
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient planar perovskite solar cells through band alignment engineering†
Juan Pablo
Correa Baena‡
*a,
Ludmilla
Steier‡
b,
Wolfgang
Tress
bc,
Michael
Saliba
c,
Stefanie
Neutzner
df,
Taisuke
Matsui
e,
Fabrizio
Giordano
b,
T. Jesper
Jacobsson
a,
Ajay Ram
Srimath Kandada
d,
Shaik M.
Zakeeruddin
b,
Annamaria
Petrozza
d,
Antonio
Abate
b,
Mohammad Khaja
Nazeeruddin
c,
Michael
Grätzel
b and
Anders
Hagfeldt
*a
aLaboratory for Photomolecular Science, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, CH-1015-Lausanne, Switzerland. E-mail: juan.correa@epfl.ch; anders.hagfeldt@epfl.ch
bLaboratory for Photonics and Interfaces, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, CH-1015-Lausanne, Switzerland
cGroup for Molecular Engineering of Functional Materials, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, CH-1015-Lausanne, Switzerland
dCenter for Nano Science and Technology@Polimi, Istituto Italiano di Tecnologia, via Pascoli 70/3 20133 Milano, Italy
eAdvanced Research Division, Panasonic Corporation, 1006, (Oaza Kadoma), Kadoma City, Osaka 571-8501, Japan
fDipartimento di Fisica, Politecnico di Milano, Piazza L. da Vinci, 32, 20133, Milano, Italy
First published on 25th August 2015
Abstract
The simplification of perovskite solar cells (PSCs), by replacing the mesoporous electron selective layer (ESL) with a planar one, is advantageous for large-scale manufacturing. PSCs with a planar TiO2 ESL have been demonstrated, but these exhibit unstabilized power conversion efficiencies (PCEs). Herein we show that planar PSCs using TiO2 are inherently limited due to conduction band misalignment and demonstrate, with a variety of characterization techniques, for the first time that SnO2 achieves a barrier-free energetic configuration, obtaining almost hysteresis-free PCEs of over 18% with record high voltages of up to 1.19 V.
Introduction
Solution processed, hybrid organic–inorganic perovskite materials were studied by Mitzi et al. in the 1990s and were recognized as excellent semiconducting materials.1 It was not, however, until Miyasaka and coworkers pioneered the work on dye-sensitized solar cell applications in 2009, that the materials were started to be recognized by the photovoltaic community.2 Since then, a myriad of studies have been published exploring different device configurations. The currently highest reported PCE value of over 20% was achieved using a thin layer of mesoporous TiO2.3 In this architecture, the perovskite material infiltrates a mesoporous TiO2 layer, which is sandwiched between a hole transporting layer (HTL, typically doped 2,2′,7,7′-tetrakis(N,N′-di-p-methoxyphenylamine)-9,9′-spirobifluorene (Spiro-OMeTAD) or polytertiary arylamine (PTAA)) and an electron selective layer (ESL, typically TiO2).From the earlier studies, it was realized that the perovskite absorber material transports both holes and electrons.4–6 Naturally, this led towards the investigation of a thin film perovskite configuration with only a compact TiO2 as the ESL.7 However, this device architecture shows pronounced hysteresis of the current–voltage (J–V) curve,8–10 especially for fast voltage sweeps and to our knowledge no PCE of over 18% in this architecture has been reported without hysteresis and the stabilized power output. Xing et al. showed that planar devices, using PCBM as the ESL and methyl ammonium lead iodide (MAPbI3) as the absorbing and transporting material, had a much improved J–V hysteretic behaviour when compared to the TiO2 ESL, which they linked to the improved interfacial charge transfer. Wojciechowski and co-workers showed that modifying the TiO2 surface with fullerene derivatives can work towards high efficiency PSCs.8 Recent studies have shown the potential of SnO2-based ESLs,11–14 but so far these devices have not shown high efficiency without hysteretic behaviour.
Using a low temperature atomic layer deposition (ALD) process to fabricate SnO2 ESLs, we demonstrate that planar PSCs can achieve almost hysteresis-free PCEs of above 18% with voltages exceeding 1.19 V. We show that this is not the case for the planar TiO2. We choose SnO2 considering the favourable alignment of the conduction bands of the perovskite materials and the ESL and show an energy mismatch in the case of TiO2. Thus, using SnO2, which has a deeper conduction band, enables us to fabricate planar devices with high efficiencies, long term air stability and improved hysteretic behaviour, while keeping the processing at low temperatures (<120 °C), which is the key for process upscaling and high efficiency tandem devices.15
Results and discussion
In Fig. 1a, we illustrate how electron injection is energetically hindered when the bands are mismatched. This is accompanied by a schematic of the planar device architecture of a typical glass/FTO/compact metal oxide/perovskite/hole transporter/gold stack. We analyse the band structure further using ultraviolet photoelectron spectroscopy (UPS) for two different perovskite materials (MAPbI3 and mixed halide/cation, i.e. (FAPbI3)0.85(MAPbBr3)0.15, referred to as the mixed perovskite throughout the text) atop TiO2 and SnO2 as shown in Fig. 1b and c derived from ESI,† Fig. S1 and S2, respectively. The ionization energy (IE), i.e. the valence band position, measurements of SnO2 and TiO2 were performed for the UV ozone-treated samples atop FTO, thus obtaining the valence band information for both substrates. It has been shown that the valence band position of the perovskite material measured by UPS has variations with respect to the substrate where they are deposited.16 Thus, we performed our measurements on perovskite films deposited on both SnO2 and TiO2 yielding IE differences of above 0.1 eV. We calculated the band diagram of the different components using the perovskite materials' valence bands as our reference. The construction of the band diagram, including bandgap estimation for the perovskite materials (thickness of ca. 400 nm), is described in ESI,† Fig. S1–S3. We found that for both perovskite materials there is a conduction band misalignment with TiO2 ESLs, in stark contrast to SnO2 where we have no such misalignment. The band diagram in Fig. 1b shows that the conduction band of MAPbI3 is ∼80 meV below that of TiO2 and about 170 meV above that of SnO2. This inhibits electron extraction by TiO2 and facilitates it using SnO2. Similarly, the conduction band of the mixed perovskite is 300 meV below compared to TiO2 and only 30 meV below compared to SnO2. Consequently, this band misalignment with TiO2 may cause undesirable consequences such as accumulation of photogenerated charges, which could hamper the device performance.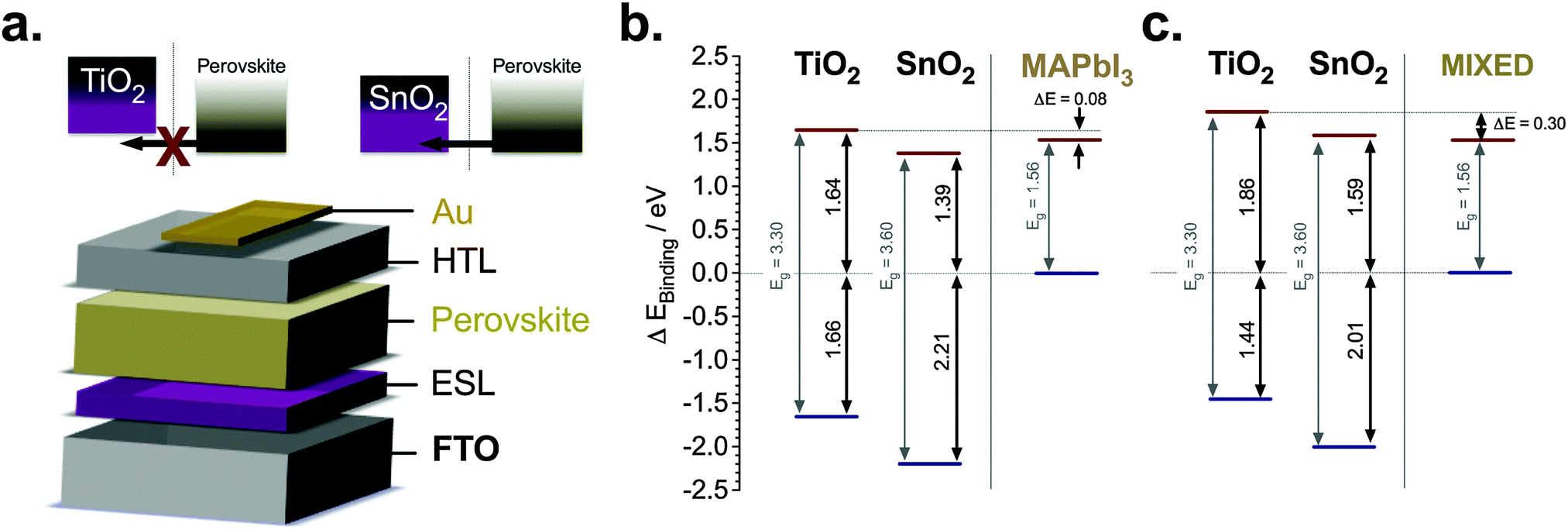 | ||
| Fig. 1 Energy level diagrams and electron injection characteristics of SnO2 and TiO2-based planar PSCs. (a) Schematic conduction band diagram of the perovskite films and the electron selective layers, TiO2 and SnO2 for (b) MAPbI3 and (c) (FAPbI3)0.85(MAPbBr3)0.15, labeled as ‘mixed’. | ||
It is important to note that the UPS measurements were carried out on perovskite films as thick as 400 nm. Since UPS is a surface measurement (measuring roughly the conditions in the first 10 nm), it is therefore a simplified picture of our device energetics. Guerrero et al. have shown that the energetics throughout the perovskite film can be different and that band bending can be induced when employing thick films.17 In addition, work by some of us has also shown that ion migration is induced in the perovskite material,18 which further complicates the energetic model in the device. Indeed, these two factors play a major role in the electronic configuration of the device and it is something that will be further investigated more in depth in future studies. However, with these measurements we elucidate that there is an intrinsic difference between the two ESLs, which lead to an understanding that there is an energetic barrier at the TiO2, but not at the SnO2/perovskite interface.
To further investigate this phenomenon, we prepared planar devices of typical stack architecture: glass/FTO/ESL/perovskite/HTL/gold contact as seen in the cross-sectional scanning electron microscopy (SEM) image in Fig. 2a. We deposited a 15 nm thick ESL of SnO2, TiO2 or Nb2O5 by ALD. The mixed perovskite layer, (FAPbI3)0.85(MAPbBr3)0.15, was spin-coated on the electrode using a similar composition as reported by Jeon et al.19 A doped spiro-MeOTAD was spin-coated as the HTL and, finally, the gold top electrode was deposited by thermal evaporation.
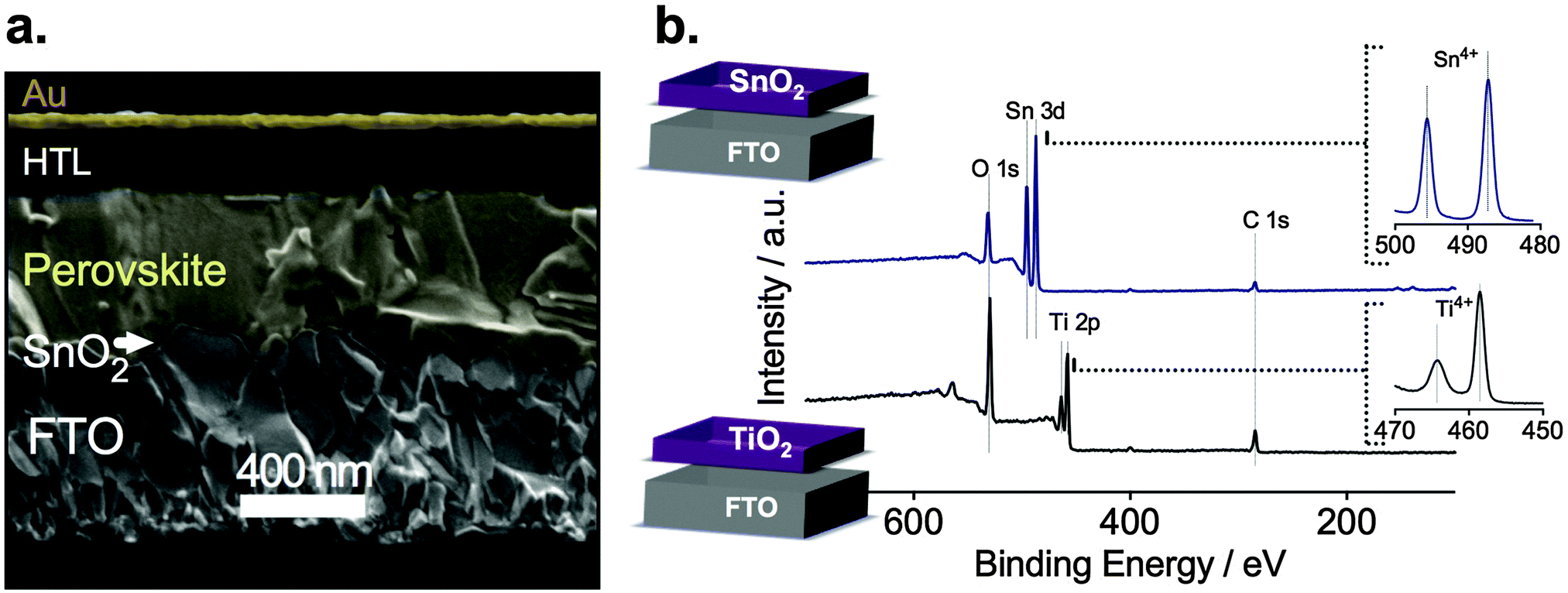 | ||
| Fig. 2 Photovoltaic device architecture and elemental composition of the electron selective layers (ESLs). (a) Cross-sectional scanning electron micrograph of a typical layered photovoltaic device composed of FTO, SnO2 as the electron selective layer (ESL), the perovskite film, a hole transporting layer (HTL, Spiro MeOTAD), and a gold top electrode. (b) X-ray photoelectron spectroscopy of TiO2 and SnO2 thin layers used as ESLs. | ||
Fig. 2b shows the X-ray photoelectron spectroscopy (XPS) of the 15 nm thick TiO2 and SnO2 layers. For TiO2, no peaks other than oxygen O 1s at 528 eV, titanium Ti 2p at 458.5 eV and Ti 2p1/2 464.2 eV were detected confirming the deposition of TiO2 without traces of cross contamination.20 We detect no signal from the underlying FTO indicating conformal and pinhole-free TiO2 coverage, which we further confirm by SEM (see ESI,† Fig. S4a). Similarly, we confirm the formation of pure SnO2 observing the oxygen peak O 1s at 530.9 eV and Sn4+ peaks at 495.6 eV as well as at 487.2 eV. The top-view SEM image also indicates a pinhole-free deposition of SnO2 (see ESI,† Fig. S4b).
In order to further understand the results by UPS in a device configuration we performed femtosecond transient absorption (TA) measurements. With this we intended to understand electron injection dynamics from the perovskite into the ESLs, and therefore, indirectly probe whether an energetic barrier exists for TiO2 or SnO2. The measurements were performed on devices with SnO2 and TiO2 and the mixed perovskite under short circuit conditions, wherein the charge injection can be resolved in time. In Fig. 3, we show the TA dynamics taken at a probe wavelength of 750 nm – the peak of the photobleach (PB) of the perovskite. The PB band, spectrally located at the onset of the absorption spectrum of the semiconductor (ESI,† Fig. S3), corresponds to the photo-induced transparency in the material due to the presence of electrons and holes in the bottom and top of the conduction and valence bands, respectively.17 Hence, the magnitude of this feature is correlated with the photo-induced carrier population and every mechanism changing the initial population, like electron/hole injection, results in its quenching. We observe a PB decay in the nanosecond timescale for both TiO2 and SnO2-based devices. However, while in the TiO2-based device the dynamics do not strongly differ from the one probed from the pristine perovskite deposited on bare glass,21 in the case of SnO2 the decay is much faster. In fact, the carrier population is reduced by approximately 60% in 1.5 ns. As both devices embody the same hole extracting layer, we conclude that the striking difference observed can be considered as the signature of different electron injection dynamics. This strongly supports our hypothesis of better electron extraction in pristine SnO2 when compared to TiO2-based devices, due to favorable energetic alignment.
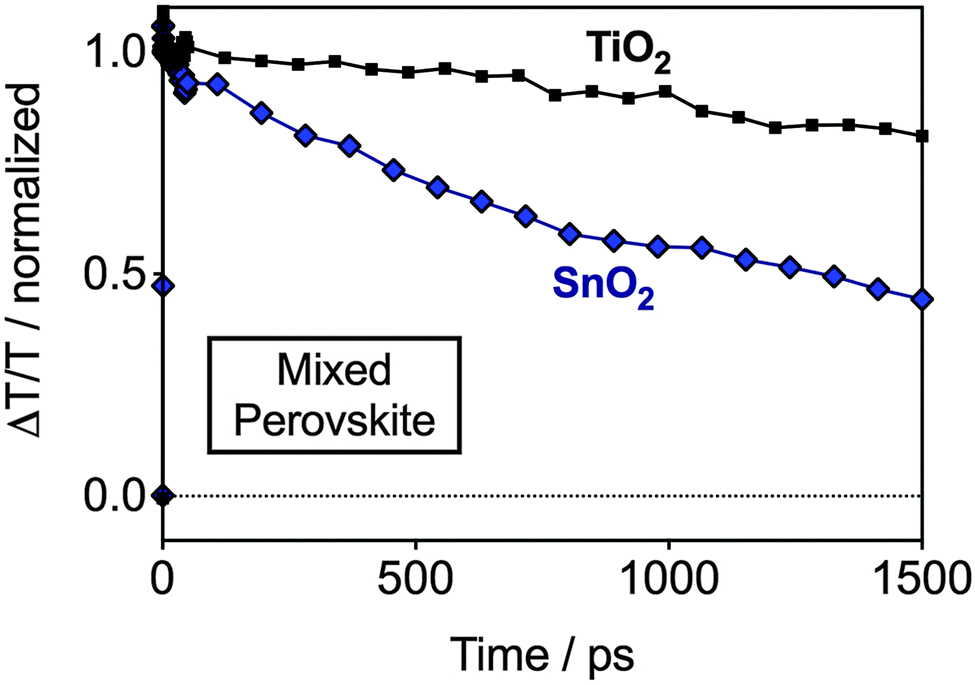 | ||
| Fig. 3 Transient absorption measurements of SnO2 and TiO2-based planar PSCs. Dynamics of the photo-bleaching bands for photo-excited perovskite measured on a typical working device employing the mixed perovskite (FAPbI3)0.85(MAPbBr3)0.15 and SnO2 or TiO2 as the ESL. The device is held at short circuit condition during the measurement. The probe wavelength is λ = 750 nm while pumping at λ = 520 nm. | ||
We note that the poor charge extraction in the TiO2 based device may appear to be surprising. However, it must be considered that, in thin film PSCs in the presence of planar TiO2 as the electron extracting layer, solar cells generally show Jsc comparable to those using a mesoporous TiO2 layer only when the device is pre-polarized.8,9,22–24 Indeed, some of us have recently demonstrated that the PB dynamics become faster when measured just after keeping the TiO2-based device at 1 V for a few seconds, suggesting that the electron transfer is suddenly activated.23 This indicates that upon polarization, the TiO2/perovskite interface is modified and such a modification is needed to allow for an efficient charge transfer, as also predicted by De Angelis et al.25
We investigated the different electronic properties of devices with TiO2 or SnO2 ESLs by analyzing the current density–voltage curves based on the mixed perovskite. In Fig. 4a, we observe a representative SnO2 device with high performance and low hysteresis between the backward and the forward scan (Table 1). This is indicative of good charge collection independent of voltage. In stark contrast, a representative TiO2-based device shows strong hysteresis and low current densities (<5 mA cm−2). This difference can also be seen in Fig. 4b where we show transient photocurrents recorded at 0.8 V resembling closely operating device conditions at the maximum power point. After ∼50 s, we observe a steady photocurrent when switching from an open circuit to 0.8 V. After switching from open circuit to 0.8 V, the current for the TiO2 device drops by 70% from 10 to a stabilized 3 mA cm−2, whereas that for the SnO2 drops by only 10% from 23 to a stabilized 20.2 mA cm−2. The stabilized current is in good agreement with the current seen in the J–V curve at 0.8 V, which is found to be 20.7 mA cm−2 (Fig. 4a). In addition, SnO2-based devices showed good long-term stability; unencapsulated devices stored in dry air were measured for over 30 days with no significant PCE variability (ESI,† Fig. S5). Small variations were found for 12 devices made in different batches with an average PCE of 16.7% (ESI,† Fig. S6). Integrating the external quantum efficiency (EQE) yielded a Jsc of 18 mA cm−2 (ESI,† Fig. S7a), which is in very good agreement with the measured Jsc in Fig. S7b (ESI†).
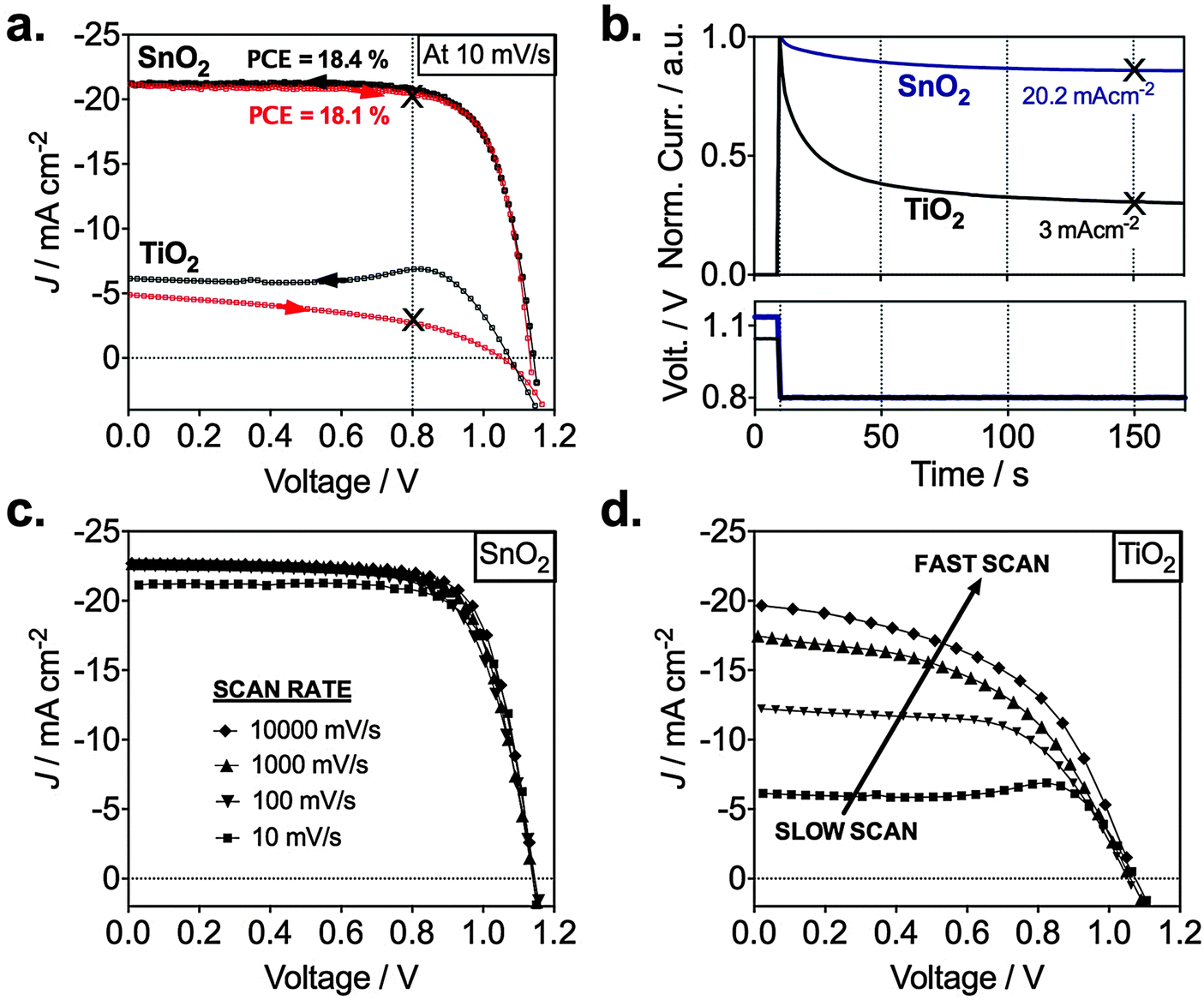 | ||
| Fig. 4 Photovoltaic characteristics of planar perovskite devices based on SnO2 and TiO2 ESLs. (a) Current–voltage properties of TiO2 and SnO2-based planar mixed halide/cation perovskite devices. Black arrows indicate the backward scan from Voc to Jsc and red arrows indicate the reversed scan. (b) Normalized transient photocurrents measured from Voc to the maximum power point voltage for both planar systems. (c) Scan rate effects on J–V characteristics of SnO2 and (d) TiO2-based devices. We note that devices showed best performance when measured after 1 week of preparation. | ||
| ESL | Scan direction | J sc (mA cm−2) | V oc (V) | FF | PCE (%) | Light intensity (mW cm−2) |
|---|---|---|---|---|---|---|
| SnO2 | Backward | 21.3 | 1.14 | 0.74 | 18.4 | 98.4 |
| Forward | 21.2 | 1.13 | 0.75 | 18.1 |
We note that for both TiO2 and SnO2, we observe open circuit voltages of around 1.14 V, which are close or even exceeds most devices prepared with mesoporous interlayers. Additionally, some of our SnO2 devices yielded stabilized voltages of over 1.19 V (ESI,† Fig. S7c) approaching the thermodynamic maximum Voc of approx. 1.32 V.26 This suggests exceptionally good charge selectivity and a low degree of charge recombination in our planar perovskite/SnO2 devices.
To understand the reason for the reduced photocurrent for the TiO2 based device, we performed current–voltage scans at various voltage sweep rates. These are shown in Fig. 4c and d, where only the backward scan is plotted which is obtained after the device was preconditioned at 1.2 V for 10 s. For the SnO2 device there is only a slight increase of the photocurrent when increasing the rate from 10 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mV s−1. Slightly enhanced sweep rates allow us to collect almost all the photogenerated charges reaching a maximum Jsc density of 23 mA cm−2. The dependence on the scan rate is much more pronounced for the TiO2-based device showing high current densities of ca. 20 mA cm−2 for the scan at 10 V s−1 with a massive drop to about 5 mA cm−2 when scanned at 10 mV s−1. This implies a low charge collection efficiency in the planar perovskite/TiO2 device at slow scan rates, though light absorption and photocurrent generation in the perovskite material is the same as for the perovskite/SnO2 configuration. The results are also in good agreement with the transient photocurrent in Fig. 4b, the electron injection characteristics in Fig. 3 and our proposed band alignment measured by UPS in Fig. 1, clearly indicating a barrier free charge transport across the perovskite/SnO2 in contrast to the perovskite/TiO2 interface. We investigated devices with ALD Nb2O5 as the ESL (ESI,† Fig. S8) which has a similar conduction band position as TiO2.27 With this, we can crosscheck if the energy level alignment is indeed critical for high hysteresis and can exclude the fact that other properties of SnO2 or TiO2 are responsible for the above results. Very similar to TiO2, the Nb2O5-based devices exhibited large hysteresis behavior and very low photocurrent densities (ESI,† Fig. S8). Several independent studies have shown similar or even more pronounced trends irrespective of the TiO2 deposition method. Spin-coating,8,9,23,28–30 sputtering30,31 and spray pyrolysis32 of TiO2 have all been demonstrated to yield highly hysteretic J–V curves in planar PSCs.
000 mV s−1. Slightly enhanced sweep rates allow us to collect almost all the photogenerated charges reaching a maximum Jsc density of 23 mA cm−2. The dependence on the scan rate is much more pronounced for the TiO2-based device showing high current densities of ca. 20 mA cm−2 for the scan at 10 V s−1 with a massive drop to about 5 mA cm−2 when scanned at 10 mV s−1. This implies a low charge collection efficiency in the planar perovskite/TiO2 device at slow scan rates, though light absorption and photocurrent generation in the perovskite material is the same as for the perovskite/SnO2 configuration. The results are also in good agreement with the transient photocurrent in Fig. 4b, the electron injection characteristics in Fig. 3 and our proposed band alignment measured by UPS in Fig. 1, clearly indicating a barrier free charge transport across the perovskite/SnO2 in contrast to the perovskite/TiO2 interface. We investigated devices with ALD Nb2O5 as the ESL (ESI,† Fig. S8) which has a similar conduction band position as TiO2.27 With this, we can crosscheck if the energy level alignment is indeed critical for high hysteresis and can exclude the fact that other properties of SnO2 or TiO2 are responsible for the above results. Very similar to TiO2, the Nb2O5-based devices exhibited large hysteresis behavior and very low photocurrent densities (ESI,† Fig. S8). Several independent studies have shown similar or even more pronounced trends irrespective of the TiO2 deposition method. Spin-coating,8,9,23,28–30 sputtering30,31 and spray pyrolysis32 of TiO2 have all been demonstrated to yield highly hysteretic J–V curves in planar PSCs.
To further confirm what is found in the literature and show that our results are not unique to the ALD technique, we prepared TiO2 by spray pyrolysis and found that the J–V curves exhibit strong hysteretic behavior (ESI,† Fig. S9). In this case, the forward scan shows an s-shaped J–V curve indicative of an unstabilized power output.33 However, the devices using spray-pyrolysed TiO2 showed an increase in the Jsc in the backward scan when compared to ALD TiO2. In order to understand the difference between these two layers, we investigated the effect of the ESLs using spiro and gold-only devices. The perovskite-free devices were investigated in reverse bias to understand whether the ESLs suffer from pinholes. Our results, summarized in ESI,† Fig. S10, show improved blocking properties for the ALD layers of both TiO2 and SnO2 when compared to spray pyrolysed TiO2. This difference likely explains the cause of increased photocurrent of the latter, which we can see in ESI,† Fig. S9.
A similar trend was found for planar devices using MAPbI3 (ESI,† Fig. S11). Here, the current densities measured are slightly higher in the backward but lower in the forward scan, suggesting the same limitation for charge extraction as noted above. This also matches our UPS results in Fig. 1b, where the conduction bands of perovskite and TiO2 are misaligned and highlights the importance of correct band alignment in all planar perovskite devices. Other studies3,19 have shown high performance at stabilized currents in thin mesoporous TiO2 based ESLs, and we note that this may be due to a proper band alignment intrinsic to the mesoporous TiO2/perovskite interface which is different from the planar configuration with the TiO2 used in this study.
We hypothesize that the preconditioning under forward bias leads to accumulation of negative charge and ion migration at the ESL–perovskite interface inducing a high electric field and/or dipole formation at this interface.10,22 An elevated electric field or possibly a reduced conduction band offset can facilitate electron injection into the ESL. After releasing the positive bias, this beneficial effect lasts for a few seconds only, which is the time needed for this charge to be removed. Sweep rates in this time range give rise to large hysteresis. For the SnO2 devices, the energy levels are already well aligned without biasing the device. Thus, charge collection is efficient showing high FF and Jsc independent of the scan rate (Fig. 4c).
Conclusions
In summary, we have demonstrated that a barrier-free band alignment between the perovskite light harvester and the charge selective contact is of great importance for an efficient PSC. We found that planar PSCs employing the compact and pinhole-free TiO2 layer made by ALD exhibit a band misalignment, leading to strong hysteresis behavior and scan rate dependent current densities, indicating capacitive effects at the interface. We chose a layer of SnO2, due to its deeper conduction band, as the electron selective contact, which achieved voltages and PCEs exceeding 1.19 V and 18%, respectively. We proved that modifying the conduction band of the ESL can result in planar, high performance PSCs with high voltages and remarkably good stability over time. Furthermore, femtosecond TA measurements clearly show that the mixed (FAPbI3)0.85(MAPbBr3)0.15 perovskite materials extract charges efficiently into SnO2 but not into TiO2 corroborating the conduction band misalignment at the TiO2/perovskite interface. From this we can conclude that a barrier-free charge transport across the SnO2/perovskite interface gives rise to the high and stable current densities – regardless of the sweep rate – which are not observed in TiO2 based devices. This study highlights the importance of a perfect band alignment for highly efficient PSCs, especially in planar devices with compact charge selective layers.Methods
Electron selective layer preparation
F:SnO2 substrates were first wiped with acetone, and then cleaned for 10 min in piranha solution (H2SO4/H2O2 = 3:1) followed by 10 min in a plasma cleaner prior to ALD deposition.
Atomic layer deposition (ALD) of semi-crystalline TiO234 was carried out in a Savannah ALD 100 instrument (Cambridge Nanotech Inc.) at 120 °C using tetrakis(dimethylamino)titanium(IV) (TDMAT, 99.999% pure, Sigma Aldrich) and H2O2. TDMAT was held at 75 °C and H2O2 at room temperature. The growth rate was 0.07 nm per cycle at a N2 flow rate of 5 sccm as measured by ellipsometry.
SnO2 was deposited at 118 °C using tetrakis(dimethylamino)tin(IV) (TDMASn, 99.99%-Sn, Strem Chemicals INC) and ozone at a constant growth rate of 0.065 nm per cycle measured by ellipsometry. TDMASn was held at 65 °C. Ozone was produced using an ozone generator (AC-2025, IN USA Incorporated) fed with oxygen gas (99.9995% pure, Carbagas) producing a concentration of 13% ozone in O2. Nitrogen was used as a carrier gas (99.9999% pure, Carbagas) with a flow rate of 10 sccm.
Nb2O5 was deposited at 170 °C and a carrier gas flow rate of 20 sccm using (tert-butylimido)bis(diethylamino)niobium (TBTDEN, Digital Specialty Chemicals, Canada) and ozone with a constant growth rate of 0.06 nm per cycle. TBTDEN was held at 130 °C.
Perovskite precursor solution and film preparation
Before perovskite deposition, the ALD layers were treated with UV ozone for 10 min to remove by-products from the deposition process. The perovskite films were deposited from a precursor solution containing FAI (1 M), PbI2 (1.1 M, TCI Chemicals), MABr (0.2 M) and PbBr2 (0.2 M, Alfa Aesar) in anhydrous DMF:DMSO 4:1 (v/v, Acros). The perovskite solution was spin-coated in a two-step program; first at 1000 for 10 s and then at 4000 rpm for 30 s. During the second step, 100 μL of chlorobenzene were poured on the spinning substrate 15 s prior to the end of the program. The substrates were then annealed at 100 °C for 1 h in a nitrogen filled glove box.
The spiro-OMeTAD (Merck) solution (70 mM in chlorobenzene) was spun at 4000 rpm for 20 s. The spiro-OMeTAD was doped at a molar ratio of 0.5, 0.03 and 3.3 with bis(trifluoromethylsulfonyl)imide lithium salt (Li-TFSI, Sigma Aldrich), tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)–cobalt(III) tris(bis(trifluoromethylsulfonyl)imide) (FK209, Dyenamo) and 4-tert-butylpyridine (TBP, Sigma Aldrich), respectively.22,35,36 As a last step 70–80 nm of gold top electrode were thermally evaporated under high vacuum.
Solar cell characterization
A ZEISS Merlin HR-SEM was used to characterize the morphology of the device cross-section. The solar cells were measured using a 450 W xenon light source (Oriel). The spectral mismatch between AM 1.5G and the simulated illumination was reduced by the use of a Schott K113 Tempax filter (Präzisions Glas & Optik GmbH). The light intensity was calibrated with a Si photodiode equipped with an IR-cutoff filter (KG3, Schott) and it was recorded during each measurement. Current–voltage characteristics of the cells were obtained by applying an external voltage bias while measuring the current response using a digital source meter (Keithley 2400). The voltage scan rate was 10 mV s−1 and no device preconditioning was applied before starting the measurement, such as light soaking or forward voltage bias applied for long time. The starting voltage was determined as the potential at which the cells furnished 1 mA in forward bias, no equilibration time was used. The cells were masked with a black metal mask limiting the active area to 0.16 cm2 and reducing the influence of the scattered light. It is important to note that the devices achieved the highest hysteresis-free efficiency after 1 week of preparation.The EQE spectra were measured under constant white light bias with an intensity of 10 mW cm−2 supplied by a LED array. The superimposed monochromatic light was chopped at 2 Hz. The homemade system comprises a 300 W Xenon lamp (ICL Technology), a Gemini-180 double-monochromator with 1200 grooves per mm grating (Jobin Yvon Ltd) and a lock-in amplifier (SR830 DSP, Stanford Research System). The EQE integration was performed according to the following equation
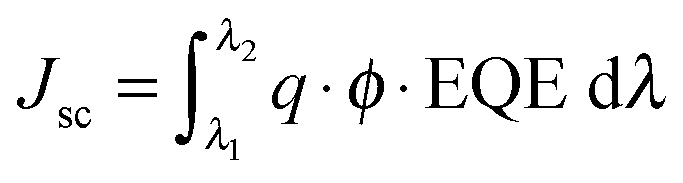
Author contributions
J.P.C.B. developed the basic concept and coordinated the project. ALD layers were prepared by J.P.C.B. and L.S. Devices were prepared by J.P.C.B., A.A., M.S., T.M. and F.G. Measurements were performed by J.P.C.B., L.S., W.T., A.A., M.S., T.M., F.G., S.N. and A.R.S.K. A.P supervised the femtosecond spectroscopy measurements. S.M.Z., A.A., M.K.N and M.G. contributed to fruitful discussions. A.H. supervised the project. All authors contributed to the writing of the paper.Competing financial interest
The authors declare no competing financial interest.Acknowledgements
UPS and XPS were performed at the Advanced Research Division, Panasonic Corporation in Japan. HR-SEM images were taken at the Centre for Electron Microscopy (CIME) at EPFL. L.S. acknowledges support from the European FP7 FET project PHOCS (no. 309223). A.A. has received funding from the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 291771. W.T., M.S., and M.K.N. thank the European Union for funding within the Seventh Framework Program (FP7/2007-2013) under grant agreement no. 604032 of the MESO project. M.G. thanks the financial support from CCEM-CH in the 9th call proposal 906: CONNECT PV, the Swiss National Science Foundation (SNF)-NRP70 (PV2050, 407040-153990/1 and 407040-153976/1) and GRAPHENE project supported by the European Commission Seventh Framework Programme under contract 604391 is gratefully acknowledged. S.N. and A.P. receive funding from the EU Horizon 2020 Research and Innovation Programme under grant agreement no. 643238 (SYNCHRONICS).References
- C. R. Kagan, D. B. Mitzi and C. D. Dimitrakopoulos, Science, 1999, 286, 945–947 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- L. Etgar, P. Gao, Z. Xue, Q. Peng, A. K. Chandiran, B. Liu, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2012, 134, 17396–17399 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- K. Wojciechowski, S. D. Stranks, A. Abate, G. Sadoughi, A. Sadhanala, N. Kopidakis, G. Rumbles, C.-Z. Li, R. H. Friend and A. K.-Y. Jen, ACS Nano, 2014, 8, 12701–12709 CrossRef CAS PubMed.
- K. Wojciechowski, T. Leijtens, S. Spirova, C. Schlueter, M. Hoerantner, J. T.-W. Wang, C.-Z. Li, A. K. Y. Jen, T.-L. Lee and H. J. Snaith, J. Phys. Chem. Lett., 2015, 6, 2399–2405 CrossRef CAS PubMed.
- Y. Zhang, M. Liu, G. E. Eperon, T. C. Leijtens, D. McMeekin, M. Saliba, W. Zhang, M. de Bastiani, A. Petrozza, L. M. Herz, M. B. Johnston, H. Lin and H. J. Snaith, Mater. Horiz., 2015, 2, 315–322 RSC.
- W. Ke, G. Fang, Q. Liu, L. Xiong, P. Qin, H. Tao, J. Wang, H. Lei, B. Li, J. Wan, G. Yang and Y. Yan, J. Am. Chem. Soc., 2015, 137, 6730–6733 CrossRef CAS PubMed.
- Q. Dong, Y. Shi, K. Wang, Y. Li, S. Wang, H. Zhang, Y. Xing, Y. Du, X. Bai and T. Ma, J. Phys. Chem. C, 2015, 119, 10212–10217 CAS.
- J. Song, E. Zheng, J. Bian, X.-F. Wang, W. Tian, Y. Sanehira and T. Miyasaka, J. Mater. Chem. A, 2015, 3, 10837–10844 CAS.
- Y. Li, J. Zhu, Y. Huang, F. Liu, M. Lv, S. Chen, L. Hu, J. Tang, J. Yao and S. Dai, RSC Adv., 2015, 5, 28424–28429 RSC.
- L. Kranz, A. Abate, T. Feurer, F. Fu, E. Avancini, J. Löckinger, P. Reinhard, S. M. Zakeeruddin, M. Grätzel, S. Buecheler and A. N. Tiwari, J. Phys. Chem. Lett., 2015, 2676–2681 CrossRef CAS PubMed.
- E. M. Miller, Y. Zhao, C. C. Mercado, S. K. Saha, J. M. Luther, K. Zhu, V. Stevanovic, C. L. Perkins and J. van de Lagemaat, Phys. Chem. Chem. Phys., 2014, 16, 22122–22130 RSC.
- A. Guerrero, E. J. Juarez-Perez, J. Bisquert, I. Mora-Sero and G. Garcia-Belmonte, Appl. Phys. Lett., 2014, 105, 133902 CrossRef PubMed.
- W. Tress, N. Marinova, T. Moehl, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Gratzel, Energy Environ. Sci., 2015, 8, 995–1004 CAS.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- H. Bender, W. D. Chen, J. Portillo, L. Van den Hove and W. Vandervorst, Appl. Surf. Sci., 1989, 38, 37–47 CrossRef CAS.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- H. J. Snaith, A. Abate, J. M. Ball, G. E. Eperon, T. Leijtens, N. K. Noel, S. D. Stranks, J. T.-W. Wang, K. Wojciechowski and W. Zhang, J. Phys. Chem. Lett., 2014, 5, 1511–1515 CrossRef CAS PubMed.
- C. Tao, S. Neutzner, L. Colella, S. Marras, A. R. Srimath Kandada, M. Gandini, M. De Bastiani, G. Pace, L. Manna, M. Caironi, C. Bertarelli and A. Petrozza, Energy Environ. Sci., 2015, 8, 2365–2370 CAS.
- G. Xing, B. Wu, S. Chen, J. Chua, N. Yantara, S. Mhaisalkar, N. Mathews and T. C. Sum, Small, 2015, 11, 3606–3613 CrossRef CAS PubMed.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 CAS.
- W. Tress, N. Marinova, O. Inganäs, M. K. Nazeeruddin, S. M. Zakeeruddin and M. Grätzel, Adv. Energy Mater., 2015, 5, 1400812 Search PubMed.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS PubMed.
- P. Docampo, F. C. Hanusch, S. D. Stranks, M. Döblinger, J. M. Feckl, M. Ehrensperger, N. K. Minar, M. B. Johnston, H. J. Snaith and T. Bein, Adv. Energy Mater., 2014, 4, 1400355 Search PubMed.
- Q. Chen, H. Zhou, Z. Hong, S. Luo, H.-S. Duan, H.-H. Wang, Y. Liu, G. Li and Y. Yang, J. Am. Chem. Soc., 2014, 136, 622–625 CrossRef CAS PubMed.
- J. Xu, A. Buin, A. H. Ip, W. Li, O. Voznyy, R. Comin, M. Yuan, S. Jeon, Z. Ning, J. J. McDowell, P. Kanjanaboos, J.-P. Sun, X. Lan, L. N. Quan, D. H. Kim, I. G. Hill, P. Maksymovych and E. H. Sargent, Nat. Commun., 2015, 6, 7081 CrossRef CAS PubMed.
- A. H. Ip, L. N. Quan, M. M. Adachi, J. J. McDowell, J. Xu, D. H. Kim and E. H. Sargent, Appl. Phys. Lett., 2015, 106, 143902 CrossRef PubMed.
- B. Wu, K. Fu, N. Yantara, G. Xing, S. Sun, T. C. Sum and N. Mathews, Adv. Energy Mater., 2015, 1500829 Search PubMed.
- E. L. Unger, E. T. Hoke, C. D. Bailie, W. H. Nguyen, A. R. Bowring, T. Heumuller, M. G. Christoforo and M. D. McGehee, Energy Environ. Sci., 2014, 7, 3690–3698 CAS.
- J. Azevedo, L. Steier, P. Dias, M. Stefik, C. T. Sousa, J. P. Araujo, A. Mendes, M. Graetzel and S. D. Tilley, Energy Environ. Sci., 2014, 7, 4044–4052 CAS.
- A. Abate, T. Leijtens, S. Pathak, J. Teuscher, R. Avolio, M. E. Errico, J. Kirkpatrik, J. M. Ball, P. Docampo, I. McPherson and H. J. Snaith, Phys. Chem. Chem. Phys., 2013, 15, 2572–2579 RSC.
- A. Abate, D. R. Staff, D. J. Hollman, H. J. Snaith and A. B. Walker, Phys. Chem. Chem. Phys., 2014, 16, 1132–1138 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee02608c |
| ‡ JPCB and LS contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |