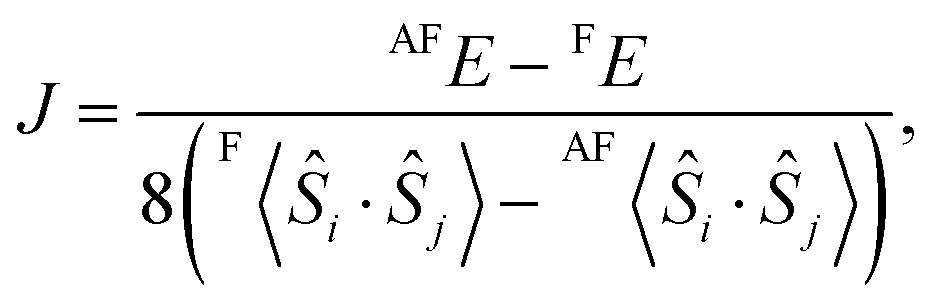DOI:
10.1039/C5QI00091B
(Research Article)
Inorg. Chem. Front., 2015,
2, 771-779
DFT and TD-DFT studies of electronic structures and one-electron excitation states of a cyanide-bridged molecular square complex†
Received
31st May 2015
, Accepted 1st July 2015
First published on 1st July 2015
Abstract
The electronic structures of a cyanide bridged Fe–Co molecular square, [Co2Fe2(CN)6(tp*)2(dtbbpy)4](PF6)2·2MeOH (1) (tp* = hydrotris (3,5-dimethylpyrazol-1-yl) borate, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine), which exhibits thermal and photo-induced two-step charge-transfer induced spin transitions (CTIST), are investigated in detail by density functional theory (DFT) and time-dependent (TD) DFT calculations. For the three phases observed in the experiment, three different model structures are constructed based on the geometries of X-ray crystallography analysis measured at low (100 K), middle (298 K) and high (330 K) temperatures. The calculated results elucidate that the ground states at the low and the high temperatures are diamagnetic [(FeIILS)2(CoIIILS)2] and ferromagnetic [(FeIIILS)2(CoIIHS)2], respectively. On the other hand, the one-electron transferred [FeIILSFeIIILSCoIIHSCoIIILS] state becomes the ground state at the intermediate temperature phase. A magnetic interaction between FeIII and CoII in the [(FeIIILS)2(CoIIHS)2] state is ferromagnetic and the most stable spin-coupling state is the all-ferromagnetic state. The TD-DFT calculation shows the two significant peaks of FeII t2g → CoIII eg around 800 nm. The results support that the experimental broad absorption peak at 770 nm is an inter-valence charge transfer (IVCT) band.
1. Introduction
Prussian blue, Fe4[Fe(CN)6]3·xH2O, is a well-known dye. The iron ions in the compound can be easily substituted by other metal ions such as Cr, Mn, Co and so on, and those are called Prussian blue analogues (PBAs). The PBAs are 3D bulk materials in which metal ions are bridged by cyanide ions. The cyanide ions mediate electronic and magnetic interactions between metal ions, therefore intriguing physical properties are found in PBAs, such as high critical temperature (Tc) magnets, spin-crossover, linkage isomerism, and ferro-electricity.1–11 One of the most important physical properties of PBAs is ferromagnetism. Babel et al. first found ferromagnetism in CsIMnII[CrIII(CN)6]·H2O (Tc = 90 K).1 About a decade later, Verdaguer and co-workers also found that Cs(I)Ni(II)[Cr(III)(CN)6]·2H2O showed ferromagnetism (Tc = 90 K).2 His group has contributed to the increase of the Tc of those systems, and has found a high Tc ferromagnet; VII0.42VIII0.58[CrIII(CN)6]0.86·2.8H2O (Tc = 315 K).3 In addition, Sato et al. revealed that the magnetic properties of K0.2Co1.4[Fe(CN)6]·6.9H2O change from paramagnetic to ferromagnetic by the irradiation of a 450 nm light, suggesting that the long-range magnetic order can be controlled by external stimuli.5 Photo-irradiation to those systems often causes inter valence charge transfers (IVCT), and it sometimes changes the spin structure, which is called a charge transfer induced spin transition (CTIST).6 These phenomena, as well as light induced spin transition and light induced excited spin state trapping (LIESST), are sometimes utilized for optical switching.7 Therefore, PBAs have attracted wide attention from both fundamental science and applied materials.8,9 On the other hand, cuban- or square-type hexacyanide complexes can be regarded as part of the PBA family of compounds. Because of their magnetic properties, these complexes have been considered promising candidates for molecular magnets.10 There have been many reports about single molecular magnets (SMM) and related complexes in the last two decades.6,11–13
In 2010, Oshio and co-workers reported that a PBA-type cyanide-bridged molecular square complex; [Co2Fe2(CN)6(tp*)2(dtbbpy)4](PF6)2·2MeOH (1) (pt* = hydrotris(3,5-dimethylpyrazol-1-yl)borate, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine), shows CTIST by thermal and photo excitations.6 The complex shows thermochromism from dark red at 330 K to dark green at 250 K. Magnetic susceptibility measurements elucidate that the complex 1 is diamagnetic at a low temperature (LT) phase (∼250 K). However the χmT value becomes 6.57 emu mol−1 K at a high temperature (HT) phase (330 K), suggesting that the complex consists of low-spin (LS) FeIII (s = 1/2) and high-spin (HS) CoII (s = 3/2). In an intermediate region between the LT and HT phases (275–310 K), there is a step in the χmT values, indicating the existence of an intermediate phase as illustrated in Fig. S1, ESI.† The X-ray crystallographic analyses at 100, 298 and 330 K reveal that the complex is a square structure consisting of Fe and Co ions. The magnetic and structural results suggest [(FeIILS)2(CoIIILS)2] and [(FeIIILS)2(CoIIHS)2] for the LT and HT phases, respectively. On the other hand, at the intermediate phase, there are still two possibilities for the electronic structure; a one-electron transferred [FeIILSFeIIILSCoIIHSCoIIILS] state or a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of [(FeIIILS)2(CoIIHS)2] and [(FeIILS)2(CoIIILS)2]. At the LS phase, the complex has a broad absorption peak around 770 nm that is considered to be the FeII → CoIII IVCT band. Significantly, 808 nm laser irradiation to the complex 1 in the LT phase causes a rapid increase of the χmT values. The results suggest that light-induced CTIST occurs in the LT phase. Although such physical properties have been reported, details about the electronic structures at each temperature, and a relationship between electronic structures and properties have not been elucidated sufficiently.
:1 mixture of [(FeIIILS)2(CoIIHS)2] and [(FeIILS)2(CoIIILS)2]. At the LS phase, the complex has a broad absorption peak around 770 nm that is considered to be the FeII → CoIII IVCT band. Significantly, 808 nm laser irradiation to the complex 1 in the LT phase causes a rapid increase of the χmT values. The results suggest that light-induced CTIST occurs in the LT phase. Although such physical properties have been reported, details about the electronic structures at each temperature, and a relationship between electronic structures and properties have not been elucidated sufficiently.
On the other hand, recent progress in computing power and density functional theory (DFT) has allowed the first principle calculations of electronic structures for real large molecules, so that a direct estimation of the physical properties becomes feasible. In addition, there have been advances in a computational scheme to obtain the J values of diradical or polyradical species to explain the magnetic behaviors.14–16 In this sense, the DFT method is a powerful tool for the analysis of the electronic structures and physical properties of the complex 1. Up to now, there have been several reports about theoretical calculations on PBAs especially for magnetic properties, however the computational models are still insufficient for the requirements of considering the properties of whole molecules.14
For the above reasons, in this paper, we perform DFT calculations to elucidate the details of the electronic structures and the relationship between electronic structures and magnetic properties of the complex 1, with sufficient model structures including all metal ions. The electronic and spin structures are calculated at low, middle and high temperature structures, and the J values between metal ions are obtained for the high temperature structure. In addition, the one-electron excitation state of the complex 1 is also examined to verify the FeII → CoIII IVCT at the low temperature structure.
2. Computational details
2.1 Construction of model structures and calculated charge and spin states
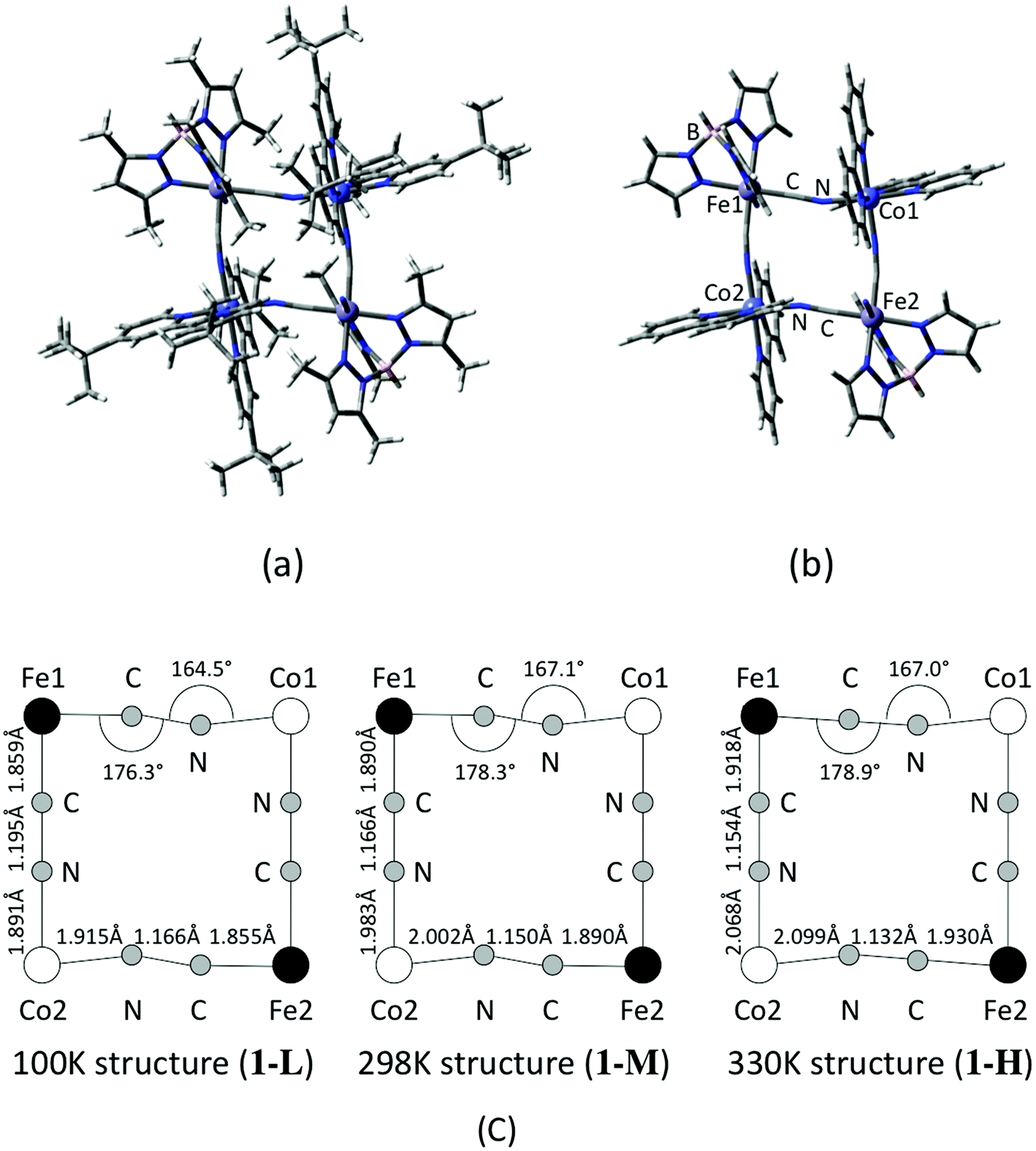
Based on the coordinate of the complex 1 measured by X-ray crystallography analyses, model structures for the DFT calculations are constructed. In order to consider a structural change with temperature, molecular geometries are taken from the data measured at three different temperatures that correspond to low, intermediate and high temperature phases, namely 100 K, 298 K and 330 K.17 A contribution of the substitution groups of the ligands is also examined by using full and reduced model structures. The full model consists of a whole structure of the complex 1, except for counter ions and MeOH in the crystal as illustrated in Fig. 1(a). On the other hand, in the case of the reduced model structure, the tert-butyl and methyl groups of dtbbpy and tp* ligands are substituted for hydrogen atoms as shown in Fig. 1(b). The model structures of the complex 1 in low (100 K), middle (intermediate) (298 K) and high (330 K) temperature phases are defined as 1-L, 1-M and 1-H, respectively, and their important structural parameters are summarized in Fig. 1(c). Unless otherwise noted, those abbreviations indicate the reduced models, and the full model is specified in the text.
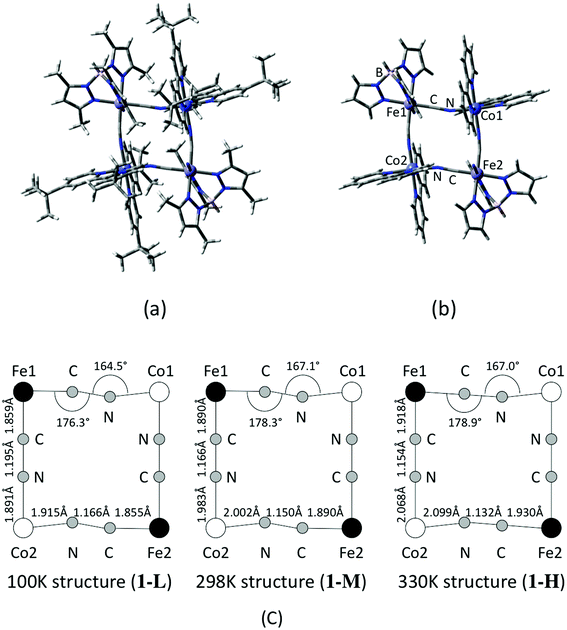 |
| | Fig. 1 Calculated model structures of complex 1. (a) Full model and (b) reduced model structures. (c) Structural parameters measured at different temperatures. | |
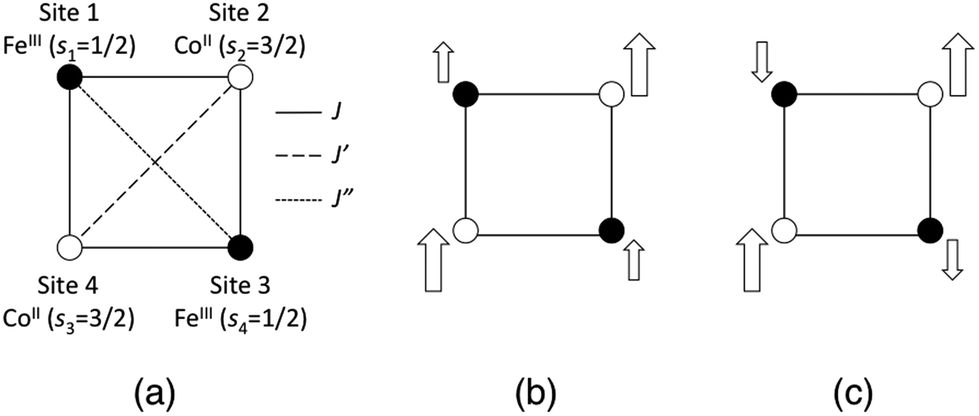
In addition to the geometric differences with the temperature, we examined two types of charge and spin states, namely [(FeIILS)2(CoIIILS)2] and [(FeIIILS)2(CoIIHS)2], for each temperature model structure. The [FeIILSFeIIILSCoIIHSCoIIILS] state is also calculated for the 1-M model structure. Because [(FeIIILS)2(CoIIHS)2] and [FeIILSFeIIILSCoIIHSCoIIILS] states have open-shell spin structures that contain FeIIILS (s = 1/2) and CoIIHS (s = 3/2) ions, both spin-coupling structures of the ferromagnetic and anti-ferromagnetic states are obtained as explained below. Note that the word “anti-ferromagnetic” used here should really be “ferri-magnetic”, however we use this expression to focus on the locally adjacent interactions. The [(FeIIILS)2(CoIIHS)2] state possesses three intra-molecular magnetic interactions (J, J′ and J′′) as illustrated in Fig. 2(a). Consequently there are five possible spin-coupling states as illustrated in Fig. S2, ESI.† Here we examine two of them as illustrated in Fig. 2(b) and (c). The spin-coupling state in Fig. 2(b) is where all adjacent spin–spin interactions are ferromagnetic (SZ = 4), while the state in Fig. 2(c) is anti-ferromagnetic (SZ = 2). Information about these spin-coupling structures (diamagnetic (D), ferromagnetic (F) or anti-ferromagnetic (AF)) is added to the abbreviations of the model structures by subscripts (see Table 1). The spin-coupling states between FeIIILS and CoIIHS in [FeIILSFeIIILSCoIIHSCoIIILS], which has a single intra-molecular magnetic interaction, are also defined as partial ferromagnetic (PF) (SZ = 2) and partial anti-ferromagnetic (PAF) (SZ = 1) interactions here. From these points of view, we carried out a total of 12 single-point calculations as summarized in Table 1.
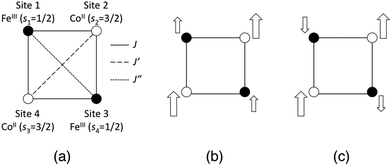 |
| | Fig. 2 (a) Illustrations of magnetic interactions between spin sites in the open-shell [(FeIIILS)2(CoIIHS)2] state. (b) The ferromagnetic (F) state. (c) The anti-ferromagnetic (AF) state. The size of the arrow expresses the size of the spin magnitude at each spin site. | |
Table 1 Classifications and abbreviations of calculated models by structure, charge and spin state
| Model size |
Charge state |
Spin state and magnetic interaction |
Temperature of X-ray crystallography |
| 100 K |
298 K |
330 K |
|
An abbreviation meaning “the reduced model” is omitted here.
|
| Full |
[(FeIILS)2(CoIIILS)2] |
Diamagnetic (D) |
Full-
1-LD
|
— |
— |
| Reduceda |
[(FeIILS)2(CoIIILS)2] |
Diamagnetic (D) |
1-LD
|
1-MD
|
1-HD
|
| [(FeIIILS)2(CoIIHS)2] |
Ferromagnetic (F) |
1-LF
|
1-MF
|
1-HF
|
| Anti-ferromagnetic (AF) |
1-LAF
|
1-MAF
|
1-HAF
|
| [FeIILSFeIIILSCoIIHSCoIIILS] |
Partial ferromagnetic (PF) |
— |
1-MPF
|
— |
| Partial anti-ferromagnetic (PAF) |
— |
1-MPAF
|
— |
2.2 Functional sets and basis sets
The electronic structures of those structural models and charge-spin states are calculated by the use of a hybrid DFT of the Becke-3–Lee–Yang–Parr (B3LYP) functional set on Gaussian 09.18 The basic functions used for all calculations are Huzinaga MIDI plus p-type orbitals for Fe and Co ions, 6-31+G* for cyanide ligands and 6-31G* for other atoms. For the diamagnetic state that consists of the low spin FeII and CoIII ions, a spin-restricted (R) calculation is applied. On the other hand, for the open-shell (F, AF, PF and PAF) states that consist of FeIII and CoII ions, the broken-symmetry (BS) calculation is employed to approximate the static correlation by the quasi-degenerate orbitals and to express localized spins on each metal site.19 We also carry out a time-dependent (TD) B3LYP calculation on 1-LD with the same functional set and basis sets to elucidate orbitals concerning the IVCT that is the electron excitation from FeII to CoIII at the low temperature diamagnetic state. The environmental condition is assumed to be in the gas phase due to the computational costs.
3. Calculated results
3.1 Electronic structure of complex 1 in the low temperature phase
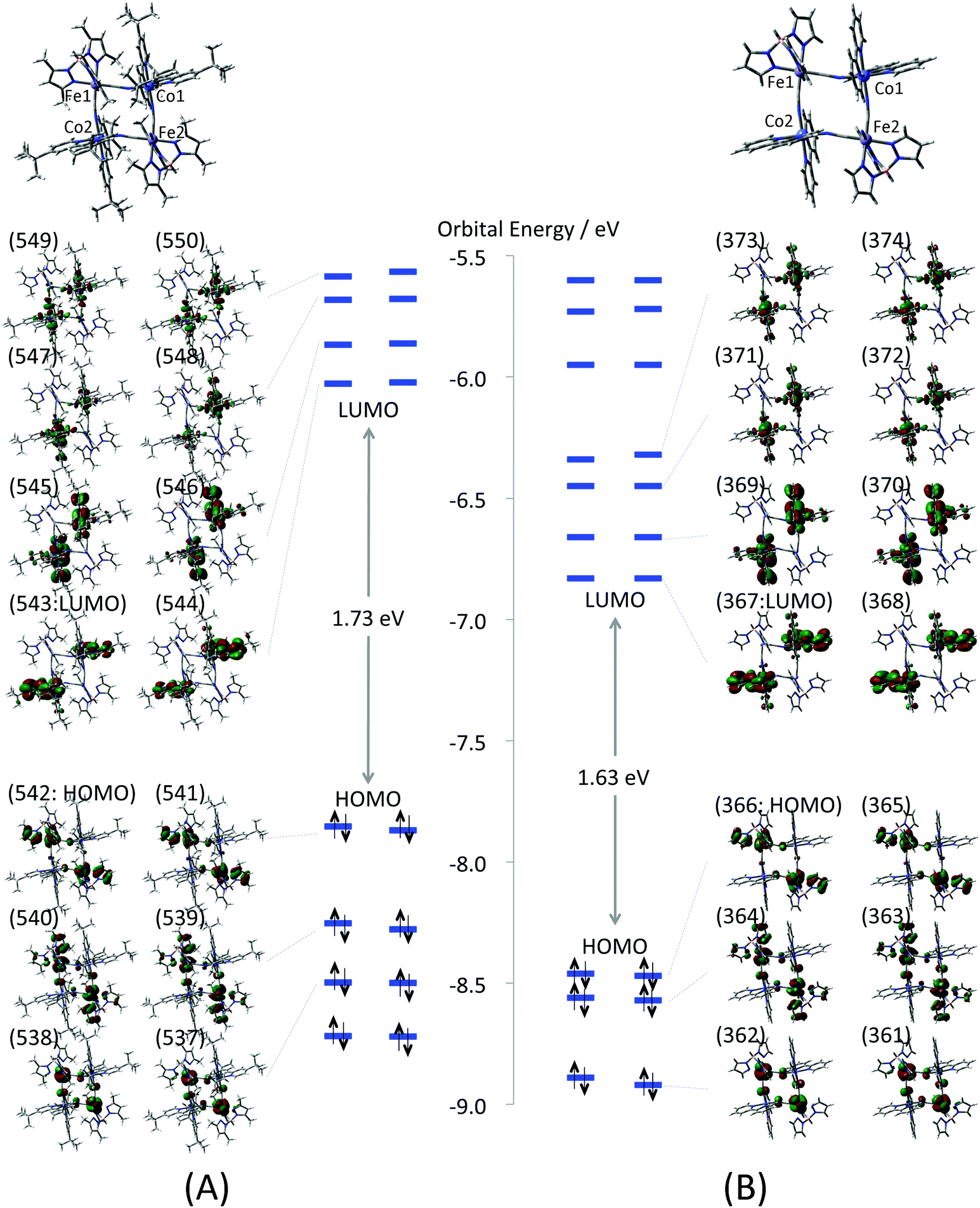
The electronic structure of the complex 1 at the low temperature phase is examined by the model structures 1-LX (X = D, F, AF). As a first step, molecular orbitals (MOs) of the full model (Full-1-LD) are compared with those of the reduced model (1-LD) to elucidate an influence of the tert-butyl and methyl groups of the dtbbpy and the tp* ligands upon the electronic structure. Their calculated MOs around the highest occupied MO (HOMO) and the lowest unoccupied MO (LUMO) are depicted in Fig. 3. All valence electron orbitals are doubly quasi-degenerated reflecting the C2 symmetry of the complex. The orbital shapes of Full-1-LD and 1-LD depicted in the figure are quite similar to each other. HOMO–HOMO−2 of both models mainly consist of Fe t2g orbitals that form π-type anti-bonding orbitals with bridging cyanide and tb* ligands. LUMO–LUMO+3 are localized at the dtbbpy ligands, and unoccupied Co eg orbitals are found in LUMO+4–LUMO+7. Those Co eg orbitals are almost localized at the Co ions but show weak σ-type anti-bonding interactions with tb* ligands.
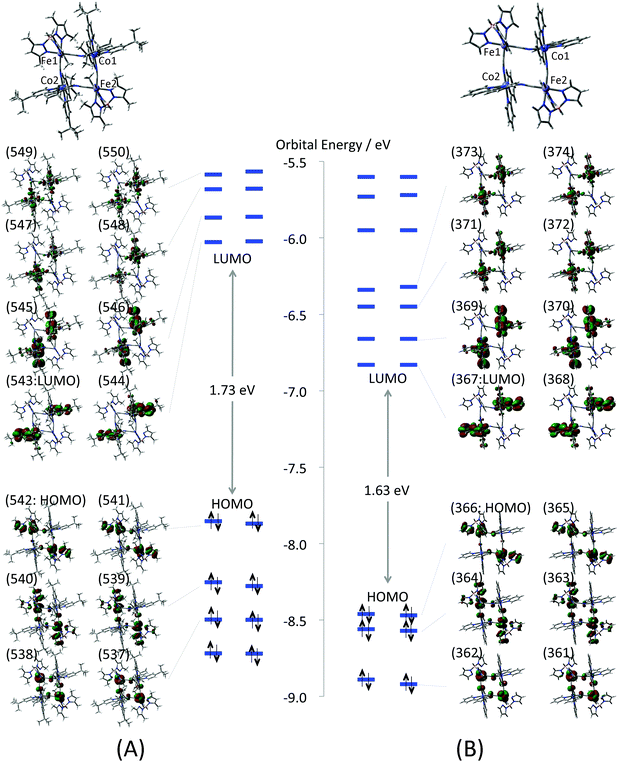 |
| | Fig. 3 Molecular orbitals of (A) full model (Full-1-LD) and (B) reduced model (1-LD). Numbers on the MOs indicate orbital numbers. Enlarged MOs are also summarized in the ESI (Fig. S3†). | |
The frontier orbital energies of 1-LD are generally stabilized about 0.7–0.8 eV in comparison with those of Full-1-LD. However there are no significant differences in orbital energy gaps. For example, the HOMO–LUMO gaps of Full-1-LD and 1-LD are 1.73 and 1.63 eV, respectively. In addition, orbital energy gaps between HOMO (Fe t2g) and unoccupied Co eg orbitals relating to the IVCT from FeII to CoIII are also 2.01 and 2.08 eV on Full-1-LD, 1-LD, respectively. This suggests that the absorption spectrum of 1-LD simulated by TD-DFT qualitatively correspond to that of Full-1-LD. The orbital analysis, in summary, indicates that orbital shapes, configurations and orbital energy gaps of the valence electrons of the full model are consistent with those of the reduced model. In other words, the tert-butyl and methyl groups of the dtbbpy the tp* ligands do not show a significant effect on the valence electron properties but are expected to contribute to the stability of the molecular structure at each temperature. From this point of view, the reduced mode structure is used for the following all calculations.
With the use of the reduced model, we also calculate the electronic structures of the open-shell [(FeIIILS)2(CoIIHS)2] i.e.1-LF and 1-LAF. Their total and relative energies are summarized in Table 2, together with data on 1-LD. The most stable state is 1-LD and the open-shell states are unstable at 2.3–2.4 eV. This result is consistent with the experimental result that the diamagnetic state [(FeIILS)2(CoIIILS)2] is the ground state at the low temperature. On the other hand, the total energy of 1-LF is slightly lower than that of 1-LAF, indicating that the magnetic interaction between FeIIILS and CoIIHS is ferromagnetic as discussed below.
Table 2 Calculated total and relative energiesa of 1-L. 〈S2〉 values are also summarized in parentheses
| Models/states |
Total energy/atomic unit |
Relative energy/eV |
|
Relative energy = E(X)−E(1-LD), X = 1-LD, 1-LF, 1-LAF.
|
|
1-LD
|
−9234.234844 (0.0000) |
0.00 |
|
1-LF
|
−9234.149165 (20.0798) |
2.33 |
|
1-LAF
|
−9234.147959 (8.0608) |
2.36 |
3.2 Electronic and spin structures of the complex 1 in the high temperature phase
As the next step, the electronic structure of the complex 1 at the high temperature phase is examined by using the X-ray geometry measured at 330 K. From the experimental results, the open-shell [(FeIIILS)2(CoIIHS)2] state is considered to be the ground state so that we examine two spin-coupling states as illustrated in Fig. 2(b) and (c) on the reduced model structure (1-HX (X = F, AF)) by BS-B3LYP. For comparison, we also examine the D state by R-B3LYP.
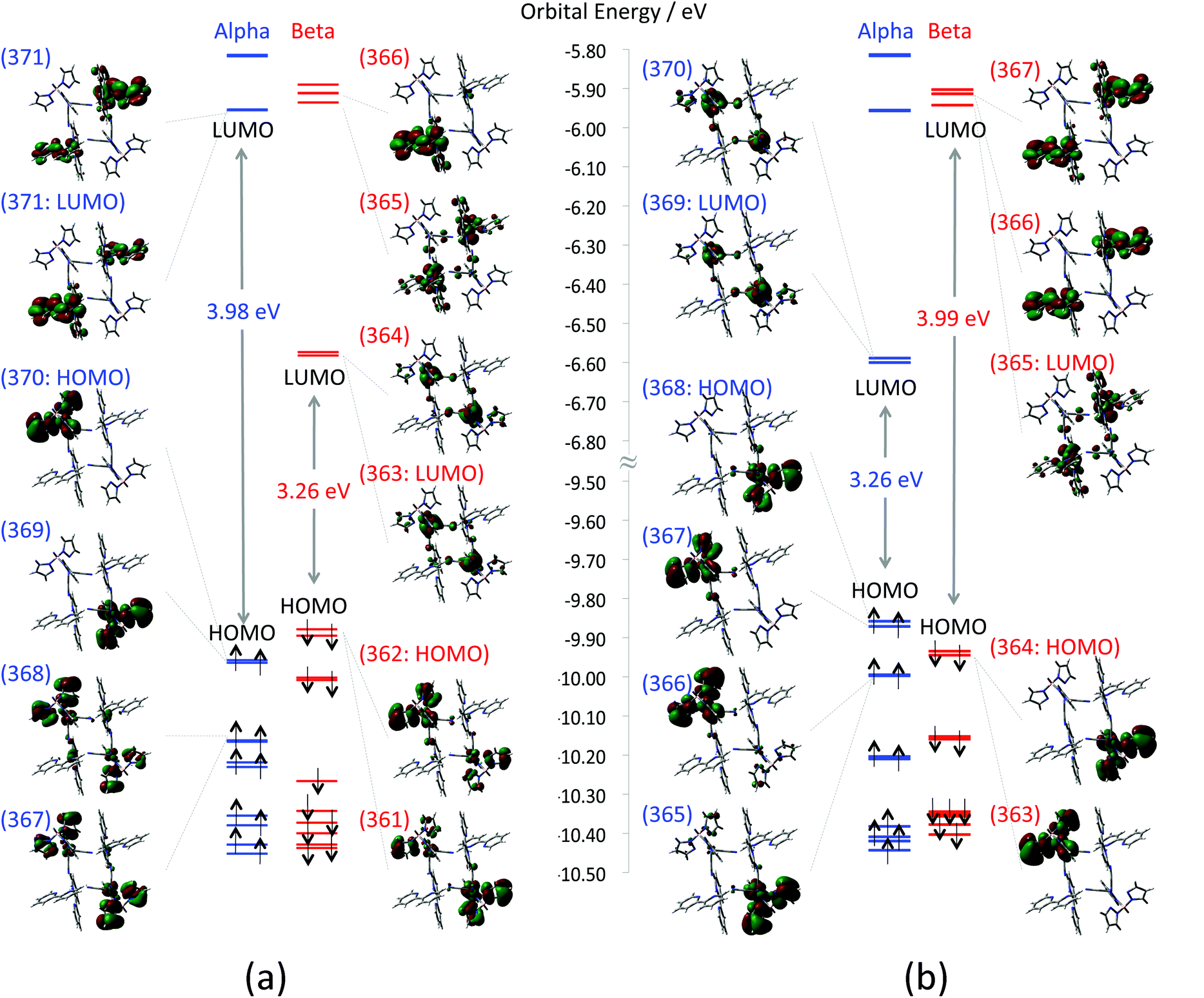
First, calculated MOs around HOMO–SOMO–LUMO of the F (FeIIIs = 1/2; CoIIs = 3/2) and the AF (FeIIIs = −1/2; CoIIs = 3/2) states are depicted in Fig. 4(a) and (b), respectively. In the case of the F state, both α and β HOMOs that are doubly quasi-degenerate consist of anti-bonding orbitals between tp* ligands (dominant contribution) and FeIII t2g orbitals. The doubly quasi-degenerate β LUMOs that consist of FeIII t2g orbitals are located 3.26 eV above the β HOMO. The β LUMO+2 orbital (orbital no. 365 in Fig. 4(a)), which is located 3.92 eV above the β HOMO, consists mainly of CoII eg orbitals. On the other hand, the AF state also has doubly quasi-degenerate HOMOs consisting of anti-bonding orbitals between tp* ligands (dominant contribution) and FeIII t2g orbitals. The unoccupied FeIII t2g orbitals of the AF state appear as α LUMOs, and the unoccupied Co eg orbital is found as a β LUMO.
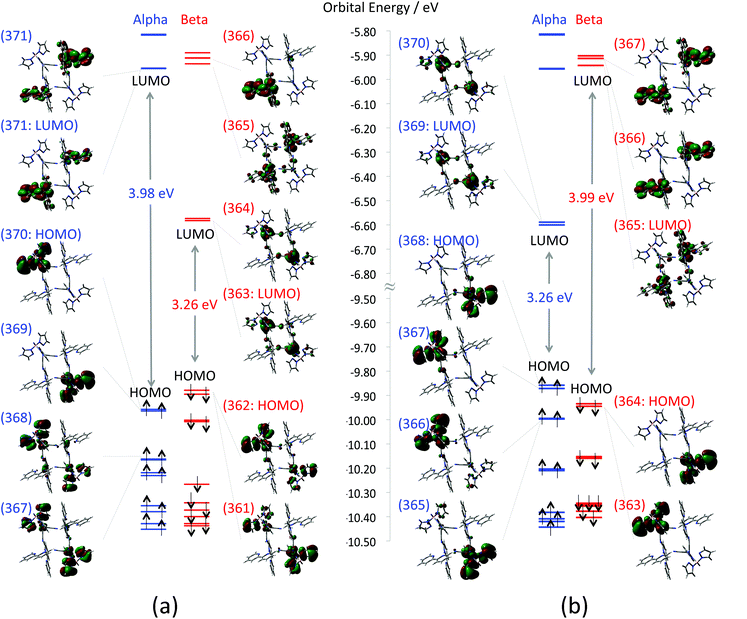 |
| | Fig. 4 Depicted molecular orbitals of (a) ferromagnetic state (1-HF) and (b) anti-ferromagnetic state (1-HAF). Orbital energy levels of α and β orbitals are marked by blue and red lines, respectively. Numbers on the MOs indicate orbital numbers. Enlarged MOs are also summarized in the ESI (Fig. S4†). | |
Next, calculated total energies, 〈S2〉 values, and Mulliken spin densities for the F, the AF and the D states are summarized in Table 2. In contrast to the low temperature models, the open-shell states i.e.1-HF and 1-HAF are stable in comparison with the D state. The energy difference between the most stable 1-HF and the unstable 1-HD is about 4 eV, which is larger in comparison with the low temperature model. The absolute values of spin density 0.95–1.0 and 2.7 on the Fe and Co ions correspond to the spins of FeIIILS and CoIIHS, respectively. The results also indicate that spins are almost localized on each metal ion. The total energy of 1-HF is slightly less stable than that of 1-HAF, suggesting that the interaction between adjacent Co and Fe ions is ferromagnetic also in the high temperature model (Table 3).
Table 3 Calculated total energies, relative energiesa 〈S2〉 values and Mulliken spin densities by BS-B3LYP for F, AF states and by R-B3LYP for D state of 1-H model structures
| |
Models/states |
|
1-HF
|
1-HAF
|
1-HD
|
|
Relative energy = E(X) − E(1-HF), X = 1-HD, 1-HF, 1-HAF.
|
| Energy/atomic unit |
−9234.084338 |
−9234.083755 |
−9233.936248 |
| (Relative energy/eV) |
(0.00) |
(0.02) |
(4.03) |
| 〈S2〉 |
20.0485 |
8.0443 |
0.0000 |
|
Mulliken spin densities
|
| Co (average) |
2.70 |
2.70 |
0.00 |
| Fe (average) |
0.95 |
−1.00 |
0.00 |
| C (bridge) |
0.06 |
0.02 |
0.00 |
| N (bridge) |
0.01 |
0.05 |
0.00 |
3.3 Possible electronic states in the intermediate phase
The calculated results that the ground states of the complex 1 at the low and high temperature models are the D and the F states, respectively, are consistent with experiment. However, the electronic structure in the intermediate phase at the middle temperature is still unclear. Two possibilities are suggested; the one-electron transferred [FeIILSFeIIILSCoIIHSCoIIILS] state and the 1:1 mixture of [(FeIIILS)2(CoIIHS)2] and [(FeIILS)2(CoIIILS2)] from the experimental results. To elucidate the possible states, the total energies of these states: [(FeIIILS)2(CoIIHS)2], [(FeIILS)2(CoIIILS)2] and [FeIILSFeIIILSCoIIHSCoIIILS] are calculated using the 1-M model structure. The results are summarized in Table 4. The most and the second most stable states are the PF and the PAF states of the [FeIILSFeIIILSCoIIHSCoIIILS], and the third one is the ferromagnetic [FeIIILS2CoIIHS2] state. On the other hand, the diamagnetic [(FeIILS)2(CoIIILS)2] state is unstable by more than 1 eV in comparison with the PF and the PAF states. Therefore, a 1:1 mixture of [(FeIIILS)2(CoIIHS)2] and [(FeIILS)2(CoIIILS)2] by averaging their total energies is still unstable in comparison with the 1-MPF state at about 0.75 eV. Based on the relative energies in the table, a population of each state at 298 K is estimated by the Boltzmann distribution. The energy gap of 0.27 eV between 1-MPF and 1-MF is too large for a thermal excitation at 298 K so that the population at the 1-MF and higher states is estimated to be almost zero. From these points of view, the electronic structure at the middle temperature is considered to be the one-electron transferred [FeIILSFeIIILSCoIIHSCoIIILS] state. Nevertheless, calculated results support an asymmetric electronic structure, and the X-ray geometry has a C2 symmetry. Hence, the experimental position of metal ions seems to be the average of FeII and FeIII or CoII and CoIII in the complex 1 in the crystal. So, if a structural asymmetry of the complex is considered, the [FeIILSFeIIILSCoIIHSCoIIILS], i.e. PF and PAF states, will be more stabilized.
Table 4 Calculated total energies, relative energies and estimated populations of each state at 298 K. 〈S2〉 values are also summarized in parentheses
| Models/states |
Total energy/atomic unit |
Relative energy/eV |
Population/% |
|
1-MD
|
−9233.994914 |
(0.0000) |
1.30 |
0.0 |
|
1-MF
|
−9234.035569 |
(20.0573) |
0.20 |
0.0 |
|
1-MAF
|
−9234.032653 |
(8.0478) |
0.27 |
0.0 |
|
1-MPF
|
−9234.042748 |
(7.1891) |
0.00 |
55.0 |
|
1-MPAF
|
−9234.042558 |
(4.1876) |
0.01 |
45.0 |
| Average of 1-MD and 1-MF |
−9234.015241 |
|
0.75 |
|
3.4 One-electron excitation from FeIII to CoII at the low temperature phase
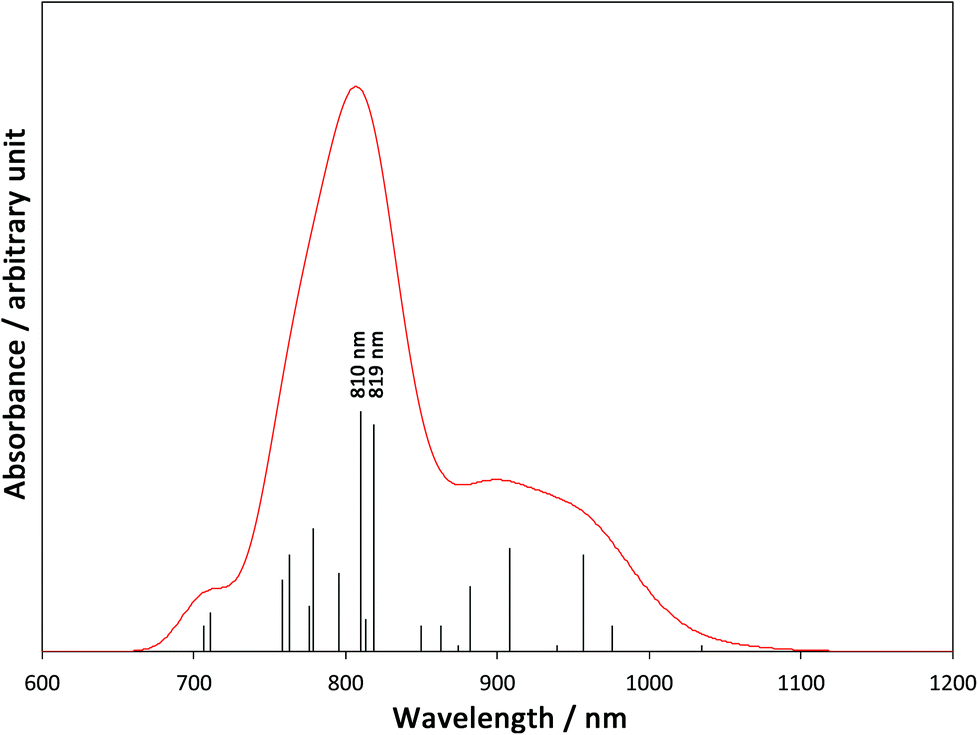
In order to elucidate the mechanism of the IVCT from FeIII to CoII in the diamagnetic [(FeIILS)2(CoIIILS)2] in the low temperature phase, we carried out the TD-B3LYP calculation on the 1-LD. A simulated spectrum is depicted in Fig. 5 and detailed data are summarized in Table S2, ESI.† There are many low excited states with zero or negligible oscillation strengths, so that 45 one-electron excited states are required to survey from the lowest excitation up to 700 nm. As summarized in Table S2,† the obtained oscillation strengths are small and almost all excitations are related to Fe → tp* MLCT. However, there are two significant peaks at 819 nm (ΔE = 1.51 eV) and 810 nm (ΔE = 1.53 eV). The dominant contributions of the peaks are excitations from Fe t2g orbitals to Co eg orbitals, which clearly indicates that those two peaks are related to Fe → Co IVCT. many MLCT excitations around the IVCT peaks will contribute to their broadening. This result explains the very broad experimental peak of IVCT at around 770 nm.
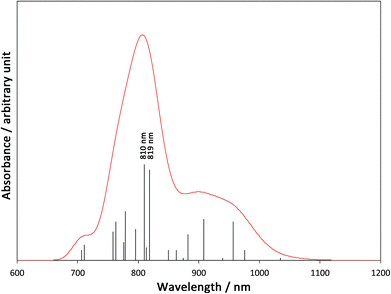 |
| | Fig. 5 A simulated absorption spectrum based on the TD-B3LYP calculation. (Peak width at half height is 0.05 eV). | |
3.5 Calculations of effective exchange coupling constant (J)
In the above sections, we obtain the result that the F state is stable in comparison with the AF state. However there are other possible spin-coupling states in the open-shell [(FeIIILS)2(CoIIHS)2] state. To elucidate the stable spin-coupling state, the effective exchange coupling constant (J) between FeIII and CoII is calculated by the energy gap between the F and the AF states of the open-shell state. The magnetic properties of the polynuclear metal complexes are often discussed by using the Heisenberg Hamiltonianwhere Jij is the orbital-averaged effective exchange integral between the spin site i and j with total spin operators Ŝi and Ŝj, respectively.10,15 Here, we only consider the isotropic spin term. As depicted in Fig. 2(a), there are three types of interactions, i.e. J, J′ and J′′. However, J′ and J′′ are considered to be small enough, so that we only consider the adjacent interaction J. A generalized scheme for calculations of J values is applied to the models. Based on the scheme, the J value between FeIII and CoII is obtained from| | 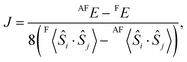 | (2) |
where X〈Ŝi·Ŝj〉 is a spin correlation function of state X (X = F or AF). A detailed derivation of eqn (2) is explained in the ESI.†
All calculated J values summarized in Table 5 are positive, meaning that the magnetic interaction between FeII and CoIII is ferromagnetic. Based on the calculated J values of 1-H, the energy levels of five spin-coupling states in Fig. S2, ESI† are estimated by solving the spin Hamiltonian. The calculated results are summarized in Table S3, ESI.† As expected, the calculated results indicate that the F and the AF states are the most stable and unstable states among the five spin-coupling states, respectively, and other states are intermediate. Similarly, the F state is more stable in comparison with the AF state in other temperature regions. This ferromagnetic interaction in the complex is consistent with other papers14 and our previous report for the cyanide-bridged [Fe–Co] chain.12 We also calculate the J values with the Ising spin model, which considers only the SZ component. The J values of the Ising model that are summarized in Table S3, ESI† thoroughly equal those of the Heisenberg model within a computational convergence. The results show that the spins on the metal sites are fully localized.
Table 5 Calculated J values of Heisenberg spin models
| Models |
J value/cm−1 |
|
1-L
|
22 |
|
1-M
|
53 |
|
1-H
|
11 |
4. Summary
In this study, we elucidate the detailed electronic and spin structures of the complex 1 that shows the CTIST. Calculated MOs of the full and the reduced model structures of the low temperature phase indicate that the shapes and orbital energy gaps of both models are quite similar to each other, suggesting that the substituent groups contribute only to the structural stability but not to the properties of valence electrons. By comparison with the total energies of the model structures, the ground states at LS and HT phases are the diamagnetic [(FeIILS)2(CoIIILS)2] and the ferromagnetic [(FeIIILS)2(CoIIHS)2] states, respectively. At the intermediate phase, the one-electron transferred [FeIILSFeIIILSCoIIHSCoIIILS] state is preferable, while the total energy of a 1:1 mixture of [(FeIIILS)2(CoIIHS)2] and [(FeIILS)2(CoIIILS)2] is too high for the thermal excitation. The calculated J values in the open-shell [(FeIIILS)2(CoIIHS)2] are positive, indicating that the ferromagnetic state is the most stable state among the other possible spin-coupling states. The two significant peaks at 800 nm by TD-B3LYP calculation support the conclusion that the experimental broad peak at 770 nm is the IVCT band.
Acknowledgements
This work has been supported by Grant-in-Aid for Scientific Research (KAKENHI) (C) (no. 26410093) from the Japan Society for the Promotion of Science (JSPS) and Grant-in-Aid for Scientific Research on Innovative Areas (“Coordination programming”, no. 24108721) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). We acknowledge Professor Hiroyuki Nojiri, the Institute for Materials Research (IMR), Tohoku University, for significant discussions. This work was also performed under the Inter-University Cooperative Research Program of the Institute for Materials Research, Tohoku University (proposal no. 14K0008, 15K0118). Finally, one of the authors (YK) acknowledges Toyota Physical and Chemical Research Institute Scholars.
References
- W.-D. Griebler and D. Babel, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1982, 37b, 832 CAS.
- V. Gadet, T. Mallah, I. Castro and M. Verdaguer, J. Am. Chem. Soc., 1992, 114, 9214 CrossRef.
- T. Mallah, S. Thiébaut, M. Verdaguer and P. Veillet, Science, 1993, 262, 1554 CAS.
- S. Ferlay, T. Mallah, R. Ouahès, P. Veillet and M. Verdaguer, Nature, 1995, 378, 701 CrossRef CAS PubMed.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704 CAS.
-
(a) M. Nihei, Y. Sekine, N. Suganami and H. Oshio, Chem. Lett., 2010, 39, 978 CrossRef CAS;
(b) M. Nihei, Y. Sekine, N. Suganami, K. Nakazawa, A. Nakao, H. Nakao, Y. Murakami and H. Oshio, J. Am. Chem. Soc., 2011, 133, 3592 CrossRef CAS PubMed;
(c) Y. Sekine, M. Nihei, R. Kumai, H. Nakao, Y. Murakami and H. Oshio, Chem. Commun., 2014, 50, 4050 RSC;
(d) Y. Sekine, M. Nihei, R. Kumai, H. Nakao, Y. Murakami and H. Oshio, Inorg. Chem. Front., 2014, 1, 540 RSC;
(e) Y. Sekine, M. Nihei and H. Oshio, Chem. Lett., 2014, 43, 1029 CrossRef CAS.
-
(a) P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed., 1994, 33, 2024 CrossRef PubMed;
(b) N. Shimamoto, S. Ohkoshi, O. Sato and K. Hashimoto, Inorg. Chem., 2002, 41, 678 CrossRef CAS PubMed;
(c) A. B. Gaspar, V. Ksenofontov, M. Seredyuk and P. Gütlich, Coord. Chem. Rev., 2005, 249, 2661 CrossRef CAS PubMed.
-
(a) W. E. Bushmann, J. Ensling, P. Gütlich and J. S. Miller, Chem. – Eur. J., 1999, 5, 3019 CrossRef;
(b) W. R. Entley and G. S. Girolami, Inorg. Chem., 1994, 33, 5165 CrossRef CAS;
(c) J. Larionova, R. Clérac, J. Sanchiz, O. Kahn, S. Golhen and L. Ouahab, J. Am. Chem. Soc., 1998, 120, 13088 CrossRef CAS;
(d) E. Coronado, M. C. Giménez-López, G. Levchenko, F. M. Romero, V. Garcia-Baonza, A. Milner and M. Paz-Pastemak, J. Am. Chem. Soc., 2005, 127, 4580 CrossRef CAS PubMed;
(e) W. Kosaka, K. Nomura, K. Hashimoto and S. Ohkoshi, J. Am. Chem. Soc., 2005, 127, 8590 CrossRef CAS PubMed;
(f) S. Ohkoshi, H. Tokoro, T. Matsuda, H. Takahashi, H. Irie and K. Hashimoto, Angew. Chem., Int. Ed., 2007, 46, 3238 CrossRef CAS PubMed.
-
(a) O. Sato, J. Tao and Y.-Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2152 CrossRef CAS PubMed;
(b) A. Bousseksou, G. Molnar, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313 RSC;
(c) A. Bleuzen, V. Marvaud, C. Mathoniere, B. Sieklucka and M. Verdaguer, Inorg. Chem., 2009, 48, 3453 CrossRef CAS PubMed;
(d) J.-D. Cafun, J. Lejeune, J.-P. Itié, F. Baudelet and A. Bleuzen, J. Phys. Chem. C, 2013, 117, 19645 CAS.
-
O. Kahn, Molecular Magnetism, VCH, New York, 1993 Search PubMed.
-
(a) C. P. Berlinguette, A. Dragulescu-Andrasi, A. Sieber, J. R. Galán-Mascarós, H.-U. Güdel, C. Achim and K. R. Dunbar, J. Am. Chem. Soc., 2004, 126, 6222 CrossRef CAS PubMed;
(b) D. Li, R. Clérac, O. Roubeau, E. Harté, C. Mathonière, R. L. Bris and S. M. Holmes, J. Am. Chem. Soc., 2008, 130, 252 CrossRef CAS PubMed;
(c) Y. Zhang, D. Li, R. Clérac, M. Kalisz, C. Mathonière and S. M. Holmes, Angew. Chem., Int. Ed., 2010, 49, 3752 CrossRef CAS PubMed;
(d) D. Li, R. Clérac, O. Roubeau, E. Harté, C. Mathonière, R. Le Bris and S. M. Holmes, J. Am. Chem. Soc., 2008, 130, 252 CrossRef CAS PubMed;
(e) G. N. Newton, M. Nihei and H. Oshio, Eur. J. Inorg. Chem., 2011, 2011, 3031 CrossRef CAS PubMed;
(f) A. Mondal, Y. Li, M. Seuleiman, M. Julve, L. Toupet, M. B.-L. Cointe and R. Lescouëzec, J. Am. Chem. Soc., 2013, 135, 1653 CrossRef CAS PubMed.
-
(a) N. Hoshino, F. Iijima, G. N. Newton, N. Yoshida, T. Shiga, H. Nojiri, A. Nakao, R. Kumai, Y. Murakami and H. Oshio, Nat. Chem., 2012, 4, 921 CrossRef CAS PubMed;
(b) M. L. Baker, Y. Kitagawa, T. Nakamura, K. Tazoe, Y. Narumi, Y. Kotani, F. Iijima, G. N. Newton, M. Okumura, H. Oshio and H. Nojiri, Inorg. Chem., 2013, 52, 13956 CrossRef CAS PubMed.
-
(a) M. Nihei, Y. Okamoto, Y. Sekine, N. Hoshino, T. Shiga, I. P.-C. Liu and H. Oshio, Angew. Chem., Int. Ed., 2012, 51, 6361 CrossRef CAS PubMed;
(b) K. Mitsumoto, E. Oshiro, H. Nishikawa, T. Shiga, Y. Yamamura, K. Saito and H. Oshio, Chem. – Eur. J., 2011, 17, 9612 CrossRef CAS PubMed;
(c) H. Oshio, N. Hoshino, T. Ito and M. Nakano, J. Am. Chem. Soc., 2004, 126, 8805 CrossRef CAS PubMed.
-
(a) M. Nishino, Y. Yoshioka and K. Yamaguchi, Chem. Phys. Lett., 1998, 297, 51 CrossRef CAS;
(b) K. Yoshizawa, F. Mohri, G. Nuspl and T. Yamabe, J. Phys. Chem. B, 1998, 102, 5432 CrossRef CAS;
(c) M. Atanasov, P. Comba and C. A. Daul, J. Phys. Chem., 2006, 110, 13332 CrossRef CAS PubMed;
(d) L. Kabalan, S. F. Matar, C. Desplanches, J. F. Létard and M. Zakhour, Chem. Phys., 2008, 352, 85 CrossRef CAS PubMed.
-
(a) A. P. Ginsberg, J. Am. Chem. Soc., 1980, 102, 111 CrossRef CAS;
(b) L. Noodleman, J. Chem. Phys., 1981, 74, 5737 CrossRef CAS PubMed;
(c) A. Bencini, F. Totti, C. A. Daul, K. Doclo, P. Fantucci and V. Barone, Inorg. Chem., 1997, 36, 5022 CrossRef CAS;
(d) M. Deumal, J. J. Novoa, M. J. Bearpark, P. Celani, M. Olivucci and M. A. Robb, J. Phys. Chem. A, 1998, 102, 8404 CrossRef CAS;
(e) R. Caballol, O. Castell, F. Illas, I. de P. R. Moreira and J. P. Malrieu, J. Phys. Chem. A, 1997, 101, 7860 CrossRef CAS;
(f) E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999, 20, 1391 CrossRef CAS;
(g) S. Vela, M. Deumal, M. Shiga, J. J. Novoa and J. Ribas-Arino, Chem. Sci., 2015, 6, 2371 RSC.
-
(a)
K. Yamaguchi, in Self-Consistent Field Theory and Applications, ed. R. Carbo and M. Klobukowski, Elsevier, Amsterdam, 1990, p. 727 Search PubMed;
(b) S. Yamanaka, T. Kawakami, H. Nagao and K. Yamaguchi, Chem. Phys. Lett., 1994, 231, 25 CrossRef CAS;
(c) T. Soda, Y. Kitagawa, T. Onishi, Y. Takano, Y. Shigeta, H. Nagao, Y. Yoshioka and K. Yamaguchi, Chem. Phys. Lett., 2000, 319, 223 CrossRef CAS;
(d) M. Shoji, K. Koizumi, Y. Kitagawa, T. Kawakami, S. Yamanaka, M. Okumura and K. Yamaguchi, Chem. Phys. Lett., 2006, 432, 343 CrossRef CAS PubMed;
(e) Y. Kitagawa, T. Matsui, N. Yasuda, H. Hatake, T. Kawakami, S. Yamanaka, M. Nihei, M. Okumura, H. Oshio and K. Yamaguchi, Polyhedron, 2013, 66, 97 CrossRef CAS PubMed.
-
(a) Low temperature phase measured at 100 K (CCDC ID: KAFXAS);
(b) Intermediate phase measured at 298 K (CCDC ID: KAFXAS01);
(c) High temperature phase measured at 330 K (CCDC ID: KAFXAS02).
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
-
(a) H. Fukutome, Prog. Theor. Phys., 1972, 47, 1156 CrossRef CAS;
(b)
A. Szabo and N. S. Ostlund, Modern Quantum Chemistry, Dover, New York, NY, 1996, p. 205 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qi00091b |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.