 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Can non-polar hydrogen atoms accept hydrogen bonds?†‡
Lixu
Yang
,
Thomas A.
Hubbard
and
Scott L.
Cockroft
*
EaStCHEM School of Chemistry, University of Edinburgh, King's Buildings, West Mains Rd, Edinburgh, EH9 3JJ, UK. E-mail: scott.cockroft@ed.ac.uk; Tel: +44(0)131 650 4758
First published on 22nd October 2013
Abstract
We examine an unusual case where a neutral hydrogen atom acts as a hydrogen-bond acceptor. The association constant between trihexylsilane and perfluoro-tert-butanol was measured as ∼0.8 M−1 in cyclohexane. Computations and experimental NMR data are consistent with a weak, but favourable Si–H⋯HO interaction.
The ubiquitous hydrogen bond continues to intrigue chemists whilst its definition continues to evolve.1 The classic H-bond acceptors nitrogen and oxygen (Fig. 1a) are now considered alongside less conventional acceptors2 such as arenes,3 halogens,4 and even the noble gases.5 While C–H⋯H–C contacts are not classified as H-bonds1 and can be attributed as arising through solvophobic6,7 and van der Waals dispersion forces,8,9 examples of favourable MH⋯H contacts (where M is an element that is less electronegative than H) have been identified that have since become known as ‘dihydrogen bonds’ (Fig. 1b).10–12 Most dihydrogen bonds identified to date involve metals or Lewis adducts that convey strong hydridic character to adjacent hydrogen atoms (Fig. 2a–c).10–13 Thus, the question remains whether non-polar, neutral hydrogen atoms are able to act as H-bond acceptors (Fig. 1c).
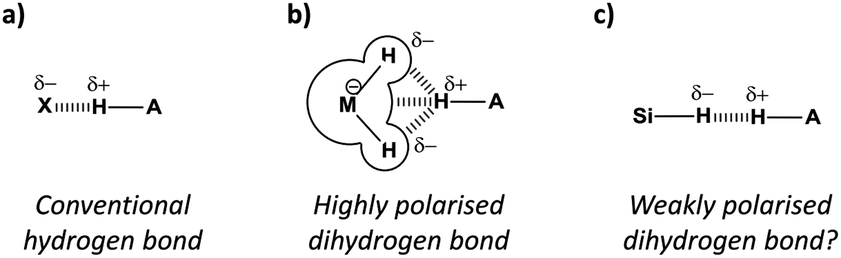 | ||
| Fig. 1 (a) Conventional hydrogen bond compared with (b) a dihydrogen bond involving hydridic hydrogen atoms bonded to a strongly polarising element M. (c) A proposed dihydrogen bond where a weakly polarised hydrogen atom acts as a hydrogen-bond acceptor. | ||
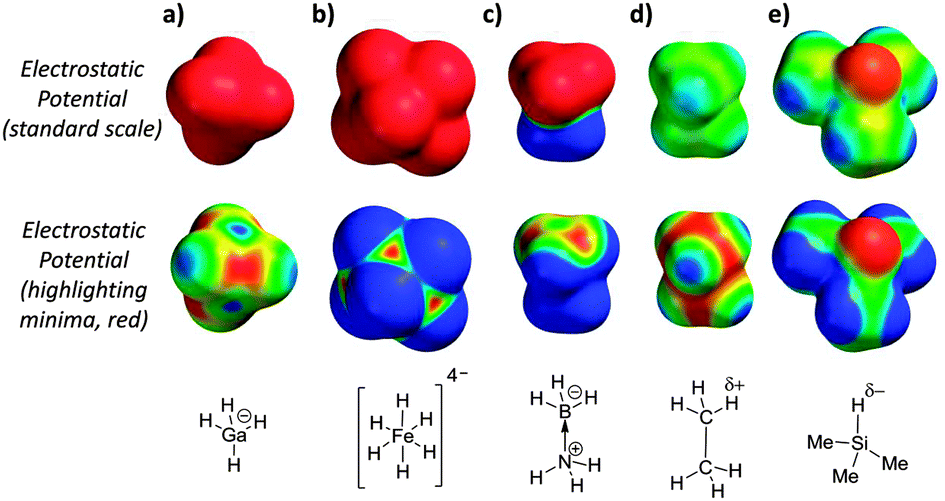 | ||
| Fig. 2 Electrostatic surface potentials (ESPs) of representative dihydrogen bond acceptors. Electrostatic potentials on the top row are scaled using a standard scale from −50 kJ mol−1 (red) to +50 kJ mol−1 (blue), while those on the bottom row are scaled to highlight the ESP minimum (ESPmin, red) to ESPmin +50 kJ mol−1 (blue). Minimised geometries and ESPs were calculated using B3LYP/LACVP. | ||
Here we present a combined solution-phase and theoretical investigation of Si–H as a fully neutral H-bond acceptor (Fig. 1c, 2e, 3–5), whilst examining the utility of electrostatic potentials for rationalising the physicochemical origins and geometric preferences of dihydrogen bonds (Fig. 1 and 2).
Where conventional H-bonds are mostly linear (Fig. 1a), most dihydrogen bonds possess bent or bifurcated geometries (Fig. 1b).12 As a result, some authors have ascribed M–H σ-bonds as being dihydrogen bond acceptors rather than the hydridic H atoms themselves,10 while in other cases there is clear evidence of interactions being formed with adjacent metal centres (Fig. 1b).12
The calculated electrostatic potential surfaces of some previously identified dihydrogen bond acceptors10–13 provide an alternative view consistent with the structural characteristics of polar dihydrogen bonds and the involvement of both M⋯H and H⋯H interactions (Fig. 2). Notably, the electrostatic minima (highlighted in red on the second row of molecular surfaces) in Fig. 2a and b, are found over the metal centres, between the hydridic hydrogen atoms, and not over the hydridic positions themselves. Similarly, although BH3·NH3 is overall neutral and often declared to be ‘isoelectronic’ with ethane,10 the electrostatic potential surface of this Lewis adduct (Fig. 2c) is quite unlike ethane (Fig. 2d); BH3·NH3 is highly polar and electrostatic minima are localised over the boron atom, which bears a formal negative charge (Fig. 2c). The magnitudes and locations of the electrostatic minima in the highly polarised examples shown in Fig. 2a–c provide a simple rationalisation for the formation of favourable electrostatic interactions with δ+ H-bond donors with geometries similar to that shown in Fig. 1b. However, the ability of much more weakly polarised hydrogen atoms to accept H-bonds in solution is yet to be examined.
Calculated electrostatic potentials reveal the trialkyl Si–H group as a potential H-bond acceptor (Fig. 2e). Since silicon has a lower Pauling electronegativity than hydrogen (1.90 vs. 2.20),14 the hydrogen atom directly bonded to the silicon centre is slightly negatively charged. In line with this prediction, recent spectroscopic studies and computations have shown that trialkyl-silanes are able to accept H-bonds in the gas-phase, but no thermodynamic data, nor the relevance of such interactions in solution have been determined to date.15 Trimethyl silane, as shown in Fig. 2e, is a gas at room temperature, and thus is not suited to the experimental investigation of its H-bond properties in solution. However, trihexylsilane (Fig. 3) has a very similar electrostatic surface potential (with a minimum of −47 kJ mol−1, Fig. S3, ESI‡), but also has a boiling point of 160 °C and good solubility in organic solvents. The anticipated weakness of the H-bond acceptor ability of the Si–H group in trihexylsilane presents a number of challenges to the measurement of such a weak interaction using a titration-based approach.
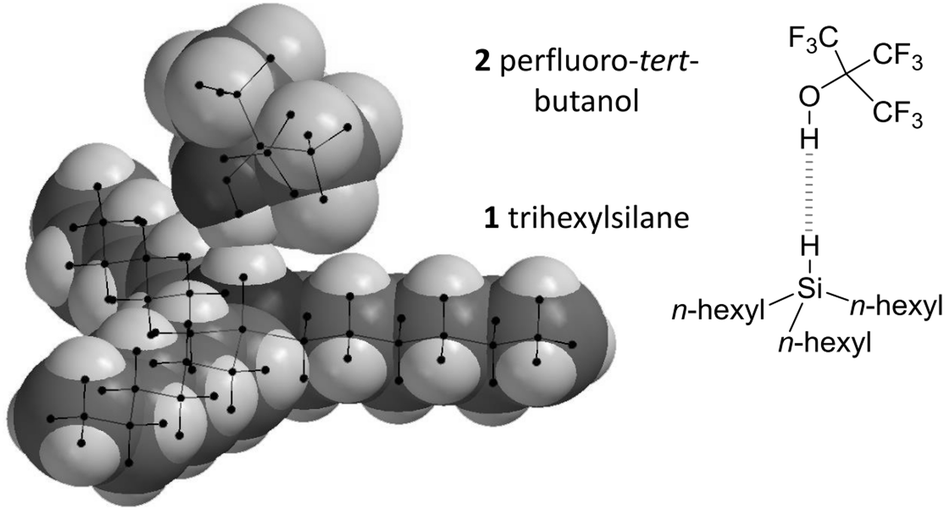 | ||
| Fig. 3 Minimised geometry of the perfluoro-tert-butanol (2) and trihexylsilane (1) complex showing overlap of the van der Waals radii of hydrogen atoms involved in the Si–H⋯HO interaction. Geometry data for the complex shown (calculated using B3LYP/cc-pVTZ) and those for other theory/basis set combinations are provided in the ESI.‡ | ||
With regards to the selection of a suitable H-bond donor, perfluoro-tert-butanol 2 (Fig. 3) is one of the strongest known single H-bond donors (with an ESP maximum of +292 kJ mol−1 Fig. S2, ESI‡) and has been employed extensively in previous studies of H-bonds by Hunter, Abraham and co-workers.16–18 Meanwhile, cyclohexane is one of the least competitive solvents known that maintains reasonable solubility of many organic molecules.17 Furthermore, potential secondary H-bonding or dispersion interactions involving the alkyl chains of trialkylsilane would cancel out due to similarity of the competitive interactions with the alkane solvent.6–8 Thus, perfluoro-tert-butanol and cyclohexane were selected as the H-bond donor and solvent respectively for examining the H-bond acceptor ability of the Si–H group found in trihexylsilane (Fig. 3).
When a hydrogen atom acts as a classic H-bond donor, a downfield shift is observed in 1H-NMR spectra. On the contrary, when a hydrogen atom acts as a H-bond acceptor then a characteristic upfield 1H-NMR shift is observed.19 An upfield shift was observed in δSi–H of trihexylsilane upon addition of perfluoro-tert-butanol in cyclohexane-d12 (Fig. 4). Although small, this upfield shift provides evidence that the Si–H is acting as a (very weak) H-bond acceptor rather than a H-bond donor. It is also worth mentioning, that in contrast to electron-rich metal hydrides (which often have 1H-NMR chemical shifts between δ −5 and −25 ppm20), the δSi–H of trihexylsilane has a chemical shift of approximately +3.8 ppm (Fig. 4), in line with its weak nucleophilicity in the absence of an activating base.21
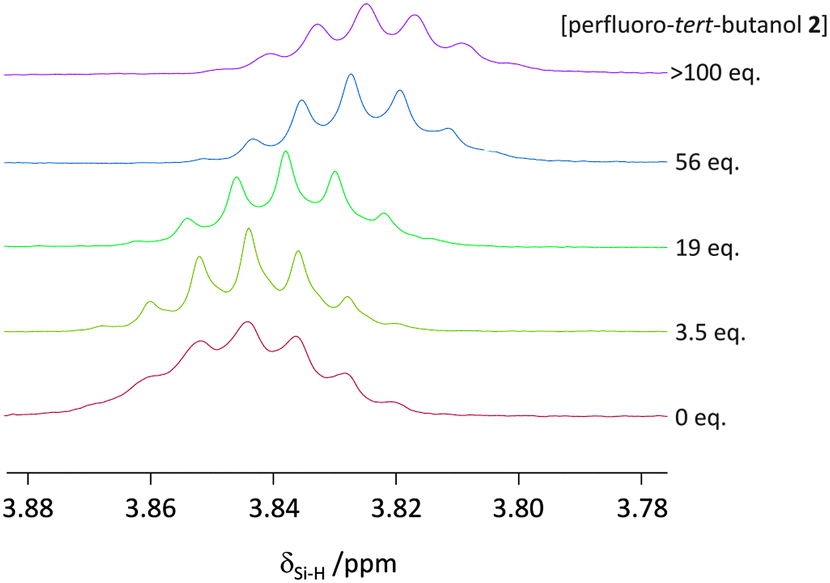 | ||
| Fig. 4 Upfield 1H-NMR chemical shift seen for the Si–H in trihexylsilane (20 mM) in cyclohexane-d12 upon increasing the concentration of perfluoro-tert-butanol at 298 K. | ||
Due to the small change in the observed 1H-NMR chemical shift of the Si–H signal upon complexation by perfluoro-tert-butanol, and since δF is more sensitive to electronic changes than δH,19F-NMR was employed for determining thermodynamic data. Perfluoro-tert-butanol shows no evidence of self-association even in very apolar solvents such as perfluorohexane.16 Thus, the low, constant concentration of perfluoro-tert-butanol means that self-association of the perfluoro-tert-butanol does not complicate the titration data. The δF of perfluoro-tert-butanol (at a constant concentration of 1 mM) moved 0.6 ppm downfield as the concentration of trihexylsilane (1) was increased in cyclohexane implicating the formation of the desired complex (Fig. 3 and 5). In contrast, a control titration in which trihexylsilane (1) was substituted with tetraethylsilane (which lacks an Si–H acceptor) gave a δF of only 0.04 ppm (Fig. S2, ESI‡) over the same concentration range. Data from three titrations with trihexylsilane (1) were found to fit a simple 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model, from which a binding constant of 0.8 ± 0.3 M−1 (95% confidence interval) was determined in cyclohexane at 298 K (Table S1, ESI‡). The small binding constant combined with the downfield δF shift and the upfield δSi–H shift indicates that Si–H is indeed a very weak H-bond bond acceptor in solution.
:1 binding model, from which a binding constant of 0.8 ± 0.3 M−1 (95% confidence interval) was determined in cyclohexane at 298 K (Table S1, ESI‡). The small binding constant combined with the downfield δF shift and the upfield δSi–H shift indicates that Si–H is indeed a very weak H-bond bond acceptor in solution.
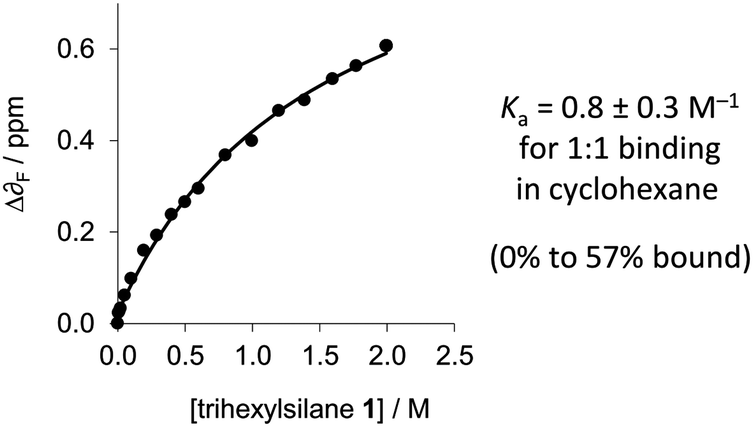 | ||
| Fig. 5 1:1 binding data for the titration of trihexylsilane (1) against a constant concentration of perfluoro-tert-butanol (2) (1 mM in cyclohexane at 298 K). Additional data are presented in the ESI.‡ | ||
Further support for the existence of a favourable dihydrogen bond between trihexylsilane and perfluoro-tert-butanol was provided by a series of HF, MP2, DFT (B3LYP), and DFT-D (M06 and ωB97X-D) geometry optimisations (Fig. 3, left and Table S2, ESI‡). Interaction energies for the trihexylsilane and perfluoro-tert-butanol complex were calculated as the difference between the energies of the complex and the isolated complex components, and were corrected for basis set superposition errors via the standard counterpoise method.22 Given that the approximate +6 kJ mol−1 entropic cost associated with biomolecular association in solution23 is not taken into account in silico, the calculated gas-phase interaction energies of −6.5 ± 2.8 kJ mol−1 for the HF and DFT calculations (Table S2, ESI‡) are remarkably consistent with our experimentally determined association constant of Ka = 0.8 ± 0.3 M−1 (i.e. ΔG in cyclohexane ≈ 0). In contrast, but in line with other findings,6 the MP2 and DFT-D methods significantly overestimate the complexation energy by several to tens of kJ mol−1. This is because gas-phase MP2 and DFT-D calculations approximate the quite significant attractive dispersion interactions between the complex components, but fail to consider the damping effects of competitive dispersion interactions with the solvent.6
Minimised SiH⋯HO distances varied between 1.69 Å and 1.99 Å (Table S2, ESI‡) depending on the level of theory, falling within the range of classical H-bond lengths (1.6–2.0 Å).24 Though such distances are well within the combined van der Waals radii of two hydrogen atoms (2.4 Å, Fig. 3),25 the use of such a metric for the identification of H-bonding interactions has been cautioned against.1 The Si–H⋯H angles observed in the calculations covered the range 153 ± 26°, though it should be added that these angles are likely to be strongly influenced by steric interactions between the bulky alkyl chains and the CF3 groups, rather than arising as a consequence of electronic effects. The O–H and Si–H bonds were also elongated in the bound state compared to the free state (+0.004 ± 0.003 Å and +0.009 ± 0.003 Å respectively), the former, but not the necessarily the latter, being concomitant with H-bond formation.1
In conclusion, the binding constant between a weakly polarised Si–H H-bond acceptor and a strong H-bond donor was determined for the first time in solution. Experimental NMR and computational data are consistent with a weak Si–H⋯HO hydrogen bond.1 Thus, the answer to the question in the title of this paper is, yes, non-polar hydrogen atoms can accept H-bonds, but only just. In addition, we show that electrostatic potential surfaces provide a simple tool for rationalising the geometric preferences of dihydrogen bonds involving hydrogen acceptors with pronounced hydridic character (Fig. 2).
We thank the EPSRC (EP/H02056X/1), Afton Chemical Ltd, MTEM and the School of Chemistry for financial support.
Notes and references
- G. R. Desiraju, Angew. Chem., Int. Ed., 2011, 50, 52–59 CrossRef CAS PubMed.
- I. Alkorta and J. Elguero, Chem. Soc. Rev., 1998, 27, 163–170 RSC.
- H. Adams, K. D. M. Harris, G. A. Hembury, C. A. Hunter, D. Livingstone and J. F. McCabe, Chem. Commun., 1996, 2531–2532 RSC.
- A. Kovács and Z. Varga, Coord. Chem. Rev., 2006, 250, 710–727 CrossRef CAS PubMed; J. D. Dunitz and R. Taylor, Chem.–Eur. J., 1997, 3, 89–98 CrossRef; J. A. K. Howard, V. J. Hoy, D. O'Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613–12622 CrossRef.
- V. Aquilanti, E. Cornicchi, M. Moix Teixidor, N. Saendig, F. Pirani and D. Cappelletti, Angew. Chem., Int. Ed., 2005, 44, 2356–2360 CrossRef CAS PubMed.
- L. Yang, C. Adam, G. S. Nichol and S. L. Cockroft, Nat. Chem., 2013 DOI:10.1038/nchem.1779.
- S. Otto, Chem. Sci., 2013, 4, 2953–2959 RSC.
- C. A. Hunter, Chem. Sci., 2013, 4, 834–848 RSC.
- P. R. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. P. Dahl, R. M. K. Carlson and A. A. Fokin, Nature, 2011, 477, 308–311 CrossRef CAS PubMed; T. Steiner and G. R. Desiraju, Chem. Commun., 1998, 891–892 RSC.
- R. Custelcean and J. E. Jackson, Chem. Rev., 2001, 101, 1963–1980 CrossRef CAS PubMed.
- R. H. Crabtree, P. E. M. Siegbahn, O. Eisenstein, A. L. Rheingold and T. F. Koetzle, Acc. Chem. Res., 1996, 29, 348–354 CrossRef CAS PubMed.
- N. V. Belkova, E. S. Shubina and L. M. Epstein, Acc. Chem. Res., 2005, 38, 624–631 CrossRef CAS PubMed.
- N. V. Belkova, O. A. Filippov, A. M. Filin, L. N. Teplitskaya, Y. V. Shmyrova, V. V. Gavrilenko, L. M. Golubinskaya, V. I. Bregadze, L. M. Epstein and E. S. Shubina, Eur. J. Inorg. Chem., 2004, 3453–3461 CrossRef CAS.
- A. L. Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
- H. Ishikawa, A. Saito, M. Sugiyama and N. Mikami, J. Chem. Phys., 2005, 123, 224309 CrossRef PubMed; E. J. Juárez-Pérez, C. Viñas, F. Teixidor and R. Núñez, J. Organomet. Chem., 2009, 694, 1764–1770 CrossRef PubMed.
- J. L. Cook, C. A. Hunter, C. M. Low, A. Perez-Velasco and J. G. Vinter, Angew. Chem., Int. Ed., 2007, 46, 3706–3709 CrossRef CAS PubMed; M. H. Abraham, P. L. Grellier, D. V. Prior, P. P. Duce, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1989, 699–711 RSC.
- R. Cabot, C. A. Hunter and L. M. Varley, Org. Biomol. Chem., 2010, 8, 1455–1462 CAS.
- R. Cabot and C. A. Hunter, Org. Biomol. Chem., 2010, 8, 1943–1950 CAS.
- E. S. Shubina, N. V. Belkova, A. N. Krylov, E. V. Vorontsov, L. M. Epstein, D. G. Gusev, M. Niedermann and H. Berke, J. Am. Chem. Soc., 1996, 118, 1105–1112 CrossRef CAS; D. Donghi, T. Beringhelli, G. D'Alfonso and M. Mondini, Chem.–Eur. J., 2006, 12, 1016–1025 CrossRef PubMed.
- P. Hrobárik, V. Hrobáriková, F. Meier, M. Repiský, S. Komorovský and M. Kaupp, J. Phys. Chem. A, 2011, 115, 5654–5659 CrossRef PubMed.
- A. D. Dilman and S. L. Ioffe, Chem. Rev., 2003, 103, 733–772 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- T. Steiner, J. Phys. Chem. A, 1998, 102, 7041–7052 CrossRef CAS.
- S. Grimme and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 12639–12642 CrossRef CAS PubMed.
Footnotes |
| † Celebrating 300 years of Chemistry at Edinburgh. |
| ‡ Electronic supplementary information (ESI) available: Experimental and computational methods and tabulated data. See DOI: 10.1039/c3cc46048g |
| This journal is © The Royal Society of Chemistry 2014 |