
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Desalting large protein complexes during native electrospray mass spectrometry by addition of amino acids to the working solution†
David J.
Clarke
* and
Dominic J.
Campopiano
School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: dave.clarke@ed.ac.uk; Tel: +44 (0)131 6504808
First published on 19th February 2015
Abstract
Native mass spectrometry is a rapidly emerging field for characterising the structure of proteins and protein assemblies. The technique relies on electrospray ionisation (ESI) to efficiently ionise the protein analyte and transmit it into the gas phase with retention of protein structure, non-covalent protein-ligand and protein–protein interactions. In native ESI, both the ionisation efficiency and the resulting mass spectral signal is adversely effected by the presence of non-volatile inorganic salts, such as sodium chloride, which form extensive adducts with the protein ions. Consequently, there is great interest in finding experimental strategies that mitigate these phenomena. Here we report our findings that the addition of 10 mM L-serine to the ESI spray solution reduces the adverse effects of sodium adduction to proteins. In the analysis of bovine serum albumin (BSA; 66 kDa), 10 mM serine increased signal to noise ratio (S/N) ∼4 fold. This increase in sensitivity was accompanied by peak narrowing (∼10 fold), which allowed more precise assignment of molecular mass. Similar effects were observed when analysing protein complexes – serine palmitoyl transferase (SPT, a 92 kDa homodimer), enolase (a 93 kDa homodimer); and alcohol dehydrogenase (ADH, a 148 kDa tetramer). Reduction in sodium ion adduction occurs with no loss of the non-covalent protein–protein interactions, and with little effect on the overall observed charge state-distribution. As a consequence of increasing signal intensity, the addition of serine to the ESI spray solution greatly improved the quality of the data obtained from native top-down electron-capture dissociation (ECD) experiments. In ECD analysis of native BSA, we observed an increasing in the S/N of all ECD fragments upon addition of 10 mM L-serine. The number of ECD fragments we observed with S/N > 1.5 increased from 15 to 44 and the number of assigned c and z ions increased from 5 to 16. Finally we show that this phenomenon is not specific to L-serine, and occurs with several amino acids such as L-alanine. Our findings suggest that desalting may occur via binding of sodium ion to the amino acid in solution. This simple and inexpensive strategy has broad utility for improving the mass spectra obtained in a range of MS-based structural proteomic studies.
Introduction
Electrospray ionisation (ESI) mass spectrometry (MS) is routinely used to measure the molecular mass of intact proteins.1 In addition, it is now well established that, by careful control of the ESI conditions and solution composition, it is possible to retain protein–ligand and protein–protein, non-covalent interactions during ionisation and transmission into the gas phase. Consequently, protein complexes and assemblies can be directly monitored by ESI MS to yield insights into the composition, stoichiometry, and dynamics of multiprotein assemblies, a process now termed native MS.2–6 This technique has matured over the last decade, and it has proven itself a valuable technique for the analysis of: protein oligomerisation,7 protein–ligand complexes,8 heteromultimeric protein complex topology,9,10 membrane proteins;11 and intact analysis of virus capsids complexes up to several million Daltons in mass.12The preservation of protein higher order structure during native ESI relies on precise control of the solution conditions, which are usually required to be aqueous and at physiological pH. Achieving these conditions is hampered by the fact that non-volatile salts are detrimental to the performance of ESI mass spectrometry, even at micromolar concentrations. Their presence in solution can cause salt clusters, adduct formation and severe ion suppression, leading to a dramatically decrease signal to noise ratio (S/N).13,14 This is particularly pronounced with metal cations, such as Na+; and ESI mass spectra recorded in the presence of Na+ salts often display multiple nonspecific Na+ adducts;15 most prominently on low charge state ions.13 Thus, many traditional biological buffer solutions (tris, phosphate, phosphate-buffered saline (PBS) etc.) are incompatible with native ESI. Therefore, volatile buffer solutions are most often employed – typically ammonium acetate or ammonium bicarbonate; and protein sample preparation for native ESI often involves stringent removal of metal ion salts by dialysis, liquid chromatography or buffer exchange prior to analysis. However, complete removal of salts is not always possible and salt-induced ion suppression in native ESI is a commonly encountered problem.
Several strategies have been proposed to suppress sodium ion addition in native ESI. Lowering the pH to ∼3 units lower than the pI of the protein has been shown to lower salt adduction.13 However, the manipulation of pH to such an extent will often affect protein higher order structure in solution, and may result in loss of physiologically relevant structural information.16 Williams and co-workers found that high concentrations (>2 M) of ammonium acetate can reduce sodium adduction,15 presumably by displacement of Na+ with NH4+ in solution, followed by loss of NH3 in the gas phase.17 However, the success of this technique has been shown to be protein-dependant, and poorly resolved mass spectra have been reported for some large proteins analysed in high concentrations of ammonium acetate.11 The most promising strategies seem to involve inclusion of specific small molecule chemical additives to reduce the formation of non-specific metal-adducts. Konermann and co-workers demonstrated that calcium adduction could be reduced by addition of the solution phase chelator L-tartrate or citrate.18 More recently, promising results have been achieved by the addition of selected low proton affinity (PA) anions in native ESI solutions.19 Flick et al. demonstrated that dramatic increases in S/N could be achieved when analysing small proteins in the presence of 25 mM ammonium bromide or ammonium iodide.20 In addition, supercharging reagents (e.g. m-nitrobenzyl alcohol (m-NBA)) have been demonstrated to reduce salt adduction.21 When added to aqueous solutions (typically at <2% v/v) prior to ESI, these high-boiling point additives can be used to increase the charge state distribution observed in protein mass spectra.22,23 The exact mechanism of this phenomenon is still being investigated.24–27 However, Cassou et al. recently reported that this supercharging phenomenon is accompanied by a decrease in salt adduction, and can effectively be used to increase S/N in native protein mass spectra. It is important to note that, using this approach, care must be taken in controlling the solution conditions and exact concentration of supercharging reagents, as labile, non-covalent interactions can be lost during this process.28 Finally, a recent report has highlighted that the exposure of aqueous nanoelectrospray droplet to organic vapours (such as volatile alcohols) can dramatically reduce Na-adduction without inducing protein denaturation.29 This phenomenon is attributed to ion evaporation of clusters of the organic species with sodium cations during the nESI process; thereby depleting the remaining (protein containing) droplet of sodium ions. This approach holds great promise, although requires specific instrument modification to allow the introduction of organic vapour into the ESI source.
Here we report a serendipitous finding that the addition of free amino acids, at low millimolar concentrations, can efficiently reduce sodium adduction to protein ions formed during native ESI. In recent studies of the pyridoxal 5′-phosphate (PLP)-dependant enzyme serine palmitoyl transferase (SPT),30–32 we noted the surprising observation that addition of the enzyme substrate, L-serine, prior to native MS analysis significantly mitigated the adverse effect of sodium adduction. Here we show that this surprising observation also occurs with a range of amino acids additives and we demonstrate these findings by analysing a range of large protein systems. This phenomenon occurs with little observable change in charge state distribution, and without any apparent dissociation of protein–protein and protein–ligand interactions. Finally, we highlight the potential of using amino acid additives, and similar ‘desalting’ additives, to improve the efficiency of native top-down fragmentation experiments.
Experimental
Materials
Yeast alcohol dehydrogenase (ADH) and enolase were purchased as lyophilized powders from Sigma-Aldrich (St. Louis, MO, USA); bovine serum albumin (BSA, ultrapure) was purchased from Gerbu Biotechnik (Gaiberg). Serine Palmitoyl Transferase (SPT) from the bacterium S. paucimobilis, was expressed and purified as described previously.31,32 Ammonium acetate and sodium chloride were obtained from Sigma-Aldrich; all amino acids were acquired from Novabiochem. Ammonium acetate was prepared in LC-MS grade water (Chromasolv; Sigma-Aldrich).Sample preparation for native ESI MS
Lyophilized proteins were dissolved in 200 mM ammonium acetate, pH 7.0. To ensure the removal of trace salts, protein samples were buffer exchanged into 200 mM ammonium acetate, pH 7.0 using Micro-biospin 6 columns (Bio-Rad) immediately prior to MS analysis (following the protocol outlined in the manufacturer's instructions). If required, sodium chloride and amino acids were added from 10 fold stock solutions prior to MS analysis. Protein samples were typically analysed at an assembly concentration of 5 μM.Mass spectrometry
Mass spectrometry data was acquired on a SolariX FT-ICR mass spectrometer equipped with a 12 Tesla superconducting magnet (Bruker Daltonics). Nanoelectrospray (nESI) was performed using a Nanomate running in positive ion infusion mode and equipped with a HD_A_0 ESI Chip (Advion Bioscience). Typically, ionisation was achieved by applying a potential of 1.5–1.7 kV to the nESI Chip and the drying gas was set to 120 °C. The ion-transmission RF frequencies and the time-of-flight were tuned based on the size of the protein assembly analysed. Native mass spectra were typically acquired between m/z 500 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to yield a broadband 1 MW time-domain transient, ion accumulation was set at 1000 ms, and each spectrum was the sum of 100 acquisitions. The resulting mass spectra were externally calibrated using ES tuning mix (Agilent) and analysed using DataAnalysis software (Bruker Daltonics).
000 to yield a broadband 1 MW time-domain transient, ion accumulation was set at 1000 ms, and each spectrum was the sum of 100 acquisitions. The resulting mass spectra were externally calibrated using ES tuning mix (Agilent) and analysed using DataAnalysis software (Bruker Daltonics).
Top-down fragmentation was performed on a SolariX FT-ICR mass spectrometer using electron capture dissociation (ECD). No quadrupole isolation was performed prior to ECD, therefore all charge states of the protein assemblies were fragmented simultaneously. Typically, 1.6 A was applied to the dispenser cathode filament (Heatwave Technologies), 12 V to the lens, 0.6 V to the bias, and a pulse of between 5 and 14 ms was employed. Fragmentation data was the sum of 1000 acquisitions and data analyses were performed using DataAnalysis software (Bruker Daltonics). The SNAP 2.0 algorithm was used for automated peak picking. The resulting top-down fragment mass lists were searched against the protein primary sequence using Prosight-PTM software packages.33 Mass error tolerances were set for all searches at 15 ppm.
Results and discussion
The effect of sodium ion adduction in native protein mass spectra
In order to ascertain the effect that sodium cations has on native mass spectra of intact proteins, we began by analysing 5 μM Bovine Serum Albumin (BSA) by nESI from a solution of 200 mM ammonium acetate, pH 7.0 with the addition of varying concentration of NaCl (Fig. 1). In similar experiments performed by Williams and co-workers on small proteins (ubiquitin, cytochrome C and myoglobin), differing sodium adducts ([M + nNa + mH](n+m)+) are easily mass resolved and each individual adducted form can observed. Thus an average number of sodium adducts on each charge state can easily be calculated.20 However, for analytes larger than 25 kDa, the isotope distributions of the individual adducted forms overlap. Consequently, salt adduction on larger proteins appears in the mass spectrum as a broadening of each charge state. Therefore we assessed the extent of salt adduction by monitoring the peak width of individual charge states. | ||
| Fig. 1 Native mass spectra of Bovine Serum Albumin (66 kDa) analysed by nESI from aqueous solution containing 200 mM ammonium acetate, pH 7.0 with (A) 0 μM NaCl (B) 200 μM NaCl (C) 500 μM NaCl (D) 1 mM NaCl. Left, the calculated charge states are indicated and monomer charge states are highlighted by red lines; dimeric charge states by blue lines. The spectra displayed are the sum of 100 scans and the intensity scales are equivalent for each spectrum. Right, expansions of the 15+ charge state of monomeric BSA; for clarity the intensities are normalised. The red bars indicate the FWHM peak width for each charge state. | ||
The native mass spectrum of BSA presents in 5 major charge states, which correspond to the +14 to +18 forms of monomeric BSA (Fig. 1A–D; highlighted in red). In addition, a second charge state distribution is observed which corresponds to the +24 to +26 charge states of a BSA dimer (Fig. 1A–D; highlighted in blue); presumably formed by non-specific association in the gas phase. From the native spectrum of BSA with no added NaCl (Fig. 1A) an average molecular mass of 66485 ± 26 Da can be measured; which is in close agreement with the calculated theoretical value of 66459 Da (the red line indicates calculated theoretical m/z for the 15+ species assuming 18 disulfide bonds, and retention of the proposed Zn2+ ligand during native ESI). Increasing the concentration of NaCl in the native ESI solution has a detrimental effect on S/N ratio (Fig. 1A–D, left hand side). In our hands addition of 1 mM NaCl (Fig. 1D) resulted in a ∼4-fold reduction in S/N. This may seem modest; however, native MS of large proteins and protein complexes is a comparatively low sensitivity technique, the user is often required to accumulate scans for several minutes to obtain adequate spectral data. S/N increases proportionally with the square root of the number of scans summed. Therefore, in order to obtain comparable data, the addition of 1 mM NaCl requires a ∼16 fold increase in the acquisition time w.r.t. a spectrum acquired in the absence of salt. Furthermore, the presence of NaCl also results in peak broadening, due to sodium adduction (Fig. 1A–D, right hand side). By estimating the full width at half maximal height (FWHM) for the 15+ charge state of BSA (red bar) the extent of peak broadening can be estimated as ∼8-fold by the addition of 1 mM NaCl. This broadening results in a reduction in the mass accuracy obtainable from the analysis; here the average molecular mass can be calculated to be 66878 ± 289 Da. In addition, peak broadening potentially obscures the presence of different proteoforms of the analyte (e.g. post translationally modified (PTM) forms, ligand-bound forms etc.).
Addition of L-serine can reduce sodium adduction in native protein mass spectra
In recent studies of the pyridoxal 5′-phosphate (PLP)-dependant enzyme serine palmitoyl transferase (SPT), we noted that addition of the enzyme substrate (L-serine), prior to native MS analysis mitigated the adverse effect of sodium adduction.32 Interestingly, this phenomenon was also observed when analysing BSA by native ESI (Fig. 2A–D). By titrating L-serine from 0–10 mM there was a clearly observable sharpening of all the peaks in the charge state distribution. The effect was dependent on the amino acid concentration, with near complete removal of Na+ adducts observed when MS analysis was performed in a solution that contained 200 mM ammonium acetate (pH 7.0), 1 mM NaCl and 10 mM L-serine (Fig. 2D). Native MS analysis of this BSA sample resulted in a spectrum with ∼4 fold increase in overall S/N and a peak-width reduction of ∼10 fold (Fig. 2D, right hand side). Thus, with the addition of 10 mM L-serine, it is possible to assign the average mass of the BSA species with far higher mass accuracy −66439 ± 32 Da (theoretical value 66459 Da) despite the presence of 1 mM NaCl. We found that a ratio of ∼10:1(L-serine:NaCl) was optimal for reduction in sodiation; and the strategy appeared to be less effective for concentrations of NaCl over 2 mM.
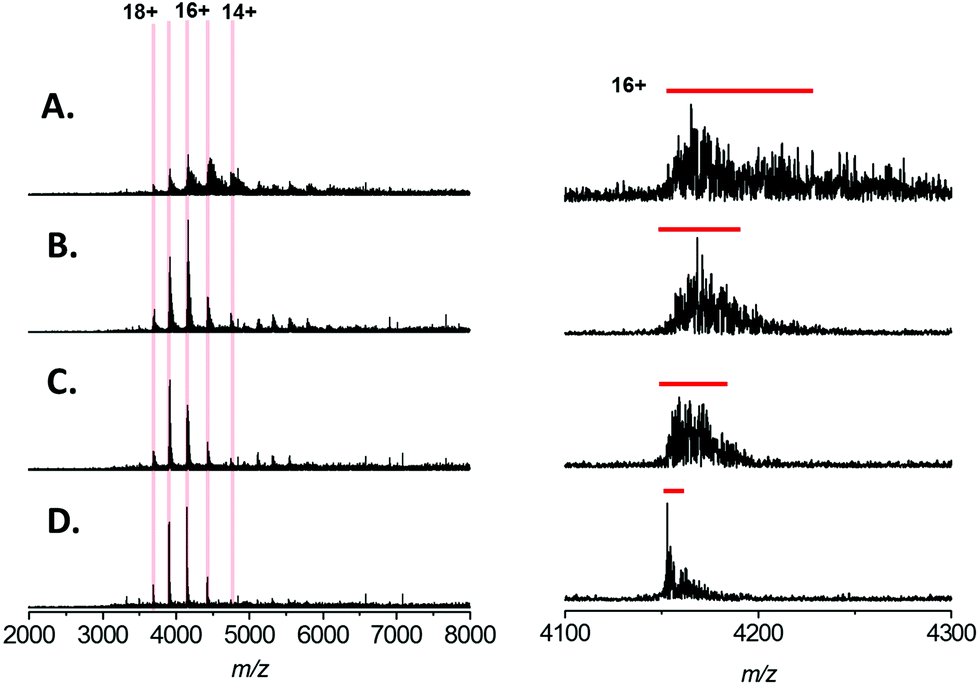 | ||
| Fig. 2 Native mass spectra of Bovine Serum Albumin (64 kDa) analysed by nESI from aqueous solution containing 200 mM ammonium acetate, pH 7.0, 1 mM NaCl with (A) 0 mM L-serine (B) 2 mM L-serine (C) 5 mM L-serine (D) 10 mM L-serine. Left, the calculated charge states for monomeric BSA are indicated and the spectra displayed are the sum of 100 scans. The intensity scales are equivalent for each spectrum. Right, expansions of the 16+ charge state of BSA; for clarity the intensities are normalised. The red bars indicate the FWHM peak width for each charge state. | ||
We subsequently extended our study to include multimeric proteins (Fig. 3A–D) – the homodimeric proteins SPT (92 kDa), yeast enolase (93 kDa); and the homotetramer yeast alcohol dehydrogenase (ADH; 148 kDa). This apparent ability of L-serine to reduce sodium adduction to protein analytes was observed for all of the native proteins analysed. On addition of L-serine, peak narrowing was evident in all of the systems studied, and S/N was increased, albeit by varying amounts (∼8 fold for SPT, ∼4 fold for enolase, ∼2 fold for ADH). Crucially, this effect occurred without dissociation of protein multimers and without evident disruption of the higher order structure of the gas phase protein assemblies. Furthermore, the charge state distribution observed for each protein assembly remained predominantly unchanged – suggesting that the addition of L-serine has no significant effect on the structural conformation of the protein multimers.34,35 Thus, it seems that this additive is a potentially powerful strategy for improving the quality of the mass spectra obtained during structural interrogations of proteins by native MS. Such studies, for example – ion mobility experiments6,36,37 or protein topology/disassembly studies by gas phase dissociation,38,39 rely on retaining solution-phase, higher order protein structure during MS analysis.
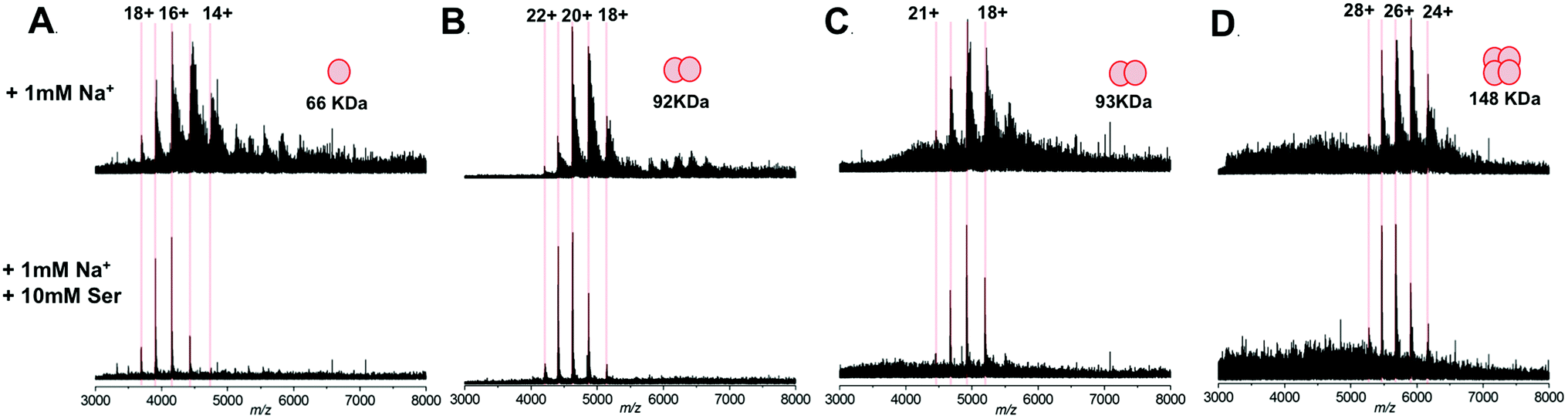 | ||
| Fig. 3 Native mass spectra of protein assemblies analysed by nESI from aqueous solution containing 200 mM ammonium acetate, pH 7.0, 1 mM NaCl; without (top) and with (bottom) the addition of 10 mM L-serine. (A) monomeric BSA (66 kDa); (B) dimeric serine palmitoyltransferase, SPT (92 kDa); (C) dimeric yeast enolase (93 kDa); (D) tetrameric yeast alcohol dehydrogenase, ADH (148 kDa). The calculated charge states for each protein assembly are indicated and the spectra displayed are the sum of 100 scans, for clarity the intensities of each spectra are normalised. | ||
Addition of L-serine increases the sequence coverage obtained during native top-down fragmentation of proteins
Native MS techniques are becomes increasing important approaches for investigating protein structure, particularly in the analysis of protein targets which prove problematic, or which are still not tractable, using traditional structural biology approaches (e.g. intrinsically disordered proteins,40 membrane proteins,11 and heterogeneous protein populations due to PTMs). For the most part, native MS studies are performed on quadrupole/time-of-flight (Q-ToF) instruments, often with ion mobility capability, due to the instruments inherent high upper mass limit – required for analysis of native proteins and large protein assemblies. However, recent studies have demonstrated that high resolution Fourier transform instruments also have great potential in this field. Heck and co-workers have developed a platform for native protein MS based on an Orbitrap instrument, and recently demonstrated that in-house modifications allowed efficient detection of capsid assemblies up to 4.5 MDa in mass.41–43 In contrast, several groups have utilised Fourier transform ion cyclotron resonance (FT-ICR) instruments for native protein MS studies (see below). Although the inherent upper mass limit of FT-ICR MS is yet not comparable to studies using Q-Tof and/or Orbitrap analysers, future developments in instrumentation and magnet design will increase their range in the upcoming years. However, the use of FT-based instrumentation has allowed protein complexes to be interrogated using the suite of top-down fragmentation techniques available on these instruments.Of all these techniques, electron-capture dissociation (ECD), developed in the McLafferty labs in 1998,44,45 has garnered the most interest; and perhaps has the most potential.46 The exact mechanism of ECD is still under investigation.47 However, it is thought to be a nonergodic process with backbone fragmentation occurring before energy randomization and without disruption of non-covalent interactions.48 Thus, it has been postulated that ECD fragmentation of native proteins and protein complexes could be utilised to report on protein higher order structure and protein–ligand binding.
Loo and co-workers first demonstrated that ECD could be used to locate ligand-binding sites when interrogating the non-covalent complex between alpha-synuclein and spermine.49 Subsequent work from our research group demonstrated that this technique could be extended to the study of protein–peptide interactions.50 More recently, a series of studies by Gross and colleagues have demonstrated ECD of native proteins and protein complexes may provide important structural information – particularly in the technique's ability to highlight regions of structural flexibility.51–53 These finding are supported by reports from the Loo group, who have demonstrated ECD of alcohol dehydrogenase (148 kDa) and aldolase (158 kDa) tetramers under native conditions.54,55 Their findings suggest that, in addition to regions of flexibility, regions located on the surface topology of the complex are prone to ECD cleavage.
It is clear therefore that ECD of native protein complexes shows great potential as a structural biology technique. However, ECD is inherently a low efficiency process. Therefore, in order to acquire ECD spectra with fragment ion intensities of sufficient S/N for accurate mass assignment, the precursor signal must be relatively high intensity (compared to top-down fragmentation using higher efficiency techniques, such as CID). Furthermore, spectra often have to be the sum of several hundred scans, resulting in long experimental acquisition times. Unfortunately, ECD efficiency is known to increase quadratically with increasing charge.44 Therefore the fact that the conditions required for native MS result in protein charge state distributions of low charge further compounds these issues. In order to mitigate these drawbacks, it is apparent that the success of native ECD experiments is reliant on the production of high intensity precursor ions. Therefore we postulated that the use of L-serine as an additive in native protein nESI solutions, to reduce the detrimental effects of Na+ adduction, could be beneficial and increase the quality of ECD fragmentation data. Thus, we performed ECD of BSA under native conditions, with and without the addition of 10 mM L-serine to the ESI solution (Fig. 4A–C). A typical ECD spectrum is shown in Fig. 4A, which clearly shows that the majority of electron capture during ECD results in no dissociation (EC no D). The positive protein ion captures an electron, reducing the ion's charge; as no backbone fragmentation occurs we observe a shift in the observed charge state distribution to higher m/z. This observation is well documented and highlights the inefficiency of native ECD experiments.52 However, after summing several hundred scans, we do observe ECD fragmentation products in the region 500–2500 m/z. Fig. 4B shows an expansion of this region of the mass spectrum and the ECD spectra are compared between nESI solutions with (bottom) and without (top) 10 mM L-serine. It is clear that addition of the L-serine results in ECD fragments of higher abundance. This is most pronounced with higher mass fragments Automated peak picking was performed using the SNAP 2.0 algorithm (Bruker Daltonics), and ECD peak lists were compiled (see ESI Tables†). These peak lists were then searched against the primary amino acid sequence of BSA using the Prosight PTM software,33 taking into account the known PTM profile of BSA (removal of the signal-peptide and propeptide sequence, aa1–24, and including 18 disulfide bonds). Although ECD fragments are present in both spectra, it is clear that the addition of L-serine improved the quality of the fragmentation data obtained under identical instrument conditions. 15 ECD peaks were observed in the spectrum analysed without addition of serine (Fig. 4B, top), and 5 of these were assigned as specific ECD c and z ions (see Table 1). In contrast, addition of 10 mM L-serine to the nESI solution resulted in 44 ECD peaks, 16 of which could be assigned as specific c and z ions (Fig. 4B, bottom, see Table 1; a full list of ECD ions can be found in ESI†). The observed S/N ratio of all fragments was higher in the spectrum acquired with L-serine in the nESI solution; and of the 5 fragment ions common in both spectra, the S/N ratio was improved by an average of 187%.
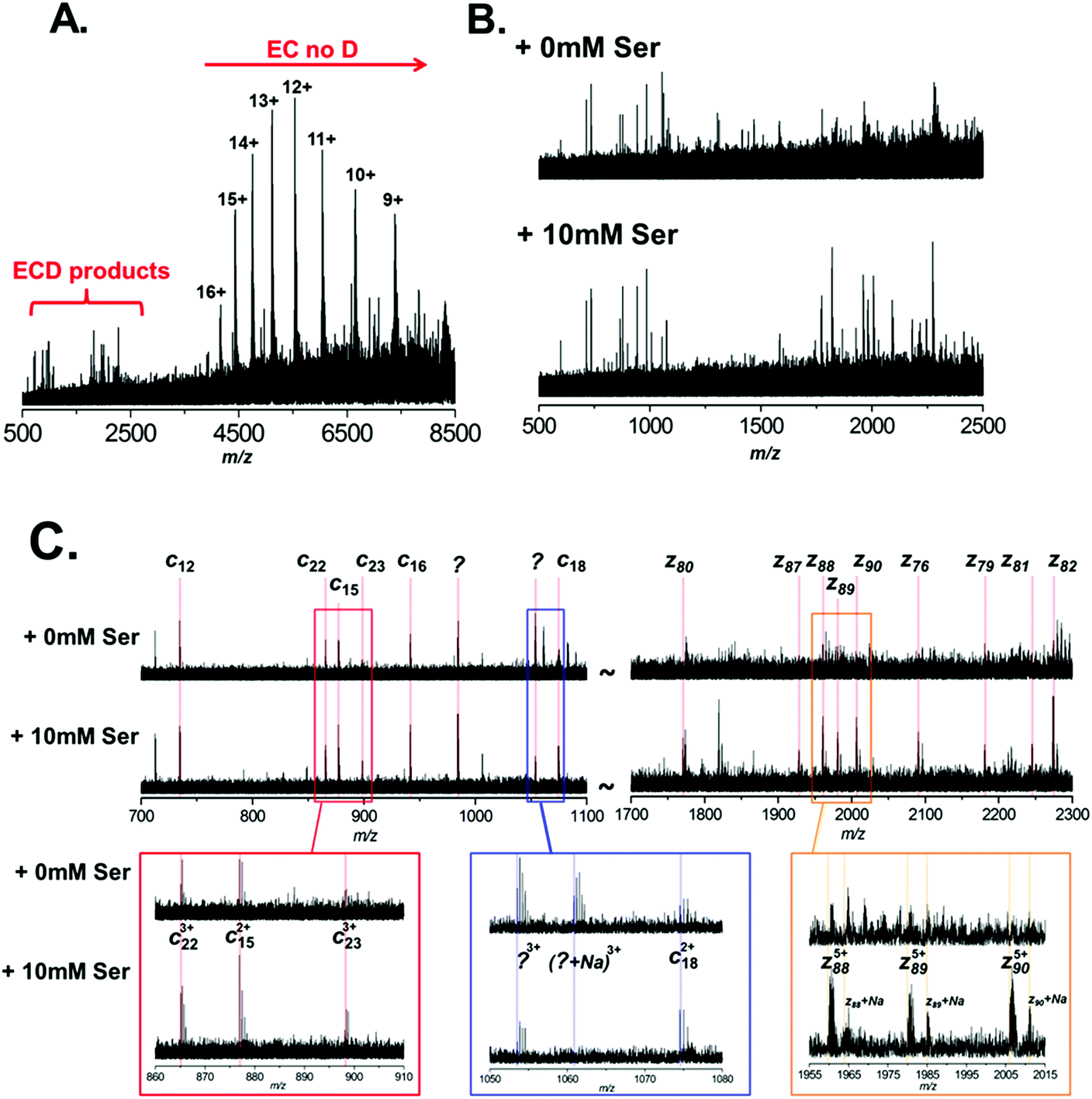 | ||
| Fig. 4 Electron-capture dissociation of BSA under native conditions. BSA was analysed from 200 mM ammonium acetate (pH 7.0), 1 mM NaCl with or without the addition of 10 mM L-serine. No precursor ion selection was performed, and entire BSA charge state was subject to ECD. (A) A typical ECD spectrum of BSA, the region where fragmentation products are observed is highlighted. (B) Expansion of the ECD spectrum in the region between m/z 500 to 2500. Here the ECD products of BSA with (B, bottom) and without (B, top) the addition of 10 mM L-serine are compared. (C) Further expansion of two regions (m/z 700 to 1100 and m/z 1700 to 2300) of the ECD spectrum. Here specific fragment ions are annotated; again ECD products of BSA with (C, bottom) and without (C, top) the addition of 10 mM L-serine are compared. | ||
| 10 mM ser | 0 mM ser | ||||
|---|---|---|---|---|---|
| Frag ion | m/z | z | Mol. mass (Da) | S/N | S/N |
| c12 | 734.3894 | 2+ | 1467.7715 | 6.7 | 4.8 |
| c15 | 876.9537 | 2+ | 1752.9001 | 5.5 | 3.6 |
| c16 | 941.4735 | 2+ | 1881.9397 | 5.8 | 3.7 |
| c18 | 1074.5235 | 2+ | 2148.0396 | 4.1 | <1.5 |
| c22 | 865.1071 | 3+ | 2593.3066 | 4.8 | 2.8 |
| c23 | 898.1285 | 3+ | 2692.3709 | 2.7 | <1.5 |
| z76 | 2089.2917 | 4+ | 8354.145 | 2.9 | N/O |
| z79 | 2179.8397 | 4+ | 8716.337 | 2.7 | N/O |
| z80 | 1769.6961 | 5+ | 8844.4513 | 2 | N/O |
| z81 | 2244.1284 | 4+ | 8973.4919 | 2.2 | N/O |
| z82 | 2272.8797 | 4+ | 9088.497 | 5.7 | <1.5 |
| z87 | 1926.7695 | 5+ | 9629.8183 | 1.8 | N/O |
| z88 | 1959.3803 | 5+ | 9792.8723 | 4.7 | 1.5 |
| z89 | 1979.5884 | 5+ | 9893.9127 | 3.2 | <1.5 |
| z90 | 2005.5986 | 5+ | 10023.9639 |
4.5 | N/O |
Fig. 4C highlights the improvement observed in S/N for several of these ions. The most striking example is observed in the region between m/z 1700 and 2300. For the spectrum acquired with L-serine, in this region a series of 9 z-ions (z76 to z90) are observed with S/N > 1.5, which allows clear determination of the charge-state for each ion and confident fragment assignment. In contrast, in the spectrum acquired without L-serine these peaks are not fully resolved and are not observed with sufficient S/N to assign with confidence. It is interesting to note that this set of z ions, used as a sequence tag, is sufficient to allow identification of the protein from the NCBI database.
As expected we also noted that Na+ adduction to fragment ions is far more prevalent in the spectrum acquired without L-serine. For example, the z885+ species is observed primarily in the [z88 + 5H]5+ form in the spectrum acquired with L-serine (Fig. 4C, lower spectrum, gold box). In contrast, the same ion is observed as several multiply-sodiated species ([z88 + 4H + Na]5+, [z88 + 3H + 2Na]5+etc.) in the spectrum without L-serine (Fig. 4C, upper spectrum, gold box).
Reduction in sodium adduction is observed upon addition of a range of amino acids and may be due to direct binding of sodium ions
Finally, we tested a range of amino acids (glycine, L-alanine, L-serine, L-histidine, and L-lysine) for their ability to reduce sodium adduction to proteins (Fig. 5A–F). It was apparent that all five amino acids tested did reduce sodium adduction to some extent, demonstrating that this phenomenon isn't specific to L-serine, and is largely independent of the side chain chemistry of the amino acid. The physical basis for this phenomenon remains unclear, and will be the basis of future investigations. However, it is interesting to note that the effectiveness of these amino acid additives at reducing sodium adduction seems to trend with the gas phase sodium affinities of the amino acids. Based on empirical findings, the amino acids have been ordered in increasing Na+-affinities as follows: Gly, Ala, Cys, Val, (Leu, lle), Ser, Met, Thr, (Phe, Pro), Asp, Tyr, (Glu, Lys), Trp, Asn, Gln, His, Arg.56 It is particularly noticeable that glycine and, to some extent, alanine (the two amino acids with the lowest sodium affinities) are less effective than L-serine as additives (Fig. 5B and C). This correlation suggests a mechanism whereby the free amino acid, which is present in the nESI solution at relatively high concentrations, directly sequesters Na+ in solution. In addition, we noted that addition of L-lysine or L-histidine resulted in a significant change in the charge state distribution of the protein, with a noticeable shift to lower charge (Fig. 5E and F). A possible explanation for this observation may be proposed based on the recent exploration of the ESI mechanism by Ogorzalek Loo et al.25 and the high proton affinities of these amino acids. As these amino acids are present in such high concentrations and are sufficiently ionisable, it may be that they consume a significant portion of the gas phase charge emitted by the ESI source; thus reducing the charge available to the protein during ESI and driving the shift in charge state distribution of the protein.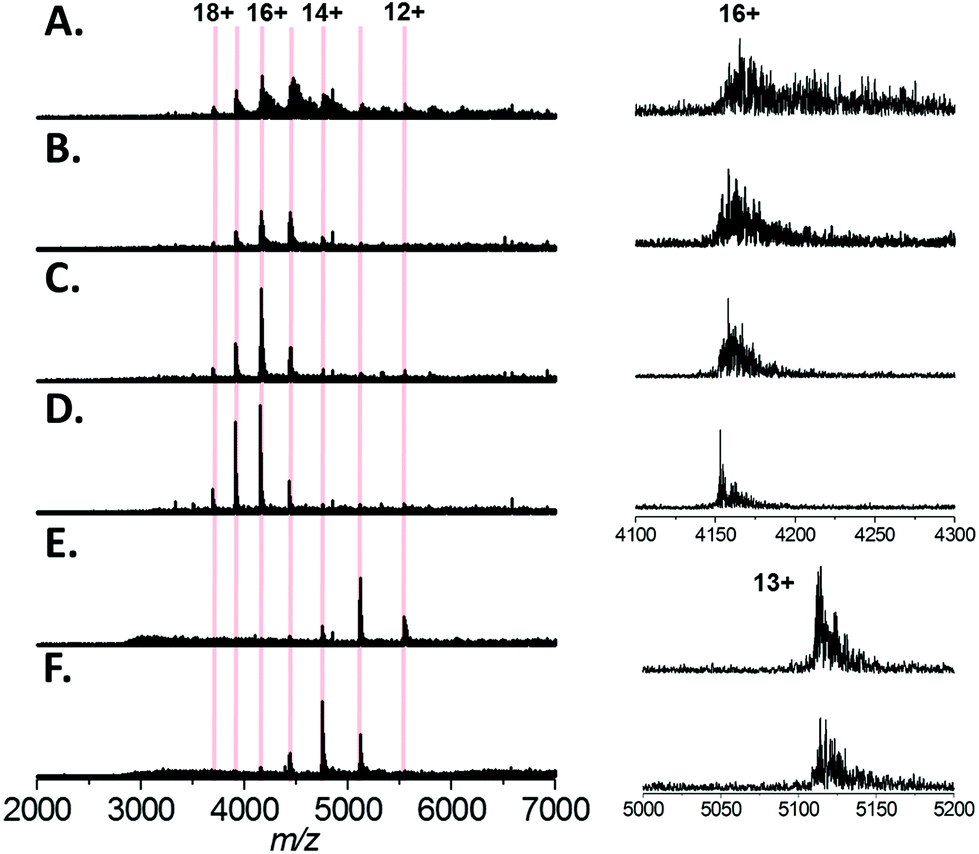 | ||
| Fig. 5 Native mass spectra of Bovine Serum Albumin (64 kDa) analysed by nESI from aqueous solution containing 200 mM ammonium acetate, pH 7.0, 1 mM NaCl with (A) no additive (B) 10 mM glycine (C) 10 mM L-alanine (D) 10 mM L-serine (E) 5 mM L-histidine (F) 5 mM L-lysine. Left, the calculated charge states for monomeric BSA are indicated. The spectra displayed are the sum of 100 scans and the intensity scales are equivalent for each spectrum. Right, the expansions of the 16+ or 13+ charge state of BSA is shown; for clarity the intensities are normalised. | ||
Conclusions
Here we report the observation that addition of millimolar concentrations of amino acids can mitigate the adverse effects of sodium adduction in native protein mass spectrometry. Addition of 10 mM L-serine to 200 mM ammonium acetate (pH 7.0) in native ESI spray solution containing 1 mM NaCl results in almost complete removal of Na+ adducts in the resulting mass spectra; enhancing the S/N for protein ions and allowing more accurate intact mass measurement of large proteins and protein assemblies. This effect occurs without disruption of non-covalent protein assemblies, and with little effect on the protein charge state distribution. We demonstrate that by using this additive to improve the signal generated in native protein MS analyses, we can increase the information gained in ECD fragmentation experiments. This strategy of removing detrimental sodium adducts in protein mass spectra by inclusion of specific additives to the ESI solution has widespread utility in MS-based structural proteomic studies; and offers a readily available, inexpensive method for improving analytical MS data without the need for instrument modification.Acknowledgements
This work was supported by the BBSRC (grant award BB/I013687/1) and a University of Edinburgh Chancellor's Fellowship awarded to D. Clarke. We wish to thank Logan Mackay for helpful discussions and technical support. Finally, we wish to thank the both peer-reviewers for helpful comments and suggestions concerning the mechanism of this reported phenomenon.References
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CAS.
- A. J. R. Heck, Nat. Methods, 2008, 5, 927–933 CrossRef CAS PubMed.
- J. L. P. Benesch, B. T. Ruotolo, D. A. Simmons and C. V. Robinson, Chem. Rev., 2007, 107, 3544–3567 CrossRef CAS PubMed.
- B. T. Ruotolo, K. Giles, I. Campuzano, A. M. Sandercock, R. H. Bateman and C. V. Robinson, Science, 2005, 310, 1658–1661 CrossRef CAS PubMed.
- J. A. Loo, Mass Spectrom. Rev., 1997, 16, 1–23 CrossRef CAS.
- A. Konijnenberg, A. Butterer and F. Sobott, Biochim. Biophys. Acta, 2013, 1834, 1239–1256 CrossRef CAS PubMed.
- L. A. Woods, S. E. Radford and A. E. Ashcroft, Biochim. Biophys. Acta, 2013, 1834, 1257–1268 CrossRef CAS PubMed.
- K. J. Pacholarz, R. A. Garlish, R. J. Taylor and P. E. Barran, Chem. Soc. Rev., 2012, 41, 4335–4355 RSC.
- H. Hernández, A. Dziembowski, T. Taverner, B. Séraphin and C. V. Robinson, EMBO Rep., 2006, 7, 605–610 Search PubMed.
- H. Hernández and C. V. Robinson, Nat. Protoc., 2007, 2, 715–726 CrossRef PubMed.
- A. Laganowsky, E. Reading, J. T. S. Hopper and C. V. Robinson, Nat. Protoc., 2013, 8, 639–651 CrossRef CAS PubMed.
- C. Uetrecht and A. J. R. Heck, Angew. Chem., Int. Ed., 2011, 50, 8248–8262 CrossRef CAS PubMed.
- P. Pan and S. A. McLuckey, Anal. Chem., 2003, 75, 5468–5474 CrossRef CAS.
- H. J. Sterling, J. D. Batchelor, D. E. Wemmer and E. R. Williams, J. Am. Soc. Mass Spectrom., 2010, 21, 1045–1049 CrossRef CAS PubMed.
- A. T. Iavarone, O. A. Udekwu and E. R. Williams, Anal. Chem., 2004, 76, 3944–3950 CrossRef CAS PubMed.
- R. R. Abzalimov, C. E. Bobst, P. A. Salinas, P. Savickas, J. J. Thomas and I. A. Kaltashov, Anal. Chem., 2013, 85, 1591–1596 CrossRef CAS PubMed.
- K. B. Turner, S. A. Monti and D. Fabris, J. Am. Chem. Soc., 2008, 130, 13353–13363 CrossRef CAS PubMed.
- J. Pan, K. Xu, X. Yang, W.-Y. Choy and L. Konermann, Anal. Chem., 2009, 81, 5008–5015 CrossRef CAS PubMed.
- T. G. Flick, S. I. Merenbloom and E. R. Williams, J. Am. Soc. Mass Spectrom., 2011, 22, 1968–1977 CrossRef CAS PubMed.
- T. G. Flick, C. a. Cassou, T. M. Chang and E. R. Williams, Anal. Chem., 2012, 84, 7511–7517 CrossRef CAS PubMed.
- C. A. Cassou and E. R. Williams, Analyst, 2014, 139, 4810–4819 RSC.
- A. T. Iavarone, J. C. Jurchen and E. R. Williams, Anal. Chem., 2001, 73, 1455–1460 CrossRef CAS.
- S. H. Lomeli, S. Yin, R. R. Ogorzalek Loo and J. A. Loo, J. Am. Soc. Mass Spectrom., 2009, 20, 593–596 CrossRef CAS PubMed.
- H. J. Sterling, M. P. Daly, G. K. Feld, K. L. Thoren, A. F. Kintzer, B. A. Krantz and E. R. Williams, J. Am. Soc. Mass Spectrom., 2010, 21, 1762–1774 CrossRef CAS PubMed.
- R. R. Ogorzalek Loo, R. Lakshmanan and J. A. Loo, J. Am. Soc. Mass Spectrom., 2014, 25, 1675–1693 CrossRef CAS PubMed.
- K. A. Douglass and A. R. Venter, J. Am. Soc. Mass Spectrom., 2012, 23, 489–497 CrossRef CAS PubMed.
- K. Chingin, N. Xu and H. Chen, J. Am. Soc. Mass Spectrom., 2014, 25, 928–934 CrossRef CAS PubMed.
- H. J. Sterling, A. F. Kintzer, G. K. Feld, C. A. Cassou, B. A. Krantz and E. R. Williams, J. Am. Soc. Mass Spectrom., 2012, 23, 191–200 CrossRef CAS PubMed.
- J. C. DeMuth and S. A. McLuckey, Anal. Chem., 2014, 87, 1210–1218 CrossRef PubMed.
- J. Lowther, B. A. Yard, K. A. Johnson, L. G. Carter, V. T. Bhat, M. C. C. Raman, D. J. Clarke, B. Ramakers, S. A. McMahon, J. H. Naismith and D. J. Campopiano, Mol. BioSyst., 2010, 6, 1682–1693 RSC.
- A. E. Beattie, D. J. Clarke, J. M. Wadsworth, J. Lowther, H.-L. Sin and D. J. Campopiano, Chem. Commun., 2013, 49, 7058–7060 RSC.
- J. M. Wadsworth, D. J. Clarke, S. A. McMahon, J. P. Lowther, A. E. Beattie, P. R. R. Langridge-Smith, H. B. Broughton, T. M. Dunn, J. H. Naismith and D. J. Campopiano, J. Am. Chem. Soc., 2013, 135, 14276–14285 CrossRef CAS PubMed.
- R. D. Leduc and N. L. Kelleher, Curr. Protoc. Bioinfo., 2007, 13.6.1–13.6.28 Search PubMed.
- I. A. Kaltashov and R. R. Abzalimov, J. Am. Soc. Mass Spectrom., 2008, 19, 1239–1246 CrossRef CAS PubMed.
- O. M. Hamdy and R. R. Julian, J. Am. Soc. Mass Spectrom., 2012, 23, 1–6 CrossRef CAS PubMed.
- C. Uetrecht, R. J. Rose, E. van Duijn, K. Lorenzen and A. J. R. Heck, Chem. Soc. Rev., 2010, 39, 1633–1655 RSC.
- B. T. Ruotolo, J. L. P. Benesch, A. M. Sandercock, S.-J. Hyung and C. V. Robinson, Nat. Protoc., 2008, 3, 1139–1152 CrossRef CAS PubMed.
- M. Zhou and V. H. Wysocki, Acc. Chem. Res., 2014, 47, 1010–1018 CrossRef CAS PubMed.
- Z. Hall, H. Hernández, J. a. Marsh, S. a. Teichmann and C. V. Robinson, Structure, 2013, 21, 1325–1337 CrossRef CAS PubMed.
- R. Beveridge, Q. Chappuis, C. Macphee and P. Barran, Analyst, 2013, 138, 32–42 RSC.
- S. Rosati, R. J. Rose, N. J. Thompson, E. van Duijn, E. Damoc, E. Denisov, A. Makarov and A. J. R. Heck, Angew. Chem., Int. Ed., 2012, 51, 12992–12996 CrossRef CAS PubMed.
- R. J. Rose, E. Damoc, E. Denisov, A. Makarov and A. J. R. Heck, Nat. Methods, 2012, 9, 1084–1086 CrossRef CAS PubMed.
- J. Snijder, M. van de Waterbeemd, E. Damoc, E. Denisov, D. Grinfeld, A. Bennett, M. Agbandje-McKenna, A. Makarov and A. J. R. Heck, J. Am. Chem. Soc., 2014, 136, 7295–7299 CrossRef CAS PubMed.
- R. A. Zubarev, D. M. Horn, E. K. Fridriksson, N. L. Kelleher, N. A. Kruger, M. A. Lewis, B. K. Carpenter and F. W. McLafferty, Anal. Chem., 2000, 72, 563–573 CrossRef CAS.
- R. Zubarev, N. L. Kelleher and F. W. McLafferty, J. Am. Chem. Soc., 1998, 120, 3265–3266 CrossRef CAS.
- H. J. Cooper, K. Håkansson and A. G. Marshall, Mass Spectrom. Rev., 2005, 24, 201–222 CrossRef CAS PubMed.
- E. A. Syrstad and F. Turecek, J. Am. Soc. Mass Spectrom., 2005, 16, 208–224 CrossRef CAS PubMed.
- R. A. Zubarev, Curr. Opin. Biotechnol., 2004, 15, 12–16 CrossRef CAS PubMed.
- Y. Xie, J. Zhang, S. Yin and J. A. Loo, J. Am. Chem. Soc., 2006, 128, 14432–14433 CrossRef CAS PubMed.
- D. J. Clarke, E. Murray, T. Hupp, C. L. Mackay and P. R. R. Langridge-Smith, J. Am. Soc. Mass Spectrom., 2011, 22, 1432–1440 CrossRef CAS PubMed.
- H. Zhang, W. Cui, J. Wen, R. E. Blankenship and M. L. Gross, J. Am. Soc. Mass Spectrom., 2010, 21, 1966–1968 CrossRef CAS PubMed.
- H. Zhang, W. Cui, J. Wen, R. E. Blankenship and M. L. Gross, Anal. Chem., 2011, 83, 5598–5606 CrossRef CAS PubMed.
- L. M. Jones, H. Zhang, W. Cui, S. Kumar, J. B. Sperry, J. a. Carroll and M. L. Gross, J. Am. Soc. Mass Spectrom., 2013, 24, 835–845 CrossRef CAS PubMed.
- H. Li, P. Wongkongkathep, S. L. Van Orden, R. R. Ogorzalek Loo and J. A. Loo, J. Am. Soc. Mass Spectrom., 2014, 25, 2060–2068 CrossRef CAS PubMed.
- H. Li, J. J. Wolff, S. L. Van Orden and J. A. Loo, Anal. Chem., 2014, 86, 317–320 CrossRef CAS PubMed.
- G. Bojesen, T. Breindahl and U. N. Andersen, Org. Mass Spectrom., 1993, 28, 1448–1452 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4an02334j |
| This journal is © The Royal Society of Chemistry 2015 |