
Synthesis, luminescence and electrochemical properties of luminescent dinuclear mixed-valence gold complexes with alkynyl bridges†
Ling-Ling
Hung
,
Wai Han
Lam
,
Keith Man-Chung
Wong
,
Eddie Chung-Chin
Cheng
,
Nianyong
Zhu
and
Vivian Wing-Wah
Yam
*
Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: wwyam@hku.hk; Fax: +(852)2857-1586; Tel: +(852)2859-2153
First published on 16th March 2015
Abstract
A series of luminescent dinuclear mixed-valence gold alkynyl complexes, [(tBuC^N^CtBu)AuIII(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)AuI(PPh3)], [(RC^N^CR)AuIII(CCCC)AuI(PPh3)] (R = H, tBu), [(RC^N^CR)AuIII(CCC6H4CC)AuI(PPh3)] (R = H, tBu), and [(RC^N^CR)AuIII(CCC6H4CC)AuI(P(C6H4R′-p)3)] (R = H; R′ = OMe, Cl) as well as the precursor complexes, [AuIII(tBuC^N^CtBu)(CCSiMe3)] and [AuIII(tBuC^N^CtBu)(CCH)], have been synthesized and their electrochemical properties have been investigated. The molecular structures of a number of dinuclear mixed-valence gold alkynyl complexes have been determined by X-ray crystallography. For complexes with a butadiynyl bridge, the emission was derived from an admixture of metal-perturbed triplet intraligand (3IL) [π–π* (RC^N^CR)] and [π–π* (CC–CC)] states while for complexes with a 1,4-diethynylbenzene bridge, the emission was assigned as derived from the excited state of the 3IL [π → π* (CCC6H4CC)] origin. Computational studies have also been performed for selected complexes to support the assignment of the spectroscopic origin and the nature of the photophysical properties.
C)AuI(PPh3)], [(RC^N^CR)AuIII(CCCC)AuI(PPh3)] (R = H, tBu), [(RC^N^CR)AuIII(CCC6H4CC)AuI(PPh3)] (R = H, tBu), and [(RC^N^CR)AuIII(CCC6H4CC)AuI(P(C6H4R′-p)3)] (R = H; R′ = OMe, Cl) as well as the precursor complexes, [AuIII(tBuC^N^CtBu)(CCSiMe3)] and [AuIII(tBuC^N^CtBu)(CCH)], have been synthesized and their electrochemical properties have been investigated. The molecular structures of a number of dinuclear mixed-valence gold alkynyl complexes have been determined by X-ray crystallography. For complexes with a butadiynyl bridge, the emission was derived from an admixture of metal-perturbed triplet intraligand (3IL) [π–π* (RC^N^CR)] and [π–π* (CC–CC)] states while for complexes with a 1,4-diethynylbenzene bridge, the emission was assigned as derived from the excited state of the 3IL [π → π* (CCC6H4CC)] origin. Computational studies have also been performed for selected complexes to support the assignment of the spectroscopic origin and the nature of the photophysical properties.
Introduction
During the last decade, there has been increasing interest in the study of transition metal σ-alkynyl complexes.1 Since the linear conjugated alkynyl units may provide the pathways for electronic communication between the metal centers, they can act as precursors for constructing carbon-rich metal-containing materials that may function as molecular wires, nonlinear optical materials and molecular electronics.2–7 It is believed that the photophysical properties of these metal alkynyl complexes can be tuned by varying the nature of the alkynyl bridges as well as the metal centers with various oxidation states, coordination geometries and coordination numbers.In the last decade, the construction of carbon-rich materials using gold(I) alkynyl systems has been an active area of research.8 This is probably due to their interesting aurophilic properties, intriguing photophysical properties as well as their ability to build supramolecular architectures based on the linear two-coordination.9 Recently, alkynylgold(I) complexes have also been explored as useful building blocks for the assembly of multinuclear mixed-metal alkynyl complexes.10 Since [Au(PR3)] is isolobal to H, mixed-metal complexes with σ-bonded alkynylgold(I) could be readily obtained by replacement of terminal hydrogen atoms on various different metal alkynyls with [Au(PR3)].9 It is envisaged that the possible communication between different metal centers might allow the heterometallic complexes to function as molecular wires and non-linear optical devices and the variations in the nature of metal centers and alkynyl ligands would provide an efficient means of tuning the photophysical and photochemical properties of the system.
On the other hand, the chemistry of mixed-valence complexes has attracted growing attention because of their potential to act as models for intramolecular electron transfer11,12 and as prototypes for molecular switches13–16 and wires.5,17–20 The study of complexes containing gold in more than one oxidation state is of particular interest.
Despite numerous reports on mixed-valence gold(I)–gold(III) complexes bearing various types of donor ligands such as thiolate21,22 and selenolate,23,24 examples of luminescent organometallic mixed-valence gold complexes containing alkynyl ligands are extremely rare. It was only recently that Laguna and co-workers reported a series of heterovalent di-, tri- and tetranuclear alkynylphosphino complexes, [AuIII(C6F5)3(PPh2CC)MI(PPh3)] (M = Au, Ag), [{AuIII(C6F5)3(PPh2CC)}2AuI][N(PPh3)2] and [{AuIII(C6F5)3(PPh2CC)AuI}2(μ-PPh2CH2CH2PPh2)].25 All the complexes were found to be emissive in the solid state at low temperatures. However, only the tetranuclear complex showed luminescence in the solid state at room temperature with emission maxima located at 445 and 455 nm, which were assigned to the metal-centered excited state arising from the short gold(I)⋯gold(I) distances. Very recently, the same group described another series of dinuclear mixed-valence gold complexes bearing alkynyl ligands, [AuI{μ-(CH2)2PPh2}2AuIII(CCR)2] (R = Ph, tBu, SiMe3).26
Nevertheless, there has been no report on the study of mixed-valence gold complexes with alkynyl moieties directly bridging the gold metal centers with different oxidation states. We recently reported the synthesis, photophysical, electrochemical and computational studies of a series of luminescent cyclometalated alkynylgold(III) complexes.27,28 Some of the gold(III) complexes have also been observed to show interesting electroluminescence29 and supramolecular assembly properties.30 These, together with our interest in gold(I) alkynyls and mixed-metal alkynyl complexes of gold(I), have prompted us to investigate the systems associated with the incorporation of the cyclometalated gold(III) alkynyl luminophore into the gold(I) phosphine system. Herein the synthesis, characterization, structures, electrochemical, electronic absorption and luminescence properties of a series of dinuclear heterovalent gold alkynyl complexes (Scheme 1) as well as their precursor cyclometalated alkynylgold(III) complexes, [AuIII(tBuC^N^CtBu)(CCSiMe3)] 1 and [AuIII(tBuC^N^CtBu)(CCH)] 2 are reported. It is envisaged that their luminescence properties would be perturbed or varied through the incorporation of two gold centers of different oxidation states to the alkynyl bridges. Through the variation in the nature of the tridentate ligands, the alkynyl ligands and the chlorogold(I) precursors, insights into the photophysical as well as the electrochemical properties of this class of complexes will be described. Density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations have also been performed for the dinuclear mixed-valence gold complexes to provide insights into the nature of the excited states involved in the absorption and emission processes.
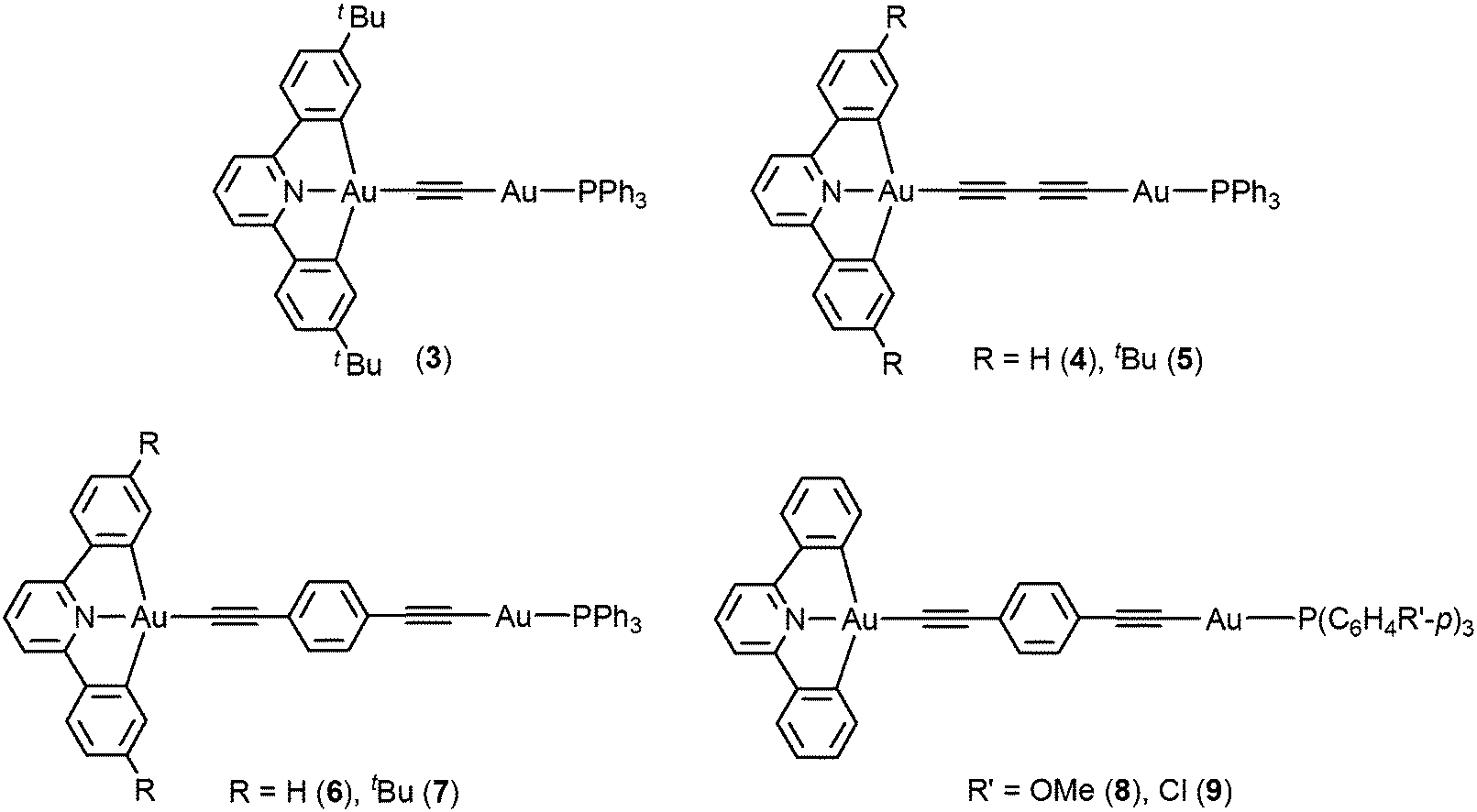 | ||
| Scheme 1 Structure of complexes 3–9. | ||
Results and discussion
Synthesis and characterization
All the dinuclear mixed-valence σ-bonded alkynylgold complexes were prepared by modification of literature procedures for the related heterometallic gold(I)–ruthenium(II) alkynyl complexes,31 in which the replacement of terminal hydrogen atoms on the corresponding alkynylgold(III) complexes with [Au(PR3)] units was achieved by the reaction of the alkynylgold(III) complexes with the chlorogold(I) phosphine precursors in the presence of a base in THF–EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). A mixed THF–EtOH (1:1 v/v) solvent system was employed in order to make the starting materials soluble in the reaction mixture. Recrystallization from slow diffusion of diethyl ether vapor into the concentrated solution of the complex resulted in the isolation of the desired product as yellow crystals or solids in reasonable yields.
:1 v/v). A mixed THF–EtOH (1:1 v/v) solvent system was employed in order to make the starting materials soluble in the reaction mixture. Recrystallization from slow diffusion of diethyl ether vapor into the concentrated solution of the complex resulted in the isolation of the desired product as yellow crystals or solids in reasonable yields.
All the newly synthesized mixed-valence alkynylgold complexes were found to be air- and thermal-stable. They have been characterized by 1H and 31P{1H} NMR spectroscopy, FAB-mass spectrometry, and IR spectroscopy, and all of them gave satisfactory results in elemental analyses. The 31P{1H} NMR spectra of the mixed-valence gold alkynyl complexes showed a singlet at δ 38.6–42.9 ppm, indicative of a single environment for the phosphine ligands. The IR spectra of most of the complexes with the exception of 1–3 showed two weak absorption bands at ca. 2101–2172 cm−1, typical of the ν(CC) stretch of the alkynyl moiety. In the case of complexes 1–3, only one weak band at 2085, 2019 and 2126 cm−1, respectively, was observed. The molecular structures of 5, 6 and 8 have also been determined by a single-crystal X-ray diffraction study.
Crystal structure determination
The perspective views and the crystal packing diagrams of complexes 5, 6 and 8 are depicted in Fig. 1–4, respectively. The crystal structure determination data are summarized in Table 1 and the selected bond distances and bond angles are tabulated in Table 2.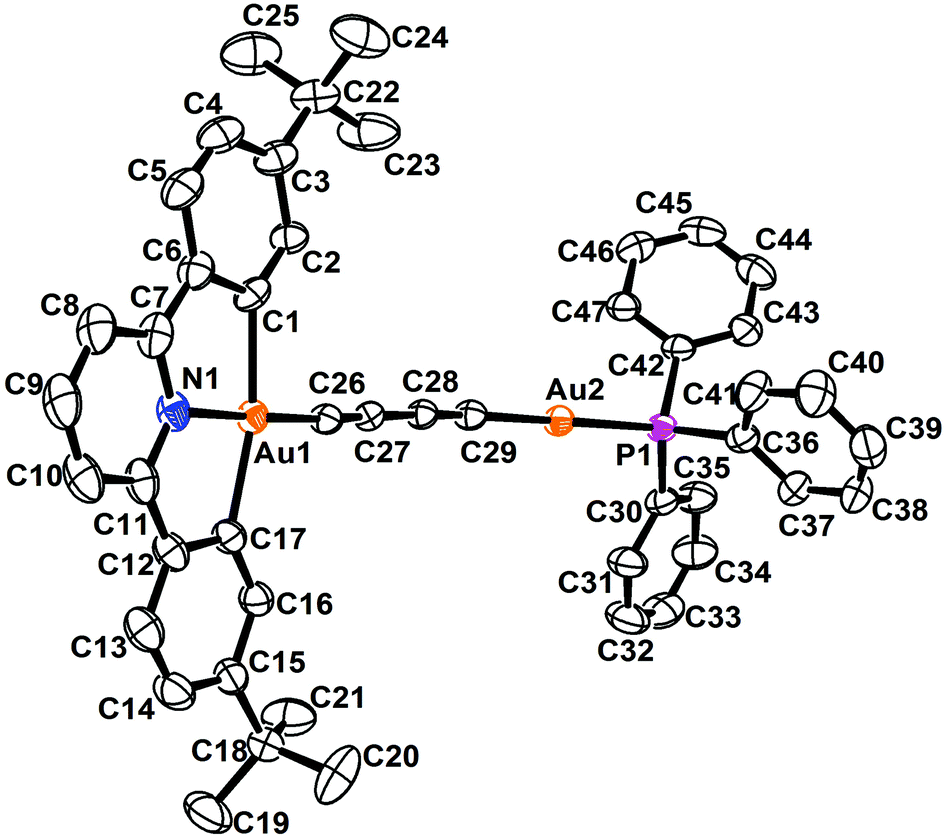 | ||
| Fig. 1 Perspective view of 5 with the atomic numbering scheme. Hydrogen atoms have been omitted for clarity. Thermal ellipsoids are drawn at the 30% probability level. | ||
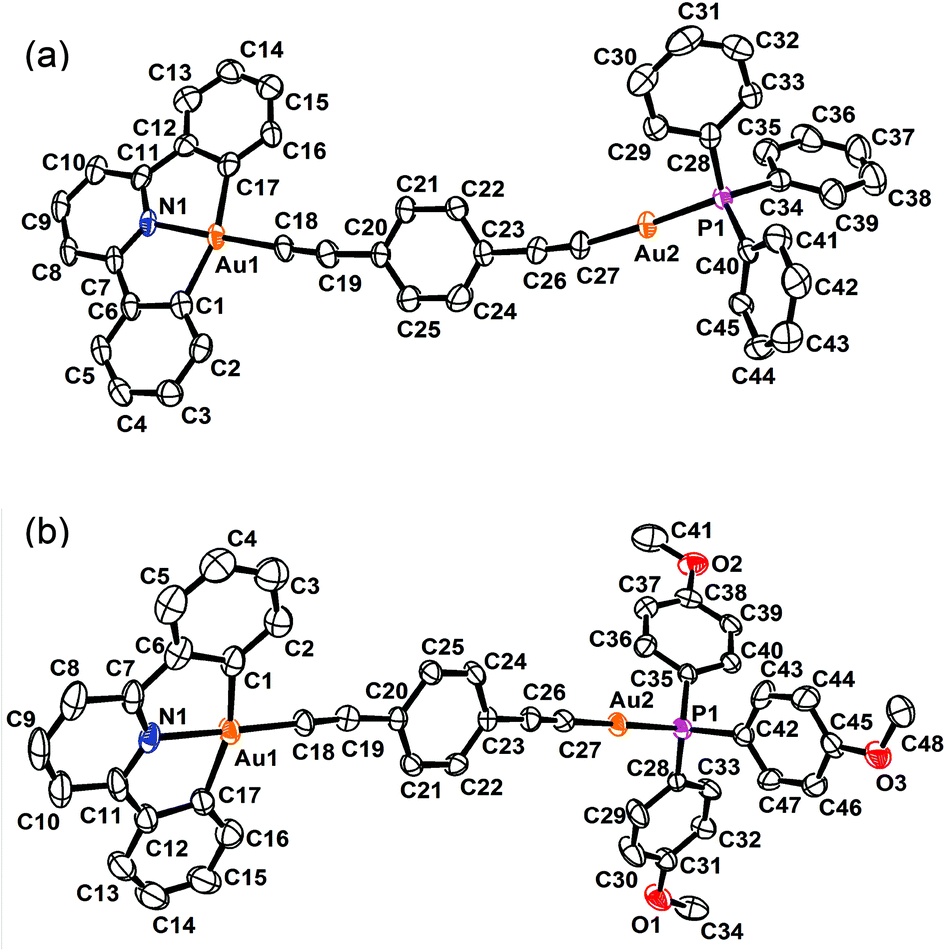 | ||
| Fig. 2 Perspective views of (a) 6 and (b) 8 with the atomic numbering scheme. Hydrogen atoms and solvent molecules have been omitted for clarity. Thermal ellipsoids are drawn at the 30% probability level. | ||
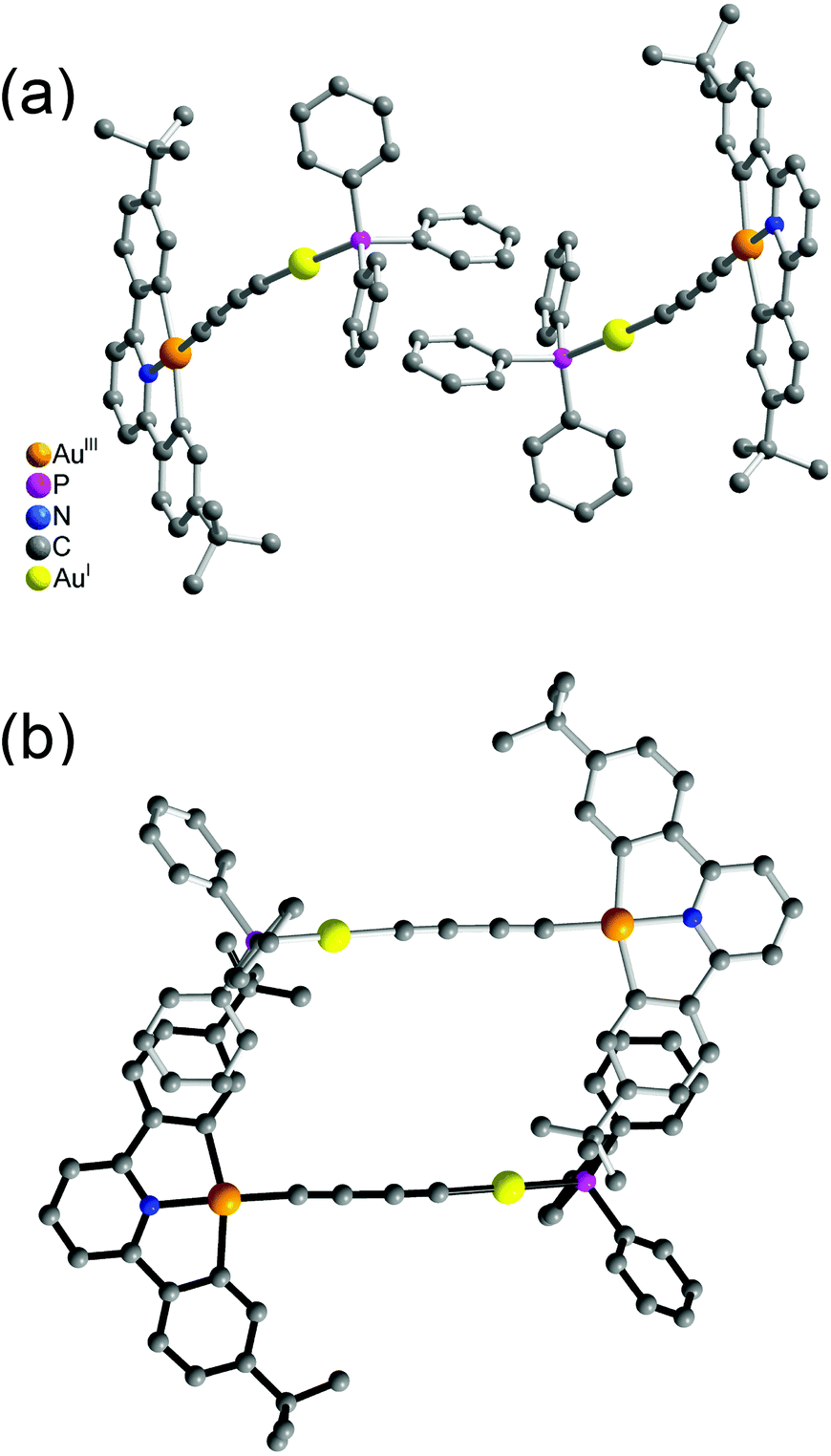 | ||
| Fig. 3 Crystal packing diagram of 5 in different views. | ||
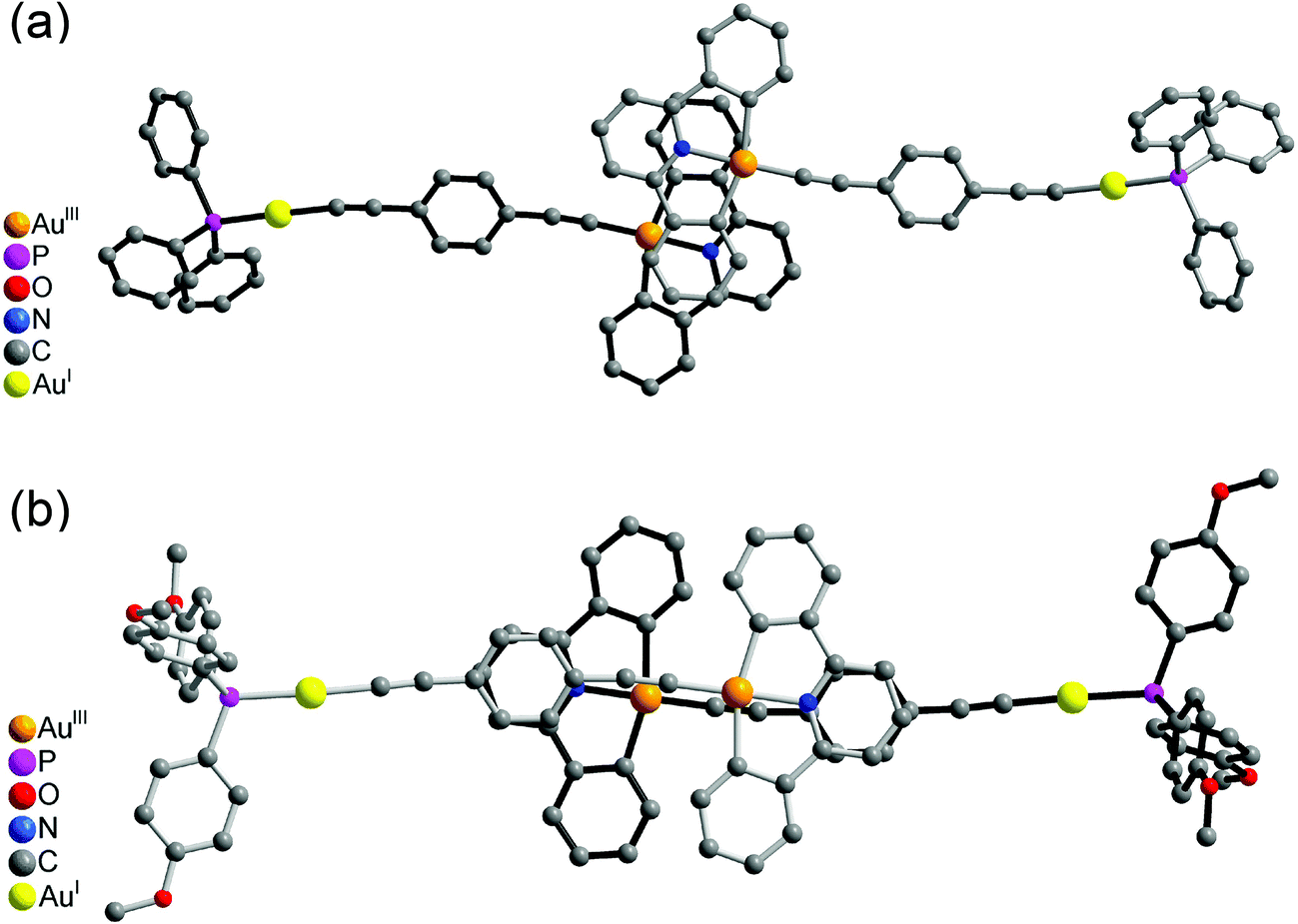 | ||
| Fig. 4 Crystal packing diagrams of (a) 6 and (b) 8, showing the head-to-head configuration. | ||
| Complex | 5 | 6 | 8 |
|---|---|---|---|
| a R int = ∑|Fo2 − Fo2 (mean)|/∑[Fo2], R1 = ∑||Fo| − |Fc||/∑||Fo| and wR2 = {[w(Fo2 − Fc2)2]/[w(Fo2)2]}1/2. | |||
| Empirical formula | C47H42NPAu2 | C47H34Cl4NPAu2 | C48H36NO3PAu2 |
| Formula weight | 1045.72 | 1179.46 | 1099.68 |
| Temperature (K) | 296(1) | 296(1) | 296(1) |
| Wavelength (Å) | 0.71073 | 0.71071 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/n | C2/c |
| a (Å) | 15.403(2) | 9.232(3) | 14.402(3) |
| b (Å) | 16.197(2) | 32.7364(11) | 14.505(3) |
| c (Å) | 17.228(3) | 14.886(5) | 39.229(9) |
| α (°) | 90 | 90 | 90 |
| β (°) | 109.552(2) | 104.784(6) | 100.194(4) |
| γ (°) | 90 | 90 | 90 |
| Volume | 4050.3(10) | 4350(2) | 8065(3) |
| Z (Å3) | 4 | 4 | 8 |
| Density (calculated) (g cm−3) | 1.715 | 1.801 | 1.811 |
| Absorption coefficient (mm−1) | 7.308 | 7.050 | 7.350 |
| F(000) | 2016 | 2256 | 4224 |
| Crystal size (mm × mm × mm) | 0.60 × 0.45 × 0.22 | 0.60 × 0.40 × 0.20 | 0.60 × 0.40 × 0.15 |
| θ range for data collection (°) | 2.7 to 25.0 | 2.6 to 25.0 | 2.7 to 25.0 |
| Index ranges | −18 ≤ h ≤ 13 | −10 ≤ h ≤ 10 | −16 ≤ h ≤ 17 |
| −19 ≤ k ≤ 19 | −38 ≤ k ≤ 27 | −15 ≤ k ≤ 17 | |
| −20 ≤ l ≤ 19 | −17 ≤ l ≤ 17 | −46 ≤ l ≤ 43 | |
| Reflections collected | 23618 |
22196 |
19849 |
| Independent reflections | 7159 | 7623 | 7097 |
| Goodness-of-fit on F2 | 1.02 | 1.16 | 1.15 |
| Final R indicesa [I > 2σ(I)] | R 1 = 0.0340 | R 1 = 0.0637 | R 1 = 0.0564 |
| wR2 = 0.0876 | wR2 = 0.1438 | wR2 = 0.1204 | |
| R indices (all data) | R 1 = 0.0499 | R 1 = 0.0818 | R 1 = 0.0768 |
| wR2 = 0.0967 | wR2 = 0.1522 | wR2 = 0.1280 | |
| Largest diff. peak and hole (e Å−3) | 1.41 and −1.10 | 1.52 and −1.82 | 2.41 and −1.47 |
| 5 | 6 | 8 | |||
|---|---|---|---|---|---|
| Bond lengths (Å) | |||||
| Au(1)–C(26) | 1.961(6) | Au(1)–C(18) | 1.957(13) | Au(1)–C(18) | 1.959(10) |
| Au(1)–N(1) | 2.001(5) | Au(1)–N(1) | 2.012(9) | Au(1)–N(1) | 2.007(8) |
| Au(1)–C(17) | 2.072(6) | Au(1)–C(17) | 2.065(13) | Au(1)–C(17) | 2.078(13) |
| Au(1)–C(1) | 2.084(6) | Au(1)–C(1) | 2.059(14) | Au(1)–C(1) | 2.042(13) |
| Au(2)–C(29) | 1.993(6) | Au(2)–C(27) | 1.995(13) | Au(2)–C(27) | 1.962(11) |
| Au(2)–P(1) | 2.2759(17) | Au(2)–P(1) | 2.283(3) | Au(2)–P(1) | 2.282(3) |
| C(26)–C(27) | 1.194(8) | C(18)–C(19) | 1.199(18) | C(18)–C(19) | 1.204(14) |
| C(28)–C(29) | 1.200(8) | C(26)–C(27) | 1.200(17) | C(26)–C(27) | 1.232(14) |
| Bond angles (°) | |||||
| C(26)–Au(1)–N(1) | 178.4(2) | C(18)–Au(1)–N(1) | 178.3(5) | C(18)–Au(1)–N(1) | 177.7(4) |
| C(17)–Au(1)–C(1) | 162.8(3) | C(1)–Au(1)–C(17) | 162.0(5) | C(1)–Au(1)–C(17) | 162.8(5) |
| N(1)–Au(1)–C(17) | 81.0(2) | N(1)–Au(1)–C(17) | 81.0(5) | N(1)–Au(1)–C(17) | 81.8(5) |
| N(1)–Au(1)–C(1) | 81.9(2) | N(1)–Au(1)–C(1) | 81.0(5) | N(1)–Au(1)–C(1) | 81.1(4) |
| C(27)–C(26)–Au(1) | 176.3(5) | C(19)–C(18)–Au(1) | 175.9(13) | C(19)–C(18)–Au(1) | 177.9(9) |
| C(29)–Au(2)–P(1) | 175.98(19) | C(27)–Au(2)–P(1) | 176.0(4) | C(27)–Au(2)–P(1) | 176.0(3) |
| C(28)–C(29)–Au(2) | 170.2(6) | C(26)–C(27)–Au(2) | 169.9(13) | C(26)–C(27)–Au(2) | 176.4(10) |
| C(26)–C(27)–C(28) | 175.6(7) | C(18)–C(19)–C(20) | 173.4(16) | C(18)–C(19)–C(20) | 173.7(12) |
| C(29)–C(28)–C(27) | 175.5(7) | C(27)–C(26)–C(23) | 173.3(15) | C(27)–C(26)–C(23) | 175.2(11) |
The X-ray crystal structure of complex 5 contained a buta-1,3-diynyl bridge linking the two heterovalent gold atoms. The AuIII–CCCC–AuI fragment was essentially linear, with angles at the gold and carbon atoms between 170.2 and 176.3°. The carbon chains showed alternating short and long C–C bond separations of 1.194, 1.383 and 1.200 Å, corresponding to the conjugated 1,3-diyne (–CCCC–) formulation and were comparable to those found in the buta-1,3-diyne free ligand (1.217 and 1.383 Å).32
On the other hand, the molecular structures of 6 and 8 consisted of a 1,4-diethynylbenzene unit bridging a square-planar-coordinated gold(III) atom and a linearly coordinated gold(I) atom. The dihedral angles between the phenyl ring of the 1,4-diethynylbenzene bridge and the [Au(C^N^C)] plane in 6 and 8 were found to be 23.7 and 18.4°, respectively. The CC bond distances of 1.199–1.232 Å were in the range typical of transition metal alkynyl systems.33–35
Similar to other gold(I) phosphine alkynyl complexes,8,36 the Au(I) coordination geometry in all the structures was virtually linear with a P–AuI–C angle of 176.0° and a AuI–CC angle of 169.9–176.4°, which were slightly deviated from the ideal 180° geometry. The observed AuI–P bond distances of 2.276–2.283 Å and AuI–C bond distances of 1.962–1.995 Å were comparable to those observed in other alkynylgold(I) phosphine systems.37–50 Due to the stronger trans influence of the alkynyl group, the Au–P bond distances were longer than those in the chlorogold(I) phosphine complexes.51,52
Similar to other related cyclometalated gold(III) systems previously reported,33–35,53,54 the coordination geometry around the gold(III) metal center in all the complexes was slightly distorted square-planar. The C–AuIII–C and C–AuIII–N angles of 162.0–162.8° and 81.0–81.9° were found to be less than 180° and 90°, respectively, as required by the bite distance exerted by the steric demand of the chelating RC^N^CR ligands. The AuIII–C (1.957–2.084 Å) and AuIII–N (2.001–2.012 Å) bond distances were comparable to those found in other related cyclometalated gold(III) complexes.33–35,53–55
It was interesting to note that the packing arrangements of the dinuclear mixed-valence gold alkynyl complexes varied from one structure to another (Fig. 3 and 4). For crystals of complexes 6 and 8, two complex molecules were packed into a dimeric structure in a head-to-head fashion. Although the AuI⋯AuI and AuIII⋯AuIII distances are too long to have significant metal⋯metal interactions (the shortest AuIII⋯AuIII distance: 5.0633 Å in 6; 4.3319 Å in 8), considerable π–π interactions were observed within the dimeric unit. In 6, two [Au(CNC)] planes were stacked and shifted laterally with respect to each other, with the interplanar angle and distance of 0.00° and 3.417 Å, respectively. In 8, π–π stacking was observed between one [Au(CNC)] plane and the ring of the 1,4-dialkynylbenzene bridging ligand in the neighboring molecule, with the interplanar angle and distance of 12.446° and 3.522 Å, respectively. In contrast, the [Au(tBuC^N^CtBu)] units in the crystal of complex 5 were found to be arranged in various manners with relatively short intermolecular π–π distances between two phenyl rings of different triphenylphosphine ligands in the neighboring molecules (3.700 Å, Fig. 3(a)) and between the phenyl ring of the [Au(tBuC^N^CtBu)] unit and the phenyl ring of the triphenylphosphine (4.055 Å, Fig. 3(b)). In contrast to most gold(I) compounds where there existed short Au⋯Au intermolecular contacts,38,52,56–64 the shortest AuI⋯AuI and AuIII⋯AuIII distances between adjacent molecules were found to be in the range of 4.3319–10.220 Å. These findings suggested insignificant aurophilic interactions in their crystal lattices, probably due to the presence of the bulky phenyl rings of the phosphine which prevented their close approach.
Electrochemistry
In general, the cyclic voltammograms of the mixed-valence gold alkynyl complexes in dichloromethane (0.1 mol dm−3 nBu4NPF6) showed one quasi-reversible reduction couple at −1.56 to −1.72 V vs. SCE and two irreversible oxidation waves in the ranges of +1.22 to +1.27 V and +1.85 to +1.92 V vs. SCE, with the exception of complexes 1–5 which showed one irreversible oxidation wave only. The cyclic voltammetric data are summarized in Table 3.| Complex | Oxidation Epa/V vs. SCEb | Reduction E1/2/V vs. SCEc |
|---|---|---|
| a In a dichloromethane solution with 0.1 M nBu4NPF6 (TBAH) as the supporting electrolyte at room temperature; scan rate 100 mV s−1. b E pa refers to the anodic peak potential for the irreversible oxidation waves. c E 1/2 = (Epa + Epc)/2; Epa and Epc are anodic and cathodic peak potentials, respectively. | ||
| 1 | +1.75 | −1.69 |
| 2 | +1.76 | −1.66 |
| 3 | +1.85 | −1.72 |
| 4 | +1.91 | −1.56 |
| 5 | +1.88 | −1.65 |
| 6 | +1.22, +1.92 | −1.59 |
| 7 | +1.27, +1.86 | −1.62 |
| 8 | +1.22, +1.85 | −1.57 |
| 9 | +1.24, +1.86 | −1.59 |
On the basis of the similar potentials observed for the reduction couple in complexes consisting of the same C^N^C chelating ligand and the previous electrochemical work on the related cyclometalated gold(III) alkynyl systems,35 the reduction process was assigned as the ligand-centered reduction of the C^N^C ligand. The electron-rich tert-butyl substituents on the tBuC^N^CtBu ligand in complexes 1, 2, 3, 5 and 7 would destabilize the π* orbital, reducing its ease of reduction to give more negative potentials.
In view of the fact that the potentials for the first irreversible anodic waves, which ranged from +1.22 to +1.27 V, were found to be similar to those with the same 1,4-diethynylbenzene bridges, the oxidation wave was assigned as the alkynyl ligand-centered oxidation. The second oxidation wave was probably due to the oxidation of the tridentate RC^N^CR ligand. No correlation was found between the values of the anodic potentials and the nature of the ligands because of their irreversible nature as well as the occurrence of decomposition after the first oxidation.
On the other hand, only one irreversible oxidation wave in the range of +1.75 to +1.91 V was observed in complexes 1–5 containing an ethynyl or buta-1,3-diynyl ligand. Due to the absence of the antibonding interaction between the CC triple bond and the phenyl ring, the ethynyl or buta-1,3-diynyl ligand was less π-donating than the 1,4-diethynylbenzene ligand. The oxidation wave was tentatively assigned to the tridentate RC^N^CR ligand-centered oxidation, probably with mixing of an alkynyl ligand-centered oxidation character. The less positive Epa value found in the tert-butyl-containing complex 5 (+1.88 V) than that in 4 (+1.91 V) was consistent with the assignment. The electron-rich tert-butyl moieties on the tBuC^N^CtBu in 5 would render the π(tBuC^N^CtBu) orbital higher-lying in energy and thus increased the ease of tBuC^N^CtBu ligand-centered oxidation.
Electronic absorption spectroscopy
In general, the electronic absorption spectra of the mixed-valence gold alkynyl complexes showed intense high-energy absorption bands at ca. 254–332 nm, with extinction coefficients on the order of 105–106 dm3 mol−1 cm−1 and moderately intense low-energy vibronic-structured absorption bands at ca. 362–412 nm with extinction coefficients on the order of 104 dm3 mol−1 cm−1. The electronic absorption spectral data in dichloromethane at ambient temperature are summarized in Table 4. With reference to previous electronic absorption studies on the free phosphines (λ = 261 and 281 nm)65 and the free alkynes (λ = 245–280 nm),41 the high-energy bands were tentatively assigned to the intraligand (IL) π → π* transitions of the phosphine and the alkynyl bridges.| Complex | Absorption | Emission | ||
|---|---|---|---|---|
| λ max /nm (ε/dm3 mol−1 cm−1) | Medium (T/K) | λ em/nm (τo/μs) | Φ em | |
|
a In dichloromethane at 298 K.
b The luminescence quantum yield, measured at room temperature using [Ru(bpy)3]2+ as a standard.
c Vibronic-structured emission band.
d In CH2Cl2–EtOH–MeOH (1:40:10 v/v).
e Double exponential decay.
f Not measured.
|
||||
| 1 | 314 (13620), 322 (12435), 376 (4010), 396 (5710), 416 (5470) |
CH2Cl2 (298)c | 486, 516, 556, 595 (<0.1) | 2.3 × 10−3 |
| Solid (298) | 501 (<0.1) | |||
| Solid (77) | 506 (27.6, 139.1)e | |||
| Glass (77)c,d | 480, 515, 550, 590 (189.6) | |||
| 2 | 312 (15685), 322 (13990), 376 (4635), 399 (6920), 416 (6600) |
CH2Cl2 (298)c | 485, 515, 550, 600 (<0.1) | 3.9 × 10−3 |
| Solid (298) | 503 (<0.1) | |||
| Solid (77) | 500 (4.9, 26.4)e | |||
| Glass (77)c,d | 478, 512, 548, 595 (315.0) | |||
| 3 | 264 (51240), 314 (17665), 326 (16690), 372 (3590), 390 (5090), 410 (4280) |
CH2Cl2 (298)c | 483, 512, 556 (<0.1) | 1.5 × 10−3 |
| Solid (298) | 495, 528, 565 (<0.1) | |||
| Solid (77) | 493, 529, 568 (11.4, 89.5)d | |||
| Glass (77)c,d | 477, 511, 548, 592 (210.9) | |||
| 4 | 262 (66680), 284 sh (24620), 314 (14820), 328 (15480), 348 sh (8985), 382 (3775), 402 (2880) |
CH2Cl2 (298)c | 475, 508, 560, 638 (0.1) | —f |
| Solid (298) | 570, 617, 648 (<0.1) | |||
| Solid (77) | 506 (10.0, 126.1)d | |||
| Glass (77)c,d | 471, 504, 539, 582 (121.4) | |||
| 5 | 264 (57860), 286 sh (35445), 314 (21010), 332 (17545), 372 (4300), 392 (5905), 412 (5180) |
CH2Cl2 (298) | 486, 515, 560, 638 (<0.1) | 4.1 × 10−3 |
| Solid (298) | 496, 528, 564, 644 (<0.1) | |||
| Solid (77) | 491, 526, 561 (8.0, 48.3)d | |||
| Glass (77)c,d | 479, 512, 549, 593 (150.5) | |||
| 6 | 268 (40260), 286 (38810), 300 (46030), 312 (50650), 318 (49950), 362 (11850), 380 (9155), 400 (4310) |
CH2Cl2 (298)c | 490, 519 (0.3) | 3.5 × 10−3 |
| Solid (298) | 567, 615 (<0.1) | |||
| Solid (77) | 515 (20.3, 101.2)d | |||
| Glass (77)c,d | 486, 515, 526, 542, 561, 576, 593, 616 (319.3) | |||
| 7 | 262 (59075), 294 (54395), 310 (59450), 320 sh (46310), 372 (10680), 390 (7830), 412 (5540) |
CH2Cl2 (298)c | 487, 517, 552 (0.3) | 7.7 × 10−3 |
| Solid (298) | 493, 527, 560 (<0.1) | |||
| Solid (77) | 492, 521, 557 (11.2, 64.7)d | |||
| Glass (77)c,d | 486, 515, 527, 542, 561, 575, 593, 616 (328.5) | |||
| 8 | 254 (79880), 284 (48770), 300 (51890), 310 (56360), 318 (54845), 362 (12040), 380 (9340), 402 (5080) |
CH2Cl2 (298)c | 488, 518 (0.4) | 1.9 × 10−3 |
| Solid (298) | 563, 610 (<0.1) | |||
| Solid (77) | 499, 535 (17.5, 157.2)d | |||
| Glass (77)c,d | 484, 505, 514, 525, 540, 560, 574, 590, 615 (341) | |||
| 9 | 268 sh (47995), 284(42510), 300 (47900), 312 (53060), 320 (54260), 362 (11695), 380 (8430), 400 (4450) |
CH2Cl2 (298)c | 488, 516 (0.3) | 1.8 × 10−3 |
| Solid (298) | 565, 610 (<0.1) | |||
| Solid (77) | 507, 545 (12.9, 75.2)d | |||
| Glass (77)c,d | 484, 505, 514, 525, 539, 559, 574, 590, 612 (319) | |||
The low-energy absorption bands in the range of ca. 362–412 nm with vibrational progressional spacings of 1302–1372 cm−1, which are typical of the skeletal vibrational progressional frequency of the RC^N^CR ligand, were ascribed to the metal-perturbed IL π → π* transition of the tridentate RC^N^CR ligand with charge transfer character from the phenyl ring to the pyridyl unit. This was further supported by the higher absorption energies of complex 4 and 6 relative to their tert-butyl analogues with the same alkynyl bridges, 5 and 7 respectively. The observation of lower energy metal-perturbed IL π–π* transitions in complexes 1–3, 5 and 7 was ascribed to the better electron-donating properties of the tert-butyl groups on the phenyl rings, which raised the energies of the π orbital localized on the phenyl ring and thus narrowed the π–π* energy separation.
Emission spectroscopy
Photo-excitation at λ ≥ 350 nm of the dinuclear mixed-valence complexes resulted in intense luminescence in both the solid state and in fluid solutions at room and low temperature. The lifetimes of the complexes were found to be in the sub-microsecond to microsecond time-scale, suggestive of the phosphorescence nature of the emission. A summary of the photophysical data in various states is tabulated in Table 4 and some representative emission spectra are depicted in Fig. 5 and 6.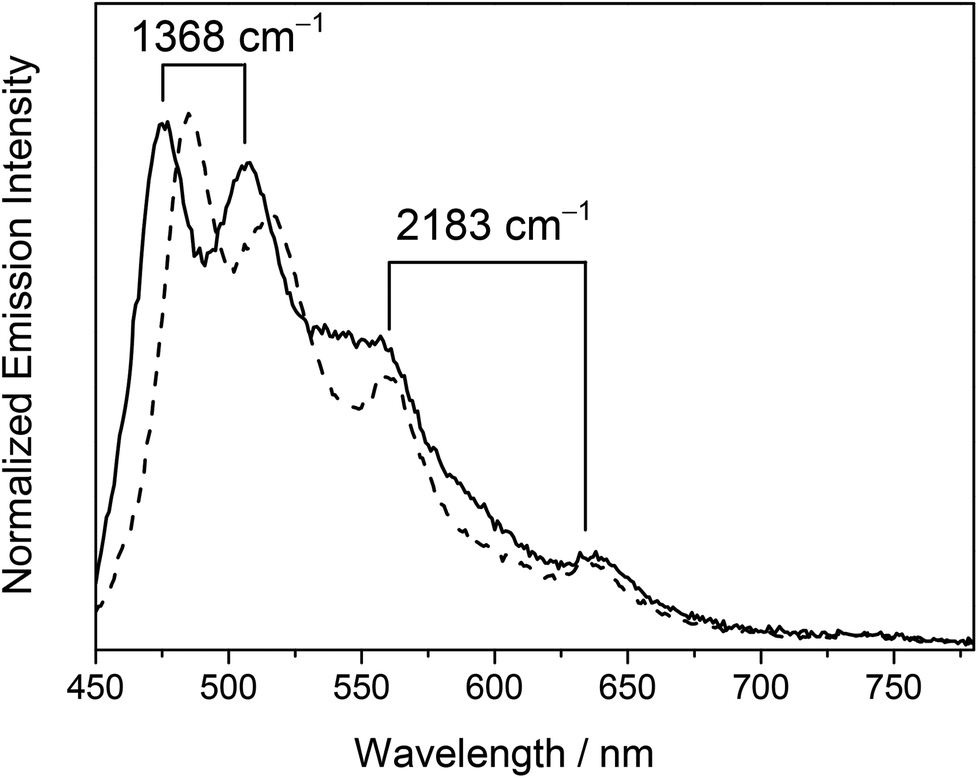 | ||
| Fig. 5 Normalized emission spectra of 4 (––) and 5 (- - -) in dichloromethane at 298 K. | ||
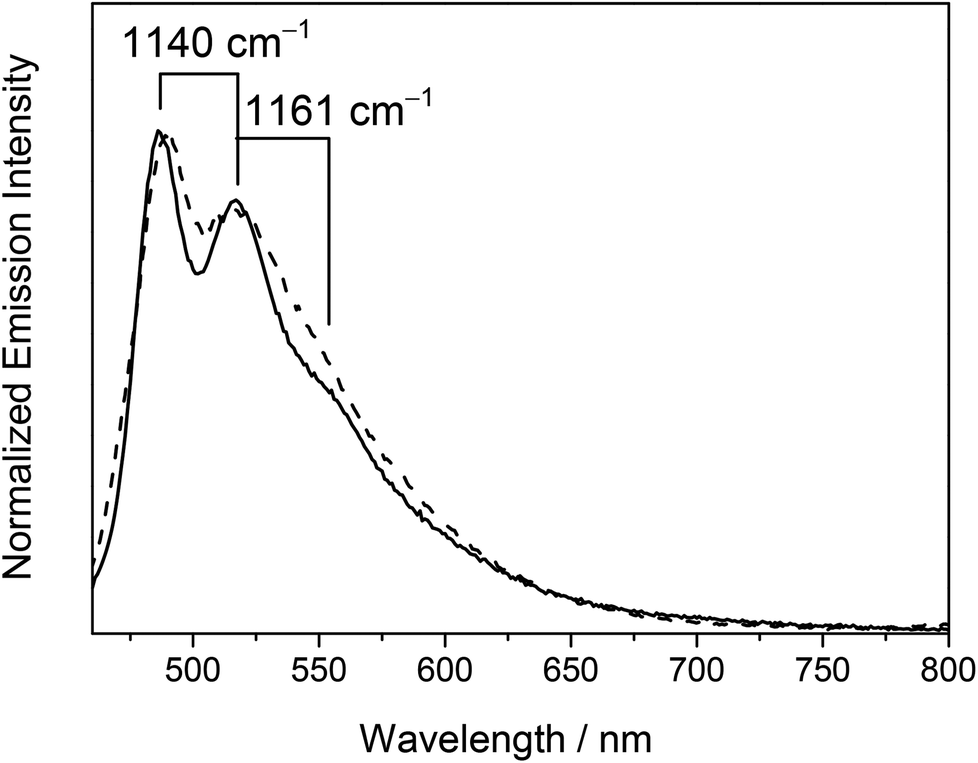 | ||
| Fig. 6 Normalized emission spectra of 6 (- - -) and 7 (––) in dichloromethane at 298 K. | ||
In a dichloromethane solution at room temperature, complexes 1–3 which contained the same tridentate tBuC^N^CtBu ligands exhibited a similar vibronically structured emission band with a band maximum at ca. 485 nm, suggesting that the emission energies of the complexes were rather insensitive to the nature of the alkynyl ligands. The vibrational progressional spacings of ca. 1300 cm−1 were characteristic of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN stretching frequencies of the tridentate ligand, indicative of the participation of the tridentate ligand in the excited state origin. The luminescence was thus assigned as originating from a metal-perturbed 3IL [π → π* (tBuC^N^CtBu)] state.
C and CN stretching frequencies of the tridentate ligand, indicative of the participation of the tridentate ligand in the excited state origin. The luminescence was thus assigned as originating from a metal-perturbed 3IL [π → π* (tBuC^N^CtBu)] state.
Complex 4 showed well-resolved vibronic-structured emission bands with peak maxima at 475, 508 (spacing of 1368 cm−1), 560 and 638 nm (spacing of 2183 cm−1) in a dichloromethane solution while complex 5 displayed vibronic-structured emission bands with peak maxima at 484, 516 (spacing of 1281 cm−1), 560 and 637 nm (spacing of 2159 cm−1). The vibrational progressional spacings of ca. 1300 and 2100 cm−1 were characteristic of the vibrational frequencies of the RC^N^CR ligand and the CC moiety, respectively. The emissions were therefore ascribed as originating from an admixture of the metal-perturbed 3IL [π → π*] state of the tridentate RC^N^CR ligand and the 3IL [π → π*] state of the buta-1,3-diynyl moiety. The higher energy vibronically structured emission bands with vibrational progressional spacings characteristic of the tridentate RC^N^CR ligand and sensitive to the nature of the substituents on the C^N^C ligand were attributed to the emissive state of the metal-perturbed 3IL [π → π* (RC^N^CR)] origin, while the lower energy structured emission bands with vibrational progressional spacings of ca. 2100 cm−1 that occurred at the same energy in complexes 4 and 5 with the same alkynyl bridge were assigned as originating from the 3IL [π → π* (CCCC)] state. Similar to the electronic absorption studies, a red shift in energy for the 3IL [π → π* (RC^N^CR)] emission was observed in the tert-butyl analogue 5 relative to 4. This was consistent with the more electron-rich nature of the tert-butyl groups on the phenyl rings of the tBuC^N^CtBu ligand, which rendered the π orbital of the RC^N^CR ligand higher-lying in energy, leading to a lower 3IL [π → π* (RC^N^CR)] emission energy.
On the other hand, the emission origin of the dinuclear heterovalent gold alkynyl complexes 6–9 was found to be fairly different from that of the previously described complexes. For complexes 6–9 bearing 1,4-diethynylbenzene bridges, a vibronic-structured emission band with a band maximum at ca. 488 nm was observed in dichloromethane at room temperature. The vibrational progressional spacings of ca. 1100 cm−1 were in line with the ground-state phenyl ring deformation, suggesting the participation of the 1,4-diethynylbenzene ligand in the excited state origin. Unlike the related alkynylgold(III) complexes reported previously33–35 where the characteristics of the tridentate C^N^C ligands played an important role in determining the emission energies, the emission energies of 6–9 were found to be independent of the nature of both the tridentate C^N^C ligand and phosphine units as revealed from their identical emission energies. As a consequence, the luminescence was tentatively assigned as originating from the 3IL [π → π*] state of the 1,4-diethynylbenzene ligand. This has further been supported by the computational study (vide infra).
The solid-state emission properties of all the complexes were studied at both room and low temperatures. All the complexes exhibited vibronically structured emission bands with peak maxima at ca. 492 nm with vibronic progression of ca. 1300 cm−1. Consequently, the emission was ascribed to the metal-perturbed 3IL [π → π* (RC^N^CR)] state.
In the low-temperature ethanol–methanol glass, complexes 1–5 with a C2 or C4 ligand exhibited vibronically structured emission bands with vibrational progressional spacings of ca. 1300 cm−1 which correlated with the skeletal vibrational frequency of the tridentate ligand. The emissive state was thus assigned as the 3IL [π → π* (RC^N^CR)] excited state.
On the other hand, the low-temperature emission spectra of 6–9 containing 1,4-diethynylbenzene bridges in the glass state displayed well-resolved highly structured bands with a peak maximum at ca. 485 nm. Three types of vibrational progressional spacings of ca. 1100, 1600 and 2100 cm−1 were identified which were ascribed to the ground-state phenyl ring deformation, symmetric phenyl ring stretch, and CC vibrational stretch respectively. The observed large Stokes’ shifts of the emissions and the long-lived emission with lifetimes in the microsecond regime indicated their triplet parentage. With reference to the findings for [Au(PPh3)(CCPh)]42 and [(Cy3P)Au(CCC6H4)nCCAu(PCy3)]41 (n = 1–4), the emission origin was assigned as derived from the 3IL [π → π*] excited state of the 1,4-diethynylbenzene moiety.
Computational studies
DFT and TDDFT calculations were performed for selected complexes 3–7 to gain further insights into the nature of the electronic transitions in the absorption and emission for the dinuclear mixed-valence gold complexes (see computational details). In general, the calculated bond distances for the ground-state optimized structures of 5 and 6 are in good agreement with those observed in their X-ray crystal structures.For 3, the lowest-energy transition with considerable oscillator strength corresponds to the excitation from the highest-energy π orbital of the C^N^C ligand to the π* orbital localized on the pyridyl unit of the C^N^C ligand, supporting the low-energy absorption band to be attributed to the IL [π → π* (RC^N^CR)] transition with some charge-transfer character from the phenyl rings to the central pyridine unit. Selected singlet–singlet and singlet–triplet transitions of the complexes are listed in Tables S1 and S2 (ESI†), respectively, and the molecular orbitals involved in the transitions are shown in Fig. 7 and Fig. S1–S4 (ESI†). The IL [π → π* (RC^N^CR)] transition is computed to be red-shifted in energy upon going from 4 and 6 (3.36 eV) to 5 and 7 (3.30 eV), which is in agreement with the trend observed in the UV-vis absorption spectra.
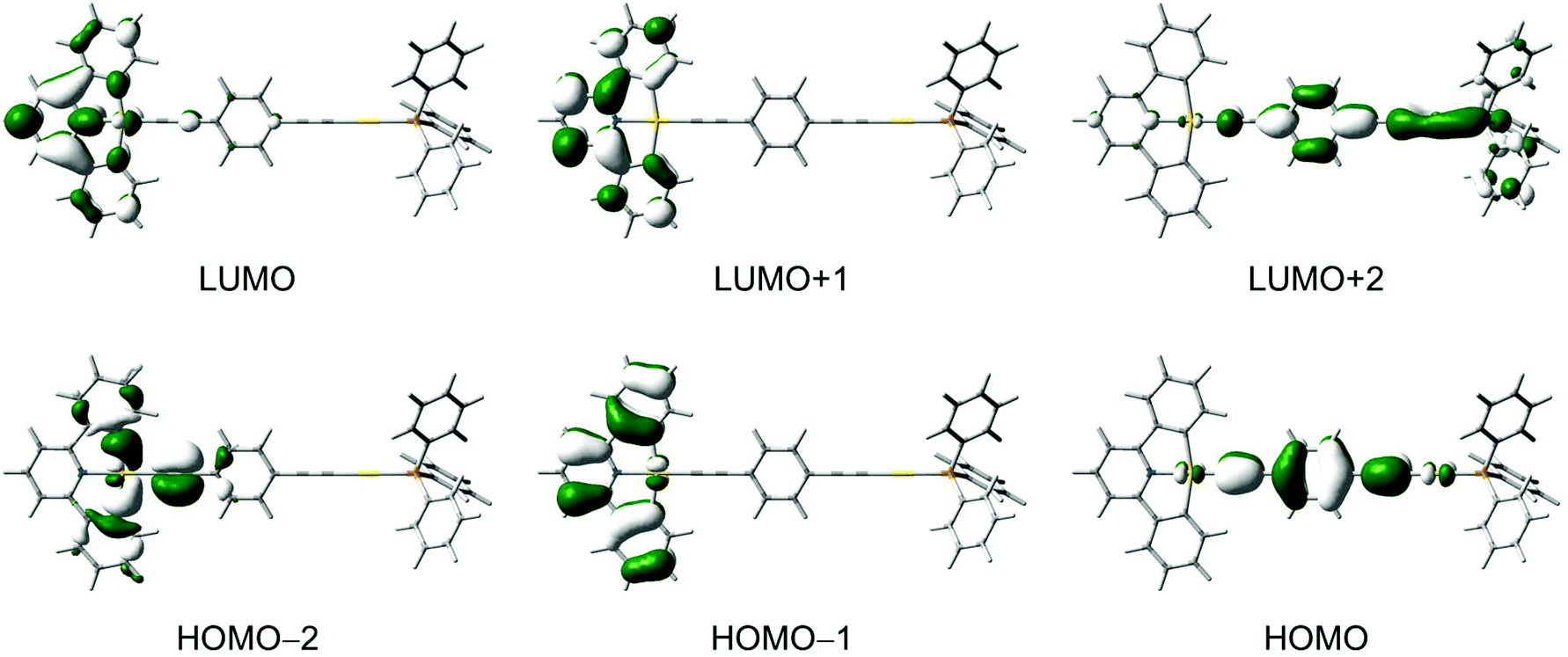 | ||
| Fig. 7 Spatial plots (isovalue = 0.03) of selected molecular orbitals of 6. | ||
An alkynyl-to-terpyridine ligand-to-ligand charge transfer (LLCT) transition is found for 3–7, which lies close in energy to the IL [π → π* (RC^N^CR)] transition. The transition corresponds to the excitation from the π orbital of the alkynyl bridges lying perpendicular to the C^N^C plane to the π* orbital of the C^N^C ligand localized on the pyridine unit. The LLCT transition is computed to be red-shifted in energy upon going from 3 (3.75 eV) to 5 (3.45 eV) to 7 (3.23 eV), which is in line with the π-electron-donating ability of the alkynyl bridge in the order of 1,4-diethynylbenzene > buta-1,3-diynyl > ethynyl.
To gain more insight into the nature of the emission for this class of complexes, the singlet–triplet transitions were computed for complexes 3–7 by using TDDFT/CPCM calculation. For 3–5, the first triplet excited state shows IL(RC^N^CR) character, while it is the 3IL(CCC6H4CC) excited state for 6 and 7. Interestingly, the lowest-energy excited state in 6 and 7 does not correspond to the triplet excited state derived from the HOMO→LUMO excitation, i.e. the alkynyl-to-terpyridine 3LLCT excited state but rather from the 3IL(CC6H4CC) excited state. It should be noted that a simple molecular orbital model does not take into account the interactions between the two unpaired electrons in the singlet or triplet excited states. Singlet excited states are higher in energy than the triplet excited states with the same configuration because of a greater electron–electron repulsion in the former case. The amount of the singlet–triplet energy splitting is a result of the electron exchange integral which is proportional to the spatial overlap of the orbitals involved in the electronic transition. The IL transitions in these complexes will have a larger energy splitting than the LLCT transitions, because the orbitals involved in the former have a larger spatial overlap. The splitting is even larger than the energy difference between the frontier molecular orbitals, which results in the lowest energy excited state being of the IL character.
Geometry optimizations of the lowest-energy triplet excited state for these complexes have been performed. The optimized structures of the triplet excited states for complexes 3–5 show that the distortion occurs mainly in one of the two phenyl units and in the central pyridine ring in the C^N^C ligand, while for 6 and 7, the distortion is localized on the 1,4-diethynylbenzene unit. Fig. 8 shows a plot of the spin densities of the lowest-energy triplet excited states. The relative energies of the lowest-energy triplet excited states with respect to the ground state [ΔE(T1–S0)] are listed in Table S3 (ESI†). A smaller ΔE(T1–S0) is found in 5 when compared with 4, which is in line with a red shift in the 3IL(RC^N^CR) emission upon going from 4 to 5. In addition, a similar ΔE(T1–S0) is calculated for 6 and 7, further supporting the 3IL(CCC6H4CC) origin of the emissive state for the complexes with the 1,4-diethynylbenzene ligand.
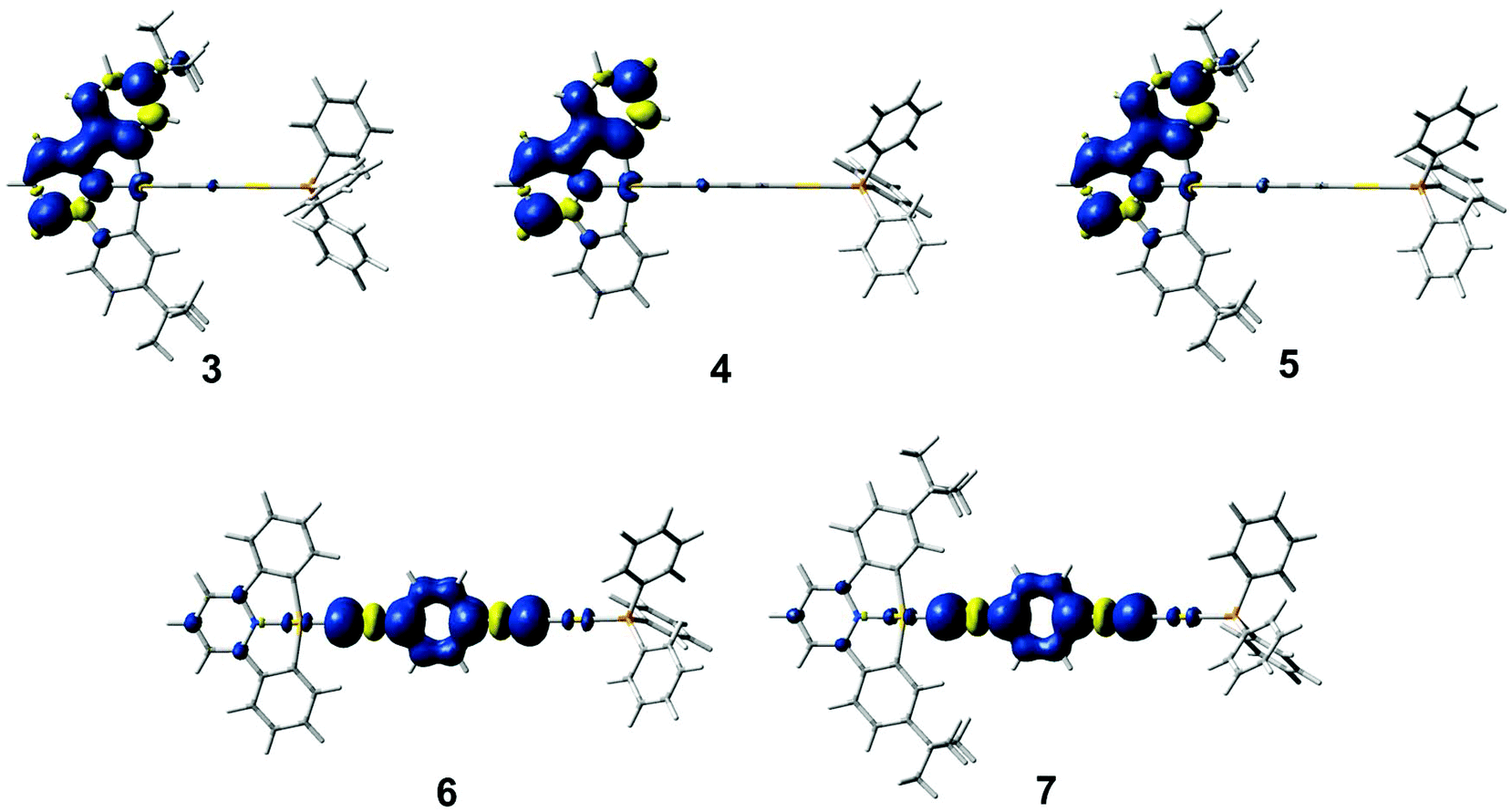 | ||
| Fig. 8 Plots of the spin density (isovalue = 0.002) of the lowest-energy triplet excited state for 3–7. | ||
Conclusion
A new class of luminescent dinuclear mixed-valence gold alkynyl complexes and the precursor complexes have been synthesized and characterized. Some of the X-ray crystal structures have been determined, in which negligible intermolecular π–π interaction between the cyclometalated C^N^C moieties was found in these complexes. The bulky triarylphosphine units were thought to have prevented the molecules from close approach, resulting in the absence of aurophilic interactions. In a dichloromethane solution at ambient temperature, the complexes with a butadiynyl bridge exhibited well-resolved vibronic-structured emission bands with peak maxima at ca. 480, 510 (vibrational progressional spacing of 1368 cm−1), 560 and 638 nm (vibrational progressional spacing of 2183 cm−1). The luminescence was ascribed as originating from an admixture of the metal-perturbed 3IL [π → π* (RC^N^CR)] and 3IL [π → π* (CCCC)] states. Interestingly, in complexes containing the 1,4-diethynylbenzene bridge, a vibronic-structured emission band with a band maximum at ca. 488 nm with vibrational progressional spacings of ca. 1100 cm−1 which was characteristic of the ground-state phenyl ring deformation was observed in the dichloromethane solution at room temperature. The emission energies were found to be insensitive to the nature of both the tridentate C^N^C ligand and phosphine units. The luminescence was thus assigned as originating from the 3IL [π → π*] state of the 1,4-diethynylbenzene moiety. Computational studies have been performed to provide insights into the electronic structures and spectroscopic properties of the complexes, which would offer a better understanding on the effect of the substituents and the nature of the alkynyl bridges on the excited state energy and allow a better design for the tuning of the excited state properties of the dinuclear mixed-valence gold alkynyl complexes.
Experimental section
Materials and reagents
All the chlorogold(I) phosphine precursors, [(R3P)AuCl], were synthesized by modification of a literature method.66 [Au(tBuC^N^CtBu)Cl]35 was synthesized by literature methods reported by our group. Tetrahydrofuran was purified and distilled using standard procedures before use.67 All other solvents were of analytical grade and were used as received. All reactions were carried out under an inert atmosphere of nitrogen using the standard Schlenk technique.Physical measurements and instrumentation
UV-visible spectra were obtained on a Hewlett-Packard 8452A diode array spectrophotometer. IR spectra were obtained using KBr discs on a Bio-Rad FTS-7 Fourier transform infrared spectrophotometer (4000–400 cm−1). 1H NMR spectra were recorded on a Bruker DPX-300 (300 MHz) or Bruker AVANCE 400 (400 MHz) Fourier transform NMR spectrometer with chemical shifts recorded relative to tetramethylsilane (Me4Si). Positive-ion FAB or EI mass spectra were recorded on a Finnigan MAT95 mass spectrometer. Elemental analyses for the metal complexes were performed on a Flash EA 1112 elemental analyzer at the Institute of Chemistry, Chinese Academy of Sciences in Beijing, P. R. China. Steady-state excitation and emission spectra were recorded on a Spex Fluorolog-2 Model F111 or Spex Fluorolog-3 Model FL3-211 fluorescence spectrofluorometer equipped with a Hamamatsu R-928 photomultiplier tube. All solutions for photophysical studies were prepared under high-vacuum in a 10 cm3 round-bottomed flask equipped with a sidearm 1 cm fluorescence cuvette and sealed from the atmosphere using a Rotaflo HP6/6 quick-release Teflon stopper. Solutions were rigorously degassed on a high-vacuum line in a two-compartment cell with no less than four successive freeze–pump–thaw cycles. Solid-state photophysical measurements were carried out with the solid sample loaded in a quartz tube inside a quartz-walled Dewar flask. Liquid nitrogen was placed into a Dewar flask for low temperature (77 K) photophysical measurements. Excited-state lifetimes of solution samples were measured using a conventional laser system. The excitation source used was the 355 nm output (third harmonic, 8 ns) of a Spectra-Physics Quanta-Ray Q-switched GCR-150 pulsed Nd-YAG laser (10 Hz). Luminescence quantum yields were measured by the optical dilution method reported by Demas and Crosby.68 A degassed aqueous solution of [Ru(bpy)3]Cl2 (Φ = 0.042, excitation wavelength at 436 nm) was used as the reference.69 Cyclic voltammetric measurements were performed by using a CH Instruments, Inc. model CHI 620A electrochemical analyzer. The electrolytic cell used was a conventional two-compartment cell. Electrochemical measurements were performed in dichloromethane solutions with 0.1 M nBu4NPF6 (TBAH) as the supporting electrolyte at room temperature. The reference electrode was a Ag/AgNO3 (0.1 M in acetonitrile) electrode, and the working electrode was a glassy carbon electrode (CH Instruments, Inc.) with a platinum wire as the counter electrode. The working electrode surface was first polished with 1 μm alumina slurry (Linde), followed by 0.3 μm alumina slurry, on a microcloth (Buehler Co.). Treatment of the electrode surfaces was as reported previously.70 The ferrocenium/ferrocene couple (FeCp2+/0) was used as the internal reference.71 All solutions for electrochemical studies were deaerated with pre-purified argon gas prior to measurements.Crystal structure determination
Single crystals of 5, 6 and 8 suitable for X-ray diffraction studies were grown by slow diffusion of diethyl ether vapor into a concentrated dichloromethane solution of the complexes. The X-ray diffraction data were collected on a Bruker SMART 1000 CCD detector using graphite monochromatized Mo-Kα radiation (λ = 0.71073 Å). Data were collected using the SMART program.72 The images were interpreted and intensities were integrated using the SAINT program.73 The structure was solved by direct methods employing the SHELXS program.74 Full-matrix least-squares refinement on F2 was used in the structure refinement using the SHELXL program.74 All of the C-bound H atoms were observable from a difference Fourier map but were all placed at geometrical positions with C–H = 0.93, 0.96 and 0.97 Å for phenyl, methyl and methylene H-atoms, respectively. All C-bound H-atoms were refined using a riding model with Uiso(H) = 1.2Ueq(carrier) for phenyl and methylene H-atoms and with Uiso(H) = 1.5Ueq(carrier) for methyl H-atoms. In the final stage of least-squares refinement, all non-hydrogen atoms were refined anisotropically.Synthesis of precursor gold(III) complexes
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CSiMe3)] (1).
A mixture of [Au(tBuC^N^CtBu)Cl] (0.25 g, 0.43 mmol) and (trimethylsilyl)acetylene (0.06 g, 0.65 mmol) in the presence of a catalytic amount of copper(I) iodide (9 mg) in triethylamine (2 mL) and dichloromethane (35 mL) was stirred at room temperature for 3 hours. After evaporation to dryness, the solid residue was purified by column chromatography on silica gel using dichloromethane as the eluent. Subsequent diffusion of n-pentane vapor into a concentrated dichloromethane solution resulted in the formation of deep yellow solids of 1. Yield: 0.22 g (82%). 1H NMR (400 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 0.29 (s, 9H, SiMe3), 1.37 (s, 18H, tBu), 7.30 (dd, J = 8.1 and 2.0 Hz, 2H, phenyl of tBuC^N^CtBu), 7.44 (d, J = 8.0 Hz, 2H, pyridyl of tBuC^N^CtBu), 7.54 (d, J = 8.1 Hz, 2H, phenyl of tBuC^N^CtBu), 7.84 (t, J = 8.0 Hz, 1H, pyridyl of tBuC^N^CtBu), 8.07 (d, J = 2.0 Hz, 2H, phenyl of tBuC^N^CtBu). Positive FAB–MS: m/z 636 [M]+. IR (KBr disc): 2085 cm−1ν(CC). Elemental analyses: found (%): C 56.78, H 5.81, N 2.12. Calcd for C30H36NSiAu: C 56.68, H 5.71, N 2.20.
CH)] (2).
To a THF (50 mL) solution of 1 (0.15 g, 0.24 mmol) was added tetra-n-butylammonium fluoride (1.0 M in THF, 0.35 mL, 0.35 mmol) and the mixture was stirred at 0 °C for one hour and then at room temperature for four hours. After evaporation to dryness, the solid residue was washed with methanol. Subsequent recrystallization from slow diffusion of n-pentane vapor into the concentrated dichloromethane solution gave 2 as yellow crystals. Yield: 0.08 g (63%). 1H NMR (300 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 1.37 (s, 18H, tBu), 2.89 (s, 1H, CCH), 7.30 (dd, J = 8.2 and 2.1 Hz, 2H, phenyl of tBuC^N^CtBu), 7.44 (d, J = 8.0 Hz, 2H, pyridyl of tBuC^N^CtBu), 7.53 (d, J = 8.2 Hz, 2H, phenyl of tBuC^N^CtBu), 7.84 (t, J = 8.0 Hz, 1H, pyridyl of tBuC^N^CtBu), 8.07 (d, J = 2.1 Hz, 2H, phenyl of tBuC^N^CtBu). Positive FAB–MS: m/z 564 [M]+. IR (KBr disc): 2019 cm−1ν(CC). Elemental analyses: found (%): C 57.05, H 5.02, N 2.55. Calcd for C27H28NAu·1/5H2O: C 57.18, H 5.05, N 2.47.
CSiMe3)] (1).
A mixture of [Au(tBuC^N^CtBu)Cl] (0.25 g, 0.43 mmol) and (trimethylsilyl)acetylene (0.06 g, 0.65 mmol) in the presence of a catalytic amount of copper(I) iodide (9 mg) in triethylamine (2 mL) and dichloromethane (35 mL) was stirred at room temperature for 3 hours. After evaporation to dryness, the solid residue was purified by column chromatography on silica gel using dichloromethane as the eluent. Subsequent diffusion of n-pentane vapor into a concentrated dichloromethane solution resulted in the formation of deep yellow solids of 1. Yield: 0.22 g (82%). 1H NMR (400 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 0.29 (s, 9H, SiMe3), 1.37 (s, 18H, tBu), 7.30 (dd, J = 8.1 and 2.0 Hz, 2H, phenyl of tBuC^N^CtBu), 7.44 (d, J = 8.0 Hz, 2H, pyridyl of tBuC^N^CtBu), 7.54 (d, J = 8.1 Hz, 2H, phenyl of tBuC^N^CtBu), 7.84 (t, J = 8.0 Hz, 1H, pyridyl of tBuC^N^CtBu), 8.07 (d, J = 2.0 Hz, 2H, phenyl of tBuC^N^CtBu). Positive FAB–MS: m/z 636 [M]+. IR (KBr disc): 2085 cm−1ν(CC). Elemental analyses: found (%): C 56.78, H 5.81, N 2.12. Calcd for C30H36NSiAu: C 56.68, H 5.71, N 2.20.
CH)] (2).
To a THF (50 mL) solution of 1 (0.15 g, 0.24 mmol) was added tetra-n-butylammonium fluoride (1.0 M in THF, 0.35 mL, 0.35 mmol) and the mixture was stirred at 0 °C for one hour and then at room temperature for four hours. After evaporation to dryness, the solid residue was washed with methanol. Subsequent recrystallization from slow diffusion of n-pentane vapor into the concentrated dichloromethane solution gave 2 as yellow crystals. Yield: 0.08 g (63%). 1H NMR (300 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 1.37 (s, 18H, tBu), 2.89 (s, 1H, CCH), 7.30 (dd, J = 8.2 and 2.1 Hz, 2H, phenyl of tBuC^N^CtBu), 7.44 (d, J = 8.0 Hz, 2H, pyridyl of tBuC^N^CtBu), 7.53 (d, J = 8.2 Hz, 2H, phenyl of tBuC^N^CtBu), 7.84 (t, J = 8.0 Hz, 1H, pyridyl of tBuC^N^CtBu), 8.07 (d, J = 2.1 Hz, 2H, phenyl of tBuC^N^CtBu). Positive FAB–MS: m/z 564 [M]+. IR (KBr disc): 2019 cm−1ν(CC). Elemental analyses: found (%): C 57.05, H 5.02, N 2.55. Calcd for C27H28NAu·1/5H2O: C 57.18, H 5.05, N 2.47.
Synthesis of mixed-valence gold alkynyl complexes
C)AuI(PPh3)] (3).
To a stirred solution of 2 (0.035 g, 0.06 mmol) in the presence of excess NaOH in EtOH–THF (10 mL, 1:1 v/v) was added [(Ph3P)AuCl] (0.029 g, 0.06 mmol), and the mixture was allowed to stir at room temperature for 6 hours. The yellow residue was filtered and washed with water, methanol and diethyl ether. Subsequent recrystallization from slow diffusion of n-pentane vapor into a concentrated dichloromethane solution of the product resulted in the formation of 3 as yellow solids. Yield: 0.032 g (53%). 1H NMR (400 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 1.38 (s, 18H, tBu), (7.29 (dd, J = 8.2 and 2.1 Hz, 2H, phenyl of tBuC^N^CtBu), 7.44 (d, J = 8.0 Hz, 2H, pyridyl of tBuC^N^CtBu), 7.47–7.55 (m, 11H, phenyl of tBuC^N^CtBu and PPh3), 7.61–7.66 (m, 6H, PPh3), 7.82 (t, J = 8.0 Hz, 1H, pyridyl of tBuC^N^CtBu), 8.27(d, J = 2.1 Hz, 2H, phenyl of tBuC^N^CtBu). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K): δ 42.91 (s, PPh3). Positive FAB–MS: m/z 1023 {M}+. IR (KBr disc): 2126 cm−1ν(CC). Elemental analyses: found (%): C 51.73, H 4.25, N 1.54. Calcd for C45H42NPAu2·H2O: C 51.98, H 4.27, N 1.35.
CCC)AuI(PPh3)] (4).
The procedure was similar to that of 3 except that [Au(C^N^C)(CCCCH)] (0.029 g, 0.06 mmol) was used in place of 2 to give 4 as yellow solids. Yield: 0.044 g (79%). 1H NMR (400 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 7.28 (dt, J = 7.3 and 1.2 Hz, 2H, phenyl of C^N^C), 7.40 (dt, J = 7.3 and 1.2 Hz, 2H, phenyl of C^N^C), 7.47–7.62 (m, 19H, PPh3 and C^N^C), 7.90 (t, J = 8.0 Hz, 1H, pyridyl of C^N^C), 7.98 (dd, J = 7.3 and 1.2 Hz, 2H, of C^N^C). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K): δ 41.62 (s, PPh3). Positive FAB–MS: m/z 934 [M]+. IR (KBr disc): 2105 and 2172 cm−1ν(CC). Elemental analyses: found (%): C 49.47, H 2.84, N 1.59. Calcd for C39H26NPAu2·H2O: C 49.23, H 2.97, N 1.47.
CCC)AuI(PPh3)] (5).
The procedure was similar to that of 3 except that [Au(tBuC^N^CtBu)(CCCCH)] (0.036 g, 0.06 mmol) was used in place of 2 to afford 5 as yellow crystals. Yield: 0.035 g (58%). 1H NMR (400 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 1.38 (s, 18H, tBu), 7.29 (dd, J = 8.0 and 2.1 Hz, 2H, phenyl of tBuC^N^CtBu), 7.41 (d, J = 8.0 Hz, 2H, pyridyl of tBuC^N^CtBu), 7.47–7.61 (m, 17H, phenyl of tBuC^N^CtBu and PPh3), 7.83 (t, J = 8.0 Hz, 1H, pyridyl of tBuC^N^CtBu), 8.05 (d, J = 2.1 Hz, 2H, phenyl of tBuC^N^CtBu). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K): δ 41.80 (s, PPh3). Positive FAB–MS: m/z 1046 [M]+. IR (KBr disc): 2101 and 2134 cm−1ν(CC). Elemental analyses: found (%): C 53.33, H 4.01, N 1.44. Calcd for C47H42NPAu2·1/2H2O: C 53.52, H 4.11, N 1.33.
CC6H4CC)AuI(PPh3)] (6).
The procedure was similar to that of 3 except that [Au(C^N^C)(CCC6H4CCH)] (0.034 g, 0.06 mmol) was used in place of 2 to afford 6 as yellow crystals. Yield: 0.029 g (50%). 1H NMR (400 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 7.17 (dt, J = 7.3 and 1.1 Hz, 2H, phenyl of C^N^C), 7.30 (dt, J = 7.3 and 1.1 Hz, 2H, phenyl of C^N^C), 7.38–7.42 (m, 4H, CCC6H4), 7.46–7.60 (m, 19H, PPh3, phenyl and pyridyl of C^N^C), 7.79 (t, J = 8.0 Hz, 1H, pyridyl of C^N^C), 7.90 (dd, J = 7.3 and 1.1 Hz, 2H, phenyl of C^N^C). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K): δ 42.31 (s, PPh3). Positive FAB–MS: m/z 1010 [M]+. IR (KBr disc): 2115 and 2147 cm−1ν(CC). Elemental analyses: found (%): C 53.36, H 2.99, N 1.62. Calcd for C45H30NPAu2: C 53.53, H 2.99, N 1.39.
CC6H4CC)AuI(PPh3)] (7).
The procedure was similar to that of 3 except that [Au(tBuC^N^CtBu)(CCC6H4CCH)] (0.041 g, 0.06 mmol) was used in place of 2 to afford 7 as yellow solids. Yield: 0.035 g (53%). 1H NMR (400 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 1.39 (s, 18H, tBu), 7.32 (dd, J = 8.2 and 2.1 Hz, 2H, phenyl of tBuC^N^CtBu), 7.38 (d, J = 8.2 Hz, 2H, CCC6H4), 7.45–7.62 (m, 21H, PPh3, phenyl and pyridyl of tBuC^N^CtBu), 7.86 (t, J = 8.0 Hz, 1H, pyridyl of tBuC^N^CtBu), 8.16 (d, J = 2.1 Hz, 2H, phenyl of tBuC^N^CtBu). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K): δ 42.29 (s, PPh3). Positive FAB–MS: m/z 1123 [M]+. IR (KBr disc): 2114 and 2153 cm−1ν(CC). Elemental analyses: found (%): C 57.21, H 4.16, N 1.53. Calcd for C53H46NPAu2·1/5C5H12: C 57.08, H 4.29, N 1.23.
CC6H4CC)AuI(P(C6H4OMe-p)3)] (8).
The procedure was similar to that of 3 except that [Au(C^N^C)(CCC6H4CCH)] (0.034 g, 0.06 mmol) and [{(C6H4OMe-p)3P}AuCl] (0.034 g, 0.06 mmol) were used in place of 2 and [(Ph3P)AuCl] respectively to give 8 as yellow crystals. Yield: 0.030 g (46%). 1H NMR (400 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 3.84 (s, 9H, OCH3), 6.99 (dd, J = 8.6 and 1.7 Hz, 6H, –PC6H4–), 7.27 (dt, J = 7.2 and 1.1 Hz, 2H, phenyl of C^N^C), 7.37–7.41 (m, 4H, phenyl of C^N^C and CCC6H4), 7.46–7.53 (m, 10H, pyridyl of C^N^C, CCC6H4 and –PC6H4–), 7.60 (dd, J = 7.2 and 1.1 Hz, 2H, phenyl of C^N^C), 7.89 (t, J = 8.0 Hz, 1H, pyridyl of C^N^C), 8.01 (dd, J = 7.2 and 1.1 Hz, 2H, phenyl of C^N^C). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K): δ 38.57 (s, P(C6H4OMe-p)3). Positive FAB–MS: m/z 1101 [M]+. IR (KBr disc): 2113 and 2149 cm−1ν(CC). Elemental analyses: found (%): C 52.21, H 3.32, N 1.41. Calcd for C48H36NPO3Au2: C 52.38, H 3.27, N 1.27.
CC6H4CC)AuI(P(C6H4Cl-p)3)] (9).
The procedure was similar to that of 3 except that [Au(C^N^C)(CCC6H4CCH)] (0.034 g, 0.06 mmol) and [{(C6H4Cl-p)3P}AuCl] (0.035 g, 0.06 mmol) were used in place of 2 and [(Ph3P)AuCl] to give 9 as yellow solids. Yield: 0.028 g (43%). 1H NMR (400 MHz, CD2Cl2, 298 K, relative to Me4Si): δ 7.27 (dt, J = 7.3 and 1.1 Hz, 2H, phenyl of C^N^C), 7.37–7.39 (m, 4H, phenyl of C^N^C and CCC6H4), 7.47–7.53 (m, 16H, pyridyl of C^N^C, CCC6H4 and –PC6H4–), 7.60 (dd, J = 7.3 and 1.1 Hz, 2H, phenyl of C^N^C), 7.89 (t, J = 8.0 Hz, 1H, pyridyl of C^N^C), 8.00 (dd, J = 7.3 and 1.1 Hz, 2H, phenyl of C^N^C). 31P{1H} NMR (162 MHz, CD2Cl2, 298 K): δ 41.07 (s, P(C6H4Cl-p)3). Positive FAB–MS: m/z 1115 [M]+. IR (KBr disc): 2110 and 2147 cm−1ν(CC). Elemental analyses: found (%): C 49.33, H 2.73, N 1.48. Calcd for C45H27NPCl3Au2·1/2(H5C2OC2H5): C 49.09, H 2.80, N 1.22.
Computational details
Calculations were carried out by using the Gaussian 09 software package.75 The ground state geometries of complexes 3–7 were fully optimized in CH2Cl2 using density functional theory with hybrid Perdew, Burke, and Ernzerhof functional (PBE0)76–78 and the conductor-like polarizable continuum model (CPCM).79,80 On the basis of the ground state optimized geometries, the time-dependent density functional theory (TDDFT) method81–83 at the same level associated with the CPCM in CH2Cl2 was employed to compute singlet–singlet and singlet–triplet transitions for the complexes. To gain more insight into the nature of emissive states, geometry optimizations of the triplet excited states were performed by using the unrestricted UPBE0/CPCM (CH2Cl2) method. Vibrational frequency calculations were performed for all stationary points to verify that each was a minimum on the potential energy surface. While an imaginary frequency was found for the ground-state (4.3 i cm−1) and triplet-excited state (3.0 i cm−1) of 4, it can be considered as a minimum as the imaginary value is very small. For all the calculations, the Stuttgart effective core potentials (ECPs) and the associated basis set were applied to describe Au84 with f-type polarization functions (ζ = 1.05),85 whereas for all other atoms, the 6-31G(d,p) basis set86–88 was used. All DFT calculations were performed with a pruned (99590) grid.
Acknowledgements
V.W.-W.Y. acknowledges support from The University of Hong Kong and the URC Strategic Research Theme on New Materials. This work was supported by the French National Research Agency (ANR)/Research Grants Council (RGC) Joint Research Scheme (A-HKU704/12), and the Research Grants Council Theme-Based Research Scheme (TBRS) (T23-713/11) of the Hong Kong Special Administrative Region. We also thank the Information Technology Services of The University of Hong Kong for providing computational resources. Dr V. K.-M. Au is also acknowledged for discussions and her technical assistance in the preparation of the manuscript, and Dr L. Szeto for her assistance in X-ray crystal structure data collection and determination.References
- V. W.-W. Yam, Acc. Chem. Res., 2002, 35, 555 CrossRef CAS PubMed.
- Materials for nonlinear optics: chemical perspectives, ed. S. R. Marder, J. E. Sohn and G. D. Stucky, ACS Symposium Series, American Chemical Society, Washington, DC, 1991, vol. 455 Search PubMed.
- Inorganic materials, ed. D. W. Bruce and D. O'Hare, John Wiley & Sons Ltd, Chichester, 1996 Search PubMed.
- N. J. Long, Angew. Chem., Int. Ed., 1995, 34, 21 CrossRef CAS.
- F. Paul and C. Lapinte, Coord. Chem. Rev., 1998, 178–180, 431 CrossRef CAS.
- N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2003, 42, 2586 CrossRef CAS PubMed.
- M. G. Humphrey and C. E. Powell, Coord. Chem. Rev., 2004, 248, 725 CrossRef.
- V. W.-W. Yam, K.-L. Cheung, S.-K. Yip and K.-K. Cheung, J. Organomet. Chem., 2003, 681, 196 CrossRef CAS.
- (a) V. W.-W. Yam and E. C.-C. Cheng, Top. Curr. Chem., 2007, 281, 269 CrossRef CAS; (b) V. W.-W. Yam and E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806 RSC.
- V. W.-W. Yam, K.-L. Cheung, E. C.-C. Cheng, N. Zhu and K.-K. Cheung, Dalton Trans., 2003, 1830 CAS.
- T. J. Meyer and H. Taube, Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. Guilard and J. A. Mclafferty, Pergamon Press, Oxford, 1987, vol. 1, ch. 7.2, p. 331 Search PubMed.
- B. N. Gordon, L. L. Williams and N. Sutin, J. Am. Chem. Soc., 1961, 83, 2061 CrossRef CAS.
- D. Astruc, Acc. Chem. Res., 1997, 30, 383 CrossRef CAS.
- P. Steenwinkel, D. M. Grove, N. Veldman, A. L. Spek and G. van Koten, Organometallics, 1998, 17, 5647 CrossRef CAS.
- M. D. Ward, Chem. Soc. Rev., 1995, 121 RSC.
- P. Laine, V. Marvaud, A. Gourdon, J. P. Launay, R. Argazzi and C. A. Bignozzi, Inorg. Chem., 1996, 35, 711 CrossRef CAS.
- F. Paul, W. E. Meyer, L. Toupet, H. J. Jiao, J. A. Gladysz and C. Lapinte, J. Am. Chem. Soc., 2000, 122, 9405 CrossRef CAS.
- S. Rigaut, L. Le Pichon, J. C. Daran, D. Touchard and P. H. Dixneuf, J. Chem. Soc., Chem. Commun., 2001, 1206 RSC.
- N. J. Long, A. J. Martin, R. Vilar, A. J. P. White, D. J. Williams and M. Younus, Organometallics, 1999, 18, 4261 CrossRef CAS.
- S. Le Stang, F. Paul and C. Lapinte, Organometallics, 2000, 19, 1035 CrossRef CAS.
- M. C. Gimeno and A. Laguna, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, New York, 2003, vol. 5, p. 911 Search PubMed.
- H. Schmidbaur, Gold: Progress in Chemistry, Biochemistry and Technology, Wiley, Chichester, U. K., 1999 Search PubMed.
- W. Eikens, C. Kienitz, P. G. Jones and C. Thöne, J. Chem. Soc., Dalton Trans., 1994, 83 RSC.
- S. Canales, O. Crespo, M. C. Gimeno, P. G. Jones and A. Laguna, Inorg. Chem., 2004, 43, 7234 CrossRef CAS PubMed.
- M. Bardají, A. Laguna and P. G. Jones, Organometallics, 2001, 20, 3906 CrossRef.
- L. A. Méndez, J. Jiménez, E. Cerrada, F. Mohr and M. Laguna, J. Am. Chem. Soc., 2005, 127, 852 CrossRef PubMed.
- K. M.-C. Wong, M. M.-Y. Chan and V. W.-W. Yam, Adv. Mater., 2014, 26, 5558 CrossRef CAS PubMed.
- (a) V. K.-M. Au, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Chem. – Eur. J., 2011, 17, 130 CrossRef CAS PubMed; (b) V. K.-M. Au, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 9076 CrossRef CAS PubMed; (c) V. K.-M. Au, W. H. Lam, W.-T. Wong and V. W.-W. Yam, Inorg. Chem., 2012, 51, 7537 CrossRef CAS PubMed.
- (a) V. K.-M. Au, K. M.-C. Wong, D. P.-K. Tsang, M.-Y. Chan, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 14273 CrossRef CAS PubMed; (b) V. K.-M. Au, D. P.-K. Tsang, K. M.-C. Wong, M.-Y. Chan, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2013, 52, 12713 CrossRef CAS PubMed; (c) M.-C. Tang, D. P.-K. Tsang, M. M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 446 CrossRef CAS PubMed; (d) M.-C. Tang, C. K.-M. Chan, D. P.-K. Tsang, Y.-C. Wong, M. M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Chem. – Eur. J., 2014, 20, 15233 CrossRef CAS PubMed.
- (a) V. K.-M. Au, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2013, 52, 558 CrossRef CAS PubMed; (b) K.-C. Yim, E. S.-H. Lam, K. M.-C. Wong, V. K.-M. Au, C.-C. Ko, W. H. Lam and V. W.-W. Yam, Chem. – Eur. J., 2014, 20, 9930 CrossRef CAS PubMed.
- M. I. Bruce, B. C. Hall, B. W. Skelton, M. E. Smith and A. H. White, J. Chem. Soc., Dalton Trans., 2002, 995 RSC.
- M. Tanimoto, K. Kuchitsu and Y. Morino, Bull. Chem. Soc. Jpn., 1971, 44, 386 CrossRef CAS.
- V. W.-W. Yam, K. M.-C. Wong, L.-L. Hung and N. Zhu, Angew. Chem., Int. Ed., 2005, 44, 3107 CrossRef CAS PubMed.
- K. M.-C. Wong, X. Zhu, L.-L. Hung, N. Zhu, V. W.-W. Yam and H.-S. Kwok, Chem. Commun., 2005, 2906 RSC.
- K. M.-C. Wong, L.-L. Hung, W. H. Lam, N. Zhu and V. W. W. Yam, J. Am. Chem. Soc., 2007, 129, 4350 CrossRef CAS PubMed.
- J. Vicente, M. T. Chicote, M. M. Alvarez-Falcón, M. D. Abrisqueta, F. J. Hernández and P. G. Jones, Inorg. Chim. Acta, 2003, 347, 67 CrossRef CAS.
- V. W.-W. Yam, S. W.-K. Choi and K.-K. Cheung, Organometallics, 1996, 15, 1734 CrossRef CAS.
- N. C. Payne, R. Ramachandran and R. J. Puddephatt, Can. J. Chem., 1995, 73, 6 CrossRef CAS.
- G. Jia, R. J. Puddephatt, J. D. Scott and J. J. Vittal, Organometallics, 1993, 12, 3565 CrossRef CAS.
- M. J. Irwin, G. Jia, N. C. Payne and R. J. Puddephatt, Organometallics, 1996, 15, 51 CrossRef CAS.
- H.-Y. Chao, W. Lu, Y. Li, M. C. W. Chan, C.-M. Che, K.-K. Cheung and N. Zhu, J. Am. Chem. Soc., 2002, 124, 14696 CrossRef CAS PubMed.
- D. Li, X. Hong, C.-M. Che, W.-C. Lo and S.-M. Peng, J. Chem. Soc., Dalton Trans., 1993, 2929 RSC.
- J. Vicente, M. T. Chicote, M. D. Abrisqueta, P. G. Jones, M. G. Humphrey, M. P. Cifuentes, M. Samoc and B. Luther-Davies, Organometallics, 2000, 19, 2968 CrossRef CAS.
- J. Vicente, M. T. Chicote, M. D. Abrisqueta and P. G. Jones, Organometallics, 1997, 16, 5628 CrossRef CAS.
- I. R. Whittall, M. G. Humphrey, S. Houbrechts, A. Persoons and D. C. R. Hockless, Organometallics, 1996, 15, 5738 CrossRef CAS.
- T. E. Müller, S. W.-K. Choi, D. M. P. Mingos, D. Murphy, D. J. Williams and V. W.-W. Yam, J. Organomet. Chem., 1994, 484, 209 CrossRef.
- S. J. Shieh, X. Hong, S.-M. Peng and C.-M. Che, J. Chem. Soc., Dalton Trans., 1994, 3067 RSC.
- M. I. Bruce, E. Horn, J. G. Matisons and M. R. Snow, Aust. J. Chem., 1984, 37, 1163 CrossRef CAS.
- M. I. Bruce and D. N. Duffy, Aust. J. Chem., 1986, 39, 1697 CAS.
- M. I. Bruce, K. R. Grundy, M. J. Liddell, M. R. Snow and E. R. T. Tieknik, J. Organomet. Chem., 1988, 344, C49 CrossRef CAS.
- K. Angermaier, E. Zeller and H. Schmidbaur, J. Organomet. Chem., 1994, 472, 371 CrossRef CAS.
- Z. Assefa, B. G. McBurnett, R. J. Staples, J. P. Fackler Jr., B. Assmann, K. Angermaier and H. Schmidbaur, Inorg. Chem., 1995, 34, 75 CrossRef CAS.
- K.-H. Wong, K.-K. Cheung, M. C.-W. Chan and C.-M. Che, Organometallics, 1998, 17, 3505 CrossRef CAS.
- C. K.-L. Li, R. W.-Y. Sun, S. C.-F. Kui, N. Zhu and C.-M. Che, Chem. – Eur. J., 2006, 12, 5253 CrossRef CAS PubMed.
- C.-W. Chan, W.-T. Wong and C.-M. Che, Inorg. Chem., 1994, 33, 1266 CrossRef CAS.
- V. W.-W. Yam, T.-F. Lai and C.-M. Che, J. Chem. Soc., Dalton Trans., 1990, 3747 RSC.
- D. Li, C.-M. Che, S.-M. Peng, S.-T. Liu, Z.-Y. Zhou and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1993, 189 RSC.
- C. King, J. C. Wang, M. N. I. Khan and J. P. Fackler Jr., Inorg. Chem., 1989, 28, 2145 CrossRef CAS.
- M. N. I. Khan, C. King, D. D. Heinrich and J. P. Fackler Jr., Inorg. Chem., 1989, 28, 2150 CrossRef CAS.
- C.-M. Che, H.-L. Kwong, C.-K. Poon and V. W.-W. Yam, J. Chem. Soc., Dalton Trans., 1990, 3215 RSC.
- C.-M. Che, H.-L. Kwong, V. W.-W. Yam and K.-C. Cho, J. Chem. Soc., Chem. Commun., 1989, 885 RSC.
- H. R. C. Jaw, M. M. Savas, R. D. Rogers and W. R. Mason, Inorg. Chem., 1989, 28, 1028 CrossRef CAS.
- H. R. C. Jaw, M. M. Savas and W. R. Mason, Inorg. Chem., 1989, 28, 4366 CrossRef CAS.
- I. J. B. Lin, J. M. Hwang, D. F. Feng, M. C. Cheng and Y. Wang, Inorg. Chem., 1994, 33, 3467 CrossRef CAS.
- G. Shaw, J. K. Becconsall, R. M. Canadine and R. Murray, Chem. Commun., 1966, 425 RSC.
- M. I. Bruce, B. K. Nicholson and O. B. Shawkataly, Inorg. Synth., 1989, 26, 324 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon, Oxford, U. K., 3rd edn, 1988 Search PubMed.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- J. van Houten and R. Watts, J. Am. Chem. Soc., 1976, 98, 4853 CrossRef CAS.
- C.-M. Che, K.-Y. Wong and F. C. Anson, J. Electroanal. Chem. Interfacial Electrochem., 1987, 226, 211 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- Bruker AXS Inc., SMART version 5.054, Madison, Wisconsin, USA, 1998 Search PubMed.
- Bruker AXS Inc., SAINT version 6.02, Madison, Wisconsin, USA, 1999 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS.
- D. Andrae, U. Häussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected singlet and triplet excited states computed by TDDFT/CPCM calculations, relative energy of the triplet excited state, selected molecular orbitals, and Cartesian coordinates of the optimized structures reported in the paper. CCDC 1046319–1046321. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00017c |
| This journal is © the Partner Organisations 2015 |