformation of norbornadiene in the presence of a photochemically active Mo(0) complexes 8 and 9. | ||
It is widely known that the most hydrosilylation catalysts bearing electron-rich transition metals such as platinum and rhodium follow the Chalk–Harrod and/or modified Chalk–Harrod mechanisms, in which Si–C bond formation occurs via the oxidative addition of the hydrosilane and the coordination of the olefin to the transition metal.29 On the other hand, Glaser and Tilley demonstrated a novel hydrosilylation reaction by using a cationic ruthenium silylene catalyst [(η5-C5Me5)(PiPr3)(H)2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SiHPh (Et2O)]+ (10), which follows a unique mechanism.30 The system attains high anti-Markovnikov regioselectivity although the system is effective only for primary hydrosilanes RSiH3. To account for the unusual properties of the ruthenium catalyst system, they proposed a novel mechanism (Glaser–Tilley mechanism) including a cationic ruthenium silylene complex as a key species, which undergoes olefin insertion into a silicon–hydrogen bond located in a position remote from the metal centre (Scheme 4). The silylene complex is regenerated in the catalytic cycle by oxidative addition of a Si–H bond, followed by 1,2-migration of a hydrogen atom from the silicon to the ruthenium.
SiHPh (Et2O)]+ (10), which follows a unique mechanism.30 The system attains high anti-Markovnikov regioselectivity although the system is effective only for primary hydrosilanes RSiH3. To account for the unusual properties of the ruthenium catalyst system, they proposed a novel mechanism (Glaser–Tilley mechanism) including a cationic ruthenium silylene complex as a key species, which undergoes olefin insertion into a silicon–hydrogen bond located in a position remote from the metal centre (Scheme 4). The silylene complex is regenerated in the catalytic cycle by oxidative addition of a Si–H bond, followed by 1,2-migration of a hydrogen atom from the silicon to the ruthenium.
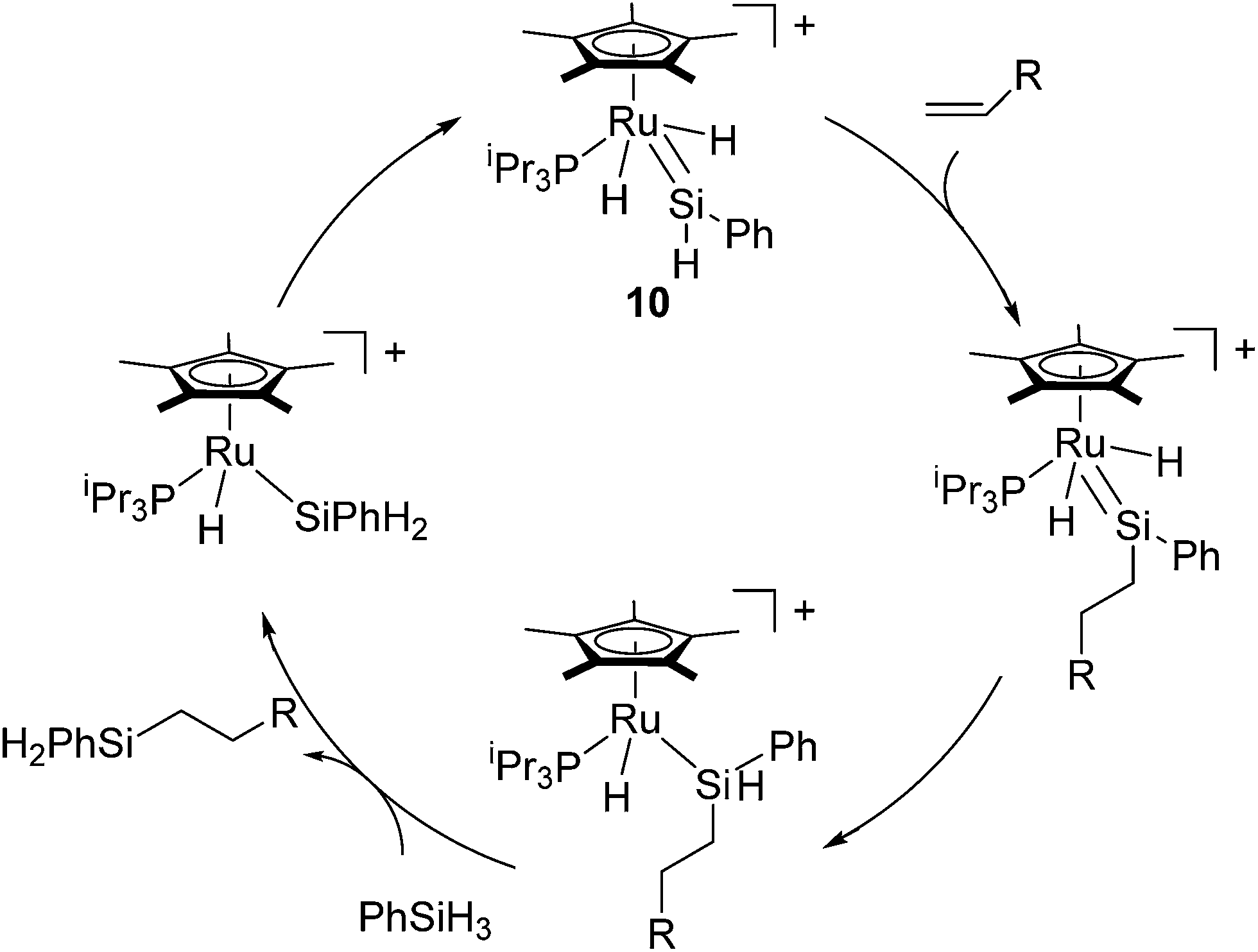 | ||
| Scheme 4 Glaser–Tilley mechanism. | ||
Detailed mechanistic information of this system was initially revealed based on DFT studies.31 The first experimental evidence for the direct formation of silylene complexes via double Si–H bond activation of a silane was reported in 2007,32 although synthesis of structurally well-defined ruthenium silylene complexes have already been reported by many groups via alternative synthetic routes.33 More recently, rather stable cationic silylene complex [(η5-C5Me5)(iPr3P)RuH2(SiHMes)]+ (11) was reported by using [CB11H6Br6]− as a counter anion (Fig. 4).34
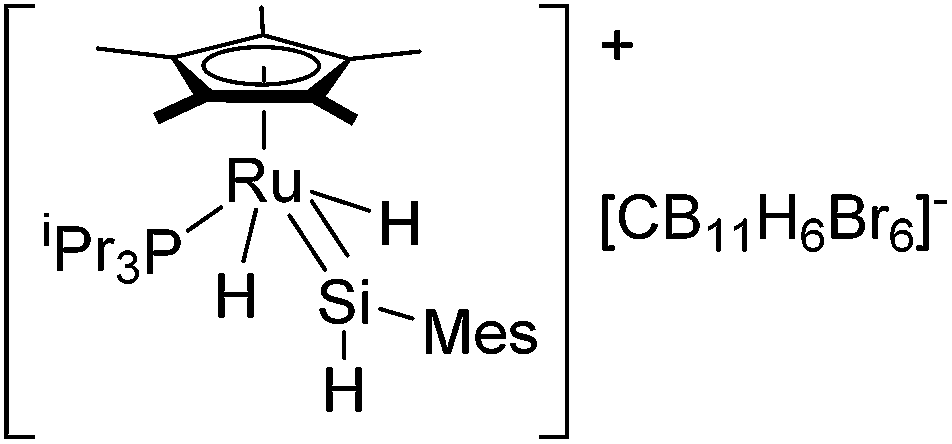 | ||
| Fig. 4 Structure of 11. | ||
Single crystal X-ray diffraction study revealed a strong Ru–H⋯Si interaction, indicating a significant electrophilicity of the silicon center, which is a key chemical property of the silylene ligand for the olefin insertion into the Si–H bond. By using 11, detailed kinetic studies have been performed to reveal that the rate-determining step is associated with the elimination of the hydrosilylated product.
3. Non precious metal catalysts
3.1. Iron catalysts
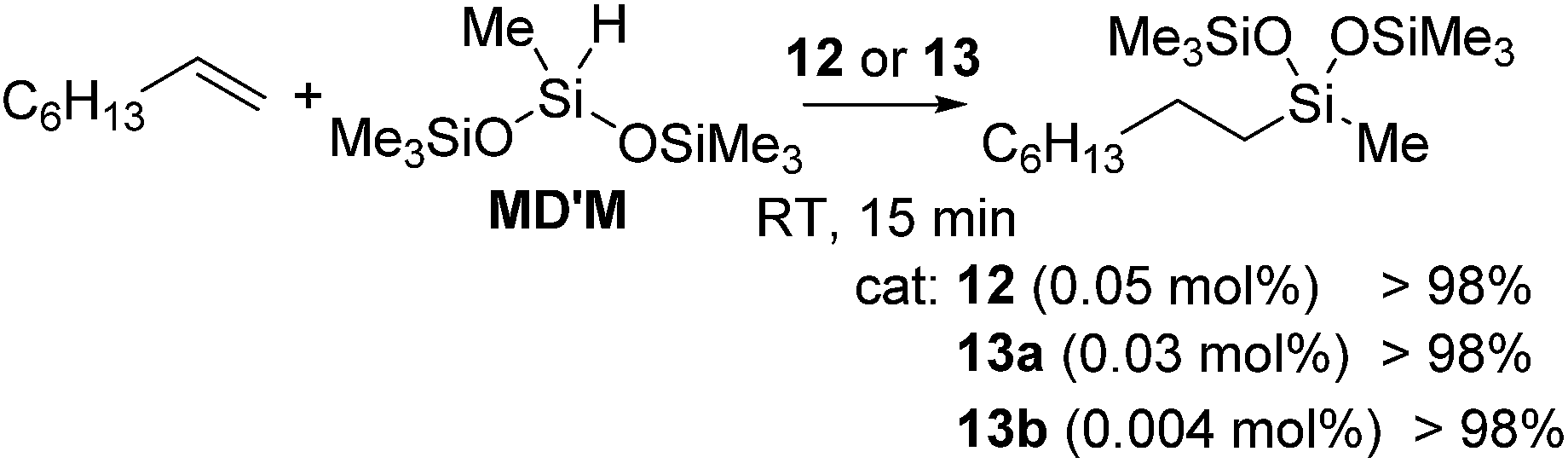
The first example of the iron hydrosilylation catalyst, Fe(CO)5, was reported in 1962 by Nesmeyanov and co-workers.35 Since then, continuous study on this field has been done over half a century, leading to the finding of several iron carbonyl systems.36 However, these systems often require high temperature thermolysis or photolysis to generate the active catalyst, and undesired side reactions, such as dehydrogenative silylation etc., compete with olefin hydrosilylation. Furthermore, iron complexes often exhibit complicated reactivity, which is difficult to control, due to their changeable electronic structures including high spin and low spin states. Therefore, examples of well-defined iron catalyst systems still remain scarce.Recently, a key breakthrough has been made by Chirik's group using iron complexes with a redox-active bis(imino)pyridine (PDI) ligand. In 2004, they initially reported the efficient hydrosilylation reaction of terminal olefins with primary and secondary hydrosilanes catalysed by [Fe(iPrPDI)(N2)2] (12) (iPrPDI = 2,6-(2,6-iPr2C6H3NCMe)2C5H3N) (Fig. 5).37 This Fe compound is also active for the more commercially relevant anti-Markovnikov addition of the tertiary silanes (EtO)3SiH and MD′M to 1-octene and allyl polyethers (eqn (7)).38 It has been revealed that reducing the size of the 2,6-aryl substituents, as in the cases of [(EtPDI)Fe(N2)]2(μ2-N2) (13a) and [(MePDI)Fe(N2)]2(μ2-N2) (13b), resulted in significant enhancement of the catalytic activity.
 | (7) |
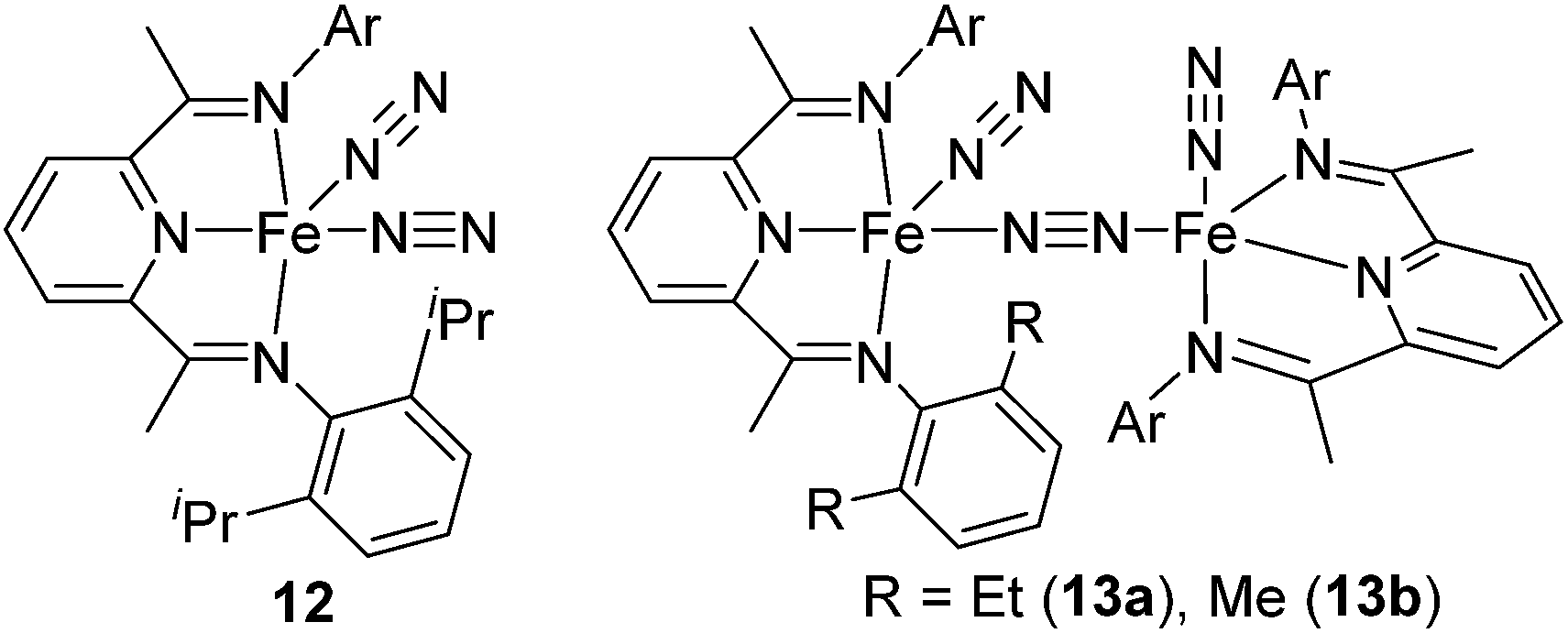 | ||
| Fig. 5 Structures of 12 and 13a,b. | ||
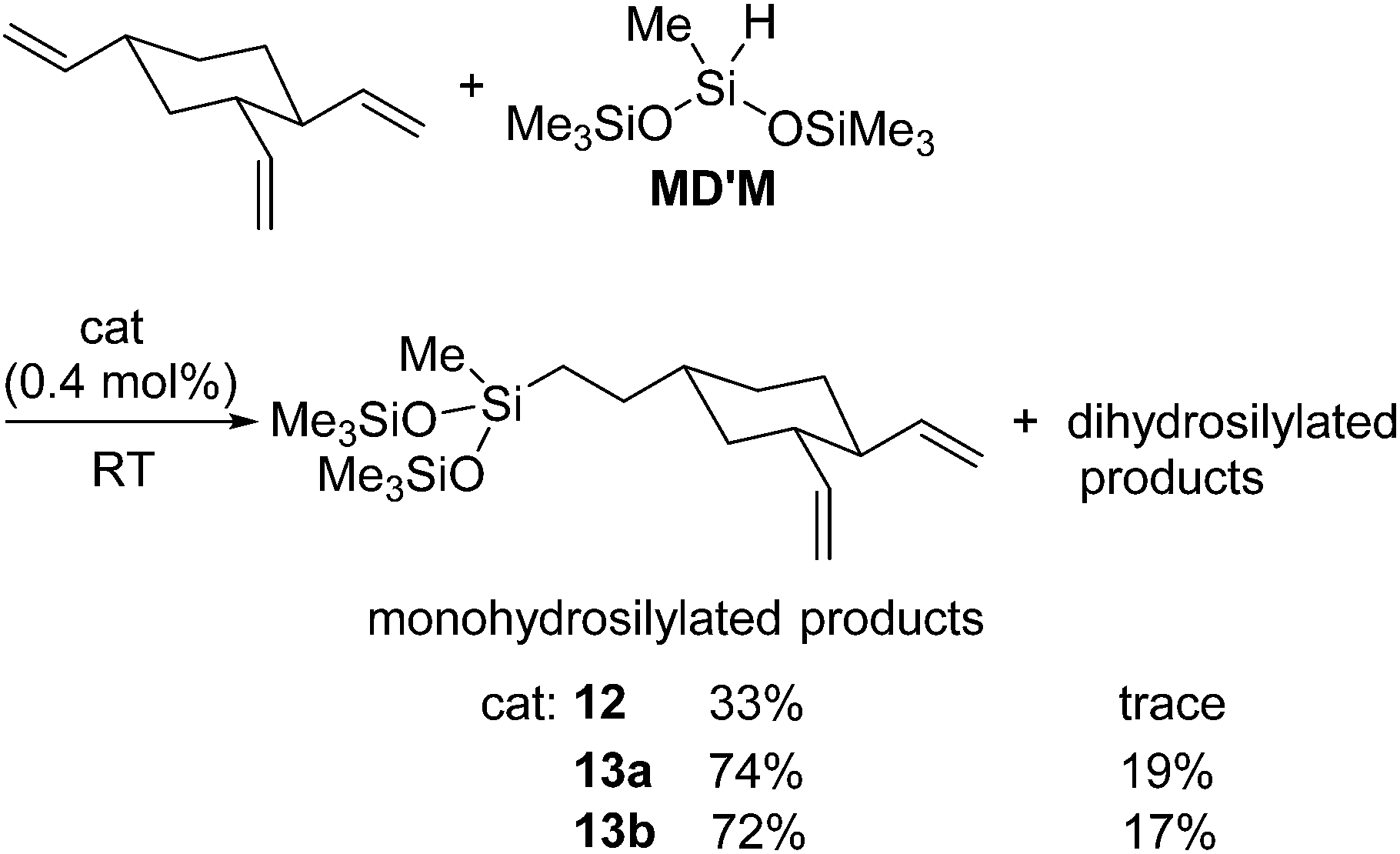
By making use of 12 and 13a,b, selective monohydrosilylation of the vinyl group at 4-position of 1,2,4-trivinylcyclohexane was also achieved (eqn (8)).39
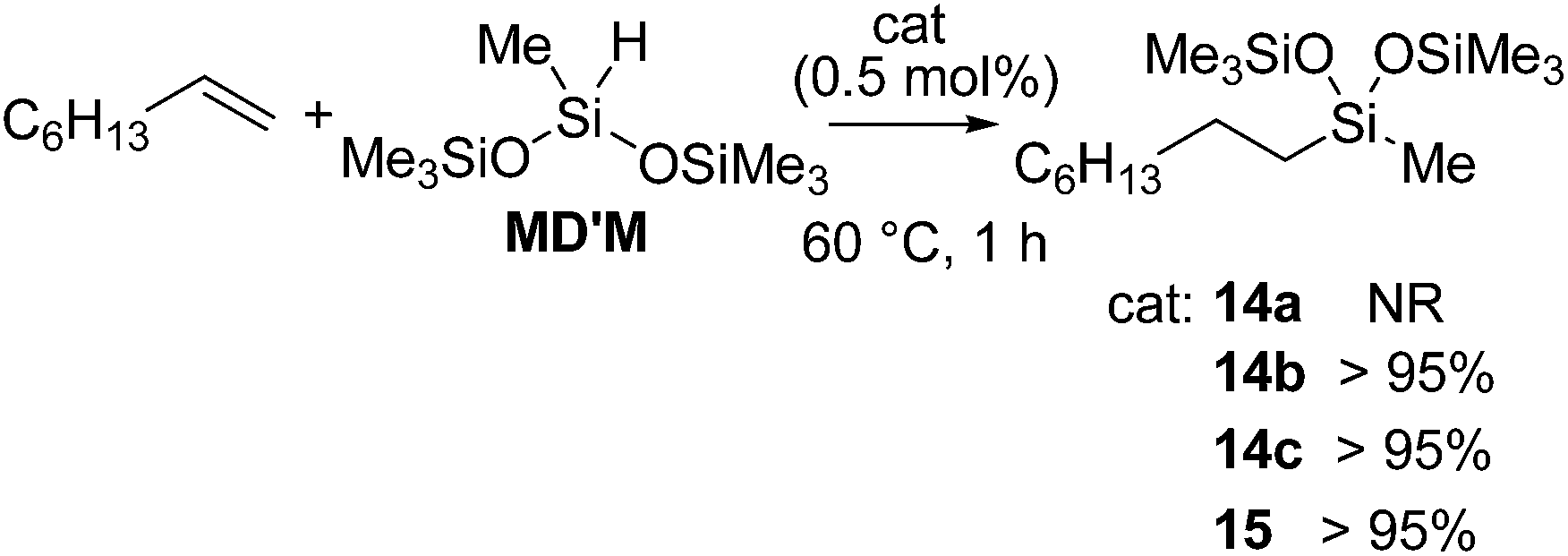
They also found out that Fe(II) dialkyl complexes bearing a bis(imino)pyridine ligand 14a–d or a terpyridine ligand 15 (Fig. 6) serve as an olefin hydrosilylation catalyst although their activity was inferior to those of bis(imino)pyridine-supported Fe(0) systems 12 and 13a,b (eqn (9)).40
 | (8) |
 | ||
| Fig. 6 Structures of 14a–d and 15. | ||
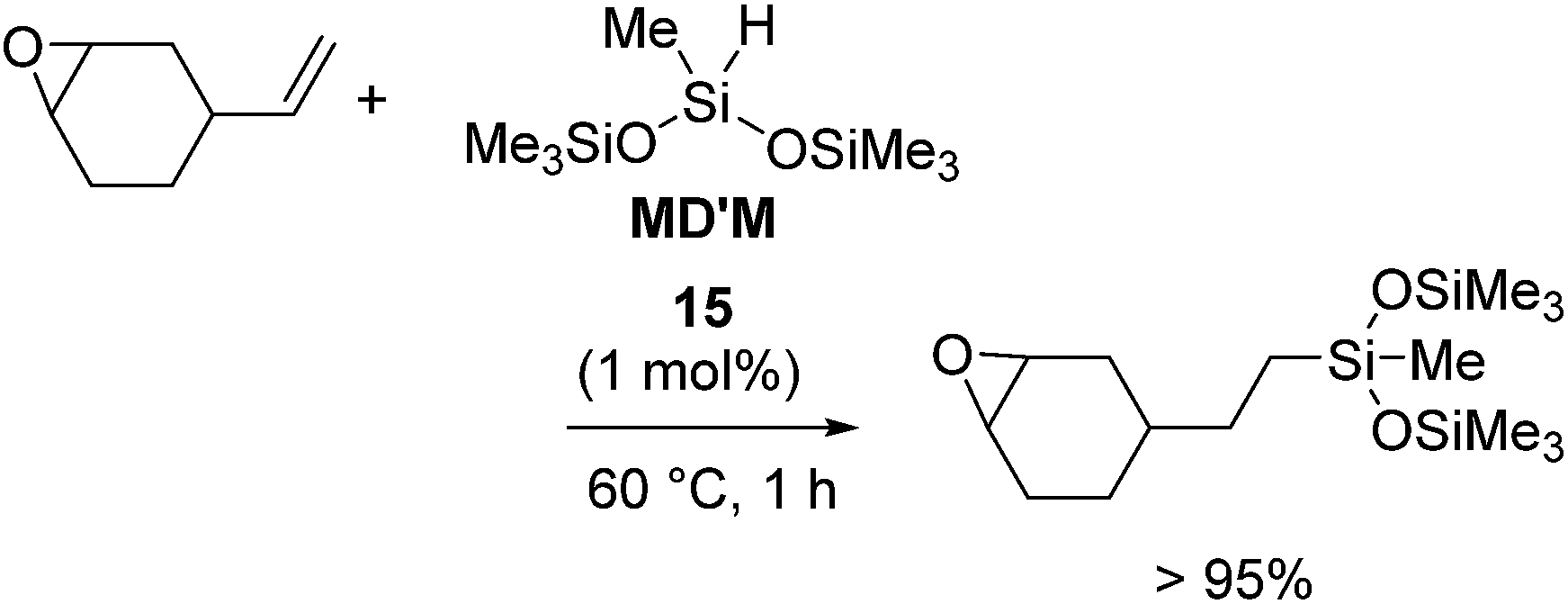
On the other hand, it should be noted that 15 selectively catalysed the hydrosilylation reaction of vinylcyclohexene oxide, whose hydrosilylated compound is utilized in UV-release coating application, textile finishes, and for preparing silicone-modified organic polymers (eqn (10)). The same hydrosilylation reaction could not be catalysed by 13a,b or 14b,c.
 | (9) |
 | (10) |
Utility of terpyridine Fe(II) complexes were also reported by Nakazawa and co-workers. They revealed that terpyridine Fe(II) dihalides 16a–f with NaBEt3H catalysed olefin hydrosilylation with primary or secondary hydrosilanes at 0.05–0.1 mol% Fe loading to give monohydrosilylated product (eqn (11)).41 Furthermore, at more than 0.3 mol% Fe loading and elevated temperature, bis-hydrosilylation of PhSiH3 with 1-octene selectively took place to give Ph(nC8H17)2SiH. It is important that the terpyridine ligand possess substituents at the 6- and 5′′-/6′′-positions to prevent forming inactive species [Fe(terpyridine)2].
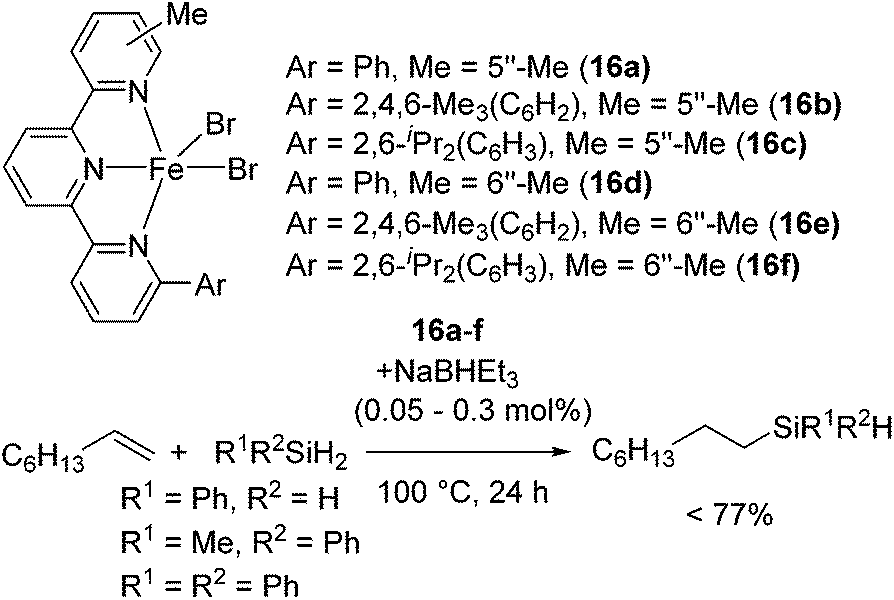 | (11) |
Recently, it was revealed that Fe(II) complexes 17 bearing an electron-donating phosphinite-iminopyridine (PNN) ligand successfully catalysed hydrosilylation of olefins with various functionalities, such as amide, ester, and keto groups. In particular, ketones are generally highly reactive and readily undergo hydrosilylation (reduction). On the other hand, in the presence of 2 mol% of 17, hydrosilylation of 5-hexene-2-one proceeded at room temperature to exclusively form olefin hydrosilylated product in >80% yield (eqn (12)).42
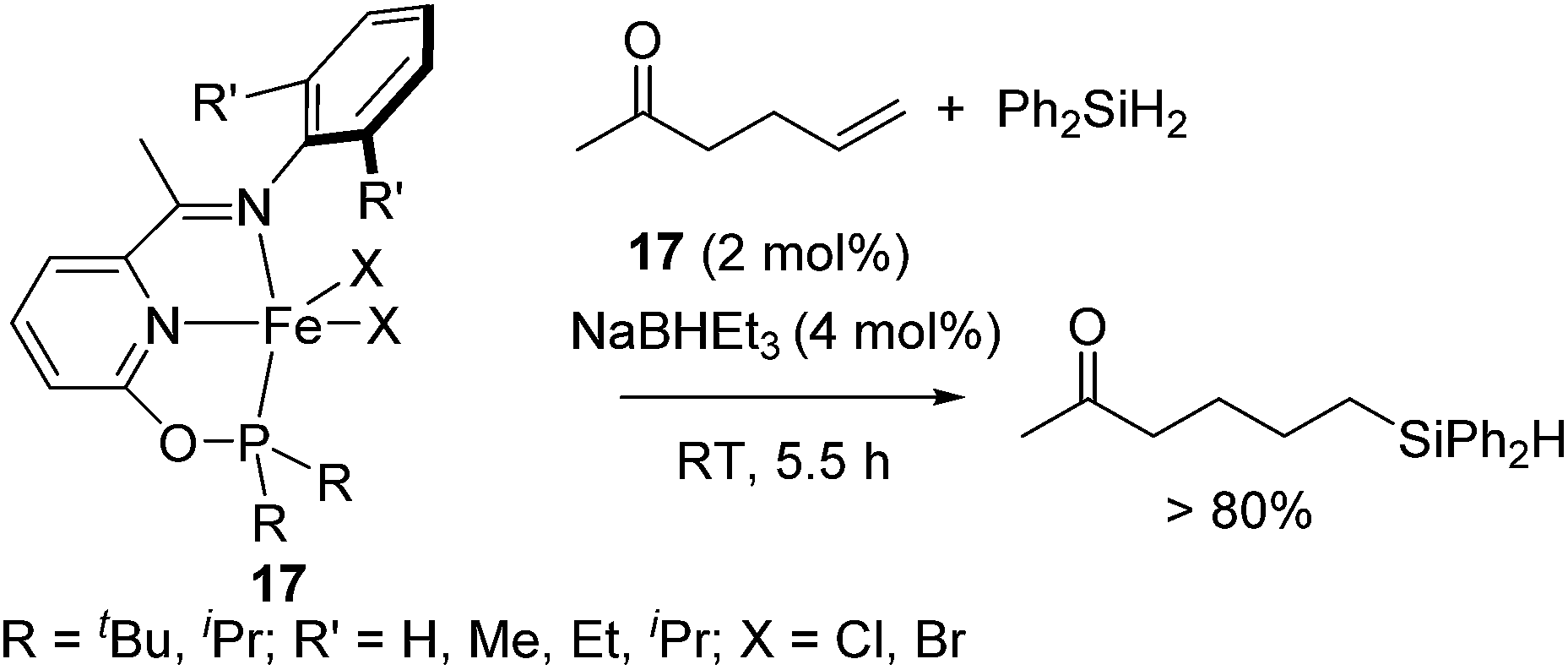 | (12) |
Ritter's group demonstrated 1,4-hydrosilylation of 1,3-diene using iron bis(iminopyridine) catalyst precursor 18 (eqn (13)), which can be easily prepared by ligand exchange between bis(aryl) Fe(II) complex 19 with the corresponding iminopyridine ligand (eqn (14)).43
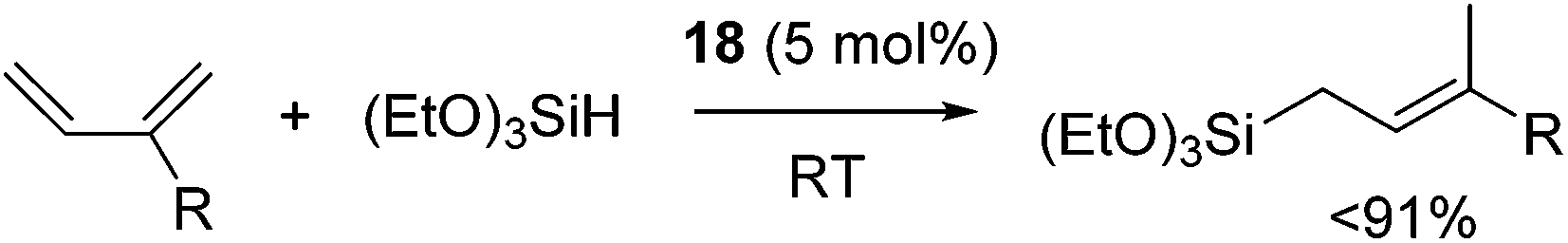 | (13) |
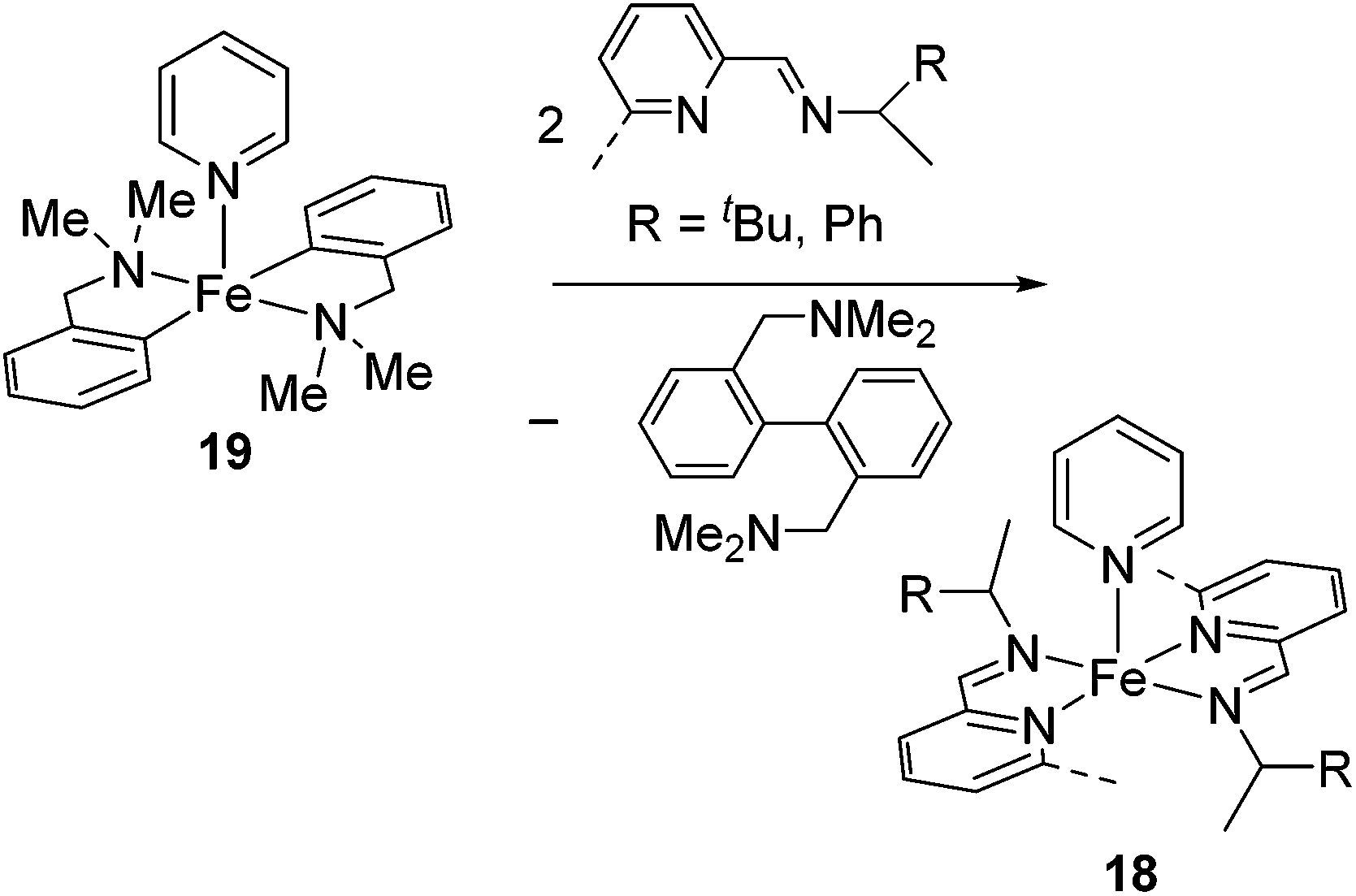 | (14) |
They found that complex 19 with 2 equiv. of the iminopyridine ligand can be used for the catalysis without the isolation of bis(iminopyridine)iron complex. It readily allowed the evaluation of various ligands to control the regioselectivity. Regioselective 1,4-hydrosilylation of dienes was accomplished by using imino pyridine ligands 20 and 21 (eqn (15)). In the presence of 5 mol% of 19 and 20/21, hydrosilylation of 1,3-dienes proceeded at room temperature to produce linear products predominantly. In these reaction systems, several functional groups, including an epoxide, an ester, and an amine, are tolerated. Double bond geometry was controlled in all cases with >99% E selectivity, possibly a consequence of the stereoselective mechanism suggested in Scheme 5. This is the first example of highly regio- and stereoselective hydrosilylation of 1,4-diene to selectively afford allylsilanes, which are useful compounds in organic synthesis.44
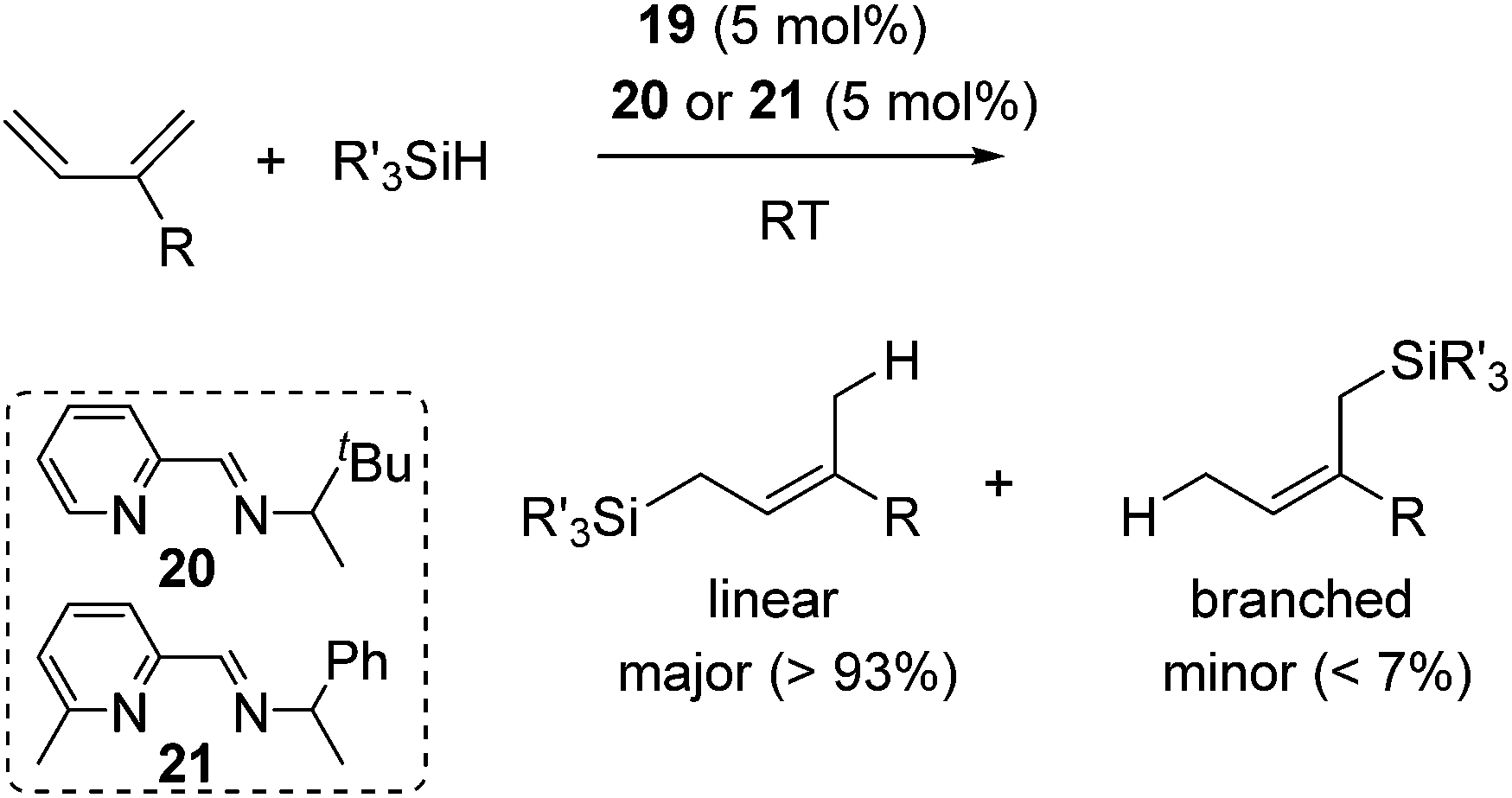 | (15) |
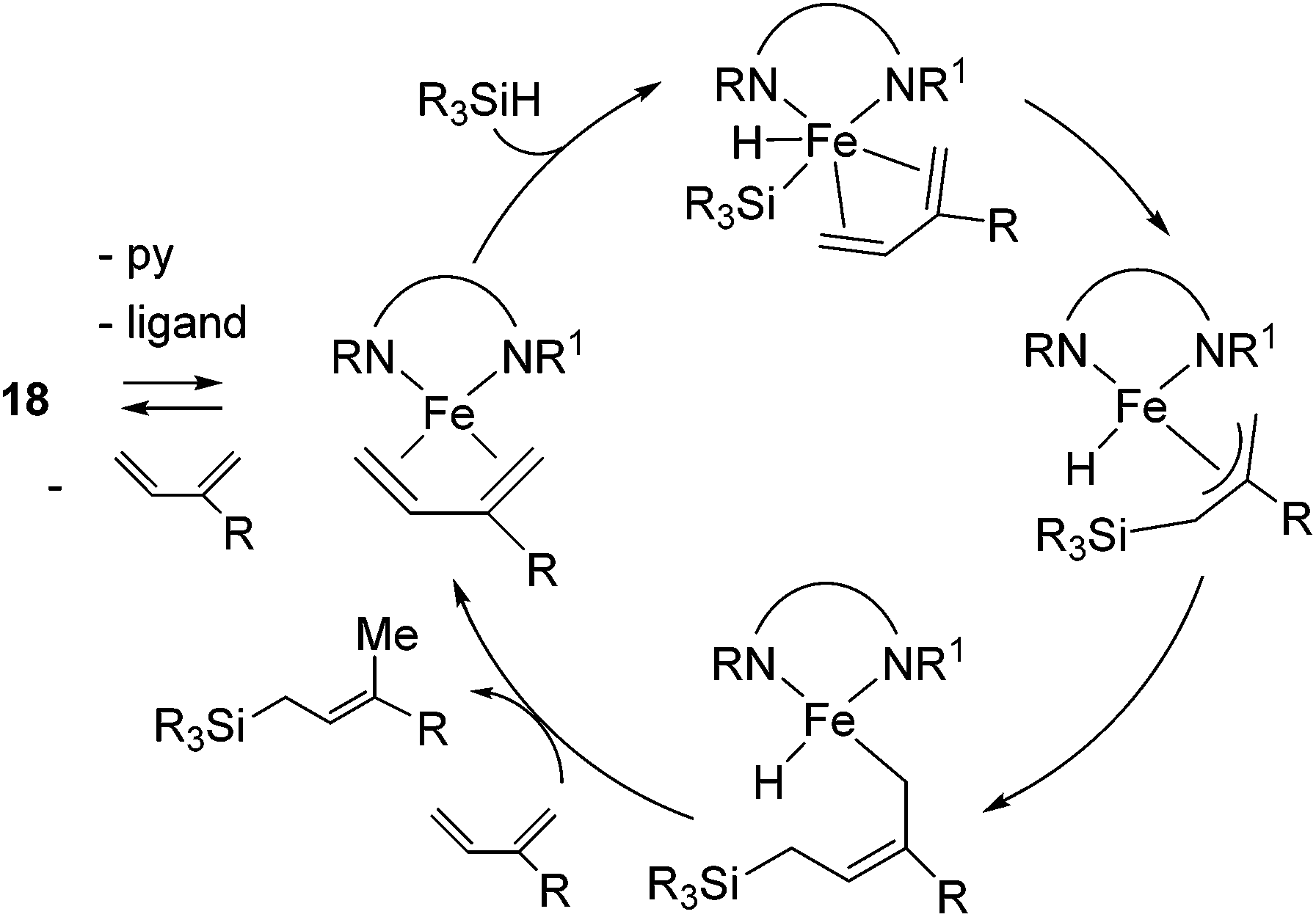 | ||
| Scheme 5 1,4-Selective hydrosilylation of diene catalysed by 18. | ||
More recently, Thomas et al. reported the utility of commercially available Fe compound and reagents, FeCl2, bis(imino)pyridine (PDI), and EtMgBr, which produce an active Fe catalyst in situ (eqn (16)).45
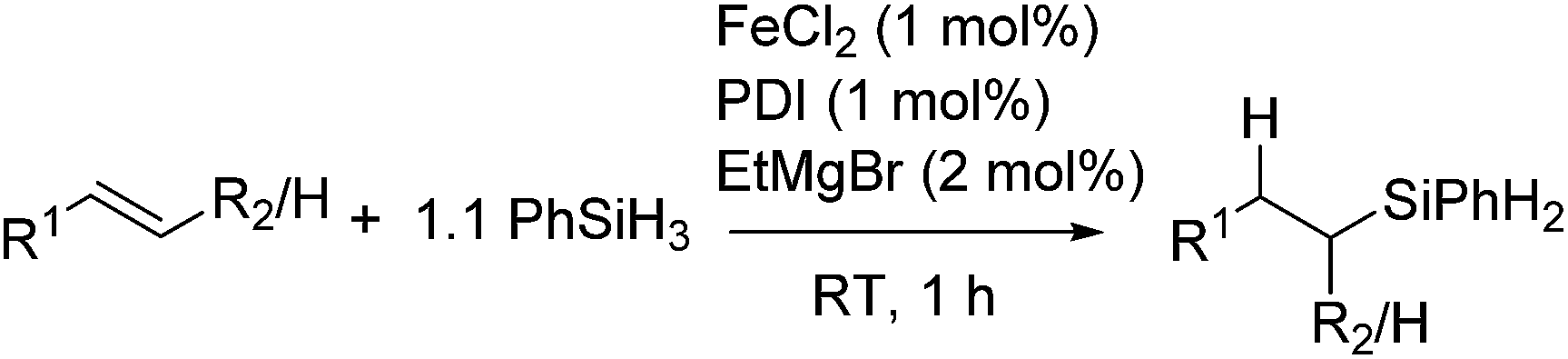 | (16) |
Using this system, olefins with various functional groups, such as halides, amino-, ester-, keto-, imino-, imino ester-groups, were hydrosilylated to give linear silane products in moderate to excellent yields with a good regioselectivity. The system is also applicable for hydrosilylation of heteroaromatic substituted olefins. The iron pre-catalyst was used at loadings as low as 0.07 mol% for the reaction of styrene with Et2SiH2, and displayed catalyst turnover frequencies approaching 60000 mol h−1. Overall, the chemo-, regio-, and stereoselective iron-catalysed hydrosilylation of olefins with good functional group tolerance is achieved. Preliminary mechanistic investigation indicated the formation of an Fe(I) species as a real active species.
3.2. Nickel catalysts
Nickel salts and its complexes have been recognized as a hydrosilylation catalyst since 1950s. In 50's and 60's, several nickel hydrosilylation catalysts were reported such as Raney nickel,46 pyridine supported nickel dichlorides,47 nickel carbonyl48 and iron pentacarbonyl-nickel chloride couple.49 In early 70's, phosphine ligated Ni(II) and Ni(0) complexes are revealed to be active towards olefin hydrosilylation with chlorinated hydrosilanes. In these systems, redistribution of hydrogen and chlorine on the silicon atom occurs during the reaction, leading to the formation of more than one hydrosilylated products.50 In 90's, it has been also revealed that phosphine coordinated Ni(II) and Ni(0) complexes preferably catalyse dehydrogenative silylation of styrene using tertiary hydrosilanes with moderate activity and selectivity.51 Simple nickel acetylacetonato (acac) salt Ni(acac)2 also act as a hydrosilylation catalyst. In the hydrosilylation of vinylsilanes, dehydrogenative silylation, hydrogenative dimerization, and substituent redistribution on the Si atom also proceed competitively.52As mentioned above, these classical nickel catalysts were of limited applicability, and require rather drastic reaction conditions. Nevertheless, nickel complexes have been long attracted enduring attention. One interesting research is the attempt to prepare the nickel equivalent of Karstedt's catalyst, [{Ni(η-CH2CHSiMe2)2O}2{μ-(η-CH2CHSiMe2)2O}] (22).53 However, complex 22 preferably catalysed dehydrogenative silylation instead of hydrosilylation (eqn (17)).
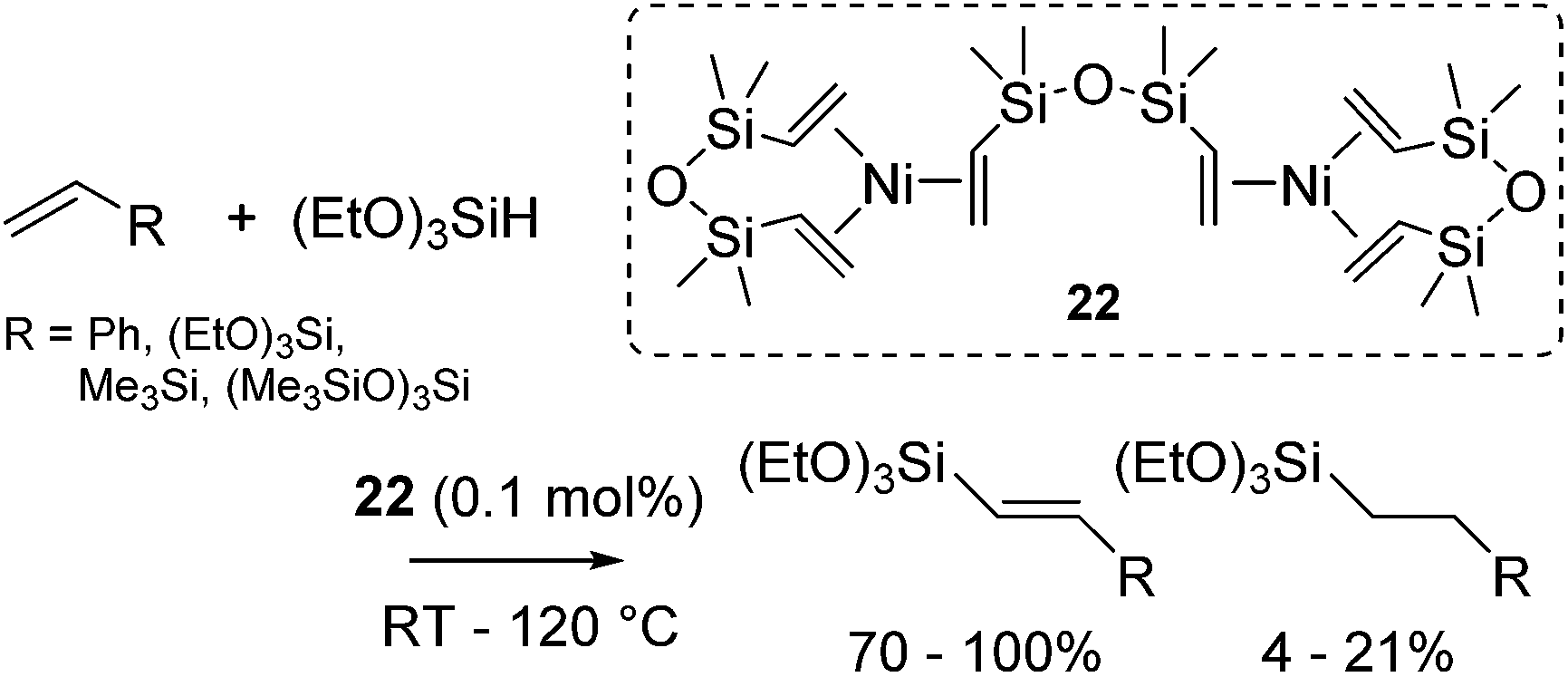 | (17) |
Recently, several highly selective catalysts have been reported. [Ni(R-Ind)(PPh3)Cl] (R-Ind = 1-Me-indenyl (23a), 1-SiMe3-indenyl (23b), 1,3-(SiMe3)2-indenyl (24) (Fig. 7) with an initiator NaBPh4 can catalyse hydrosilylation of styrene with PhSiH3 to produce PhCH(Me)SiPhH2 (eqn (18)).54 Since dehydrogenative homocoupling of PhSiH3 also proceeds in these reactions, addition of 1.5 equiv. PhSiH3 is necessary to obtain the hydrosilylated product in a good yield.
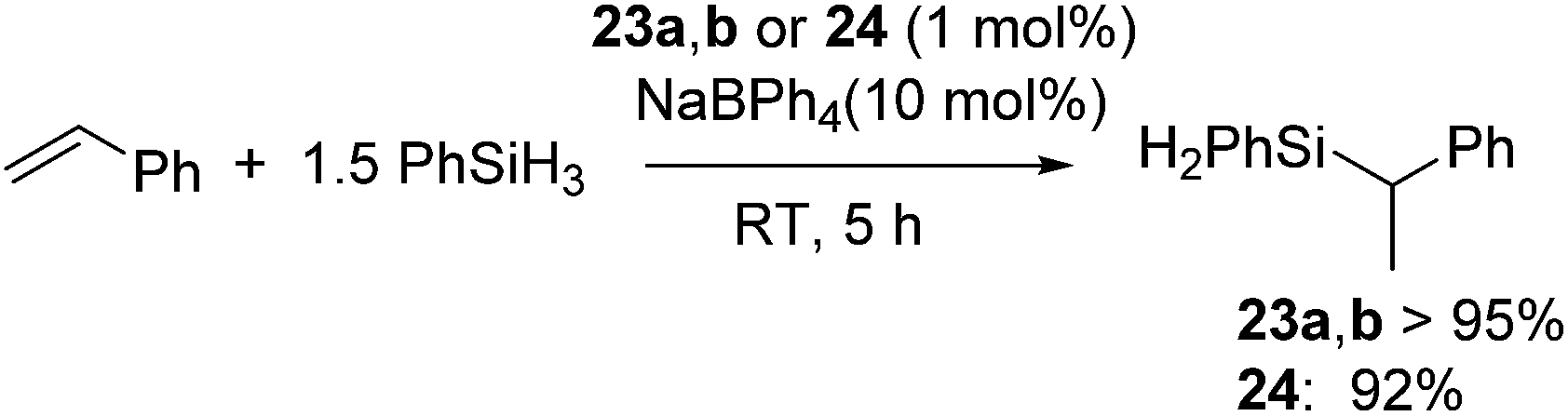 | (18) |
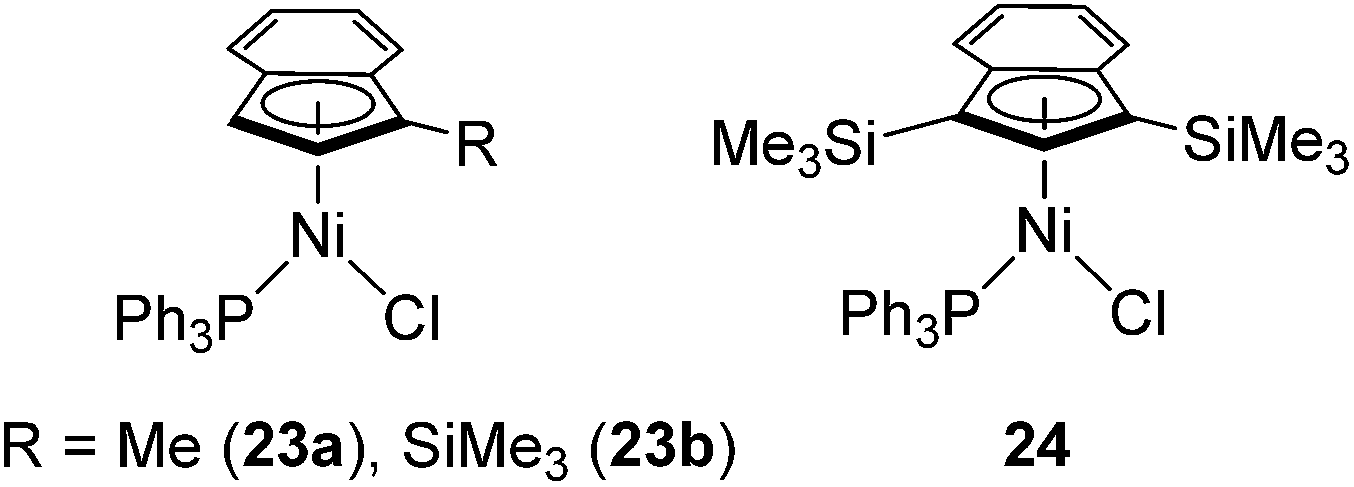 | ||
| Fig. 7 Structures of 23a,b and 24. | ||
Complexes 23 and 24 exhibit comparable catalytic activities under these conditions, showing poor effects of the substituents on the R-Ind ligand. Complex 24 achieves the same product yield when the reaction was carried out in the absence of NaBPh4, while 23a and 23b gave much lower yield under the same reaction conditions. It was suggested that the results reflected the greater PPh3 dissociation occurring with 24 to produce coordinatively unsaturated active species, which is presumably a function of the two big SiMe3 groups on the R-Ind ligand. The proposed mechanism is shown in Scheme 6. The common active species [Ni(R-Ind)(H)L] (L = PPh3 or substrates) is proposed in both reactions with or without NaBPh4. In the presence of NaBPh4, a highly Lewis acidic cationic intermediate is in situ formed, which successively abstract H from PhSiH3 to form the active hydride species as well as silylium ion [PhSiH2][BPh4]. In the absence of NaBPh4, phosphine-dissociated [Ni(R-Ind)Cl] reacts with PhSiH3 to form the hydride species with the co-production of PhSiH2Cl.
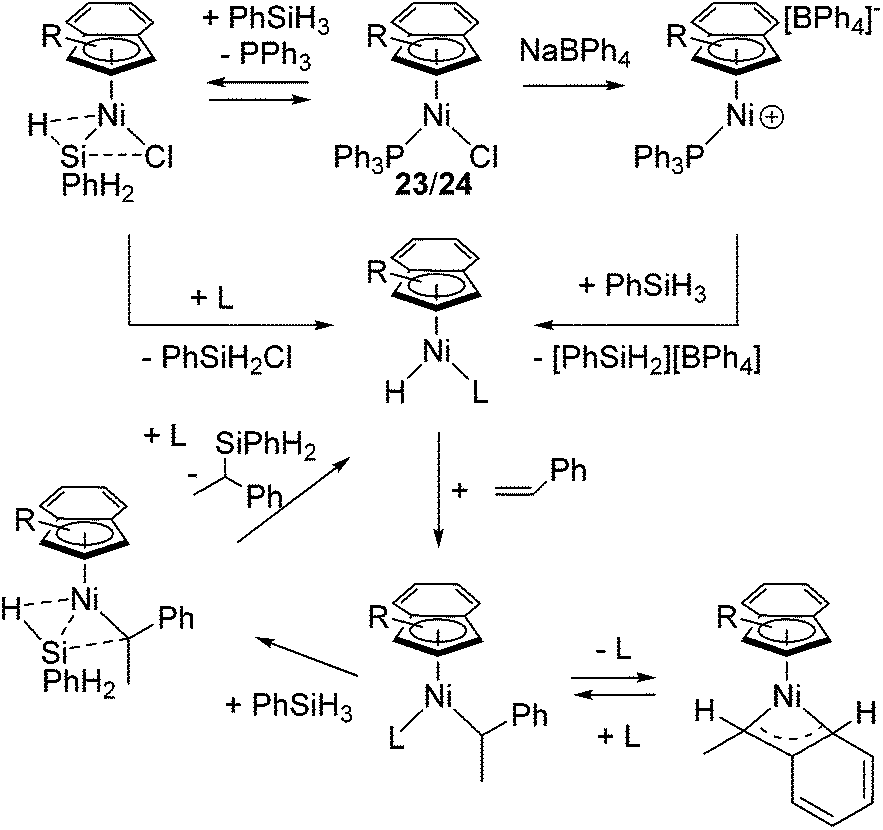 | ||
| Scheme 6 Hydrosilylation mechanism catalysed by 23/24. | ||
Several cationic allyl nickel complexes [Ni(η3-allyl)(κ1P-PR2CH2CHCH2)2][BAr′4] (R = Ph (25a), iPr (25b)) and [Ni(η3-allyl)(NHC)] [NHC = 1-(2-picolyl)-3-(R)-imidazol-2-ylidene (R = Me (26a), iPr (26b), nBu (26c), Ph (26d)), 1-(2-picolyl)-3-methylbenzoimidazol-2-ylidene (27), 1-(2-picolyl)-4,5-dichloro-3-methylimidazolylidene (28)] also catalyse Markovnikov selective hydrosilylation of styrene with PhSiH3 to give PhCH(Me)SiPhH2 as a major product (Fig. 8, eqn (19)).55 When the reaction was performed using [Ni(η3-allyl)(NHC)] and monitored by 1H NMR, distinctive signals were observed in the metal-hydride region, strongly indicating the involvement of Ni-hydride species in the catalytic cycle.
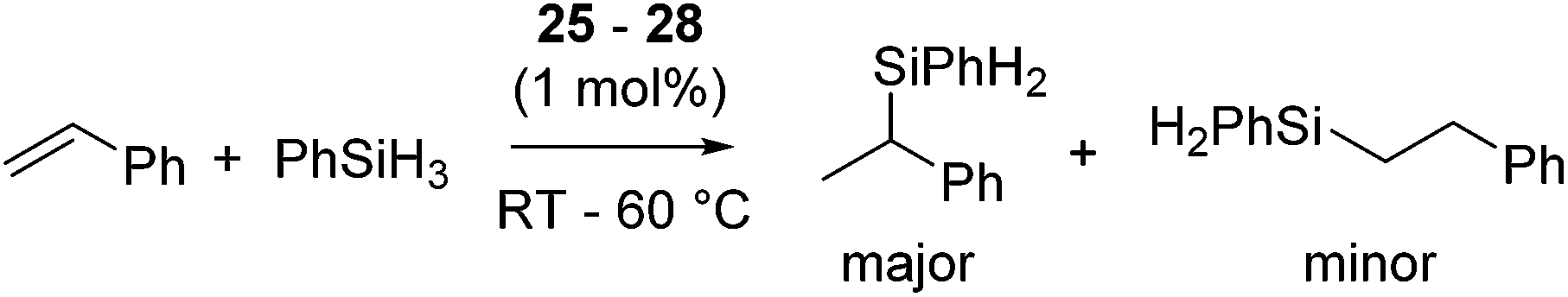 | (19) |
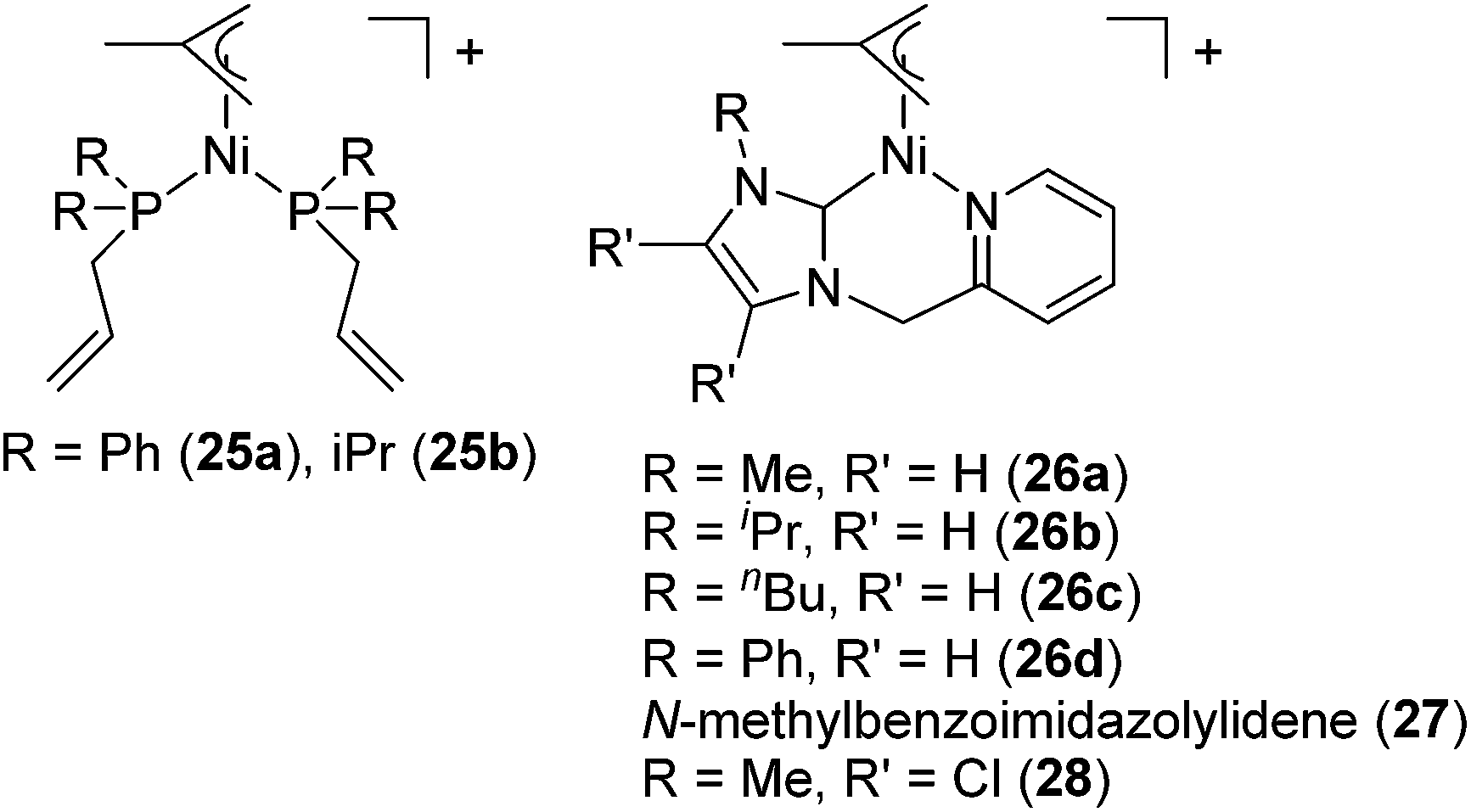 | ||
| Fig. 8 Structures of 25–28. | ||
In contrast to the above examples, the phosphine coordinated Ni(II) system, NiBr2 + 2PPh3, catalyses anti-Markovnikov selective hydrosilylation of styrenes with Ph2SiH2 (eqn (20)).56 It has also been found that the resulting hydrosilylated products can be further transformed into dihydrobenzosiloles via intramolecular dehydrogenative cyclization in the presence of the Ir catalyst. The series of reactions proceed in a one-pot manner to give the final products in good overall yields.
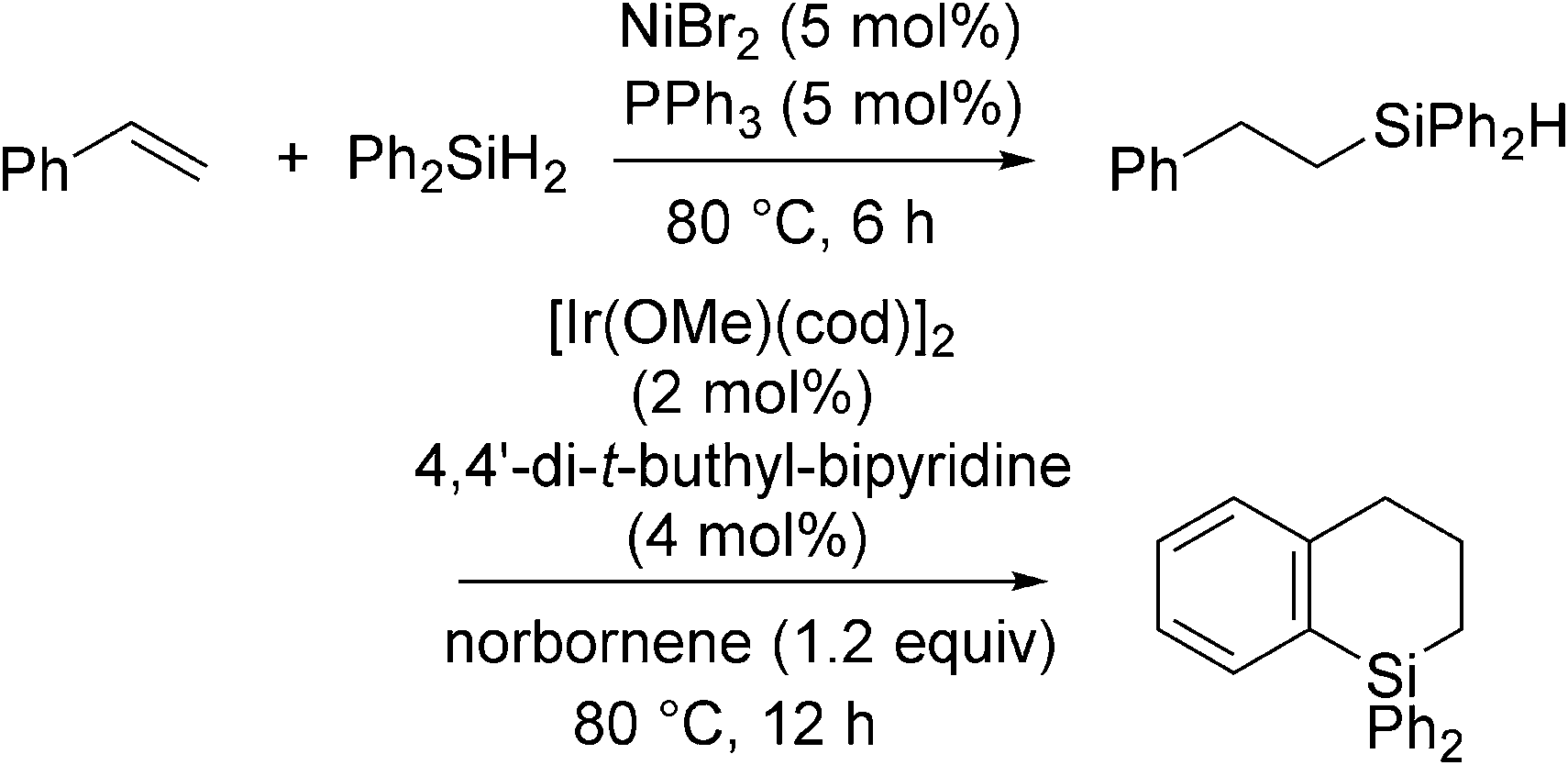 | (20) |
Tilley and co-workers demonstrated hydrosilylation of 1-octene with Ph2SiH2 using two-coordinate nickel bis(amido) complex [Ni{N(SiMe3)(2,6-(iPr)2(C6H3))}2] (29) (eqn (21)).57
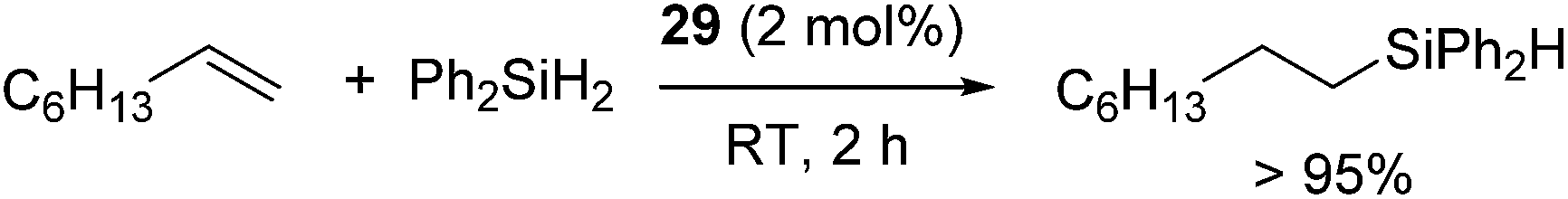 | (21) |
3.3. Early transition metal catalyst
Early transition metals (Ti, Zr, Hf),58 as well as lanthanide and actinides59 are also known to promote the hydrosilylation of olefins, although the history of this field is much shorter compared to that of late transition metal catalysts.Two kinds of mechanism are normally proposed for the hydrosilylation reaction catalysed by a d0 metal complex, which are different from the well-known Chalk–Harrod mechanism; (i) olefin insertion into the M–H bond followed by Si–C bond formation via σ-bond metathesis of the resulting metal–alkyl with a hydrosilane, (ii) Si–C bond formation via olefin insertion into the M–Si bond, followed by σ-bond metathesis (Scheme 7).
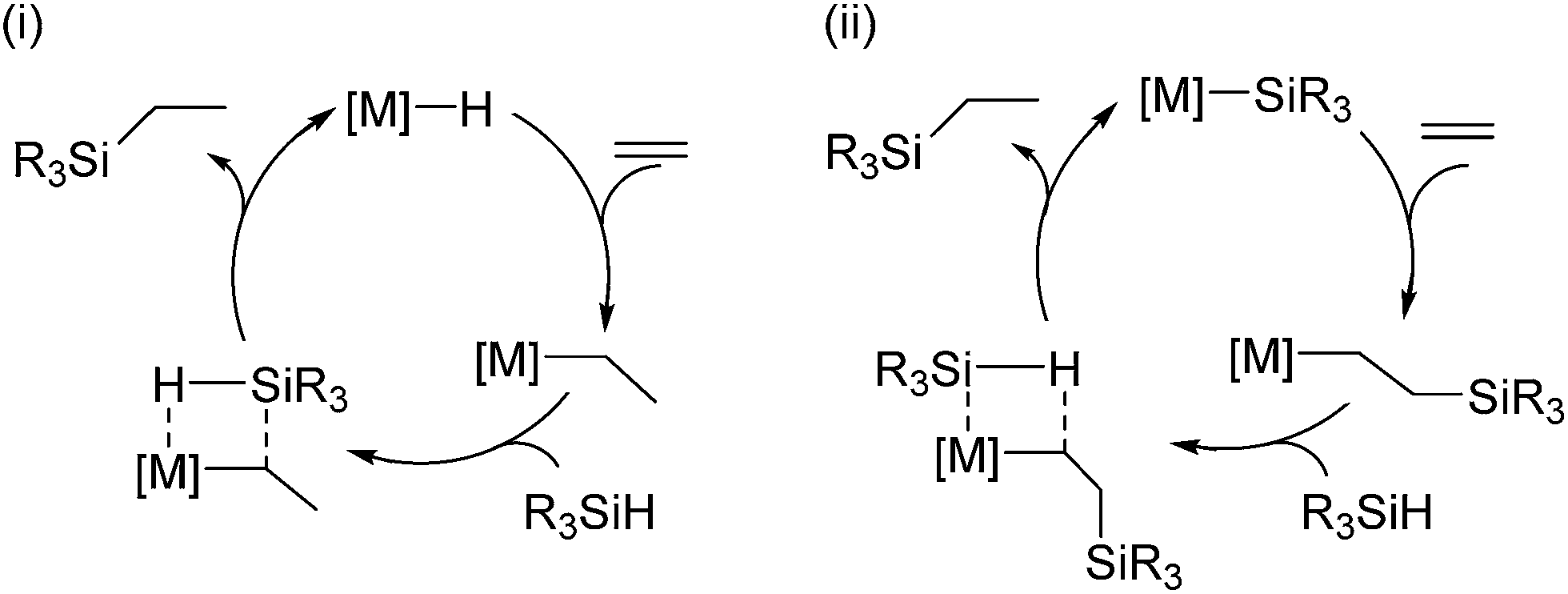 | ||
| Scheme 7 Hydrosilylation mechanism catalysed by d0 metals via metathesis. | ||
Lanthanide and actinide catalysts exhibit good reaction selectivity and are applicable to regio- and enantioselective hydrosilylations, although only limited substrate scope was disclosed. Compared to these catalysts, group 4 metal catalysts exhibit rather low activity and selectivity. Recently, on the other hand, significant progress has been made to achieve efficient and selective hydrosilylation using a titanium catalyst.
In 2005, Gendre, Möise and co-workers reported the utility of Cp2TiF2 (30) (Cp = η5-C5H5) as a cheap and stable catalyst precursor for the hydrosilylation of 1,3-dienes.60 Activation of the precatalyst Cp2TiF2 (3 mol%) is required by simply heating Cp2TiF2 with the reactant, PhSiH3 and isoprene, at 45 °C for a few minutes, showing the color change from yellow to dark purple. Further reaction at room temperature for 20 min gave bis-hydrosilylated products in high yields via selective anti-1,4-hydrosilylation of 1,3-dienes (eqn (22)). This reaction is also applicable to secondary and tertiary hydrosilanes.
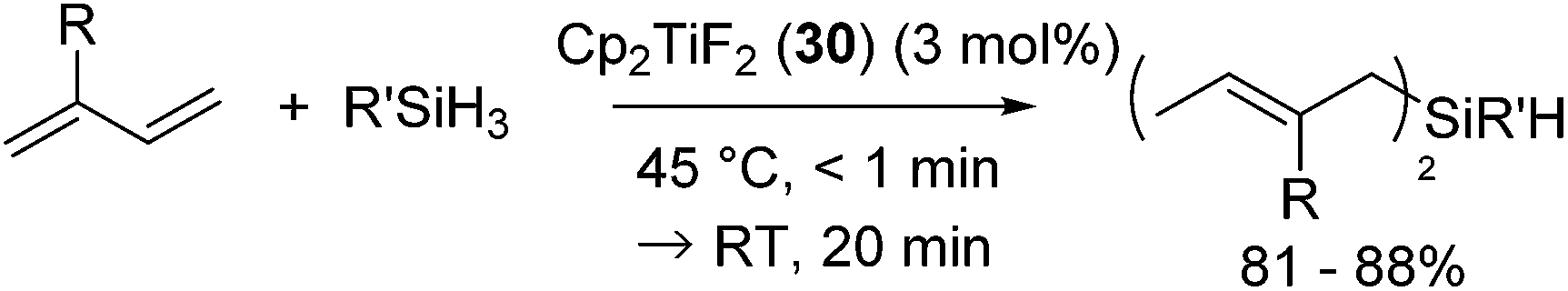 | (22) |
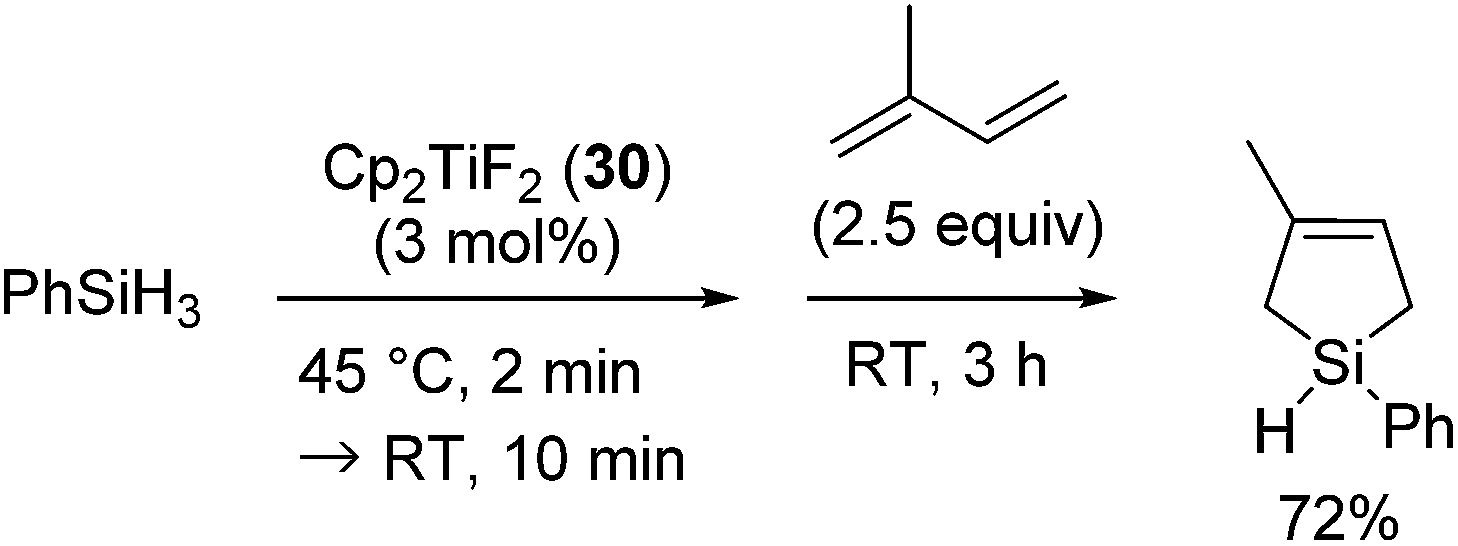
Different from the above results, when 30 was activated with PhSiH3 in the absence of 1,3-diene, the reaction of PhSiH3 with isoprene afforded 1-phenyl-3-methyl-3-silacyclopentene in 72% yield (eqn (23)).
 | (23) |
In these reactions, two different mechanisms were proposed (Scheme 8). Cp2TiF2 reacts with PhSiH3 to give [Cp2TiH(SiH2Ph)]. In the presence of 1,3-diene, the Ti species further follows successive insertion of the diene into the Ti–H bond forming a π-allyltitanium species, Si–C bond formation via reductive elimination to give the hydrosilylated product and “Cp2Ti”, and oxidative addition of PhSiH3 to regenerate [Cp2TiH(SiH2Ph)]. On the other hand, in the absence of 1,3-diene, [Cp2TiH(SiH2Ph)] is converted to [Cp2Ti(SiH2Ph)]. The resulting silyl complex successively undergoes olefin insertion into the Ti–Si bond to provide a π-allyltitanium species, Si–C bond formation via intramolecular σ-bond metathesis to form the silacyclopentene and Cp2TiH, and regeneration of [Cp2Ti(SiH2Ph)] by the reaction of Cp2TiH with PhSiH3.
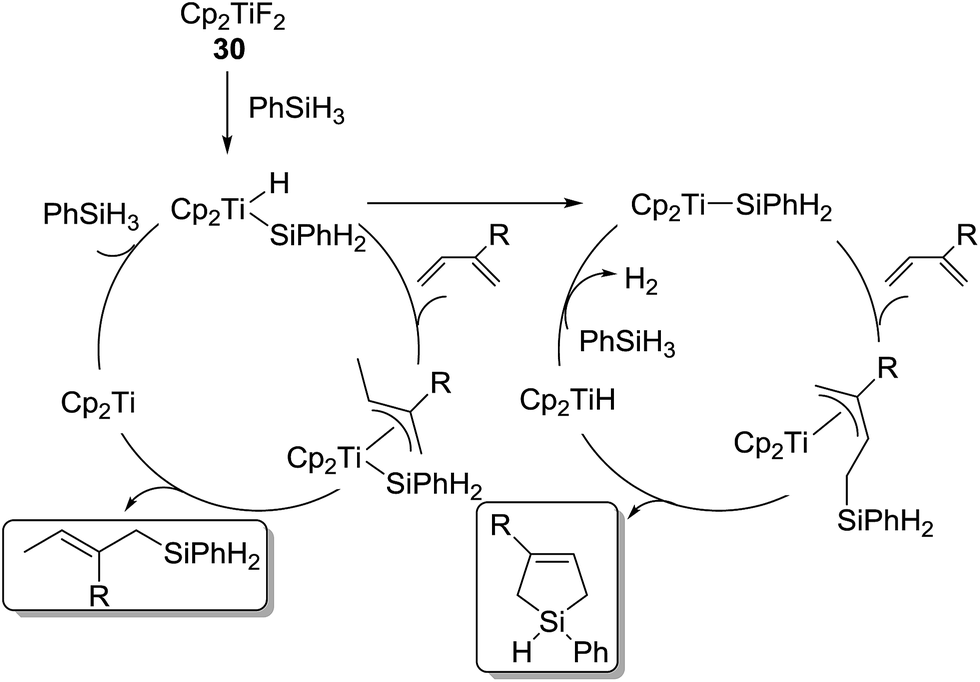 | ||
| Scheme 8 Possible mechanisms for the reaction of PhSiH3 with 1,3-dienes catalysed by 30. | ||
It has been revealed that [Cp2Ti(OPh)2] (31) also acts as a good precursor for the hydrosilylation of olefins.61 In the reaction of PhSiH3 with trimethoxyvinylsilane in the presence of 31 (3 mol%), simultaneous dehydrocoupling of PhSiH3 and hydrosilylation reaction took place to give hydrosilylated oligosilanes.
3.4. Alkali and alkaline earth metal catalysts
Group 1 and 2 metal complexes have recently been proved to act as hydrosilylation catalysts. The first example is introduced by Harder and co-workers in 2006. They performed hydrosilylation of conjugated double bonds using Ca, Sr, and K complexes as a catalyst.62 In the case of hydrosilylation of 1,1-diphenylethene, regiochemistry can be completely controlled by the choice of the metals. For example, hydrosilylation of 1,1-diphenylethene with PhSiH3 in the presence of a calcium catalyst 32/33 or strontium catalyst 34 proceeded at 50 °C to give a Markovnikov adduct, whereas the reaction resulted in the selective formation of anti-Markovnikov adduct in the presence of a potassium catalyst 35 (Fig. 9, Scheme 9).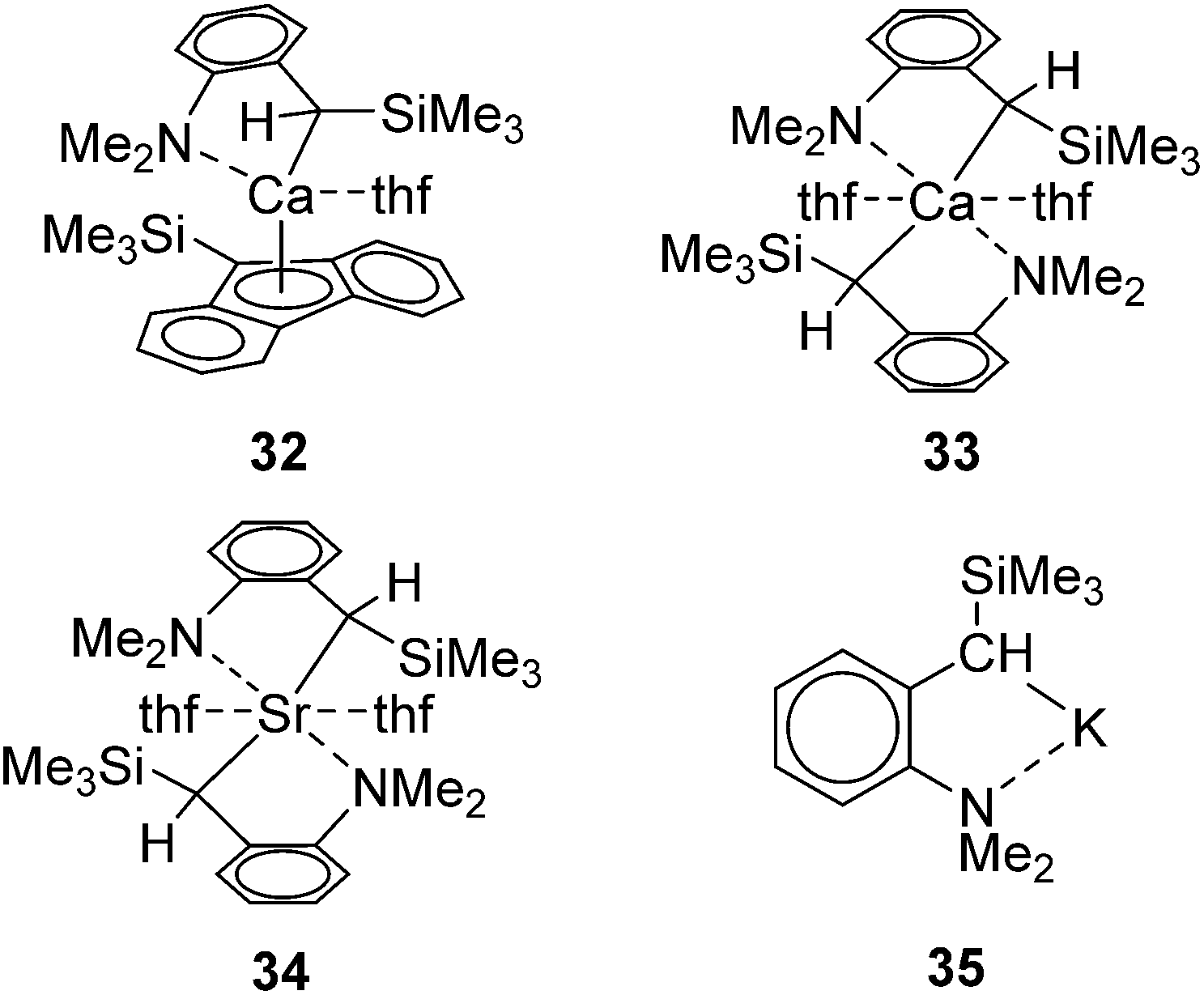 | ||
| Fig. 9 Structures of 32–35. | ||
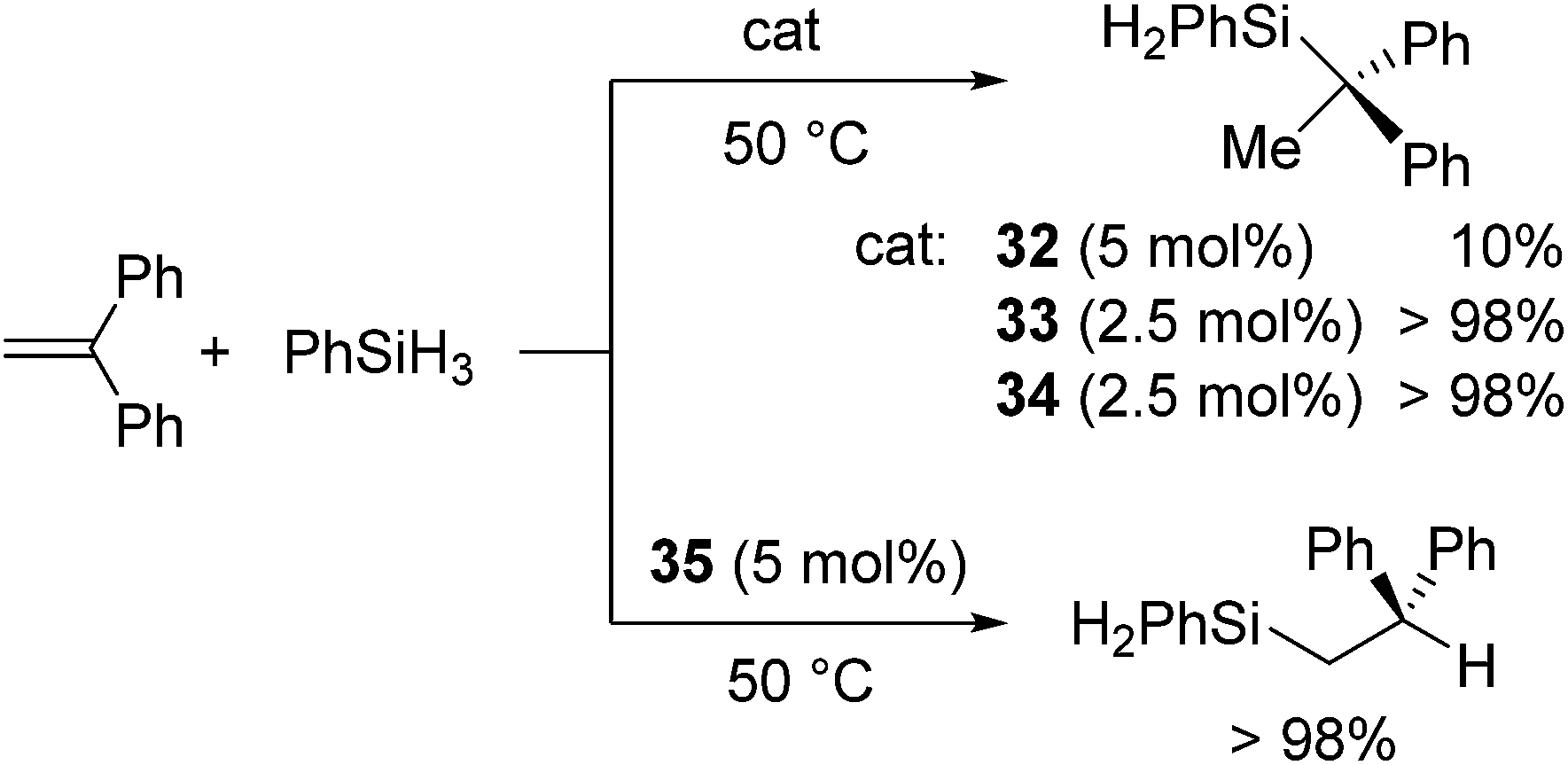 | ||
| Scheme 9 Hydrosilylation of diphenylethylene catalysed by 32–35. | ||
Alkali and alkaline earth metals are known to exhibit similar reactivity to organolanthanides. Therefore, possible catalytic cycle could be analogous to that established for the hydrosilylation with lanthanide catalysts including a metal hydride as a key intermediate (Scheme 7(i)).
This could be true for the formation of Markovnikov product. On the other hand, it is unlikely that anti-Markovnikov product obtained in Scheme 9 is formed by the hydride cycle through a 1,2-insertion which produces the resonance unstabilized Ph2CHCH2− ion. Thus, the authors suggested two possible mechanisms for the formation of the anti-Markovnikov product, cycles A and B in Scheme 10. Cycle A includes the silanide intermediate [PhSiH4]− formed by the reaction of PhSiH3 with the metal hydride, which undergoes concerted insertion reaction of the olefin. Owing to steric repulsion between Ph groups in 1,1-diphenylsilane and the silanide anion, anti-Markovnikov adduct is exclusively formed. This mechanism is also proposed in the KF-catalysed hydrosilylation of ketones.63 Cycle B includes the metal silanide intermediate, which is formed by the loss of H2 from the pentavalent PhSiH4−. Addition of the silyl group to 1,1-diphenylethene gives the resonance-stabilized intermediate [PhSiH2CH2CPh2]−[M]+, which gives anti-Markovnikov adduct after σ-bond metathesis with the silane.
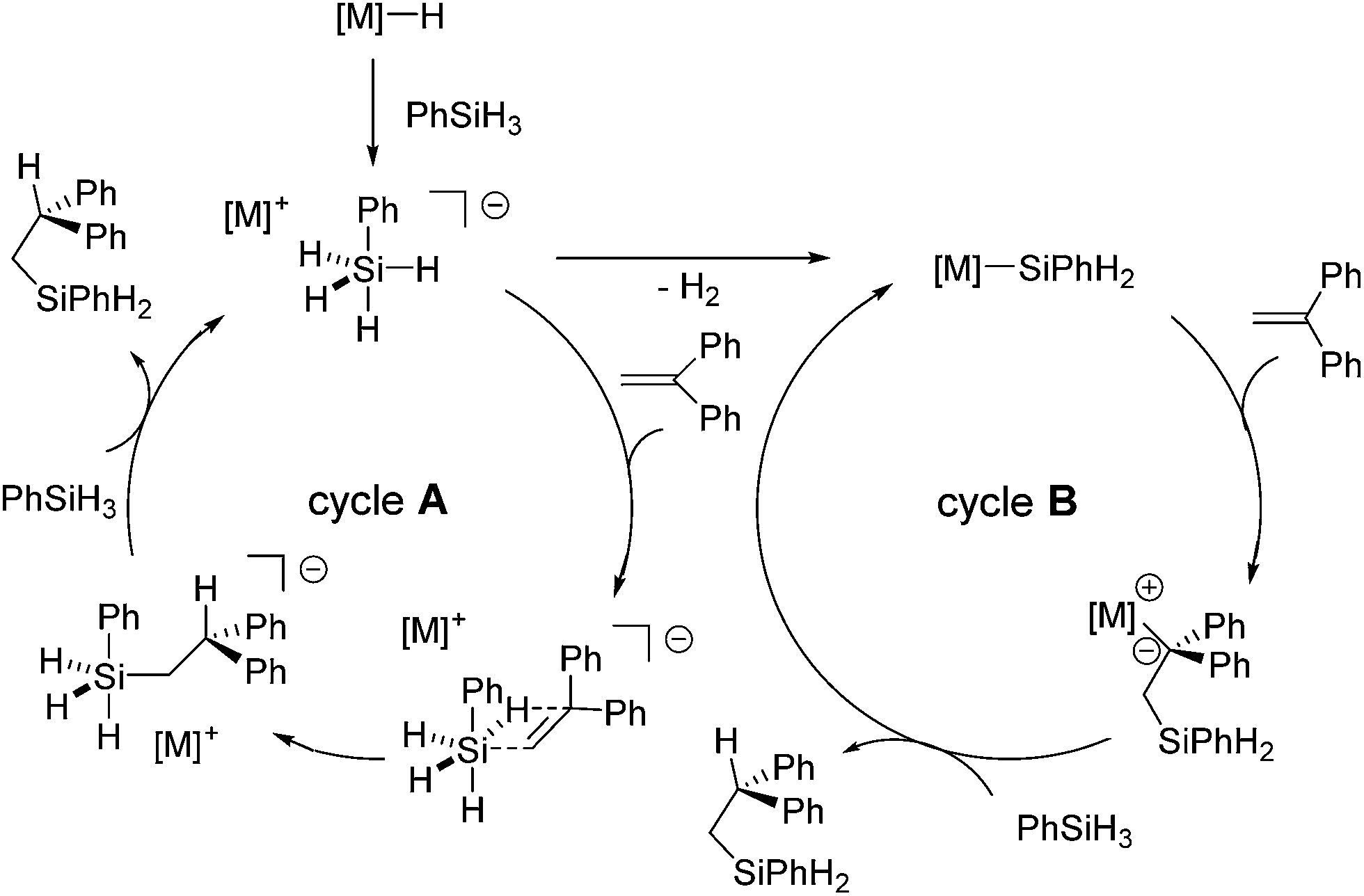 | ||
| Scheme 10 Hydrosilylation mechanism catalysed by alkaline earth metals. | ||
Related to the above proposed mechanism, Okuda and co-workers recently reported a well-defined alkali and alkaline earth metal silyl complexes, [Li(12-crown-4)SiPh3](thf)0.5 (36), [Na(15-crown-5)SiPh3](thf)0.5 (37), [K(18-crown-6)SiPh3(thf)] (38)64 and [Ca(SiPh3)2(thf)4] (39).65
These complexes catalyse hydrosilylation of 1,1-diphenylethene with Ph3SiH, Ph2SiH2, or PhSiH3 to exclusively form anti-Markovnikov adduct (eqn (24)), supposingly following catalytic cycle A in Scheme 10. This is thus far the first example of well-defined metal silyl hydrosilylation catalysts in the field of early main group metal chemistry.
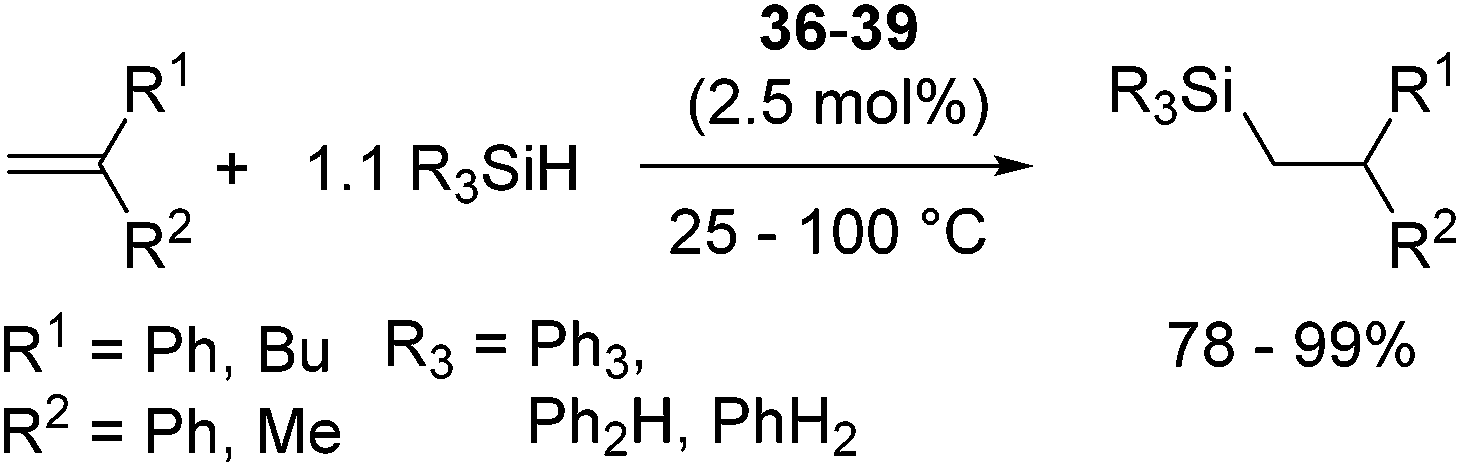 | (24) |
4. Non-metal catalysts
As a non-metal catalyst for hydrosilylation reactions, radical initiators,66 tertiary amines67 and Lewis acids68–71 are known. Among them, this section mainly deals with Lewis acid catalysed hydrosilylation reactions, which recently significantly developed. Aluminum Lewis acid catalysed hydrosilylation has already been described in the patent submitted in 1978.68 Thus, this section initially introduces aluminum catalysed hydrosilylation systems. In this patent, it was described that AlCl3, AlBr3 and aluminum oxychloride catalysed the hydrosilylation of various olefins and acetylene with chlorodimethylsilane. In 1985, Oertle and Wetter demonstrated hydrosilylation of various olefins with chlorinated hydrosilanes and suggested the mechanism initiated by the formation of a hydroalane species from AlCl3 and the hydrosilane, followed by hydroalumination of the olefin and transmetallation of the resulting alkyl alane with the hydrosilane (Scheme 11a).69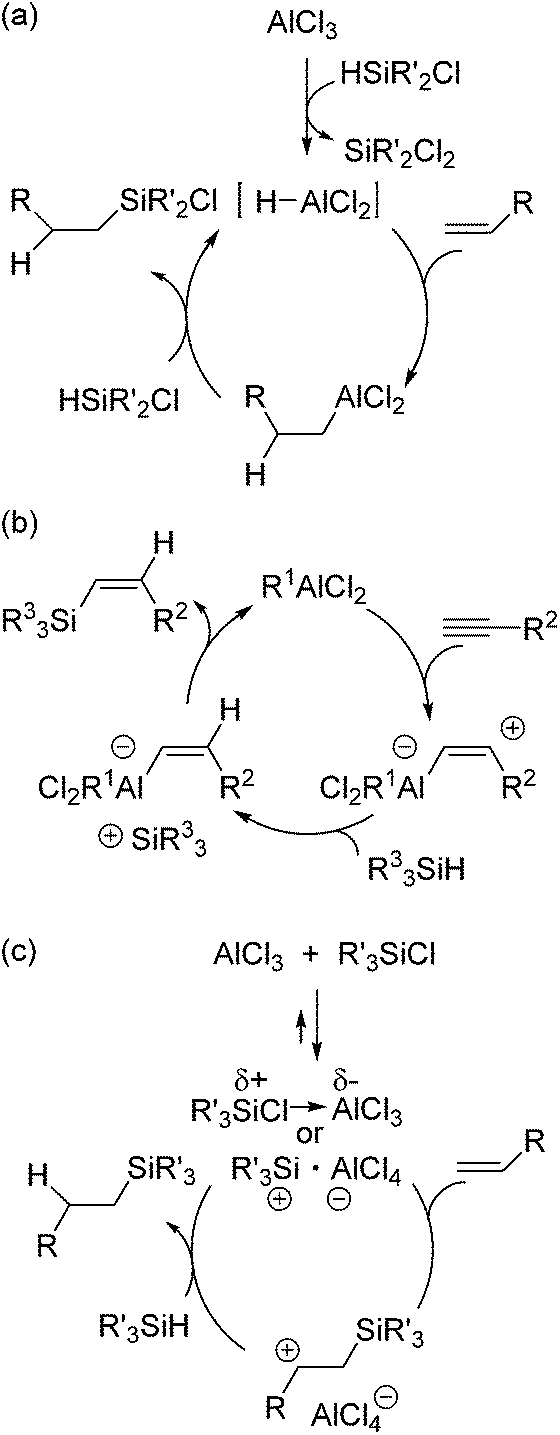 | ||
| Scheme 11 Hydrosilylation mechanism catalysed by aluminium catalysts. | ||
Yamamoto and co-workers reported the hydrosilylation of alkynes with trialkylsilanes catalysed by AlCl3 or EtAlCl2.72 For these reactions, they proposed a different mechanism including addition of a Lewis acid to a C–C triple bond, trapping of the formed carbenium ion of the zwitterionic intermediate with the hydrosilane, and coupling of the silylcation and the vinyl group (Scheme 11b).
A few years later, Jung and co-workers reported the hydrosilylation of cyclic alkenes and linear alkenes with trialkylsilanes by using various Lewis acid catalysts.71 They found out that the reaction rate drastically increased when trialkylchlorosilane was used as an activator. Based on these facts, they suggested a new mechanism in which a trialkylsilylium ion is formed as the key intermediate (Scheme 11c). At the initial stage of the catalytic cycle, silylium cation (R3Si+AlCl4−) or polarized donor–acceptor complex (Et3Siδ+Cl → AlCl3δ−) is formed, which successively reacts with the olefin to give a carbenium intermediate and then with the hydrosilane to form the hydrosilylated product and regenerate the active intermediate (Scheme 11c).
Related to the above studies, chemistry of silylium cations have been closely investigated by Lambert and co-workers. They successfully isolated a silylium cation [Et3Si(benzene)]+ by using B(C6F5)4− as a stable non-coordinating counter anion and demonstrated the stoichiometric reaction between the silylium cation and an olefin, resulting in the formation of a carbenium ion.73 Inspired by these findings, Gevorgyan and co-workers developed B(C6F5)3 (40) catalysed hydrosilylation reactions using various hydrosilanes possessing aryl and/or alkyl groups (eqn (25)).74
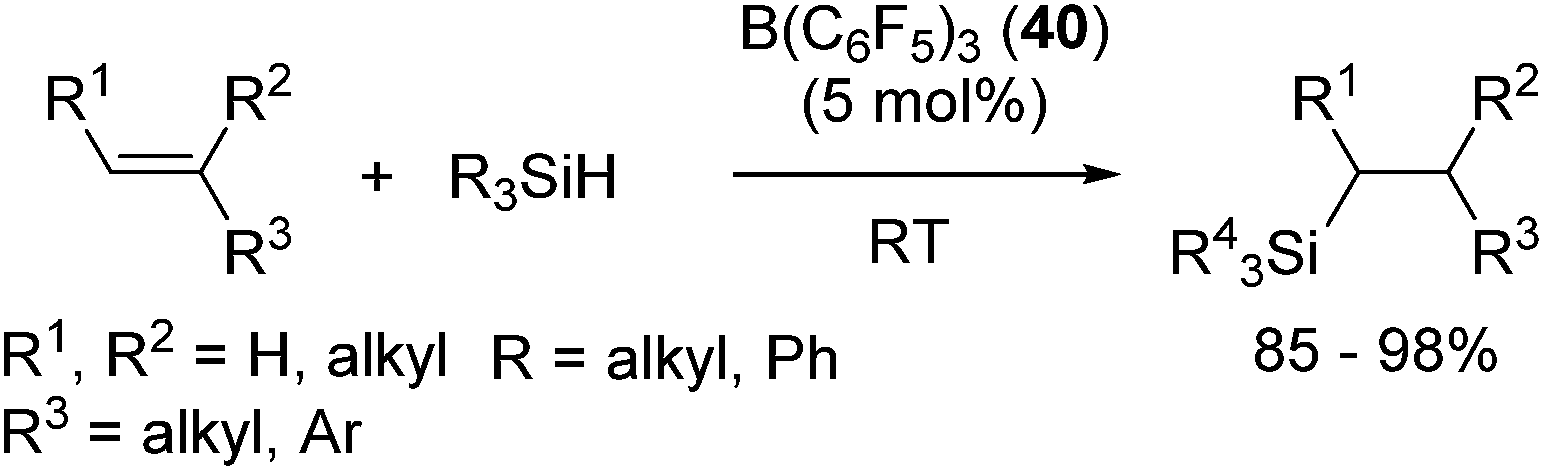 | (25) |
It should be noted that no polymerization proceeded in this system, which is a typical side-reaction for the former Lewis acid-catalysed hydrosilylation reactions. In this study, the mechanism involving addition of a silylium cation across the CC double bond followed by trapping of the resulting carbenium cation with a boron-hydride (or hydrosilane) was confirmed (Scheme 12).72
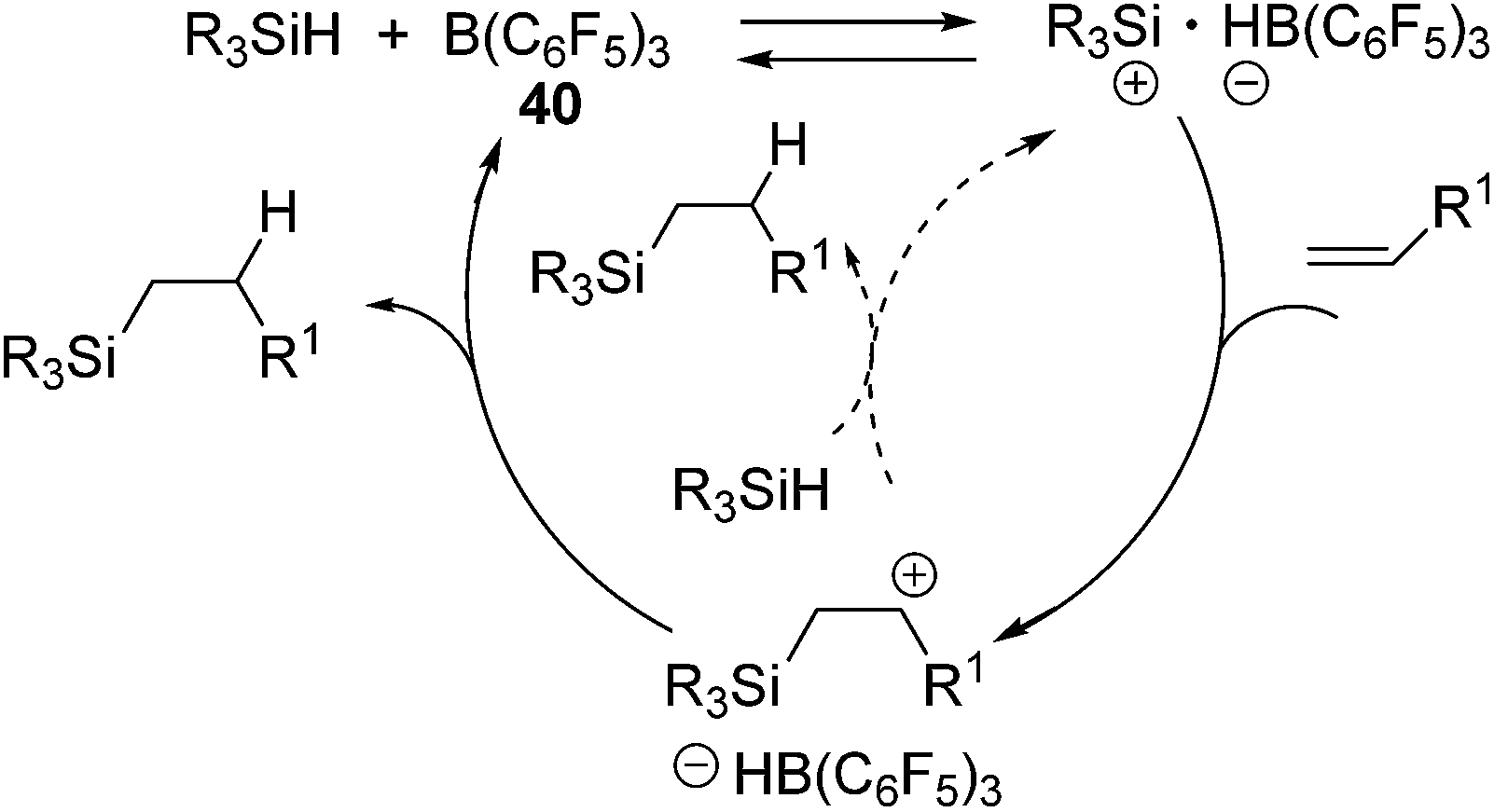 | ||
| Scheme 12 Hydrosilylation mechanism catalysed by 40. | ||
More recently, Stephan and co-workers focused on the Lewis acidity of organophosphonium salts [(C6F5)3−xPhxPF][B(C6F5)4] (x = 0 (41a), 1 (41b)) and [(SIMes)PFPh2][B(C6F5)4]2 (42) (SIMes = 1,3-dimethylimidazolidin-2-ylidene) and demonstrated their utility in hydrosilylation reactions.75 Although the catalyst loading is rather high (ca. 2 mol%) in these systems, these catalysts exhibit tolerance to some functional groups such as chloro and silyl-ether functionalities (eqn (26)).
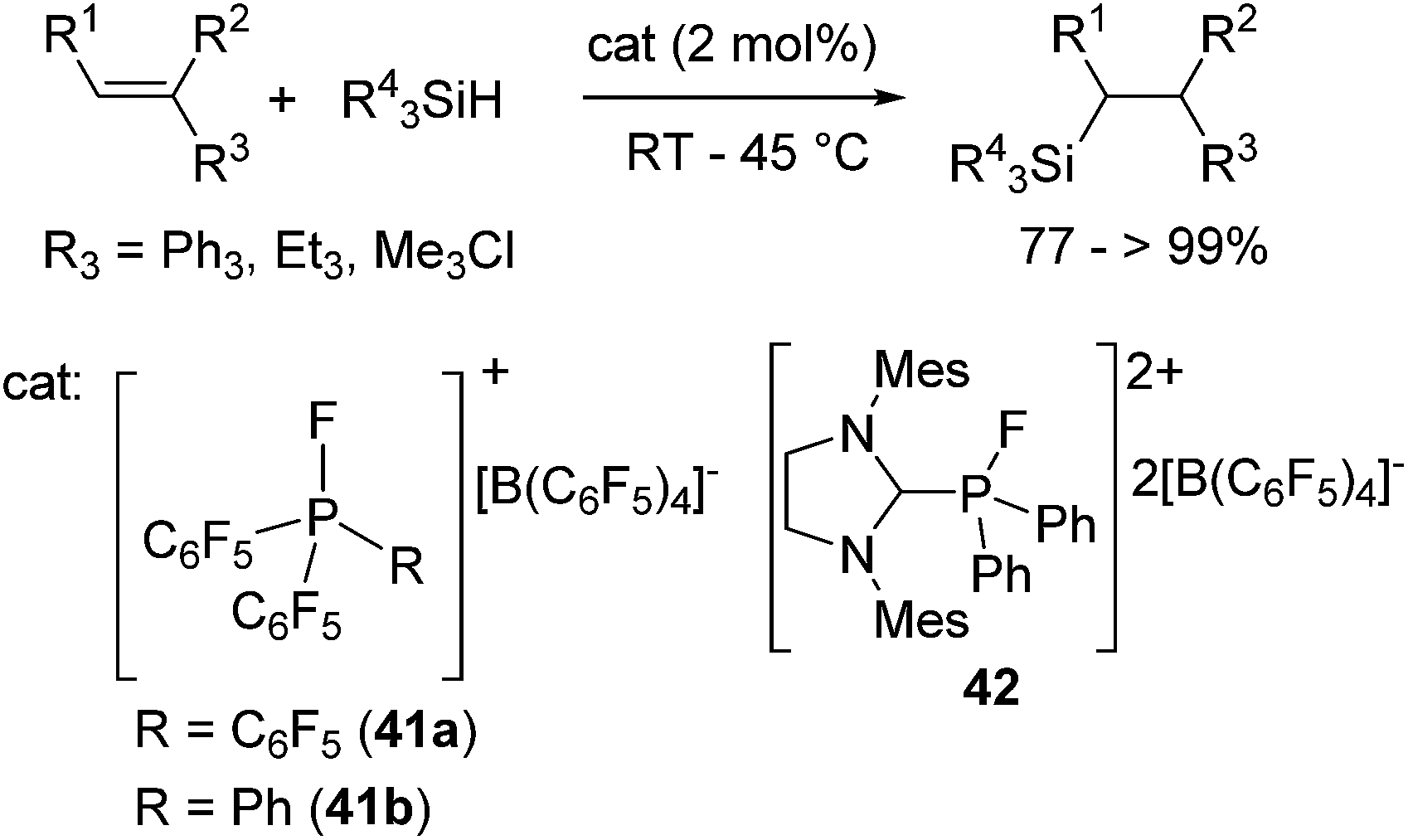 | (26) |
For monocationic phosphonium [(C6F5)3PF][B(C6F5)4] (41a) system, they proposed a plausible mechanism which includes direct activation of the Si–H bond of the hydrosilanes by the phosphonium P atom (Scheme 13). It is to be noted that these studies demonstrated for the first time the utilization of new Lewis acids, organo phosphonium salts, as an organocatalyst.
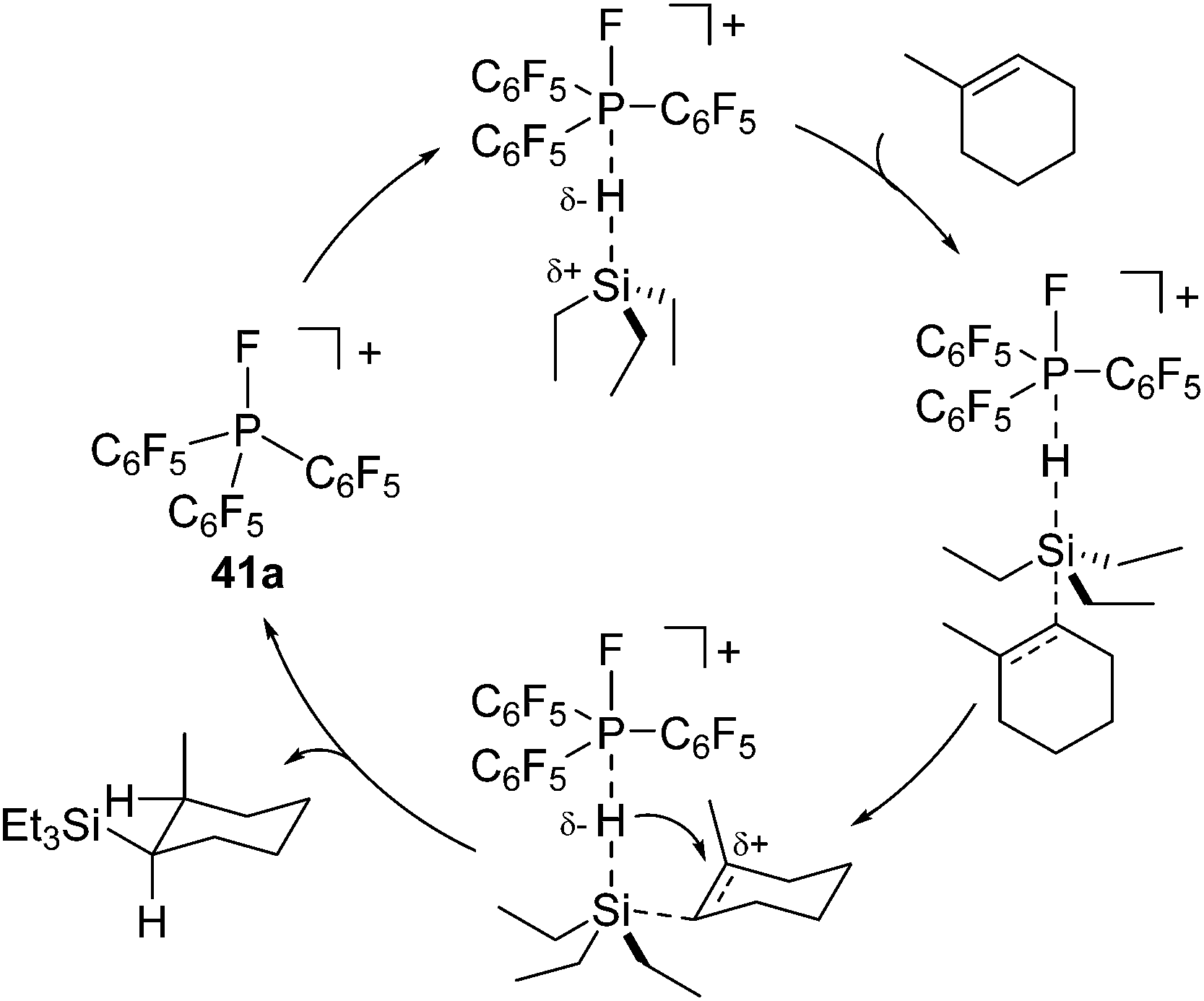 | ||
| Scheme 13 Hydrosilylation mechanism catalysed by 41a. | ||
Recently, Oestreich and co-workers reported hydrosilylation of olefins with Me3SiH and Me2SiH2, which are rarely applied for hydrosilylation due to their highly volatile, flammable and potentially explosive nature, by using 3-silylated-cyclohexa-1,4-dienes as hydrosilane precursors.76 The key points of the reaction are shown in Scheme 14. They utilized the strong hydride abstraction ability of B(C6F5)3 (40) for 3-silylated cyclohexa-1,4-dienes to form transient intermediate A. Complete hydride abstraction from A lead to the formation of B, which is further transformed to the benzene-stabilized sililyum cation [R3Si(benzene)]+ via aromatization of benzene ring. The silylium ion further reacts with an olefin to give the corresponding hydrosilylated product following the same process shown in Scheme 12.
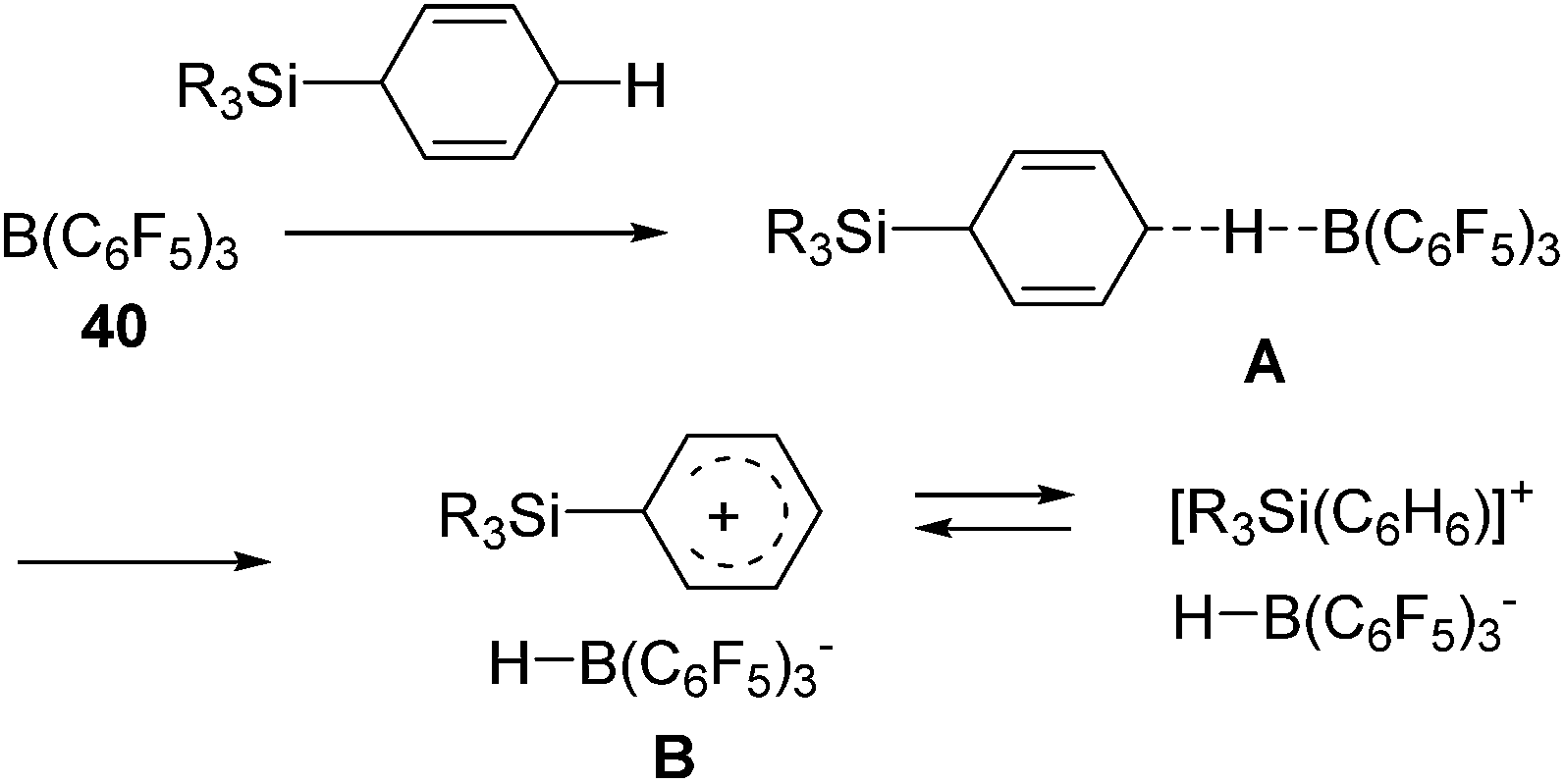 | ||
| Scheme 14 Reaction path for formal hydrosilylation of olefins with Me3SiH/Me2SiH2. | ||
By using this system, various kinds of terminal, internal, cyclic alkenes and styrene derivatives were successfully hydrosilylated at room temperature in the presence of 5 mol% of 40 (eqn (27)).
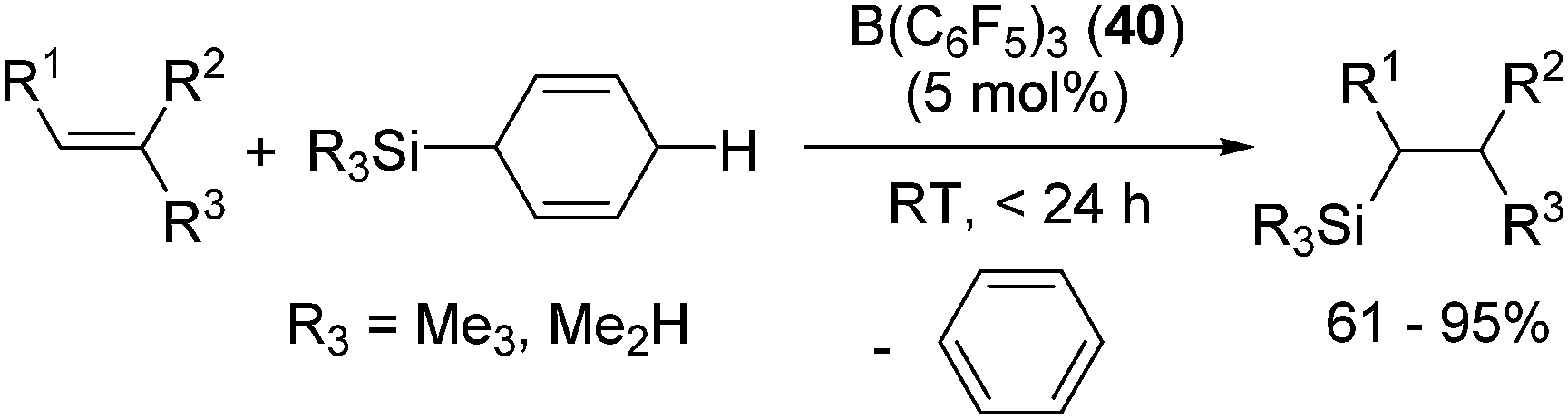 | (27) |
As a non-metal catalyst, N-heterocyclic carbenes (NHC) 43–46 (Fig. 10) are also known to catalyse the hydrosilylation reaction following the reaction paths shown in Scheme 15. By using this method, chemoselective reduction of olefins and alkynes containing α-hydroxy substituent were attained (eqn (28)).77
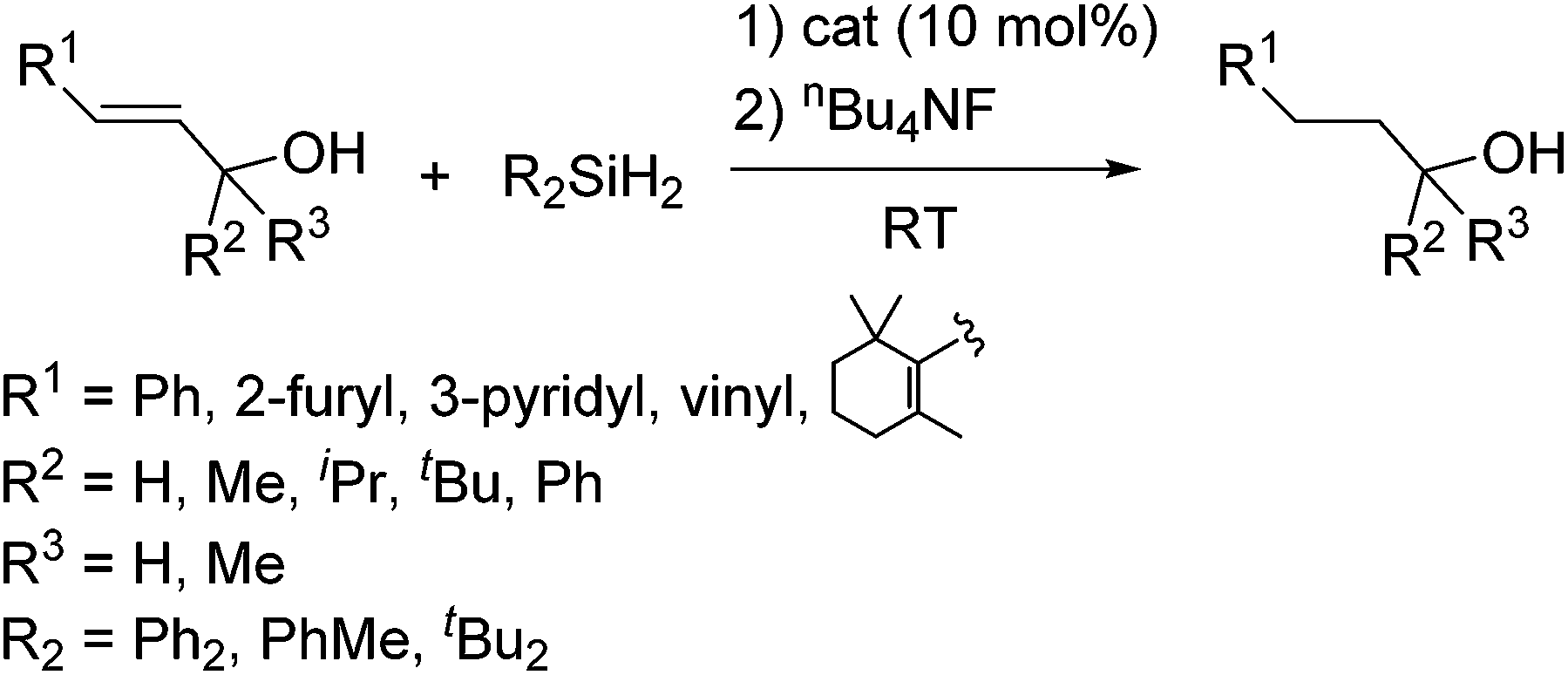 | (28) |
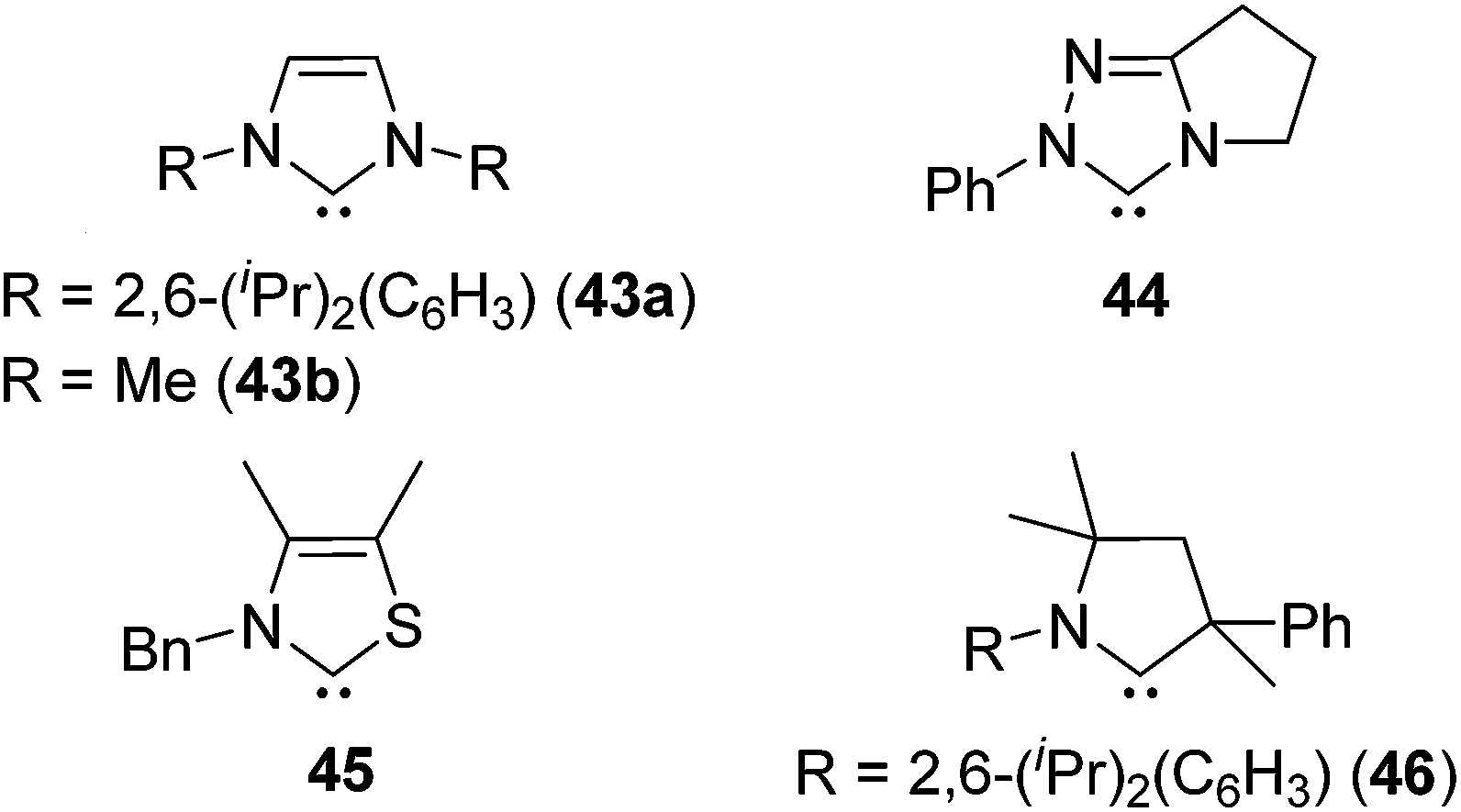 | ||
| Fig. 10 Structures of 43–46. | ||
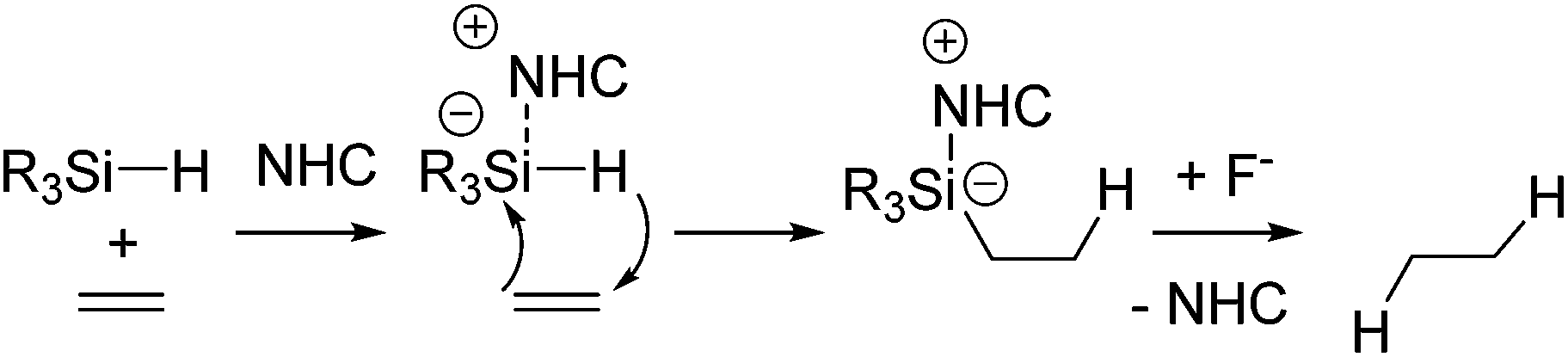 | ||
| Scheme 15 NHC-catalysed reduction of olefins. | ||
It is to be noted that styryl and propargyl alcohols, which often undergo the dehydrogenative coupling with hydrosilanes to give siloxy compounds in the presence of precious metal catalysts, selectively undergo the reduction of the multiple bonds through the hydrosilylation with Ph2SiH2 by using 44 as a catalyst (eqn (29)).
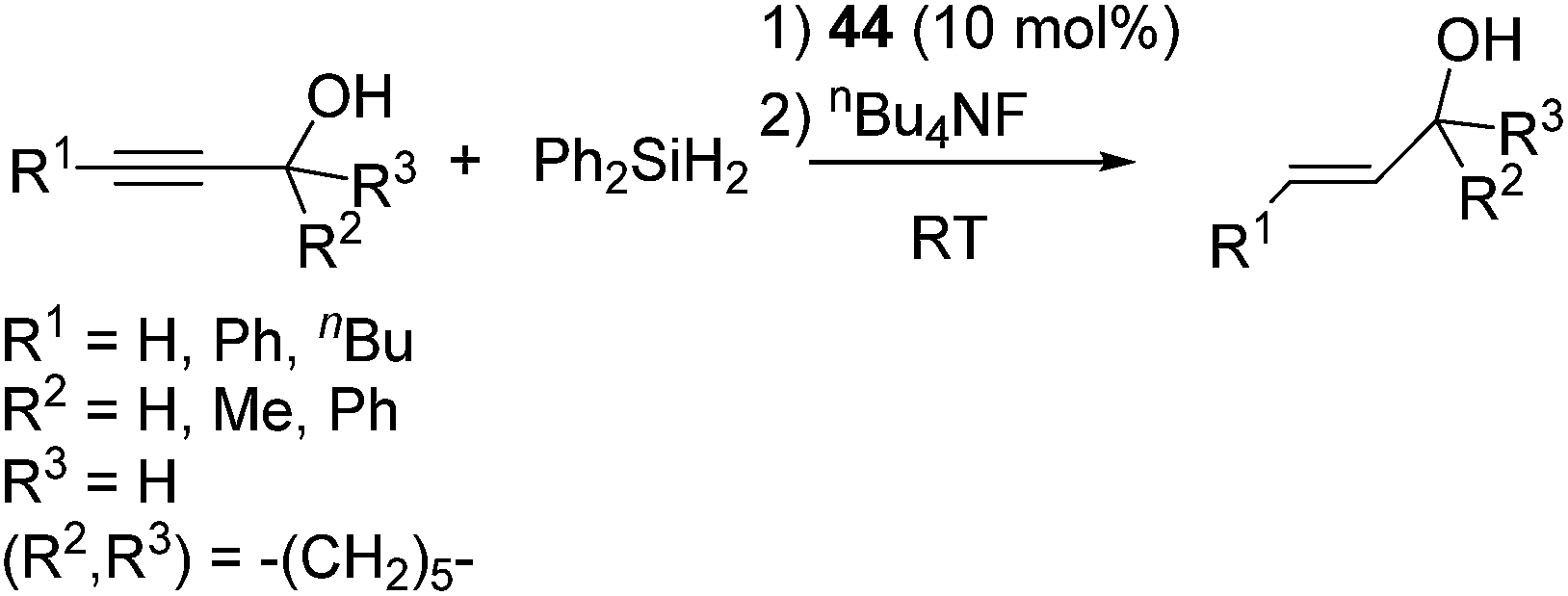 | (29) |
5. Conclusions
During the last decade, a variety of new approaches have been pursued using various kinds of catalysts in academia as well as in industry. In the platinum catalysis systems, suppression of undesired side reactions has been achieved to some extent and the catalytic activity has been slightly improved as compared to the conventional Karstedt's catalyst in some cases. By using other late transition metals such as iridium and rhenium, unique chemo- and regioselective hydrosilylations have been attained. Significant progress has been achieved in the hydrosilylation reactions utilizing cheaper metals (Fe, Ni, Ti, etc.). In particular, precise design of iron catalysts established the systems exhibiting high efficiency and high compatibility towards various functionalized olefins containing epoxide, amide, ester, keto groups etc. The use of non-transition metal catalysts including early main group metals, Lewis acids and free carbenes has also been intensively studied. Since they follow unique reaction mechanisms, which are different from the well-known mechanism for the transition metal catalysts, it is expected that the systems could afford a new avenue to the hydrosilylation reactions.However, even with these progresses, most of hitherto mentioned catalysts are still far away from an industrial application, and there are still many problems to be solved. Further improvement of catalytic activity, selectivity and catalyst stability is required in order to replace current platinum catalyst systems, in particular for the curing of silicone materials. For the production of highly sophisticated organosilicon materials with wider variety of functions, issues facing silicone industry are, for example, (1) to find better methods for suppressing deactivation by coordination with sulphur or nitrogen compounds, which are often used in the rubber industry, (2) to attain highly selective hydrosilylation of allyl compounds (CH2CHCH2X) with Y3SiH (Y = OR, Cl) for the production of γ-substituted propylsilanes, which are used as silane coupling agents for various applications.8,78 The latter reaction is a well-known example of a hydrosilylation process with rather imperfect selectivity with generation of many by-products including R3SiX, propene and propylsilane.79 Considering these problems, hydrosilylation chemistry is still a challenging research field.
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research from MEXT Japan.Notes and references
- L. N. Lewis, J. Stein, Y. Gao, R. E. Colborn and G. Hutchins, Platinum Met. Rev., 1997, 41, 66 CAS
.
- K. Tamao, N. Ishida, T. Tanaka and M. Kumada, Organometallics, 1983, 2, 1694 CrossRef CAS
- Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918 CrossRef CAS
- I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063 CrossRef CAS PubMed
- L. H. Sommer, E. W. Pietrusza and F. C. Whitmore, J. Am. Chem. Soc., 1947, 69, 188 CrossRef CAS
- J. L. Speier, J. A. Webster and G. H. Barnes, J. Am. Chem. Soc., 1957, 79, 974 CrossRef CAS
- B. D. Karstedt, General Electric Company, US3775452A, 1973
- B. Marciniec, J. Gulinski, W. Urbaniac and Z. W. Kornetka, Comprehensive Handbook on Hydrosilylation, Pergamon, Oxford, UK, 1992 Search PubMed
- B. Marciniec and J. Guliński, J. Organomet. Chem., 1993, 446, 15 CrossRef CAS
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440 CrossRef CAS PubMed
- A. J. Holwell, Platinum Met. Rev., 2008, 52, 243 CrossRef
- I. E. Markó, S. Stérin, O. Buisine, G. Mignani, P. Branlard, B. Tinant and J.-P. Declercq, Science, 2002, 298, 204 CrossRef PubMed
- O. Buisine, G. Berthon-Gelloz, J.-F. Brière, S. Stérin, G. Mignani, P. Branlard, B. Tinant, J.-P. Declercq and I. E. Markó, Chem. Commun., 2005, 3856 RSC
- J. W. Sprengers, M. J. Mars, M. A. Duin, K. J. Cavell and C. J. Elsevier, J. Organomet. Chem., 2003, 679, 149 CrossRef CAS
- J. J. Dunsford, K. J. Cavell and B. Kariuki, J. Organomet. Chem., 2011, 696, 188 CrossRef CAS PubMed
- M. A. Taige, S. Ahrens and T. Strassner, J. Organomet. Chem., 2011, 696, 2918 CrossRef CAS PubMed
- B. Marciniec, K. Posała, I. Kownacki, M. Kubicki and R. Taylor, ChemCatChem, 2012, 4, 1935 CrossRef CAS
- C. M. Downing and H. H. Kung, Catal. Commun., 2011, 12, 1166 CrossRef CAS PubMed
- J. Li, C. Niu, J. Peng, Y. Deng, G. Zhang, Y. Bai, C. Ma, W. Xiao and G. Lai, Appl. Organomet. Chem., 2014, 28, 454 CrossRef CAS
- M. Xue, J. Li, J. Peng, Y. Bai, G. Zhang, W. Xiao and G. Lai, Appl. Organomet. Chem., 2014, 28, 120 CrossRef CAS
- B. J. Truscott, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2013, 270 RSC
- J. Li, J. Peng, G. Zhang, Y. Bai, G. Lai and X. Li, New J. Chem., 2010, 34, 1330 RSC
- J. Li, J. Peng, Y. Bai, G. Zhang, G. Lai and X. Li, J. Organomet. Chem., 2010, 695, 431 CrossRef CAS PubMed
- J. A. Muchnij, F. B. Kwaramba and R. J. Rahaim, Org. Lett., 2014, 16, 1330 CrossRef CAS PubMed
- M. Igarashi, T. Matsumoto, T. Kobayashi, K. Sato, W. Ando, S. Shimada, M. Hara and H. Uchida, J. Organomet. Chem., 2014, 752, 141 CrossRef CAS PubMed
- H. Dong, Y. Jiang and H. Berke, J. Organomet. Chem., 2014, 750, 17 CrossRef CAS PubMed
- M. A. Schroeder and M. S. Wrighton, J. Am. Chem. Soc., 1974, 96, 6235 CrossRef
- M. Stosur and T. Szymańska-Buzar, J. Mol. Catal. A: Chem., 2008, 286, 98 CrossRef CAS PubMed
- A. J. Chalk and J. F. Harrod, J. Am. Chem. Soc., 1965, 87, 16 CrossRef CAS
- P. B. Glaser and T. D. Tilley, J. Am. Chem. Soc., 2003, 125, 13640 CrossRef CAS PubMed
- C. Beddie and M. B. Hall, J. Am. Chem. Soc., 2004, 126, 13564 CrossRef CAS PubMed
- M. A. Rankin, D. F. MacLean, G. Schatte, R. MacDonald and M. Stradiotto, J. Am. Chem. Soc., 2007, 129, 15855 CrossRef CAS PubMed
- H. Yoo, P. J. Carroll and D. H. Berry, J. Am. Chem. Soc., 2006, 128, 6038 CrossRef CAS PubMed
- M. E. Fasulo, M. C. Lipke and T. D. Tilley, Chem. Sci., 2013, 4, 3882 RSC
- A. N. Nesmeyanov, R. K. Freidlina, E. C. Chukovskaya, R. G. Petrova and A. B. Belyavsky, Tetrahedron, 1962, 17, 61 CrossRef CAS
- M. A. Schroeder and M. S. Wrighton, J. Organomet. Chem., 1977, 128, 345 CrossRef CAS
- S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794 CrossRef CAS PubMed
- A. M. Tondreau, C. C. H. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis and P. J. Chirik, Science, 2012, 335, 567 CrossRef CAS PubMed
- C. C. H. Atienza, A. M. Tondreau, K. J. Weller, K. M. Lewis, R. W. Cruse, S. A. Nye, J. L. Boyer, J. G. P. Delis and P. J. Chirik, ACS Catal., 2012, 2, 2169 CrossRef
- A. M. Tondreau, C. C. H. Atienza, J. M. Darmon, C. Milsmann, H. M. Hoyt, K. J. Weller, S. A. Nye, K. M. Lewis, J. Boyer, J. G. P. Delis, E. Lobkovsky and P. J. Chirik, Organometallics, 2012, 31, 4886 CrossRef CAS
- K. Kamata, A. Suzuki, Y. Nakai and H. Nakazawa, Organometallics, 2012, 31, 3825 CrossRef CAS
- D. Peng, Y. Zhang, X. Du, L. Zhang, X. Leng, M. D. Walter and Z. Huang, J. Am. Chem. Soc., 2013, 135, 19154 CrossRef CAS PubMed
- J. Y. Wu, B. N. Stanzl and T. Ritter, J. Am. Chem. Soc., 2010, 132, 13214 CrossRef CAS PubMed
- C. E. Masse and J. S. Panek, Chem. Rev., 1995, 95, 1293 CrossRef CAS
- M. D. Greenhalgh, D. J. Frank and S. P. Thomas, Adv. Synth. Catal., 2014, 356, 584 CrossRef CAS
- A. D. Petrov, V. F. Mironov, V. M. Vdovin and S. I. Sakykh-Zade, Izv. Akad. Nauk SSSR, Otd. Khim. Nauk, 1956, 256 (Chem. Abstr., 1956, 50, 13726) CAS
- S. Nozakura and S. Konotsune, Bull. Chem. Soc. Jpn., 1956, 29, 326 CrossRef CAS
- E. T. Chukovskaya and R. K. Freidlina, Izv. Akad. Nauk SSSR, Otd. Khim. Nauk, 1963, 761 (Chem. Abstr., 1963, 59, 7551) CAS
- A. N. Nesmeyanov, R. K. Freidlina, E. C. Chukovskaya, R. G. Petrova and A. B. Belyavsky, Tetrahedron, 1962, 17, 61 CrossRef CAS
- Y. Kiso, M. Lumada, K. Maeda, K. Sumitani and K. Tamao, J. Organomet. Chem., 1973, 50, 311 CrossRef CAS
- B. Marciniec, H. Maciejewski and I. Kownacki, J. Mol. Catal. A: Chem., 1998, 135, 223 CrossRef CAS
- B. Marciniec, H. Maciejewski and J. Mirecki, J. Organomet. Chem., 1991, 418, 61 CrossRef CAS
- H. Maciejewski, B. Marciniec and I. Kownacki, J. Organomet. Chem., 2000, 597, 175 CrossRef CAS
- Y. Chen, C. Sui-Seng, S. Boucher and D. Zargarian, Organometallics, 2005, 24, 149 CrossRef CAS
- I. Hyder, M. Jiménez-Tenorio, M. C. Puerta and P. Valerga, Dalton Trans., 2007, 3000 RSC
- A. Kuznetsov and V. Gevorgyan, Org. Lett., 2012, 14, 914 CrossRef CAS PubMed
- M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2012, 48, 7146 RSC
- J. F. Harrod and S. S. Yun, Organometallics, 1987, 6, 1381 CrossRef CAS
- G. A. Molander and J. A. C. Romero, Chem. Rev., 2002, 102, 2161 CrossRef CAS PubMed
- L. Bareille, S. Becht, J. L. Cui, P. L. Gendre and C. Moïse, Organometallics, 2005, 24, 5802 CrossRef CAS
- J. Garcia, D. J. M. Meyer, D. Guillaneux, J. J. E. Moreau and M. W. C. Man, J. Organomet. Chem., 2009, 694, 2427 CrossRef CAS PubMed
- F. Buch, J. Brettar and S. Harder, Angew. Chem., Int. Ed., 2006, 45, 2741 CrossRef CAS PubMed
- J. Boyer, R. Corriu, R. Perz and C. Reye, Tetrahedron, 1981, 37, 2165 CrossRef CAS
- V. Leich, K. Lamberts, T. P. Spaniol and J. Okuda, Dalton Trans., 2014, 14315 RSC
- V. Leich, T. P. Spaniol, L. Maron and J. Okuda, Chem. Commun., 2014, 50, 2311 RSC
- T. Dohmaru, Y. Nagata and J. Tsurugi, Chem. Lett., 1973, 1031 CrossRef CAS
- S. Nozakura and S. Konotsune, Bull. Chem. Soc. Jpn., 1956, 29, 322 CrossRef CAS
- U. Finke and H. Moretto, DE2804204, 1978
- K. Oertle and H. Wetter, Tetrahedron Lett., 1985, 26, 5511 CrossRef CAS
- K. Yamamoto and M. Takemae, Synlett, 1990, 259 CrossRef CAS PubMed
- Y.-S. Song, B. R. Yoo, G.-H. Lee and I. N. Jung, Organometallics, 1999, 18, 3109 CrossRef CAS
- N. Asao, T. Sudo and Y. Yamamoto, J. Org. Chem., 1996, 61, 7654 CrossRef CAS
- J. B. Lambert and Y. Zhao, J. Am. Chem. Soc., 1996, 118, 7867 CrossRef CAS
- M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936 CrossRef CAS PubMed
- M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308 CrossRef CAS PubMed
- A. Simonneau and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 11905 CrossRef CAS PubMed
- Q. Zhao, D. P. Curran, M. Malacria, L. Fensterbank, J.-P. Goddard and E. Lacôte, Chem.–Eur. J., 2011, 17, 9911 CrossRef CAS PubMed
- U. Deschler, P. Kleinschmit and P. Panster, Angew. Chem., Int. Ed., 1986, 25, 236 CrossRef
- P. Gigler, M. Drees, K. Riener, B. Bechlars, W. A. Herrmann and F. E. Kuehn, J. Catal., 2012, 295, 1 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |