Radical vinylation of dioxolanes and N-acylpyrrolidines using vinyl bromides†‡
Takashi
Kippo
,
Yuki
Kimura
,
Ayami
Maeda
,
Hiroshi
Matsubara
,
Takahide
Fukuyama
and
Ilhyong
Ryu
*
Department of Chemistry, Graduate School of Science, Osaka Prefecture University, Sakai, Osaka 599-8531, Japan. E-mail: ryu@c.s.osakafu-u.ac.jp; Fax: +81(72)254-9695
First published on 19th June 2014
Abstract
Radical vinylation was investigated using vinyl bromides with an electron-withdrawing substituent at the β-position. The vinylation of 1,3-dioxolanes proceeded well to give 2-vinyl-1,3-dioxolanes in good yields. The α-vinylation of N-acylpyrrolidines also proceeded well to give 2-vinyl-N-acylpyrrolidines.
The addition/fragmentation reaction of carbon-centered radicals with hetero-substituted alkenes provides a potentially useful method for vinylation (Scheme 1),1 and thus far a variety of vinylating agents have proven useful for this purpose. These include vinyltins,2 vinylsulfides,3 vinyl sulfones,4 β-nitrostyrenes,5 vinyl indiums,6 vinyl galliums,6b and vinyl chlorides.7
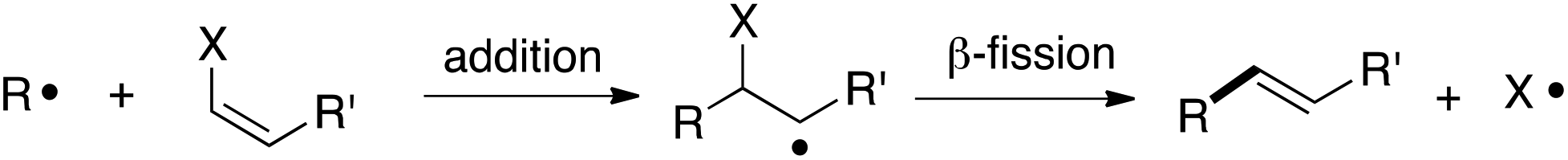 | ||
| Scheme 1 Radical vinylation using vinylating agents. | ||
Since allyl bromides can serve as useful radical allylating reagents via radical addition/fragmentation reactions,8–10 we naturally thought that vinyl bromides would also serve as potential radical vinylating reagents, although previous research examples are rare. In 1993, Singleton and Huval reported a radical vinylation reaction of alkyl bromides with 3-bromo-2-methylacrylonitrile in the presence of stoichiometric hexabutylditin.11 In this reaction, ditin played a critical role in the conversion of a bromine radical to a tin radical, which was capable of abstracting a bromine atom from alkyl bromides to generate alkyl radicals. More recently, Heinrich and coworkers reported a non-chain radical addition of aryl radicals to ethyl 3-bromoacrylate, in which an aryl radical was formed by the one-electron reduction of an aryl diazonium salt by an iron(II) salt.12 In a recent report by Liang and coworkers the α-vinylation of THF with 1,2-dibromostyrene took place in the presence of sodium fluoride as a base. This reaction suffered from poor chain propagation and therefore harsh conditions (120 °C, 27 h) were employed.13 We reasoned that the less-effective chain propagation of vinyl bromides compared with that of allyl bromides may have been due to the steric hindrance of vinyl carbon due to an α-bromine atom substituent slowing the radical addition step. In order to achieve effective radical vinylation with vinyl bromides, we thought that two items were crucial: (i) the judicious choice of easily broken C–H bonds and (ii) the introduction of a polar factor into vinyl bromides in order to encourage addition. In this work, we report that the vinylation of 1,3-dioxolanes 1, readily available from aldehydes and 1,2-ethylene glycol, with vinyl bromides 2 having an electron-deficient substituent at the 2-position proceeded smoothly to give α-vinylated products in good yields (Scheme 2, eqn (1)). We also found that the α-vinylation of N-acylpyrrolidines 5 was successful using a similar vinylation reaction (eqn (2)).
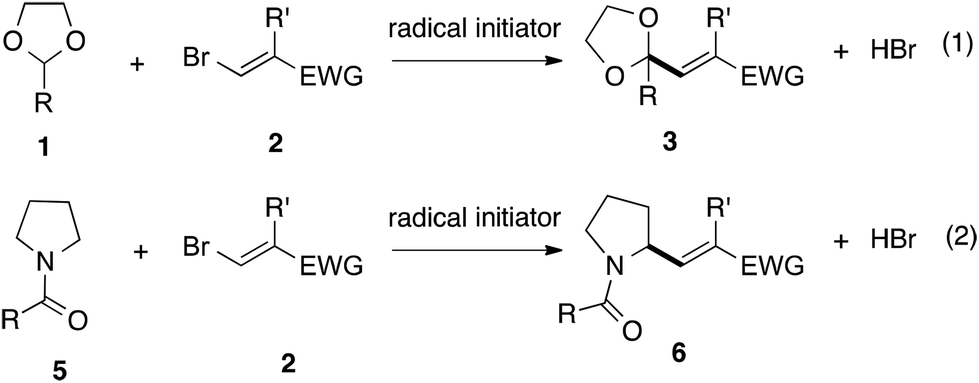 | ||
| Scheme 2 This work: radical vinylation of 1,3-dioxolanes 1 and N-acylpyrrolidines 5 using vinyl bromides 2. | ||
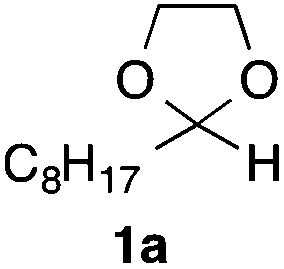
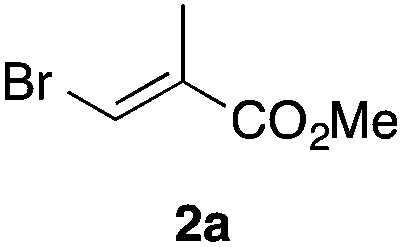
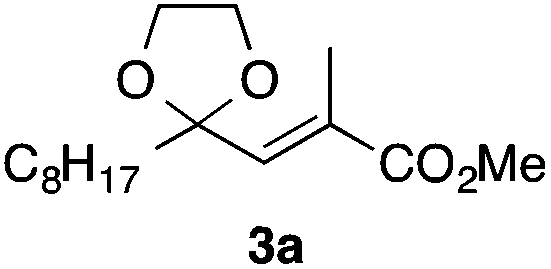
In the initial study, we examined the reaction of 2-octyl-1,3-dioxolane (1a) derived from 1-nonanal with (E)-methyl 3-bromo-2-methylacrylate (2a) under several reaction conditions (Scheme 3). With acetonitrile solutions of 1a and 2a (3-fold excess) in the presence of di-tert-butylhyponitrite (DTBHN)14 as a radical initiator, the envisaged α-vinylation product, (E)-2-vinyl-1,3-dioxolane 3a, was formed, albeit in 20% yield. The product yield of 3a was increased in a system employing large excess amounts of 1a. Thus, a 5-fold excess of 1a gave 3a in 61% yield. The addition of two portions of DTBHN (10 mol% + 10 mol% (after 3 h)) resulted in a further increase in the yield of 3a to 68%.
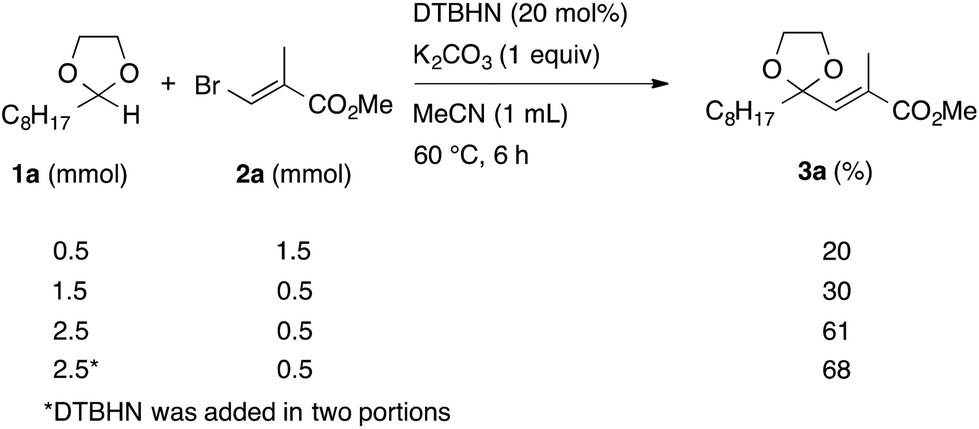 | ||
| Scheme 3 Optimization of reaction conditions. | ||
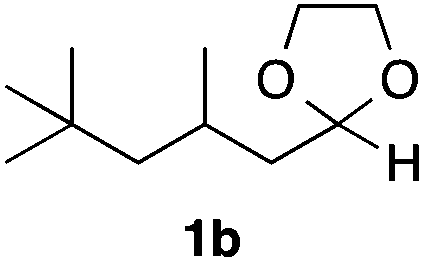
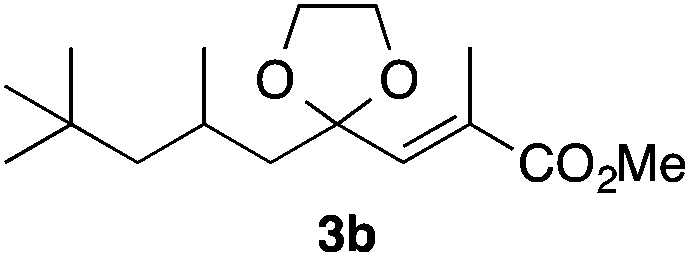
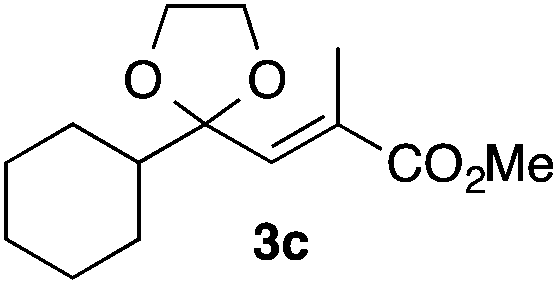
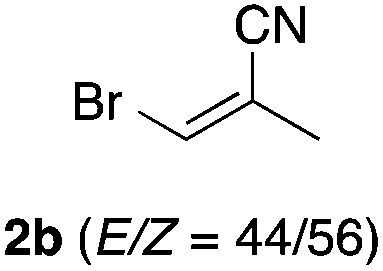
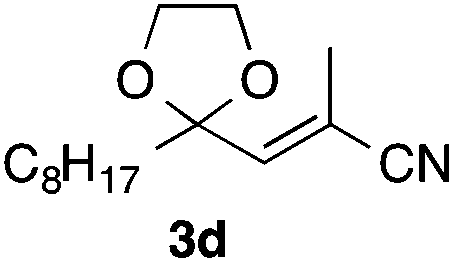
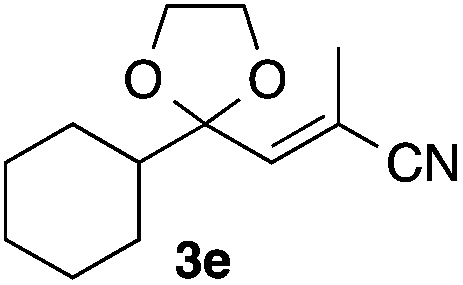
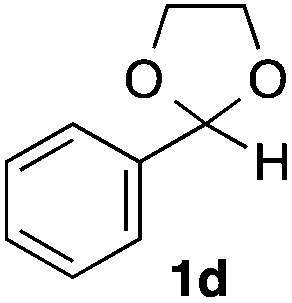
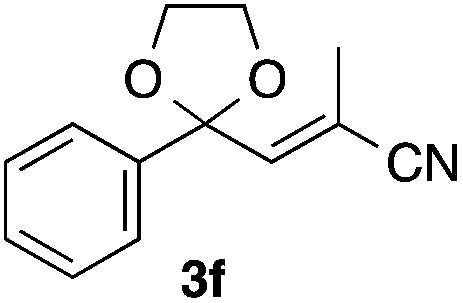
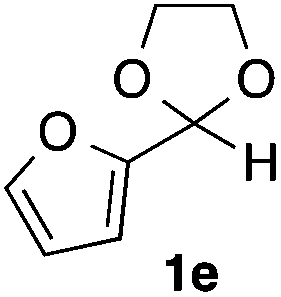
With the optimized conditions in hand, we studied the scope and limitations of the present vinylation method using a variety of 1,3-dioxolanes 1 and vinyl bromides 2 (Table 1). Larger scale reaction using 2 mmol of 2a gave 3a in 69% yield after isolation by silica gel chromatography (entry 1). Dioxolane 1b was reacted with 2a to give 2-vinyl-1,3-dioxolane 3b in 79% yield (entry 2). The reaction of 2-cyclohexyl-1,3-dioxolane (1c) with 2a gave 3c in 56% yield (entry 3). Also, 3-bromo-2-methacrylonitrile (2b, E/Z = 44/56) was reacted with 1a and 1c to give 3d and 3e in 79 and 56% yields, respectively (entries 4 and 5), and 2-aryl-1,3-dioxolanes 1d and 1e were reacted with 2b to give 3f and 3g in 85 and 64% yields, respectively (entries 6 and 7). In these reactions, products 3d–3g were given as an E/Z mixture that was enriched by an E isomer. It is noteworthy that unlike the case of 2a, the regiochemistry of the reaction using 3-bromoacrylate (2c) was not perfect, in which a mixture of the desired 1,2-disubstituted alkene 3h and the bromine-containing regioisomer 3h′ was obtained in 51 and 19% yields, respectively (entry 8). The use of E-form bromide 2d gave similar results (entry 9). It was also interesting that essentially no reaction took place when we carried out the reaction of 1,3-dioxane 4, derived from nonanal and 1,3-propanediol, with 2a (entry 10). The obtained vinylated 1,3-dioxolanes 3 can be converted to unsaturated 1,4-dicarbonyl compounds by standard acid treatment. For example, when dioxolane 3h was treated with p-TsOH in acetone at r.t.,15 γ-keto-α,β-unsaturated ester 5 was obtained in 82% yield (eqn (3)).
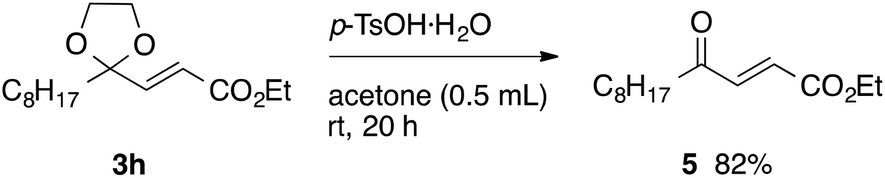 | (3) |
|
|
||||
|---|---|---|---|---|
| Entry | 1 | 2 | 3 | Yieldc (%) |
| a Conditions: 1 (2.5 mmol), 2 (0.5 mmol), K2CO3 (0.5 mmol), DTBHN (20 mol%), MeCN (1 mL), 60 °C, 6 h. b At the beginning of the reaction, DTBHN (10 mol%) was used and after 3 h additional DTBHN (10 mol%) was added. c Isolated yield after column chromatography on SiO2. d Reaction was performed on a 2 mmol scale of 2a with 30 mol% of DTBHN (three portion addition) at 60 °C for 12 h. For detailed procedure, see ESI. e E/Z ratio was determined by 1H-NMR analysis of the crude reaction mixture. f C6H6 was used as a solvent. g Detected by GC-MS. | ||||
| 1 |
|
|
|
68 (69)d only E |
| 2 |
|
2a |
|
79 only E |
| 3 |
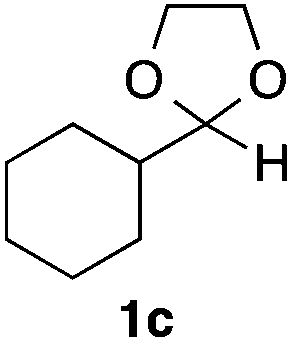
|
2a |
|
56 only E |
| 4 | 1a |
|
|
79 E/Z = 93/17e |
| 5 | 1c | 2b |
|
56 E/Z = 74/26e |
| 6 |
|
2b |
|
85 E/Z = 79/21e |
| 7 |
|
2b |
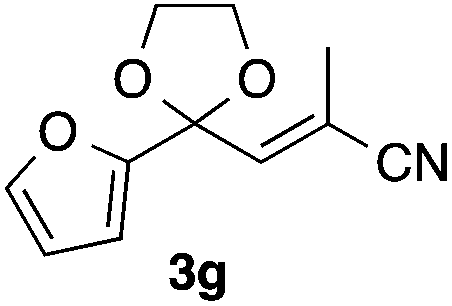
|
64 E/Z = 78/21e |
| 8f | 1a |
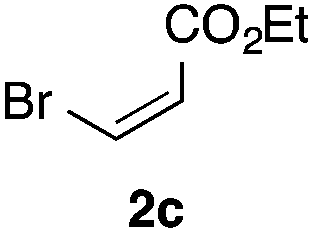
|
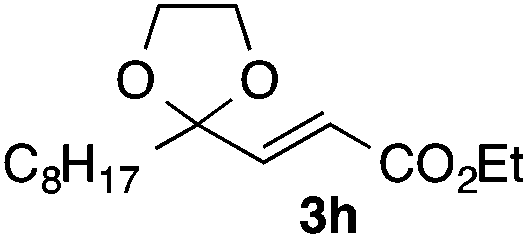
|
51 only E |
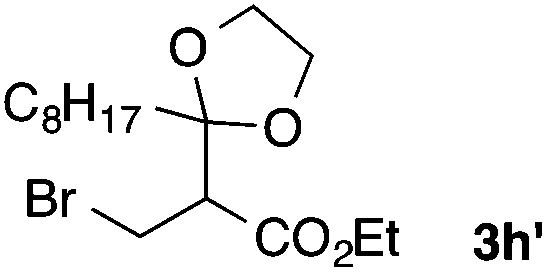
|
19 | |||
| 9f | 1a |
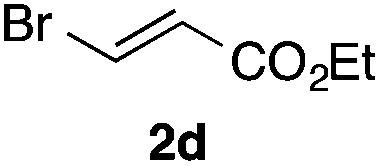
|
3h | 53 |
| 3h′ | 20 | |||
| 10 |
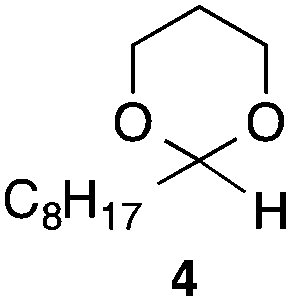
|
2a | Traceg | |
Next, we tried to extend this protocol to the α-vinylation of N-heterocycles using N-acetylpyrrolidine (6a) as a model. When a mixture of 6a, 2a, DTBHN, and K2CO3 in MeCN was heated at 60 °C for 6 h, an α-vinylation reaction took place efficiently to give the corresponding vinylated amide 7a in 80% yield (Scheme 4). In a similar manner, the vinylation of N-propanoylpyrrolidine (6b) with 2a gave 7b in 88% yield. As we experienced in the reaction of 1,3-dioxanes, the reactivity of N-acetylpiperidine, six-membered substrate, was low, giving a low yield of the corresponding vinylated product along with a large amount of the starting material recovered.
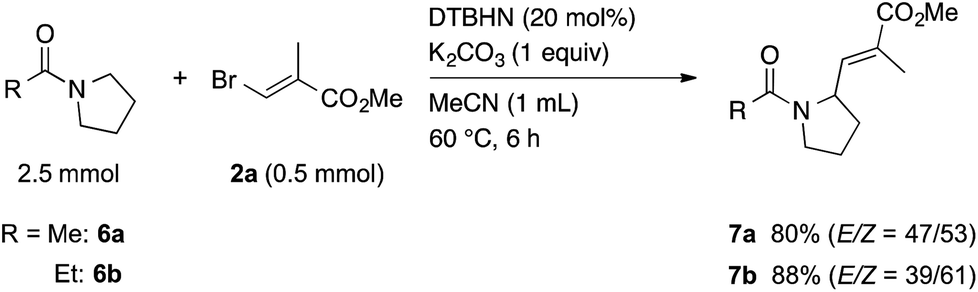 | ||
| Scheme 4 Vinylation of N-acylpyrrolidines 6 with vinyl bromide 2a. | ||
The reactivity difference that varied in the ring size led us to examine MO calculations to elucidate each transition state for the hydrogen abstraction by a bromine radical. Calculated reaction profiles of α-hydrogen abstraction are shown in ESI.‡ At the HF/6-311G**(C,H,O,N) + LanL2DZdp(Br) level,16 activation energies for the α-hydrogen abstraction of 2-methyl-1,3-dioxolane and 2-methyl-1,3-dioxane were calculated to be 69.4 and 75.8 kJ mol−1 respectively, while those of N-acetylpyrrolidine and N-acetylpiperidine were predicted to be 71.8 and 75.3 kJ mol−1, respectively. These results indicated that abstraction of α-hydrogens in five-membered rings with a bromine radical proceeds more easily than that in six-membered rings.17
A proposed reaction mechanism for the present radical vinylation of 1,3-dioxolane 1 was outlined in Scheme 5, which consists of one radical initiation step and three chain propagation steps: (i) the formation of an α,α-dioxy radical via the hydrogen abstraction of a tert-butoxy radical generated from the radical initiator DTBHN; (ii) the addition of the resulting α,α-dioxy radical to vinyl bromide 2; (iii) the β-elimination of the bromine radical to give 2-vinyl-1,3-dioxolane 3; and (iv) the regeneration of the α,α-dioxy radical by hydrogen abstraction of the liberated bromine radical. The selective formation of E-isomer can be rationalized by an equilibrium between two rotamers A and B, in which steric repulsion between the dioxolanyl group and the methoxy carbonyl group would destabilize B, rendering E-isomer as a major product.
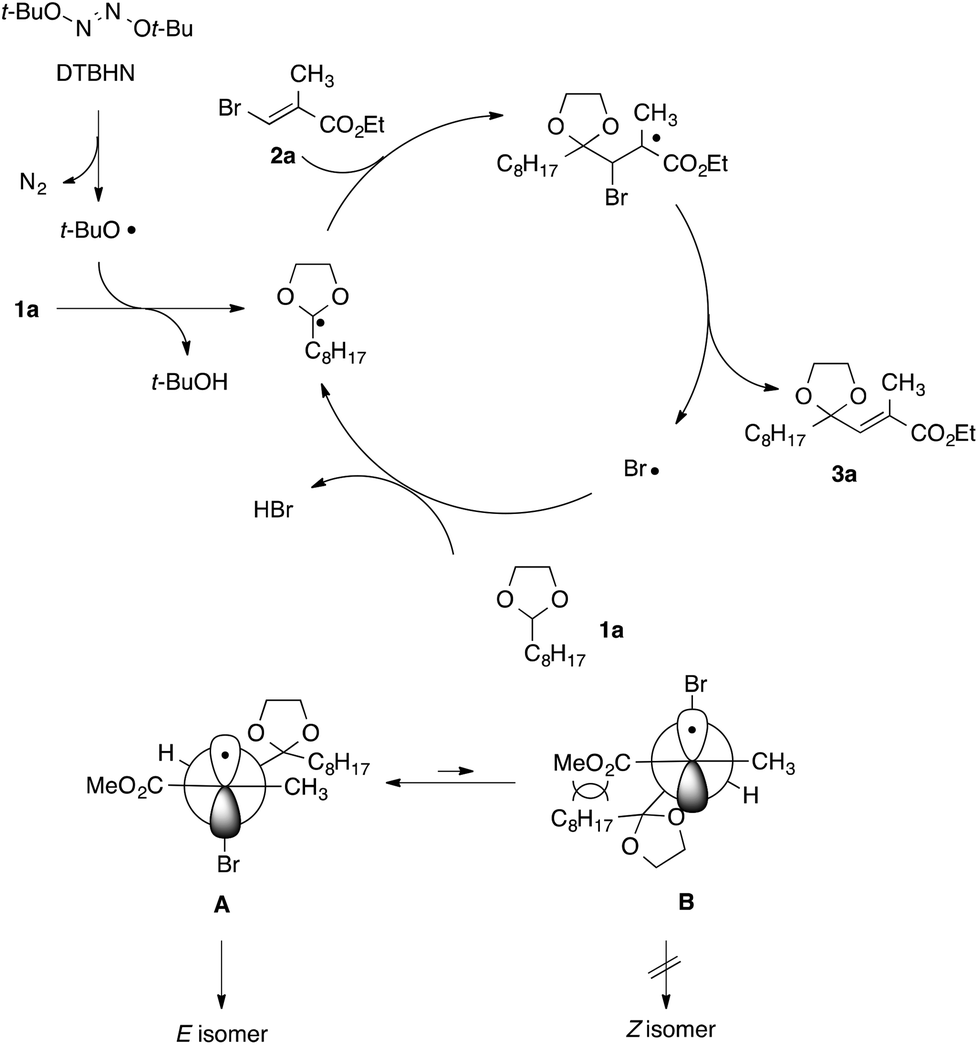 | ||
| Scheme 5 Proposed reaction mechanism. | ||
In conclusion, we have demonstrated that a radical vinylation of 2-alkyl- and 2-aryl-1,3-dioxolanes using vinyl bromides proceeded well to give 2-vinyl-1,3-dioxanes in good yields. We also developed a radical vinylation of N-acylpyrrolidines using vinyl bromides. Chain propagation of the vinylation was quite sensitive to the ring-size of the substrates, which presumably affected the H-abstraction step by the bromine radical. Further applications of the present vinylation method are currently in progress in our laboratory.
Experimental section
Typical procedure for radical vinylation of 1,3-dioxolanes 2
To a 20 mL screw-capped test tube, di-tert-butylhyponitrite (DTBHN, 8.7 mg, 0.05 mmol), potassium carbonate (69.1 mg, 0.5 mmol), 2-octyl-1,3-dioxolane (1a, 465.7 mg, 2.5 mmol), methyl (E)-3-bromo-2-methylacrylate (2a, 89.5 mg, 0.5 mmol), and degassed MeCN (0.5 mL) were added. The test tube was purged with argon and sealed. Then, the mixture was stirred at 60 °C. After 3 h, an additional portion of a solution of DTBHN (8.7 mg, 0.05 mmol) in MeCN (0.5 mL) was added and the resulting mixture was stirred at 60 °C for 3 h. The reaction mixture was filtered through a short plug of Celite and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography on SiO2 (hexane–EtOAc = 100/1 to 30/1) to give the vinylated product 3a as a single E diastereomer (96.7 mg, 68%).Typical procedure for radical vinylation of 1-acylpyrrolidines 6
To a 20 mL screw-capped test tube, di-tert-butylhyponitrite (DTBHN, 17.4 mg, 0.1 mmol), potassium carbonate (69.1 mg, 0.5 mmol), 1-acetylpyrrolidine (6a, 282.9 mg, 2.5 mmol), methyl (E)-3-bromo-2-methylacrylate (2a, 89.5 mg, 0.5 mmol), and degassed (Ar bubbling for 15 min before use) MeCN (1.0 mL) were added, and the test tube was purged with argon and sealed. The mixture was stirred at 60 °C for 6 h. The reaction mixture was filtered through a short plug of Celite, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography on SiO2 (hexane–EtOAc = 25/75 to 0/100) to give methyl 3-(1-acetylpyrrolidin-2-yl)-2-methylacrylate (7a) (84.5 mg, E/Z = 47/53 (by 1H NMR), 80%).Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from the MEXT and the JSPS.References
- For an overview, see: I. J. Rosenstein, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001, vol. 1, pp. 50–71 Search PubMed.
- G. E. Keck, J. H. Byers and A. M. Tafesh, J. Org. Chem., 1988, 53, 1127 CrossRef CAS.
- G. A. Russell, H. Tashtoush and P. Ngoviwatchai, J. Am. Chem. Soc., 1984, 106, 4622 CrossRef CAS.
- (a) F. Bertrand, B. Quiclet-Sire and S. Z. Zard, Angew. Chem., Int. Ed., 1999, 38, 1943 CrossRef CAS; (b) J. Xiang, W. Jiang, J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1997, 119, 4123 CrossRef CAS.
- C.-F. Yao, C.-M. Chu and J.-T. Liu, J. Org. Chem., 1998, 63, 719 CrossRef CAS PubMed.
- (a) K. Takami, H. Yorimitsu and K. Oshima, Org. Lett., 2004, 6, 4555 CrossRef CAS PubMed; (b) K. Takami, S. Usugi, H. Yorimitsu and K. Oshima, Synthesis, 2005, 824 CAS.
- (a) A. F. A. Reijnhart, Recl. Trav. Chim. Pays-Bas, 1927, 46, 72 CrossRef; (b) S. G. Cohen, H. T. Wolosinski and P. J. Scheuer, J. Am. Chem. Soc., 1950, 72, 3952 CrossRef CAS.
- (a) M. S. Kharasch and M. Sage, J. Org. Chem., 1949, 14, 79 CrossRef CAS; (b) M. S. Kharasch and G. Büchi, J. Org. Chem., 1949, 14, 84 CrossRef CAS.
- (a) J. M. Tanko and M. Sadeghipour, Angew. Chem., Int. Ed., 1999, 38, 159 CrossRef CAS; (b) J. A. Struss, M. Sadeghipour and J. M. Tanko, Tetrahedron Lett., 2009, 50, 2119 CrossRef CAS PubMed.
- (a) T. Kippo, T. Fukuyama and I. Ryu, Org. Lett., 2010, 12, 4006 CrossRef CAS PubMed; (b) T. Kippo, T. Fukuyama and I. Ryu, Org. Lett., 2011, 13, 3864 CrossRef CAS PubMed; (c) T. Kippo, K. Hamaoka and I. Ryu, J. Am. Chem. Soc., 2013, 135, 632 CrossRef CAS PubMed; (d) T. Kippo and I. Ryu, Chem. Commun., 2014, 50, 5993 RSC.
- C. C. Huval and D. A. Singleton, Tetrahedron Lett., 1993, 34, 3041 CrossRef CAS.
- (a) M. R. Heinrich, O. Blank, D. Ullrich and M. Kirschstein, J. Org. Chem., 2007, 72, 9609 CrossRef CAS PubMed; (b) H. Jasch, Y. Landais and M. R. Heinrich, Chem. – Eur. J., 2013, 19, 8411 CrossRef CAS PubMed.
- Y. Yang, H. Huang, X. Zhang, W. Zeng and Y. Liang, Synthesis, 2013, 3137 CAS.
- H. Kiefer and T. G. Traylor, Tetrahedron Lett., 1966, 7, 6163 CrossRef.
- Y. Matsushita, H. Furusawa, T. Matsui and M. Nakayama, Chem. Lett., 1994, 1083 CrossRef CAS.
- No transition states were found on other levels of theory than HF.
- In contrast, methyl or ethyl radical has been shown to effectively abstract α-hydrogen of tetrahydropyran or N-methylpiperidine, see: (a) T. Akindele, K. Yamada and K. Tomioka, Acc. Chem. Res., 2009, 42, 345 CrossRef CAS PubMed; (b) T. Yoshimitsu, K. Matsuda, H. Nagaoka, K. Tsukamoto and T. Tanaka, Org. Lett., 2007, 9, 5115 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00138a |
| This journal is © the Partner Organisations 2014 |