
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and kinetic resolution of N-Boc-2-arylpiperidines†
Edward J.
Cochrane
a,
Daniele
Leonori
a,
Lorraine A.
Hassall
b and
Iain
Coldham
*a
aDepartment of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. E-mail: i.coldham@sheffield.ac.uk
bAstraZeneca, Alderley Park, Macclesfield, Cheshire SK10 4TG, UK
First published on 11th July 2014
Abstract
The chiral base n-BuLi/(−)-sparteine or n-BuLi/(+)-sparteine surrogate promotes kinetic resolution of N-Boc-2-arylpiperidines by asymmetric deprotonation. The enantioenriched starting material was recovered with yields 39–48% and ers up to 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. On lithiation then electrophilic quench, 2,2-disubstituted piperidines were obtained with excellent yields and enantioselectivities.
:3. On lithiation then electrophilic quench, 2,2-disubstituted piperidines were obtained with excellent yields and enantioselectivities.
Piperidines substituted in the 2-position with at least one aryl group are a class of molecules often possessing high biological activities and are therefore of interest to synthetic organic, medicinal and process chemists.1 For example, piperidine 1 is a poly ADP ribose polymerase inhibitor (Fig. 1).2 Piperidine 2 is a lead compound for orally active NK1 antagonists.3 Piperidine 2 was synthesised by Xiao and co-workers as a mixture of diastereomers via the THF-mediated lithiation-substitution of (±)-N-Boc-2-phenylpiperidine.3a The need for a stereoselective synthesis is exemplified by the ∼50-fold difference in activity between the HCl salt of the (R,R) and the (R,S) diastereomers (IC50 0.3 nM and >15 nM, respectively).4
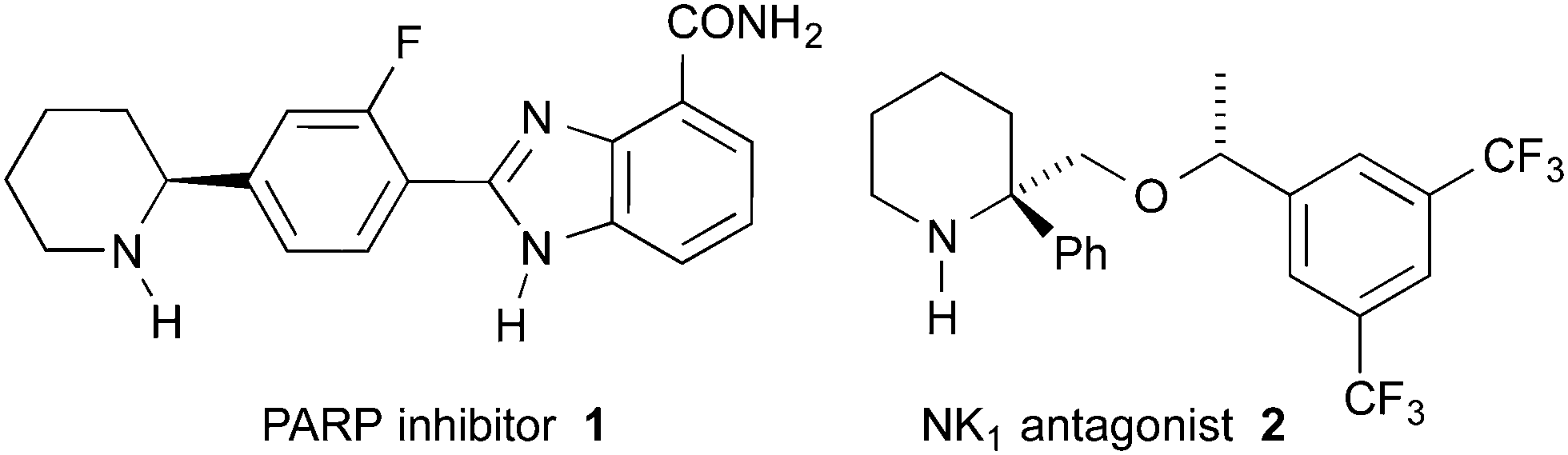 | ||
| Fig. 1 Some biologically active 2-arylpiperidines. | ||
A general approach to the asymmetric synthesis of 2,2-disubstituted piperidines continues to pose a problem for synthetic chemists. Building on their expertise of lithiated nitrogen-containing heterocycles, Coldham and O'Brien and co-workers reported that the Boc group in N-Boc-2-phenylpiperidine rotates rapidly, even at −78 °C, to allow high yields of lithiation at the benzylic position and that the organolithium is configurationally stable at low temperature.5 This led to the synthesis of enantiomerically enriched 2-alkyl-2-phenylpiperidines, such as piperidine 4a, using enantiomerically enriched N-Boc-2-phenylpiperidine 3a (Scheme 1).
 | ||
| Scheme 1 Lithiation-substitution of piperidine 3a. | ||
The enantioenriched N-Boc-2-phenylpiperidine (S)-3a was obtained by an asymmetric reduction of the corresponding cyclic imine.5a An alternative approach is by Negishi reaction using the chiral organozinc species formed either from the 2-tributylstannane, itself formed by dynamic resolution of 2-lithiated N-Boc-piperidine,6 or directly by ‘catalytic dynamic resolution’,7 although the latter method was unsuccessful in our hands. We therefore sought a simpler method and wondered if racemic N-Boc-2-arylpiperidines were amenable to kinetic resolution. There is growing interest in the kinetic resolution of amines, mostly by N-acylation.8 In contrast, the kinetic resolution of amines by deprotonation has received very little study.9 The closest example is that from Beak and co-workers, who showed that the chiral base n-BuLi/(−)-sparteine can effect a kinetic resolution of an acyclic N-Boc-α-methylbenzylamine, albeit with fairly low selectivity (er up to 81:19 for 12% yield of recovered starting material).9a In this paper, we report that N-Boc-2-arylpiperidines are excellent substrates for highly selective kinetic resolution by asymmetric deprotonation.
Racemic N-Boc-2-phenylpiperidine and related 2-aryl or 2-heteroaryl derivatives can be prepared by several different methods.10 We selected to extend the method used for racemic 3a,5a for the preparation of a variety of N-Boc-2-arylpiperidines (Scheme 2). Addition of the aryllithium, obtained by bromine–lithium exchange, to 5-bromovaleronitrile, followed by reduction of the intermediate cyclic imine with sodium borohydride and addition of Boc2O gave the desired racemic piperidines 3a–g. Reasonable yields for the three-step procedure were obtained, except with p-MeOC6H4Li which gave significant amounts of methoxybenzene, perhaps due to its propensity to abstract a proton alpha to the nitrile. The corresponding organomagnesium reagent was no more successful.
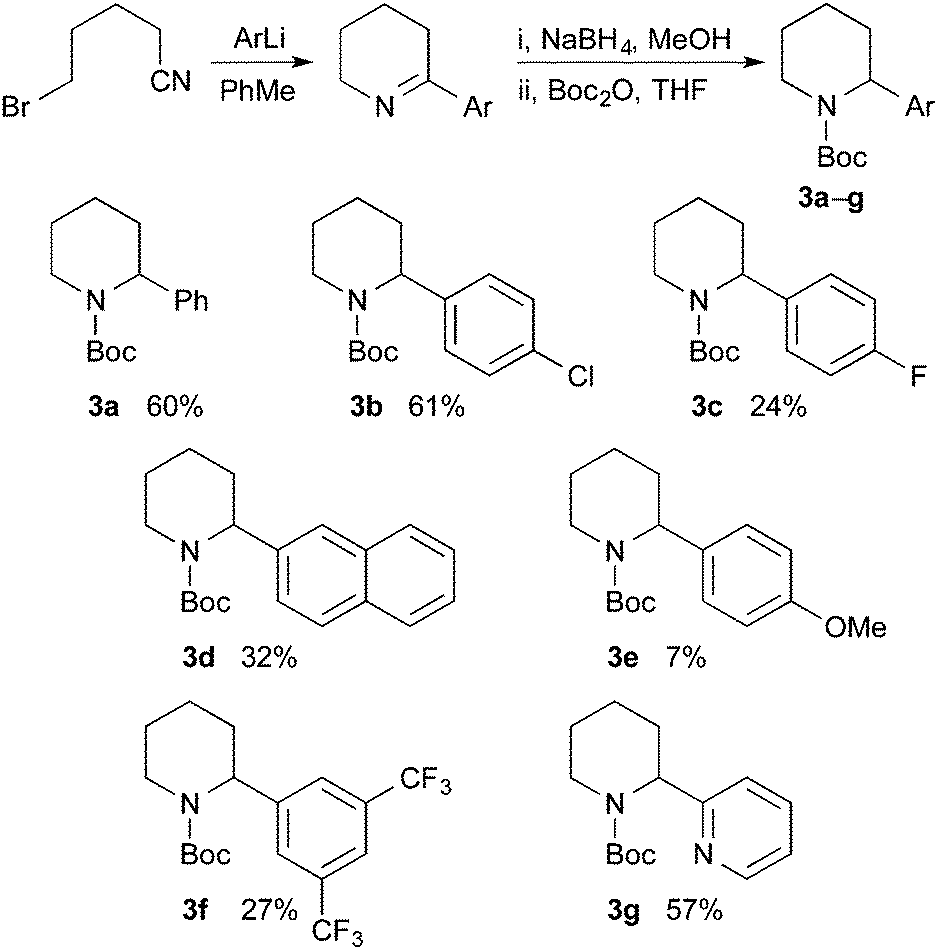 | ||
| Scheme 2 Synthesis of (±)-N-Boc-2-arylpiperidines 3a–g. | ||
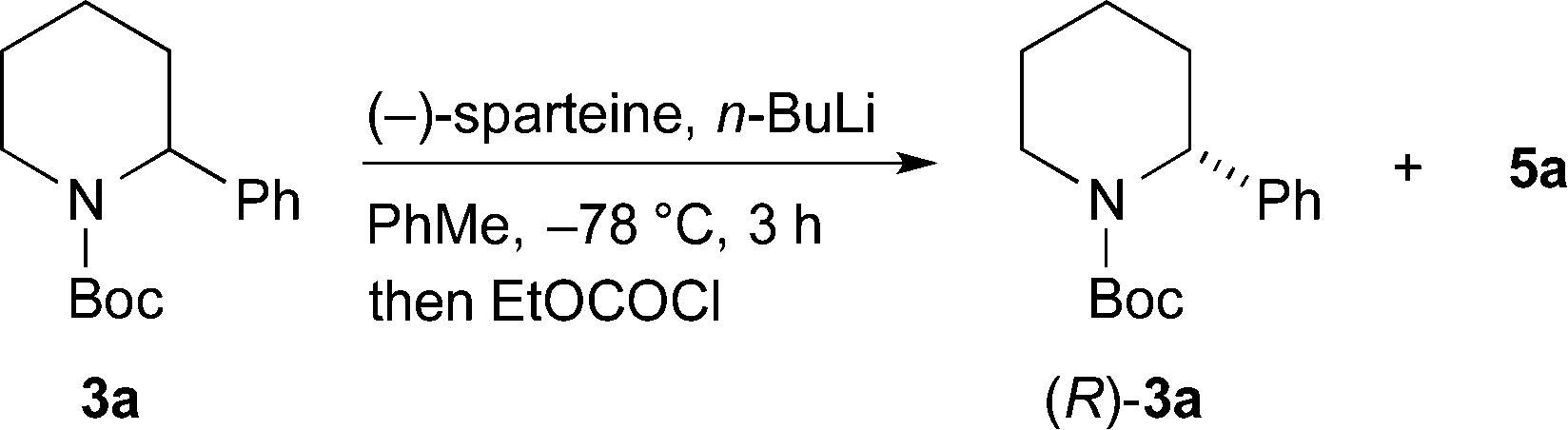
For the kinetic resolution of these substrates, we initially focused on developing conditions using the substrate 3a. A mixture of 0.55 equiv. of n-BuLi and (−)-sparteine (pre-mixed for 5 min) in PhMe was used for the lithiation of N-Boc-2-phenylpiperidine 3a (Scheme 3). After 5 min, addition of iodomethane gave the product 4a in 30% yield with a promising er (79:21). With the electrophile ethyl chloroformate, both the yield of the product 5a and the er were improved. By changing the solvent to Et2O, a similar er (87:13) of 5a was obtained but in reduced yield (see ESI† for yield and er of recovered 3a). The er was determined by chiral stationary phase (CSP) HPLC and the optical rotation of 4a (see ESI†) matched that reported for the (S) enantiomer.5a These data predict, as expected based on the lithiation of N-Boc-piperidine,11 that the enantiomer (S)-3a is preferentially lithiated by n-BuLi/(−)-sparteine.
 | ||
| Scheme 3 Initial study of the kinetic resolution with pre-mixed n-BuLi/(−)-sparteine. | ||
As an alternative, we investigated a different method of addition, whereby n-BuLi and (−)-sparteine were not premixed. n-BuLi was added to a solution containing the piperidine 3a and (−)-sparteine in PhMe. After different time periods the electrophile EtOCOCl was added (Table 1). A marked improvement in the er of the product 5a was obtained, and this was associated with a significant decrease in yield. By increasing the reaction time to 3 h, it was possible to obtain the product 5 with er 92:8 and a yield of 42% (entry 5).
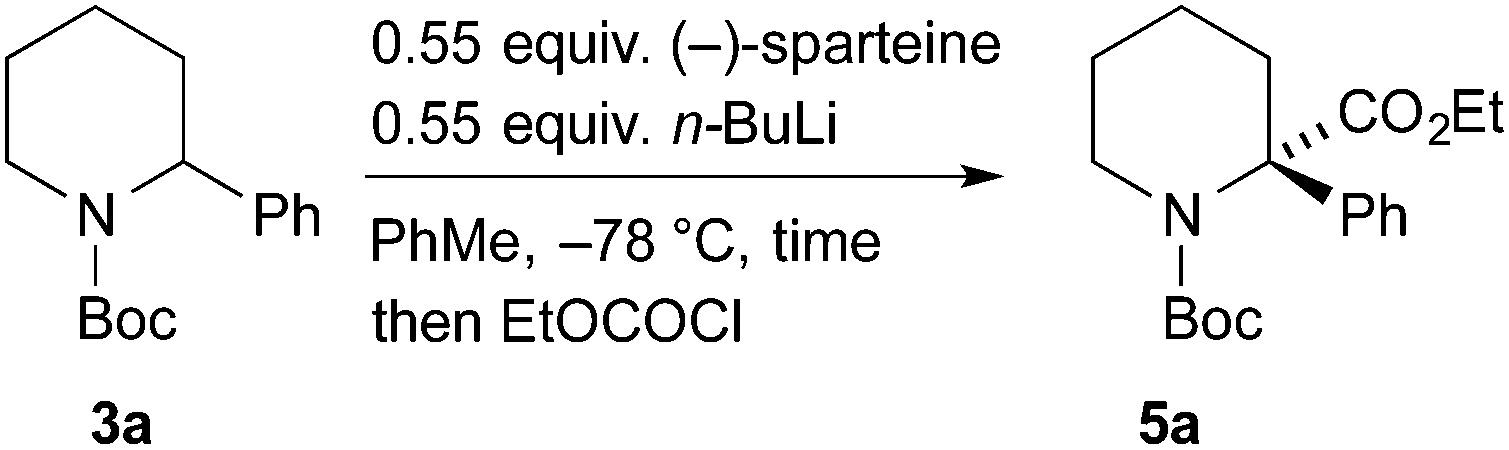
To investigate the differences in reaction times for these methods, we performed in situ IR spectroscopy to monitor the stretching frequency of the carbonyl group (Fig. 2). When using pre-mixed n-BuLi/(−)-sparteine (Fig. 2a), partial lithiation occurs over about 15 min, as judged by the steady but partial loss of νc![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) o 1691 cm−1 (CO stretch for 3a) and formation of a new peak at 1640 cm−1 (assigned to νco in the lithiated intermediate). However, without pre-mixing, the lithiation required several hours (Fig. 2b). The method chosen clearly impacts on the selectivity, with the slower lithiation allowing a more selective reaction. The faster reaction occurs with the pre-mixed n-BuLi/(−)-sparteine complex that will have a higher concentration of the dimeric aggregation state.12
o 1691 cm−1 (CO stretch for 3a) and formation of a new peak at 1640 cm−1 (assigned to νco in the lithiated intermediate). However, without pre-mixing, the lithiation required several hours (Fig. 2b). The method chosen clearly impacts on the selectivity, with the slower lithiation allowing a more selective reaction. The faster reaction occurs with the pre-mixed n-BuLi/(−)-sparteine complex that will have a higher concentration of the dimeric aggregation state.12
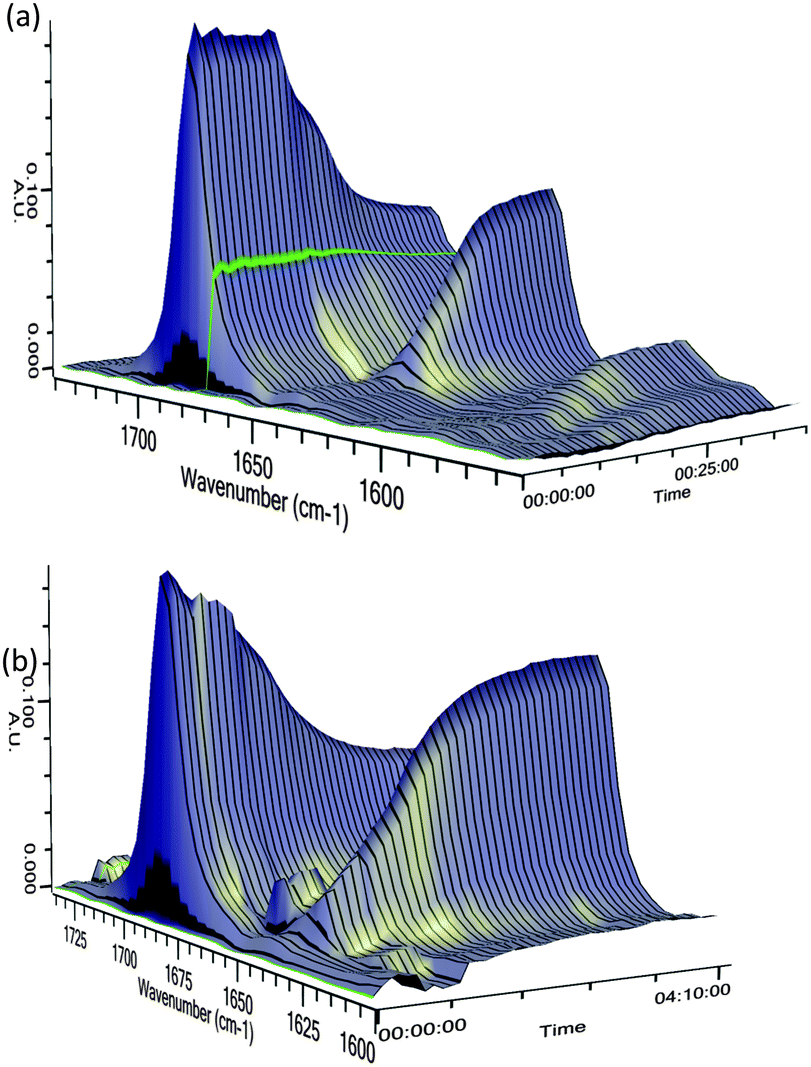 | ||
| Fig. 2
In situ IR plots of the lithiation of 3a with n-BuLi/(−)-sparteine at −78 °C in PhMe with time in h:min:s. (a) νco3a (1691 cm−1), pre-mixed n-BuLi/(−)-sparteine added at time 16 min, νco lithiated 3a (1640 cm−1); (b) νco3a (1691 cm−1) and (−)-sparteine then n-BuLi added at time 16 min, νco lithiated 3a (1640 cm−1). | ||
With optimised conditions in-hand, we studied the use of other electrophiles but these gave lower yields and ers (37% yield, er 82:18 using allyl bromide; 24% yield, er 74:26 using MeI). The lower selectivities may be due to partial racemisation of the lithiated intermediate prior to slower electrophilic quench.
Due to the variation in er dependent on the choice of electrophile, we decided to focus on obtaining a good yield and er for the recovered starting material. This was investigated by varying the number of equivalents of the chiral base (Table 2). The best conditions that we found used 0.7 equiv. of n-BuLi and (−)-sparteine and gave an excellent yield and er of recovered 3a (entry 2). A yield of 45% with er 96:4 (R:S) corresponds to a relative rate of reaction for the two enantiomers krel ∼ 22.13
The kinetic resolution was then tested with the different 2-arylpiperidines that were prepared as shown in Scheme 2. We were pleased to find that high levels of selectivity could be achieved with many of these compounds (Scheme 4). The yields and er values of the recovered starting materials are given in Scheme 4 for the different aromatic derivatives and it was found that both electron-poor (p-chloro 3b, p-fluoro 3c) and electron-rich (p-methoxy 3e) substituents are tolerated in the kinetic resolution. However, some loss in selectivity occurs with the 3,5-bis-trifluoromethyl 3f and 2-pyridyl 3g derivatives. This may be due to a faster and therefore less selective lithiation with these more acidic substrates.
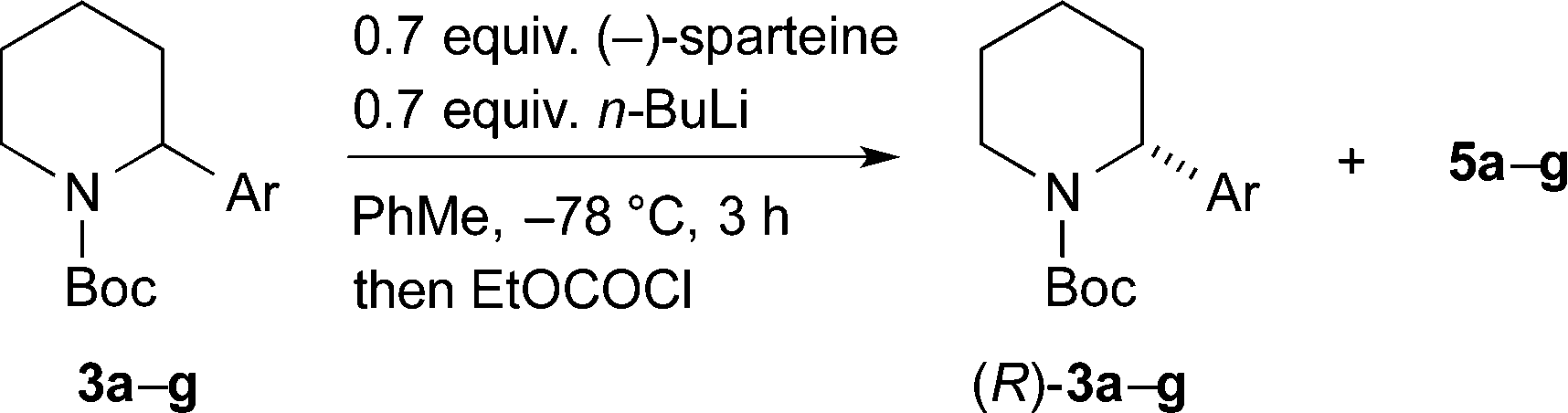 | ||
| Scheme 4 Kinetic resolution of various N-Boc-2-arylpiperidines. | ||
In addition, we have demonstrated that the other enantiomer of the 2-arylpiperidine can be formed by changing the chiral ligand to O'Brien's (+)-sparteine surrogate.14 This resulted in the formation of (S)-3a (41% yield, er 91:9) and (S)-3b (42% yield, er 84:16).
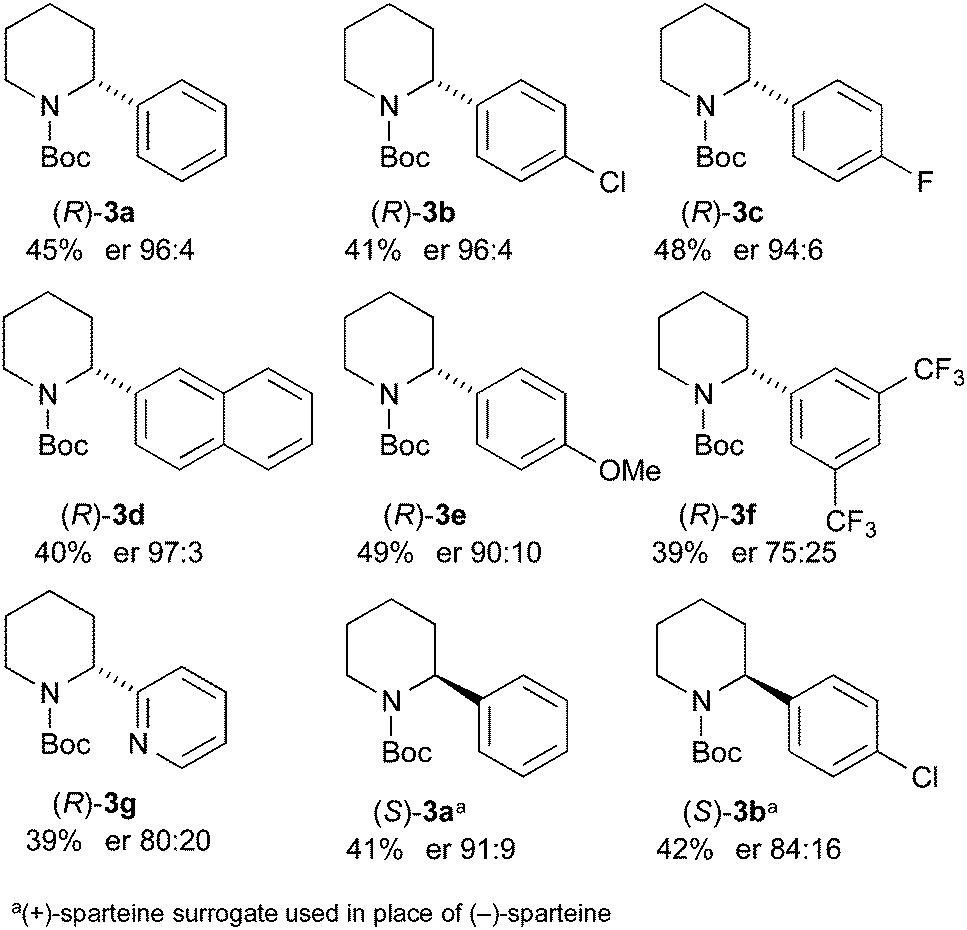
This methodology is reliant on using ethyl chloroformate as a sacrificial electrophile, consuming the undesired enantiomer. The kinetic resolution was tested with Me3SnCl as the electrophile (Scheme 5). This could enable a subsequent tin–lithium exchange to recover the starting material after proton quench. The kinetic resolution performed well using this electrophile to give the recovered piperidine 3a in 42% yield and er 94:6. The er of the stannane 6a was not determined but this product was treated with n-BuLi in THF followed by addition of acetic acid. When this reaction was conducted at −78 °C, the piperidine (S)-3a was isolated with er 82:18, suggesting that tin–lithium exchange and protonation occurs with retention of configuration. However, when the exchange and protonation was conducted at room temperature, racemic 3a was recovered in high yield. Therefore this approach allows recycling of the undesired product to allow a further kinetic resolution.
 | ||
| Scheme 5 Kinetic resolution of N-Boc-2-phenylpiperidine with Me3SnCl quench. | ||
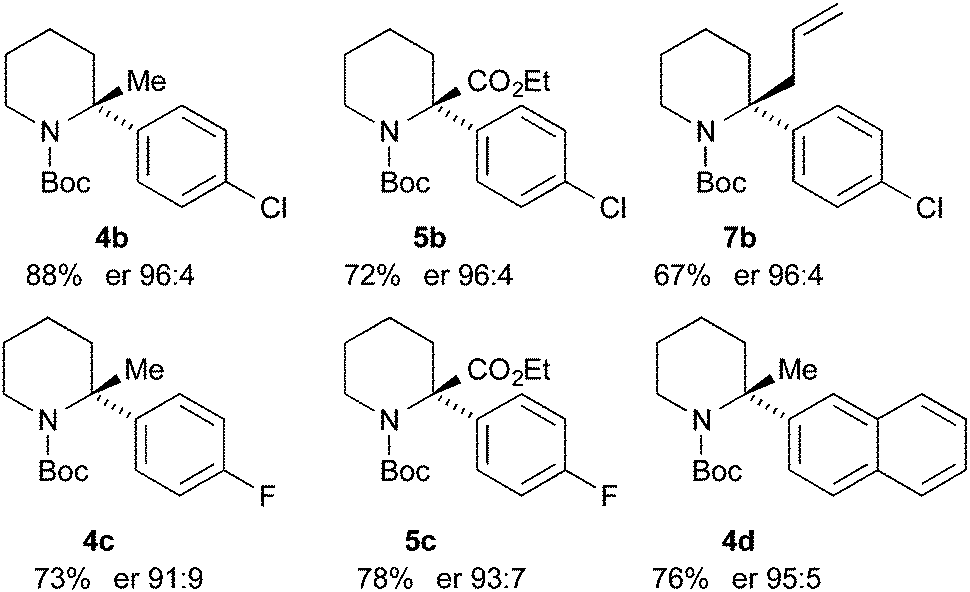
Finally, we have performed THF-mediated lithiation then substitution on the enantioenriched recovered starting materials to show that these are amenable to the formation of 2,2-disubstituted piperidines without loss of enantioselectivity.5,15 Treatment of the piperidine (R)-3b (er 96:4) with n-BuLi followed by MeI, EtOCOCl or allyl bromide gave the products 4b, 5b, and 7b with good yields and with no loss of enantioselectivity (Scheme 6). Similarly, piperidines (R)-3c (er 94:6) and (R)-3d (er 97:3) gave the products 4c, 5c, and 4d with good yields and very little reduction in er. Based on related chemistry using piperidine 3a,5 we assume that the absolute configuration of these products is as shown, in which lithiation and electrophilic quench occur with retention of configuration.
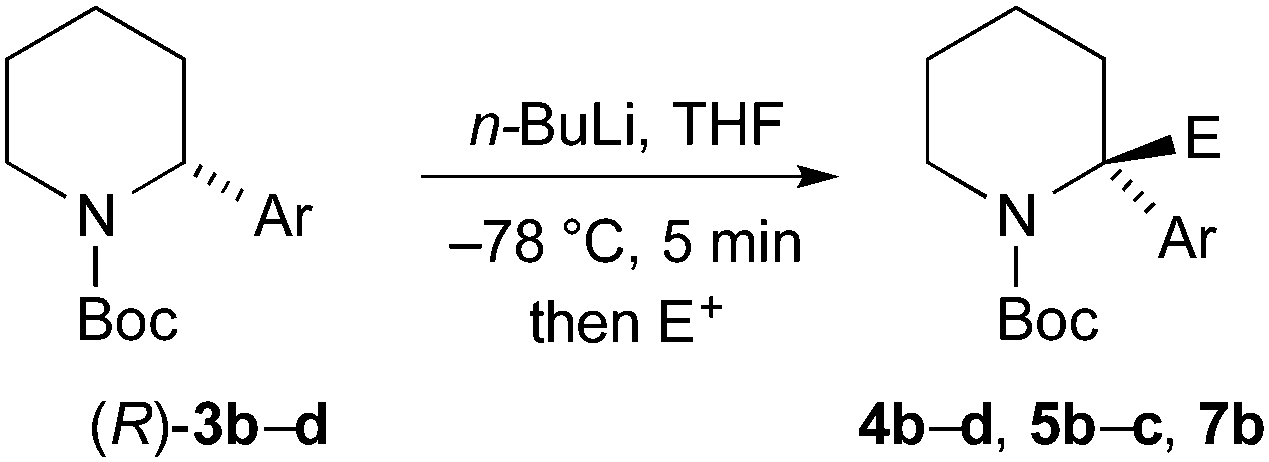 | ||
| Scheme 6 Lithiation-substitution of various N-Boc-2-arylpiperidines. | ||
The chiral base n-BuLi with (−)-sparteine or the (+)-sparteine surrogate is effective for the kinetic resolution of racemic N-Boc-2-arylpiperidines in PhMe. The recovered starting material can be isolated with high enantiomer ratios, especially if n-BuLi and the chiral ligand are not pre-mixed. Use of in situ IR spectroscopy helped to optimise the conditions for the kinetic resolution. By using a trialkyltin halide electrophile, the quenched product can be recycled by tin–lithium exchange and protonation. After kinetic resolution, the recovered enantioenriched N-Boc-2-arylpiperidines can be deprotonated with n-BuLi in THF at −78 °C and quenched with electrophiles without loss of enantiopurity to provide highly enantioselective syntheses of 2,2-disubstituted piperidine products.
We thank the EPSRC, the University of Sheffield and AstraZeneca for funding.
Notes and references
- See, for example, (a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed; (b) S. Hardy and S. F. Martin, Org. Lett., 2011, 13, 3102 CrossRef CAS PubMed.
- T. D. Penning, G.-D. Zhu, J. Gong, S. Thomas, V. B. Gandhi, X. Liu, Y. Shi, V. Klinghofer, E. F. Johnson, C. H. Park, E. H. Fry, C. K. Donawho, D. J. Frost, F. G. Buchanan, G. T. Bukofzer, L. E. Rodriguez, V. Bontcheva-Diaz, J. J. Bouska, D. J. Osterling, A. M. Olson, K. C. Marsh, Y. Luo and V. L. Giranda, J. Med. Chem., 2010, 53, 3142 CrossRef CAS PubMed.
- (a) D. Xiao, B. J. Lavey, A. Palani, C. Wang, R. G. Aslanian, J. A. Kozlowski, N.-Y. Shih, A. T. McPhail, G. P. Randolph, J. E. Lachowicz and R. A. Duffy, Tetrahedron Lett., 2005, 46, 7653 CrossRef CAS; (b) T. Harrison, B. J. Williams and C. J. Swain, Bioorg. Med. Chem. Lett., 1994, 4, 2733 CrossRef CAS.
- (a) D. Xiao, C. Wang, A. Palani, H.-C. Tsui, G. Reichard, S. Paliwal, N.-Y. Shih, R. Aslanian, R. Duffy, J. Lachowicz, G. Varty, C. Morgan, F. Liu and A. Nomeir, Bioorg. Med. Chem. Lett., 2010, 20, 6313 CrossRef CAS PubMed; (b) D. Xiao, C. Wang, H.-C. Tsui, A. Palani, R. Aslanian and A. V. Buevich, Tetrahedron Lett., 2013, 54, 6199 CrossRef CAS.
- (a) N. S. Sheikh, D. Leonori, G. Barker, J. D. Firth, K. R. Campos, A. J. H. M. Meijer, P. O'Brien and I. Coldham, J. Am. Chem. Soc., 2012, 134, 5300 CrossRef CAS PubMed ; for further examples, see; (b) T. K. Beng, J. S. Woo and R. E. Gawley, J. Am. Chem. Soc., 2012, 134, 14764 CrossRef CAS PubMed.
- I. Coldham, S. Raimbault, D. T. E. Whittaker, P. T. Chovatia, D. Leonori, J. J. Patel and N. S. Sheikh, Chem. – Eur. J., 2010, 16, 4082 CrossRef CAS PubMed.
- T. K. Beng and R. E. Gawley, Org. Lett., 2011, 13, 394 CrossRef CAS PubMed.
- For some reviews, see (a) V. P. Krasnov, D. A. Gruzdev and G. L. Levit, Eur. J. Org. Chem., 2012, 1471 CrossRef CAS; (b) H. Pellissier, Adv. Synth. Catal., 2011, 353, 1613 CrossRef CAS . For some recent examples with 2-substituted piperidines, see; (c) M. Binanzer, S.-Y. Hsieh and J. W. Bode, J. Am. Chem. Soc., 2011, 133, 19698 CrossRef CAS PubMed; (d) S.-Y. Hsieh, M. Binanzer, I. Kreituss and J. W. Bode, Chem. Commun., 2012, 48, 8892 RSC; (e) I. Kreituss, Y. Murakami, M. Binanzer and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10660 CrossRef CAS PubMed; (f) S.-Y. Hsieh, B. Wanner, P. Wheeler, A. M. Beauchemin, T. Rovis and J. W. Bode, Chem. – Eur. J., 2014, 20, 7228 CrossRef CAS PubMed.
- (a) N. C. Faibish, Y. S. Park, S. Lee and P. Beak, J. Am. Chem. Soc., 1997, 119, 11561 CrossRef CAS; (b) S. V. Kessar, P. Singh, K. N. Singh, P. Venugopalan, A. Kaur, P. V. Bharatam and A. K. Sharma, J. Am. Chem. Soc., 2007, 129, 4506 CrossRef CAS PubMed . For some other kinetic resolutions by deprotonation, see; (c) J. Becker, R. Froehlich, K. Salorinne and D. Hoppe, Eur. J. Org. Chem., 2007, 3337 CrossRef CAS; (d) J. Granander, F. Secci, P. O'Brien and B. Kelly, Tetrahedron: Asymmetry, 2009, 20, 2432 CrossRef CAS.
- See, for example, (a) I. Coldham and D. Leonori, Org. Lett., 2008, 10, 3923 CrossRef CAS PubMed; (b) S. Seel, T. Thaler, K. Takatsu, C. Zhang, H. Zipse, B. F. Straub, P. Mayer and P. Knochel, J. Am. Chem. Soc., 2011, 133, 4774 CrossRef CAS PubMed; (c) K. D. Hesp, D. P. Fernando, W. Jiao and A. T. Londregan, Org. Lett., 2014, 16, 413 CrossRef CAS PubMed; (d) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef CAS PubMed . See also; (e) D. Stead, G. Carbone, P. O'Brien, K. R. Campos, I. Coldham and A. Sanderson, J. Am. Chem. Soc., 2010, 132, 7260 CrossRef CAS PubMed; (f) G. Barker, J. L. McGrath, A. Klapars, D. Stead, G. Zhou, K. R. Campos and P. O'Brien, J. Org. Chem., 2011, 76, 5936 CrossRef CAS PubMed; (g) Z. Cui, H.-J. Yu, R.-F. Yang, W.-Y. Gao, C.-G. Feng and G.-Q. Lin, J. Am. Chem. Soc., 2011, 133, 12394 CrossRef CAS PubMed; (h) A. Millet, P. Larini, E. Clot and O. Baudoin, Chem. Sci., 2013, 4, 2241 RSC; (i) C.-V. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809 CrossRef CAS PubMed.
- W. F. Bailey, P. Beak, S. T. Kerrick, S. Ma and K. B. Wiberg, J. Am. Chem. Soc., 2002, 124, 1889 CrossRef CAS PubMed.
- (a) J. L. Rutherford, D. Hoffmann and D. B. Collum, J. Am. Chem. Soc., 2002, 124, 264 CrossRef CAS PubMed; (b) C. Strohmann, K. Strohfeldt and D. Schildbach, J. Am. Chem. Soc., 2003, 125, 13672 CrossRef CAS PubMed.
- Using the equation krel = ln[(1 − C)(1 − ee)]/ln[(1 − C)(1 + ee)] according to H. B. Kagan and J. C. Fiaud, Top. Stereochem., 1988, 18, 249 CAS.
- P. O'Brien, Chem. Commun., 2008, 655 RSC.
- X. Li and I. Coldham, J. Am. Chem. Soc., 2014, 136, 5551 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/c4cc04576a |
| This journal is © The Royal Society of Chemistry 2014 |